

Abstract
In site-directed spin labeling, a covalently attached nitroxide probe containing a chemically inert unpaired electron is utilized to obtain information on the local environment of the parent macromolecule. Studies presented here examine the feasibility of probing local DNA structural and dynamic features using a class of nitroxide probes that are linked to chemically substituted phosphorothioate positions at the DNA backbone. Two members of this family, designated as R5 and R5a, were attached to eight different sites of a dodecameric DNA duplex without severely perturbing the native B-form conformation. Measured X-band electron paramagnetic resonance (EPR) spectra, which report on nitroxide rotational motions, were found to vary depending on the location of the label (e.g., duplex center vs termini) and the surrounding DNA sequence. This indicates that R5 and R5a can provide information on the DNA local environment at the level of an individual nucleotide. As these probes can be attached to arbitrary nucleotides within a nucleic acid sequence, they may provide a means to “scan” a given DNA molecule in order to interrogate its local structural and dynamic features.
DNA is a universal carrier of genetic information and is a key to gene maintenance and expression in most life forms. The linear sequence of a DNA molecule, constituted with four basic building blocks (four nucleotides), defines a host of physical attributes, such as shape (three-dimensional structure), dynamics, and electrostatics. These properties govern DNA interactions with proteins, other nucleic acid molecules, and small molecule ligands, thus dictating biological functions. Many studies have been devoted to understand DNA structure, and a large amount of information is available. Various spectroscopic measurements, including solution (1, 2) and solid-state NMR (3, 4), EPR1 (5, 6), and fluorescence (7–9), have revealed complex DNA dynamics that involve correlated and uncorrelated processes occurring on time scales ranging from picoseconds to milliseconds. It has been suggested that site-specific DNA dynamics are linked to functions. For example, sequence-dependent mobility variations of cytosine nucleotides have been linked to site-specific DNA methylation, which is a mechanism of gene regulation (3, 4, 10). Furthermore, altered flexibility at DNA lesion sites has been proposed to play a key role in DNA recognition by repair proteins (11–13). However, current knowledge on sequence dependent DNA dynamics is primitive, and methods for probing site-specific dynamic features in large DNA and DNA/protein complexes are lacking.
In SDSL, a chemically inert nitroxide radical is covalently linked to a specific site of a macromolecule, and EPR spectroscopy is used to obtain information on structural and dynamic features of the labeling site. This technique requires only a small amount of a labeled sample (~500 pmol), and is capable of studying high molecular weight complexes under physiological conditions. SDSL has matured as a tool for studying protein structure and dynamics (14). In nucleic acid studies, SDSL has been applied to obtain distance constraints, to probe local and global dynamics, and to monitor conformational changes (15).
We have reported a phosphorothioate scheme where a nitroxide is attached to a phosphorothioate group that is chemically substituted at a specific location of a nucleic acid (Figure 1A) (16, 17). The phosphorothioate scheme allows efficient nitroxide attachment at an arbitrary nucleotide within a desired sequence. It opens up the possibility of “nitroxide scanning”, where a nitroxide is systematically moved along a stretch of primary sequence in order to obtain information at the level of an individual nucleotide. Figure 1A shows two nitroxide probes, R5 and R5a, which are attached using the phosphorothioate scheme and differ only in the 4-substituent of the pyrroline ring. R5 has been used to monitor changes in global molecular tumbling associated with RNA/RNA interactions (16). More recently, R5 and R5a have been employed to study nanosecond dynamics of an RNA element within a large folded ribozyme (18). In addition, distance measurements using a pair of R5’s have been reported (19, 20).
Figure 1.

(A) The phosphorothioate labeling scheme. During the solid phase chemical synthesis, a phosphorothioate is introduced at a specific nucleotide. The modified oligonucleotide is further reacted with 2,2,5,5-tetramethyl-1-oxylpyrroline derivatives (HO368 orHO1820), resulting in covalently linked R5 or R5a, respectively. Three torsion angles of the DNA–nitroxide linker are t1(O5′–P–S–CS), t2(P–S–CS–C3), t3(S–CS–C3–C2). (B) Sequence and secondary structure of the CS DNA duplex. Phosphorothioate groups labeled with nitroxides in this study are shown in green. (C) NMR structure of the CS DNA (33).
Work reported here explores the feasibility of probing local DNA environment, defined as structural and dynamic features at the labeling site, by monitoring rotational diffusive motion of R5 or R5a. Nitroxide rotational motions (equivalent to nitroxide dynamics) are reported by cw-EPR spectroscopy, in which the spectral line shape is dictated by rotational averaging of anisotropic Zeeman and hyperfine tensors of a nitroxide. At the commonly used X-band, the cw-EPR spectrum line shape changes drastically as the nitroxide rotational correlation time vary between 0.1 and 50 ns. If nitroxide dynamics in this regime are affected by the parent molecule, the observed EPR spectrum will vary according to structural and dynamic features at the labeling site. In such cases, the EPR spectrum serves as a reporter on the local environment. EPR line shape analysis is one of the most commonly used sources of information in protein SDSL studies (21). It has also been successfully applied to extract information from nitroxide probes attached to base and sugar positions of nucleic acids (15). Particularly, a number of research groups, including Bobst (22), Robinson (5, 23, 24), and more recently Sigurdsson (6), have studied DNA duplexes using nitroxides attached to specific base positions, and their work has revealed complex DNA dynamic behaviors that include both collective and local modes of motion.
Coupling between an EPR line shape and the local macromolecule environment depends on the chemical scheme of nitroxide labeling. While R5 and R5a have been employed in a number of nucleic acid SDSL studies, there were no prior reports on whether dynamics of these probes can provide local, site-specific information on DNA or RNA. Compared with other classes of nitroxides, R5 and R5a may have two potentially problematic issues. First, the pyrroline ring of the nitroxide is linked to the DNA phosphorus atom by rotatable bonds (Figure 1A). Internal motion of the nitroxide, defined as rotations about these bonds, may dominate nitroxide dynamics. This may lead to uniform nitroxide spectra at different DNA sites, thus preventing studies of site-specific features. In addition, a chemically introduced phosphorothioate adopts either one of two chiral configurations (Rp or Sp). Nitroxides attached to Rp and Sp diastereomers are sterically different, which may cause differences in their EPR spectra. Separations of Rp and Sp diastereomers have been demonstrated on a number of short oligonucleotides using laborious experimental procedures (25–27). Such separations have been reported only for oligonucleotides that are shorter than 15 nucleotides, and their success/failure depends heavily on sample specific features, such as the oligonucleotide sequence and the location of the phosphorothioate modification site. Due to the complexity in diastereomer separation, in many SDSL studies one may prefer to use mixed phosphorothioate diastereomers, in which an observed EPR spectrum is a sum of those obtained from either diastereomer. Therefore, it is important to establish what information on the local DNA environment can be obtained using mixed phosphorothioate diastereomers.
Here, we placed R5 and R5a to multiple positions within a model dodecameric DNA duplex (Figure 1B), and measured the corresponding X-band EPR spectra without separating nitroxide diastereomers. BothR5andR5a gave spectra that vary according to the location of the label (e.g., duplex center vs termini) and the surrounding DNA sequence. The trend of spectral variations is consistent between the two probes. The results establish that R5 andR5a can be used to probe DNA local environment. This may lead to a new way for studying DNA structure and dynamics.
MATERIALS AND METHODS
Materials
All oligonucleotides were obtained from Integrated DNA Technologies (Coralville, IA). Reactive nitroxide derivatives 3-methanesulfonyloxymethyl-2,2,5,5-tetramethyl-1-oxylpyrroline (HO346, precursor for R5) and 4-bromo-3-bromomethyl-2,2,5, 5-tetramethyl-1-oxylpyrroline (HO1820, precursor for R5a) were synthesized as reported previously (28, 29).
