

Abstract
A protein structure should provide the information needed to understand its observed properties. Significant progress has been made in developing accurate calculations of acid/base and oxidation/reduction reactions in proteins. Current methods and their strengths and weaknesses are discussed. The distribution and calculated ionization states in a survey of proteins is described, showing that a significant minority of acidic and basic residues are buried in the protein and that most of these remain ionized. The electrochemistry of heme and quinones are considered. Proton transfers in bacteriorhodopsin and coupled electron and proton transfers in photosynthetic reaction centers, 5-coordinate heme binding proteins and cytochrome c oxidase are highlighted as systems where calculations have provided insight into the reaction mechanism.
Keywords: Electrostatics, Electrochemistry, pKa, E m , Bacteriorhodopsin, Simulation, Bioenergetics
Redox and protonation reactions represent the simplest chemistry, involving only transfer of electrons and/or protons. Acid/base and oxidation/reduction reactions play important roles in biology. The source of the pH dependence of protein stability is the changing ionization state of protein residues [1]. Protonation changes are essential for protein function. For example, ATP is synthesized by the F0/F1 ATPase, which uses changes in protonation of a buried residue to generate mechanical work [2–4]. The proton gradient that drives the ATPase is derived from proton-coupled electron transfers through proteins embedded in membranes [5,6]. Charged groups within proteins modify electrostatic fields at protein active sites [7], and provide proton conduction pathways [8,9]; while charges on protein surfaces are essential for protein–protein [10,11] and protein–lipid [12] recognition.
Significant effort has been made to understand the free energy of ionization of residues, cofactors, and substrates within proteins. Computational methods try to match, then explain, and predict measured results. There have been a number of recent reviews describing various simulation methods, along with their strengths and weaknesses [1,13–19]. The structure is a rich data-set that is too often used only for a qualitative analysis of the active site geometry. In contrast, calculations can provide a detailed, quantitative analysis of the protein structure. By extracting from the structure values that are measured experimentally simulations improve our understanding of how the protein works. Tested predictions then provide a more stringent test of these ideas. In the end, trusted calculations can explore possibilities in silico which are difficult to test experimentally.
The same computational tools are used to determine how proteins tune pKas and Ems. The electrostatic energy terms are the most important, because both reactions represent a change in the net charge. The in situ free energy is determined by interactions with solvent water, local hydrogen bonds, longer-range charge–charge or charge–dipole interactions, and conformational changes triggered by the reaction. In addition, calculations of Ems and pKas are often inseparable. Redox reactions are usually coupled to some redistribution of protons within the protein, as the charge change at the redox center modifies the pKas of the surrounding residues. In the same way, modifications of protonation states with pH influence redox site Ems [20–22].
1. Thermodynamic analysis of pKas and Ems in proteins
The thermodynamics of pKas and Ems in proteins can be broken down into a series of transfer steps (Fig. 1). The thermodynamic box starts with characterization of the reaction in isolation from the protein. The Gas Phase (line 1) and Aqueous Solution (line 2) reactions consider ionization of a single isolated group. Transfer into Protein (line 3) introduces interactions with other dipoles and charges and the possibility of conformational changes (line 4), proton transfers between residues and coupled electron and proton transfer reactions (not shown).
Fig. 1.
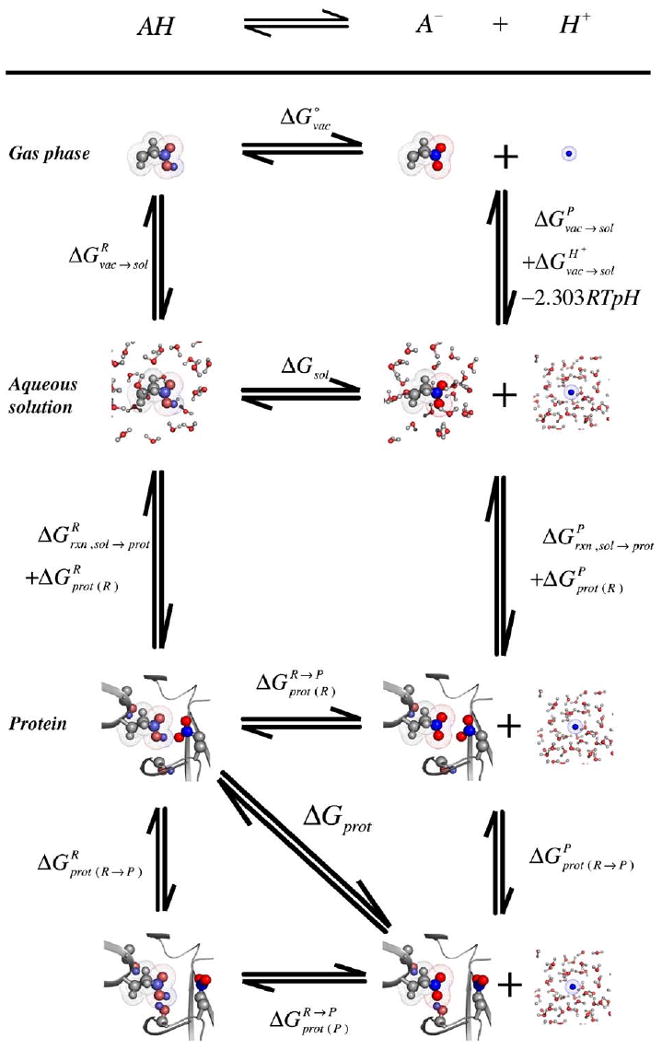
Thermodynamic cycle for calculating the in situ free energy of an acid–base reaction in protein. The reactant (R) is AH and A− and H+ the products (P). Line 1: The cycle starts with the reaction in gas phase with the energy for proton dissociation in vacuum (Section 1.1). Line 2: ΔGsol: the free energy of losing a proton, is shifted from by the energy for transferring reactant product and proton into solvent (Section 1.2). The proton transfer from vacuum gives the reaction a pH dependence of −2.303RTpH. Line 3: The reaction is moved into the protein shifting the free energy of deprotonation from ΔGsol to ΔGprot (Section 1.3). There are changes in reaction field energy of reactant and product and . These are assumed to be the same in the protein equilibrated around the reactants (prot(R)) or products (prot(P)) (Section 1.3.1). and give the protein and solute–protein interactions in the protein equilibrated around the reactant and product (Section 1.3.2). There are 2 paths from reactant to product in the protein (Section 1.3.2). In one the protein moves into the product conformation while still binding the reactant; in the other the product is formed with the protein still equilibrated around the reactant after which the protein relaxes . The measured reaction ΔGprot is the energy difference between reactant and product, each in the equilibrated protein. The two acids in the protein are in the position of Glu 194 and 204 in bacteriorhodopsin with Glu204 as the reacting species while Glu194 remains ionized (see Figs. 2 and 3). Top left (reactant in reactant equilibrated protein): the protonated Glu204 makes a hydrogen bond to the ionized Glu194. This is the lowest energy state with one acid protonated; Top right (product in the reactant equilibrated protein) Glu204 is ionized but Glu194 is still in a position to make a hydrogen bond. This state is at high energy because of the repulsion between the two anions; bottom left (reactant in product equilibrated protein) the neutral Glu204 and ionized Glu 194 have already moved into the conformation they will take when 204 is ionized breaking the favorable hydrogen bond; bottom right (product in product equilibrated protein) is the lowest energy conformation with both acids ionized.
1.1. Reaction free energy in a vacuum
Electrostatic calculations generally treat the neutral form in an acid/base reaction as the reference or reactant state. For an acid releasing a proton (AHm→A−+mH+), the reaction free energy in vacuum (ΔGvac) is the difference in energy of the product , including the released proton and the reactant (First line Fig. 1):
| (1a) |
m is the number of protons released on the reaction, positive for acids and negative for bases. An analogous equation can be written for reduction of an oxidized reactant in a redox reaction :
| (1b) |
n is the number of electrons gained, so is positive for reduction. The reaction free energy in vacuum can be calculated using a quantum mechanical analysis (Section 2.1.2) [23–26].
1.2. Reaction free energy in the solution
The solvation (reaction field or Born) (ΔGrxn) is the energy of favorable electrostatic interactions of a solute with a polar solvent such as water (see 2.2) [27]. The product, reactant, and proton gain solvation energies, , , and when they are transferred from vacuum to solvent (Second line Fig. 2). The standard state free energy in solution differs from ΔGvac because the transfer energies are not the same for all species in the reaction:
Fig. 2.
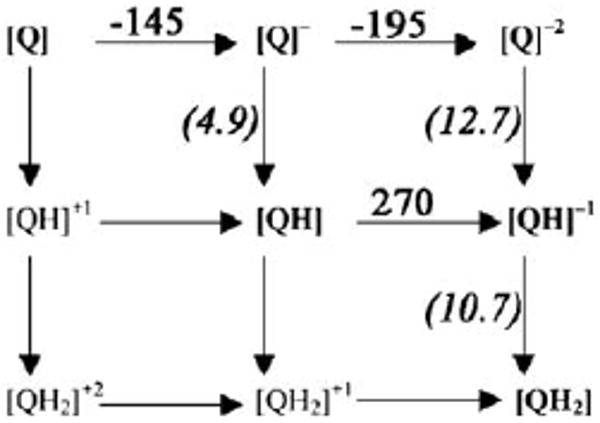
Ubiquinone redox and protonation states. Em,sol and pKa,sol values for reactions in water [30]. The ΔGsol for proton transfer (vertical lines) can be obtained from Eq. (3a), for electron transfer (horizontal lines) from Eq. (3b) and the coupled electron and proton transfers (diagonal lines) from Eq. (3c).
| (2a) |
R is the gas constant, and T is the temperature. At 25 °C kcal/mol. For a redox reaction:
| (2b) |
F is the Faraday constant. At 25 °C meV. The transfer or solvation energy arises from (a) changes in the electronic polarization and conformational preferences of the reactant and product, (b) the energy to reorganize the solvent around them, and (c) pair-wise interactions between solute and equilibrated solvent.
The free energy for an acid to lose a proton is dependent on the pH and the concentration of reactant and product:
| (3a) |
For a redox reaction ΔGsol depends on Eh, the solution redox potential:
| (3b) |
For a reaction where both electrons and protons are transferred, ΔGsol depends on both pH and Eh. With equal concentration of reactant and product:
| (3c) |
The pKa,sol and Em,sol obtained from Eq. (3) can be compared with measured values.
1.3. Reaction free energy in a protein
The reaction free energy in the protein (ΔGprot), differs from ΔGsol because the energy to transfer reactant and product into the protein will be different (Third line Fig. 1). The differences in electrostatic interactions are the primary source of the pKa and Em shifts. The process of transfer can be divided into 3 steps.
1.3.1. Removing the solvent
Some or all waters are stripped away from the reactant or product when it moves into the protein, which causes it to loose solvation energy (ΔGrxn). The ΔΔGrxn is the difference between the transfer energy of the product and reactant :
| (4) |
Any charged or dipolar group loses favorable interactions moving out of water; consequently both and are positive [16]. However, this term is larger for charged species; therefore transfer always favors the neutral form in a protonation or redox reaction (Section 2.2). All groups retain some interaction with the solvent and ΔΔGrxn remains position dependent, even when residues appear to be deeply buried in the protein [28].
1.3.2. Carrying out the reaction in “frozen” protein
While the reactant and product loose interactions with the solvent, they gain new interactions within the protein. In contrast to the solvation energy, which as a “self-” energy term only depends on the ionization state of one group, the ‘pair-wise’ interactions within the proteins depend on the position and ionization state of other residues. These terms are considered first with the ionization and conformation of the protein held fixed. For the protein equilibrated around the reactant, (prot(R)) is the difference in the interactions of the product and reactant with the protein (see Section 2.3):
| (5) |
Since only a proton or electron separates the reactant and product in the reactions considered here, there are generally only small differences in the van der Waals energy. is thus mostly contributed by changes in electrostatic interactions.
1.3.3. Equilibrating the protein around the product
The protein is then relaxes into the form equilibrated around the product (prot(P)), with a change in energy of (line 4 in Fig. 1):
| (6) |
where is the energy of the protein equilibrated around the reactant, with the active group already in the product state and the energy of the protein equilibrated around the product.
There are two paths for this conformational change (Fig. 1). In the first, as just described, the reaction occurs in the protein equilibrated around the reactant and then the system relaxes, Alternately, the protein first moves to the conformation equilibrated around the product, with the reactant bound , followed by the chemical reaction. Both paths yield the same total energy (ΔGprot), which will be the measured value [29].
| (7) |
It takes energy to rearrange the protein and solvent equilibrated around the reactant (prot(R)) to be equilibrated around the product (prot(P)) in the presence of reactant. Once the product has been formed the energy is paid back by favorable interactions with the product, making the prot(P) the lowest energy state of the system as a whole. Thus, successful calculation of the pKas and Ems must be able to determine the direct interaction of the reactant with prot(R), and the product with prot(P) (Eqs. (5) and (6)) as well as the cost of transformation between prot(R) and prot(P).
1.4. pKa and Em shifts moving from solution into the protein
Overall, the free energy of the reaction in the protein, starting from a reference state in solution, is:
| (8) |
where ΔΔGprotein is the shift in the reaction free energy due to differences between product and reactant loss of solvation energy and interactions with the protein as well as the energy needed to move the solvent and protein from the conformation equilibrated around the reactant to that equilibrated around the product. Thus,:
| (9) |
For an acid–base reaction the in situ pKa is:
| (10a) |
The pKa within the protein depends on (a) the intrinsic chemistry of the titrating site in water encapsulated in the pKa,sol and the shift in the energy of ionization by the protein given by ΔΔGprotein. ΔΔGprotein is also a function of the pH and Eh, because the electrostatic environment for each residue depends on the ionization state of all of the others (Section 3.3). An analogous expression can be written for a redox reaction where the in situ Em is:
| (10b) |
The free energy of a coupled electron and proton transfer reaction is:
| (10c) |
For pure proton transfers n=0 and the pKa shift moving into the protein (ΔpKa,protein) is −ΔΔGprotein/1.36m kcal/mol. For proton independent half reactions m=0, so the shift in the Em in the protein (ΔEm,protein) is −ΔΔGprotein/n meV [29]. For proton coupled electron transfers, ΔΔGprotein yields changes in both pKa,protein and Em,protein [30].
1.5. Free energy of protonation or redox changes at an arbitrary pH or Eh
The pKa is the pH at which the free energy of ionized and neutral states are the same. In solution, the energy difference between the two forms changes as 2.303 RTm(pKa,sol−pH) (Eq. (3a)). In the protein ΔΔGprotein is added to the energy gap (Eq. (9)) and this term is pH and/or Eh dependent since it depends on the ionization state of all residues in the protein. There are many reasons why it is useful to know the energy gap between the two ionization states at an arbitrary pH and Eh as well as the in situ pKa or Em. This can provide the energy of transient protonation changes along a proton transfer pathway [9], of an active site transition state [31], of electron transfer reactions carried out in a frozen media [7] or measured on a fast time scale [32]. The free energy of ionization at a given pH (ΔGpH) is:
| (11a) |
and it is related to the solution pKa,sol by:
| (11b) |
here represents the Boltzmann distribution of the ionization and conformation states of all other residues in the protein relaxed around the reactant at the pH of interest. Eq. (11) is a mean field approximation of Eq. (9) that lacks the energy due to changes in the protein coupled to the ionization of the site of interest . Thus, this is the energy of changing protonation before protein relaxation. An analogous expression can be written for the free energy of changing the redox state of a cofactor in a frozen protein (Eq. (10b)).
2. Challenges in the calculation of the Ems and pKas
Fig. 1 provides the standard framework for calculating pKas and Ems in proteins. However, there remain challenges in calculating each needed energy term.
2.1. The reaction chemistry needs to be characterized in a well-defined solvent
The analysis of how reactions are modified by the protein starts with understanding the basic reaction chemistry in isolation (Fig. 1). Only then can the perturbation of the thermodynamics of transferring a reaction into protein (ΔGprotein) be determined to obtain the in situ pKas and Ems.
2.1.1. Quantum mechanical calculations of Em,sol and pKa,sol
The reaction free energy in vacuum (Eq. (1)) can be calculated using ab initio or more frequently, density functional theory (DFT) [33,34] methods. The Ems and pKas for metal clusters can also be determined by the same methods, although, these require the consideration of more complex transition metal chemistry [35,36]. Despite the fact that there are attempts to do simulations on larger systems [37,38], calculations are still largely limited to <100 atoms, representing a very small region of a protein.
To test the calculation of reaction free energy in vacuum, the system must be transferred into a solvent in which the pKa,sol or Em,sol can be measured (Eq. (4)). Experimental [39] and computational [40] studies help establish the effects of different solvents on Ems and pKas. The transfer energy may be as difficult to calculate accurately as the vacuum ionization free energy change. Water is the preferred reference solvent [41,42]. An implicit continuum model is usually used, rather than an atomistic explicit solvent (see Section 2.2). It is always more reliable to calculate relative values of pKa,sol and Em,sol for a series of compounds rather than absolute values. Here the absolute transfer free energy for the proton or electron is not needed (Eq. (2)). Values have been calculated for many small molecules where the calculated can differ from experimental pKa,sol and Em,sol by less than 1 ΔpK unit, or 60 meV [23,24,43–47].
2.1.2. Measuring the Em,sol and pKa,sol
There are a number of good experimental model systems to study pKa,sol and Em,sol of biologically interesting molecules in water. As long as the protonated and deprotonated forms are stable in water, a pKa,sol can be measured [48–50]. The pKas of amino acids are obtained for isolated groups [51] or for sidechains in short capped peptides [52–54]. Measured pKas for a given type of functional group can vary by 0.1–0.9 pH [51]. The pKas in short polypeptides or in unfolded proteins are lower then found for the isolated functional group [55]. This may be a result of the propensity of sidechains to orient into the positive end of the adjacent amide group [56].
Obtaining Em,sol for redox reactions can be more problematic. For example, the redox cofactors, such as flavins and quinones, bind 2 electrons going from fully oxidized to fully reduced species (Fig. 2) [20–22,57]. To get a complete picture of the thermodynamics connecting all 9 possible species, the Em must be measured from a pH below the pKa,sol of the most oxidized species to a pH above the pKa,sol of the most reduced species. In addition, proteins often favor single electron reactions, while the resultant free radical species have limited stability in solution. This can make it difficult to measure the Em,sol for the biologically important redox couples.
2.1.3. Examples of measured Em,sol and pKa,sol
Hemes and quinones are well-studied redox cofactors used by many proteins. Hemes in cytochromes (Section 5.1) transfer a single electron and no protons while quinones in different binding sites (Section 5.2) can transfer 1 or 2 electrons, with or without coupled proton transfers.
2.1.3.1. Measured Em,sol and associated pKa,sol for hemes
Six-coordinate hemes with 2 axial ligands generally change oxidation states between a neutral, ferrous FeII and a cationic, ferric FeIII, species [58]. The macrocycle itself retains a −2 charge. The heme is not a protonatable group, although it does have two attached propionic acids, which are not conjugated into the ring system so can be considered independently (Section 5.2). Proteins, such as cytochromes use hemes to shuttle electrons between sites within the protein or between different proteins [22,59]. The microperoxidases, fragments of cytochrome c that retain 8–12 amino acids, provide a well-studied heme model system [60–66]. The heme remains attached, via the two Cys ligands, and keeps its axial His ligand and two propionic acids as peripheral ligands. The Em,sol of bis-His MPs is ≈−220 mV (vs. S.H.E.) [63,64]. His-Met MPs have Em,sol of −70 mV [62–64], 150 mV more positive than that of a bis-His MP. The microperoxidase Ems are pH dependent due to the titration of the liganding His, the propionic acids, and a water/hydroxide that can be the second axial ligand making them a less then ideal model system [67].
Many proteins bind hemes with only a single amino acid as an axial ligand [58]. The open 6th ligand position can be used for transporting ligands such as the oxygen in hemoglobin. Other proteins with 5-coordinate hemes, such as peroxidases, oxidases, and P-450s, carry out chemistry at the open position. All these hemes can also bind water as the 6th ligand. The Em,sol for the microperoxidase-8 (MP8) His-water is −140 mV [68,69], and is −205 mV for the His-hydroxyl measured at high pH [68]. The hydroxyl in the ferric His-aquo-heme MP8 has a measured pKa,sol of 9.6 [65] while it is 10.9 in the oxidized MP8 [70]. Thus, the formal charge on the metal shifts the water pKa,sol by only 1.3 pH units. The protonation of the water bound heme is functionally important for Heme A3 in the Heme-Cu oxidase, where it helps determine the number of protons coupled to the anaerobic reduction of these proton-pumping proteins [31,71].
Modifications of the heme porphyrin ring and its connection to the protein can also change the heme redox chemistry [72]. The b and c type hemes differ by the latter having a covalent attachment to the protein via two Cys. This linkage may lower the Em by 50 mV or less [73]. The c-type hemes have His–Met ligands, which have a 150 mV more positive Em,sol than the Bis–His hemes. This Em,sol shift indicates that compared with the Bis–His hemes the oxidized His–Met heme is bound ≈300 times less tightly than the reduced species [30]. Thus, the c-type covalent linkage may help keep the oxidized His–Met heme from dissociating from its axial ligands and then the protein [72].
Heme o and a are found in heme-Cu terminal oxidases such as bo3 and aa3 [74]. An o-heme differs from a b-heme by the substitution of a hydroxy-ethyl farnesyl side chain for a vinyl group. In model systems this increases the affinity of the heme for the protein without changing the midpoint [75]. An a-heme differs from an o-heme by the oxidation of a methyl to a more electron withdrawing formyl group. The a-type heme has an Em,sol 100 [72] to 160 [76,77] mV more positive than the c-type MPs with the same axial ligands. The oxidized a-type heme binds its ligands ≈2500 less tightly then the b-type heme, with little difference in the reduced heme affinity [75]. Thus, proteins that use an a-type heme raise the Em by adding a formyl group reducing the affinity of the oxidized heme. The added farnesyl group then increases the affinity without changing the Em by favoring binding of both redox species [75,78].
2.1.3.2. Measured Em,sol and pKa,sol for quinones
Flavins [57,79] and quinones [21,22,80] have 9 different redox states with 0, 1 or 2 electrons and protons [30]. The doubly reduced, doubly protonated, or the fully oxidized, deprotonated species tend to be the most stable forms in water at pH 7. For ubiquinone (UQ) the Em,sol for reduction to the semiquinone is lower than for the formation of the fully protonated dihydroquinone (QH2). Thus, in water at the physiological pH, UQ is reduced in an n=2 reaction to QH2 [21,81]. However, the physiological reactions with quinones generally involve single electron transfer steps [30,82,83]. Estimates of the Em,sol for one electron reduction and pKa,sol for the semiquinone have been made for a small number of quinones in water [30,84–88].
In the absence of protons, the semiquinone is stable in solution. The Em,sol for Q/Q− has been determined in the aprotic solvent dimethylformamide (DMF) for a large number of quinones [89–92]. For UQ it was measured to be −360 mV [91], which is significantly lower than the best estimates of −150 mV in water [30]. This large decrease of Em,sol in DMF has been found for other quinones [87]. A pure continuum electrostatics analysis of the change in the reaction field energy moving the Q to Q− reaction from DMF (ε=37) to water (ε=80), only predicts a shift stabilizing the anionic semiquinone by ≈40 mV, shifting the Em,sol to −320 mV [93] (see Section 2.2). This discrepancy shows that more specific interactions need to be considered to determine the changes in ΔGsol in different solvents [39,40].
