

Abstract
Hybrid quantum mechanics/molecular mechanics calculations using Austin Model 1 system-specific parameters were performed to study the SN2 displacement reaction of chloride from 1,2-dichloroethane (DCE) by nucleophilic attack of the carboxylate of acetate in the gas phase and by Asp-124 in the active site of haloalkane dehalogenase from Xanthobacter autotrophicus GJ10. The activation barrier for nucleophilic attack of acetate on DCE depends greatly on the reactants having a geometry resembling that in the enzyme or an optimized gas-phase structure. It was found in the gas-phase calculations that the activation barrier is 9 kcal/mol lower when dihedral constraints are used to restrict the carboxylate nucleophile geometry to that in the enzyme relative to the geometries for the reactants without dihedral constraints. The calculated quantum mechanics/molecular mechanics activation barriers for the enzymatic reaction are 16.2 and 19.4 kcal/mol when the geometry of the reactants is in a near attack conformer from molecular dynamics and in a conformer similar to the crystal structure (DCE is gauche), respectively. This haloalkane dehalogenase lowers the activation barrier for dehalogenation of DCE by 2–4 kcal/mol relative to the single point energies of the enzyme's quantum mechanics atoms in the gas phase. SN2 displacements of this sort in water are infinitely slower than in the gas phase. The modest lowering of the activation barrier by the enzyme relative to the reaction in the gas phase is consistent with mutation experiments.
Haloalkane dehalogenases catalyze the hydrolytic cleavage of carbon–halogen bonds in aliphatic and aromatic halogenated compounds. The haloalkane dehalogenase from the nitrogen-fixing hydrogen bacterium Xanthobacter autotrophicus GJ10 (DhlA) prefers 1,2-dichloroethane (DCE) as substrate and converts it to 2-chloroethanol and chloride (1). A catalytic triad consisting of Asp-124, His-289, and Asp-260 is the central residue in the dehalogenation reaction (Scheme S1). On binding DCE in the predominantly hydrophobic active site, it undergoes SN2 displacement of chloride by nucleophilic attack of Asp-124-COO− to form an ester intermediate at the rate of 50 ± 10 s−1 (1, 2). The ester intermediate is subsequently hydrolyzed by an activated water molecule. The dyad of His-289 and Asp-260 is thought to be responsible for activating the water molecule (3). This enzyme functions most efficiently at pH 8.2, likely because the imidazole NE2 of His-289 needs to be unprotonated for the hydrolysis reaction to proceed. Dehalogenases are of great interest because they are able to react with halogenated molecules under mild conditions (4). Many halogenated molecules are pollutants, and bioremediation is a highly desirable method for removing these harmful molecules from the environment. Dehalogenases have not existed in nature for a long time and have not yet evolved into optimal enzymes. For example, the catalytic efficiency for DhlA is 4,550 M−1⋅s−1 for DCE (5), as compared with 5.6 × 107 M−1⋅s−1 for decarboxylation of orotidine 5′-monophosphate by orithine 5′-monophosphate decarboxylase from yeast (6). A better understanding of the factors that influence the dehalogenation reaction in these proteins could eventually lead to an enzyme with improved catalytic properties.
Scheme 1.

DhlA consists of 310 residues and is a α/β hydrolase protein. The enzyme contains two domains: the main domain consists of an eight-stranded β-sheet surrounded by α-helices and contains the active site; the cap domain consists of five α-helices. This enzyme is an excellent candidate for computational study because it is monomeric and the crystal structure has been solved at high resolution (2). A hybrid quantum mechanics/molecular mechanics (QM/MM) (7, 8) study was performed to determine the activation barrier for the SN2 displacement reaction of chloride from 1,2-dichloroethane in this haloalkane dehalogenase. We find that the enzyme makes only a small contribution in lowering the activation barrier in the dehalogenation reaction relative to the reaction in the gas phase when taking the lowest energy path to the transition state (TS) from the near attack conformer (NAC) of the ground state (9, 10). The reaction barrier in the enzyme, however, is expected to be considerably lower than that in water.