DNA Labeling and Purification
R5 labeling was carried out as described previously (17). For R5a labeling, crude oligonucleotides containing one phosphorothioate modification (~7 nmol) were treated with 1.2 mg of HO1820 in 4 μL of MES (1 M, pH = 5.8), 16 μL of water and 20 μL of acetonitrile. After incubation at room temperature for 24 h, the labeled product was first purified by anion-exchange HPLC, and then desalted using a homemade G-25 Sephadex column. Details of purification procedures have been previously described (17). Desalted oligonucleotides were lyophilized, resuspended in 10–15 μL of water and stored at −20 °C.
The final concentration of labeled oligonucleotides was determined by UV absorption at 260 nm. Extinction coefficients of the unmodified DNA (108,200 and 125,800 M−1 cm−1 for the left and right strands shown in Figure 1B, respectively) were used, as neither the phosphorothioate modification nor the nitroxide alters absorbance at 260 nm.
DNA Thermal Denaturation
DNA duplexes labeled with a single R5a were prepared in buffer A (100 mM NaCl, 50 mM Tris-HCl, pH = 7.5). The final duplex concentrations were 1–2 μM. Melting transition curves were obtained using a DU800 UV–Vis spectrometer (Beckman Coulter, Fullerton, CA). Temperature was increased from 5 to 75 °C with a rate of 1 °C/min outside the transition region and 0.5 °C/min within the transition region. Absorbance at 260 nm was recorded every 0.5 or 0.2 °C accordingly. Meting curves were analyzed as previously described to obtain thermodynamic parameters of duplex formation (30). Each DNA sample was melted 4–7 times and averaged thermodynamic parameters are reported. In control studies, labeled duplexes were subjected to repeated heating/cooling cycles in which temperature was varied with the same average heating rate as in the melting experiments. After seven cycles less than 6% of R5 or R5a were found to detach from the DNA. This amount of probe detachment does not affect the measured thermodynamic parameters beyond the range of reported errors.
EPR Measurements
To prepare an EPR sample, approximately 1 nmol of a spin-labeled oligonucleotide was annealed with a 10%molar excess of an unlabeled complementary strand. Annealing was carried out in buffer A with overnight incubation at room temperature. Formation of double-stranded DNA was confirmed using native gel electrophoresis (data not shown). To remove excess unannealed single-stranded DNA and a small amount of detached probe, the mixture was diluted with 600 μL of buffer A, and then concentrated to a final volume of 4–7 μL at 15 °C using an Ultrafree centrifugal filter (MWCO 5 kDa, Millipore, Inc.). The annealed duplex was then used to prepare an EPR sample, which contained approximately 20–40 μM of a labeled DNA duplex, 100mMNaCl, 50mMTris-HCl (pH = 7.5), and 34% (w/w) sucrose. Finally, EPR spectra were measured with approximately 10 – 15 μL of samples individually placed in a glass capillary (1.0 mm i.d. × 1.2 mm o.d. Vitrocom, Inc., Mountain Lakes, NJ) that was sealed at one end.
We note that a cosolvent (e.g., sucrose) was added to samples in order to minimize spectral effects due to uniform global tumbling of the model DNA duplex (Figure 1B, MW = 7,290 Da). Using the hydrodynamic model of Tirado and de la Torre (31), in an aqueous solution at 20 °C the global tumbling of the model duplex was estimated to have rotational correlation times of 3.5 ns (τ||) and 6.9 ns (τ⊥) for rotations that are parallel and perpendicular to the DNA helical axis, respectively. This is expected to significantly average nitroxide magnetic tensors, thus largely masking site-dependent spectral variations at X-band. Presence of 34% (w/w) sucrose increases τ|| and τ⊥ to 14 and 28 ns, respectively. The observed EPR spectra are dictated by motions faster than 14 ns and presumably are more adept to report site-specific features.
X-band EPR spectra were acquired on a Bruker EMX spectrometer using a high sensitivity cavity (ER 4119HS, Bruker Biospin, Inc.). The incident microwave power was 2mW, and the field modulation was 1–2 G at a frequency of 100 kHz. Sample temperature was maintained at 5 °C during spectral acquisitions using a liquid nitrogen variable temperature setup. All EPR spectra were baseline corrected and normalized to the same number of spins using software kindly provided by the Hubbell group at UCLA.
Modeling of Sterically Accessible Nitroxide Rotamer Space
NASNOX program was previously reported for identifying ensembles of sterically allowable R5 conformers (17, 19, 32), and was updated to allow modeling of R5a. The studies used the NMR structure of the model DNA duplex (33), with the nitroxide (R5 or R5a) modeled at either the Rp or the Sp phosphorothioate of a desired nucleotide. Discrete searches of torsion angles t1, t2 and t3 (Figure 1A) between 0° and 360° were carried out to select sterically allowable conformers. For R5, t1, t2, and t3 search steps were 5°, 30°, and 30°, respectively. For R5a, t1 was varied in a 5° step while t2 and t3 were fixed (t2 = 180°, t3 = 100° or −100°, values obtained based on MD simulations, unpublished data). For each ensemble, a quantity S was computed according to
where γ is the angle between an individual NO vector (defined by the nitroxide nitrogen and oxygen atoms) and the average NO vector of the ensemble.
RESULTS
Model DNA System and Nitroxide Labeling
The model DNA used in this work is a dodecameric duplex with nonself- complementary strands (Figure 1B). The DNA was designated as CS, following its PDB ID 1CS2. The NMR structure of CS (Figure 1C) shows a near canonical B-duplex geometry, Popova et al. although deviations from standard B-form parameters were observed (33). Particularly, the minor groove narrows along the T8T9T10/A15A16A17 stretch and widens at the T10A11 step. Previously, we measured distances between pairs of R5 attached to the CS DNA, with the results showing good agreement between the SDSL measured distances and those predicted based on the NMR structure (19).
Using the phosphorothioate scheme, nitroxides were attached, one at a time, to eight different positions within the CS duplex (Figure 1B). The nonsymmetric nature of the CS DNA ensures that one and only one nitroxide is present in each labeled duplex. In the following text, a given site of the duplex is referred to as CSx, while a specific nitroxide labeled duplex and the associated EPR spectrum are named according to the labeling site and the nitroxide identity. For example, “CS2” refers to site 2 of the duplex (i.e., the local environment at the phosphate group of nucleotide 2), while “CS2_R5” designates a duplex with an R5 attached at the phosphate group of nucleotide 2 or its EPR spectrum. All data reported here were obtained with R5 or R5a attached to mixed phosphorothioate diastereomers at each labeling site.
Consistent with previous reports (17, 19), R5 labeling was found to be efficient and site-specific. Similarly high efficiency and specificity were observed for R5a. Labeling efficiency of R5a was estimated to be >95% based on anion-exchange HPLC. Mass spectrometry measurements showed that one and only one R5a is attached to a DNA strand (Figure S1 in the Supporting Information). For R5a labeling, the 3-bromomethyl precursor (HO1820, Figure 1A) is sufficiently reactive and can be used directly without being converted to the 3-iodomethyl derivative.
DNA Structure Perturbation
Previously, when a pair of R5 was attached to the CS duplex, onlyminor perturbation to the native B-form configuration was detected using CD spectroscopy and thermal denaturation measurements (19). MD simulation studies also indicated that R5 is accommodated within the groove of the helix and does not induce severe helical distortion ((32) and unpublished data). R5a was attached to the DNA using the same chemical scheme, and therefore is not expected to drastically perturb CS duplex based on the R5 results. To test this, thermal denaturation studies were carried out on single R5a labeled CS duplexes (Figure S2 in the Supporting Information). The data confirm that a single R5a label hasminor effects on DNA duplex stability, with the unlabeled and labeled duplexes showing small differences in the measured thermodynamic parameters of duplex formation (Table 1). For example, the differences in the free energy of duplex formation between unlabeled and labeled duplexes (ΔΔG°37°C) are all less than 1.0 kcal/mol (Table 1). The destabilizing effect is most prominent at the interior site CS9 (+0.6 kcal/mol) and become smaller at the termini. We also note that at the terminal site of the mutant DNA(CS2_1A, Table 1B), ΔΔG°37°C was measured to be −0.3 kcal/mol, indicating that R5a stabilizes the duplex at this site.