There are several different quinones with different Em,sol used in biology. Rhodoquinone (RQ) and menaquinone (MQ) have Em,sol 150 mV lower then UQ, making them better electron donors and biology makes use of this. For example, some eukaryotes use the UQ containing succinate dehydrogenase to reduce quinones under aerobic conditions. Under oxygen stress they switch to quinonol furmarate reductase, which oxidize MQ or RQ [94,95]. The electron transfer from reduced MQ or RQ to fumarate is more favorable then for UQ, while UQ is more easily reduced by succinate. Some organisms make do with a single quinone. Thus, mammals use only UQ in all their membrane electron transfer proteins. Each protein modulates its quinone Em as needed by changing the local environment. Sometimes homologous proteins in related organisms use different quinones. For example, B. viridis photosynthetic reaction centers (RCs) use MQ in the QA site while Rb. sphaeroides RCs use UQ [96,97]. Both use UQ in the QB site. Despite the lower Em,sol for MQ their Ems in the QA site differ by only 20 mV [98]. Thus, while substitution of quinones with different Em,sol can be used to change the in situ energetics, biology can also modify the bound quinone Ems to obtain the needed in situ electrochemistry.
2.1.4. Building simple protein model systems
Often interesting bio-inorganic reactions in proteins have no ready analogues in solution. There is a considerable interest in designing small complexes to serve as models for these biological reactions. For example, Mn-clusters have been assembled [99–101] to model the oxygen-evolving complex of PSII [102–104]. Heme–Cu complexes are designed to reduce oxygen to water [105,106] as in the terminal oxidases [8,107]. Other examples include metal clusters designed to model blue-copper proteins [108] and iron–sulfur proteins [109].
Another approach is to build unique cofactors into small model proteins. This method is exemplified by the construction of a double Cu binding site in azurin to model CuA in cytochrome c oxidase [110], or a Cu being added to myoglobin to model the CuB–Heme binuclear center [111,112].
2.2. The interaction of reactant and product with water
The interaction of the reactant and product with water is important for a number of reasons. The solvation energy loss, ΔΔGrxn (Eq. (9)) is a key determinant of the in situ reaction thermodynamics, always stabilizing the state with the smallest charge [113–118]. Also, proteins are of finite size so even deeply buried reactants retain significant interactions with the surrounding water. Accurate calculations can be made with explicit water added [119–121]. However these calculations need many extra atoms to correctly model long range interactions [122], require correction for long-range electrostatic effects given the modest numbers of waters that can be included [123], require a good model for water [124], and must be run for a long time to sample many water positions so that the system energies with reactant and product are accurate [125]. Most calculations make use of an implicit solvent. Here the single parameter of the dielectric constant is used to average all the effects of the distribution of solvent conformations around the reaction site [16,42,121,126–128].
Dielectric continuum theory approximates the electrostatic interactions of charged and polar solutes with a solvent whose ability to respond to a charge is summarized in its dielectric constant [24,27,113,114,129]. The Poisson or Poisson–Boltzmann (PB) equation is considered the most accurate way to calculate these energies [130,131]. The Poisson–Boltzmann equations allow the solvent ionic strength to be included in the analysis [132]. The energy of transferring a spherical charge (q) with a radius r from a solvent with dielectric constant ε1 to one with ε2 is [27,129]:
| (12) |
Here, C is 331.5 kcal/mol or 14.4 eV, r is in Ångstroms, and q is in multiples of the charge on a proton. It is always favorable to transfer a charge into a medium with a higher dielectric constant. The transfer energy increases steeply with the net charge, and becomes smaller as the size of the sphere increases. The protein has a lower dielectric constant than water, but there is considerable variation in the value used for protein in simulations (Section 2.4).
When the reactants are not spheres or other simple shapes the PB equation must be solved numerically [130,131,133]. To calculate the transfer energy for molecules, the atomic coordinates, partial charges and radii must be assigned to all atoms. The radii define the solvent accessible surface, which may be different than the van der Waals terms used in molecular mechanics force fields. PARSE [134], CHARMM [135], AMBER [136], OPLS [137] charges and radii are often used to calculate the solvation energy [130]. The Generalized–Born (GB) method provides a faster way to obtain the transfer energy [130,138,139]. This technique parameterizes radii at each position in the protein, allowing the Born equation (Eq. (12)) to be extended to the calculation of arbitrarily assemblies of spheres [125,139–141]. However, results calculated with the PB equation still provide the benchmark for GB studies, [130,141,142].
2.3. The interactions with the large number of charges and dipoles in the protein
Interactions of charges and dipoles in the protein with the reactant and product are an important contributor to the Em and pKa shifts in proteins (Eq. (5)). In a classical, electrostatic model the interaction between the reactant and the protein equilibrated around the reactant (prot(R)) is [16]
| (13) |
where Ψij is the electrostatic potential at atom i of the reactant due to an atomic partial charge on atom j of the protein, qi is the atomic partial charge on each reactant atom, the sum r runs over all of the atoms in the reactant, and the sum prot(R) runs over all non-reacting atoms in the protein. C is 331.5 kcal/mol or 14.4 eV. and differ because the charges, qi, on the reactant and product atoms will certainly be different. In addition, the charges on the protein atoms and the distance to the reactant (rij) can change, modifying Ψij if other residues in the protein change ionization state or conformation when the reaction occurs.
When the response of the medium to charges is uniform so the system can be treated with a single dielectric constant, Ψij can be calculated analytically with Coulomb's Law:
| (14) |
where ε is the dielectric constant and rij the distance between each pair of atoms. Eq. (13), with ε=1, is generally used when solvent is modeled by explicit, moving water molecules. Here interactions with each atom of the solvent needs to be enumerated and averaged over a long trajectory [119,121]. Implicit solvent methods retain the benefits described in Section 2.2. Solutions of the PB equation [133] provides Ψij for an arbitrary distribution of dielectric constants [16]. GB [125,130] and other implicit solvent methods [142–144] are also used to calculate Ψij. The PB equation treats the solvent as a medium with a high dielectric constant, and it allows the pair-wise interactions to be appropriately screened by the high dielectric solvent surrounding the protein. The electrostatic pair-wise interactions are now highly position-dependent. Groups on the surface have very little impact on reactions occurring more than a few Ångstroms away due to the screening by water with its high dielectric constant. In contrast, electrostatic interactions can be important at distances of 10–15 Å for groups buried in large proteins, especially when they are embedded in the membrane [145,146]. The main problem with this approach is an appropriate protein dielectric constant must be assigned (Section 3.2).
2.4. Intra-protein interactions need to take into account regions of the protein with different flexibility and polarity
To determine the free energy of a reaction in the protein it is necessary to calculate the energy of protein re-equilibration around reactant and product (Eq. (7)). Electronic polarization, backbone or sidechain motions, changes in ionization of nearby groups, and ion binding all contribute to the response to changes in charge. Each of the techniques to be described in Section 3 deals with these changes differently.
Continuum electrostatics based methods use a dielectric response for the protein (εp) to average changes in the protein so they need not be included explicitly [16,128,147]. The Boltzmann term in the PB equation adds the screening due to ions in the solution, equivalent to the Debye–Huckel effects in a medium with a uniform dielectric constant. A dielectric constant of ≈2 accounts for the electronic polarization of any condensed medium. The dielectric response of dried proteins is ≈4 [148]. Calculations show this is arises from small-scale microdipole motions [149]. The motion calculated with Molecular Dynamics around introduced charges in the protein core are equivalent to a medium with a small effective dielectric constant [150]. In contrast, a large effective dielectric constant of ≈30 is found near the protein surface and near mobile charged groups [151–156].
2.5. Ionization states of residues are interdependent
Ionizable residues make up 25% of an average protein [28]. Since electrostatic interactions can be felt at long-range within the protein, protonation states of distant residues influence and are influenced by a reaction at the active site. Thus, the ΔΔGprotein is sensitive to the ionization state of all the surrounding residues (Eq. (13)). A complete analysis of the acid/base thermodynamics of a protein needs to consider 2N different ionization states, where N is the number of residues with 2 ionization states. For small proteins it can be possible to enumerate combinations of ionization states for the subset of residues that titrate in the protein in the same pH or Eh range [157–159]. However, Monte Carlo sampling is generally used to recover the Boltzmann distribution of all ionization states at each pH [160]. The pH at which a group has an equal probability of being ionized and neutral provides the calculated pKa.
The pKas of groups in clusters are the most difficult to calculate [161–163]. Residues are in a cluster if they are closely with strong interactions and have similar pKas so they titrate near the same pH. Coupled group ionization is common in protein active sites (Section 5.2) [146,164]. Clusters play important roles in proton-coupled electron transfers [30,165]. One example is the two acidic residues, GluL212 and AspL213, in the QB site of photosynthetic reaction centers. These play an important role in delivering protons when the secondary quinone, QB, is reduced [96,97]. If the Glu is ionized first (near pH 4), then the negative charge raises the Asp pKa to above 9. If the Asp is ionized at the lower pH, then the Glu pKa becomes high. The calculated pKa for these individual group vary by more than 5 pH units in simulations that analyze either different structures with the same method, or the same structure with different techniques [30,93,165–168]. Since, the net charge on the two acids remains the same, with only one being ionized between pH 4 and 9, the precise distribution of cluster protonation has only modest effects on the equilibrium pKas and Ems for the important surrounding groups [30]. However, the two acids have different accessibility to the protein surface so which is ionized may influence the kinetics of proton uptake coupled to electron transfer [169]. Experimental results strongly favor GluL212 being the residue that binds a proton when QB is reduced [170,171].
2.6. Good benchmarks are needed to test and refine calculation methods
Calculations need to be tested against measured values. Residue pKas are measured by NMR [172,173], by the difference in the pH dependence of denaturation energies with and without specific ionizable residues [174,175], by potentiometric titration [176], and by FTIR [170,177–179]. Each method has limitations. NMR measurements cannot routinely be carried out on proteins larger than 50 kDa, while FTIR requires assignment of bands to a particular residue. A change in the pH dependence of a protein, following site-directed mutation, is often used to assign the reaction pKa to a particular residue. However, other residues can change ionization in response to mutation, creating ambiguity in the interpretation of the data [180].
Benchmark analyses are best done when there are several pKas known in a protein, or the electrochemistry of a given cofactor can be compared in a number of proteins. Heme electrochemistry can be studied in many cytochromes with a wide range of measured Ems [29,72,181] some with more than one heme in the protein [182–185]. Quinone electrochemistry can be compared at different binding sites in the same protein and in different proteins [30,93,186]; as can iron–sulfur cluster [187], and blue-copper center [188,189] reactions.
There are ≈20 proteins with ≈200 measured values commonly used as benchmarks for calculation [161,173]. Most pKa calculation methods are optimized to fit this small dataset. In the end, most methods report a similar match between calculated and experimental data [24,161,190–199]. This may be because these sites are poorly chosen. For example, many such sites are of surface residues, which are not very perturbed by the protein [128]. Additionally, each technique has partially hidden variables that can be adjusted. There are few studies that directly compare different programs in unbiased tests [200]. It may be necessary to develop an analogue to the CASP challenge for protein structure prediction [201], or CAPRI challenge for docking [202] for calculations of reaction thermodynamics in proteins to be truly tested.
3. Calculating Ems and pKas in proteins
As described above calculation of a pKa or Em in a protein relies on an accurate assessment of the total free energy of the reactant and product in the equilibrated, solvated protein. Analysis of the intrinsic electron or proton affinity, ΔGvac or ΔGosol, requires a fundamental quantum mechanical analysis (Section 3.5). Classical methods (Sections 3.1 3.2 3.3 3.4) can only calculate the shift in the reaction free energy moving into the protein (Eqs. (8) (9) (10)). An in situ pKa or Em differs from the solution value because the loss of solvation energy always favors the neutral form of a residue; the surrounding charges and dipoles then favor the charged or dipolar state. Each method of calculation uses different approximations to obtain the needed energies. In addition, the methods differ in whether they consider all possible ionization microstates in the protein or only the properties of a few sites of interest.
3.1. Empirical techniques
As with other knowledge-based techniques, empirical methods are the fastest but provide the least physical insight into how the protein modulates an in situ pKa or Em. Empirical methods use purely geometric measurements, such as surface exposure [125], to provide a value for the desolvation energy (Eq. (4)). Then, the pair-wise interactions with the protein charges and dipoles are added (e.g. Eq. (13)). A screening function, or distance dependent dielectric constant must be included to account for the solvent around the protein. These methods do not account for any specific atomic motion coupled to electron or proton transfers. Any protein reorganization must be included implicitly by a larger screening function. These methods allow calculation of the ionization states of all residues in the protein at one time, so coupling between them can be correctly included (Section 2.5).
A wide variety of empirical techniques have been developed. (a) Mehler's Screened Coulomb Potential shifts pKas with a factor derived from the hydrophobicity of surrounding residues [203]. The method provides a simple way to use the information that hydrophilic groups which partition into water [204] have dipoles or charges that tend to favor ionization of nearby groups. (b) A generic algorithm has been used to develop parameters describing the importance of specific atom types near a titrating group [205]. (c) Jensen [198] has developed a purely geometric search to find residues that account for local hydrogen bonding and buried charge–charge interactions; it uses the number of atoms surrounding a group to assess its burial. (d) Another method divides the protein into different regions with different effective dielectric constants. Here analytic functions provide the desolvation penalty and a dielectric constant that is region and distance dependent is used to obtain pair-wise interactions [206]. With a good training set that includes both buried and surface ionizable residues and enough parameters, these empirical approaches can often match the experimental pKas better than methods based on a more detailed physical picture of the reaction.
3.2. Methods using energies from classical continuum electrostatics with Monte Carlo sampling of states
Techniques based on continuum electrostatics use a physics based, classical analysis method [1,15,147, 157,207–212]. They start with a reference pKa,sol or Em,sol, preferably in water, and then calculate the energy of transferring the reactant and product into the protein (Eq. (9)). They assume that the reaction free energy is shifted from that found in the water by changes in solvation energy and the electrostatic pair-wise interactions with charges and dipoles in the protein. The Poisson–Boltzmann (PB) equation [133] is generally used to calculate these energy terms [15,16,18]. The whole protein can be included without cutoffs, so the free energy of residue ionization changes with the ionization state of all of the other groups in the proteins. Monte Carlo sampling allows determination of the Boltzmann distribution of all 2N ionization states as a function of pH and Eh even for a protein with many ionizable sites [157,160,211]. The process samples microstates, which define the ionization state of all residues. The energy of microstate x (ΔGx) is:
| (15) |
where RT is 0.59 kcal/mol (25.8 meV), N is the number of ionizable residues, is 1 for residues that are ionized in the state and 0 for all others. Each ΔG term represents the difference between the energy of the ionized and neutral form of the residue. ΔΔGrxn represents the double difference (Grxn,ionized−Grxn,neutral)protein−(Grxn,ionized−Grxn,neutral)solution (Eq. (4)). ΔGpol represents the pair-wise interactions with the groups in the protein that do not change ionization such as non-titrating sidechains and the backbone dipoles and ΔGij is the difference in interaction of ionized and neutral forms of residue i with all other residues that are ionized in microstate x. Changes in Lennard–Jones energy are usually ignored since states only differ in the number of electrons and protons. The limits on the summation of the inter-residue terms ensure that each interaction is counted once. Monte Carlo sampling establishes the Boltzmann distribution of the different ionization states of each residue at a given solution redox potential (Eh) and pH. Usually, a pair-wise interaction matrix that includes every ionization state of all of the residues is precalculated [213]. Only self-energy terms such as the desolvation penalty and pair-wise interactions are included. Higher order terms that would arise from the interactions between residues being dependent on the ionization state of another site are not considered [199,213].
The PB equation allows the dielectric response to vary over space. This approach accounts for the impact of the surrounding water using just the dielectric constant, encapsulating a very complex set of interactions in a single parameter. However, the method needs to assign a dielectric constant to the protein (εp). This factor accounts for the different energies of prot(R) and prot (P), in the same way that the use of ε=80 treats the rearrangement of water around the reactant and product [16,128,147]. The use of a single dielectric response for protein, has limited the accuracy of continuum electrostatics. A single εp cannot account for the large difference in the rigidity of a protein, the distribution of polar sidechains, or of cavities that can bind water throughout a protein. Values as low as 4, especially inside membrane proteins [30,93,146,165,186,214–216], to 8 [161] to 20 for smaller proteins [208,212], to as high as 80 [192] have been used.
For many sites, a change in εp affects the calculated pKa or Em by a relatively small amount [199]. An increase in εp diminishes the loss in solvation energy (Eq. (12)), making it easier to ionize buried groups, and at the same time it makes the pairwise interactions smaller (Eq. (13)). As long as most pairwise interactions in proteins are favorable, these two changes can roughly cancel. Generally, a large εp weakens the influence of the protein, and so can hide errors. Thus, methods with larger values of εp can look impressive in benchmark calculations [212]. However, more complete analysis of the role of the dielectric constant in calculating electron transfer reorganization energies, as well as the site pKas or Ems suggest that a lower value of εp is more physical [217].
Calculations with a large protein dielectric constant, such as 20, can be less successful in calculating the pKas of active sites, because they underestimate strong local pair-wise interactions, such as hydrogen bonds [198]. However, a low dielectric constant, such as 4, underestimates the effect of conformational flexibility. For example, crystal structures often show surface charges making salt bridges. The calculations with a rigid structure with a low dielectric constant yield lower pKas for the acidic partner and higher pKas for the basic group than found experimentally [161–163]. A high protein dielectric constant [190], or the use of artificial screening terms [161], are needed to obtain good matches to experimental results. However, methods where the sidechains can adopt a range of conformations yield good results with a low εp without using additional terms [199]. Likewise, hydrogen bonds will reorient to remain in equilibrium with the ionization states throughout the protein [163,165]. Freezing their orientation around ionizable residues over-stabilizes the initial state, leading to significant errors [213].
3.2.1. Conformational flexibility in continuum electrostatics
PB methods for calculating electrostatic energies in proteins have been modified to incorporate non-uniform dielectric constants [218–222], averaged results in multiple protein structures [162,223–226], and added explicit conformational degrees of freedom to optimize hydrogen bond networks [194,209,227].
Multi-Conformational Continuum Electrostatics (MCCE) is a software package, which calculates the equilibrium conformation and ionization states of protein side chains, buried waters, ions, and ligands as a function of pH and Eh; while maintaining a rigid backbone [161,199,213]. This represents a hybrid approach combining Poisson–Boltzmann calculations of electrostatic interactions with a complete molecular mechanics force field. The current program (MCCE2) [199] (available online at http://www.sci.ccny.cuny.edu/∼mcce), does full rotamer sampling of all sidechains and samples ligand occupancy, and orientation in binding sites. This differs from methods that average the results from different structures [162,223–226], in that conformational and ionization changes are treated consistently being sampled in the same calculation. Furthermore, the complete rotamer search allows for larger conformational changes than methods that simply optimize hydrogen bond networks. MCCE2 provides good matches to the benchmark pKas using a protein dielectric constant as low as 4 [199]. Thus, the conformational changes added to the analysis provide accurate pKas or Ems with a small εp, while explicitly showing changes in the protein structure on site ionization. In addition, continuum electrostatics calculations assume a linear response of the medium to changes in charge, which is not a good description of proteins. In contrast, explicit conformational changes show how the response can saturate. For example, in the photosynthetic reaction centers a quinone in the QB site is reduced first to the semiquinone then to the fully reduced quinone. The first reduction reorganizes a hydrogen bond network in the binding site stabilizing the semiquinone [30,216]. However, once this has occurred the system has no groups that can rearrange to stabilize the Q−2 and the quinone binds a proton before the second reduction [30,228].
3.3. PDLD based techniques
The Protein Dipoles Langevin dipoles method (PDLD) provides a semi-microscopic view of the protein and solvent response [128,191,229–231]. It does not use a dielectric constant to account for the response of water and protein. Rather, the protein atoms are associated with explicit polarizable dipoles; while a lattice of Langevin dipoles is used for the solvent. The PDLD technique is able to incorporate changes in the protein structure in Molecular Dynamic simulations, thereby treating the heterogeneous protein response to charges more easily than standard, single conformation continuum electrostatics techniques [191].
3.4. MD based techniques
Molecular dynamics (MD) is the technique most often used to explore the trajectories of proteins under different conditions [232]. MD based methods allow the protein to move freely so the heterogeneous response of the protein can be fully incorporated into the analysis [154]. However, the Newtonian mechanics used to define the rules for motion do not allow the method to directly explore chemical reactions. MD simulations must assign specific charges to each residue so cannot easily account for the system behavior at a pH near a residue pKa, where different molecules in the ensemble have different protonation states. In standard methods the forces on the protein must be recalculated every fraction of a femtosecond so MD methods have difficulty reaching equilibrium for reactions that take microseconds or milliseconds. The use of implicit solvent removes the many atoms for the solvent waters from the analysis allowing the system to reach equilibrium more rapidly. The Generalized Born (GB) [125,130] or other formulations [143,193,203,233] provide fast and reasonably accurate estimates of the effect of the solvent water on the free energy of a given distribution of charged groups in a protein. Most methods to solve the PB equation are too slow to be solved at each time step. Methods are being developed that can incorporate energies obtained by solution of the more accurate PB equation into MD [234–236].
MD methods have begun to be adapted for calculation of pKas within proteins [19]. A relatively simple approach runs a continuum electrostatics based Monte Carlo pKa analysis on ensembles of MD simulated conformations [223,237]. Here MD simulations are not run with equilibrium charges assigned to each residue. This simplification introduces systematic errors, since simulations with a particular charge set will cause the trajectory to equilibrate around that charge distribution, resulting in it being over stabilized [238,239]. Approaches where partial ionization is accounted for by scaling the residue charges have been developed [240]. In these, a residue which is 50% ionized in the ensemble interacts with its environment, as if it only has a 0.5 charge. This analysis is comparable to the classical mean field Tanford–Roxby approach used in MC analysis [241]. These methods can work if the titrating groups are isolated, but will fail if the groups are in a cluster with interdependent ionization states [157].
Another group of methods run MC protonation sampling at intervals within an MD trajectory, allowing residues to change between neutral and ionized [242]. The simulations can calculate the electrostatic energies using an implicit solvent with PB [243,244] or GB [196,245] techniques, or with explicit water [195,246]. The latter is very expensive, but it uses a consistent set of variables for the MD and MC phases of simulation. The protonation changes can sample the whole protein, or single sites of interest [246]. Alternately, the decision to switch protonation states in the MC steps can be determined by thermodynamic integration that evaluates the relative energy of protonated and unprotonated forms [247,248]. Other methods use short periods of a simulation where the trajectory has fractional ionization states, with an imposed potential along the titration coordinate. MD free energy simulations have also been used to calculate the pKas in proteins [119]. All of these methods report reasonable matches to data, but the more detailed methods have problems converging; even with nanosecond trajectories for small molecules [19].
3.5. QM and QM/MM based methods
Advances have been made in incorporating quantum mechanical analyses of pKas and Ems into a protein environment using quantum mechanical–molecular mechanics (QM/MM) methods [24,25,44,249–251]. Here perhaps 100 active site atoms are treated quantum mechanically while the rest of the protein moves using Newtonian Physics in an MD simulation. This method alleviates many of the key limitations of classical MD or CE methods (Sections 3.1–3.4), which assume that the intrinsic ionization chemistry of a residue remains the same in solution and protein, and that the partial charges and polarizability of individual residues are independent of their context. The change in ionization equilibrium with reactant conformation can also accounted for by QM/MM methods [252]. Molecular charge densities are used for the atoms in the QM region. They maintain hydrogen bond directionality [253] and quadrupole moments that are lost in the atom centered partial charges used in most classical methods. For example, aromatic hydrogen bonds [254,255] will influence the pKas in a QM/MM calculation, but would not be seen with atom centered charges where there are no off-atom charges representing the π electrons. QM/MM allows the dynamics of the surrounding residues to be treated in detail. However, the long-range electrostatic energies must now be calculated using a classical analysis [256]. It can be challenging to connect the electrostatic interactions between the QM and MM portions of the simulation [257,258]. In addition, the treatment of the molecular mechanics region has the same weaknesses as the MD methods described above. Thus, while these MM regions can be assigned different positions, they cannot undergo protonation or redox chemistry. Thus, these methods cannot model any ionization reactions that couple QM and MM regions of the simulation.
Pure QM methods can be used to calculate the pKas and Ems considering only a small region of the protein with a higher level analysis than used in QM/MM [33–36]. The simulation region for QM analysis must be chosen carefully to maintain the correct long-range electrostatic potentials from the rest of the protein, as well as the nearby hydrogen bonds [259–261]. Recent studies suggest that a simple PB based calculation can help choose a simulation region where the potential at the reactants is equivalent to that contributed by the protein as a whole [31].