Methods
The accuracy of our QM/MM results depends on how well the chosen Hamiltonian (energy function) reproduces the experimental heats of formation of the molecules along the reaction coordinate. To this end, the semiempirical Austin Model 1 (AM1) Hamiltonian (11) was recalibrated to reproduce the energetics and geometries of molecules along the SN2 reaction path of the nucleophilic attack of acetate to 1,2-dichloroethane (Table 1). The AM1 parameters were fitted to heats of formation, dipole moments, and minimum energy structures. The dipole moments and structures were calculated by using gaussian 98 (12) at the MP2/6–31G(d) for the molecules and MP2/6–31+G(d,p) for the complexes. The complexes were partially optimized with the O⋅⋅⋅C distance fixed, and the nonreacting carboxylate oxygen is between the hydrogens of the reacting carbon of DCE (Fig. 1A). This geometry for the reactants is similar to that found in DhlA for a NAC. An optimization of the AM1 parameters by using the Levenberg–Marquet method was performed (13). Fifty minimization steps were performed on the target molecules and complexes before each optimization cycle. A set of AM1-system specific parameters [AM1-SSP; see supplementary material (www.pnas.org)], which reasonably reproduced the heats of formation, dipole moments, and geometries along the SN2 displacement of chloride from DCE by acetate was obtained (Table 1).
Table 1.
Heats of formation for reactants, complexes, and products for the dehalogenation reaction of DCE by acetate, kcal/mol
| Molecule | AM1 | AM1-SSP | Target |
|---|---|---|---|
| Reactants | |||
| Acetate | −115.47 | −122.36 | −120.70* |
| DCE | −33.12 | −31.34 | −29.98† |
| Complexes‡ | |||
| 1.9 Å | −135.50 | −150.71 | −152.36 |
| TS | −135.59 | −150.86 | −150.15 |
| 2.4 Å | −158.56 | −164.52 | −163.63 |
| 2.8 Å | −160.55 | −167.18 | −167.74 |
| 3.0 Å | −159.87 | −166.47 | −167.26 |
| 3.2 Å | −159.00 | −165.44 | −166.20 |
| 4.0 Å | −155.73 | −161.39 | −161.46 |
| 5.0 Å | −153.73 | −158.70 | −157.58 |
| Products | |||
| 2-Chloroethylacetate | −110.08 | −111.25 | −111.12§ |
| Chloride anion | −37.66 | −54.93 | −54.36¶ |
Ref. 31.
Ref. 32.
The heat of formation for these complexes was obtained by adding the ab initio interaction energy for the complex to the experimental heats of formation of acetate and DCE. The first column list the distance between the attacking carboxylate oxygen to the reacting carbon (see Fig. 1A).
The heat of formation was obtained from a CBS4 calculation (Ref. 33).
Ref. 34.
Figure 1.

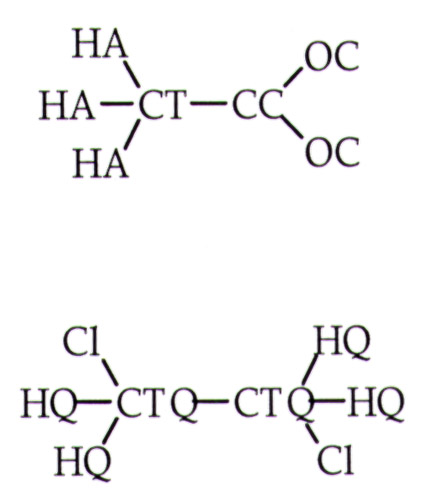
A shows the geometry of acetate and DCE as found in enzyme NAC and used in the ab initio calculations of the interaction energies. B shows the relative ab initio optimized geometries of the reactants obtained without angular contraints.
