

Abstract
Measuring enzymatic activities in biological fluids is a form of activity-based proteomics and may be utilized as a means of developing disease biomarkers. Activity-based assays allow amplification of output signals, thus potentially visualizing low-abundant enzymes on a virtually transparent whole-proteome background. The protocol presented here describes a semi-quantitative in vitro assay of proteolytic activities in complex proteomes by monitoring breakdown of designer peptide-substrates using robotic extraction and a MALDI-TOF mass spectrometric read-out. Relative quantitation of the peptide metabolites is done by comparison with spiked internal standards, followed by statistical analysis of the resulting mini-peptidome. Partial automation provides reproducibility and throughput essential for comparing large sample sets. The approach may be employed for diagnostic or predictive purposes and enables profiling of 96 samples in 30 hours. It could be tailored to many diagnostic and pharmaco-dynamic purposes, as a read-out of catalytic and metabolic activities in body fluids or tissues.
INTRODUCTION
It is often stated that, from a functional or biomarker point-of-view, the analysis of proteins trumps that of mRNA (e.g., using DNA micro-arrays) since `activities' are what ultimately matter in a cell. However, systematic measurement and quantitation of enzymatic activities in a complex biological matrix, while clearly in the realm of proteomics, is a largely unpracticed specialty at the moment. Enzymatic activity is bringing about chemical change in a target, ranging from simple modifications to anabolic or catabolic reactions. In the literal sense, activity-based or functional proteomics would therefore entail profiling based on genuine activity readouts. Yet, in a typical mass spectrometry (MS)-based proteomic analysis, enzymes are simply looked at as `proteins' that may or may not be physically present in certain locations and that may undergo temporal changes in concentration. Furthermore, many enzymes are present below the MS detection limits in a given sample and are therefore `invisible' in traditional screens. As an alternative, the case could be made that, given enough substrate and time, and optimal assay conditions, catalytic or synthetic products may accumulate to levels that become readily detectable using some type of analytical technique. This may be particularly true for proteolytic enzymes as they generate peptide products that are tailor-made for MS-based proteomic read-outs.
Human cells produce well over 500 proteases 1, 2. Although they all share one principal functionality, the lysis of peptide bonds, their biological roles differ greatly, ranging from digestion of food and discarded cellular proteins to more specific functions such as basic control mechanisms in the cell, orderly progression of physiological pathways and response to trauma, to name a few 1, 2. Proteases have also been implicated in disease, most notably in cancer where they may both promote tumor progression or suppression 3-5. Typical assays monitor substrate conversion via simple colorometric or fluorophoric read-outs 6, 7 that are easily performed in parallel but not so easily multiplexed. It is also difficult to measure `compound' proteolytic activities that degrade a substrate, either in unison or sequentially. Examples of sequential peptide degradation have unexpectedly been observed in recent oncopeptidomics studies 8-10. In these studies, subsets of serum peptides provided class discrimination between patients with different types of solid tumors and control individuals without cancer. Nearly all relevant peptides sorted into about a dozen nested sets of sequences, each the result of exopeptidase activities that confer cancer-type-specific differences superimposed on the proteolytic events of the ex vivo coagulation and complement degradation pathways 9.
Based on those findings, we reasoned that isotopically labeled (13C and/or 15N) peptides such as FPA or C3f, or any synthetic peptide containing a known endo-or exopeptidase target sequence for that matter, would allow to accurately track degradation over time when added to serum or plasma, or to any complex matrix containing active proteases. The kinetics and/or resulting patterns could then perhaps be used to compare defined peptidase activities between individual proteomes of two or more (groups of) biological samples. Ideally, such a test should allow straightforward analysis in a highly reproducible and inter-sample comparable manner. To address this need, we have developed a protocol to measure de novo peptide breakdown in large numbers of biological samples using MALDI-TOF MS, with relative quantitation of the metabolites by comparison with double-labeled, non-degradable reference peptides (consisting of D-amino acids only), spiked into the samples at the same time as the substrate(s) to reflect adsorptive and processing-related losses 11. The full array of metabolites is then quantitated (C.V.'s ≤15% over multiple replicates) and the results subjected to kinetic analysis and univariate or multivariate statistical analysis 11.
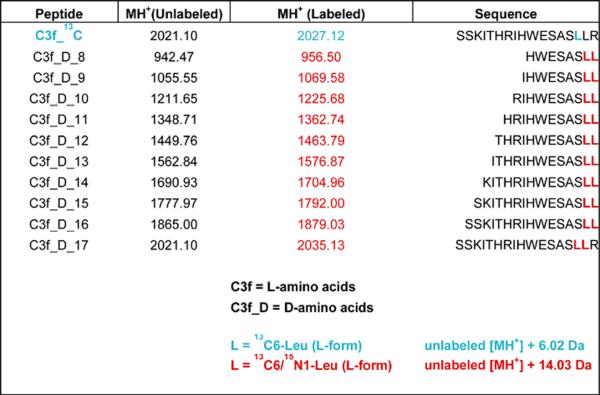
As shown in Figure 1, the workflow of the protocol presented herein consists of eight interlocking modules. Modules 1 and 2 involve pre-assay activities, such as sample collection and handling (module 1; steps 17-35), before parallel incubation with exopeptidase substrate(s). Samples can be bodily fluids, tissue or cell extracts, secreted proteins, or fractions obtained from non-denaturing column chromatography of any of the above complex proteomes. The peptide substrate(s), plus reference peptides that correspond to each of the anticipated degradation products (see, for example, the reference peptide series for C3f degradation products in Table 1), should have been synthesized, quantitated and tested in advance (module 2; steps 6-7 and Box 2). These peptide sets can be generic or specifically designed for a particular assay; they can be modeled after existing peptide sequences (e.g., C3f) or derived from artificial peptide library screens. After incubation for a predetermined period of time in the sample(s) under study (module 3; steps 8-16), newly generated degradation products and undegraded substrate are extracted, mixed with MALDI matrix and deposited on metal targets, using an automated liquid handler (`robot'), in preparation of MALDI-TOF MS analysis (module 4; steps 36-63). After normalization, baseline subtraction and peak alignment of all spectra (module 5: signal processing; steps 64-67), the resulting data can be visually inspected in the form of spectral overlays (module 6; step 68) and/or used for relative quantitation (module 7: calculation of ion intensity ratios for every degradation product / reference pair; step 69). The numerical data may then be taken for statistical analysis if so required, for instance to evaluate putative diagnostic potential of selected degradome patterns in subsequent verification studies (module 8; steps 70-77). Modules 1 and 4 plus box 3 are basically the same as described in our earlier protocol 12 ; modules 5, 6 and 8 have been described in our second earlier protocol 13,with modifications.
Figure 1. Exopeptidase Assay Operation.

Workflow showing the various steps of the exopeptidase assay approach for activity biomarker discovery described in this protocol. Steps are organized into 8 modules as described in the Introduction.
Table 1.
C3f degradation ladder (mono-isotopic masses of singly protonated peptides are listed).
 |
BOX 2 | PEPTIDE QUANTITATION STEPS.
1. Reconstitute the newly synthesized peptides based on the estimated amount and concentration in 30% acetonitrile, 0.1% FA to obtain approximately 10 μg / μL.
2. Transfer a 1-μL aliquot to a 0.5-mL eppendorf tube and dry using a speed vacuum.
3. Send the sample to a service laboratory for amino acid analysis.
4. Recalculate the concentration of the peptide stock based on the amino acid analysis quantitation result.
5. Adjust some of the stock solution to 100 pmol / μL in 30% acetonitrile, 0.1 % FA. Store both stocks at -20°C.
6. Prepare a mixture of all the reference peptides with a final concentration of 10 pmol / μL for each peptide (10 peptides in the C3f-ladder).
BOX 3 | EXOPROTEASE ASSAY TECAN PROGRAM.
1. Mix 75 μL suspension of Dynabeads RPC18 magnetic particles (previously placed in an eight-well strip of 0.2-mL tubes) by pipetting it ten times up and down (50 μL) to obtain homogeneous dispersion.
2. Aspirate 60 μL per tip from the eight-well strip of 0.2-mL tubes.
3. Discard 15 μL of beads for each tip to avoid having air bubbles in the tip when the beads are dispensed and mixed with the serum.
4. Dispense 5 μL of the bead suspension to each of the 48 wells (8 tips × 6 columns).
5. Discard the remaining 15 μL of beads back into the eight-well strip of 0.2-mL tubes.
6. Mix 5 μL of beads with 40 μL of sample in each of the 48 wells by slowly pipetting five times up and down.
7. Wash tips after each column is complete.
8. Repeat steps 6 & 7 for each of the 6 columns.
9. Pull the magnetic particles to the side by transferring the 96-well plate to the side-magnet rack (Figure 2b).
10. Remove and discard the supernatant of each of the 48 wells while the microtiter plate is in the side-magnet rack position.