4. Continuum electrostatics analysis of the distribution of buried charged groups in proteins
The Born reaction field (solvation) energy stabilizes charges in water. This favorable interaction is diminished when they are moved into the protein interior (Eq. (12)). For example, a continuum electrostatic analysis estimates an ionized Asp will loose ≈17 kcal/mol when it is moved into a medium with a dielectric constant of 4 (Table 1). Destabilizing terms of this magnitude are larger than the total stability of most proteins, meaning that uncompensated charge burial would lead to protein unfolding. This has led to the expectation that there will be very few buried changes in proteins. However, proteins are not a simple low dielectric medium. Each residue is linked by an amide bond, which has a dipole moment larger than that of water. In addition, 23% of the residues are polar (Asn, Gln, Ser, Thr, and Tyr), while 27% are ionizable (Asp, Glu, Arg, Lys, His). The concentration of polar moieties inside proteins has been estimated as being on the order of 25M [262]. Therefore, a protein is different from a high dielectric solvent, such as water, not by being much less polar, but by being less polarizable [16,263,264].
Table 1.
Survey of buried ionizable resides in 490 proteins
| All ionizable | Asp | Glu | Arg | Lys | ||
|---|---|---|---|---|---|---|
| Isolated side-chains | ||||||
| pKa,sol | 3.9 | 4.1 | 12.5 | 10.8 | ||
|
|
8.9 | 9.1 | 8.5 | 5.8 | ||
| % ionizedB | 1% | 1% | 97% | 6% | ||
| Max ΔΔGrxn (kcal/mol) | 17.4 | 17.5 | 15.9 | 18.5 | ||
| Total database 490 proteins | ||||||
| # Res | 36,192 | 8976 | 10,232 | 7707 | 9277 | |
| % ionized | 93.5% | 93.6% | 89.3% | 96.6% | 95.5% | |
| Count of favorable interactions with polar residues C | ||||||
| Ser+Thr | 413 | 263 | 143 | 7 | 0 | |
| Tyr | 141 | 45 | 40 | 47 | 9 | |
| Asn+Gln | 225 | 85 | 41 | 77 | 22 | |
| Buried Residues (ΔΔGrxn >6.8 kcal/mol) | ||||||
| # buried | 6106 | 1731 | 1326 | 2483 | 566 | |
| % buried | 16.9% | 19.3% | 13.0% | 32.2% | 6.1% | |
| # ionized | 5225 | 1498 | 1017 | 2282 | 428 | |
| % ionized | 85.6% | 86.5% | 76.7% | 91.9% | 75.6% | |
|
|
7.1±1.8 | 7.5±1.9 | 7.3±1.9 | 6.8±1.5 | 7.1±1.9 | |
|
|
−2.5±3.8 | −5.1±3.7 | −3.7±3.1 | −0.3±2.6 | −1.7±4.0 | |
|
|
−6.9±5.7 | −7.2±6.5 | −7.4±5.4 | −6.6±5.1 | −6.4±5.8 | |
| % ≥1 salt bridgeF | 73.3% | 84.2% | 80.9% | 72.3% | ||
All data is from [28]. A: pKa,sol+mΔΔGrxn would be the pKa with 6.8 kcal/mol desolvation energy (5 ΔpK units) and no other interactions; m is the number of protons lost, +1 for acids; ΔΔGrxn is the difference between the transfer energy for ionized and neutral forms (Eq. 4). B: % ionized at pH 7 with 5 ΔpK units desolvation energy. With a desolvation of 12.7 kcal/mol (7.5 ΔpK units) only 1% of the Arg would be ionized at pH 7 in the absence of stabilizing interactions; 8.8% of the Arg are this deeply buried and 86% of them are >90% ionized at pH 7. Max ΔΔGrxn: transfer energy for isolated side-chain with an interior ε of 4 from water (ε=80) to a solvent with ε=4 (Eq. (4)). PARSE charges and radii are used [134]. C: number of examples of interactions where the polar side-chain stabilizes ionization of the acid or base by ≥3.4 kcal/mol. D: The average loss of solvation energy for all residues with ΔΔGrxn>6.8 kcal/mol. E: The sum of the interactions with the whole backbone (ΔΔGbkbn), or all side-chains (ΔΔGres), for each of the buried residues (Eq. 13), averaged over all of the buried residues. F: Fraction of residues that are >90% ionized with at least one a group of opposite charge stabilizing the charged state by −3.4 kcal/mol (2.5 ΔpK units). The data on all proteins can be found in a searchable database at www.sci.ccny.cuny.edu/∼mcce.
Proteins can stabilize charges in particular locations by interactions with specific charged or polar groups; so there is often little cost for burying native charges within a protein interior. However, the lack of flexibility can produce large penalties for changing charge, either through removing native charges or introducing new ones. Experimental and computational studies have shown that specific charged residues can either stabilize [265–268] or destabilize [269–273] proteins, depending on their context [270,274,275]. Proteins involved in redox chemistry or proton-pumping are designed to accommodate charge changes during their reaction cycle (Section 5). The finding that many proteins unfold only at extreme pHs indicates that it is not easy to change the ionization state of buried residues [1]. Finally is should be noted that proteins are not designed to maximize stability. Thus, even if charged residues are moderately destabilizing, they still may be accommodated. Thus, stability can be increased in other ways such as by burying more hydrophobic surface.
One way to estimate how destabilizing are buried charges is to look at how often they are found in native proteins. Surveys of active site ionizable residues show that 70% are <5% solvent exposed when the substrate is bound [276]. However, these active site residues could be exceptional, being buried at significant cost to the protein. Early surveys suggested that buried ionizable residues are rare [277,278]. Then again, only a few small proteins with little internal volume were analyzed. More recent surveys of the solvent exposure of ionizable groups [268,279], or their desolvation energy [28,56], show that as many as 30% are buried and most of them are ionized.
MCCE was used to calculate the degree of burial, and predict the ionization state of all acidic and basic residues, in 490 proteins selected to include a wide range of protein folds and sizes (Table 1). There are several criteria for the degree of side-chain burial. The solvent exposure of the terminal atoms that have most of the accumulated charge in ionized residues [280– 282] gives a qualitative measure. The loss of solvation energy, ΔΔGrxn, calculated with the Poisson–Boltzmann equation provides a quantitative measure of the energy change on burial. The desolvation penalty, which is the (solvation energy in water) − (solvation energy in the protein), roughly correlates with the solvent exposure of a group [28]. However, residues with little solvent exposure can maintain some residual solvation energy. Thirty five percent of the Asp, Glu, Arg and Lys residues have lost 4.08 kcal/mol ΔΔGrxn, sufficient to shift a pKa by 3 pH units in the absence of other interactions, and 17% have lost 6.8 kcal/mol solvation energy (5 ΔpK units) (Table 1). Using a threshold of 6.8 kcal/mol for a residue to be buried, there are on average 3.95 buried ionizable residues per 100 amino acids. Smaller proteins have less internal space, so on average there are 1.9±2.3 buried charges per 100 residues (proteins <100 amino acids) and 4.5±2.0 per 100 residues (proteins >300 residues). Thus, a significant minority of the ionizable residues are deeply buried in the proteins. The key question is whether these buried residues are ionized. With a ΔΔGrxn of 6.8 kcal/mol (5 ΔpK units) the acids would be 1% ionized, Lys is 6% ionized, and Arg is 97% ionized at pH 7 if there were no other interactions with the protein (Table 1). Overall the protonatable residues in these proteins are calculated to be 93.5% ionized, while 85.6% of the buried residues remain more than 90% ionized at pH 7. His, which are important residues for protein function, are found to be 23% buried. Since they have a pKa,sol of 6.5 they will only be 24% ionized in solution at pH 7. Thus, the protein would need to stabilize their charge to see them predominantly ionized and only 6% of all His are calculated to be >90% ionized at pH 7.
The prediction that most buried Asp, Glu, Arg or Lys are ionized implies that favorable interactions stabilizing the charged state are of the same magnitude as the loss of solvation energy. The factors that keep the residues ionized were compared for the acids and bases [28]. Most of the buried, ionized residues have at least 1 significant interaction with a oppositely charged group (Table 1). As shown previously, the backbone is much more likely to stabilize acids than bases [56]. The larger size of the oxygen relative to the hydrogen in an amide bond is the primary reason that the potential from the backbone dipoles is predominantly positive inside proteins. This size difference means that in the allowable regions in Ramachadran space, side-chains come off their backbones towards the positive end of each amide dipole. The two neighboring amides to the N- and C-terminal always raise the potential of a side-chain with shorter side-chains, such as Asp, feeling a larger affect. In addition, the side-chains shield the positive end of the dipole from the solvent. A negative C=O end of the amide pointing outward is more likely to be solvent exposed than an outward directed H–N amide dipole. This raises the potential throughout the protein interior. Thus, in the group of buried residues with an average ΔΔGrxn of 7.1 kcal/mol, ionization of the acids is stabilized by the backbone, on average, by more then 4 kcal/mol, while Arg and Lys are, on average, stabilized by less then 2 kcal/mol (Table 1). Surprisingly, the polar interactions are also different for acids and bases. While Asn and Gln stabilize all charges, Ser and Thr stabilize only acids, and Tyr rarely stabilizes Lys. Thus, hydroxyls are found to be better hydrogen bond donors than acceptors. Pairwise interactions with other ionized residues provide stabilization of many buried, ionized residues, but are especially important for keeping bases ionized within a protein.
5. Examples of how proteins modulate in situ pKas and Ems
5.1. Heme Ems
Extensive studies have explored how ligand type [29,72, 283,284], orientation [285–287], electrostatic interactions [29,118,288], and the protein scaffold [289] affect in situ heme properties [72]. For example, six-coordinate bis-His-hemes have Ems ranging from −410 to +360 mV. Since these have the same ligand, the redox differences are predominately due to the intra-protein electrostatic environment [29,72,181,182]. For each heme the loss of solvation energy [114,115,118], interactions with the protein backbone and other residues [28,29,118,181,182], and protein conformation changes on heme ionization [29,290] affect the Ems. Proteins can also modulate heme electrochemistry by changing the heme axial ligands (Section 2.1.3). When the Met ligand is replaced by a His the Em is lowered by 200 mV in cytochrome c [291] or 150 mV in microperoxidases [72].
The large range of cytochrome Ems has been subject to theoretical analysis by PDLD [292,293], continuum electrostatics [16,29,182,294], and other techniques [185,240,295–299]. The pKas and Ems of the aquo-Heme a3 and CuB in cytochrome c oxidase have been analyzed by continuum electrostatics [71,300,301], and density functional theory (DFT) [302]. In cytochrome P450, DFT and QM/MM calculations have been used to explain the unusual low-spin state of ferric aquo-heme [303–305], and to study the hydroxylation mechanism [306–309].
5.2. Hemes and their propionic acids
Each heme has two covalently attached propionic acid groups with a pKa,sol of 4.9 [50,310]. An ionized propionic acid will always lower the heme Em. The coupling between the acids and hemes is largely through-space rather than through-bond, as the protonatable groups are not conjugated to the redox active heme. This view is supported by comparison of the perturbation of the propionic acid pKa by heme reduction in DFT and in PB calculations [311]. DFT treats the heme and its acids as a single molecule; while the continuum electrostatic analysis views them as separable, in the same way that different amino acid side-chains on a single polypeptide are viewed as distinct units. The changes in the propionic acid pKa upon heme reduction, are quite similar in these two methods of calculation [31].
While the acids cannot be changed by mutation, esterifying the acids in b5 increases the Em by 60 mV and significantly decreases the cytochrome stability as hydrogen bonds to the acid are lost [312]. Calculations indicate that propionic acids can lower the heme Em by as little as 20 mV to more than 150 mV in different proteins [29,182]. In general, most propionic acids have their CG carbon ≈8 Å from the heme with iron so their interactions with the heme are not modified by changes in distance. There are two important factors that determine the impact of the acid: the degree of solvent exposure, and the in situ pKa of the acid. In different proteins the acids range from being fully buried to being largely exposed. For an ionized propionic acid, the electrostatic interaction with the buried heme is inversely proportional to the desolvation energy of the acid [264]. In addition, the acids in different cytochromes range from being partially to fully ionized at pH 7, even if they are deeply buried [29]. The more ionized the acid is, the larger its affect on the Em. In rare cases, such as c556 in Rb. viridis reaction centers, the two propionic acids are within hydrogen bonding distance of each other [182]. In this case, one acid is the proton donor while the other acts as the proton acceptor, and the total charge is maintained at −1. The high Em of 310 mV for this heme can be primarily attributed to the loss of one negative charge near the heme.
As the propionic acids shift the heme Em, heme oxidation shifts the pKa of the acids. Thus, the same ΔΔGprotein that shifts the Em by 60 mV will shift the pKa by 1 pH unit (Fig. 2). The heme propionic acid pKas and their influence on the pH-dependence of cytochrome Ems, have also been studied by continuum electrostatics analysis [29,313,314]. If the acids are fully ionized in the reduced state, as found in many proteins, they cannot have their ionization shifted on heme oxidation. In this case, the acid serves to lower the Em without making it pH dependent. However, if the propionic acids are not fully ionized when the cytochrome is reduced, they become an important contributor to the proton release coupled to heme oxidation. The extent of coupling depends on the interaction with the heme. For example, the heme redox reaction shifts the pKa of the largely exposed propionic acid on the porphyrin A ring in cytochrome b5 by less than 0.5 unit [315], but shifts the pKa of the buried propionic acid on the D ring in c551 by 2.5 pH units [316]. Calculation of these pKa shifts yield values in good agreement with those found experimentally [29].
It has been proposed that the surface exposure of the heme group is a major determinant of the heme Em, with exposure favoring oxidation, lowering the Em [317–319]. Electrostatic analysis of heme electrochemistry shows that there is only a small variation in the surface exposure, or of the ΔΔGrxn, for different cytochromes with vastly dissimilar Ems. No correlation between the heme Ems and the exposure of the heme ring and axial ligands has been found [29]. However, if the propionic acids are considered as part of the heme, there is some correlation between the exposure of the acids and the Em [29]. The hemes with deeply buried propionic acids tend to have higher Ems. This is because buried propionic acids are either neutral, or kept ionized by forming a salt bridge with a positively charged basic residue.
5.3. Quinones in bacterial photosynthetic reaction centers (RCs)
RCs have provided an important system for study of how proteins modify quinone electrochemistry. RCs from Blastochloris viridis (previously identified as Rhodopseudomonas viridis) were the first membrane protein known to atomic resolution [320]. The reactions in RCs from Rhodobacter sphaeroides have been very well studied [96,97]. There are well worked out methods to measure the Ems of QA [98] and QB [321,322] in situ, even down to cryogenic temperature [323,324]. Knowledge of the in situ ΔGo for electron transfer allowed much of the data underlying the ‘Dutton Ruler’, which connects electron tunneling rates to the distance between redox centers, to be measured in RCs [325–327].
The overall reaction in RCs uses the energy of a photon to take electrons off of 2 cytochromes c, reducing ubiquinone (UQ) to the dihydroquinone UQH2. There are two Q binding sites: QA and QB. The protein modifies the UQ behavior to differentiate them. Only the oxidized QA and anionic semiquinone are found. QA does not dissociate from the protein. QB serves as the two-electron gate [328], found in three relatively stable redox states: unreduced quinone (Q), anionic semiquinone (Q•−) and fully reduced and protonated dihydroquinone (QH2). The anionic semiquinone is tightly bound to the protein, while the Q and QH2 freely exchange with the quinone pool in the membrane [329–331]. The pathway for the second reduction indicates that of the two possible intermediates, the high energy is easier to form than , so proton binding occurs prior to electron transfer [30,228]. Thus, of the nine possible redox states for QB five are found on the reaction pathway (Fig. 2). There are two binding sites for QB, distal and proximal, as seen in the crystal structures [332,333]. Kinetic measurements find no evidence for quinone reduction in the outer, distal site [334–338] and simulation suggests the Em of the quinone in this site is very low so it cannot be reduced [30].
With the wealth of experimental information about the redox chemistry in wild-type and mutant proteins, RCs provide an excellent system to test simulation techniques. MD has been used to study QB movements [339], conformational gating [340] and changes in protonation states of amino acids GluL212 and AspL213 on the first electron transfer [341]. The electron transfers from QA to QB have been studied by various PB methods using both Bl. viridis and Rb. sphaeroides RC structures [93,165-168,180,342]. Multi-conformation continuum electrostatics (MCCE) has been used to study the energy of 7 of the 9 different quinone redox states in the QA, and active and inactive QB sites [30].
5.4. Proton transfer in bacteriorhodopsin
Bacteriorhodopsin is a transmembrane proton pump that transfers protons from the cell interior to the low pH extracellular space, generating a proton gradient (Figs. 3 and 4) [343–346]. The study of bacteriorhodopsin has benefited from a wealth of crystal structures. By late 2005 there were 33 models of the ground state in the protein databank [345]. In addition there are structures trapped in the K, L, M1, M2, N′ and O states available [345,347,348]. These structures reveal specific changes as the protein goes around the reaction cycle. The structures show changes in the Schiff base orientation [348], which drives the repositioning of the G and F helices [347,349], changes in water and polar side chain positions in the active site central cluster, [347], reorientation of an Arg that bridges the central and exit clusters [350,351], and changes in orientation of the residues in the exit cluster [351]. In addition, FTIR has been used to assign ionization changes during the photocycle of the Schiff base [352], Asp 85 [352–354], Asp 212 [354,355], central cluster water [354,356–358], and Arg 82 [359–361].
Fig. 3.
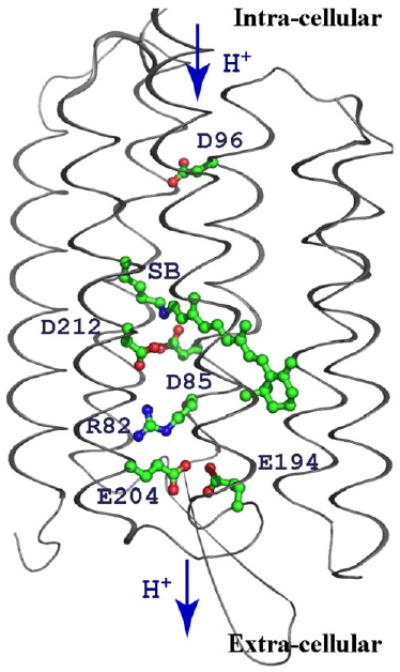
The functionally important residues in bacteriorhodopsin (PDB:1C3W [367]). Protons are transferred from intra-cellular side of the membrane to extracellular. Essential buried ionizable residues contributing to proton pumping are represented in sticks and spheres.
Fig. 4.
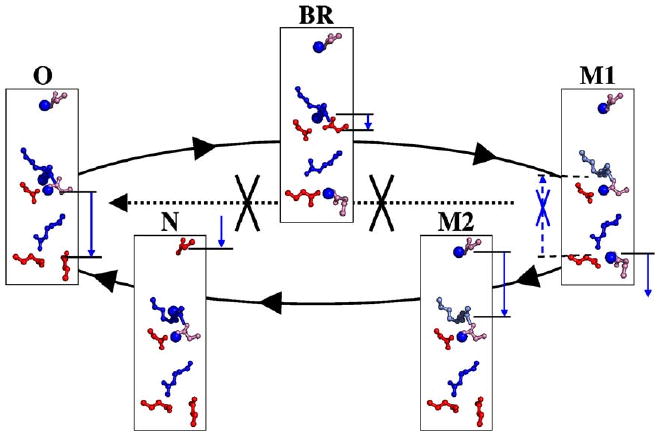
Reaction cycle of bacteriorhodopsin. Only those intermediates contributing to proton transfer are shown. The mobile protons are shown by blue spheres. The cycle BR→M1→M2→N→O→BR (solid line) transfers one proton, loosing a proton to the periplasm in M1 and gaining a proton from the cytoplasm in N. In state M1 (early M), if the proton moves from exit to central cluster rather than to the periplasm so the protein moves to O instead of M2 (late M) (dashed line), proton pumping would be short-circuited. The residues shown are Asp 96 (top); central cluster: SB (top), Asp 85 (left), Asp 212 (right); exit cluster: Glu 204 (left), Glu 194 (right) (see Fig. 2). The blue arrows show the proton motions moving to the next state. Ionized acids are red and bases are blue. Neutral acids or bases are pink or light blue.
Three clusters of residues have been identified, which change their protonation during the reaction cycle (Fig. 3). The isolated Asp96 is the proton input site [362–366] near the intracellular side of the protein [367–369]. The central cluster includes the Schiff base, which covalently attaches retinal to Lys216 [370,371], Asp85 [362,372], and Asp212 [373–377]. It is surrounded by a number of important polar resides including: Thr 89 [378,379], Tyr 57 [380,381], and 185 [374,382–390], and buried waters [367]. Lastly, there is an exit cluster composed of Glu194 and Glu204, as well as a number of buried waters [214,351,391–396]. Arg82 lies between the central and exit cluster [367–369,397].
Given this wealth of structural and biochemical information, bacteriorhodopsin has proved very amenable to analysis by calculation. Comparing the structures trapped in different states provides deeper understanding of how the observed proton shifts are driven by the structural changes. The calculated in situ pKas in crystal structures of trapped intermediates, the proton transfer pathways and the mechanism used to maintain directional proton transfer have all been studied. PB calculations [146,214,215,398] have been used to quantitatively explain the importance of residues identified experimentally.
5.4.1. Ground state
The bacteriorhodopsin ground state has a neutral Asp 96, protonated Schiff base (SBH+), an anionic Asp85 and 212, and a Glu194 and 204 cluster with one proton bound (Figs. 3 and 4) [343]. Continuum electrostatics was used to analyze the earliest cryo-electron microscope structures of bacteriorhodopsin [210,399,400]. More recent PB and MCCE calculations using high-resolution structures have shown good agreement with the experimentally observed proton distribution for the key groups [146,215]. The analysis shows how these three deeply buried residues remain charged. Both Asps in the cluster need to be ionized to stabilize the SB charge. Asp ionization is stabilized by the positive SB, as well as the neighboring Arg82, Thr 89, the backbone dipoles, and nearby waters. The calculations also find a single proton bound to the exit cluster. The exit cluster may adopt a mixture of ionization states. Some calculations find that one of the two glutamic acids can be protonated [146], or exist in a mixture of microstates with some having 194 protonated, and others having 204 protonated [401]. Calculations which include show both glutamic acids to be deprotonated with the extra proton in the water cluster [214] as suggested by FTIR spectroscopy [396]. Overall, in the ground state the net charge on the input residue is 0, with −.1 on both central and exit clusters.
5.4.2. Ground state→K→L state
The bacteriorhodopsin reaction cycle is initiated by absorption of a photon by the retinal, causing it to change from all-trans to 13-cis. Through the K and L states the SB nitrogen moves into the extracellular side of the retinal, away from Asp 85 and 212. This stage of the reaction does not involve changes in residue protonation. QM [402–405], QM/MM [406–412], and MD [413–415] methods have addressed the questions of whether the kinetics of retinal photo-isomerization are governed by two electronic states or three, of what structural changes occur, and of the contributions made by the protein to shifts in the spectrum. Calculations conclude that there are three electronic states contributing to the kinetics of retinal isomerization [403,406], although the second excited state may not be important in the presence of a counter ion [405]. Buried waters are shown to stabilize cluster ionization [404] and to complete the hydrogen bond network favoring proton transfer [412,413]. The photoisomerization into the 13-cis configuration has been shown to be a result of the constraints from the retinal binding pocket, which forces the retinal to twist around these double bonds [411,415]. Calculations have also shown that the spectral shift during isomerization arises from changes of interactions with the nearby residues [408,409]. A PB analysis of the SB ionization in K and L states indicates the SB pKa decreases by 4∼5 pH units when it is isomerized, which in turn prepares the it to lose its proton in the next stages of the reaction cycle [398].