The AM1-SSP were inputted into the program charmm (Ver. 27a2) for QM/MM calculations (14). Two-dimensional potential energy surfaces for the SN2 chloride displacement from DCE were calculated by using the AM1-SSP Hamiltonian for acetate interacting with DCE as a function of the bond making (O⋅⋅⋅C) and bond breaking (C⋅⋅⋅Cl) in the gas phase. All atoms were treated quantum mechanically. The lengths of these two bonds were constrained by using harmonic constraints with force constants of 5,000 kcal⋅mol−1⋅Å−2 for each constraint. Two harmonic angular constraints were placed between DCE and acetate. A constraint was placed on the angle formed by the two carbons of DCE and the attacking oxygen of acetate (C—C⋅⋅⋅O) to have an equilibrium value of 90° (see Fig. 1A). The other angular constraint was on the angle formed by the reacting carbon in DCE and the attacking oxygen and carboxylate carbon of acetate (C⋅⋅⋅O—C) to have an equilibrium value of 120°. Both angle constraints used a force constant of 25 kcal⋅ mol−1⋅degree−2. These angular constraints were necessary to ensure that the constrained O⋅⋅⋅C distance was the shortest between acetate and DCE (see Fig. 1B). The potential energy of the system was minimized by using a combination of steepest descents (200 steps) and adopted basis Newton–Raphson methods (500 steps). O⋅⋅⋅C was varied from 3.2 to 1.4 Å, and C⋅⋅⋅Cl was varied from 1.8 to 2.6 Å. The TS was determined by locating the saddle point on the potential energy surface.
For QM/MM calculations, the van der Waals parameters (supplementary material; see www.pnas.org) of the AM1-SSP atoms were adjusted to reproduce ab initio interaction energies (15) of DCE and acetate interacting with a water molecule over a series of distances and geometries calculated at the MP2/aug-cc-pVDZ level of theory and corrected for basis set superposition error by using the counterpoise method (16). In addition, a specific van der Waals interaction between the leaving chloride of DCE and the NE1 of Trp-125 and Trp-175 was used in the calculation (ɛ = −3.50 kcal/mol, σ = 3.50 Å). The energy of this interaction was linearly scaled from 0 to −3.50 kcal/mol between the C—Cl bond length of the leaving chloride in DCE from 1.8 to 2.2 Å, respectively. This energy term was necessary to reproduce the ab initio interaction energy between the transition-state structure and indole used in calibrating the van der Waals parameters.
For QM/MM calculations in the enzyme active site, two coordinate sets with DCE in different positions were used (Fig. 2). The final coordinates from a previous molecular dynamics (MD) simulation of the haloalkane dehalogenase containing the transition-state structure with His-289 protonated at ND1 was used for one set of coordinates (17). This coordinate set will be denoted COMF1 in the text and corresponds to a NAC. For the other conformer, the final coordinates from the above-mentioned MD simulation were initially used. The DCE in the active site was rotated by ≈90°, placing the molecule in the same position found in the crystal structure. This coordinate set is denoted COMF2 in the text. DCE is in a gauche conformer for both starting structures (17). DCE is in the trans conformation in the crystal structure (2). The trans conformation for DCE would likely make nucleophilic attack by Asp-124 difficult because a hydrogen from DCE would be directly between the attacking carboxylate oxygen and the reacting carbon. A sphere of TIP3P water 20 Å in radius centered on the carboxylate of Asp-124 was overlaid with the enzyme coordinates (18). Any oxygen of a water molecule within 2.8 Å of an enzyme atom was deleted. A sphere 20 Å in radius centered on the carboxylate group of Asp-124 was removed from the enzyme. DCE and the —CH2COO− sidechain of Asp-124 were treated as QM atoms. The QM/MM boundary was between CB (QM)-CA (MM) in Asp-124. A link atom was placed on CB of Asp-124 to fill out the valency (8). The final system consisted of 15 QM atoms and 4,237 MM atoms (222 waters). Quantum mechanical atoms here treated by using AM1-SSP and MM atoms were treated by using charmm parameters (19). Electrostatic interactions were cut off at 12 Å by using a shifting function, and the Lennard–Jones term was cut off by using a switching function between 10 and 11 Å (14). Residues not attached to their preceding and following residues were constrained by using harmonic constraints. The methodology used for preparing the active site of the enzyme was the same as used by Brooks and Karplus for their stochastic boundary molecular dynamics study of lysozyme (20). Two-dimensional QM/MM energy surfaces were calculated for the SN2 displacement reaction in the enzyme active site. The QM/MM energy surface was generated as a function of two distances; the attacking carboxylate oxygen of Asp-124 to the carbon bonded to the leaving chloride (OD2⋅⋅⋅C2) and the bond length of the carbon chloride bond (C2⋅⋅⋅Cl2). Harmonic constraints with force constants of 5,000 kcal⋅ mol−1⋅Å−2 were used to vary the distances. OD2⋅⋅⋅C2 was varied from 3.2 to 1.4 Å, and C2⋅⋅⋅Cl2 was varied from 1.8 to 2.6 Å. Typically, the potential energy was minimized by using 100 steps of steepest descents followed by 500 steps of adopted-basis Newton–Raphson method.
