

Abstract
A sequential intramolecular-intermolecular enantioselective alkene difunctionalization reaction has been developed which is thought to proceed through Pd-catalyzed quinone methide formation. The synthesis of new chiral heterocyclic compounds with adjacent chiral centers is achieved in enantiomeric ratios up to 99:1 and diastereomeric ratios up to 10:1.
The formation of two carbon-heteroatom bonds across an alkene is a process which rapidly increases molecular complexity. The osmium-catalyzed Sharpless dihydroxylation1 is the epitome of enantioselective alkene difunctionalization, though recent research has included other metal catalysts and expanded to the formation of functional groups other than 1,2-diols.2 Currently, significant focus has been on palladium catalysis, likely due to the efficiency with which palladium activates olefins for nucleophilic attack.3 However, in order to achieve the second bond construction, β-hydride elimination from a Pd-alkyl intermediate A, which leads to a Wacker-type monofunctionalized alkene product 1,4 must be prevented (Scheme 1). The alkene dialkoxylation reaction developed in our laboratory is believed to accomplish this through the formation of a quinone methide intermediate, which allows for attack by a second equivalent of alcohol.5 Based on this mechanistic proposal, we envisioned an alkene difunctionalization reaction where a sequential intra-intermolecular process would allow for the selective formation of two distinct carbon-heteroatom bonds by employing substrates which contain a nucleophile tethered to the alkene (Scheme 1). We hypothesized initial intramolecular nucleopalladation to form heterocyclic intermediate B. Subsequent formation of a quinone methide intermediate C allows for attack by an exogenous nucleophile to form the product and release Pd0, which is reoxidized using molecular oxygen. Herein we report a highly enantioselective addition of two distinct nucleophiles across alkenes capable of quinone methide formation to access oxygen-based heterocycles with contiguous chiral centers.
Scheme 1.

Avoiding β-hydride elimination to develop Pd-catalyzed alkene difunctionalization reactions.
In exploring the possibility of sequential intra-intermolecular alkene functionalization reactions, we examined 2 as a substrate6 with methanol as the exogenous nucleophile. Under the standard conditions for enantioselective intermolecular alkene dialkoxylation,5b using (S)-iPrQuinox as the chiral ligand, a low yield of the desired product was observed with promising enantioselectivity, but low diastereoselectivity (Table 1, entry 1).7 Addition of a catalytic amount of base and removal of molecular sieves led to modest improvements in product yield (entry 2). Reflecting on our intermolecular asymmetric dialkoxylation reaction, where copper improved yield and chemoselectivity but was removed in order to achieve high enantioselectivity, we revisited the use of copper as an additive to improve the reaction.5b We hypothesized that the reason for decreased enantioselectivity with increased copper loading was that Cu sequesters the chiral ligand from Pd via ligand substitution, resulting in an achiral palladium catalyst. Thus we added 20 mol% CuCl2 along with enough chiral ligand to coordinate both metals (32 mol%). A significant improvement in yield was observed without a detrimental effect on enantiomeric ratio (entry 3). Based on the accelerated rate of the reaction, we were able to decrease the metal and ligand loadings (entry 4). In order to find conditions that would allow for the use of nucleophiles other than methanol, other solvents were evaluated. We were encouraged to find that both THF and toluene led to increased enantioselectivity, albeit with a decrease in rate and product yield (entries 5 and 6). A 1:1 mixture of THF: toluene was also found to be a viable solvent system (entry 7). Improvements in rate and yield were observed when using CuCl instead of CuCl2, potentially due to a decrease in the [Cl−] or a change in oxidation potential (entry 9).8
Table 1.
Optimization of reaction conditions.
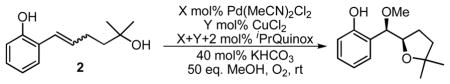 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | X | Y | solvent | time | %conva | %yielda | erb | drb |
| 1c | 10 | -- | MeOH | 15 h | 100 | 2 | 88:12 | 2.4:1 |
| 2 | 10 | -- | MeOH | 15 h | 74 | 8 | 92:8 | 2.7:1 |
| 3 | 10 | 20 | MeOH | 10 min | 100 | 95 | 92:8 | 9.4:1 |
| 4 | 4 | 8 | MeOH | 30 min | 100 | 87 | 92:8 | 9.6:1 |
| 5 | 4 | 8 | THF | 2 h | 79 | 54 | 98:2 | 7.8:1 |
| 6 | 4 | 8 | toluene | 2 h | 96 | 68 | 97:3 | 5.1:1 |
| 7 | 4 | 8 | 1:1 THF:toluene | 5 h | 100 | 67 | 97:3 | 6.7:1 |
| 8d | 4 | 8 | 1:1 THF:toluene | 2 h | 99 | 80 | 98:2 | 8.9:1 |
Reactions run on 0.1 mmol scale with [2]=0.1 M.