5.4.3. L state→early M→late M state
As the M state is formed the proton is left behind as the retinal twists, protonating Asp85. The ionization state of the central cluster moves from SB+85−212− to SB0850212−. A variety of studies have simulated the proton transfer pathway from the SB to Asp85, tracing the important hydrogen bond network [404,410,412,416]. PB calculations of structures in the ground and M states show the pH for half ionization of the SB has shifted down from >14 in the ground state to 5.5 in the M state, while that for Asp85 shifts up from 3 to >14 [146]. However, the net charge of the cluster remains −1, so no protons are lost to or gained from solution. Rather than assigning pKas to each residue, a pKa can be assigned to the cluster as a whole [146]. There are 8 microstates for the ionization of SB, Asp85, and Asp 212: one with a charge of +1, three with 0 charge, three with −1 charge, and one with a −2 charge. In the ground and M states, the microstates with −1 charge have the lowest energy and are the most occupied. The pKa for the cluster moving from 0 to −1 changes from 3 in the ground state to 5.5 in the M state. However, on isomerization of the SB, the energy of the SB+85−212− state has been calculated to move up ≈2 kcal/mole, while the SB0850212− moves down ≈2 kcal/mole. This shifts the order of the two states transferring the proton, stoichiometrically, from the SB to Asp85. The proton transfer requires only modest changes in the structure of the protein. In contrast, to change an isolated pKa by >8.5 pH units with the bulk solution as proton acceptor would require a change of >11.6 kcal/mol in the energy of ionization. This could not be accomplished without significant structural rearrangements, which are not found.
As the M state evolves, changes occur in the exit cluster ionization, which have been followed by MCCE calculations [146]. In the ground and early M states, this cluster has one excess proton bound. Arg 82 lies between the two clusters. By the late M state, the two Glutamic acids have moved apart and now the proton is released from the cluster, changing the net charge from −1 in the ground state to −2. In the ground state structure the pair-wise interactions between the two Glutamic acids keep the proton bound. By the late M state the protein has carried out half of the physiologically important reaction by releasing a proton into the extracellular space.
The importance of the motion of the acids can be seen in a trapped M state analogue with Glu 204 replaced by a Gln [417]. This crystal structure cannot have a charge of −2 since there is only one acid and the Glu–Gln hydrogen bond is never broken. After restoring the Glu to the structure in silico the calculated proton distribution shows the exit cluster still retains one proton because the two acids remain too close together, even though the proton transfer in the central cluster has occurred and the Arg has shifted downward [146].
5.4.4. M state→N state
In the next stage, the proton is transferred from Asp96, which is protonated in the ground and M states, to reprotonate the Schiff base 12 Å away [418]. This forms an ionized Asp96, a protonated SB and Asp85, with Asp212 remaining ionized. Asp96 is in a very hydrophobic region of the protein with few polar residues nearby that can stabilize its ionization, so it has been calculated to have a very high pKa in the ground state [146]. PB calculations suggest that ionization is only slightly uphill in the N state, due to an increase in solvent accessibility and the reorientation of Thr46 [215]. The deprotonated Asp96 need only be accessible as a transient intermediate for it to function in proton transfer. For example, Glu286 plays an important role in proton transfer in cytochrome c oxidase [419–421], despite having a pKa over 10 [31,421]. The deprotonated Asp 96 has never been trapped experimentally, consistent with its high calculated pKa. In the ground state there are no cavities in the cytoplasmic region to connect Asp 96 and the SB; thus the SB is not accessible to the cytoplasm [422]. QM/MM calculations suggest that a water chain can be formed in the hydrophobic region between the Asp 96 and the Schiff base in the M state, [423].
5.4.5. N→O state→ground state
Following the transient formation of the N state, the formation of O involves reprotonation of Asp96 from the intracellular space. Removing a proton from the high pH cell interior completes the second half of the pumping reaction for the protein. Finally, the protein returns to the ground state, ready to restart the reaction cycle. This requires the transfer of the proton from the Asp85 in the central cluster to the exit cluster. It has been proposed that Arg82 acts as a proton shuttle, leaving its proton on the exit cluster and moving towards the central cluster to be reprotonated by the Asp [424].
5.4.6. Ensuring the pump does not short-circuit
For bacteriorhodopsin to function as a proton pump, the early M→O and O→ground state transitions must adopt different proton transfer pathways. The early to late M transition is key to proton pumping (Fig. 4). Proton transfer from the exit to central clusters, formally the reverse of the O→ground state transfer would take the protein directly from early M to O. This would bypass the proton transfer from Asp96 to the SB, proton uptake from the intracellular space and release into the extracellular space. The key question is why the proton is released from the exit cluster in the M state to the solvent not to the central cluster.
The late M state is destabilized by the charges on the central and exit clusters. Even though they are 12 Å apart a negative charge on one cluster raises the other cluster pKa by 2.5 pH units [146]. The effective dielectric constant for this interaction is only ≈8 despite each cluster being surrounded by charged and polar residues. The long-range interaction favors a total charge of −2 on the two clusters. In the ground and early M states, both central and proton release clusters have a net charge of −1. In the N and O states, the central cluster is neutral, with a charge of −2 on the proton release cluster. However the late M state has a total charge of −3 with a central cluster charge of −1 (SB0850212−) and an exit cluster charge of −2. Calculations using late M structures show the equilibrium protonation state is a mixture of O and late M, with both states accessible in Monte Carlo sampling [146,215]. Thus, once bacteriorhodopsin reaches late M, structural changes stabilize the proton loss on the exit cluster and the proton gain on the central cluster. However, despite the larger net charge in the late M state this state is similar in energy to the O state, permitting its formation.
Because late M and O have similar energies, the kinetics of proton transfer must ensure that proton release is faster than transfer from the exit cluster to the central cluster for late M to be formed in high yield. The charged and polar residues including Asp 212, Tyr 83 and 185, and Arg 82 appear to form a hydrogen bond network, which would allow easier proton transfer between the central and exit clusters. In contrast, protonation of the SB by Asp96 requires structural changes in the cytoplasmic region to open a cavity and form a hydrogen bond network between the two groups [349]. However, the position of the SB on the cytoplasmic side of the retinal closer to Asp 96 does favor the correct proton transfer. In addition, the short-circuiting transfer from the exit cluster, would need to go through the protonated Asp85 or 212 to the SB which is still facing Asp 96. Thus, the short-circuiting pathway in the M state with the cis-SB is longer than the O→ground state transition with the trans-SB where the proton can be transferred directly from Asp 85 to the exit cluster.
Both PB calculations [215] and experiments [424] suggest that Arg82 can help to gate the proton transfer, closing the exit-to-central-cluster path in the M state. In the O→ground state transition Arg 82 appears to release its proton to the exit cluster, and subsequently moves to be reprotonated by the central cluster. While a neutral Arg will be a high energy intermediate [215], this proton transfer mechanism avoids the proton passing near the positively charged Arg. Proteorhodopsin, a homologous proton pump, which conserves the Arg but not the exit cluster, can carry out rapid proton release [425]. In the M state the Arg motion towards the extra-cellular space is triggered by the redistribution of the central cluster charge, and not the −2 charge on the exit cluster (see discussion of Glu204 to Gln mutant above). After the Arg 82 motion occurs, the hydrogen bond between Arg 82 and central cluster Asp 212 breaks and waters in central cluster cavity rearrange [351]. Thus, in the M state it is not as easy for Arg 82 to release a proton to the central cluster to facilitate the proton transfer as it is in the O→ground state transition. Lastly, in the M state the positive charge on the outward pointing Arg helps to expel the proton to the outside.
Acknowledgments
We would like to thank Gail Schneider and Gennady Khirich for careful reading of the manuscript and NIH RO1-GM64540 for financial support and the RCMI infrastructure grant RR03060.
References
- 1.Garcia-Moreno EB, Fitch CA. Structural interpretation of pH and salt-dependent processes in proteins with computational methods. Methods Enzymol. 2004;380:20–51. doi: 10.1016/S0076-6879(04)80002-8. [DOI] [PubMed] [Google Scholar]
- 2.Abrahams JP, Leslie AGW, Lutter R, Walker JE. Structure at 2.8 Å resolution of F1-ATPase brom bovine heart mitochondria. Nature. 1994;370:621–628. doi: 10.1038/370621a0. [DOI] [PubMed] [Google Scholar]
- 3.Rastogi VK, Girvin ME. Structural changes linked to proton translocation by subunit c of the ATP synthase. Nature. 1999;402:263–268. doi: 10.1038/46224. [DOI] [PubMed] [Google Scholar]
- 4.Oster G, Wang H. ATP synthase: two motors, two fuels. Structure. 1999;7:67–71. doi: 10.1016/s0969-2126(99)80046-x. [DOI] [PubMed] [Google Scholar]
- 5.Saraste M. Oxidative phosphorylation at the fin de siecle. Science. 1999;283:1488–1493. doi: 10.1126/science.283.5407.1488. [DOI] [PubMed] [Google Scholar]
- 6.Shurki A, Strajbl M, Schutz CN, Warshel A. Electrostatic basis for bioenergetics. Methods Enzymol. 2004;380:52–84. doi: 10.1016/S0076-6879(04)80003-X. [DOI] [PubMed] [Google Scholar]
- 7.Gunner MR, Nicholls A, Honig B. Electrostatic potentials in Rhodopseudomonas viridis reaction center: implications for the driving force and directionality of electron transfer. J Phys Chem. 1996;100:4277–4291. [Google Scholar]
- 8.Ferguson-Miller S, Babcock GT. Heme/copper terminal oxidases. Chem Rev. 1996;96:2889–2907. doi: 10.1021/cr950051s. [DOI] [PubMed] [Google Scholar]
- 9.Sham Y, Muegge I, Warshel A. Simulating proton translocations in proteins: probing proton transfer pathways in the Rhodobacter sphaeroides reaction center. Proteins: Struct Funct Genet. 1999;36:484–500. [PubMed] [Google Scholar]
- 10.Tiede DM, Vashishta AC, Gunner MR. Electron transfer kinetics and electrostatic properties of the Rhodobacter sphaeroides reaction center and soluble c-cytochromes. Biochemistry. 1993;32:4515–4531. doi: 10.1021/bi00068a006. [DOI] [PubMed] [Google Scholar]
- 11.Sims PA, Wong CF, McCammon JA. Charge optimization of the interface between protein kinases and their ligands. J Comp Chem. 2004;25:1416–1429. doi: 10.1002/jcc.20067. [DOI] [PubMed] [Google Scholar]
- 12.McLaughlin S, Murray D. Plasma membrane phosphoinositide organization by protein electrostatics. Nature. 2005;438:605–611. doi: 10.1038/nature04398. [DOI] [PubMed] [Google Scholar]
- 13.Beroza P, Case DA. Calculations of proton-binding thermodynamics in proteins. Method Enzymol. 1998;295:170–189. doi: 10.1016/s0076-6879(98)95040-6. [DOI] [PubMed] [Google Scholar]
- 14.Warshel A, Papazyan A. Electrostatic effects in macromolecules: fundamental concepts and practical modeling. Curr Opin Struct Biol. 1998;8:211–217. doi: 10.1016/s0959-440x(98)80041-9. [DOI] [PubMed] [Google Scholar]
- 15.Ullmann GM, Knapp EW. Electrostatic models for computing protonation and redox equilibria in proteins. Eur Biophys J. 1999;28:533–551. doi: 10.1007/s002490050236. [DOI] [PubMed] [Google Scholar]
- 16.Gunner MR, Alexov E. A pragmatic approach to structure based calculation of coupled proton and electron transfer in proteins. Biochim Biophys Acta. 2000;1458:63–87. doi: 10.1016/s0005-2728(00)00060-8. [DOI] [PubMed] [Google Scholar]
- 17.Neves-Petersen MT, Petersen SB. Protein electrostatics: a review of the equations and methods used to model electrostatic equations in biomolecules—Applications in biotechnology. Biotechnol Annu Rev. 2003;9:315–395. doi: 10.1016/s1387-2656(03)09010-0. [DOI] [PubMed] [Google Scholar]
- 18.Bashford D. Macroscopic electrostatic models for protonation states in proteins. Front Biosci. 2004;9:1082–1099. doi: 10.2741/1187. [DOI] [PubMed] [Google Scholar]
- 19.Mongan J, Case DA. Biomolecular simulations at constant pH. Curr Opin Struct Biol. 2005;15:157–163. doi: 10.1016/j.sbi.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 20.Clark WM. Oxidation-Reduction Potentials on Organic Systems. Waverly Press; Baltimore: 1960. [Google Scholar]
- 21.Rich PR, Bendall DS. A mechanism for the reduction of cytochromes by quinols in solution and its relevance to biological electron transfer reactions. FEBS Lett. 1979;105:189–194. doi: 10.1016/0014-5793(79)80608-0. [DOI] [PubMed] [Google Scholar]
- 22.Cramer WA, Knaff DB. Energy Transduction in Biological Membranes: A Textbook of Bioenergetics. Springer-Verlag; New York: 1991. [Google Scholar]
- 23.Lim C, Bashford L, Karplus M. Absolute pKa calculations with continuum dielectric methods. J Phys Chem. 1991;95:5610–5620. [Google Scholar]
- 24.Jensen JH, Li H, Robertson AD, Molina PA. Prediction and rationalization of protein pKa values using QM and QM/MM methods. J Phys Chem A. 2005;109:6634–6643. doi: 10.1021/jp051922x. [DOI] [PubMed] [Google Scholar]
- 25.Li H, Hains AW, Everts JE, Robertson AD, Jensen JH. The prediction of protein pKa's using QM/MM: the pKa of lysine 55 in turkey ovomucoid third domain. J Phys Chem B. 2002;106:3486–3494. [Google Scholar]
- 26.Soriano E, Serdan S, Ballesteros P. Computational determination of pKa values. A comparison of different theroetical approaches and a novel procedure. J Mol Struct THEOCHEM. 2004;684:121–128. [Google Scholar]
- 27.Bockris JOM, Reddy AKN. Modern Electrochemistry. Vol. 1. Plenum; New York: 1973. [Google Scholar]
- 28.Kim J, Mao J, Gunner MR. Are acidic and basic groups in buried proteins predicted to be ionized? J Mol Biol. 2005;348:1283–1298. doi: 10.1016/j.jmb.2005.03.051. [DOI] [PubMed] [Google Scholar]
- 29.Mao J, Hauser K, Gunner MR. How cytochromes with different folds control heme redox potentials. Biochemistry. 2003;42:9829–9840. doi: 10.1021/bi027288k. [DOI] [PubMed] [Google Scholar]
- 30.Zhu Z, Gunner MR. Energetics of quinone-dependent electron and proton transfers in Rhodobacter sphaeroides photosynthetic reaction centers. Biochemistry. 2005;44:82–96. doi: 10.1021/bi048348k. [DOI] [PubMed] [Google Scholar]
- 31.Song Y, Michonova-Alexova E, Gunner MR. Calculated proton uptake on anaerobic reduction of cytochrome c oxidase: Is the reaction electroneutral? Biochemistry. 2006;45:7959–7975. doi: 10.1021/bi052183d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reece SY, Stubbe J, Nocera DG. pH Dependence of charge transfer between tryptophan and tyrosine in dipeptides. Biochim Biophys Acta. 2005;1706:232–238. doi: 10.1016/j.bbabio.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 33.Gogonea V, Suarez D, van der Vaart A, Merz KM., Jr New developments in applying quantum mechanics to proteins. Cur Opin Struct Biol. 2001;11:217–223. doi: 10.1016/s0959-440x(00)00193-7. [DOI] [PubMed] [Google Scholar]
- 34.Friesner RA. Ab initio quantum chemistry: methodology and applications. Proc Natl Acad Sci U S A. 2005;102:6648–6653. doi: 10.1073/pnas.0408036102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Siegbahn PE, Blomberg MR. Transition-metal systems in biochemistry studied by high-accuracy quantum chemical methods. Chem Rev. 2000;100:421–437. doi: 10.1021/cr980390w. [DOI] [PubMed] [Google Scholar]
- 36.Noodleman L, Lovell T, Han WG, Li J, Himo F. Quantum chemical studies of intermediates and reaction pathways in selected enzymes and catalytic synthetic systems. Chem Rev. 2004;104:459–508. doi: 10.1021/cr020625a. [DOI] [PubMed] [Google Scholar]
- 37.Friesner RA, Dunietz BD. Large-scale ab initio quantum chemical calculations on biological systems. Acc Chem Res. 2001;34:351–358. doi: 10.1021/ar980111r. [DOI] [PubMed] [Google Scholar]
- 38.Ishida T, Fedorov DG, Kitaura K. All electron quantum chemical calculation of the entire enzyme system confirms a collective catalytic device in the Chorismate mutase reaction. J Phys Chem B. 2006;110:1457–1463. doi: 10.1021/jp0557159. [DOI] [PubMed] [Google Scholar]
- 39.Svith H, Jensen H, Almstedt J, Andersson P, Lundback T, Daasbjerg K, Jonsson M. On the nature of solvent effects on redox properties. J Phys Chem A. 2004;108:4805–4811. [Google Scholar]
- 40.Chipman DM. Computation of pKa from dielectric continuum theory. J Phys Chem A. 2002;106:7413–7422. [Google Scholar]
- 41.Cramer CJ, Truhlar DG. Implicit solvation models: equilibria, structure, spectra, and dynamics. Chem Rev. 1999;99:2161–2200. doi: 10.1021/cr960149m. [DOI] [PubMed] [Google Scholar]
- 42.Orozco M, Luque FJ. Theoretical methods for the description of the solvent effect in biomolecular systems. Chem Rev. 2000;100 doi: 10.1021/cr990052a. [DOI] [PubMed] [Google Scholar]
- 43.Toth AM, Liptak MD, Phillips DL, Shields GC. Accurate relative pKa calculations for carboxylic acids using complete basis set and Gaussian-n models combined with continuum solvation methods. J Chem Phys. 2001;114:4595–4606. doi: 10.1021/ja010534f. [DOI] [PubMed] [Google Scholar]
- 44.Riccardi D, Schaefer P, Cui Q. pKa calculations in solution and proteins with QM/MM free energy perturbation simulations: a quantitative test of QM/MM protocols. J Phys Chem B. 2005;109:17715–17733. doi: 10.1021/jp0517192. [DOI] [PubMed] [Google Scholar]
- 45.Chen JL, Noodleman L, Case DA, Bashford D. Incorporating solvation effects into density functional electronic structure calculations. J Phys Chem. 1994;98:11059–11068. [Google Scholar]
- 46.Li J, Fisher CL, Chen JL, Bashford D, Noodleman L. Calculation of redox potentials and pKa values of hydrated transition metal cations by a combined density functional and continuum dielectric theory. Inorg Chem. 1996;35:4694–4702. [Google Scholar]
- 47.Klicic JJ, Friesner RA, Liu SY, Guida WC. Accurate prediction of acidity constants in aqueous solution via density functional theory and self-consistent reaction field methods. J Phys Chem A. 2002;106:1327–1335. [Google Scholar]
- 48.Tanford C. The interpretation of hydrogen ion titration curves of proteins. Adv Protein Chem. 1962;17:69–165. [Google Scholar]
- 49.Martell AE, Smith RM. Critical Stability Constants. 1–IV. Plenum Press; New York: 1974. [Google Scholar]
- 50.Lide DR. CRC Handbook of Chemistry and Physics. CRC Press; Boca Raton: 1990. [Google Scholar]
- 51.Kyte J. Structure in Protein Chemistry. Gaarland; New York: 1995. [Google Scholar]
- 52.Keim P, Vigna RA, Nigen AM, Morrow JS, Gurd FRN. Carbon 13 nuclear magnetic resonance of pentapeptides of glycine containing central residues of methionine, proline, arginine, and lysine. J Biol Chem. 1974;249:4149–4156. [PubMed] [Google Scholar]
- 53.Richarz R, Wüthrich K. Carbon-13 NMR chemical shifts of the common amino acid residues measured in aqueous solutions of the linear tetrapeptides H–Gly–Gly–X–L–Ala–OH. Biopolymers. 1975;17:2133–2141. [Google Scholar]
- 54.Matthew JB, Gurd FRN, Garcia-Moreno B, Flanagan MA, March KL, Shire SJ. pH-dependent processes in proteins. CRC Crit Rev Biochem. 1985;18:91–197. doi: 10.3109/10409238509085133. [DOI] [PubMed] [Google Scholar]
- 55.Oliveberg M, Arcus VL, Fersht AR. pKa values of carboxyl groups in the native and denatured states of barnase. The pKa values of the denatured state are on average 0.4 units lower than those of model compounds. Biochemistry. 1995;34:9424–9433. doi: 10.1021/bi00029a018. [DOI] [PubMed] [Google Scholar]
- 56.Gunner MR, Saleh MA, Cross E, ud-Doula A, Wise M. Backbone dipoles generate positive potentials in all proteins: origins and implications of the effect. Biophys J. 2000;78:1126–1144. doi: 10.1016/S0006-3495(00)76671-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mayhew SG. The effects of pH and semiquinone formation on the oxidation–reduction potentials of flavin monoculeotide: A reappraisal. Eur J Biochem. 1999;265:698–702. doi: 10.1046/j.1432-1327.1999.00767.x. [DOI] [PubMed] [Google Scholar]
- 58.Reddy MR, Erion MD. Calculation of relative solvation free energy differences by thermodynamic perturbation method dependence of free energy results on simulation length. J Comput Chem. 1999;20:1018–1027. [Google Scholar]
- 59.Scott RA, Mauk AG. University Science Books. Sausalito: 1996. [Google Scholar]
- 60.Harbury HA, Loach PA. Oxidation-linked proton functions in heme octa- and undecapeptides from mammalian cytochrome c. J Biol Chem. 1960;255:3640–3645. [PubMed] [Google Scholar]