11. Add 200 μL of a 0.1% TFA solution to each of the 48 wells (while the plate is in the side-magnet rack position).
12. Wash the magnetic beads by moving the 96-well plate five times back and forth over the long magnets, one row to the left and to the right at a time. The movement causes a lateral jarring motion that will pull the particles from left to right and back, by alternately positioning the rows of the plate to either side of the magnets.
13. Aspirate and discard the washing solution.
14. Repeat the wash procedure (Steps 11-13).
15. Aspirate slowly and discard 120 μL of the washing solution after the second wash.
16. Move the microtiter plate to the plate holder 1 position.
17. Resuspend the magnetic particles with the remaining washing solution by pipetting up and down five times.
18. Move the 96-well plate to the bottom-magnet rack position and wait 160 s to allow the beads to collect at the bottoms of the tubes (Figure 2b).
19. Very carefully, remove the rest of the washing solution and discard it.
20. Add 6 μL of 50% acetonitrile (from column 1 of the cooler rack plate) to each of the 48 wells.
21. Resuspend the magnetic particles by pipetting the beads up and down quickly ten times.
22. Move the microtiter plate to side-magnet rack position.
23. Transfer 5 μL of eluate from each of the 48 wells from the first half of the 96-well plate to the second half of the plate (i.e., eluates in column1 go to column 7, those in 2 go to 8 and so forth).
24. Add 10 μL of matrix solution to each of the 48 wells containing 5 μL of the eluate on the second half of the plate (columns 7-12).
25. Mix the matrix with the eluate by pipetting up and down.
26. Aspirate 1 μL of the mixture (matrix plus eluate) and spot it on the MALDI plate.
27. After completion of this program, prepare calibrants for continuation of the program.
100 μL Calibrants (see REAGENTS SETUP) + 100 μL matrix
Dispense 20 μL of matrix and calibrant mix in each well in column A of a clean non-skirted microtiter plate in microtiter plate holder 2.
28. Aspirate 8 μL and spot 1 μL per calibrant spot on the MALDI plate. Note: even though every calibrant spot on the MALDI plate is from the same mixture, each has a different designation depending on where it is located on the plate.
The protocol / workflow as outlined in Figure 1 has been successfully used to assay serum samples obtained from 48 patients with metastatic thyroid cancer and 48 gender- and age-matched healthy controls, utilizing 3 different peptide substrates in separate analyses. The full array of peptide metabolites (a de facto 26-member `mini-peptidome') was quantitated and subjected to multivariate statistical analysis and machine learning methods to yield class predictions with 94% sensitivity and 90% specificity 11. While providing proof-of-principle for discovery of blood-based, `compound' activity-biomarkers (i.e., based on panels of exopeptidases), this study did not yet address measuring activities of individual exopeptidases and of panels or single endopeptidases, which therefore will need to be included in future applications. This will depend on designing and developing MALDI-TOF MS-compatible peptide substrates that can be cleaved by only one or a few functionally related proteases out of the more than 500. Similar to many existing fluorescence-based substrates, they may consist of a non-degradable, highly ionizable component to which 1, 2 or 3 cleavable amino acids have been attached, the removal of which could be readily measured by even the most simple of mass spectrometry platforms. Endopeptidase substrates, on the other hand, may contain a multi-amino acid, cleavable linker that corresponds to a know target sequence (e.g., from P3 to P3') of the protease under study. We speculate that degradation of such substrates will be easier to quantify, in multiplexed assays, and may allow examining key elements of fundamental biological processes such as development, cell-cycle progression, apoptosis, clotting and wound healing; as well as monitoring disease (e.g., cancer) and the presence of infectious disease agents in bodily fluids; and evaluating efficacy of novel protease inhibitors.
MATERIALS
REAGENTS
α-Cyano-4-hydroxycinnamic acid (Agilent cat. no. G2037A) ! CAUTION Supplied in 36% methanol and 56% acetonitrile; both methanol and acetonitrile are flammable and toxic.
Calibration standards (Bruker Daltonics cat. nos. 206195 (peptide calibration standard: Angiotensin II, Angiotensin I, Substance P, Bombesin, ACTH clip 1-17, ACTH clip 18-39, Somatostatin 28; covered mass range: ~ 1,000 Da - 3,200 Da) and 206355 (protein calibration standard I: Insulin, Ubiquitin I, Cytochrome C, Myoglobin; covered mass range: ~ 5,000 Da - 17,000 Da))
Synthetic peptide `3879' (YVPLPNVPQPGRRPFPTFPGQGPFNPKIKWPQGY) 12
Custom synthetic peptides (an example — C3f — is given in Table 1)
Dynabeads RPC-18 (12.5 mg / mL, Dynal cat. no. 102.01) ! CAUTION Supplied in a 20% ethanol solution; ethanol is highly flammable and an irritant to eyes.
Acetonitrile (Burdick & Jackson cat. no. 015-1) ! CAUTION Highly flammable and toxic.
Trifluoroacetic acid (TFA) (Pierce cat. no. 28903) ! CAUTION Highly corrosive.
Formic acid (FA) (Fluka cat. no. 06440) ! CAUTION Highly corrosive.
Human serum (Sigma cat. no. S7023) ! CAUTION For all blood and blood derived samples, always observe precautions: handle all biological samples as a potential source of pathogens, use the appropriate protective attire (lab coats, safety glasses, latex gloves, etc.) and dispose properly of all biohazardous materials.
Milli-Q (MQ) water (USFilter Purelab Plus System)
PBS (Bio-Rad 10 concentrate, cat. no. 161-0780)
Isopropanol (J.T. Baker cat. no. 9095-03) ! CAUTION Highly flammable and an irritant to eyes.
REAGENTS SETUP
10pmol/μl peptide stock solution
Dilute 100 pmol / μL peptide stock solution in MQ water to yield 10 pmol / μL.
! CRITICAL Prepare and keep on wet ice for no longer than ~8 h.
0.2 pmol/μl non-degradable reference peptide
Dilute 10 pmol / μL non-degradable reference peptide in MQ water to yield 0.2 pmol / μL.
! CRITICAL Prepare and keep on wet ice for no longer than ~8 h.
50 % (vol/vol) acetonitrile
Dilute acetonitrile to 50% with MQ water.
▲CRITICAL Prepare the day of the experiment.
•
0.1% TFA
Make up 50 mL 0.1 % TFA in MQ water.
▲CRITICAL Prepare and keep at room temperature (RT: 19-24°C) for no longer than one week.
50mg/ml Dynabeads
Concentrate the Dynabeads to 50 mg / mL. Do this by first washing the Dynabeads twice with 0.1% TFA and then resuspending in 0.1% TFA to make the final concentration 50 mg / mL. This removes the ethanol from the stock solution. The remainder of this stock can be used in future experiments.
PREPARATION OF CALIBRANTS FOR 48 SAMPLES
Dilute synthetic peptide `3879' (10 mg / mL) 1:40 in 0.1% TFA, then 1:10 in 50% acetonitrile, resulting in a final dilution of 1:400. Dilute Bruker peptide set 1:3 in 0.1% TFA (Refer to Bruker protocol). Combine one volume of the `pep-3879' dilution and one volume of the `Bruker-pep' dilution. Dilute this mixture 1:5 in 50% acetonitrile; this gives the `Peptide mix'. Dilute Bruker protein set 1:1 in 50% acetonitrile; this gives the `Protein mix'. Combine 51 μL peptide mix + 8.5 μL protein mix + 1.4 μL cytochrome c + 7.1 μL 50% acetonitrile; this gives the `Master mix' (total 68 μL). The 'Master mix' is the final solution that contains the proper concentrations of all calibrants. Mix 0.5 μL `Master Mix' and 0.5 μL of the Matrix. Spot 1 μL onto calibrant spot on the MALDI plate to ensure proper calibrant signal peaks.
EQUIPMENT
BD Vacutainer SST (`tiger-top') tube (Becton Dickinson cat. no. 367988)
BD Vacutainer `red-top' tube (Becton Dickinson cat. no. 366430)
Cryovials (source vials; Fisher cat. no. 0566966)
Cryobox aluminum rack for box storage (VWR cat. no. 55710-171)
Microcentrifuge plastic racks, 96-tube (Fisher cat. no. 05-541-44)
Nunc CryoStore polyethylene-coated boxes (Fisher cat. no. 12-565-182)
Z4M bar-code printer (Zebra Technologies)
Transcode labels (Digitrax cat. no. EP-TUF-05)
Genesis Freedom 100 liquid handler (Tecan)
Robot tips (Tecan cat. no. 71-700S)
Long magnets, epoxy-coated, neodymium-iron-boron (NdFeB), 2.75 inch long 0.25 inch wide 0.125 inch deep (K&D Magnetics)
Disk magnets, NdFeB, 0.25 inch diameter 0.25 inch thick (Forcefield)
Custom aliquoting rack (Figure 2a; see EQUIPMENT SETUP)
Custom side-magnet rack (Figure 2b; see EQUIPMENT SETUP)
Custom bottom-magnet rack (Figure 2b; see EQUIPMENT SETUP)
Foam insets for sample storage (New England Foam Products)
Template III PCR half-skirted plate (USA Scientific cat. no. 1402-9700)
0.2-mL eight-tube strips with eight-dome cap strips (USA Scientific cat. no. 1402-2700)
Self-adhesive aluminum foil (USA Scientific cat. no. 2923-0100)
Soft rubber roller for adhesive foil (USA Scientific cat. no. 9127-2940)
Microcentrifuge tubes with attached caps (VWR cat. no. 20170-310)
Speed Vacuum (Savant SC110A)
PCR cooler starter set (USA Scientific cat. no. 4051-0509)
Autoflex MALDI-TOF mass spectrometer (Bruker)
Bruker 384-spot MALDI target plates (Bruker cat. no. 55420/26755)
Gemini software (Tecan)
FlexControl software (Bruker)
FlexAnalysis software (Bruker)
MALDI automation execution file v 2.1.exe: Microsoft Windows custom-written software used to randomize the serum samples and generate the file to automatically run the mass spectrometer (this software can be down loaded at: http://cbio.mskcc.org/tempst).