Figure 2.

Starting conformations of DCE in the active site of DhlA. A has the DCE is the same position as found in the crystal structure of the enzyme⋅substrate complex (COMF2). Structure in B is the last coordinate set from a molecular dynamics simulation of DhlA starting with the same crystal structure (COMF1). The conformation of DCE in both structures is gauche. The dashed lines show the interaction between OD2 of Asp-124 and C2 of DCE (OD2⋅⋅⋅C2).
Results and Discussion
The activation energies for DCE undergoing nucleophilic attack by acetate in two different attack geometries in the gas phase were determined with the AM1-SSP Hamiltonian. One complex was energy minimized with the angular and distance constraints described in Methods (Fig. 1B). The other complex had additional dihedral constraints placed between acetate and DCE, which orients the nonreacting oxygen of acetate to reside between the two hydrogens of the reacting carbon of DCE (Fig. 1A). Having the dihedral constraints between the acetate and DCE forces the conformation of the attacking acetate to mimic the reacting geometries of DCE and Asp-124 in the haloalkane dehalogenase when going from NAC to TS. For both geometries, the amount of bond breaking and bond making is the same in the TS structure. The carboxylate oxygen of acetate to the carbon bonded to the leaving chloride of DCE distance is 1.95 Å, and the carbon chloride distance in DCE is 2.25 Å (Fig. 3). The lowest energy complexes differ between these two geometries. In the calculations using only angular constraints between the acetate and DCE, the energy difference between the lowest energy structure and the TS is 26.0 kcal/mol. An activation barrier of 26.0 kcal/mol is in good agreement with the value of 23.2 kcal/mol from MP2/6–31+G(d)//HF-6–31+G(d) calculations of the reaction of acetate with DCE in the gas phase from this laboratory (21). The energy difference between the lowest energy structure (NAC in this case) and TS when dihedral constraints are included in the calculation is 16.9 kcal/mol. The large difference in activation energy between the former and latter conformation is because of the ability of the second carboxylate oxygen of acetate in the former conformation to interact with the other carbon of DCE (Fig. 3). The separation between these two atoms is 2.80 Å. This additional interaction provides a greater stabilization energy for the complex.
Figure 3.

Gas-phase structures of DCE undergoing nucleophilic attack by acetate. Structures in A have no dihedral constraints. Structures in B have dihedral constraints to restrict the attacking geometry of acetate to one similar to the enzyme. Parenthetical values are in angstroms and correspond to the distance between the attacking carboxylate oxygen of acetate to the carbon of DCE to the carbon of DCE and the leaving chloride (C⋅⋅⋅O, C⋅⋅⋅Cl).
The large difference in the activation energies when starting with the most stable (gas phase-optimized) geometry and that seen in the reactive conformer of the enzymatic reaction has been seen in ab initio calculations of model compounds (catecholate and trimethylsulfonium) representing the SN2 transmethylation reaction occurring between catecholate and S-adenosyl-l-methionine in catechol O-methyltransferase (COMT) from rat liver (22, 23). In the model compound reaction, one methyl group is transferred from trimethyl sulfonium to the ionized oxygen of catecholate to form dimethyl sulfide and O-methylcatechol (Scheme S2). Geometry optimization of catecholate with trimethylsulfonium formed a complex with all three methyl groups of trimethyl sulfonium in close contact with the ionized oxygen of catecholate (24). The associated TS for this structure has the two nonreacting methyl groups above the phenyl group of catecholate. The activation energy from this geometry is 22 kcal/mol. When the geometry of the reactants is constrained to that found in COMT, the activation barrier decreases significantly (22). In the pseudo enzyme reaction, only one methyl group from trimethyl sulfonium is interacting with the ionized oxygen of catecholate, and the other two methyl groups are directed away from the phenyl group of catecholate in the ground-state and transition-state structures. The activation energy from this geometry is ≈3 kcal/mol. The large difference in activation energy between the structures is because of having a more stable ground state in the complex without constraints. COMT significantly reduces the activation barrier for the transmethylation reaction by directing the methylene groups bonded to the sulfur of S-adenosyl-l-methionine away from the catecholate, thereby avoiding the most stable structure. In addition, Zheng and Ornstein have pointed out the ab initio calculated gas-phase ion-molecule complexes obtained for model compounds used to study the reaction in glutathione S-transferase play little role in the enzymatic reaction (25).