- 61.Harbury HA, Loach PA. Interaction of nitrogenous ligands with heme peptides from mammalian cytochrome c. J Biol Chem. 1960;235:3646–3653. [PubMed] [Google Scholar]
- 62.Harbury HA, Cronin JR, Fanger MW, Hettinger TP, Murphy AJ, Meyer YP, Vinogradov SN. Complex formation between methionine and a heme peptide from cytochrome c. Proc Natl Acad Sci U S A. 1965;54:1658–1664. doi: 10.1073/pnas.54.6.1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wilson GS. Electrochemical studies of porphyrin redox reactions as cytochromes models. Bioelectrochem Bioenerg. 1974;1:172–179. [Google Scholar]
- 64.Battistuzzi G, Borsari M, Cowan JA, Ranieri A, Sola M. Control of cytochrome c redox potential: axial ligation and protein environment effects. J Am Chem Soc. 2002;124:5315–5324. doi: 10.1021/ja017479v. [DOI] [PubMed] [Google Scholar]
- 65.Munro OQ, Marques HM. Heme-peptide models for hemoproteins. 1. solution chemistry of N-acetylmicroperoxidase-8. Inorg Chem. 1996;35:3752–3767. doi: 10.1021/ic9502842. [DOI] [PubMed] [Google Scholar]
- 66.Marques HM, Perry CB. Hemepeptide models for hemoproteins: the behavior of N-acetylmicroperoxidase-11 in aqueous solution. J Inorg Biochem. 1999;75:281–291. doi: 10.1016/s0162-0134(99)00100-2. [DOI] [PubMed] [Google Scholar]
- 67.Adams PA, Baldwin DA, Marques HM. Hemepeptides from cytochrome c: preparation, physical and chemical properties, and their use as a model compounds for the hemoproteins. In: Scott RA, Mauk AG, editors. Cytochrome c Sourcebook. University Science Books; Mill Valley: 1996. pp. 635–692. [Google Scholar]
- 68.Marques HM, Cukrowski I, Vashi PR. Co-ordination of weak field ligands by N-acetylmicroperoxidase-8 (NAcMP8), a ferric haempeptide from cytochrome c, and the influence of the axial ligand on the reduction potential of complexes of NAcMP8. J Chem Soc. 2000;2000:1335–1342. [Google Scholar]
- 69.Santucci R, Reinhard H, Brunori M. Direct electrochemistry of the undacapeptide from cytochrome c (microperoxidase) at a glassy carbon electrode. J Am Chem Soc. 1988;110:8536–8357. [Google Scholar]
- 70.Vashi PR, Marques HM. The coordination of imidazole and substituted pyridines by the hemeoctapeptide N-acetyl-ferromicroperoxidase-8 (FeIINAcMP8) J Inorg Biochem. 2004;98:1471–1482. doi: 10.1016/j.jinorgbio.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 71.Song Y, Mao J, Gunner MR. Electrostatic environment of hemes in proteins: pKas of hydroxyl ligands. Biochemistry. 2006;45:7949–7958. doi: 10.1021/bi052182l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Reedy CJ, Gibney BR. Heme protein assemblies. Chem Rev. 2004;104:617–649. doi: 10.1021/cr0206115. [DOI] [PubMed] [Google Scholar]
- 73.Shifman JM, Gibney BR, Sharp RE, Dutton PL. Heme redox potential control in de novo designed four-alpha–helix bundle proteins. Biochemistry. 2000;39:14818–14821. doi: 10.1021/bi000927b. [DOI] [PubMed] [Google Scholar]
- 74.Garcia-Horsman JA, Barquera B, Rumbley J, Ma J, Gennis RB. The superfamily of heme-copper respiratory oxidases. J Bacteriol. 1994;176:5587–5600. doi: 10.1128/jb.176.18.5587-5600.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhuang J, Reddi AR, Wang Z, Khodaverdian B, Hegg EL, Gibney BR. Evaluating the roles of the heme a side chains in cytochrome c oxidase using designed heme proteins. Biochemistry. 45 doi: 10.1021/bi060565t. in press. [DOI] [PubMed] [Google Scholar]
- 76.Vanderkooi G, Stotz E. Reductive alteration of heme a hemochromes. J Biol Chem. 1965;240:3418–3424. [PubMed] [Google Scholar]
- 77.Vanderkooi G, Stotz E. Oxidation–reduction potentials of heme a hemochromes. J Biol Chem. 1966;241:3316–3323. [PubMed] [Google Scholar]
- 78.Zhuang J, Amoroso JH, Kinloch R, Dawson JH, Baldwin MJ, Gibney BR. Evaluation of electron-withdrawing group effects on heme binding in designed proteins: implications for heme a in cytochrome c oxidase. Inorg Chem. 2006;45:4685–4694. doi: 10.1021/ic060072c. [DOI] [PubMed] [Google Scholar]
- 79.Anderson RF. Energetics of the one-electron reduction steps of riboflavin, FMN, and FAD to their fully reduced forms. Biochim Biophys Acta. 1983;722:158–162. doi: 10.1016/0005-2728(83)90169-x. [DOI] [PubMed] [Google Scholar]
- 80.Rich PR, Bendall DS. The kinetics and thermodynamics of the reduction of cytochrome c by substituted p-benzoquinols in solution. Biochim Biophys Acta. 1980;592:506–518. doi: 10.1016/0005-2728(80)90095-x. [DOI] [PubMed] [Google Scholar]
- 81.Rich PR. The quinone chemistry of bc complexes. Biochem Biophys Acta. 2004;1658:165–171. doi: 10.1016/j.bbabio.2004.04.021. [DOI] [PubMed] [Google Scholar]
- 82.Wraight CA. The role of quinones in bacterial photosynthesis. Photochem Photobiol. 1979;30:767–776. [Google Scholar]
- 83.Osyczka A, Moser CC, Dutton PL. Fixing the Q cycle. Trends Biochem Sci. 2005;30:176–182. doi: 10.1016/j.tibs.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 84.Bishop CA, Tong LKJ. Equilibria of Substituted Semiquinones at High pH. J Am Chem Soc. 1965;18:501–505. [Google Scholar]
- 85.Swallow AJ. Physical chemistry of semiquinones. In: Trumpower BL, editor. Function of Quinones in Energy Conserving Systems. Academic Press; New York: 1982. pp. 59–72. [Google Scholar]
- 86.Morrison LE, Schelhorn JE, Cotton TE, Bering CL, Loach PA. Electrochemical and Spectral Properties of Ubiquinone and Synthetic Analogs: Relevance to Bacterial Photosynthesis. In: Trumpower BL, editor. Function of Quinones in Energy Conserving Systems. Academic Press; New York: 1982. pp. 35–58. [Google Scholar]
- 87.Wraight CA. Functional linkage between the QA and QB sites of photosynthetic reaction centers. In: Garab G, editor. Proceedings of the XIth International Photosynthesis Congress. Kluwer; Dordrecht: 1998. pp. 693–698. [Google Scholar]
- 88.Gordillo GJ, Schiffrin DJ. The electrochemistry of ubiquinone-10 in a phospholipid model membrane. Faraday Discuss. 2000;116:89–107. doi: 10.1039/b004379f. [DOI] [PubMed] [Google Scholar]
- 89.Prince RC, Gunner MR, Dutton PL. Quinones of value to electron-transfer studies: Oxidation–reduction potentials of the first reduction step in an aprotic solvent. In: Trumpower BL, editor. Function of Quinones in Energy Conserving Systems. Academic Press; New York: 1982. pp. 29–33. [Google Scholar]
- 90.Ashnagar A, Bruce M, Dutton PL, Prince R. One- and two-electron reduction of hydroxy-1,4-naphthoquinone and hydroxy-9,10-anthraquinones. The role of internal hydrogen bonding and its bearing on the redox chemistry of the anthracycline antitumour quinones. Biochim Biophys Acta. 1984;801:351–359. doi: 10.1016/0304-4165(84)90138-7. [DOI] [PubMed] [Google Scholar]
- 91.Prince RC, Dutton PL, Bruce JM. Electrochemistry of ubiquinones. FEBS Lett. 1983;160:273–276. [Google Scholar]
- 92.Prince RC, Lloyd-Williams P, Bruce JM, Dutton PL. Voltammetric measurements of quinones. Methods Enzymol. 1986;125:109–119. doi: 10.1016/s0076-6879(86)25010-7. [DOI] [PubMed] [Google Scholar]
- 93.Rabenstein B, Ullmann GM, Knapp EW. Energetics of electron-transfer and protonation reactions of the quinones in the photosynthetic reaction center of Rhodopseudonas viridis. Biochemistry. 1998;37:2488–2495. doi: 10.1021/bi971921y. [DOI] [PubMed] [Google Scholar]
- 94.Van Hellemond JJ, Klockiewicz M, Gaasenbeek CP, Roos MH, Tielens AG. Rhodoquinone and complex II of the electron transport chain in anaerobically functioning eukaryotes. J Biol Chem. 1995;270:31065–31070. doi: 10.1074/jbc.270.52.31065. [DOI] [PubMed] [Google Scholar]
- 95.Wallace BJ, Young IG. Role of quinones in electron transport to oxygen and nitrate in Escherichia coli. Studies with a ubiA- menA-double quinone mutant. Biochim Biophys Acta. 1977;461:84–100. doi: 10.1016/0005-2728(77)90071-8. [DOI] [PubMed] [Google Scholar]
- 96.Wraight CA. Proton and electron transfer in the acceptor quinone complex of photosynthetic reaction centers from Rhodobacter sphaeroides. Front Biosci. 2004;9:309–337. doi: 10.2741/1236. [DOI] [PubMed] [Google Scholar]
- 97.Paddock ML, Feher G, Okamura MY. Proton transfer pathways and mechanism in bacterial reaction centers. FEBS Lett. 2003;555:45–50. doi: 10.1016/s0014-5793(03)01149-9. [DOI] [PubMed] [Google Scholar]
- 98.Woodbury NW, Parson WW, Gunner MR, Prince RC, Dutton PL. Radical-pair energetics and decay mechanisms in reaction center containing anthraquinones or benzoquinones in place of ubiquinone. Biochim Biophys Acta. 1986;851:6–22. doi: 10.1016/0005-2728(86)90243-4. [DOI] [PubMed] [Google Scholar]
- 99.Chen H, Collomb MN, Duboc C, Blondin G, Riviere E, Faller JW, Crabtree RH, Brudvig GW. New linear high-valent tetranuclear manganese-oxo cluster relevant to the oxygen-evolving complex of photosystem II with oxo, hydroxo, and aqua coordinated to a single Mn (IV) Inorg Chem. 2005;44:9567–9573. doi: 10.1021/ic051462m. [DOI] [PubMed] [Google Scholar]
- 100.Carrell G, Tyryshkin M, Dismukes C. An evaluation of structural models for the photosynthetic water-oxidizing complex derived from spectroscopic and X-ray diffraction signatures. J Biol Inorg Chem. 2002;7:2–22. doi: 10.1007/s00775-001-0305-3. [DOI] [PubMed] [Google Scholar]
- 101.Sproviero EM, Gascon JA, McEvoy JP, Brudvig GW, Batista VS. Characterization of synthetic oxomanganese complexes and the inorganic core of the O2-evolving complex in photosystem II: Evaluation of the DFT/B3LYP level of theory. J Inorg Biochem. 2006;100:786–800. doi: 10.1016/j.jinorgbio.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 102.Ferreira KN, Iverson TM, Maghlaoui K, Barber J, Iwata S. Architecture of the photosynthetic oxygen-evolving center. Science. 2004;303:1831–1838. doi: 10.1126/science.1093087. [DOI] [PubMed] [Google Scholar]
- 103.Barber J. Water, water everywhere, and its remarkable chemistry. Biochim Biophysica Acta. 2004;1655:123–132. doi: 10.1016/j.bbabio.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 104.Loll B, Kern J, Saenger W, Zouni A, Biesiadka J. Towards complete cofactor arrangement in the 3.0 A resolution structure of photosystem II. Nature. 2005;438:1040–1044. doi: 10.1038/nature04224. [DOI] [PubMed] [Google Scholar]
- 105.Chang CJ, Loh ZH, Shi C, Anson FC, Nocera DG. Targeted proton delivery in the catalyzed reduction of oxygen to water by bimetallic pacman porphyrins. J Am Chem Soc. 2004;126:10013–10020. doi: 10.1021/ja049115j. [DOI] [PubMed] [Google Scholar]
- 106.Collman JP, Boulatov R, Sunderland CJ, Fu L. Functional analogues of cytochrome c oxidase, myoglobin, and hemoglobin. Chem Rev. 2004;104:561–588. doi: 10.1021/cr0206059. [DOI] [PubMed] [Google Scholar]
- 107.Babcock G. How oxygen is activated and reduced in respiration. Proc Natl Acad Sci U S A. 1999;96:12971–12973. doi: 10.1073/pnas.96.23.12971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Solomon EI, Szilagyi RK, George SD, Basumallick L. Electronic structures of metal sites in proteins and models: Contributions to function in blue copper proteins. Chem Rev. 2004;104:419–458. doi: 10.1021/cr0206317. [DOI] [PubMed] [Google Scholar]
- 109.Rao PV, Holm RH. Synthetic analogues of the active sites of iron–sulfur proteins. Chem Rev. 2004;104:527–560. doi: 10.1021/cr020615+. [DOI] [PubMed] [Google Scholar]
- 110.Hay MT, Lu Y. Metal-binding properties of an engineered purple CuA center in azurin. J Biol Inorg Chem. 2000;5:699–712. doi: 10.1007/s007750000158. [DOI] [PubMed] [Google Scholar]
- 111.Sigman JA, Kim HK, Zhao X, Carey JR, Lu Y. The role of copper and protons in heme-copper oxidases: kinetic study of an engineered heme-copper center in myoglobin. Proc Natl Acad Sci U S A. 2003;100:3629–3634. doi: 10.1073/pnas.0737308100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhao X, Yeung N, Wang Z, Guo Z, Lu Y. Effects of metal ions in the CuB on the redox properties of heme in heme-copper oxidases: spectroelectrochemical studies of an engineered heme-copper center in myoglobin. Biochemistry. 2005;44:1210–1214. doi: 10.1021/bi0479151. [DOI] [PubMed] [Google Scholar]
- 113.Parsegian A. Energy of an ion crossing a low dielectric membrane: solutions to four relevant electrostatic problems. Nature. 1969;221:844–846. doi: 10.1038/221844a0. [DOI] [PubMed] [Google Scholar]
- 114.Kassner RJ. Effects of nonpolar environments on the redox potentials of heme complexes. Proc Natl Acad Sci U S A. 1972;69:2263–2267. doi: 10.1073/pnas.69.8.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kassner RJ. A theoretical model for the effects of local nonpolar heme environments on the redox potentials in cytochromes. J Am Chem Soc. 1973;95:2674–2676. doi: 10.1021/ja00789a044. [DOI] [PubMed] [Google Scholar]
- 116.Kassner RJ, Yang W. A theoretical model for the effects of solvent and protein dielectric on the redox potentials of iron–sulfur clusters. J Am Chem Soc. 1977;99:4351–4355. doi: 10.1021/ja00455a024. [DOI] [PubMed] [Google Scholar]
- 117.Warshel A, Russell ST. Calculations of electrostatic interactions in biological systems and in solutions. Q Rev Biophys. 1984;17:283–422. doi: 10.1017/s0033583500005333. [DOI] [PubMed] [Google Scholar]
- 118.Churg AK, Warshel A. Control of the redox potential of cytochrome c and microscopic dielectric effects in proteins. Biochemistry. 1986;25:1675–1681. doi: 10.1021/bi00355a035. [DOI] [PubMed] [Google Scholar]
- 119.Simonson T, Carlsson J, Case DA. Proton binding to proteins: pKa calculations with explicit and implicit solvent models. J Am Chem Soc. 2006;126:4167–4180. doi: 10.1021/ja039788m. [DOI] [PubMed] [Google Scholar]
- 120.Wagoner J, Baker NA. Solvation forces on biomolecular structures: A comparison of explicit solvent and Poisson–Boltzmann models. J Comp Chem. 2004;25:1623–1629. doi: 10.1002/jcc.20089. [DOI] [PubMed] [Google Scholar]
- 121.Koehl P. Electrostatics calculations: latest methodological advances. Curr Opin Struct Biol. 2006;16:142–151. doi: 10.1016/j.sbi.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 122.Kasenholz MA, Hunenberger PH. Influence of artificial periodocity and ionic strength in molecular dynamics simulations of charged biomolecules employing lattice-sum methods. J Phys Chem B. 2004;108:774–788. [Google Scholar]
- 123.Sagui C, Darden TA. Molecular dynamics simulations of biomolecules: long-range electrostatic effects. Annu Rev Biophys Biomol Struct. 1999;28:155–179. doi: 10.1146/annurev.biophys.28.1.155. [DOI] [PubMed] [Google Scholar]
- 124.Ren P, Ponder JW. Polarizable atomic multipole water model for molecular mechanics simulations. J Phys Chem B. 2003;107:5933–5947. [Google Scholar]
- 125.Feig M, Brooks CL., III Recent advances in the development and application of implicit solvent models in biomolecule simulations. Curr Opin Struct Biol. 2004;14:217–214. doi: 10.1016/j.sbi.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 126.Tomasi J, Persico M. Molecular interactions in solution: An overview of methods based on continuous distributions of the solvent. Chem Rev. 1994;94:2027–2094. [Google Scholar]
- 127.Roux B, Simonson T. Implicit solvent models. Biophys Chem. 1999;78:1–20. doi: 10.1016/s0301-4622(98)00226-9. [DOI] [PubMed] [Google Scholar]
- 128.Schutz CN, Warshel A. What are the “dielectric constants” of proteins and how to validate electrostatic models? Proteins. Struct Funct Genet. 2001;44:400–417. doi: 10.1002/prot.1106. [DOI] [PubMed] [Google Scholar]
- 129.Rashin AA, Honig B. Reevaluation of the Born model of ion hydration. J Phys Chem. 1985;89:5588–5593. [Google Scholar]
- 130.Feig M, Onufriev A, Lee MS, Im W, Case DA, Brooks CL., III Performance comparison of Generalized Born and Poisson methods in the calculation of electrostatic solvation energies for protein structures. J Comput Chem. 2004;30:265–284. doi: 10.1002/jcc.10378. [DOI] [PubMed] [Google Scholar]
- 131.Baker NA. Improving implicit solvent simulations: a Poisson-centric view. Cur Opin Struct Biol. 2005;15:137–143. doi: 10.1016/j.sbi.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 132.Sharp KA, Honig B. Calculating total electrostatic energies with the nonlinear Poisson–Boltzmann equation. J Phys Chem. 1990;94:7684–7692. [Google Scholar]
- 133.Nicholls A, Honig B. A rapid finite difference algorithm utilizing successive over-relaxation to solve the Poisson–Boltzmann equation. J Comput Chem. 1991;12:435–445. [Google Scholar]
- 134.Tannor DJ, Marten B, Murphy R, Friesner RA, Sitkoff D, Nicholls A, Ringnalda M, Goddard WA, III, Honig B. Accurate first principles calculation of molecular charge distributions and solvation energies from ab initio quantum mechanics and continuum dielectric theory. J Am Chem Soc. 1994;116:11875–11882. [Google Scholar]
- 135.MacKerell AD, Jr, Bashford D, Bellott M, Dunbrack RL, Jr, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, III, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. All-atom emprical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 136.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz J, Ferguson DM, Spellman DC, Fox T, Caldwell JW, Kollman PA. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- 137.Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;118:11225–11236. [Google Scholar]
- 138.Jorgensen WL, Ulmschneider JP, Tirado-Rives J. Free energies of hydration from a generalized Born model and all-atom force field. J Phys Chem B. 2004;108:16246–16270. [Google Scholar]
- 139.Onufriev A, Case DA, Bashford D. Effective Born radii in the generalized Born approximation: the importance of being perfect. J Comput Chem. 2002;23:1297–1304. doi: 10.1002/jcc.10126. [DOI] [PubMed] [Google Scholar]
- 140.Qui D, Shenkin PS, Hollinger FP, Still WC. The GB/SA continuum model for solvation. A fast analytical method for the calculation of approximate Born radii. J Phys Chem A. 1997;101 [Google Scholar]
- 141.Bashford D, Case DA. Generalized Born models of macromolecular solvation effects. Annu Rev Phys Chem. 2000;51:129–152. doi: 10.1146/annurev.physchem.51.1.129. [DOI] [PubMed] [Google Scholar]
- 142.Mallik B, Masunov A, Lazaridis T. Distance and exposure dependent effective dielectric function. J Comput Chem. 2002;23:1090–1099. doi: 10.1002/jcc.10104. [DOI] [PubMed] [Google Scholar]
- 143.Lazaridis T. Implicit solvent simulations of peptide interactions with anionic lipid membranes. Proteins: Struct Funct Genet. 2005;58:518–527. doi: 10.1002/prot.20358. [DOI] [PubMed] [Google Scholar]
- 144.Hassan SA, Mehler EL, Zhang D, Weinstein H. Molecular dynamics simulations of peptides and proteins with a continuum electrostatic model based on screened Coulomb potentials. Proteins. 2003;51:109–125. doi: 10.1002/prot.10330. [DOI] [PubMed] [Google Scholar]
- 145.Gunner MR, Nicholls A, Honig B. Electrostatic potentials in Rhopseudomonas viridis reaction center: Implications for the driving force and directionality of electron transfer. J Phys Chem. 1996;100:4277–4291. [Google Scholar]
- 146.Song Y, Mao J, Gunner MR. Calculation of proton transfers in bacteriorhodopsin bR and M intermediates. Biochemistry. 2003;42:9875–9888. doi: 10.1021/bi034482d. [DOI] [PubMed] [Google Scholar]
- 147.Simonson T. Macromolecular electrostatics: continuum models and their growing pains. Curr Opin Struct Biol. 2001;11:243–252. doi: 10.1016/s0959-440x(00)00197-4. [DOI] [PubMed] [Google Scholar]
- 148.Harvey S. Treatment of electrostatic effects in macromolecular modeling, Proteins. Struct Funct Genet. 1989;5:78–92. doi: 10.1002/prot.340050109. [DOI] [PubMed] [Google Scholar]
- 149.Gilson MK, Honig BH. The dielectric constant of a folded protein. Biopolymers. 1986;25:2097–2119. doi: 10.1002/bip.360251106. [DOI] [PubMed] [Google Scholar]
- 150.Mertz EL, Krishtalik LI. Low dielectric response in enzyme active site. Proc Natl Acad Sci U S A. 2000;97:2081–2086. doi: 10.1073/pnas.050316997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Simonson T, Perahia D. Dielectric properties of proteins from simulations: tools and techniques. Comp Phys Comm. 1995;1995:291–303. [Google Scholar]
- 152.Simonson T, Perahia D. Internal and interfacial dielectric properties of cytochrome c from molecular dynamics in aqueous solution. Proc Natl Acad Sci U S A. 1995;92:1082–1086. doi: 10.1073/pnas.92.4.1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Simonson T, Brooks CL. Charge Screening and the dielectric constant of proteins: Insights from molecular dynamics. J Am Chem Soc. 1996;118:8452–8458. [Google Scholar]
- 154.Simonson T. Dielectric constant of cytochrome c from simulations in a water droplet including all electrostatic interactions. J Am Chem Soc. 1998;120:4873–4878. [Google Scholar]
- 155.Krol M. Comparison of various implicit solvent models in molecular dynamics simulations of immunoglobulin G light chain dimer. J Comput Chem. 2003;15:531–546. doi: 10.1002/jcc.10186. [DOI] [PubMed] [Google Scholar]
- 156.Archontis G, Simonson T. Dielectric relaxation in an enzyme active site: molecular dynamics simulations interpreted with a macroscopic continuum model. J Am Chem Soc. 2001;123:10056–11047. doi: 10.1021/ja010716+. [DOI] [PubMed] [Google Scholar]
- 157.Yang AS, Gunner MR, Sampogna R, Sharp K, Honig B. On the calculation of pKa's in proteins. Proteins: Struct Funct Genet. 1993;15:252–265. doi: 10.1002/prot.340150304. [DOI] [PubMed] [Google Scholar]
- 158.Gilson MK. Multiple-Site titration and molecular modeling: Two rapid methods for computing energies and forces for ionizable groups in proteins. Proteins: Struct Funct Genet. 1993;15:266–282. doi: 10.1002/prot.340150305. [DOI] [PubMed] [Google Scholar]