Table 1.
Thermodynamic Parameters of Duplex Formation for R5a Labeled CS Duplexes
| labeled position | ΔH°b (kcal/mol) | ΔS°b (cal/(mol·K)) | ΔG°37°cc (kcal/mol) | ΔΔG°37°cd (kcal/mol) |
|---|---|---|---|---|
| (A) Wild-Type CS Duplexa | ||||
| none | −69 ± 2 | −192 ± 8 | −9.4 | |
| CS7 | −68 ± 5 | −191 ± 17 | −9.2 | 0.2 |
| CS9 | −63 ± 5 | −174 ± 15 | −8.8 | 0.6 |
| CS12 | −67 ± 2 | −186 ± 7 | −9.2 | 0.2 |
| CS14 | −64 ± 2 | −176 ± 5 | −9.1 | 0.3 |
| (B) Mutant CS Duplexe | ||||
| none | −75 ± 3 | −214 ± 8 | −9.1 | |
| CS2_1A | −80 ± 1 | −227 ± 5 | −9.4 | −0.3 |
| CS24_12T | −78 ± 3 | −222 ± 9 | −9.0 | 0.1 |
DNA sequence as shown in Figure 1A.
Averages and errors were obtained based on results from multiple sets of melting measurements.
ΔG°37°C = ΔH° − 310 K × ΔS°. Averages of ΔG°37°C value from multiple melting measurements are reported, with errors estimated to be less than ± 0.1 kcal/mol.
ΔΔG°37°C = ΔG°37°C (labeled) − ΔG°37°C (unlabeled).
DNA sequence modified at the duplex terminus: C1/G24 to A1/T24.
Site-Dependent Variations in R5 Spectra
X-band EPR spectra of R5 attached to eight sites within the CS DNA were obtained in the presence of 34%(w/w) sucrose at 5 °C (Figure 2). All R5 spectra have a common feature: the center line is sharp and the hyperfine splitting is not observed. This suggests that R5 is undergoing fast and relatively unrestricted rotation at these sites. This is consistent with the expected large degree of freedom in R5 internal motion, where all three single bonds connecting the pyrroline ring to the DNA may rotate at the nanosecond time scale ((32) and unpublished data).
Figure 2.

EPR spectra of R5 labeled CS DNA duplexes.
Nonetheless, line width variations were observed between differentR5 spectra, indicating thatR5mobility changes between different DNA sites. The normalized spectra of CS2_R5 and CS14_R5 are nearly identical (Figure 3A), and the resulting difference spectrum is similar to those obtained in control studies where duplexes labeled at certain sites were prepared and measured multiple times (Figure S3 in the Supporting Information). This indicates that CS2 and CS14 have a very similar local environment. Interestingly, CS2_R5 and CS14_R5 show clearly narrower lines with larger amplitude compared to those at other sites (Figures 3B, 3C), with the corresponding difference spectra varying significantly from those obtained in control measurements (Figure S3 in the Supporting Information). This indicates that CS2 and CS14, which are located at duplex 5′ termini, have higher R5 mobility as compared to sites at the duplex interior (sites 5, 7, 9, and 19) and those at the 3′ termini (sites 12 and 24). For the remaining six sites (5,7,9,12,19, and 24), slight line shape variations can be observed (Figure 2). However, the differences between these sites are smaller than those between this group and CS2/CS14 (Figures 3B, 3C). We also note that EPR spectra at the 3′ terminal sites (sites 12 and 24) are nearly identical (Figure 3D), with R5mobility comparable to that of the interior sites (e.g., sites 5 and 9, Figure 2) and clearly lower than that at the 5′ termini (sites 2 and 14) (Figure 3C).
Figure 3.
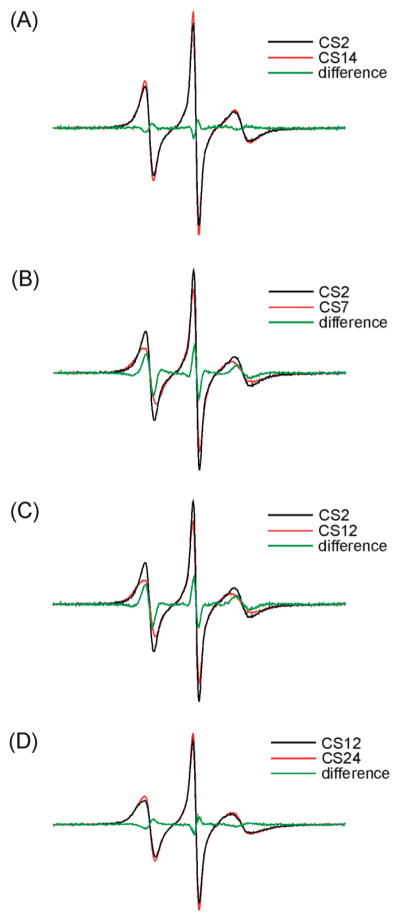
Comparisons of R5 spectra.
To further quantify variations in R5 mobility, rotational correlation time (τ) was estimated from EPR spectral line width and amplitudes according to a formalism that treats nitroxide motion as a fast isotropic rotation (16, 34). The plot of τ values at different sites (Figure 4) reveals nitroxide mobility variations that are consistent with conclusions drawn from direct line shape comparisons (Figures 2 and 3). The 5′ terminal sites (CS2 and CS14) have the smallest τ (2.13 and 2.03 ns, respectively, with errors estimated to be <5%) and therefore the highest rate of nitroxide motion, again indicating that these two sites have higher mobility as compared to those at the interior and the 3′ termini. The τ values of 3′ terminal sites (CS12 and CS24, 2.81, and 2.65 ns, respectively) are comparable to those of the interior sites (2.57–2.89 ns), indicating a similar mobility between the 3′ terminal and duplex interior sites. Finally, among interior sites, duplex centers (CS7 and CS19) show the highest τ and therefore the lowest mobility.
Figure 4.

Estimated R5 rotational correlation time (τ). R5 location is indicated next to the corresponding data point. Squares and triangles represent sites belonging to the right and left DNA strands shown in Figure 1B, respectively. Errors in τ were determined to be <5% based on analysis of EPR spectra obtained from several R5 labeled duplexes that were prepared and measured multiple times.
Overall, the data reveal thatR5mobility is different at different sites, indicating site-specific couplings between R5 motion and local DNA environment. Furthermore, local environments at both 5′ terminal sites (CS2 and CS14) are distinct from the interior sites, while those at both 3′ terminal sites (CS12 and CS24) are comparable to the interior sites. Similar results were observed using R5a.
Site-Dependent Variations of R5a Spectra
The X-band EPR spectra of R5 show characteristics of fast motion at all CS sites due to the large degree of freedom in nitroxide internal motion (see preceding section). If nitroxide internal motions are reduced, one may expect to observe larger site-dependent spectral variations and better sensitivity to local DNA environment. Such a strategy, which has been successfully implemented previously in protein studies (35), was tested here by investigating R5a labeled DNA. R5a has a bromine atom at theC4 position of the nitroxide pyrroline ring (Figure 1A), which restricts two out of the three torsional rotations (t2 and t3, Figure 1A) in the nitroxide internal motion according to MD simulations (unpublished data). R5a was attached to eight positions of the CS duplex, and spectra were measured at 5 °C (Figure 5). At each site, R5a spectrum is indeed broader than the corresponding R5 spectrum, indicating slower motion of R5a as compared to R5.