Scheme 2.

To study the dehalogenation reaction in the active site of haloalkane dehalogenase, we generated two-dimensional QM/MM energy surfaces. The QM/MM energy surfaces were created as a function of two distances; the attacking carboxylate oxygen of Asp-124 to carbon bonded to the leaving chloride in DCE (OD2⋅⋅⋅C2) and the leaving chloride to its associated carbon of DCE (C2⋅⋅⋅Cl2). Two different coordinate sets were used to calculate the energy difference from going from the ground state to the transition state (COMF1 and COMF2, see Fig. 2). The lowest energy structures for these two conformers differ in their location on the energy surface. For COMF1, the lowest energy structure is at OD2⋅⋅⋅C2 = 2.60 Å and C2⋅⋅⋅Cl2 = 1.80 Å. The lowest energy structure for COMF2 is at OD2⋅⋅⋅C2 = 3.20 Å and C2⋅⋅⋅Cl2 = 1.80 Å. There is minimal difference between the active site of DhlA when containing the lowest QM/MM energy structure and the TS structure (Table 2). The largest change between these two structures is in the distance between the leaving chloride and Trp-125 and Trp-175. In the lowest energy structure of COMF1, the leaving chloride is hydrogen bonding only to one tryptophan (Trp-175). HE1 of Trp-125 is 3.42 Å from the leaving chloride, consistent with the crystal structure and PM3 calculations of the enzyme's active site (2, 26). In the molecular dynamics simulation of DhlA, the leaving chloride is hydrogen bonded to HE1 of Trp-125 but not Trp-175 in the ground state (17). In the ground state, the charge on the chlorines of DCE is small (approximately −0.20 a.u.), which likely allows the hydrogen bond between either tryptophan to form and break easily. As charge builds up on the leaving chloride in the TS, Trp-125 forms an additional hydrogen bond to the chloride. The distance between HE1 of Trp-125 and the chloride reduces to 2.72 Å. In the TS structure, the leaving chloride to HE1 distances of Trp-125 and Trp-175 are almost equivalent. This is consistent with the high-resolution crystal structure of DhlA containing chloride and the molecular dynamics study of DhlA containing the transition state (17, 27). When DCE is in the x-ray position (COMF2), interactions between the enzyme with DCE and Asp-124 are very similar in the ground-state and the transition-state structures. For DCE in either position in the active site, the nonreacting carboxylate oxygen of Asp-124 (OD1) is hydrogen bonding with the backbone amides (HN) for Glu-56 and Trp-125 (Table 2). The distances between OD1 to HN are almost identical in the ground-state and transition-state structures for both conformers. These hydrogen bonds prevent OD1 from interacting with DCE.
Table 2.
Interactions between DhlA and DCE and side chain of Asp-124, Å
| Interaction | COMF1
|
COMF2
|
||
|---|---|---|---|---|
| ES | TS | ES | TS | |
| Trp-125 HN—Asp-124 OD1 | 1.80 | 1.80 | 1.74 | 1.77 |
| Glu-56 HN—Asp-124 OD1 | 1.91 | 1.94 | 1.89 | 1.90 |
| Trp-125 HE1—DCE Cl2 | 2.80 | 2.88 | 3.42 | 2.72 |
| Trp-175 HE1—DCE Cl2 | 2.71 | 2.48 | 2.47 | 2.53 |
| Trp-125 NE1—Trp-175 NE1 | 6.88 | 6.84 | 7.07 | 6.68 |
The enzyme causes an earlier TS relative to the gas phase for DCE in either position in the active site. In the enzyme, the TS is reached when the OD2⋅⋅⋅C2 separation is 2.05 Å (Fig. 4). The TS for acetate and DCE in the gas phase has a O⋅⋅⋅C separation of 1.95 Å. A similar result was observed in an AM1 (standard) study of the dehalogenation of DCE in the haloalkane dehalogenase (28). Damborsky et al. found that having only Trp-125 and Trp-175 in their calculations was enough to induce an earlier transition state for DCE and Asp-124. The C2⋅⋅⋅Cl2 distance in the TS structure in the enzyme has contracted relative to the gas-phase structure, 2.15 Å and 2.25 Å, respectively. The calculated activation barrier for the SN2 displacement of the chloride from DCE is 16.2 kcal/mol for COMF1 and corresponds to going from the NAC to TS (Table 3). The activation energy is higher for COMF2, 19.4 kcal/mol.