ConvertDirectoryToASCII.obm: a custom-written script that translates all the Bruker raw spectra from a given directory into text files; script should be located in the `FlexAnalysisMacromodules' folder (this software can be down loaded at: http://cbio.mskcc.org/tempst).
Matlab software. Use instructions available from Mathworks website (http://www.mathworks.com) to install MATLAB.
QPeaks software (Spectrum Square Associates)
Entropycal software (Spectrum Square Associates)
Signal Processing & Preview (SPP) software (MSKCC) (this software can be down loaded at: http://cbio.mskcc.org/tempst).
MassSpectraViewer (MSV) software (MSKCC) (this software can be down loaded at: http://cbio.mskcc.org/tempst).
Qcealign (MSKCC). This script does the spectra processing in Matlab calling the Qpeaks and the Entropycal algorithms (this software can be down loaded at: http://cbio.mskcc.org/tempst).
Laboratory Information Management System (LIMS), see EQUIPMENT SETUP.
Figure 2. Automation of sample processing for serum peptidome analysis.

A. Robot layout for sample aliquotting. Two different racks are used for the aliquotting step: a source vial rack (for 5-mL tubes) to contain the serum vials (step 3 of the protocol); and an aliquoting rack (for 0.5-mL tubes) to hold the bar-coded aliquot tubes.
B. Robot layout for serum peptide processing. Various containers for magnetic beads, washing and elution buffers, a wash station and a waste receptacle are pictured. Five different, 96-position plates for various sample processing steps and the MALDI target plate are shown, as well as the positioning of the magnets in the 96-well plates (insets). The plate with the `side-magnets' is used to remove supernatants after peptide binding and washing, and to transfer eluates. The plate with the `bottom-magnets' is used to pull down beads to the tip of the tube before adding a minimal volume of elution solvent.
▲CRITICAL STEP It is crucial to use the exact same suppliers and catalog numbers specified for each reagent and equipment.
EQUIPMENT SETUP
Memorial Sloan-Kettering Cancer Center clinical proteomics laboratory information management system (LIMS)
This protocol is based on the availability of a custom-made LIMS as described in an earlier protocol 12. Any LIMS can be used to carry out this protocol provided that it has some basic capabilities: (i) A sample accession module must be available so that new samples arriving to the lab can be entered into the LIMS. Subject and sample information will be entered and linked through this module. Other information will also be stored relevant to the accession event. This module must also be able to calculate aliquots and generate bar code labels. (ii) We recommend the following fields for sample accession: site, contact, project, cancer type, disease stage, specimen type, number of vials received, print number of labels, tube identifier, volume, date drawn, date received and comments. (iii) A bar-code printer module must be part of the LIMS so that bar-code labels can be printed on a local bar-code printer. The bar-code labels will be used for the source vials and the aliquots. (iv) An inventory system module must be available to perform the different sample management activities. This module will allow the management of sample storage, providing a way of accessioning samples into the inventory module in an automated manner.
Custom aliquoting rack
Designed to hold 16 rows of nine 0.5-mL Eppendorf tubes for use with a liquid handler robot of variable pipette separation (Tecan Genesis Freedom 100). At the end of each row is a sample source vial held in a separate block. The top parts are placed on the base, the latter having inserts that match the work surface of the liquid handler. All parts should be machined in aluminum and clear anodized (Figure 2 and Supplementary Material 1).
Custom side-magnet rack
Designed to have seven long magnets (see EQUIPMENT) at the sides of the vials of the 96-well plate. Parts should be machined in clear Lexan (Figure 2 and Supplementary Material 2).
Custom bottom-magnet rack
Designed to have 96 disk magnets (see EQUIPMENT) at the bottoms of the vials of the 96-well plate. The parts should be machined in clear Lean plastic with standard 96-well plate dimensions (Figure 2 and Supplementary Material 3).
PROCEDURE
Testing new candidate exoprotease substrates TIMING ~2-14 h
1| Thaw an aliquot of human serum from Sigma (250 μL) on wet ice for an hour.
2| During this time, prepare fresh reagents (see REAGENTS SETUP).
3| Analyze substrate peptides before proteolysis (zero time point) using 100 pmol of the peptide which will be used in a particular assay (e.g., C3f_13C) to verify the molecular weight (m/z value) (Box 1).
BOX 1 - Manual solid-phase extraction.
A `time-zero' data point is taken using 100 pmol of the peptide under study to verify the MH+ value.
1. Transfer 20 μL of thawed serum (or plasma) to a well of a 0.2-mL eight-tube strip.
2. To the sample, add 10 μL of 10 pmol / μL substrate (labeled / unlabeled) solution to yield 100 pmol.
▲ CRITICAL STEP Change tips in between each step to avoid contamination.
3. Immediately, add 5 μL of the pre-washed Dynabeads and mix it three times using your pipette tip.
4. Place the eight-tube strip on a magnetic plate to separate the peptide bound Dynabeads from the sample.
5. Remove and discard the supernatant.
6. Wash the Dynabeads by adding 200 μL of 0.1% TFA and mixing it by moving the eight-tube strip back and forth eight times.
7. Aspirate and discard the washing solution.
8. Repeat the wash step (steps 6 and 7) one more time.
9. After removing the wash solution, resuspend the beads with 6 μL of 50% acetonitrile by pipetting the beads up and down quickly ten times.
10. Transfer 5 μL of eluate into a new well.
11. Add 10 μL of matrix solution to the well containing 5 μL of the eluate.
12. Mix the matrix with the eluate by pipetting up and down.
13. Aspirate 1 μL of the mixture and spot on a MALDI plate.
14. After the spot has crystallized, insert the plate into a MALDI-TOF mass spectrometer.
15. Select the method `ExoproteaseAssay.par' Box 4. (Reflectron mode method created in house) in the FlexControl program.
16. Acquire a spectrum and save it in a new created folder to be stored for future reference.
17. Open the FlexAnalysis program and choose your newly created file.
18. You should be able to see the parent peptide peak among the endogenous serum peptides. ! CAUTION If 100 pmol is insufficient, you must adjust the amount of peptide and repeat the above steps until the proper concentration is established. This may vary among peptides.
? TROUBLESHOOTING
4| Select preliminary incubation time points to test. Generally, two preliminary incubation time points are selected, for instance 15 minutes (min) and 1 hour (h).
? TROUBLESHOOTING
5|Prepare Samples for the preliminary test time points, as described in Box 1, and evaluate the selected timepoints. Based on the results (extent of degradation or lack thereof) after these incubations, a decision should be made about which additional time points to select. For example, if many or all rungs of the expected ladder are observed after 15 min, incubations of the peptide in serum will also be done for 5 and 30 min. If there was degradation only after 1 h, additional time points at 30 min and 2 h time will be investigated. Lastly, if no degradation was observed after 1 h, incubation periods of 3 h, 6 h and overnight should be tested (Figure 3).
Figure 3. C3f peptide degradation ladder.

A degradable, isotopically-labeled substrate (C3f_13C) was incubated for 30 min in human serum at RT (19-24°C). The reaction was stopped, and subjected to automated solid-phase peptide extraction and MALDI-TOF MS analysis. MS signals for all the degradation products derived from C3f_13C are marked with colored dots.
▲ CRITICAL STEP Cover the samples in the eight-tube strip with its eight-dome cap strip to avoid evaporation and precipitation.
▲ CRITICAL STEP Incubate reactions at room temperature (RT: 19-24 °C)
6|After the best time point(s) are established, synthesize isotopically labeled versions of the peptides judged to be optimal substrates, as well as synthesize a series of doubly-labeled, non-degradable D-peptides (reference peptides consisting of D-amino acids) that correspond to each rung in the degradation ladder.