Figure 5.

EPR spectra of R5a labeled CS DNA duplexes. Black arrows indicate the resolved hyperfine extrema atCS7 andCS19. Red arrows indicate the presence of a second spectral component at CS2 and CS14. Spectra of CS9, 12, 14, 19, and 24 were corrected for the residual free nitroxide (<3%).
With reduced internal motion, R5a is more prominently influenced by the local DNA environment, and larger spectral variations are observed between the labeled sites (Figure 5). However, higher sensitivity of the probe renders line shape analysis more complex, and simple line width measurements or τ estimations no longer fully capture features of R5a dynamics. As a first step in analyzing site-dependent R5a motions, R5a spectra were categorized into three major groups based on differences and similarities in their line shapes.
The first group includes positions 7 and 19, which have nearly identical spectra with a split low-field peak (Figure 5, indicated by black arrows). Splitting of a high-field peak is also present, although harder to observe due to its intrinsically low amplitude (Figure 5, black arrows). These features arise from partially resolved parallel and perpendicular components of the hyperfine tensor due to incomplete averaging, and are a signature of a nitroxide undergoing anisotropic rotational diffusion. This class of lineshapes has been observed in previous studies of proteins (36) and nucleic acids (37).
Group 2 includes CS5, CS9, CS12 and CS24 (Figure 5). These spectra show broad low-field peaks. Splittings of low- and high-field peaks can also be observed, although they are less prominent than those in the group 1 spectra. These features indicate that the nitroxide is undergoing anisotropic rotation, but with a higher degree of hyperfine tensor averaging and therefore higher mobility than that of group 1. Within the group 2 sites, CS12 and CS24 are located at the 3′ termini of the DNA duplex, while CS5 and CS9 are located within the duplex interior and have different adjacent nucleotides. The similarity in their spectra indicates that R5a is experiencing a similar local environment despite variations in the nucleotide sequence and labeling position. This trend is consistent with the R5 results, where CS12_R5 and CS24_R5 (3′ terminal sites) give similar spectra to CS5_R5 and CS9_R5 (interior sites) (Figure 2).
Group 3 includes CS2 and CS14, which are at the 5′ termini of the DNA duplex. Both spectra show multiple components, indicating the presence of at least two nitroxide populations with distinct modes of motion. In the CS2 spectrum, a small bump is clearly present at the shoulder of the low-field peak (Figure 5, red arrow), reflecting a subpopulation of a nitroxide with significantly reduced mobility. Due to the immobilized subpopulation, the center and high field lines in the CS2 spectrum are lower in their amplitudes and broader when compared to other sites. The CS14 spectrum is dominated by a high mobility nitroxide population, and has the highest degree of hyperfine tensor averaging among the R5a spectra (Figure 5). The immobilized subpopulation is much less visible in CS14 than that in CS2 at 5 °C (Figure 5, red arrow).
Consistent with R5 data, R5a indicates that 5′ terminal sites (CS2 and CS14) are different from the interior sites and the 3′ terminal sites. While R5 gives highly similar spectra at the 5′ terminal sites, R5a has distinct spectra with different proportion of spectral subcomponents. As detailed in the Discussion, the immobilized component may arise from nitroxide/DNA interactions that are captured by the R5a probe. These results, therefore, highlight the sensitivity differences between R5 and R5a.
DNA Base Mutations Affect R5a Spectra
To further demonstrate that R5a reports on DNA local environment, the terminal C1/G24 base pair of the CS duplex was substituted with A1/T24 (Figure 6), and the effect on R5a spectra was studied. Two factors governed the choice of this particular mutation. First, the mutations were placed at the duplex terminus, so that only one nearest neighbor unit is affected (C1T2/A23G24). Second, within the context of one nearest neighbor unit, the C1/G24 to A1/T24 substitution gives the biggest possible change in the empirical thermodynamic parameters (ΔH° (CT/GA) = −7.8 kcal/mol vs ΔH° (AT/TA)= −7.2 kcal/mol, (38)). Assuming thermodynamic parameters are linked to DNA local environment, larger differences in thermodynamic parameters may suggest larger local environment variations, thus maximizing probability of observing R5a spectral changes.
Figure 6.

EPR spectra of R5a at positions 2 and 24 in the mutant (top) and wild type (bottom) CS duplexes. Arrows indicate immobilized spectral components. CS2_1A, CS24 and CS24_24T spectra were corrected for the presence of the residual free nitroxide (<3%).
EPR spectra were obtained at 5 °C for a single R5a attached to either position 2 or position 24 of the mutant duplex (CS2_1A and CS24_24T, respectively; Figure 6). The mutant CS24_24T_R5a spectrum has a narrower low-field peak as compared to that of CS24_R5a, and no splitting is observed (Figure 6). This reports a higher degree of hyperfine tensor averaging due to increased nitroxide mobility in the mutant. At position 2, the mutant DNA (CS2_1A_R5a) gives a multiple component spectrum, similar to that observed at the wild type CS2 site. The bump at the shoulder of the low-field peak is more pronounced in the mutant spectrum (Figure 6, marked by arrows), which indicates a higher fraction of an immobilized population. Overall, the mutant studies revealed that rotational diffusion of R5a is affected by variations in the nucleotide sequence at the labeling site.
Effect of a Cosolvent: Sucrose vs Ficoll 70
In data presented above, EPR spectra were recorded in 34% (w/w) sucrose solution to reduce Brownian diffusion of the entire DNA molecule (see Materials and Methods). While this strategy has been commonly used in SDSL studies, it is not clear whether sucrose differentially affects nitroxide behavior at different DNA sites, thus interfering with the analysis of site-dependent spectral variations. To address this question, R5a spectra at three CS sites were measured in the presence of Ficoll 70 (MW = 70,000 Da) instead of sucrose. Ficoll 70 is a copolymer of sucrose with epichlorohydrin. It increases solution viscosity, but minimally affects its osmolality (39).
The results reveal that spectra obtained in ~30%(w/w) Ficoll 70 maintain all the site-dependent features as those measured in sucrose. As shown in Figure 7, CS2_1A_R5a preserves a two component spectrum, while CS7_R5a retains a clear splitting of the low-field peak (indicated by arrows). Furthermore, in Ficoll 70, CS12_R5a yields a spectrum with a broad low-field peak and an unresolved splitting, indicating higher nitroxide mobility as compared toCS7_R5a. The same trend is observed using sucrose. We also note that all spectra measured in Ficoll 70 have narrower lines and therefore report higher nitroxide mobility than the corresponding sucrose spectra. Sucrose and Ficoll are drastically different in their molecular weight and chemical composition, and likely affect macromolecular Brownian motion via different mechanisms (40).
Figure 7.
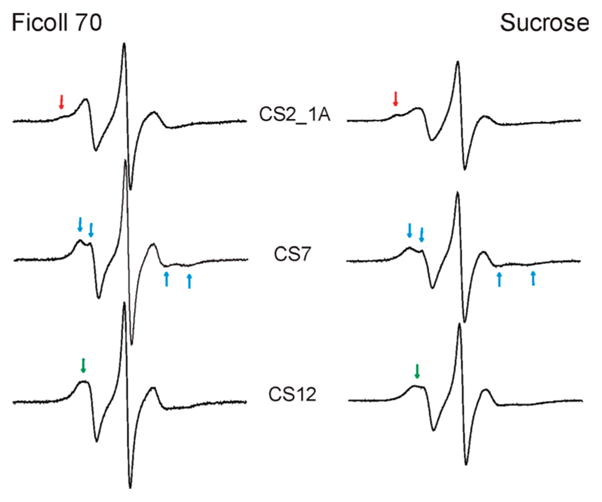
R5a spectra obtained in different cosolvents. Spectra recorded in ~30% (w/w) Ficoll 70 are shown on the left, while the corresponding spectra recorded in 34%sucrose (w/w) are shown on the right. Arrows in the same color indicate spectral features that are maintained at the same site in two cosolvents.