Figure 4.

Transition-state structure for COMF1 in the active site of DhlA. Dashed lines indicate hydrogen bonds formed between the enzyme and DCE and side chain of Asp-124. The hydrogen- bonding arrangement in COMF2 is similar to that depicted.
Table 3.
QM/MM and AM1-SSP for the enzymatic and gas-phase dehalogenation reaction, respectively, kcal/mol
| ES | Enzyme TS | Difference | |
|---|---|---|---|
| COMF1 | |||
| Enzyme | −202.8 | −186.6 | 16.2 |
| Gas phase | −171.2 | −151.2 | 20.0 |
| COMF2 | |||
| Enzyme | −203.6 | −184.2 | 19.4 |
| Gas phase | −174.3 | −152.9 | 21.4 |
To determine the extent to which DhlA lowers the activation barrier of the dehalogenation reaction, the coordinates of the reactants (DCE and sidechain of Asp-124) were removed from each energy-minimized enzyme coordinate set. The energies of the Asp-124 sidechain (acetate) and DCE were determined from single-point AM1-SSP calculations in the gas phase with the conformation of the reactants in exactly the same position as in the enzyme (Table 3). The energy difference of the COMF1 reactants from the lowest energy structure to the enzymatic TS is 20.0 kcal/mol in the gas phase. The activation barrier in the enzyme (16.2 kcal/mol) is 3.8 kcal/mol lower than the energy difference between reactants and the enzymatic TS in the gas phase. The amount of TS stabilization is less for COMF2. The energy difference between the lowest energy structure and enzymatic transition-state structure is 21.4 kcal/mol in the gas phase. The energy difference of the reactants is lower by 2.1 kcal/mol in the enzyme relative to the gas phase.
The amount of stabilization energy provided by the enzyme is not too surprising given the results of mutational studies of Trp-125 and Trp-175 (5, 29). Single mutations of these two tryptophans to glutamines showed that the catalytic efficiency (kcat/Km) of DhlA is ≈100-fold lower for W175Q relative to W125Q for DCE. There is only a ≈40-fold difference in the catalytic efficiency between these two mutants for 1,2-dibromoethane (5). In addition, the binding constant (Km) is affected more by mutation to Trp-175 than to Trp-125. DhlA shows only a small decrease in activity when Trp-125 is replaced with phenylalanine. There is only a 3-fold decrease in kcat for DCE in the W125F mutant. Replacement of either tryptophan with an aromatic residue shows no more than a 60-fold decrease in catalytic efficiency for DCE. Interestingly, kcat for the wild-type enzyme and the W175Y mutant is the same for 1,2-dibromoethane. It is not necessary for the aromatic residue at position 125 or 175 to be dipolar. The specific activity of mutants W125F and W175F of DhlA are no more than 20-fold less than the wild-type enzyme for DCE (29). A usual number of aromatic residues line the active site, and Trp-125 interacts with Phe-128 and Phe-222 and Trp-175 interacts with Phe-190 and Trp-194 (Fig. 5). It was pointed out by Krooshof et al. that the need for aromatic residues at positions 125 and 175 could be necessary to retain the structural integrity of the active-site cavity of DhlA (29). Mutants with short polar sidechains likely disrupt these aromatic interactions and change the active site. Replacement of Trp-125 or Trp-175 with arginine leads to a nonactive enzyme (5, 29) Comparisons of wild-type and W175Y DhlA containing an acetate in the active site at pH 5 (Protein Data Bank nos. 1BE0 and 1BEZ, respectively) indicate the aromatic residues in the active site exhibit greater mobility in the mutant even though their positions are essentially unchanged with the wild type (29). The average B-factor for residues Trp-125, Phe-128, Phe-190, Trp-194, and Phe-222 is 5.40 ± 2.03 Å2 for the wild-type enzyme and 8.32 ± 2.72 Å2 for the W175Y mutant. The acetate in the active-site cavity also has a much higher average B-factor in the mutant enzyme (27.0 Å2) relative to the wild-type enzyme (15.8 Å2).