7|Quantitate newly synthesized peptides by amino acid analysis, as described in Box 2)
Testing new labeled exoprotease substrates and reference peptides in serum pools TIMING ~ 2-15 h
8|Prepare pools of the clinical groups you will be studying. First transfer an equal amount from each individual sample type into a large vial. Then mix the new created pool by pipetting up and down couple of times. Make 20 μL aliquots of the pool and freeze samples in -80 °C.
▲ CRITICAL STEP Pools must be prepared on wet ice.
9|Thaw an aliquot of the pool on wet ice.
! CAUTION Leftover sample should not be frozen for further use.
10|Repeat steps 2 - 4 with the thawed aliquots (exactly as performed for the unlabeled peptide) to confirm that the same degradation time and concentration conditions apply to the labeled version.
Incorporating the corresponding reference D-peptides into your assay
11|Transfer 20 μL of thawed pool of reference D-peptides (table 1) to a well of a 0.2-mL eight-tube strip.
12|Add 10 μL of 0.2 pmol / μL reference peptide mix (see REAGENTS SETUP) to yield a total amount of 2 pmol.
! CAUTION Change tips in between each step to avoid contamination.
13|Add 10 μL of 10 pmol / μL substrate (degradable labeled peptide) (see REAGENTS SETUP) to yield a total amount of 100 pmol.
! CAUTION Change tips in between each step to avoid contamination.
14|Cap the eight-tube strip with its eight-dome cap strips and then start incubation at RT (19-24°C).
15|After incubation, follow Box 1.
16|Check that MS results show the presence of the `exogenous' ladder with its corresponding reference D-peptides (see mass spectrometry section for details). If this is not the case, more time points should be added to the pilot studies and /or the quantitation of the D-peptides double-checked.
Blood collection and Serum preparation and storage TIMING ~ 21 h
17|Collect venous blood into one BD Vacutainer SST tube.
▲ CRITICAL STEP Fill blood collection tubes to the top (8.5 mL) to avoid hemolysis.
! CAUTION For all blood and blood derived samples, always observe precautions: handle all biological samples as a potential source of pathogens, use the appropriate protective attire (lab coats, safety glasses, latex gloves, etc.) and dispose properly of all biohazardous materials.
? TROUBLESHOOTING
18|Gently invert the tube five times to mix the clot activator with the blood. Allow blood to clot for 1 h at RT (19-24°C) with the tube vertical.
19|Keep the SST tubes on wet ice in vertical position until transport. Then place the tubes in an appropriately coded bag. Keep below 4°C during transport.
20|Spin SST tubes in a clinical centrifuge at 1,400-2,000g for 10 min at RT.
21|Label 4-mL cryovials.
22|Transfer the serum (upper phase) to the appropriately labeled 4-mL cryovials. The volume of serum per cryovial should be approximately 1 mL. These cryovials will be called `source vials' throughout this protocol.
23|Immediately store all serum samples at -80°C. Avoid freeze-thawing cycles. Transport serum samples to the MS laboratory on dry ice.
24|Enter all sample information into the LIMS before the samples arrive at the lab. Fill out the necessary information for each specimen (see EQUIPMENT SETUP). Bar-code labels for the original source vial tubes should be created automatically every time sample information is submitted. Keep labels organized in the same order as the samples to make the process easier. Generate a source vial list for verification while labeling the source vials.
25|Label the source vials while they are frozen on dry ice. Note that the barcode labels are designed for small 0.5-mL microcentrifuge tubes and not for bigger vials. Labels will not overlap on the back of larger tubes, so it is difficult for the labels to stay attached, especially when the frozen vials have water condensation on the surface. For those reasons, it may be necessary to wipe the tubes dry with a piece of paper tissue, prior to attaching the label and adding a piece of tape on top to secure the label effectively.
▲ CRITICAL STEP Always double-check that sample IDs on source vials match the sample information associated with their bar-code labels in the database.
26|Store the labeled source vials in the -80 °C freezer in cardboard freezer boxes with temporary labels indicating the project and date, and marked `source vials to be aliquoted.' If more than one source vial is received for each specimen, then use only one of them for aliquoting. Place the remaining source vials in separate boxes temporarily labeled with tape with the project name, date and marked `source vials for permanent storage.' These vials will be entered later into bar-coded boxes and those boxes will be added to the inventory.
▲ PAUSE POINT Serum source vials can be kept frozen for at least 5 years at -80 °C.
27|Take the boxes containing the source vials to be aliquoted out of the freezer and place the vials on dry ice.
▲CRITICAL STEP The samples should remain frozen while the bar-codes for the aliquots are being generated.
28|Scan the bar-code of the first source vial to be aliquoted. A `Specimen ID' will be generated if the database recognizes the sample correctly. Select the volume of serum in the source vial. Indicate the volume of the aliquots (50 μL) and select the number of aliquots to be created based on the volume of sample available. New labels will be printed for all of the aliquots created. If sufficient sample is available, generate nine aliquots of 50 μL each per specimen (source vial). Keep adding specimens until the list has been filled.
29|Label clean 0.5-mL microcentrifuge tubes with the aliquot bar-code labels and organize them according to the order of the original source vials. If available, use 12×8-hole plastic racks for 0.5-mL tubes or place the labeled tubes directly in the aliquot racks of the Tecan liquid handler 12. Generally, tubes have to be organized in eight rows of nine tubes. From each set of tubes, eight will be placed in temporary `aliquots for permanent storage' boxes and one aliquot (normally, aliquot no.1) will be placed in a separate box marked `to be run with Tecan' and dated.
30|Place the source vials on wet ice once all the aliquot tubes have been labeled, organized and checked. Use big-flat ice buckets to place the samples in the same order they will be aliquoted and let thaw for 60-90 min.
▲ CRITICAL STEP Verify that the source vials and their associated aliquots match before proceeding with the aliquoting process to avoid thawing the serum of source vials that were not planned to be aliquoted.
31|Load the clean, newly bar-coded aliquot tubes in the aliquot racks (16 rows of nine aliquots of each sample). Tubes have to be placed so the lids rest against the back lip of the rack, allowing the tubes to be closed easily once they have been aliquoted with serum. Place the aluminum aliquoting racks in the robot carriers in the layout as indicated in Figure 2a and then load the rack with the source vial samples in the same order as the aliquot tubes. Keep the source vial racks on wet ice to maintain the serum as cold as possible before the aliquoting process begins.
32|Open the Gemini program by clicking on the icon located on the PC Desktop. Run the Gemini aliquot program or manually perform the aliquoting using microcentrifuge micropipettes. Aliquoting process can be done automatically or manually, as long as all the tubes are carefully organized.
33|Close the tubes and put them in their corresponding foam inserts and boxes on dry ice, to be temporarily stored at -80°C in the prelabeled temporary boxes. The boxes' numbers and dates will allow the samples to be correctly identified during later steps. This process must be kept as short as possible to avoid exposure of the samples to room temperature.
▲ PAUSE POINT. Serum aliquots can be kept frozen for at least 5 years at -80 °C.
34|Create labels for the boxes for permanent storage of the aliquots. Calculate how many boxes are needed to store all the created aliquots, dividing the total number of aliquots by 128 (maximum number of aliquots per box) and rounding up to the next whole number. For this purpose new `Containers' will be created in the `Inventory' section of the LIMS. During this process, bar-code labels for the upper and lower foam inserts, for the entire freezer box, and for the freezer racks will be created. Place the label on the center of the outer part of the box. The label for the upper foam insert goes in the upper left corner of the inner part of the box and that for the lower foam insert at the lower right corner of the inner part.
35|Scan the aliquots to be placed in their permanent locations in the prelabeled boxes, using one temporary box at a time. Enter the aliquot's permanent location in the inventory section of the LIMS. Scan the samples and place them in the physical location assigned by the database. (A1 is the top left spot in the foam inserts.) In a similar manner, fill the other containers: place aliquots inside foam inserts, foam inserts inside cardboard boxes, boxes inside freezer racks, racks inside freezer shelves, shelves inside freezer and freezer inside room. Keep all aliquots frozen on dry ice at all times.
▲ CRITICAL STEP Make sure that all aliquots go to their proper permanent storage box. From this time on, information on where the specimens are will be stored only in the LIMS system.
▲ PAUSE POINT Serum aliquots can be kept frozen for at least 5 years at -80 °C.
Set up of the robot run TIMING ~ 4 h
36|Scan the bar-codes of the aliquots to be run and make a list of their bar-code identifiers. Save the list with the project name and date as a plain text file (.txt in NotePad or TextEdit). Keep all aliquots frozen on dry ice at all times. These aliquots (normally aliquot no. 1 of each sample) should be in temporary `to be run' boxes after the aliquoting step.
37|Open the randomization software, MALDI autoexecute generator v 3.0.exe 12, and indicate the number of samples to be run. The maximum number of serum aliquots that can be run for each MALDI plate is 96. If more than 96 samples are added, the program will create two or more MALDI plates.