Consistency between the sucrose and Ficoll 70 data suggests that in the current study the observed spectral variations report differences in local DNA environment and are not artifacts due to the use of a particular cosolvent.
DISCUSSION
Data reported above clearly demonstrate that X band cw-EPR spectra of both R5 and R5a may vary according to local DNA environment. This establishes that R5 and R5a can be used to examine site-specific features in DNA at the level of an individual nucleotide. In the following sections, we discuss possible probe perturbation to DNA and potential modes of nitroxide/DNA coupling that may influence the observed EPR spectra.
Perturbations Due to R5 and R5a Nitroxide Probes
A number of experiments have collectively shown that R5 and R5a, which are attached using the same chemical scheme, do not significantly alter the native conformation of the CS duplex. Thermal melting studies on R5a single-labeled (this work) and R5 double-labeled (19) DNA revealed that the probe(s) minimally affects duplex stability, with ΔΔG°37°C < 1.0 kcal/mol in all cases studied. Circular dichroism spectroscopy measurements indicated that R5 double-labeled DNA does not alter its B-form conformation (19). Distance measurements using a pair of R5 yielded results that are in good agreement with the NMR determined DNA structure (19). We conclude that, within the sensitivity of these measurements, neither R5 nor R5a drastically alters the DNA duplex conformation.
As extrinsic probes, R5 and R5a do impose a number of changes onto the DNA. The two most noticeable ones are the presence of the pyrroline ring and the neutralization of one negative charge at the phosphate group. With the labeling scheme used, there is sufficient linker flexibility to allow the pyrroline ring to adjust to the groove of the DNA. In addition, pyrroline/DNA interactions are not expected to have sufficient energy to drastically alter the native DNA structure. Instead, these interactions may give rise to unique spectral features, such as immobilized components (Figure 6, CS2 and CS2_1A, and see later discussion).
Effects of charge neutralization are not clear. A number of studies have examined the effect of nonsymmetric charge neutralization of the DNA backbone using alkylphosphonate modifications that are conceptually very similar to R5 and R5a. DNA containing varying lengths of consecutive or alternating alkylphosphonate modifications have been studied using gel electrophoresis assays (41), NMR(42), EPR (43), and MD simulations (44, 45). The results revealed various degrees of DNA helix bending, as well as possible changes in helical parameters and deoxyribose flexibility (41, 42, 45). However, we are aware of only one report, where solution NMR was used to examine DNA duplexes containing a single alkylphosphonate (46). The study revealed a small bending of the DNA helical axis and changes in sugar puckering and helical parameters that are localized near the modification site. These results were attributed to neutralization of the phosphate charge and steric/hydrophobic effects of the alkyl moiety. Within the scope of R5 and R5a SDSL studies, further investigations are needed to examine how neutralization of a single charge perturbs native DNA conformations and affects data interpretation.
Comparison between R5 and R5a
R5 and R5a differ by one chemical group (4-Hvs 4-Br at the pyrroline ring, Figure 1A) that primarily affects internal motions of the nitroxides. In R5, torsional rotations that dictate nitroxide internal motions (t1, t2, t3 in Figure 1A) are relatively unrestricted on the nanosecond time scale ((32) and unpublished data), resulting in a high degree of averaging of magnetic tensors and thus spectra with narrow lines (Figure 2 and Results). In R5a, MD simulations indicate that t2 and t3 torsional rotations are restricted (unpublished data), thus reducing averaging of magnetic tensors. This gives rise to broader R5a spectra with more pronounced site-dependent spectral features. Similar observations have been reported in protein SDSL studies (35).
In most CS sites studied, the same trend of nitroxide mobility variations is observed between R5 and R5a, indicating that similar features of the DNA local environment are reported by these probes. For instance, both R5 and R5a show that duplex center sites (CS7, CS19) are the most restrictive in terms of nitroxide motion. At the 3′ terminal sites (CS12 and CS24), both probes give almost identical spectra, indicating that at 5 °C, the local environment at the 3′ termini is comparable to that of the duplex interior (CS5 and CS9) and dissimilar to that at the 5′ termini (CS2 and CS14).
Furthermore, both R5 and R5a report that at the 5′ termini (CS2 and CS14) the local DNA environment is different from that at other sites. However, CS2_R5 and CS14_R5 have almost identical spectra (Figure 2 and 3A), while CS2_R5a and CS14_R5a show different proportions of immobilized subpopulations (Figure 5). As discussed later, the R5a spectral differences may arise from subtle changes in site-dependent DNA/nitroxide contacts. We also note that the CS14_R5a spectrum, even with the presence of the low mobility component, shows the highest overall R5a mobility compared to other sites. This is consistent with data showing that the 5′ termini have the highest R5 dynamics.
While neither R5 norR5a significantly perturbs the DNA, R5a has more restricted internal motions as compared to R5 and therefore its overall motions are more strongly coupled to local DNA environment. Probing a given site with R5 and R5a simultaneously is more informative: it may distinguish DNA structural and dynamic features that affect both probes similarly, as well as those that influence the probes differently. This aids SDSL data interpretation.
Coupling between Nitroxide Dynamics and DNA: Modulation of Nitroxide Internal Motions
Site-dependent R5 and R5a spectral variations clearly demonstrate a coupling between DNA local environment and nitroxide dynamics. However, a detailed understanding of the rules governing such coupling is required in order to use a nitroxide spectrum to deduce information about a given DNA site. DNA may modulate R5 and R5a dynamics by two modes. Each DNA site may differentially influence nitroxide internal motions. In addition, DNA motions, which encompass all dynamic modes except the uniform global tumbling, may be transferred to the pyrroline ring via motions of the phosphorus atom. In this section we consider DNA modulations of nitroxide internal motions. Possible effects due to DNA local motions are discussed in the next section.
Nitroxide internal motions are dictated by DNA three-dimensional structure, which imposes restrictions on a nitroxide “rotamer” space. Two factors may dictate the rotamer space at each labeling site. One is the allowable volume that the pyrroline ring can occupy without steric collision with the DNA. The other is nitroxide/DNA interactions, such as hydrophobic contacts or hydrogen bonding, which may modify the pattern of occupancy within the sterically allowed volume.
To assess the sterically allowable volume, the NASNOX program was used to generate ensembles of R5a or R5 at each CS site. Each ensemble was selected based on steric exclusion between the DNA and the nitroxide, and was characterized by a quantity S that was computed based on the angular distribution of the nitroxide N–O bond vector (see Materials and Methods). The S value, which ranges from 0 to1, is analogous to an order parameter. A large S indicates small directional variations thus a limited allowable volume for the nitroxide, while a small S suggests large directional variations and an expanded allowable volume.
Computed S values for R5a are shown in Table 2. For Sp diastereomers, identical S values were obtained, suggesting the same steric constrictions at various CS sites. Sp diastereomers direct the pyrroline ring away from the DNA and toward the solvent. This may account for the lack of site-dependent variations in S values. R5a Rp diastereomers, which direct the pyrroline ring toward the DNA major groove, exhibit small variations in S values (±0.022 from the average, Table 2), again indicating similar steric constrictions. R5 modeling showed similar results, with deviations from the averaged S being less than 0.011 (data not shown).
Table 2.