- 159.Spassov VZ, Bashford D. Multiple site ligand binding to flexible macromolecules separation of global and local conformational change and an iterative mobile clustering approach. J Comput Chem. 1999;20:1091–1111. [Google Scholar]
- 160.Beroza P, Fredkin DR, Okamura MY, Feher G. Protonation of interacting residues in a protein by a Monte Carlo method: application to lysozyme and the photosynthetic reaction center of Rhodobacter sphaeroides. Proc Natl Acad Sci U S A. 1991;88:5804–5808. doi: 10.1073/pnas.88.13.5804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Georgescu RE, Alexov EG, Gunner MR. Combining conformational flexibility and continuum electrostatics for calculating pKas in proteins. Biophys J. 2002;83:1731–1748. doi: 10.1016/S0006-3495(02)73940-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Alexov E. Role of the protein side-chain fluctuations on the strength of pair-wise electrostatic interactions: comparing experimental with computed pKas. Proteins: Struct Funct Genet. 2003;50:94–103. doi: 10.1002/prot.10265. [DOI] [PubMed] [Google Scholar]
- 163.Nielsen JE, McCammon JA. On the evaluation and optimization of protein X-ray structures for pKa calculations. Protein Sci. 2003;12:313–326. doi: 10.1110/ps.0229903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ondrechen M, Clifton J, Ringe D. THEMATICS: a simple computational predictor of enzyme function from structure. Proc Natl Acad Sci U S A. 2001;98:12473–12478. doi: 10.1073/pnas.211436698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Alexov EG, Gunner MR. Calculated protein and proton motions coupled to electron transfer: electron transfer from QA- to QB in bacterial photosynthetic reaction centers. Biochemistry. 1999;38:8253–8270. doi: 10.1021/bi982700a. [DOI] [PubMed] [Google Scholar]
- 166.Beroza P, Fredkin DR, Okamura MY, Feher R. Electrostatic calculations of amino acid titration electron transfer, in the reaction center. Biophys J. 1995;68:2233–2250. doi: 10.1016/S0006-3495(95)80406-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Rabenstein B, Ullmann GM, Knapp EW. Electron transfer between the quinones in the photosynthetic reaction center and its coupling to conformational changes. Biochemistry. 2000;39:10487–10496. doi: 10.1021/bi000413c. [DOI] [PubMed] [Google Scholar]
- 168.Ishikita H, Morra G, Knapp EW. Redox potential of quinones in photosynthetic reaction centers from Rhodobacter sphaeroides: dependence on protonation of Glu-L212 and Asp-L213. Biochemistry. 2003;42:3882–3892. doi: 10.1021/bi026781t. [DOI] [PubMed] [Google Scholar]
- 169.Gunner MR, Zhu Z. Protons forge new paths. Structure (Camb) 2004;12:518–519. doi: 10.1016/j.str.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 170.Hienerwadel R, Grzybek S, Fogel C, Kreutz W, Okamura MY, Paddock ML, Breton J. Protonation of Glu L212 following QB-formation in the photosynthetic reaction center of Rhodobacter sphaeroides: Evidence from time-resolved infrared spectroscopy. Biochemistry. 1995;34:2832–2843. doi: 10.1021/bi00009a013. [DOI] [PubMed] [Google Scholar]
- 171.Nabedryk E, Breton J, Okamura MY, Paddock ML. Proton uptake by carboxylic acid groups upon photoreduction of the secondary quinone (QB) in bacterial reaction centers from Rhodobacter sphaeroides: FTIR studies on the effects of replacing GLU H173. Biochemistry. 1998;37:14457–14462. doi: 10.1021/bi981139d. [DOI] [PubMed] [Google Scholar]
- 172.Edgcomb SP, Murphy KP. Variability in the pKa of histidine side-chains correlates with burial within proteins. Proteins: Struct Funct Genet. 2002;49:1–6. doi: 10.1002/prot.10177. [DOI] [PubMed] [Google Scholar]
- 173.Forsyth WR, Antosiewiez JM, Robertson AD. Empirical relationships between protein structure and carboxyl pKa values in proteins. Proteins: Struct Funct Genet. 2002;48:388–403. doi: 10.1002/prot.10174. [DOI] [PubMed] [Google Scholar]
- 174.Shortle D, Stites WE, Meeker AK. Contributions of the large hydrophobic amino acids to the stability of staphylococcal nuclease. Biochemistry. 1990;29:8033–8041. doi: 10.1021/bi00487a007. [DOI] [PubMed] [Google Scholar]
- 175.Dwyer JJ, Gittis AG, Karp DA, Lattman EE, Spencer DS, Stites WE, Garcia-Moreno B. High apparent dielectric constants in the interior of a protein reflect water penetration. Biophys J. 2000;79:1610–1620. doi: 10.1016/S0006-3495(00)76411-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Bolen DW, Santoro MM. Unfolding free energy changes determined by the linear extrapolation method. 2. Incorporation of ΔG°N-U values in a thermodynamic cycle. Biochemistry. 1988;27:8069–8074. doi: 10.1021/bi00421a015. [DOI] [PubMed] [Google Scholar]
- 177.Fahmy K, Weidlich O, Engelhard M, Sigrist H, Siebert F. Aspartic acid-212 of bacteriorhodopsin is ionized in the M and N photocycle intermediates: an FTIR study on specifically 13C-labeled reconstituted purple membranes. Biochemistry. 1993;32:5862–5869. doi: 10.1021/bi00073a020. [DOI] [PubMed] [Google Scholar]
- 178.Rich PR, Meunier B, Mitchell R, Moody AJ. Coupling of charge and proton movement in cytochrome c oxidase. Biochim Biophys Acta. 1996;1275:91–95. [Google Scholar]
- 179.Gennis RB. Some recent contributions of FTIR difference spectroscopy to the study of cytochrome oxidase. FEBS Lett. 2003;555:2–7. doi: 10.1016/s0014-5793(03)01150-5. [DOI] [PubMed] [Google Scholar]
- 180.Alexov E, Miksovska J, Baciou L, Schiffer M, Hanson DK, Sebban P, Gunner MR. Modeling the effects of mutations on the free energy of the first electron transfer from to QB in photosynthetic reaction centers. Biochemistry. 2000;39:5940–5952. doi: 10.1021/bi9929498. [DOI] [PubMed] [Google Scholar]
- 181.Voigt P, Knapp EW. Tuning heme redox potentials in the cytochrome c subunit of photosynthetic reaction centers. J Biol Chem. 2003;278:51993–52001. doi: 10.1074/jbc.M307560200. [DOI] [PubMed] [Google Scholar]
- 182.Gunner MR, Honig B. Electrostatic control of midpoint potentials in the cytochrome subunit of the Rhodopseudomonas viridis reaction center. Proc Natl Acad Sci U S A. 1991;88:9151–9155. doi: 10.1073/pnas.88.20.9151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Oliveira AS, Teixeira VH, Baptista AM, Soares CM. Reorganization and conformational changes in the reduction of tetraheme cytochromes. Biophys J. 2005;89:3919–3930. doi: 10.1529/biophysj.105.065144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Teixeira VH, Soares CM, Baptista AM. Studies of the reduction and protonation behavior of tetraheme cytochromes using atomic detail. J Biol Inorg Chem. 2002;7:200–216. doi: 10.1007/s007750100287. [DOI] [PubMed] [Google Scholar]
- 185.Martel PJ, Soares CM, Baptista AM, Fuxreiter M, Naray-Szabo G, Louro RO, Carrondo MA. Comparative redox and pKa calculations on cytochrome c3 from several Desulfovibrio species using continuum electrostatic methods. J Biol Inorg Chem. 1999;4:73–86. doi: 10.1007/s007750050291. [DOI] [PubMed] [Google Scholar]
- 186.Ishikita H, Loll B, Biesiadka J, Saenger W, Knapp EW. Redox potentials of chlorophylls in photosystem II reaction center. Biochemistry. 2005;44:4118–4124. doi: 10.1021/bi047922p. [DOI] [PubMed] [Google Scholar]
- 187.Langen R, Jensen GM, Jacob U, Stephens PJ, Warshel A. Protein control of iron–sulfur cluster redox potentials. J Biol Chem. 1992;267:25625–25627. [PubMed] [Google Scholar]
- 188.Gray HB, Malmstrom BG, Williams RJ. Copper coordination in blue proteins. J Biol Inorg Chem. 2000;5:551–559. doi: 10.1007/s007750000146. [DOI] [PubMed] [Google Scholar]
- 189.Li H, Webb SP, Ivanic J, Jensen JH. Determinants of the relative reduction potentials of type-1 copper sites in proteins. J Am Chem Soc. 2004;126:8010–8019. doi: 10.1021/ja049345y. [DOI] [PubMed] [Google Scholar]
- 190.Antosiewicz J, McCammon JA, Gilson MK. Prediction of pH-dependent properties in proteins. J Mol Biol. 1994;238:415–436. doi: 10.1006/jmbi.1994.1301. [DOI] [PubMed] [Google Scholar]
- 191.Sham YY, Chu ZT, Warshel A. Consistent calculations of pKa's of ionizable residues in proteins: Semi-microscopic and microscopic approaches. J Phys Chem. 1997;101:4458–4472. [Google Scholar]
- 192.Sandberg L, Edholm O. A fast and simple method to calculate protonation states in proteins. Proteins: Struct Funct Genet. 1999;36:474–483. doi: 10.1002/(sici)1097-0134(19990901)36:4<474::aid-prot12>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 193.Mehler EL, Guarnieri F. A self-consistent, microenvironment modulated screened coulomb potential approximation to calculate pH-dependent electrostatic effects in proteins. Biophys J. 1999;77:3–22. doi: 10.1016/S0006-3495(99)76868-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Nielsen JE, Vriend G. Optimizing the hydrogen-bond network in Poisson–Boltzmann equation-based pKa calculations. Proteins: Struct Funct Genet. 2001;43:403–412. doi: 10.1002/prot.1053. [DOI] [PubMed] [Google Scholar]
- 195.Burgi R, Kollman PA, Gunsteren WFV. Simulating proteins at constant pH: An approach combining molecular dynamics and Monte Carlo simulation. Proteins. 2002;47:469–480. doi: 10.1002/prot.10046. [DOI] [PubMed] [Google Scholar]
- 196.Lee MS, Salsbury FR, Brooks CL., III Constant-pH molecular dynamics using continuous titration coordinates. Proteins: Struct Funct Genet. 2004;56:738–752. doi: 10.1002/prot.20128. [DOI] [PubMed] [Google Scholar]
- 197.Li H, Robertson AD, Jensen JH. The determinants of carboxyl pKa values in turkey ovomucoid third domain. Proteins: Struct Funct Genet. 2004;55:689–704. doi: 10.1002/prot.20032. [DOI] [PubMed] [Google Scholar]
- 198.Li H, Robertson AD, Jensen JH. Very fast empirical prediction and rationalization of protein pKa values. Proteins. 2005;61:704–721. doi: 10.1002/prot.20660. [DOI] [PubMed] [Google Scholar]
- 199.Mao J, Song Y, Gunner MR. Better rotamer packing for continuum electrostatics pKa calculations: MCCE2. J Comp Chem. in preperation. [Google Scholar]
- 200.Lee CF, Makhatadze GI, Wong KB. Effects of charge-to-alanine substitutions on the stability of ribosomal protein l30e from Thermococcus celer. Biochemistry. 2005;44:16817–16825. doi: 10.1021/bi0519654. [DOI] [PubMed] [Google Scholar]
- 201.Moult J. A decade of CASP: progress, bottlenecks and prognosis in protein structure prediction. Curr Opin Struct Biol. 2005;15:285–289. doi: 10.1016/j.sbi.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 202.Mendex R, Leplae R, Lensink MF, Wodak SJ. Assessment of CAPRI predictions in rounds 3–5 shows progress in docking procedures. Proteins. 2005;60:150–169. doi: 10.1002/prot.20551. [DOI] [PubMed] [Google Scholar]
- 203.Mehler EL, Fuxreiter M, Simon I, Garcia-Moreno B. The role of hydrophobic microenvironments in modulating pKa shifts in proteins. Proteins: Struct Funct Genet. 2002;48:283–292. doi: 10.1002/prot.10153. [DOI] [PubMed] [Google Scholar]
- 204.Radzicka A, Wolfenden R. Comparing the polarities of the amino acids: Side chain distribution coefficients between the vapor phase cyclohexane, 1-octonal, and neutral aqueous solution. Biochemistry. 1988;27:1664–1670. [Google Scholar]
- 205.Godoy-Ruiz R, Perez-Jimenez R, Garcia-Mira MM, Plaza del Pino IM, SR JM. Empirical parametrization of pK values for carboxylic acids in proteins using a genetic algorithm. Biophys Chem. 2005;115:263–266. doi: 10.1016/j.bpc.2004.12.028. [DOI] [PubMed] [Google Scholar]
- 206.Wisz MS, Hellinga HW. An empirical model for electrostatic interactions in proteins incorporating multiple geometry-dependent dielectric constants. Proteins. 2003;51:360–377. doi: 10.1002/prot.10332. [DOI] [PubMed] [Google Scholar]
- 207.Bashford D, Karplus M. The pKas of ionizable groups in proteins: atomic detail from a continuum electrostatic model. Biochemistry. 1990;29:10219–10225. doi: 10.1021/bi00496a010. [DOI] [PubMed] [Google Scholar]
- 208.Antosiewicz J, McCammon JA, Gilson MK. The determinants of pKas in proteins. Biochemistry. 1996;35:7819–7833. doi: 10.1021/bi9601565. [DOI] [PubMed] [Google Scholar]
- 209.You TJ, Bashford D. Conformation and hydrogen ion titration of proteins: a continuum electrostatic model with conformational flexibility. Biophys J. 1995;69:1721–1733. doi: 10.1016/S0006-3495(95)80042-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Bashford D, Gerwert K. Electrostatic calculations of the pKa values of ionizable groups in bacteriorhodopsin. J Mol Biol. 1992;224:473–486. doi: 10.1016/0022-2836(92)91009-e. [DOI] [PubMed] [Google Scholar]
- 211.Bashford D, Karplus M. Multiple-site titration curves of proteins: An analysis of exact and approximate methods for their calculation. J Phys Chem. 1991;95:9556–9561. [Google Scholar]
- 212.Antosiewicz J, McCammon JA, Gilson MK. Prediction of pH-dependent properties of proteins. J Mol Biol. 1994;238:415–436. doi: 10.1006/jmbi.1994.1301. [DOI] [PubMed] [Google Scholar]
- 213.Alexov EG, Gunner MR. Incorporating protein conformational flexibility into the calculation of pH-dependent protein properties. Biophys J. 1997;72:2075–2093. doi: 10.1016/S0006-3495(97)78851-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Spassov VZ, Luecke H, Gerwert K, Bashford D. pKa calculations suggest storage of an excess proton in a hydrogen- bonded water network in bacteriorhodopsin. J Mol Biol. 2001;312:203–219. doi: 10.1006/jmbi.2001.4902. [DOI] [PubMed] [Google Scholar]
- 215.Onufriev A, Smondyrev A, Bashford D. Proton affinity changes driving unidirectional proton transport in the bacteriorhodopsin photocycle. J Mol Biol. 2003;332:1183–1193. doi: 10.1016/s0022-2836(03)00903-3. [DOI] [PubMed] [Google Scholar]
- 216.Ishikita H, Knapp EW. Variation of Ser-L223 hydrogen bonding with the QB redox state in reaction centers from Rhodobacter sphaeroides. J Am Chem Soc. 2004;126:8059–8064. doi: 10.1021/ja038092q. [DOI] [PubMed] [Google Scholar]
- 217.Archontis G, Simonson T. Proton binding to proteins: a free-energy component analysis using a dielectric continuum model. Biophys J. 2005;88:3888–3904. doi: 10.1529/biophysj.104.055996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Demchuk E, Wade RC. Improving the continuum dielectric approach to calculating pKas of ionizable groups in protein. J Phys Chem. 1996;100:17373–17387. [Google Scholar]
- 219.Krishtalik LI, Kuznetsov AM, Mertz EL. Electrostatics for proteins: description in terms of two dielectric constants simultaneously. Proteins: Struct Funct Genet. 1997;28:174–182. [PubMed] [Google Scholar]
- 220.Voges D, Karshikoff A. A model of a local dielectric constant in proteins. J Chem Phys. 1998;108:2219–2227. [Google Scholar]
- 221.Rocchia W, Alexov E, Honig B. Extending the applicability of the nonlinear Poisson–Boltzmann Equation: multiple dielectric constants and multivalent ions. J Phys Chem B. 2001;105:6507–6514. [Google Scholar]
- 222.Teixeira VH, Cunha CA, Machuqueiro M, Oliveira ASF, Victor BL, Soares CM, Baptista AM. On the use of different dielectric constants for computing individual and pairwise terms in Poisson–Boltzmann studies of protein ionization equilibrium. J Phys Chem B. 2005:14691–14706. doi: 10.1021/jp052259f. [DOI] [PubMed] [Google Scholar]
- 223.Ripoll DR, Vorobjev YN, Liwo A, Vila JA, Scheraga HA. Coupling between folding and ionization equilibria: Effects of pH on the conformational preferences of polypeptides. J Mol Biol. 1996;264:770–783. doi: 10.1006/jmbi.1996.0676. [DOI] [PubMed] [Google Scholar]
- 224.Van Vlijmen HWT, Schaefer M, Karplus M. Improving the accuracy of protein pKa calculations: conformational averaging versus the average structure. Proteins: Struct Funct Genet. 1998;33:145–158. doi: 10.1002/(sici)1097-0134(19981101)33:2<145::aid-prot1>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 225.Koumanov A, Karshikof A, Friis EP, Borchert TV. Conformational averaging in pKa calculations: improvement and limitations in prediction of ionization properties of proteins. J Phys Chem B. 1999;103:9339–9344. [Google Scholar]
- 226.Gorfe AA, Ferrara P, Caflisch A, Marti DN, Bosshard HR, Jelesarov I. Calculation of protein ionization equilibria with conformational sampling: pKa of a model leucine zipper, GCN4 and barnase. Proteins: Struct Funct Genet. 2002;46:41–60. doi: 10.1002/prot.10027. [DOI] [PubMed] [Google Scholar]
- 227.Beroza P, Case D. Including side chain flexibility in continuum electrostatic calculations of protein titration. J Phys Chem. 1996;100:20156–20163. [Google Scholar]
- 228.Graige MS, Paddock ML, Bruce JM, Feher G, Okamura MY. Mechanism of proton-coupled electron transfer for quinone (QB) reduction in reaction centers of Rb. Sphaeroides. J Am Chem Soc. 1996;118:9005–9016. [Google Scholar]
- 229.Lee FS, Chu ZT, Warshel A. Microscopic and semimicroscopic calculations of electrostatic energies in proteins by the POLARIS and ENZYMIX programs. J Comput Chem. 1993;14:161–185. [Google Scholar]
- 230.Sham YY, Chu ZT, Tao H, Warshel A. Examining methods for calculations of binding free energies: LRA, LIE, PDLD-LRA, and PDLD/S-LRA calculations of ligands binding to an HIV protease. Proteins: Struct Funct Genet. 2000;39:393–407. [PubMed] [Google Scholar]
- 231.Sham YY, Muegge I, Warshel A. The effect of protein relaxation on charge–charge interactions and dielectric constant of proteins. Biophys J. 1998;74:1744–1753. doi: 10.1016/S0006-3495(98)77885-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.McCammon JA, Karplus M. Molecular dynamics simulations of biomolecules. Nat Struct Biol. 2002;9:646–652. doi: 10.1038/nsb0902-646. [DOI] [PubMed] [Google Scholar]
- 233.Lazaridis T, Karplus M. Effective energy functions for protein structure prediction. Cur Opin Struct Biol. 2000;10:139–145. doi: 10.1016/s0959-440x(00)00063-4. [DOI] [PubMed] [Google Scholar]
- 234.Luo R, David L, Gilson MK. Accelerated Poisson–Boltzmann calculations for static and dynamic systems. J Comput Chem. 2002;23:1244–1253. doi: 10.1002/jcc.10120. [DOI] [PubMed] [Google Scholar]
- 235.Hofinger S. Solving the Poisson–Boltzmann equation with the specialized computer chip MD-GRAPE-2. J Comput Chem. 2005;26 doi: 10.1002/jcc.20250. [DOI] [PubMed] [Google Scholar]
- 236.Lwin TZ, Zhou R, Luo R. Is Poisson–Boltzmann theory insufficient for protein folding simulations? J Chem Phys. 2006;124:034902. doi: 10.1063/1.2161202. [DOI] [PubMed] [Google Scholar]
- 237.Kuhn B, Kollman PA, Stahl M. Prediction of pKa shifts in proteins using a combination of molecular mechanical and continuum solvent calculations. J Comput Chem. 2004;25:1865–1872. doi: 10.1002/jcc.20111. [DOI] [PubMed] [Google Scholar]
- 238.Varma S, Chiu SW, Jakobsson E. The influence of amino acid protonation states on molecular dynamics simulations of the bacterial porin OmpF. Biophys J. 2006;90:112–123. doi: 10.1529/biophysj.105.059329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Wlodek ST, Antosiewicz J, McCammon JA. Prediction of titration properties of structures of a protein derived from molecular dynamics trajectories. Protein Sci. 1997;6:373–382. doi: 10.1002/pro.5560060213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Baptista AM, Martel PJ, Peterson SB. Simulation of protein conformational freedom as a function of pH: constant-pH molecular dynamics using implicit titration. Proteins: Struct Funct Genet. 1997;27:523–544. [PubMed] [Google Scholar]
- 241.Tanford C, Roxby R. Interpretation of protein titration curves. Application to lysozyme. Biochemistry. 1972;11:2192. doi: 10.1021/bi00761a029. [DOI] [PubMed] [Google Scholar]
- 242.Baptista AM, Teixeira VH, Soares CM. Constant-pH molecular dynamics using stocastic titration. J Chem Phys. 2002;117:4184–4200. [Google Scholar]
- 243.Dlugosz M, Antosiewicz JM, Robertson AD. Constant-pH molecular dynamics of protonation–structure relationship in a hexapeptide derived from ovomucoid third domain. Phys Rev E. 2004;69:021915. doi: 10.1103/PhysRevE.69.021915. [DOI] [PubMed] [Google Scholar]
- 244.Dlugosz M, Antosiewicz JM. Constant-pH molecular dynamics simulations: a test case for succinic acid. Chem Phys. 2004;302:161–170. [Google Scholar]
- 245.Luo R, Head MS, Moult J, Gilson MK. pKa shifts in small molecules and HIV protease: Electrostatics and conformation. J Am Chem Soc. 1998;120:6138–6146. [Google Scholar]
- 246.Eberini I, Baptista AM, Gianazza E, Fraternali F, Beringhelli T. Reorganization in apo- and holo-β-lactoglobulin upon protonation of Glu89: molecular dynamics and pKa calculations. Proteins: Struct Funct Genet. 2004;54:744–758. doi: 10.1002/prot.10643. [DOI] [PubMed] [Google Scholar]
- 247.Borjesson U, Hunenberger PH. pH-dependent stability of a decalysine a-helix studied by explicit-solvent molecular dynamics simulations at constant pH. J Phys Chem B. 2004;104:13551–13559. [Google Scholar]
- 248.Borjesson U, Hunenbergera PH. Explicit-solvent molecular dynamics simulation at constant pH: Methodology and application to small amines. J Chem Phys. 2001;114:9706–9719. [Google Scholar]
- 249.Shurki A, Warshel A. Structure/function correlations of proteins using MM, QM/MM, and related approaches: methods, concepts, pitfalls, and current progress. Adv Prot Chem. 2003;66:249–313. doi: 10.1016/s0065-3233(03)66007-9. [DOI] [PubMed] [Google Scholar]
- 250.Friesner RA, Guallar V. Ab initio quantum chemical and mixed quantum mechanics/molecular mechanics (QM/MM) methods for studying enzymatic catalysis. Annu Rev Phys Chem. 2005;56:389–427. doi: 10.1146/annurev.physchem.55.091602.094410. [DOI] [PubMed] [Google Scholar]