38) Press the `begin' button and the program will ask to choose the file containing the file names (the recently created plain text file). Once you have selected the file, a layout of the samples' positions on the MALDI plate in the randomized order will be generated, together with the calibrant locations. The layout generated by the randomization software does not represent the 96-well plate where the serum samples will be placed for the solid-phase extraction. Instead, it represents the 384 spots corresponding to the MALDI target plate, showing where the 96 serum samples plus the calibrants will be spotted for mass spectrometry analysis.
39) Next, the program will ask where to save the output file (Autoexecute file), which contains the set of instructions for the FlexControl program to run the mass spectrometer automatically.
40) Print the target layouts (generated in step 38), as they will be needed during the organization of the randomized serum aliquots and will later be filed in the corresponding record's folder.
41| Locate and organize the aliquots in 96-well plastic racks (precooled on dry ice for 10 min) according to the randomized order shown in the target layouts. Always verify the sample list and the layouts to make sure that listed samples, information in the layout and tube identifications match. Take the boxes with the aliquots already organized out of the -80°C freezer and put them in big-flat ice buckets containing dry ice. Then, take aliquots from the boxes and put them in 96-well plastic racks following the randomized order. Keep all aliquots frozen on dry ice at all times.
▲ CRITICAL STEP This is a very tedious but important step. Any error in properly organizing the samples in the randomized order will affect the whole experiment.
▲ PAUSE POINT Serum aliquots can be kept frozen at least 5 years at -80°C.
42| Transfer output files (Autoexecute files) to the MALDI-TOF PC (for instance: C:\data\methods\autoexecute files).
▲CRITICAL STEP Print Autoexecute files to keep them in the records.
43| Schedule the runs of clinical samples. Check the following: Have you randomized all the aliquots to be run, organized them in plastic racks, double-checked to ensure each aliquot is in the correct position on the correct plate and placed them in the -80°C freezer?; Have you generated Autoexecute files and printouts and transferred them to the appropriate folders?; Are all the reagents and materials available for the assay?
44| Start setting up the robot during the morning of the day of the runs by cleaning the Tecan liquid handler, degassing the water that the robot will use for the runs, ensuring the waste container has enough room to receive what the assay will discard. Also, flush the system twice.
▲CRITICAL STEP There should be no visible bubbles traveling the lines throughout the flushes. Flush until no bubbles remain.
? TROUBLESHOOTING
45|Perform an system performance verification experiment (see BOX 5) on the morning of, or the day before, analysis of each sample set submitted for activity assaying.
BOX 5 | EXOPEPTIDASE ASSAY PERFORMANCE VERIFICATION.
1. Thaw an aliquot of frozen human serum (Sigma) on wet ice for 1 h.
2. Prepare the Tecan robot (step 44) and reagents (enough for the run; 10 wells) (see REAGENTS SETUP) while the samples thaw on wet ice. Prepare extra solution in case needed.
3. After the human serum (Sigma) has thawed, transfer 20 μL to the designated wells of 0.2-mL polypropylene “Template III PCR” half-skirted, 96-well microtiter plate. To each serum sample, add 10 μL of 0.2 pmol / μL reference peptide mix (final amount = 2 pmol). And then add 10 μL of 10 pmol / μL C3f_13C (final amount = 100 pmol)
▲ CRITICAL STEP Sample preparation is done on wet ice.
4. Add 40 μL PBS to remainder of empty wells.
5. Cover the 96-well plate with aluminum foil and incubate at RT (19-24 °C) for 3 h.
6. Place plate in the `plate holder 1' position (Figure 2b).
7. Run the Gemini program (identical to the program that is run for the first half of the plate during the runs with clinical samples) (Box 3).
8. Remove the target and take it to the MALDI-TOF mass spectrometer.
9. Insert the MALDI target to be analyzed and select the method `ExoproteaseAssay.par.'
10. Open the quality control Autoexecute template and edit the file (change the date and directory folder for saving the spectra). After editing the file, save a newly named file in the quality control Autoexecute file directory.
11. Open the edited Autoexecute file in the FlexControl program and run it.
12. Open the FlexAnalysis program after the run has finished. Open all the just-created quality control spectra using the `open multiple spectra' function.
13. Overlay the ten mass spectra and make sure that the overlay has a similar peptide complexity to that shown in Figure 5a. In addition, zoom in on the areas to see the degradation ladder (Figure 5b).
14. Calculate the ratios of the ion intensities of selected `exogenous' degradation products (i.e., those derived from labeled, exogenous substrate) over the ion intensities of the corresponding reference peptides (sequences listed in Table 1) for each replicate:
C3f_13C_8 / C3f_D_8;
C3f_13C_10 / C3f_D_10;
C3f_13C_11 / C3f_D_11;
C3f_13C_12 / C3f_D_12;
C3f_13C_16 / C3f_D_16.
Coefficients of variation (C.V.'s) of these ratios, achieved during a quality control analysis of 10 technical replicates, should be <15% for each of the 5 peptide pairs 11.
Automated solid-phase extraction TIMING ~ 6 h
46| Thaw the 48 serum samples, taking the first plastic rack (containing aliquots in the randomized order) from the -80°C freezer and transferring the samples to wet ice for about 60 min, carefully keeping the same order of the samples as in the plastic racks.
47| Prepare reagents and the calibrants (see REAGENTS SETUP).
▲CRITICAL STEP Calibrants and some of the reagents must be prepared the day of the assay.
48| Mix the Dynabeads gently until completely dispersed. Using an 8-well strip of 0.2-mL thin-wall tubes, put 75 μL of magnetic beads in each of the 8 wells and place in the proper holder on the robot deck (Figure 2b).
? TROUBLESHOOTING
49| Add fresh 0.1 % TFA (See REAGENTS) to the TFA trough on the robot (Figure 2b).
50| Prepare a 96-well half-skirted microtiter plate: the first column should contain 100 μL 50% acetonitrile in each well; the second column should contain 100 μL of matrix.
▲ CRITICAL STEP 50% acetonitrile should be made fresh on the day of experiment.
51|Tightly seal the wells of the 96-well half-skirted microtiter plate with a piece of self-adhesive foil using a rubber roller. Attach the plate to the cooler rack with a piece of Parafilm wrapped around the microtiter plate and the inner part of the cooler rack, so the plate will remain in place when the robot tips pierce the foil. Also, use 4 mini binder clips attaching the cooling rack to the robot to prevent it from falling out of position. The cooler rack should be kept at -20°C for several hours before it can be used for a clinical run. Note that the rack changes color with temperature making it easier to know whether it can be used for a run.
? TROUBLESHOOTING
52| Place a clean MALDI target plate in position (Figure 2b).
53| Transfer 20 μL from each aliquot (50 μL) to the proper well of a half-skirted 96-well microtiter plate. Note that only the first half of the microtiter plate contains serum samples. The second half will be used during the SPE automatic process. If prepared properly, the aliquots should have thawed in the same positions as they will be placed in the microtiter plate. Check corresponding randomization layout to make sure
▲CRITICAL STEP Reactions must be prepared on wet ice.
54| Add 10 μL of the non-degradable reference peptide mix (see REAGENTS SETUP) to yield total of 2 pmol per reaction.
▲CRITICAL STEP Change pipette tips in between each sample to avoid cross contamination.
55| Add 10 μL of your substrate (for example, C3f-13C) (see REAGENTS SETUP) to yield a total of 100 pmol per reaction.
▲CRITICAL STEP Change pipette tips in between each sample to avoid cross contamination.
56| Cover the 96-well plate with aluminum foil and incubate at RT (19-24°C) for an appropriate time which has already been determined in steps 4 and 5 (for example, C3f-13C requires 3 h incubation time).
? TROUBLESHOOTING
57| Place plate in the `plate holder 1' position (Figure 2b).
58| After the incubation of reactions is complete, flush the robot one more time.
59| Run the TECAN program. The robot will carry out the steps described in Box 3.
? TROUBLESHOTTING
▲ CRITICAL STEP This protocol can also be done manually using 96-well plate holders, magnet racks and eight-channel multipipettes. However, the manual procedure is not recommended because of a lack of reproducibility.
Mass Spectrometry TIMING ~ 2 h
60| Insert the target plate to be analyzed in the MALDI-TOF mass spectrometer.
61| Go to FlexControl, load the correct Autoexecute file and start the automatic run, this will perform the steps described in Box 4. The method referred to in this Autoexecute file should be updated to use the proper laser energy. The method is a Reflectron mode.
BOX 4 | `ExoproteaseAssay.par' FILE CREATION STEPS.
1. Go to the first spot in the MALDI plate.
2. Start collecting the spectrum for the 0.7- 4 kDa m/z: average 400 laser shots, delivered in four sets of 100 shots (at 50 Hz) to each of four different locations on the surface of the matrix spot (performing a spiral movement from one set to the next one).