Sterically allowed conformational space of R5a as estimated by NASNOX
|
S value |
||
|---|---|---|
| Labeled position | Rp | Sp |
| CS2 | 0.537 | 0.496 |
| CS5 | 0.554 | 0.495 |
| CS7 | 0.576 | 0.496 |
| CS9 | 0.546 | 0.495 |
| CS12 | 0.510 | 0.495 |
| CS14 | 0.513 | 0.498 |
| CS19 | 0.546 | 0.495 |
| CS24 | 0.522 | 0.494 |
| av ± std deviation | 0.538 ± 0.022 | 0.496 ± 0.001 |
Overall, the NASNOX results indicate very similar sterically allowable volumes at each labeling site of the CS duplex. This likely stems from isostericity of Watson–Crick base pairs and regularity of the B-form DNA duplex geometry: each base-pair unit displays very similar C1′–C1′ distances and glycosidic bond orientations, and can replace one another (e.g., A/T → C/G) without significant alterations of the phosphate–sugar backbone configuration. Since CS DNA contains all Watson–Crick base pairs, each backbone site has a similar configuration, resulting in similar sterically allowable volumes experienced by the nitroxide. However, it should be emphasized that NASNOX provides only a zero-order estimation. To achieve efficiency, NASNOX uses a fixed DNA structure and employs a stepwise search algorithm without involving a force field. These simplifications may limit its accuracy. More sophisticated computation approaches, such as MD simulations, may provide more detailed understanding of how DNA affects nitroxide conformations.
With a nearly constant sterically allowable volume along the CS duplex, spectral variations may arise from site-specific nitroxide/DNA interactions. Simple modeling using the reported NMR structure indicates that, for the Rp diastereomer at CS2, either the 2-methyl groups or the 4-bromine atom of the R5a pyrroline ring may be positioned at approximately 3 A ° to the methyl group of the 3′ neighboring thymine (T2 of CS DNA) (Figure 8). This may give rise to interactions that significantly immobilize a subpopulation of the nitroxide, resulting in a two-component CS2_R5a spectrum (Figure 5). Similarly, immobilized spectral components observed in CS14_R5a (Figure 5) and CS2_1A_R5a (Figure 6) likely originate from local R5a/DNA interactions.
Figure 8.

Proposed R5a/DNA interactions at the 5′ terminal site CS2. The models were generated using WebLab ViewerPro 3.7 (Molecular Simulations, Inc.), with R5a attached to the Rp phosphorothioate of the target site in the NMR structure of the CS duplex. The 4-Br atom of R5a is shown as a dark red ball, the C2 methyls of R5a are shown as purple balls, and the C5-methyl of T2 is shown as a green ball. In the modeling, the torsion angle t1 was varied, while t2 and t3 were set based on MD simulation data, which show t2 ≡ 180°; and t3 = +100° or −100°. (A) R5a modeled with t1 = −60°, t2 = 180°, and t3 = +100°. This particular t1 value gives the closest distance between the 4-Br atom of R5a and the C5-methyl of T2 without any steric collision between R5a and DNA. (B) R5a modeled with t1 = −50°, t2 = 180°, and t3 = −100°. This configuration gives the closest distance between one of the 2-methyl groups of R5a and the C5-methyl of T2 without any steric collision.
Modeling studies predict that nitroxide/DNA interactions described above are favored only for the Rp diastereomer of CS2_R5a, but not for the Sp diastereomer, which will be tested in the future. This analysis highlights a need to further investigate diastereomer specific behaviors, which is a common issue facing R5 and R5a. While data reported here clearly demonstrate the feasibility of probing DNA environment using mixed diastereomers, studies using individual diastereomers may expand the scope of information obtainable from these nitroxide probes.
In summary, different sites in the CS duplex present a similar sterically allowable space to the nitroxide, but may vary in terms of nitroxide/DNA contacts that may depend on factors such as the surrounding sequence or diastereomeric characteristics of the label. The variable contacts may partially account for the observed EPR spectral features, particularly the presence of immobilized second components. Studies are underway to elucidate the correlation between EPR spectral features and site-specific nitroxide/DNA interactions.
Coupling between Nitroxide Dynamics and DNA: Correlation to DNA Local Motions
Here we examine how R5/R5a mobility varies depending on the labeling position (duplex termini vs interior) and the flanking nucleotide sequence. The analysis suggests a positive correlation between R5/R5amobility and expected DNA local flexibility. This provides evidence that these probes can report on certain modes of DNA local motion.
The first example comes from CS7 and CS19. Based on the labeling position (duplex center) and thermodynamic stability of the flanking sequence (-GC- dinucleotide step) (38), these two sites are expected to exhibit lower DNA local flexibility as compared to other sites. Indeed, at CS 7 and CS 19, R5/R5a spectra are similar at 5 °C, and the nitroxide mobility is lower than that at other sites (Figure 4 and 5).
Another example of positional dependence of nitroxide mobility comes from comparisons between 5′ terminal sites CS2, CS14 and the interior site CS5. Although these sites share the same -CT- dinucleotide sequence, a much higher R5 mobility is observed at CS2 and CS14 (Figure 2). Furthermore, overall R5a mobility at CS14, in spite of the presence of an immobile component, is higher than that at CS5 (see Figure 5 and Results). This trend of nitroxide mobility may be accounted for by increased DNA local flexibility at CS2 and CS14 due to the reduction of stacking constraints at the 5′ termini.
R5 and R5a mobility also changes according to the neighboring nucleotide sequence. The 3′ terminal sites (CS24 and CS12) are located across from the 5′ terminal sites (CS2 and CS14), yet R5 mobility at both 3′ termini is lower than that at the 5′ termini (Figure 2), andR5amobility at CS12 and CS24 is lower than that of CS14 (Figure 5). In the CS duplex, labels at the 5′ terminal sites are sandwiched by -CT- dinucleotide step, while those at the 3′ termini are flanked by -AG- step. Energetics of stacking at these two steps is different: gas phase ab initio calculations have shown that -AG- dinucleotide is more stable (lower stacking energy) than that of -CT- (47); calculations using Langevin dipoles also concluded that in water the order of stacking stability is pyrimidine/pyrimidine (e.g., -CT-)<purine/pyrimidine<purine/purine (e.g., -AG-) (48). The more favorably stacked sites (i.e., 3′ termini in CS DNA) likely have lower local flexibility, thus exhibiting lower nitroxide mobility.
A second example of sequence dependence comes from mutation studies (Figure 6). Mutating the C1/G24 base pair to A1/T24 leads to a less favorably stacked dinucleotide (-AG- vs -AT-) and a loss of one hydrogen bond at the terminal base-pair. This reduces the thermodynamic stability at site 24, giving rise to higher DNA local flexibility and consequently increased R5a mobility (Figure 6).
As R5/R5a mobility positively correlates with expected DNA local flexibility, our data indicate that flexibility (or dynamics) at the 3′ termini of the CS duplex is similar to that of the interior sites, but noticeably lower than that at the 5′ termini. This phenomenon was reported previously, where 13C NMR relaxation experiments revealed that order parameters of sugar C–H bond vectors at the 3′ termini of a self-complementary duplex were comparable with average order parameters at central nucleotides (49). Data obtained from R5 and R5a thus argue against a commonly used description of DNA flexibility, where duplex termini are always more dynamic than internal sites due to the lack of stacking constraints.
R5 and R5a assess DNA local flexibility from the perspective of the phosphodiester group, thus providing EPR probes for studying local motions at the DNA backbone. This differs from previously reported nitroxides that were attached to various base positions (5, 6, 22, 23). While DNA backbone dynamics may be important in a number of events such as DNA–protein interactions, experimental studies in this area are very limited. We are aware of work from only two groups: Redfield and coworkers examined picosecond dynamics of 31P atoms in DNA by field-cycling NMR relaxation (50); and Drobny and coworkers investigated sequence dependent nanosecond–microsecond dynamics of deuterated 5′-methylenes using solid-state NMR (4, 51). In particular, the Drobny group has reported reduction in dynamics of deoxycytosine 5′-methylene upon cytosine methylation within certain sequences. It was proposed that the decrease in backbone flexibility is due to methylation induced base stacking enhancement (4, 51). The notion that local flexibility at the DNA backbone depends on nucleotide sequence and is correlated with base stacking stability is consistent with conclusions drawn from R5 and R5a data reported here.