Figure 5.

Picture of aromatic residues in contact with Trp-125 and Trp-175 in the active site of DhlA.
Conclusions
The QM/MM energetics of this enzymatic dehalogenation reaction in DhlA illustrate the importance of positioning of the reactants in enzyme catalysis. The enzyme takes the lowest energy path to the transition state by having the nonreacting carboxylate oxygen of Asp-124 hydrogen bonded to the backbone amides of Glu-56 and Trp-125, thus this oxygen is not able to interact with DCE to form a more stable complex and increase the activation barrier. The importance of positioning the reactants is central in the NAC concept (9, 10). Computational studies of the ring closure reaction of cyclic anhydrides have shown the number of NACs formed in the molecule is linearly related to the rate enhancement observed experimentally (30). The enzyme is also able to lower the activation energy further by 2–4 kcal/mol by forming hydrogen bonds with Trp-125 and Trp-175 to the leaving chloride of DCE in the transition state. A few kcal/mol lowering of the activation barrier in the enzyme compared with the gas phase can be compared with the reaction barrier in water of ≈9 kcal/mol greater than in the gas phase (21). Desolvating the reactants in the enzymatic reaction results in a substantial energetic advantage.
Supplementary Material
Acknowledgments
This study was funded by a grant from the National Science Foundation (NSF) (MCB-9727937). We gratefully acknowledge computer time on University of California Santa Barbara's SGI Origin 2000, which is supported by grants from NSF (CDA96–01954) and Silicon Graphics, Inc.
Abbreviations
- DhlA
haloalkane dehalogenase from Xanthobacter autotrophicus GJ10
- AM1
Austin Model 1
- AM1-SSP
AM1-system specific parameters
- QM/MM
quantum mechanics/molecular mechanics
- DCE
1,2-dichloroethane
- NAC
near attack conformer
- TS
transition state
References
- 1.Schanstra J P, Kingma J, Janssen D B. J Biol Chem. 1996;271:14747–14753. doi: 10.1074/jbc.271.25.14747. [DOI] [PubMed] [Google Scholar]
- 2.Verschueren K H G, Seljee F, Rozenboom H J, Kalk K H, Dijkstra B W. Nature (London) 1993;363:693–698. doi: 10.1038/363693a0. [DOI] [PubMed] [Google Scholar]
- 3.Pries F, Kingma J, Krooshof G H, Jeronimus-Stratingh C M, Bruins A P, Janssen D B. J Biol Chem. 1995;270:10405–10411. doi: 10.1074/jbc.270.18.10405. [DOI] [PubMed] [Google Scholar]
- 4.Stucki G, Thuer M. Appl Microbiol Biotechnol. 1994;42:167–172. doi: 10.1007/BF00170241. [DOI] [PubMed] [Google Scholar]
- 5.Kennes C, Pries F, Krooshof G H, Bokma E, Kingma J, Janssen D J. Eur J Biochem. 1995;228:403–407. [PubMed] [Google Scholar]
- 6.Radzicka A, Wolfenden R. Science. 1995;267:90–93. doi: 10.1126/science.7809611. [DOI] [PubMed] [Google Scholar]
- 7.Warshel A, Levitt M. J Mol Biol. 1976;103:227–249. doi: 10.1016/0022-2836(76)90311-9. [DOI] [PubMed] [Google Scholar]
- 8.Field M J, Bash P A, Karplus M. J Comp Chem. 1990;6:700–733. [Google Scholar]
- 9.Bruice T C, Lightstone F C. Acc Chem Res. 1999;32:127–136. [Google Scholar]