3. Acquire spectra in reflectron mode geometry under 20 kV (16.45 kV during delayed extraction) of ion accelerating and -1.4 kV multiplier potentials and with gating of mass ions set to m/z 500. Delayed extraction is maintained for 80 ns to give time lag focusing after each laser shot. The effective laser energy delivered to the target is carefully controlled to be 16 μJ (± 10%) per shot. The entire irradiation program is controlled using the instrument's `Autoexecute' function, which automates the following steps: loading the correct spectrum acquisition method (with the optimized instrument settings), going to the right spot on the MALDI plate, and delivering the four sets of 100 shots.
4. Keep collecting mass spectra for all of the samples and calibrants.
? TROUBLESHOOTING
▲ CRITICAL STEP The spectra can also be collected manually, going spot-to-spot and delivering sets of laser shots using the acquisition method optimized for the mass range (0.7 - 4 kDa). However, the manual procedure is very tedious and subject to bias as the operator aims the laser beam at an approximate location of choice.
62| Open the FlexAnalysis program after the run has been completed. Open all the spectra that have just been generated using the `open multiple spectra' function and check that all the spectra have been created (by comparing the spectra with the Autoexecute file of the run).
63| Perform spectral analysis. This can be done through the `Process' functions in FlexAnalysis. Alternatively, spectra can be translated to tab-separated text files that contains both x and y coordinates for each spectrum as separate columns using a custom-written macro (see EQUIPMENT). Once the spectra are in text format, they can be analyzed using different software.
Signal Processing and Quantitation
64| Convert mass spectra from binary format to ASCII files, containing two columns of data (x: m/z; y: intensity), by a custom-written macro in FlexAnalysis (Bruker Daltonics, Billerica, MA).
65| Transfer data to MATLAB. Do additional data processing with a custom script, “qcealign” invoking qpeaks, a commercial program, to do smoothing, base-line subtraction, and peak labeling (see EQUIPMENT) 13.
66| Use Signal Processing & Preview (SPP), a custom-built graphical viewer for spectra in ASCII format, to plot raw and processed spectra side by side to review the outcome of signal processing and to optimize parameters for Qpeaks 13. The singlet width parameter should be set to 1500, thereby specifying the resolution for processing. After processing, a peak table with normalized intensities, smoothed curve, and baseline is created for each spectrum before alignment.
67| Use the custom algorithm, “Entropycal”, to align sample data files to a reference file (a spectrum sum of all the sample files) using a minimum entropy algorithm by taking unsmoothed (“raw”), baseline-corrected data 13. All peaks in the rows within (m/z) of the strongest peak at a given m/z value are binned together, and a spreadsheet containing the normalized aligned data is created for further data analysis.
68| Analyze this spreadsheet in conjunction with a custom visual interface for processed spectra, “Mass Spectra Viewer” (see EQUIPMENT), to select only those peaks that correspond to the peptide ladder(s) resulting from peptide substrate degradation, and to the spiked reference peptides 12.
69| Calculate the ratios of the normalized ion intensities of the DEGR peaks over the normalized ion intensities of the corresponding REF peptide peaks for each rung of the ladder (this can be completed in Excel). The spreadsheet resulting from the replicate analyses should be subjected to the same process, and the DEGR/REF ratios for each of the rungs of the same peptide ladder from the replicate experiments averaged.
Importing data and creating experiments in GeneSpring
70| In the same location as the parameter file, Qcealign will have created a “processed01.out” file; check this is present This is the binned, aligned peaklist for all samples.
71| Launch GeneSpring. From the File Menu, select “Import Data”. In the resulting dialog window, navigate to and select the “processed01.out” file. This is the binned, aligned peaklist for all samples of the project; it is in the same location as the parameter file and it is created by Qcealign 13. A dialog will appear asking for the name. Click “Create a New Genome” and enter a name for the dataset. For this example, we will use “CancerStudy” which is the name of the parent folder where the spectra are stored. Then click “Next”. Another window appears. The first column should be set to “Gene Identifier”. The remaining columns should be set to Signal. Then, click next to dismiss this window. Another window will appear asking to import more data. Since we have none, click next again. A window will appear, saying how many samples are created. Click “Yes” to continue. Then GeneSpring will create an experiment. When you hit “Next” to continue to create an experiment, a warning may appear saying that a column lacks a title. This means that there is an empty column. Scroll through and locate the empty column (usually the last column) and change the column designation from “Signal” to “Unused”.
72| To create an experiment, first type a name for the Experiment and hit “Save.” We will call it “CancerStudy” for our data set. Next, a window appears to set the different statistical properties of the experiment. Click on “Normalizations.” Since Qcealign already normalized the data, we need no normalizations. Remove all default normalizations using the “delete” button and press “OK.” Then, click on the “Parameters” button. In the appearing window, enter the parameters you want to study. Click on “New Parameter” and enter the clinical information for each sample. Enter “Parameter1” as the name for the Column, accepting the default column properties. If the Parameter is Numeric (i.e. body weight), then set “Numeric” to “Yes”, otherwise leave it as “No.” Similarly, set the value for “Logarithmic” to Yes or No, depending on whether the data is in log scale. One can enter many parameters with which to analyze the data. Copy the parameter information from the spreadsheet obtained in step 8. Press “Save” when done.
73| Next, click on “Experiment Interpretation”. In the resulting dialog, for the Interpretation named “Default Interpretation,” set Mode to “Ratio (signal/control)”. Also set the clinical Parameters. Make sure “Use Cross-Gene Error Model in this Interpretation” is unchecked. No conditions should be excluded. Press “Save” to continue. Since we don't use the Error Model, skip the “Error Model” button and press “Close.”
Statistical Analysis
74|Depending on the number of samples available two different strategies can be used to do the statistical analysis. In option A, training and test set are generated if enough samples are available. The training set is used to build a statistical model that later can be validated using the test set. In option B, the statistical analysis is done on the entire dataset using a cross-validation approach.
Option A
Setting up Training and Test Sets
(i) From the Experiment Menu, select “Create New Experiments”. Select the “Filter on Parameter” tab. One of the added Parameters contains information specifying whether samples were part of the “Training” or the “Test” set. In that parameter, the training set samples are labeled as “Training” while the additional test set samples are marked as “test.”
(ii) From the “Filter On Parameter,” we select the Parameter Value “Training” and click on “Add All.” This adds all the samples from the training set to a new experiment. Click “Next”. A window appears. Click “Import Parameter.” Select the previous experiment. All the parameters from that experiment will appear. Click “Select All” and then “OK.” The values for the training samples will be moved into this experiment. Press “Next.” A window asking about Normalizations appears. Remove all normalizations as before and press “Next.” A window asking for a name for the Experiment appears. We name this set “Training.” All dialogs disappear and the main window reappears.
(iii) With the new “Training” experiment selected, from the Experiment window, select “Experiment Interpretation”. Change Mode to “Ratio (signal/control)”.
(iv) Repeat Steps (i) to (iii) to create a new Experiment called “Test” where the samples labeled “Test” are moved in with their corresponding parameter information.
(v) Differential expression: From the Tools Menu, select “Statistical Analysis (ANOVA)”. Make sure it indicates “all genes” next to the “Choose Gene list” button. Choose the default Interpretation for the Training experiment just created. Then click “Choose Experiment” and the name should appear next to the button. Mode should be “Ratio (signal/control)”. Make sure that Cross-Gene Error Model is inactive (Click next to dismiss this window). Click on the “Parameter to Test” pull-down menu and select the desired parameter. The settings should be the following:
-Test Type should be set to “Non-parametric” test.
-False Discover Rate should be set to .05
-Multiple Testing Correction should be set to “Benjamini and Hochberg False Discovery Rate.”
- There should be no Post Hoc Tests?
▲ CRITICAL STEP If the Experiment and Cross-Gene Error Model values are not correct, click “Close”. Then go back to the Experiment Interpretation window and set the proper values. One can do this be selecting “Experiment Interpretation” from the “Experiment” Menu.
(vi) Click “Start” and Save your results (called a gene list) with an appropriate name. We will call ours “CancerStudy_p05” and hit “Save” to record the results. The p-value can be changed to a more stringent value as needed. We generally save a gene list from the results of p<0.00001.
(vii) Class prediction: From the “Tools” Menu, select “Class Prediction.” Set the Training Experiment to be the Training Set. Working in the K-Nearest Neighbors tab, select the “Parameter to Predict.” We select “Parameter 1.” Set Gene Selection Method to be “All Genes from Selected List.” Select the Gene List saved from step v. Set the decision cutoff for p-value ratio to 1. Press “Start.” To save the results from the pop-up window, copy and paste into an Excel spreadsheet. To optimize the results with the training set, repeat by varying the number of neighbor from 3 to 9. Once optimal conditions (as judged by the lowest prediction errors) are found through iterative cross-validation using the training set, select the “Test” experiment and set it as the “Test Set”. Change Function from “Crossvalidate Training Set” to “Predict Test Set.” Keeping the values for optimal conditions found from the Training Set, press “Start.” Save the Results in an Excel spreadsheet.