Conclusions
We have demonstrated that R5 andR5a are able to report differences in local DNA environment without severely perturbing the native conformation of the parent molecule. These probes can be easily attached to arbitrary DNA sites and provide a unique view on the local environment through the phosphodiester group. Consistent with studies on other nitroxides, R5 and R5a motions were found to be influenced by a number of factors, including the mode of linker motion, the location of the labeling site, and the surrounding nucleotide sequence. Further studies, such as analysis of R5/R5a spectral variations according to diastereomer identity, DNA base sequence, secondary structure elements (mispaired or modified bases, abasic sites, etc.), and environmental conditions (salt, temperature, etc.), may reveal more detailed correlations between R5 and R5a spectra and local DNA structural and dynamic features. This will provide a means for studying local environment in large DNA molecules by scanning them in a nucleotide by nucleotide fashion.
Supplementary Material
Footnotes
Research supported by grants from the National Institutes of Health (GM069557, P.Z.Q.), National Science Foundation (MCB 054652, P.Z. Q.), and the Hungarian Research Fund (OTKA T048334, K.H.).
Abbreviations: EPR, electron paramagnetic resonance; cw-EPR, continuous-wave EPR; SDSL, site-directed spin labeling; MES, 2-(N-morpholino) ethanesulfonic acid; Tris, 2-amino-2-(hydroxymethyl)-1,3-propanediol; MD, molecular dynamics.
SUPPORTING INFORMATION AVAILABLE
Additional figures as noted in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Gueron M, Leroy JL. Studies of base pair kinetics by NMR measurement of proton exchange. Methods Enzymol. 1995;261:383–413. doi: 10.1016/s0076-6879(95)61018-9. [DOI] [PubMed] [Google Scholar]
- 2.Shajani Z, Varani G. NMR studies of dynamics in RNA and DNA by 13C relaxation. Biopolymers. 2007;86:348–359. doi: 10.1002/bip.20650. [DOI] [PubMed] [Google Scholar]
- 3.Meints GA, Miller PA, Pederson K, Shajani Z, Drobny G. Solid-state nuclear magnetic resonance spectroscopy studies of furanose ring dynamics in the DNA HhaI binding site. J Am Chem Soc. 2008;130:7305–7314. doi: 10.1021/ja075775n. [DOI] [PubMed] [Google Scholar]
- 4.Pederson K, Meints GA, Shajani Z, Miller PA, Drobny GP. Backbone dynamics in the DNA HhaI protein binding site. J Am Chem Soc. 2008;130:9072–9079. doi: 10.1021/ja801243d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Okonogi TM, Reese AW, Alley SC, Hopkins PB, Robinson BH. Flexibility of duplex DNA on the submicrosecond timescale. Biophys J. 1999;77:3256–3276. doi: 10.1016/S0006-3495(99)77157-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cekan P, Smith AL, Barhate N, Robinson BH, Sigurdsson ST. Rigid spin-labeled nucleoside C: a nonperturbing EPR probe of nucleic acid conformation. Nucleic Acids Res. 2008;36:5946–5954. doi: 10.1093/nar/gkn562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Orden AV, Jung J. Review fluorescence correlation spectroscopy for probing the kinetics and mechanisms of DNA hairpin formation. Biopolymers. 2008;89:1–16. doi: 10.1002/bip.20826. [DOI] [PubMed] [Google Scholar]
- 8.Shirude PS, Balasubramanian S. Single molecule conformational analysis of DNA G-quadruplexes. Biochimie. 2008;90:1197–1206. doi: 10.1016/j.biochi.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Altan-Bonnet G, Libchaber A, Krichevsky O. Bubble dynamics in double-stranded DNA. Phys Rev Lett. 2003;90:138101. doi: 10.1103/PhysRevLett.90.138101. [DOI] [PubMed] [Google Scholar]
- 10.Dornberger U, Leijon M, Fritzsche H. High base pair opening rates in tracts of GC base pairs. J Biol Chem. 1999;274:6957–6962. doi: 10.1074/jbc.274.11.6957. [DOI] [PubMed] [Google Scholar]
- 11.Isaacs RJ, Spielmann HP. A model for initial DNA lesion recognition by NER and MMR based on local conformational flexibility. DNA Repair. 2004;3:455–464. doi: 10.1016/j.dnarep.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 12.Yang W. Poor base stacking at DNA lesions may initiate recognition by many repair proteins. DNA Repair. 2006;5:654–666. doi: 10.1016/j.dnarep.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 13.Maillard O, Camenisch U, Clement FC, Blagoev KB, Naegeli H. DNA repair triggered by sensors of helical dynamics. Trends Biochem Sci. 2007;32:494–499. doi: 10.1016/j.tibs.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 14.Fanucci GE, Cafiso DS. Recent advances and applications of site-directed spin labeling. Curr Opin Struct Biol. 2006;16:644–653. doi: 10.1016/j.sbi.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 15.Sowa GZ, Qin PZ. Site-directed spin labeling studies on nucleic acid structure and dynamics. Prog Nucleic Acid Res Mol Biol. 2008;82:147–197. doi: 10.1016/S0079-6603(08)00005-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qin PZ, Butcher SE, Feigon J, Hubbell WL. Quantitative analysis of the isolated GAAA tetraloop/receptor interaction in solution: a site-directed spin labeling study. Biochemistry. 2001;40:6929–6936. doi: 10.1021/bi010294g. [DOI] [PubMed] [Google Scholar]
- 17.Qin PZ, Haworth IS, Cai Q, Kusnetzow AK, Grant GP, Price EA, Sowa GZ, Popova A, Herreros B, He H. Measuring nanometer distances in nucleic acids using a sequence-independent nitroxide probe. Nat Protoc. 2007;2:2354–2365. doi: 10.1038/nprot.2007.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grant GPG, Boyd N, Herschlag D, Qin PZ. Motions of the Substrate Recognition Duplex in a Group I Intron Assessed by Site-Directed Spin Labeling. J Am Chem Soc. 2009;131:3136–3137. doi: 10.1021/ja808217s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cai Q, Kusnetzow AK, Hubbell WL, Haworth IS, Gacho GP, Van Eps N, Hideg K, Chambers EJ, Qin PZ. Site-directed spin labeling measurements of nanometer distances in nucleic acids using a sequence-independent nitroxide probe. Nucleic Acids Res. 2006;34:4722–4730. doi: 10.1093/nar/gkl546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cai Q, Kusnetzow AK, Hideg K, Price EA, Haworth IS, Qin PZ. Nanometer distance measurements in RNA using site-directed spin labeling. Biophys J. 2007;93:2110–2117. doi: 10.1529/biophysj.107.109439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Columbus L, Hubbell WL. A new spin on protein dynamics. Trends Biochem Sci. 2002;27:288–295. doi: 10.1016/s0968-0004(02)02095-9. [DOI] [PubMed] [Google Scholar]
- 22.Keyes RS, Bobst AM. Spin Labeling: The next Millennium. Vol. 14. Plenum Press Corp; New York: 1998. Spin-Labeled Nucleic Acids, in Biological Magnetic Resonance; pp. 283–338. [Google Scholar]
- 23.Robinson BH, Mailer C, Drobny G. Site-specific dynamics in DNA: experiments. Annu Rev Biophys Biomol Struct. 1997;26:629–658. doi: 10.1146/annurev.biophys.26.1.629. [DOI] [PubMed] [Google Scholar]