- 251.Li G, Cui Q. pKa calculations with QM/MM free energy perturbations. J Phys Chem B. 2003;107:14521–14528. [Google Scholar]
- 252.Hudaky P, Perczel A. Conformation dependence of pKa: Ab initio and DFT investigation of Histidine. J Phys Chem A. 2004;108:6195–6205. [Google Scholar]
- 253.Dixon RW, Kollman PA. Advancing beyond the atom-centered model in additive and nonadditive molecular mechanics. J Comput Chem. 1997;18:1632–1646. [Google Scholar]
- 254.Levitt M, Perutz MF. Aromatic rings act as hydrogen bond acceptors. J Mol Biol. 1988;201:751–754. doi: 10.1016/0022-2836(88)90471-8. [DOI] [PubMed] [Google Scholar]
- 255.Meyer EA, Castellano RK, Diederich F. Interactions with aromatic rings in chemical and biological recognition. Angew Chem Int Ed Engl. 2003;42:1210–1250. doi: 10.1002/anie.200390319. [DOI] [PubMed] [Google Scholar]
- 256.Mongan J, Case DA, McCammon JA. Constant pH molecular dynamics in generalized Born implicit solvent. J Comput Chem. 2004;25:2038–2048. doi: 10.1002/jcc.20139. [DOI] [PubMed] [Google Scholar]
- 257.Schaefer P, Riccardi D, Cui Q. Reliable treatment of electrostatics in combined QM/MM simulation of macromolecules. J Chem Phys. 2005;123:14905. doi: 10.1063/1.1940047. [DOI] [PubMed] [Google Scholar]
- 258.Vreven T, Byun KS, Komaromi I, Dapprich S, Montgomery JA, Morokuma K, Firsch MJ. Combining quantum mechanics methods with molecular mechanics methods in ONIOM. J Chem Theory Comput. 2006;2:815–826. doi: 10.1021/ct050289g. [DOI] [PubMed] [Google Scholar]
- 259.Blomberg MR, Siegbahn PE, Wikström M. Metal-bridging mechanism for O–O bond cleavage in cytochrome c oxidase. Inorg Chem. 2003;42:5231–5243. doi: 10.1021/ic034060s. [DOI] [PubMed] [Google Scholar]
- 260.Blomberg MRA, Siegbahn PEM. A quantum chemical study of tyrosyl reduction and O–O bond formation in photosystem II. Mol Phys. 2003;101:323–333. [Google Scholar]
- 261.Blomberg M, Siegbahn PEM, Babcock G, Wikström M. O–O bond splitting mechanism in cytochrome oxidase. J Inorg Biochem. 2000;80:261–269. doi: 10.1016/s0162-0134(00)00080-5. [DOI] [PubMed] [Google Scholar]
- 262.Wimley WC, Gawrisch K, Creamer TP, White SH. Direct measurement of salt-bridge solvation energies using a peptide model system: implications for protein stability. Proc Natl Acad Sci U S A. 1996;93:2985–2990. doi: 10.1073/pnas.93.7.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Sharp KA. In: Computational Approaches to Enzymatic Reactivity. G NSa, Warshel A., editors. Kluwer Academic; 1995. [Google Scholar]
- 264.Gunner MR, Alexov E, Torres E, Lipovaca S. The importance of the protein in controlling the electrochemistry of heme metalloproteins: methods of calculation and analysis. J Biol Inorg Chem. 1997;2:126–134. [Google Scholar]
- 265.Anderson DE, Becktel WJ, Dahlquist FW. pH-induced denaturation of proteins: A single salt bridge contributes 3–5 kcal/mol to the free energy of folding of T-4 lysozyme. Biochemistry. 1990;29:2403–2408. doi: 10.1021/bi00461a025. [DOI] [PubMed] [Google Scholar]
- 266.Spassov VZ, Atanasov BP. Spatial optimization of electrostatic interactions between the ionized groups in globular proteins. Proteins: Struct Funct Genet. 1994;19:222–229. doi: 10.1002/prot.340190306. [DOI] [PubMed] [Google Scholar]
- 267.Waldburger CD, Schildbach JF, Sauer RT. Are buried salt-bridges important for protein stability and conformational specificity? Nat Struct Biol. 1995;2:122–128. doi: 10.1038/nsb0295-122. [DOI] [PubMed] [Google Scholar]
- 268.Kumar S, Nussinov R. Salt bridge stability in monomeric proteins. J Mol Biol. 1999;293:1241–1255. doi: 10.1006/jmbi.1999.3218. [DOI] [PubMed] [Google Scholar]
- 269.Novotny J, Sharp K. Electrostatic fields in antibodies and antibody/ antigen complexes. Prog Biophys Mol Biol. 1992;58:203–224. doi: 10.1016/0079-6107(92)90006-r. [DOI] [PubMed] [Google Scholar]
- 270.Hendsch Z, Tidor B. Do salt bridges stabilize proteins? A continuum electrostatic analysis. Protein Sci. 1994;3:211–226. doi: 10.1002/pro.5560030206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Elcock AH, McCammon JA. Electrostatic contributions to the stability of halophilic proteins. J Mol Biol. 1998;280:731–748. doi: 10.1006/jmbi.1998.1904. [DOI] [PubMed] [Google Scholar]
- 272.Sheinerman FB, Norel R, Honig B. Electrostatic aspects of protein–protein interactions. Curr Opin Struct Biol. 2000;10:153–159. doi: 10.1016/s0959-440x(00)00065-8. [DOI] [PubMed] [Google Scholar]
- 273.Lee LP, Tidor B. Optimization of binding electrostatics: charge complementarity in the barnase–barstar protein complex. Protein Sci. 2001;10:362–377. doi: 10.1110/ps.40001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Giletto A, Pace CN. Buried, charged, non-ion-paired aspartic acid 76 contributes favorably to the conformational stability of ribonuclease T1. Biochemistry. 1999;38:13379–13384. doi: 10.1021/bi991422s. [DOI] [PubMed] [Google Scholar]
- 275.Karshikoff A, Ladenstein R. Ion pairs and the thermotolerance of proteins from hyperthermophiles: a ‘traffic rule’ for hot roads. TIBS. 2001;26:550–556. doi: 10.1016/s0968-0004(01)01918-1. [DOI] [PubMed] [Google Scholar]
- 276.Bartlett GJ, Porter CT, Borkakoti N, Thornton JM. Analysis of catalytic residues in enzyme active sites. J Mol Biol. 2002;324:105–211. doi: 10.1016/s0022-2836(02)01036-7. [DOI] [PubMed] [Google Scholar]
- 277.Rashin AA, Honig B. On the environment of ionizable groups in globular proteins. J Mol Biol. 1984;173:515–521. doi: 10.1016/0022-2836(84)90394-2. [DOI] [PubMed] [Google Scholar]
- 278.Barlow DJ, Thornton JM. The distribution of charged groups in proteins. Biopolymers. 1986;25:1717–1733. doi: 10.1002/bip.360250913. [DOI] [PubMed] [Google Scholar]
- 279.Kajander T, Kahn PC, Passila SH, Cohen DC, Lehtio L, Adolfsen W, Warwicker J, Schell U, Goldman A. Buried charged surface in proteins. Structure Fold Des. 2000;8:1203–1214. doi: 10.1016/s0969-2126(00)00520-7. [DOI] [PubMed] [Google Scholar]
- 280.Richards FM. Areas, volumes, packing, and protein structure. Ann Rev Biophys Bioeng. 1977;6:151–176. doi: 10.1146/annurev.bb.06.060177.001055. [DOI] [PubMed] [Google Scholar]
- 281.Richmond TJ. Solvent accessible surface area and excluded volume in proteins. Analytical equations for overlapping spheres and implications for the hydrophobic effect. J Mol Biol. 1984;178:63–89. doi: 10.1016/0022-2836(84)90231-6. [DOI] [PubMed] [Google Scholar]
- 282.Richmond TJ, Richards FM. Packing of a-helices: geometrical constraints and contact areas. J Mol Biol. 1978;119:537–555. doi: 10.1016/0022-2836(78)90201-2. [DOI] [PubMed] [Google Scholar]
- 283.Wallace CJA, Clark-Lewis I. Functional role of heme ligation in cytochrome c: effects of replacement of methionine 80 with natural and non-natural residues by semisynthesis. J Biol Chem. 1992;1992:3852–3861. [PubMed] [Google Scholar]
- 284.Wallace CJA, Clark-Lewis I. Functional role of heme ligation in cytochrome c. J Biol Chem. 1992;267:3852–3861. [PubMed] [Google Scholar]
- 285.Poulos TL. Heme enzyme crystal structure, Heme Proteins. Struct Funct Genet. 1988;7:2–36. [Google Scholar]
- 286.Rietjens I. The role of the axial ligand in heme-based catalysis. J Biol Inorg Chem. 1996;1:355. [Google Scholar]
- 287.Galstyan AS, Zaric SD, Knapp EW. Computational studies on imidazole heme conformations. J Biol Inorg Chem. 2005;10:343–354. doi: 10.1007/s00775-005-0642-8. [DOI] [PubMed] [Google Scholar]
- 288.Stellwagen E. Haem exposure as the determinate of oxidation–reduction potential of haem proteins. Nature. 1978;275:73–74. doi: 10.1038/275073a0. [DOI] [PubMed] [Google Scholar]
- 289.Martin ACR, Orengo CA, Hutchinson EG, Jones S, Karmir-antzou M, Laskowski RA, Mitchell JBO, Taroni C, Thornton JM. Protein folds and function. Structure. 1998;6:875–884. doi: 10.1016/s0969-2126(98)00089-6. [DOI] [PubMed] [Google Scholar]
- 290.Muegge I, Qi PX, Wand AJW, Chu ZT, Warshel A. The reorganization energy of cytochrome c revisited. J Phys Chem. 1997;101:825–836. [Google Scholar]
- 291.Raphael AL, Gray HB. Axial ligand replacement in horse heart cytochrome c by semisynthesis. Proteins: Struct Funct Genet. 1989;6:338–340. doi: 10.1002/prot.340060316. [DOI] [PubMed] [Google Scholar]
- 292.Churg AK, Weiss RM, Warshel A, Takano T. On the action of cytochrome c: correlating geometry changes upon oxidation with activation energies of electron transfer. J Phys Chem. 1983;87:1683–1694. [Google Scholar]
- 293.Cutler RL, Davies AM, Creighton S, Warshel A, Moore GR, Smith M, Mauk AG. Role of arginine-38 in regulation of the cytochrome c oxidation–reduction equilibrium. Biochemistry. 1989;28:3188–3197. doi: 10.1021/bi00434a012. [DOI] [PubMed] [Google Scholar]
- 294.Warwicker J, Watson HC. Calculation of the electric potential in the active site cleft due to a a-helix dipoles. J Mol Biol. 1982;157:671–679. doi: 10.1016/0022-2836(82)90505-8. [DOI] [PubMed] [Google Scholar]
- 295.Zhou HX. Control of reduction potential by protein matrix: lesson from a spherical protein model. J Biol Inorg Chem. 1997;2:109–113. [Google Scholar]
- 296.Warshel A, Papazyan A, Muegge I. Microscopic and semimacroscopic redox calculations: what can and can not be learned from continuum models. J Biol Inorg Chem. 1997;2:143–152. [Google Scholar]
- 297.Soares CM, Martel PJ, Mendes J, Carrondo MA. Molecular dynamics simulation of cytochrome c3: studying the reduction processes using free energy calculations. Biophys J. 1998;74:1708–1721. doi: 10.1016/S0006-3495(98)77882-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Baptista A, Martel PJ, Soares CM. Simulation of electron-proton coupling with a Monte Carlo method: application to cytochrome c3 using continuum electrostatics. Biophys J. 1999;76:2978–2998. doi: 10.1016/S0006-3495(99)77452-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Zaric SD, Popovic DM, Knapp EW. Factors determining the orientation of axially coordinated imidazoles in heme proteins. Biochemistry. 2001;40:7914–7928. doi: 10.1021/bi010428q. [DOI] [PubMed] [Google Scholar]
- 300.Kannt A, Lancaster CRD, Michel H. The coupling of electron transfer and proton translocation: electrostatic calculations on Paracoccus denitrificans cytochrome c oxidase. Biophys J. 1998;74:708–721. doi: 10.1016/S0006-3495(98)73996-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Popovic DM, Stuchebrukhov AA. Electrostatic study of the proton pumping mechanism in bovine heart cytochrome c oxidase. J Am Chem Soc. 2004;126:1858–1871. doi: 10.1021/ja038267w. [DOI] [PubMed] [Google Scholar]
- 302.Siegbahn PE. The catalytic cycle of tyrosinase: peroxide attack on the phenolate ring followed by O–O cleavage. J Biol Inorg Chem. 2003;8:567–576. doi: 10.1007/s00775-003-0449-4. [DOI] [PubMed] [Google Scholar]
- 303.Green MT. Role of the axial ligand in determining the spin state of resting cytochrome P450. J Am Chem Soc. 1998;120:10772–10773. [Google Scholar]
- 304.Guallar V, Friesner RA. Cytochrome P450CAM enzymatic catalysis cycle: a quantum mechanics/molecular mechanics study. J Am Chem Soc. 2004;126:8501–8508. doi: 10.1021/ja036123b. [DOI] [PubMed] [Google Scholar]
- 305.Schoneboom JC, Thiel W. The resting state of P450cam: A QM/MM study. J Phys Chem B. 2004;108:7468–7478. [Google Scholar]
- 306.Ogliaro F, Harris N, Cohen S, Filatov M, deVisser DP, Shaik S. A model “rebound” mechanism of hydroxylation by cytochrome P450: Stepwise and effectively converted pathways and their reactivity patterns. J Am Chem Soc. 2000;122:8977–8989. [Google Scholar]
- 307.Kamachi T, Yoshizawa K. A theoretical study on the mechanism of camphor hydroxylation by compound I of cytochrome P450. J Am Chem Soc. 2003;125:4652–4661. doi: 10.1021/ja0208862. [DOI] [PubMed] [Google Scholar]
- 308.Guallar V, Baik MH, Lippard SJ, Friesner RA. Peripheral heme substituents control the hydrogen-atom abstraction chemistry in cytochromes P450. Proc Natl Acad Sci U S A. 2003;100:6998–7002. doi: 10.1073/pnas.0732000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Schoneboom JC, Cohen S, Lin H, Shaik S, Thiel W. Quantum mechanical/molecular mechanical investigation of the mechanism of C–H hydroxylation of camphor by cytochrome P450cam: theory supports a two-state rebound mechanism. J Am Chem Soc. 2004;126:4017–4034. doi: 10.1021/ja039847w. [DOI] [PubMed] [Google Scholar]
- 310.Kanicky J, Shah D. Effect of degree, type, and position of unsaturation on the pKa of long-chain fatty acids. J Colloid Interface Sci. 2002;256:201–207. doi: 10.1006/jcis.2001.8009. [DOI] [PubMed] [Google Scholar]
- 311.Blomberg MRA, Siegbahn PEM, Babcock GT, Wikström M. Modeling cytochrome oxidase: a quantum chemical study of the O–O bond cleavage mechanism. J Am Chem Soc. 2000;22:12848–12858. [Google Scholar]
- 312.Banci L, Bertini I, Branchini BR, Hajieva P, Spyroulias GA, Turano P. Dimethyl propionate ester heme-containing cytochrome b5: structure and stability. J Biol Inorg Chem. 2001;6:490–503. doi: 10.1007/s007750100217. [DOI] [PubMed] [Google Scholar]
- 313.Rogers NK, Moore GR, Sternberg MJE. Electrostatic interactions in globular proteins: Calculation of the pH dependence of the redox potential of cytochrome c551. J Mol Biol. 1985;182:613–616. doi: 10.1016/0022-2836(85)90248-7. [DOI] [PubMed] [Google Scholar]
- 314.Rogers NK, Moore GR. On the energetics of conformational changes and pH dependent redox behaviour of electron transfer proteins. FEBS Lett. 1988;228:69–73. doi: 10.1016/0014-5793(88)80587-8. [DOI] [PubMed] [Google Scholar]
- 315.Das DK, Medhi OK. The role of heme propionate in controlling the redox potential of heme: Square wave volammetry of protoporphyrinato IX iron (III) in aqueous surfactant micelles. J Inorg Biochem. 1998;70:83–90. doi: 10.1016/s0162-0134(98)10002-8. [DOI] [PubMed] [Google Scholar]
- 316.de Silva D, Sykes A. Interpretation of effects of pH on rate constants for the oxidation of three ferrocytochromes c-551 with [Fe (CN) 6] 3 - and [Co (phen) 3]3+, and assignment of pKa values. Biochim Biophys Acta. 1988;952:334–341. doi: 10.1016/0167-4838(88)90135-5. [DOI] [PubMed] [Google Scholar]
- 317.Tezcan FA, Winkler JR, Gray HB. Effects of ligation and folding on reduction potentials of heme proteins. J Am Chem Soc. 1998;120:13383–13388. [Google Scholar]
- 318.Rivera M, Seetharaman R, Girdhar D, Wirtz M, Zhang X, Wang X, White S. The reduction potential of cytochrome b5 is modulated by its exposed heme edge. Biochemistry. 1998;37:1485–1494. doi: 10.1021/bi972390g. [DOI] [PubMed] [Google Scholar]
- 319.Fantuzzi A, Sadeghi S, Valentti F, Rossi GL, Gilardi G. Tuning the reduction potential of engineered cytochrome c-553. Biochemistry. 2002;41:8718–8724. doi: 10.1021/bi025759x. [DOI] [PubMed] [Google Scholar]
- 320.Deisenhofer J, Epp O, Miki R, Michel H. Structure of the protein subunits in the photosynthetic reaction center of Rhodopseudomonas viridis at 3 Å resolution. Nature. 1985;318:618–624. doi: 10.1038/318618a0. [DOI] [PubMed] [Google Scholar]
- 321.Mancino LJ, Dean DP, Blankenship RE. Kinetics and thermodynamics of the reaction in isolated reaction centers from the photosynthetic bacterium Rhodopseudomonas sphaeroides. Biochim Biophys Acta. 1984;764:46–54. [Google Scholar]
- 322.Kleinfeld D, Okamura MY, Feher G. Electron transfer in reaction centers of Rhodopseudomonas sphaeroides: I. Determination of the charge recombination pathway of and free energy and kinetic relations between and . Biochim Biophys Acta. 1984;766:126–140. doi: 10.1016/0005-2728(84)90224-x. [DOI] [PubMed] [Google Scholar]
- 323.Xu Q, Gunner MR. Temperature dependence of the free energy, enthalpy and entropy of charge recombination in photosynthetic reaction centers. J Phys Chem B. 2000;104:8035–8043. [Google Scholar]
- 324.Xu Q, Gunner MR. Exploring the energy profile of the to QB electron transfer reaction in bacterial photosynthetic reaction centers: pH dependence of the conformational gating step. Biochemistry. 2002;41:2694–2701. doi: 10.1021/bi011834c. [DOI] [PubMed] [Google Scholar]
- 325.Gunner MR, Dutton PL. Temperature and −ΔG° dependence of the electron transfer from BPh− to QA in reaction center protein from Rhodobacter sphaeroides with different quinones as QA. J Am Chem Soc. 1989;111:3400–3412. [Google Scholar]
- 326.Gunner MR, Robertson DE, Dutton PL. Kinetic studies on the reaction center protein from Rhodopseudomonas sphaeroides: The temperature and free energy dependence of electron transfer between various quinones in the QA site and the oxidized bacteriochlorophyll dimer. J Phys Chem. 1986;90:3783–3795. [Google Scholar]
- 327.Moser CC, Keske JM, Warncke K, Farid R, Dutton PL. Nature of biological electron transfer. Nature. 1992;355:796–802. doi: 10.1038/355796a0. [DOI] [PubMed] [Google Scholar]
- 328.Wraight CA. Electron acceptors of bacterial photosynthetic reaction centers II. H+ binding coupled to secondary electron transfer in the quinone acceptor complex. Biochim Biophys Acta. 1979;548:309–327. doi: 10.1016/0005-2728(79)90138-5. [DOI] [PubMed] [Google Scholar]
- 329.Diner BA, Schenck CC, DeVitry C. Effect of inhibitors, redox state and isoprenoid chain length on the affinity of ubiquinone for the secondary acceptor binding site in the reaction centers of photosynthetic bacteria. Biochim Biophys Acta. 1984;766:9–20. [Google Scholar]
- 330.Okamura MY, Paddock ML, Graige MS, Feher G. Proton and electron transfer in bacterial reaction centers. Biochim Biophys Acta. 2000;1458:148–163. doi: 10.1016/s0005-2728(00)00065-7. [DOI] [PubMed] [Google Scholar]
- 331.Madeo J, Gunner MR. Modeling binding kinetics at the QA site in bacterial reaction centers. Biochemistry. 2005;44:10994–11004. doi: 10.1021/bi050544j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Lancaster CRD. Ubiquinone reduction and protonation in photosynthetic reaction centres from Rhodopseudomonas viridis: X-ray structures and their functional implications. Biochim Biophys Acta. 1998;1365:143–150. [Google Scholar]
- 333.Stowell MHB, McPhillips TM, Rees DC, Soltis SM, Abresch E, Feher G. Light-induced structural changes in photosynthetic reaction center: implications for mechanism of electron-proton transfer. Science. 1997;276:812–816. doi: 10.1126/science.276.5313.812. [DOI] [PubMed] [Google Scholar]
- 334.Remy A, Gerwert K. Coupling of light-induced electron transfer to proton uptake in photosynthesis. Nat Struct Biol. 2003;10:637–644. doi: 10.1038/nsb954. [DOI] [PubMed] [Google Scholar]
- 335.Breton J, Boullais C, Mioskowski C, Sebban P, Bacious L, Nabedryk E. Vibrational spectroscopy favors a unique QB binding site at the proximal position in wild-type reaction centers and in the Pro-L209> Tyr mutant from Rhodobacter sphaeroides. Biochemistry. 2002;41:12921–12927. doi: 10.1021/bi026565k. [DOI] [PubMed] [Google Scholar]
- 336.Xu Q, Baciou L, Sebban P, Gunner MR. Exploring the energy landscape for to QB electron transfer in bacterial photosynthetic reaction centers: Effect of substrate position and tail length on the conformational gating step. Biochemistry. 2002;41:10021–10025. doi: 10.1021/bi025573y. [DOI] [PubMed] [Google Scholar]
- 337.Breton J. Absence of large-scale displacement of quinone QB in bacterial photosynthetic reaction centers. Biochemistry. 2004;43:3318–3326. doi: 10.1021/bi049811w. [DOI] [PubMed] [Google Scholar]
- 338.Pokkuluri PR, Laible PD, Crawford AE, Mayfield JF, Yuosef MA, Ginell SL, Hanson DK, Schiffer M. Temperature and cryoprotectant influence secondary quinone binding position in bacterial reaction centers. Febs Lett. 2004;43:9909–9917. doi: 10.1016/j.febslet.2004.06.042. [DOI] [PubMed] [Google Scholar]
- 339.Zachariae U, Lancaster CR. Proton uptake associated with the reduction of the primary quinone QA influences the binding site of the secondary quinone QB in Rhodopseudomonas viridis photosynthetic reaction centers. Biochim Biophys Acta. 2001;1505:280–290. doi: 10.1016/s0005-2728(01)00174-8. [DOI] [PubMed] [Google Scholar]
- 340.Walden SE, Wheeler RA. Protein conformational gate controlling binding site preference and migration for ubiquinone-B in the photosynthetic reaction center of Rhodobacter sphaeroides. J Phys Chem B. 2002;106:3001–3006. [Google Scholar]
- 341.Grafton AK, Wheeler RA. Amino acid protonation states determine binding sites of the secondary ubiquinone and its anion in the Rhodobacter sphaeroides photosynthetic reaction center. J Phys Chem. 1999;103:5380–5387. [Google Scholar]
- 342.Lancaster CRD, Michel H, Honig B, Gunner MR. Calculated coupling of electron and proton transfer in the photosynthetic reaction center of Rhodopseudomonas viridis. Biophys J. 1996;70:2469–2492. doi: 10.1016/S0006-3495(96)79820-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Luecke H. Atomic resolution structures of bacteriorhodopsin photocycle intermediates: the role of discrete water molecules in the function of this light-driven ion pump. Biochim Biophys Acta. 2000;1460:133–156. doi: 10.1016/s0005-2728(00)00135-3. [DOI] [PubMed] [Google Scholar]
- 344.Balashov SP. Protonation reactions and their coupling in bacteriorhodopsin. Biochim Biophys Acta. 2000;1460:75–94. doi: 10.1016/s0005-2728(00)00131-6. [DOI] [PubMed] [Google Scholar]
- 345.Hirai T, Subramaniam S. Structural insights into the mechanism of proton pumping by bacteriorhodopsin. FEBS Lett. 2003;545:2–8. doi: 10.1016/s0014-5793(03)00386-7. [DOI] [PubMed] [Google Scholar]