- 10.Bruice T C, Benkovic S J. Biochemistry. 2000;39:6267–6274. doi: 10.1021/bi0003689. [DOI] [PubMed] [Google Scholar]
- 11.Dewar M J S, Zoebisch E G, Healy E F, Stewart J J P. J Am Chem Soc. 1985;107:3902–3909. [Google Scholar]
- 12.Frisch M J, Trucks G W, Schlegel H B, Scuseria G E, Robb M A, Cheeseman J R, Zakrzewski V G, Montgomery J A, Stratmann R E, Burant J C, et al. gaussian 98. Pittsburgh, PA: Gaussian; 1998. , Rev. A.6. [Google Scholar]
- 13.Belward J A. In: Numerical Algorithms. Mohamed J L, Walsh J E, editors. Oxford: Clarendon; 1986. pp. 275–292. [Google Scholar]
- 14.Brooks B R, Bruccoleri R E, Olafson B D, States D J, Swaminathan S, Karplus M. J Comp Chem. 1983;4:187–217. [Google Scholar]
- 15.Bash P A, Ho L L, MacKerell A D, Jr, Levine D, Hallstrom P. Proc Natl Acad Sci USA. 1996;93:3698–3703. doi: 10.1073/pnas.93.8.3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boys S F, Bernardi F. Mol Phys. 1970;19:553–566. [Google Scholar]
- 17.Lightstone F C, Zheng Y-J, Bruice T C. J Am Chem Soc. 1998;120:5611–5621. [Google Scholar]
- 18.Jorgensen W L, Chandrasekhar J, Madura J D, Impey R W, Klein M L. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 19.MacKerell A D J, Bashford D, Bellott M, Dunbrack R L, Jr, Evanseck J D, Field M J, Fischer S, Gao J, Guo H, Ha S, et al. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 20.Brooks C L I, Karplus M. J Mol Biol. 1989;208:159–181. doi: 10.1016/0022-2836(89)90093-4. [DOI] [PubMed] [Google Scholar]
- 21.Maulitz A H, Lightstone F C, Zheng Y-J, Bruice T C. Proc Natl Acad Sci USA. 1997;94:6591–6595. doi: 10.1073/pnas.94.13.6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kahn K, Bruice T C. J Am Chem Soc. 2000;122:46–51. [Google Scholar]
- 23.Kuhn B, Kollman P A. J Am Chem Soc. 2000;122:2586–2596. [Google Scholar]
- 24.Zheng Y-J, Bruice T C. J Am Chem Soc. 1997;119:8137–8145. [Google Scholar]
- 25.Zheng Y-J, Ornstein R L. J Am Chem Soc. 1997;119:648–655. [Google Scholar]
- 26.Lightstone F C, Zheng Y-J, Maulitz A H, Bruice T C. Proc Natl Acad Sci USA. 1997;94:8417–8420. doi: 10.1073/pnas.94.16.8417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ridder I S, Rozeboom H J, Dijkstra B W. Acta Crystallogr. 1999;D55:1273–1290. doi: 10.1107/s090744499900534x. [DOI] [PubMed] [Google Scholar]
- 28.Damborsky J, Kuty M, Nemec M, Koca J. J Chem Inf Comput Sci. 1997;37:562–568. [Google Scholar]
- 29.Krooshof G H, Ridder I S, Tepper A W J W, Vos G J, Rozeboom H J, Kalk K H, Dijkstra B W, Jannsen D B. Biochemistry. 1998;37:15013–15023. doi: 10.1021/bi9815187. [DOI] [PubMed] [Google Scholar]
- 30.Lightstone F C, Bruice T C. J Am Chem Soc. 1996;118:2595–2605. [Google Scholar]
- 31.Wenthold P G, Squires R R. J Am Chem Soc. 1994;116:11890–11897. [Google Scholar]
- 32.Cox J D, Pilcher G. Thermochemistry of Organic and Organometallic Compounds. New York: Academic; 1970. [Google Scholar]
- 33.Ochterski J W, Petersson G A, Montgomery J A., Jr J Chem Phys. 1996;104:2598–2619. [Google Scholar]
- 34.Martin J D D, Hepburn J W. J Chem Phys. 1998;109:8139–8142. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}