- 24.Smith AL, Cekan P, Brewood GP, Okonogi TM, Alemayehu S, Hustedt EJ, Benight AS, Sigurdsson ST, Robinson BH. Conformational Equilibria of Bulged Sites in Duplex DNA Studied by EPR Spectroscopy. J Phys Chem B. 2009;113:2664–2675. doi: 10.1021/jp808260b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kanehara K, Mizuguchi M, Makino K. Isolation of oligodeoxynucleoside phosphorothioate diastereomers by the combination of DEAE ion-exchange and reversed-phase chromatography. Nucleosides Nucleotides. 1996;15:399–406. [Google Scholar]
- 26.Subach FB, Miuller S, Tashlitskii VN, OV PI, Gromova ES. Preparation of DNA-duplexes, containing internucleotide thiophosphate groups in various positions of the recognition site for the EcoRII restriction endonuclease. Bioorg Khim. 2003;29:623–631. doi: 10.1023/b:rubi.0000008898.42854.7c. [DOI] [PubMed] [Google Scholar]
- 27.Grant GP, Popova A, Qin PZ. Diastereomer characterizations of nitroxide-labeled nucleic acids. Biochem Biophys Res Commun. 2008;371:451–455. doi: 10.1016/j.bbrc.2008.04.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hankovszky HO, Hideg K, Lex L. Nitroxyls; VII. Synthesis and Reactions of Highly Reactive 1-Oxyl-2,2,5,5-tetramethyl-2,5-dihydro-pyrrole-3-ylmethyl Sulfonates. Synthesis. 1980:914–916. [Google Scholar]
- 29.Kálai T, Balog M, Jekö J, Hideg K. 3-Substituted 4-Bromo-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-1-yloxyl Radicals as versatile Synthons for Synthesis of New Paramagnetic Heterocycles. Synthesis. 1998:1476–1482. [Google Scholar]
- 30.Qin PZ, Iseri J, Oki A. A model system for investigating lineshape/structure correlations in RNA site-directed spin labeling. Biochem Biophys Res Commun. 2006;343:117–124. doi: 10.1016/j.bbrc.2006.02.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tirado MM, Garciadelatorre J. Rotational-Dynamics of Rigid, Symmetric Top Macromolecules - Application to Circular-Cylinders. J Chem Phys. 1980;73:1986–1993. [Google Scholar]
- 32.Price EA, Sutch BT, Cai Q, Qin PZ, Haworth IS. Computation of nitroxide-nitroxide distances in spin-labeled DNA duplexes. Biopolymers. 2007;87:40–50. doi: 10.1002/bip.20769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leporc S, Mauffret O, Tevanian G, Lescot E, Monnot M, Fermandjian S. An NMR and molecular modelling analysis of d(CTACTGCTTTAG). d(CTAAAGCAGTAG) reveals that the particular behaviour of Tp A steps is related to edge-to-edge contacts of their base-pairs in the major groove. Nucleic Acids Res. 1999;27:4759–4767. doi: 10.1093/nar/27.24.4759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Knowles PF, Marsh D, Rattle HWE. Magnetic Resonance of Biomolecules. John Wiley & Sons, Ltd; New York: 1976. [Google Scholar]
- 35.Columbus L, Kalai T, Jeko J, Hideg K, Hubbell WL. Molecular motion of spin labeled side chains in alpha-helices: analysis by variation of side chain structure. Biochemistry. 2001;40:3828–3846. doi: 10.1021/bi002645h. [DOI] [PubMed] [Google Scholar]
- 36.McHaourab HS, Lietzow MA, Hideg K, Hubbell WL. Motion of spin-labeled side chains in T4 lysozyme. Correlation with protein structure and dynamics. Biochemistry. 1996;35:7692–7704. doi: 10.1021/bi960482k. [DOI] [PubMed] [Google Scholar]
- 37.Qin PZ, Hideg K, Feigon J, Hubbell WL. Monitoring RNA base structure and dynamics using site-directed spin labeling. Biochemistry. 2003;42:6772–6783. doi: 10.1021/bi027222p. [DOI] [PubMed] [Google Scholar]
- 38.SantaLucia J. A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc Natl Acad Sci USA. 1998;95:1460–1465. doi: 10.1073/pnas.95.4.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ficoll PM 70, Ficoll PM 400, data file 18-1158-27 ed., pp 1–6, Amersham Biosciences.
- 40.Lavalette D, Hink MA, Tourbez M, Tetreau C, Visser AJ. Proteins as micro viscosimeters: Brownian motion revisited. Eur Biophys J. 2006;35:517–522. doi: 10.1007/s00249-006-0060-z. [DOI] [PubMed] [Google Scholar]
- 41.Strauss-Soukup JK, Vaghefi MM, Hogrefe RI, Maher LJ., III Effects of neutralization pattern and stereochemistry on DNA bending by methylphosphonate substitutions. Biochemistry. 1997;36:8692–8698. doi: 10.1021/bi9705467. [DOI] [PubMed] [Google Scholar]
- 42.Thiviyanathan V, Vyazovkina KV, Gozansky EK, Bichenchova E, Abramova TV, Luxon BA, Lebedev AV, Gorenstein DG. Structure of hybrid backbone, methylphosphonate DNA heteroduplexes: Effect of R and S stereochemistry. Biochemistry. 2002;41:827–838. doi: 10.1021/bi011551k. [DOI] [PubMed] [Google Scholar]
- 43.Okonogi TM, Alley SC, Harwood EA, Hopkins PB, Robinson BH. Phosphate backbone neutralization increases duplex DNA flexibility: A model for protein binding. Proc Natl Acad Sci USA. 2002;99:4156–4160. doi: 10.1073/pnas.072067799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hamelberg D, Williams LD, Wilson WD. Effect of a neutralized phosphate backbone on the minor groove of B-DNA: molecular dynamics simulation studies. Nucleic Acids Res. 2002;30:3615–3623. doi: 10.1093/nar/gkf472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kosikov KM, Gorin AA, Lu XJ, Olson WK, Manning GS. Bending of DNA by asymmetric charge neutralization: All-atom energy simulations. J Am Chem Soc. 2002;124:4838–4847. doi: 10.1021/ja0125218. [DOI] [PubMed] [Google Scholar]
- 46.Soliva R, Monaco V, Gomez-Pinto I, Meeuwenoord NJ, Marel GA, Boom JH, Gonzalez C, Orozco M. Solution structure of a DNA duplex with a chiral alkyl phosphonate moiety. Nucleic Acids Res. 2001;29:2973–2985. doi: 10.1093/nar/29.14.2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sponer J, Jurecka P, Marchan I, Luque FJ, Orozco M, Hobza P. Nature of base stacking: Reference quantum-chemical stacking energies in ten unique B-DNA base-pair steps. Chem Eur J. 2006;12:2854–2865. doi: 10.1002/chem.200501239. [DOI] [PubMed] [Google Scholar]
- 48.Florian J, Sponer J, Warshel A. Thermodynamic parameters for stacking and hydrogen bonding of nucleic acid bases in aqueous solution: Ab initio/Langevin dipoles study. J Phys Chem B. 1999;103:884–892. [Google Scholar]
- 49.Spielmann HP. Dynamics of a bis-intercalator DNA complex by 1H-detected natural abundance 13C NMR spectroscopy. Biochemistry. 1998;37:16863–16876. doi: 10.1021/bi980789e. [DOI] [PubMed] [Google Scholar]
- 50.Roberts MF, Cui Q, Turner CJ, Case DA, Redfield AG. High-resolution field-cycling NMR studies of a DNA octamer as a probe of phosphodiester dynamics and comparison with computer simulation. Biochemistry. 2004;43:3637–3650. doi: 10.1021/bi035979q. [DOI] [PubMed] [Google Scholar]
- 51.Geahigan KB, Meints GA, Hatcher ME, Orban J, Drobny GP. The dynamic impact of CpG methylation in DNA. Biochemistry. 2000;39:4939–4946. doi: 10.1021/bi9917636. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.