- 346.Edmonds BW, Luecke H. Atomic resolution structures and the mechanism of ion pumping in bacteriorhodopsin. Front Biosci. 2004;9:1556–1566. doi: 10.2741/1264. [DOI] [PubMed] [Google Scholar]
- 347.Neutze R, Pebay-Peyroula E, Edman K, Royant A, Navarro J, Landau EM. Bacteriorhodopsin: a high-resolution structural view of vectorial proton transport. Biochim Biophys Acta. 2002;1565:144–167. doi: 10.1016/s0005-2736(02)00566-7. [DOI] [PubMed] [Google Scholar]
- 348.Lanyi JK, Schobert B. Local–global conformational coupling in a heptahelical membrane protein: transport mechanism from crystal structures of the nine states in the bacteriorhodopsin photocycle. Biochemistry. 2004;43:3–8. doi: 10.1021/bi035843s. [DOI] [PubMed] [Google Scholar]
- 349.Schobert B, Brown LS, Lanyi JK. Crystallographic structures of the M and N intermediates of bacteriorhodopsin: assembly of a hydrogen-bonded chain of water molecules between Asp-96 and the retinal Schiff base. J Mol Biol. 2003;330:553–570. doi: 10.1016/s0022-2836(03)00576-x. [DOI] [PubMed] [Google Scholar]
- 350.Scharnagl C, Fischer SF. Conformational flexibility of arginine-82 as source for heterogeneous and pH- dependent kinetics of the primary proton transfer step in the bacteriorhodopsin photocycle: An electrostatic model. Chem Phys. 1996;212:231–246. [Google Scholar]
- 351.Luecke H, Schobert B, Richter HT, Cartailler JP, Lanyi JK. Structural changes in bacteriorhodopsin during ion transport at 2 Ångstrom resolution. Science. 1999;286:255–261. doi: 10.1126/science.286.5438.255. [DOI] [PubMed] [Google Scholar]
- 352.Ormos P. Infrared spectroscopic demonstration of a conformational change in bacteriorhodopsin involved in proton pumping. Proc Natl Acad Sci U S A. 1991;88:473–477. doi: 10.1073/pnas.88.2.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Braiman MS, Dioumaev AK, Lewis JR. A large photolysis-induced pKa increase of the chromophore counterion in bacteriorhodopsin: implications for ion transport mechanisms of retinal proteins. Biophys J. 1996;70:939–947. doi: 10.1016/S0006-3495(96)79637-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Shibata M, Kandori H. FTIR studies of internal water molecules in the Schiff base region of bacteriorhodopsin. Biochemistry. 2005;44:7406–7413. doi: 10.1021/bi050122+. [DOI] [PubMed] [Google Scholar]
- 355.Kandori H, Yamazaki Y, Sasaki J. Water-mediated proton transfer in proteins: an FTIR study of bacteriorhodopsin. J Am Chem Soc. 1995;117:2118–2119. [Google Scholar]
- 356.Hatanaka M, Kandori H, Maeda A. Localization and orientation of functional water molecules in bacteriorhodopsin as revealed by polarized Fourier transform infrared spectroscopy. Biophys J. 1997;73:1001–1006. doi: 10.1016/S0006-3495(97)78133-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Tanimoto T, Furutani Y, Kandori H. Structural changes of water in the Schiff base region of bacteriorhodopsin: proposal of a hydration switch model. Biochemistry. 2003;42:2300–2306. doi: 10.1021/bi026990d. [DOI] [PubMed] [Google Scholar]
- 358.Kandori H. Hydration switch model for the proton transfer in the Schiff base region of bacteriorhodopsin. Biochim Biophys Acta. 2004;1658:72–79. doi: 10.1016/j.bbabio.2004.03.015. [DOI] [PubMed] [Google Scholar]
- 359.Hatanaka M, Sasaki J, Kandori H, Ebrey TG, Needleman R, Lanyi JK, Maeda A. Effects of arginine-82 on the interactions of internal water molecules in bacteriorhodopsin. Biochemistry. 1996;35:6308–6312. doi: 10.1021/bi952973v. [DOI] [PubMed] [Google Scholar]
- 360.Hutson MS, Alexiev U, Shilov SV, Wise KJ, Braiman MS. Evidence for a perturbation of arginine-82 in the bacteriorhodopsin photocycle from time-resolved infrared spectra. Biochemistry. 2000;39:13189–13200. doi: 10.1021/bi000426q. [DOI] [PubMed] [Google Scholar]
- 361.Tanimoto T, Shibata M, Belenky M, Herzfeld J, Kandori H. Altered hydrogen bonding of Arg82 during the proton pump cycle of bacteriorhodopsin: a low-temperature polarized FTIR spectroscopic study. Biochemistry. 2004;43:9439–9447. doi: 10.1021/bi049368p. [DOI] [PubMed] [Google Scholar]
- 362.Braiman MS, Mogi T, Stern LJ, Khorana HG, Rothschild KJ. Vibrational spectroscopy of bacteriorhodopsin mutants: light-driven proton transport involves protonation changes of aspartic acid residues 85, 96, and 212. Biochemistry. 1988;27:8516–8520. doi: 10.1021/bi00423a002. [DOI] [PubMed] [Google Scholar]
- 363.Gerwert K, Hess B, Soppa J, Oesterhelt D. Role of aspartate-96 in proton translocation by bacteriorhodopsin. Proc Natl Acad Sci U S A. 1989;86:4943–4947. doi: 10.1073/pnas.86.13.4943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Otto H, Marti T, Holz M, Mogi T, Lindau M, Khorana HG, Heyn MP. Aspartic, acid-96 is the internal proton donor in the reprotonation of the schiff base of bacteriorhodopsin. Proc Natl Acad Sci. 1989;86:9228–9232. doi: 10.1073/pnas.86.23.9228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Holz M, Drachev LA, Mogi T, Otto H, Kaulen AD, Heyn MP, Skulachev VP, Khorana HG. Replacement of aspartic acid-96 by asparagine in bacteriorhodopsin slows both the decay of the M intermediate and the associated proton movement. Proc Natl Acad Sci U S A. 1989;86:2167–2171. doi: 10.1073/pnas.86.7.2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Miller A, Oesterhelt D. Kinetic optimization of bacteriorhodopsin by aspartic acid-96 as an internal proton donor. Biochim Biophys Acta. 1990;1020:57–64. [Google Scholar]
- 367.Luecke H, Schobert B, Richter HT, Cartailler JP, Lanyi JK. Structure of bacteriorhodopsin at 1.55 A resolution. J Mol Biol. 1999;291:899–911. doi: 10.1006/jmbi.1999.3027. [DOI] [PubMed] [Google Scholar]
- 368.Belrhali H, Nollert P, Royant A, Menzel C, Rosenbusch JP, Landau EM, Pebay-Peyroula E. Protein, lipid and water organization in bacteriorhodopsin crystals: a molecular view of the purple membrane at 1.9 A resolution. Structure Fold Des. 1999;7:909–917. doi: 10.1016/s0969-2126(99)80118-x. [DOI] [PubMed] [Google Scholar]
- 369.Facciotti MT, Rouhani S, Burkard FT, Betancourt FM, Downing KH, Rose RB, McDermott G, Glaeser RM. Structure of an early intermediate in the M-state phase of the bacteriorhodopsin photocycle. Biophys J. 2001;81:3442–3455. doi: 10.1016/S0006-3495(01)75976-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Oesterhelt D, Stoeckenius W. Rhodopsin-like protein from the purple membrane of Halobacterium halobium. Nature. 1971;233:149–152. doi: 10.1038/newbio233149a0. [DOI] [PubMed] [Google Scholar]
- 371.Stoeckenius W, Lozier RH, Bogomolni RA. Bacteriorhodopsin and the purple membrane of halobacteria. Biochim Biophys Acta. 1979;505:215–278. doi: 10.1016/0304-4173(79)90006-5. [DOI] [PubMed] [Google Scholar]
- 372.Metz G, Siebert F, Engelhard M. Asp85 is the only internal aspartic acid that gets protonated in the M intermediate and the purple-to-blue transition of bacteriorhodopsin. A solid-state 13C CP-MAS NMR investigation. FEBS Lett. 1992;303:237–241. doi: 10.1016/0014-5793(92)80528-o. [DOI] [PubMed] [Google Scholar]
- 373.Otto H, Marti T, Holz M, Mogi T, Stern LJ. Substitution of amino acids asp-85, asp-212, and arg-82 in bacteriorhodospin affects the proton release phase of the pump and the pk of the schiff base. Proc Natl Acad Sci U S A. 1990;87:1018–1022. doi: 10.1073/pnas.87.3.1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Rothschild KJ, Braiman MS, He YW, Marti T, Khorana HG. Vibrational spectroscopy of bacteriorhodopsin mutants. Evidence for the interaction of aspartic acid 212 with tyrosine 185 and possible role in the proton pump mechanism. J Biol Chem. 1990;265:16985–16991. [PubMed] [Google Scholar]
- 375.Needleman R, Chang M, Ni B, Varo G, Fornes J, White SH, Lanyi JK. Properties of Asp212–Asn bacteriorhodopsin suggest that Asp212 and Asp85 both participate in a counterion and proton acceptor complex near the Schiff base. J Biol Chem. 1991;266:11478–11484. [PubMed] [Google Scholar]
- 376.Cao Y, Varo G, Klinger AL, Czajkowsky DM, Braiman MS, Needleman R, Lanyi JK. Proton transfer from Asp-96 to the bacteriorhodopsin Schiff base is caused by a decrease of the pKa of Asp-96 which follows a protein backbone conformational change. Biochemistry. 1993;32:1981–1990. doi: 10.1021/bi00059a015. [DOI] [PubMed] [Google Scholar]
- 377.Brown LS, Varo G, Hatanaka M, Sasaki J, Kandori H, Maeda A, Friedman N, Sheves M, Nedleman R, Lanyi JK. The complex extracellular domain regulates the deprotonation and reprotonation of the retinal Schiff base during the bacteriorhodopsin photocycle. Biochemistry. 1995;34:12903–12911. doi: 10.1021/bi00039a053. [DOI] [PubMed] [Google Scholar]
- 378.Marti T, Otto H, Mogi T, Rosselet SJ, Heyn MP, Khorana HG. Bacteriorhodopsin mutants containing single substitutions of serine or threonine residues are all active in proton translocation. J Biol Chem. 1991;266:6919–6927. [PubMed] [Google Scholar]
- 379.Russell TS, Coleman M, Rath P, Nilsson A, Rothschild KJ. Threonine-89 participates in the active site of bacteriorhodopsin: evidence for a role in color regulation and Schiff base proton transfer. Biochemistry. 1997;36:7490–7497. doi: 10.1021/bi970287l. [DOI] [PubMed] [Google Scholar]
- 380.Fischer WB, Sonar S, Marti T, Khorana HG, Rothschild KJ. Detection of a water molecule in the active-site of bacteriorhodopsin: hydrogen bonding changes during the primary photoreaction. Biochemistry. 1994;33:12757–12762. doi: 10.1021/bi00209a005. [DOI] [PubMed] [Google Scholar]
- 381.Govindjec R, Kono M, Balashov SP, Imasheva E, Sheves M, Ebrey TG. Effects of substitution of tyrosine 57 with asparagine and phenylalanine on the properties of bacteriorhodopsin. Biochemistry. 1995;34:4828–4838. doi: 10.1021/bi00014a040. [DOI] [PubMed] [Google Scholar]
- 382.Braiman MS, Mogi T, Stern LJ, Hackett NR, Chao BH, Khorana HG, Rothschild KJ. Vibrational spectroscopy of bacteriorhodopsin mutants: I. Tyrosine-185 protonates and deprotonates during the photocycle. Proteins. 1988;3:219–229. doi: 10.1002/prot.340030403. [DOI] [PubMed] [Google Scholar]
- 383.Ahl PL, Stern LJ, During D, Mogi T, Khorana HG, Rothschild KJ. Effects of amino acid substitutions in the F helix of bacteriorhodopsin. Low temperature ultraviolet/visible difference spectroscopy. J Biol Chem. 1988;263:13594–13601. [PubMed] [Google Scholar]
- 384.Ahl PL, Stern LJ, Mogi T, Khorana HG, Rothschild KJ. Substitution of amino acids in helix F of bacteriorhodopsin: effects on the photochemical cycle. Biochemistry. 1989;28:10028–10034. doi: 10.1021/bi00452a022. [DOI] [PubMed] [Google Scholar]
- 385.Dunach M, Berkowitz S, Marti T, He YW, Subramaniam S, Khorana HG, Rothschild KJ. Ultraviolet-visible transient spectroscopy of bacteriorhodopsin mutants. Evidence for two forms of tyrosine-185—Phenylalanine. J Biol Chem. 1990;265:16978–16984. [PubMed] [Google Scholar]
- 386.Jang DJ, el-Sayed MA, Stern LJ, Mogi T, Khorana HG. Effect of genetic modification of tyrosine-185 on the proton pump and the blue-to-purple transition in bacteriorhodopsin. Proc Natl Acad Sci U S A. 1990;87:4103–4107. doi: 10.1073/pnas.87.11.4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Dunach M, Marti T, Khorana HG, Rothschild KJ. UV-visible spectroscopy of bacteriorhodopsin mutants: substitution of Arg-82, Asp-85, Tyr-185, and Asp-212 results in abnormal light–dark adaptation. Proc Natl Acad Sci U S A. 1990;87:9873–9877. doi: 10.1073/pnas.87.24.9873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Ames JB, Ros M, Raap J, Lugtenburg J, Mathies RA. Time-resolved ultraviolet resonance Raman studies of protein structure: application to bacteriorhodopsin. Biochemistry. 1992;31:5328–5334. doi: 10.1021/bi00138a012. [DOI] [PubMed] [Google Scholar]
- 389.Liu XM, Sonar S, Lee CP, Coleman M, RajBhandary UL, Rothschild KJ. Site-directed isotope labeling and FTIR spectroscopy: assignment of tyrosine bands in the bR→M difference spectrum of bacteriorhodopsin. Biophys Chem. 1995;56:63–70. doi: 10.1016/0301-4622(95)00016-q. [DOI] [PubMed] [Google Scholar]
- 390.Ludlam CF, Sonar S, Lee CP, Coleman M, Herzfeld J, RajBhandary UL, Rothschild KJ. Site-directed isotope labeling and ATR-FTIR difference spectroscopy of bacteriorhodopsin: the peptide carbonyl group of Tyr 185 is structurally active during the bR→N transition. Biochemistry. 1995;34:2–6. doi: 10.1021/bi00001a001. [DOI] [PubMed] [Google Scholar]
- 391.Balashov SP, Govindjee R, Imasheva ES, Misra S, Ebrey TG, Feng Y, Crouch RK, Menick DR. The two pKa's of aspartate-85 and control of thermal isomerization and proton release in the arginine-82 to lysine mutant of bacteriorhodopsin. Biochemistry. 1995;34:8820–8834. doi: 10.1021/bi00027a034. [DOI] [PubMed] [Google Scholar]
- 392.Balashov SP, Imasheva ES, Ebrey TG, Chen N, Menick DR, Crouch RK. Glutamate-194 to cysteine mutation inhibits fast light-induced proton release in bacteriorhodopsin. Biochemistry. 1997;36:8671–8676. doi: 10.1021/bi970744y. [DOI] [PubMed] [Google Scholar]
- 393.Dioumaev AK, Richter HT, Brown LS, Tanio M, Tuzi S, Saito H, Kimura Y, Needleman R, Lanyi JK. Existence of a proton transfer chain in bacteriorhodopsin: participation of Glu-194 in the release of protons to the extracellular surface. Biochemistry. 1998;37:2496–2506. doi: 10.1021/bi971842m. [DOI] [PubMed] [Google Scholar]
- 394.Essen L, Siegert R, Lehmann WD, Oesterhelt D. Lipid patches in membrane protein oligomers: crystal structure of the bacteriorhodopsin–lipid complex. Proc Natl Acad Sci U S A. 1998;95:11673–11678. doi: 10.1073/pnas.95.20.11673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Brown LS, Sasaki J, Kandori H, Maeda A, Needleman R, Lanyi JK. Glutamic acid 204 is the terminal proton release group at the extracellular surface of bacteriorhodopsin. J Biol Chem. 1995;270:27122–27126. doi: 10.1074/jbc.270.45.27122. [DOI] [PubMed] [Google Scholar]
- 396.Rammelsberg R, Huhn G, Lubben M, Gerwert K. Bacteriorhodopsin's intramolecular proton-release pathway consists of a hydrogen-bonded network. Biochemistry. 1998;37:5001–5009. doi: 10.1021/bi971701k. [DOI] [PubMed] [Google Scholar]
- 397.Grigorieff N, Ceska TA, Downing KH, Baldwin JM, Henderson R. Electron-crystallographic Refinement of the Structure of Bacteriorhodopsin. J Mol Biol. 1996;259:393–421. doi: 10.1006/jmbi.1996.0328. [DOI] [PubMed] [Google Scholar]
- 398.Engels M, Gerwert K, Bashford D. Computational studies of the early intermediates of the bacteriorhodopsin photocycle. Biophys Chem. 1995;56:95–104. doi: 10.1016/0301-4622(95)00020-x. [DOI] [PubMed] [Google Scholar]
- 399.Sampogna RV, Honig B. Environmental effects on the protonation states of active site residues in bacteriorhodopsin. Biophys J. 1994;66:1341–1352. doi: 10.1016/S0006-3495(94)80925-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Sampogna R, Honig B. Electrostatic coupling between retinal isomerization and the ionization state of Glu-204: a general mechanism for proton release in bacteriorhodopsin. Biophys J. 1996;71:1165–1171. doi: 10.1016/S0006-3495(96)79320-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Song Y, Gunner MR. Subtle structural change of the bacteriorhodopsin stabilize the proton transfer between different intermediates. in preperation. [Google Scholar]
- 402.Nonella M. Effect of charge distribution on electrostatic chromophore–protein interactions in Bacteriorhodopsin. J Comput Chem. 1997;18:677–693. [Google Scholar]
- 403.Gonzalez-Luque R, Garavelli M, Bernardi F, Merchan M, Robb MA, Olivucci M. Computational evidence in favor of a two-state, two-mode model of the retinal chromophore photoisomerization. Proc Natl Acad Sci U S A. 2000;97:9379–9384. doi: 10.1073/pnas.97.17.9379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Murata K, Fujii Y, Enomoto N, Hata M, Hoshino T, Tsuda M. A study on the mechanism of the proton transport in bacteriorhodopsin: The importance of the water molecule. Biophys J. 2000;79:982–991. doi: 10.1016/S0006-3495(00)76352-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Nonella M. Electrostatic protein-chromophore interactions promote the all-trans→13-cis isomerization of the protonated retinal Schiff base in bacteriorhodopsin: An ab initio CASSCF/MRCI study. J Phys Chem B. 2000;104:11379–11388. [Google Scholar]
- 406.Humphrey W, Lu H, Logunov I, Werner HJ, Schulten K. Three electronic state model of the primary phototransformation of bacteriorhodopsin. Biophys J. 1998;75:1689–1699. doi: 10.1016/S0006-3495(98)77611-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Hayashi S, Ohmine I. Proton transfer in bacteriorhodopsin: Structure, excitation, IR spectra, and potential energy surface analyses by an ab initio QM/MM method. J Phys Chem B. 2000;104:10678–10691. [Google Scholar]
- 408.Hayashi S, Tajkhorshid E, Pebay-Peyroula E, Royant A, Landau EM, Navarro J, Schulten K. Structural Determinants of Spectral Tuning in Retinal Proteins-Bacteriorhodopsin vs Sensory Rhodopsin II. J Phys Chem B. 2001;105:10124–10131. [Google Scholar]
- 409.Rajamani R, Gao J. Combined QM/MM study of the opsin shift in bacteriorhodopsin. J Comput Chem. 2002;23:96–105. doi: 10.1002/jcc.1159. [DOI] [PubMed] [Google Scholar]
- 410.Hayashi S, Tajkhorshid E, Schulten K. Structural changes during the formation of early intermediates in the bacteriorhodopsin photocycle. Biophys J. 2002;83:1281–1297. doi: 10.1016/S0006-3495(02)73900-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Hayashi S, Tajkhorshid E, Schulten K. Molecular dynamics simulation of bacteriorhodopsin's photoisomerization using ab initio forces for the excited chromophore. Biophys J. 2003;85:1440–1449. doi: 10.1016/S0006-3495(03)74576-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Hayashi S, Tajkhorshid E, Kandori H, Schulten K. Role of hydrogen-bond network in energy storage of bacteriorhodopsin's light-driven proton pump revealed by ab initio normal-mode analysis. J Am Chem Soc. 2004;126:10516–10517. doi: 10.1021/ja047506s. [DOI] [PubMed] [Google Scholar]
- 413.Edholm O, Berger O, Jahnig F. Structure and fluctuations of bacteriorhodopsin in the purple membrane: a molecular dynamics study. J Mol Biol. 1995;250:94–111. doi: 10.1006/jmbi.1995.0361. [DOI] [PubMed] [Google Scholar]
- 414.Xu D, Martin C, Schulten K. Molecular dynamics study of early picosecond events in the bacteriorhodopsin photocycle: dielectric response, vibrational cooling and the J, K intermediates. Biophys J. 1996;70:453–460. doi: 10.1016/S0006-3495(96)79588-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Tajkhorshid E, Baudry J, Schulten K, Suhai S. Molecular dynamics study of the nature and origin of retinal's twisted structure in bacteriorhodopsin. Biophys J. 2000;78:683–693. doi: 10.1016/S0006-3495(00)76626-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Bondar AN, Fischer S, Smith JC, Elstner M, Suhai S. Key role of electrostatic interactions in bacteriorhodopsin proton transfer. J Am Chem Soc. 2004;126:14668–14677. doi: 10.1021/ja047982i. [DOI] [PubMed] [Google Scholar]
- 417.Luecke H, Schobert B, Cartailler JP, Richter HT, Rosengarth A, Needleman R, Lanyi JK. Coupling photoisomerization of retinal to directional transport in bacteriorhodopsin. J Mol Biol. 2000;300:1237–1255. doi: 10.1006/jmbi.2000.3884. [DOI] [PubMed] [Google Scholar]
- 418.Bousche O, Sonar S, Krebs MP, Khorana HG, Rothschild KJ. Time-resolved Fourier transform infrared spectroscopy of the bacteriorhodopsin mutant Tyr-185→Phe: Asp-96 reprotonates during O formation; Asp-85 and Asp-212 deprotonate during O decay. Photochem Photobiol. 1992;56:1085–1095. doi: 10.1111/j.1751-1097.1992.tb09732.x. [DOI] [PubMed] [Google Scholar]
- 419.Junemann S, Meunier B, Fisher N, Rich PR. Effects of mutation of the conserved glutamic acid-286 in subunit I of cytochrome c oxidase from Rhodobacter sphaeroides. Biochemistry. 1999;38:5248–5255. doi: 10.1021/bi9830112. [DOI] [PubMed] [Google Scholar]
- 420.Wikstrom M, Jasaitis A, Backgren C, Puustinen A, Verkhovsky MI. The role of the D- and K-pathways of proton transfer in the function of the haem-copper oxidases. Biochim Biophys Acta. 2000;1459:514–520. doi: 10.1016/s0005-2728(00)00191-2. [DOI] [PubMed] [Google Scholar]
- 421.Ådelroth P, Karpeforsa M, Gildersona G, Tomsonc FL, Gennis RB, Brzezinski P. Proton transfer from glutamate 286 determines the transition rates between oxygen intermediates in cytochrome c oxidase. Biochim Biophys Acta. 2000;1459:533–539. doi: 10.1016/s0005-2728(00)00194-8. [DOI] [PubMed] [Google Scholar]
- 422.Calimet N, Ullmann GM. The influence of a transmembrane pH gradient on protonation probabilities of bacteriorhodopsin: the structural basis of the back-pressure effect. J Mol Biol. 2004;339:571–589. doi: 10.1016/j.jmb.2004.03.075. [DOI] [PubMed] [Google Scholar]
- 423.Lee YS, Krauss M. Dynamics of proton transfer in bacteriorhodopsin. J Am Chem Soc. 2004;126:2225–2230. doi: 10.1021/ja036115v. [DOI] [PubMed] [Google Scholar]
- 424.Xiao Y, Hutson MS, Belenky M, Herzfeld J, Braiman MS. Role of arginine-82 in fast proton release during the bacteriorhodopsin photocycle: a time-resolved FT-IR study of purple membranes containing 15N-labeled arginine. Biochemistry. 2004;43:12809–12818. doi: 10.1021/bi049238g. [DOI] [PubMed] [Google Scholar]
- 425.Krebs RA, Alexiev U, Partha R, DeVita AM, Braiman MS. Detection of fast light-activated H+ release and M intermediate formation from proteorhodopsin. BMC Physiol. 2002;2:5. doi: 10.1186/1472-6793-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]