(viii) For Support Vector Machine, select the second tab in the “Class Prediction” window. Keeping the same training set, change function to “Crossvalidate Training set.” Set “Parameter to Predict” to the appropriate parameter. We select “Parameter 1”. The Gene list created in step v should be the one used as before. Gene Selection Method remains “All genes from Selected List,” unchanged. Press “Start.” Save the results in an Excel spreadsheet by copying and pasting. Optimize the conditions for the training set by varying the kernel function. Occasionally, it might be necessary to change the scaling factor from 0 to 1 or 2 (that depends on how balanced the number of samples are in the groups tested). Once optimal values are found, then set the Test to the “Test” Experiment. Change the Function to “Predict Test Set.” Save the results in an Excel Spreadsheet. Press “Close” to exit Class Prediction.
Option B
Class prediction with Leave-one-out-crossvalidation (LOOCV)
(i) From the “Tools” Menu, select “Class Prediction.” Set the Training Experiment to be the Training Set. Working in the K-Nearest Neighbors tab, select the “Parameter to Predict.” We select “Parameter 1.” Set Gene Selection Method to be “All Genes from Selected List.” Select the Gene List saved. Set the decision cutoff for p-value ratio to 1. Press “Start.” To save the results from the pop-up window, copy and paste into an Excel spreadsheet. To optimize the results with the training set, repeat by varying the number of neighbor from 3 to 9. Once optimal conditions (as judged by the lowest prediction errors) are found though iterative cross-validation using the training set, select the “Test” experiment and set it as the “Test Set”. Change Function from “Crossvalidate Training Set” to “Predict Test Set.” Keeping the values for optimal conditions found from the Training Set, press “Start.” Save the Results in an Excel spreadsheet.
(ii) For Support Vector Machine, select the second tab in the “Class Prediction” window. Keeping the same training set, change function to “Crossvalidate Training set.” Set “Parameter to Predict” to the appropriate parameter. We select “Parameter 1”. The Gene list created should be the one used as before. Gene Selection Method remains “All genes from Selected List,” unchanged. Press “Start.” Save the results in an Excel spreadsheet by copying and pasting. Optimize the conditions for the training set by varying the kernel function. Occasionally, it might be necessary to change the scaling factor from 0 to 1 or 2 (that depends on how balanced the number of samples are in the groups tested). Once optimal values are found, then set the Test to the “Test” Experiment. Change the Function to “Predict Test Set.” Save the results in an Excel Spreadsheet. Press “Close” to exit Class Prediction.
Visual inspection of processed spectra (MassSpectra Viewer -- MSV)
75| Create a viewer definition file. This tells the viewer which samples are in which clinical group (see Supplementary material 4). The definition file needs a minimum of three columns and looks like the example below. The basefile name column does not use “_1” or “_2” - just the name. The MSV automatically adds the suffix when looking for the ASCII files. The next columns (zero to an unlimited number) list various demographic or clinical parameters, such as gender, age, etc… No spaces are to be used anywhere. The columns are tab-separated:
| # | Basefile name | Group | Parameter 1 (gender) | Parameter 2 (age) | Parameter 3 (xyz) |
| 1 | 000ZG60005DVA | Cancer1 | Female | 56 | |
| 2 | 000ZG70007DCK | Cancer1 | Male | 61 | |
| 3 | 000ZG80003VKD | Control | Male | 58 | |
| 4 | 000ZG90002EGO | Control | Female | 52 |
76| Go to MATLAB and type “masspectraviewer” in the command window 12. Click on “Folder Setting”. A window appears. Press “Browse” next to the “Folder of ASCII files” and select the directory holding the processed ASCII data files. It should be called “Final_ASCII_Spectra” according to our data structure and located inside the Processed01 folder. Press the second “Browse” button and select the “Matrix” folder, also in the Processed01 folder. Press the last “Browse” button and select the viewer definition file, which was created in the previous step. Then press “Save & Return.” Then press “Make Matrix.” Once that is done, press “Group Color.” A dialog box will appear. The clinical subgroups will appear in the left box. Select each item. Pick a color by scrolling up and down the color bar. Once the appropriate color is picked, press the “Select” button to assign that color to that subgroup. Repeat for each subgroup. Once all the groups are given a color, press “Save and Return.” Once the main screen returns, press Update. Zoom in on peaks selected for the clinical parameter and verify the results.
▲ TROUBLESHOOTING
77| Repeat steps 75-76 for each additional clinical parameter that was used during the statistical analysis.
TIMELINE
Testing new candidate exoprotease substrates (Steps 1-5), ~2-14 h
Synthesizing labeled/unlabeled exopeptidase substrates and reference peptides (Step 6). Timing depends on the turnaround times of the peptide synthesis facility; typically ~2 to 6 weeks
Quantitate newly synthesized peptides by amino acid analysis (Step 7).
Timing depends on the turnaround times of the amino acid analysis facility; typically ~1 to 4 weeks
Testing new labeled exoprotease substrates and reference peptides in serum pools (Steps 8-16), ~2-15 h
Blood collection and Serum preparation and storage (Steps 17-35), ~21 h
Set up of the robot run (Steps 36-45), ~4 h
Automated solid-phase extraction (Steps 46-59), ~6 h
Mass Spectrometry (Steps 60-63), ~2 h
Data Analysis (Steps 64-77), ~8 h
TROUBLESHOOTING
Table 2: Troubleshooting Table
Table 2.
Troubleshooting Table
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| Testing new peptide candidate (1-6) | Not able to see the parent peak | Concentration too low (or too high) | Try different concentrations |
| Not able to see the degradation ladder | Wrong concentration / incubation time | Adjust concentration and incubation times | |
| Blood collection (12) | Serum is red | Blood hemolysis | Fill blood collection tubes to the top. Also, make sure that clotting times and other preceding steps such as centrifugation speeds and time are strictly followed |
| Robot Set-up (BOX 3) | No sample spotted on the MALDI plate | Air bubbles in the robot lines | Extensively flush all of the lines and make sure that the water intake is properly sparged |
| Reagents preparation for automated solid phase extraction (BOX 3) | Inconsistent MS signals among samples | Insufficient mixing of the magnetic beads | Make sure the beads are in a homogeneous suspension before they are placed in the 8-well strip to avoid clumping |
| Low signal-to-noise of peptide ion signals | Acetonitrile evaporation due to either a problem with the aluminum foil sealing of the cooler rack plate or with the cooler not being cold enough | Make sure that the aluminum foil perfectly seals the 96-well plate and that the cooler has been at -20° C for several hours before it is used for a clinical run. Check that the rack has the correct color before using it | |
| Peptide ion signal is suppressed by matrix signal | Evaporation in the original matrix solution container (concentration of matrix is too high) | Make sure that the original matrix container is perfectly capped at all times, and at 4° C | |
| Sample evaporation | Long incubation at room temperature | Cover the samples with aluminum foil | |
| Tecan Run (BOX 3) | The robot ROMA arm crashes (i.e., not aligned properly) when it transfers the 96-well plate to the “side-magnet rack” | The “side-magnet rack”has two possible orientations on the deck. The present orientation may not be able to receive the microtiter plate from the ROMA | Make sure that the Gemini program and the actual orientation of the rack on the Tecan layout agree |
| Low signal-to-noise of peptide signals | Timing delays in bottom magnet rack are not sufficient to collect the magnetic beads in the bottom of the wells. OR the final eluate is diluted because there is wash buffer remaining after the wash removal steps | Increase the delay times to collect the magnetic beads in the bottom of the wells and/or reset the heights of the tips when removing the washing buffer | |
| Non-Uniform or overlapping spots on MALDI plate | Tips are touching the MALDI plate during spotting, so the spotting movements become erratic and spots are not homogenous, or the tips are not close enough to the plate during spotting so there is no proper spreading of the sample | Recalibrate the heights of the Tecan tips to make them all uniform | |
| Mass Spectrometry (53) | Low signal-to-noise of peptide ion signals | Laser power is too low to desorp/ionize the peptides. | Increase the laser power in 2% increments until the peptide signals appear |
| Inconsistent MS signals among samples | Sample crystallization is not homogeneous because there is not enough organic solvent | Make sure that the original matrix container is tightly capped at all time at 4° C | |
| Converting raw data to ASCII (56) | Macro does not appear in the tools menu | Macro is installed in the wrong folder | Find where the other macros are installed. Typically they are in “C:\Methods\FlexAnalysisMacroModules”. But setup may vary. But if other items are appearing under the Tools menu, this macro should be placed in the same location as the other items |
| Processed spectra overlay (MSV) (57) | Typing “masspectraviewer” does not launch the viewer | Matlab does not know where the viewer is installed | Follow directions in the setup to reinstall the MSV and its associated files 12 |
| The Folder field shows “0” instead of the path to the folder in the “Folder Setting” dialog | The original folder was moved No folder was selected. The “cancel” button in the Browse button was pressed | Reselect the folder by pressing the “Browse” button | |
| “Group Color” does not show the clinical subgroups | The viewer definition file could not be found or is the wrong format | Create a viewer definition file as described in Step 29 (see also Supplementary Method 1) | |
| The viewer definition file is the wrong format | Check the definition file to make sure that tabs are used instead of spaces Make sure that all comments are preceded by the “#” character Make sure that there is no extra carriage return until the last data set is defined |
||
| Clinical subgroup is of the wrong color or not displayed in the legend | Subgroup has not been selected | Be sure to click on the subgroup and set the color. Then hit the “Select” button |
ANTICIPATED RESULTS
Although protease assays that use peptide substrates and a MALDI-TOF MS read-out may be employed to answer a variety of questions in the health and life sciences (see Introduction), the only applications so far of the unique platform/method combination described here have been to screen for proteolytic activities in serum and plasma 8, 11, and to monitor purification of individual exopeptidases from cancer cell secretomes (unpublished). In contrast to C3f, a surprisingly large number of `other' serum peptides that have been tested as candidate assay substrates didn't degrade in serum or plasma, not even after prolonged incubations (unpublished observations). Only fibrinopeptide A (FPA: ADSGEGDFLAEGGGVR), FPA without the N-terminal Ala (FPA(-A): DSGEGDFLAEGGGVR), a peptide mapping to the C-terminus of the clusterin beta-chain (Clus2: RPHFFFPKSRIV), and a peptide derived from alpha-1-antitrypsin (A1AT: LMIDQNTKSPLFMGKVVNPTQK) have proven utility as assay substrates at this time 11. Substantial degradation of 100 pmoles of any of the five aforementioned substrates can be anticipated within 15 min (FPA) to 15 h (FPA(-A)) after addition to 20 μL human control serum or plasma 11. More specifically, the C3f-based system performance verification test described in Box 5 should yield the indicated results after 1-3 h incubation, with C.V.'s of normalized MS peptide-ion intensities of 5 selected metabolites (see Box 5) all below 15% over 10 technical replicates at fixed incubation times. This also applies to replicate assays using Clus2, FPA(-A) and A1AT peptides as substrates (2 h incubation for Clus2; 6-15 h for the others). FPA degrades within minutes after addition to serum, which makes accurate timing and reproducibility exceedingly difficult, especially when dealing with large sample sets, typically resulting in C.V.'s >30%.
The current protocol has also been used to assay serum samples obtained from 48 patients with metastatic thyroid cancer and 48 gender-and age-matched healthy controls, and utilizing 3 different peptide substrates (C3f; FPA(-A); Clus2) in separate analyses. The full array of peptide metabolites (a de facto 26-member `mini-peptidome') was then quantitated and subjected to multivariate statistical analysis and machine learning methods to ultimately yield class predictions with 94% sensitivity and 90% specificity 11. It should be noted, however, that none of these 26 individual peptides comprising the assay degradome could, by itself, be used as a quantitative `biomarker' (in the form of a MALDI-TOF MS-generated ion intensity signal) to completely distinguish all the cancer patients from healthy controls. At best, the ion intensities of certain peptide peaks, as for instance for the three C3f degradation products in the color-coded spectral overlays shown in Figure 6, are on average higher in one particular group than in the other, but without being completely separated. In our hands, no such perfect separation has been observed until now between any two cancer patient / control groups, for any of the degradation products of any substrate peptide tested (unpublished observations). Considering the complexity of blood protease panels, and the unique concentrations and activities of each of the individual members acting in concert, and in view of the biological variability, it is perhaps unrealistic to anticipate discovery of a single peptide metabolite that will have bona fide diagnostic capacity. Instead, the metabolites must either be interpreted as diagnostic `patterns' (as is common practice at the moment) and/or specific assays should be developed for the individual enzymes.
Figure 6. MALDI-TOF MS overlays of selected ion-peaks corresponding to C3f_13C degradation products in sera from thyroid cancer patients and healthy controls.

Selected degradation products derived from 13C-labeled C3f (40 pmoles starting material), following a 3-h incubation in separate 20-μL aliquots of each of the individual samples, are shown. Spectra were obtained, aligned and normalized as described under `Signal Processing and Quantitation' (steps 64-69), and are displayed using the Mass Spectra Viewer. Each of the three overlays contains 96 spectra with normalized intensities: 48 controls (in blue) and 48 thyroid cancer patients (red). The `C-13' isotope from the isotopic envelope is shown for each peptide-ion peak. Substrate sequence, position of the label (first Leu -- in blue) and mono-isotopic molecular mass is listed in Table 1.
Supplementary Material
Figure 4. Exopeptidase assay: C3f isotope-labeling design.

MALDI-TOF MS showing the isotopic envelopes for each of the three iso-peptides having an identical sequence (RIHWESASLL), derived from C3f, but different molecular masses: endogenous, exogenous (degradation product), and reference (i.e., internal standard). Note that the exogenous peptide was singly labeled by incorporation of one [13C6]leucine; each all-D reference peptide was doubly labeled with two [13C6,15N1]leucines, hence the 14-Da mass difference between the endogenous and reference peptide.
Figure 5. Assay Performance Experiment.

A. Overlay of the ten mass spectra of a QC experiment. The spectral overlay shows the result of the weekly quality control experiment performed to assess the reproducibility of the liquid handler and the MALDI-TOF mass spectrometer. Ten of the 48 sample wells in half of a 96-well microtiter plate are filled with 20 μL of human serum, 10 μL of 0.2 pmol / μL reference peptide mix and, 10 μL of 10 pmol / μL substrate (C3f-13C). Samples are incubated for 30 min in RT and then processed using the same automated solid-phase extraction protocol.
B. Sections of the spectral overlay shown in panel A. Four mass range windows for the peptides with m/z= 1865.00, 1211.65, 1690.93 and 1449.76 peptides are shown. The mono-isotopic mass (m/z) is shown for each peptide-ion peak.
ACKNOWLEDGMENTS
This work was supported by US National Institutes of Health grants R33 CA111942 and U24 CA126485. It is part of NCI's Clinical Proteomic Technologies Initiative (http://proteomics.cancer.gov) and Clinical Proteomic Technology Assessment for Cancer (CPTAC) consortium (Broad Institute of MIT and Harvard; Memorial Sloan-Kettering Cancer Center; Purdue University; University of California, San Francisco; and Vanderbilt University School of Medicine).
Footnotes
Competing Financial Interests The authors declare that they have no competing financial interests.
REFERENCES
- 1.Lopez-Otin C, Bond JS. Proteases: multifunctional enzymes in life and disease. J Biol Chem. 2008;283:30433–30437. doi: 10.1074/jbc.R800035200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Overall CM, Blobel CP. In search of partners: linking extracellular proteases to substrates. Nature reviews. 2007;8:245–257. doi: 10.1038/nrm2120. [DOI] [PubMed] [Google Scholar]
- 3.Palermo C, Joyce JA. Cysteine cathepsin proteases as pharmacological targets in cancer. Trends in pharmacological sciences. 2008;29:22–28. doi: 10.1016/j.tips.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 4.Lopez-Otin C, Matrisian LM. Emerging roles of proteases in tumour suppression. Nat Rev Cancer. 2007;7:800–808. doi: 10.1038/nrc2228. [DOI] [PubMed] [Google Scholar]
- 5.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 6.Leiting B, et al. Catalytic properties and inhibition of proline-specific dipeptidyl peptidases II, IV and VII. Biochem J. 2003;371:525–532. doi: 10.1042/BJ20021643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martinez JM, et al. Aminopeptidase activities in breast cancer tissue. Clin Chem. 1999;45:1797–1802. [PubMed] [Google Scholar]
- 8.Villanueva J, et al. Serum peptide profiling by magnetic particle-assisted, automated sample processing and MALDI-TOF mass spectrometry. Anal Chem. 2004;76:1560–1570. doi: 10.1021/ac0352171. [DOI] [PubMed] [Google Scholar]
- 9.Villanueva J, et al. Differential exoprotease activities confer tumor-specific serum peptidome patterns. J Clin Invest. 2006;116:271–284. doi: 10.1172/JCI26022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Villanueva J, et al. Serum peptidome patterns that distinguish metastatic thyroid carcinoma from cancer-free controls are unbiased by gender and age. Mol Cell Proteomics. 2006;5:1840–1852. doi: 10.1074/mcp.M600229-MCP200. [DOI] [PubMed] [Google Scholar]
- 11.Villanueva J, et al. A sequence-specific exopeptidase activity test (SSEAT) for “functional” biomarker discovery. Mol Cell Proteomics. 2008;7:509–518. doi: 10.1074/mcp.M700397-MCP200. [DOI] [PubMed] [Google Scholar]
- 12.Villanueva J, Lawlor K, Toledo-Crow R, Tempst P. Automated serum peptide profiling. Nature protocols. 2006;1:880–891. doi: 10.1038/nprot.2006.128. [DOI] [PubMed] [Google Scholar]
- 13.Villanueva J, Philip J, DeNoyer L, Tempst P. Data analysis of assorted serum peptidome profiles. Nature protocols. 2007;2:588–602. doi: 10.1038/nprot.2007.57. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.