Determined by GC analysis using an internal standard.
Determined by GC with a column equipped with a chiral stationary phase.
With 50 mg 3 Å MS and without KHCO3.
CuCl was used in place of CuCl2
With these optimized conditions, the scope of the alkene difunctionalization reaction was evaluated. In addition to methanol, n-butanol can be used as exogenous nucleophile (Table 2, 3b) where the use of toluene as solvent was found to give slightly improved yields.9 Alcohols containing a functional group are also well tolerated, including 3-butenol, 2-methoxyethanol, and 2-chloroethanol (entries 3c-3e). Ethers with the potential for deprotection are formed using benzyl alcohol and trimethylsilylethanol, with the latter giving an excellent er of 99:1 (3f & 3g). A relatively complex chiral alcohol, (−)-myrtenol, was employed successfully (3h), demonstrating the potential to couple two chiral partners. To enhance miscibility, tert-amylalcohol was employed for the addition of water, yielding the free secondary alcohol product with excellent enantioselectivity (3i). This solvent was found to be preferred for other polar nucleophiles, such as ethylene glycol (3j). Excitingly, the use of sodium azide demonstrates that an exogenous nitrogen nucleophile is viable (3k). While low diastereoselectivity is observed, the diastereomers are readily separable. In examining what other ring systems can be accessed, it was found that both tertiary and primary alcohol substrates cyclize to yield tetrahydrofuran (3l) and tetrahydropyran (3m) containing compounds. A 1,4-dioxane is formed (3n) in modest yield and good enantioselectivity.
Table 2.
Evaluation of scope.
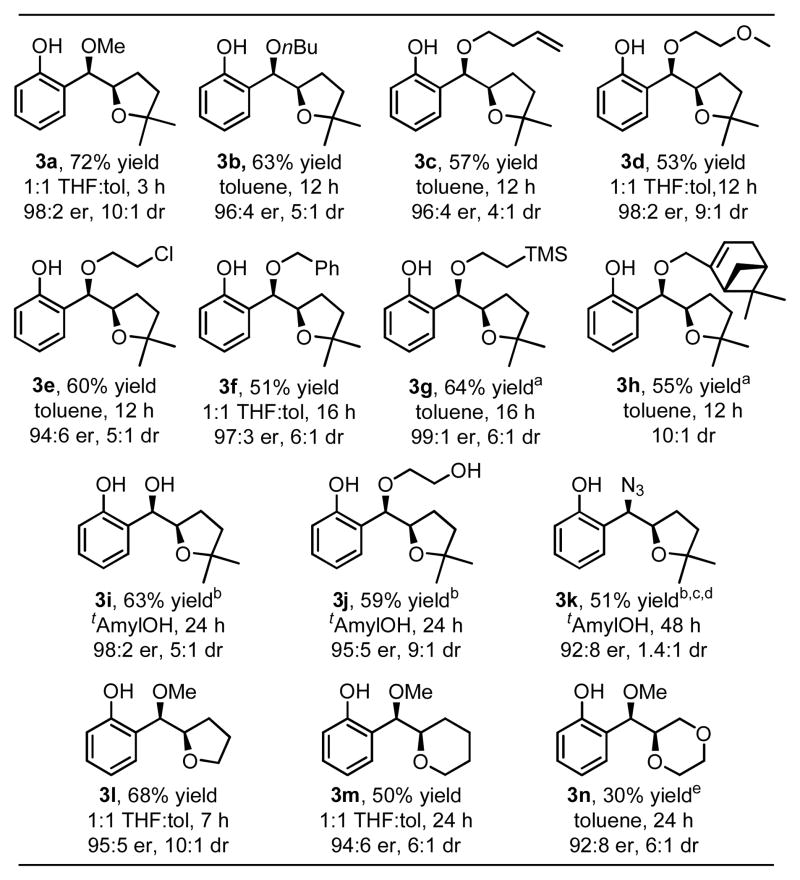 |
Reaction conditions: 4 mol% Pd(MeCN)2Cl2, 8 mol% CuCl, 14 mol% (S)-iPrQuinox, 40 mol% KHCO3, 50 eq. NuH, balloon O2, rt, 0.1 M, 0.50 mmol scale. er for major diastereomer determined by GC, HPLC or SFC using a column equipped with a chiral stationary phase. dr determined by GC or 1H NMR. Major diastereomer confirmed by single crystal X-ray analysis of entry 1 and absolute configuration determined by Mosher ester analysis of entry 9.
25 eq. of NuH.
4 mol% Pd[(S)-iPrQuinox]Cl2, 8 mol% Cu[(S)-iPrQuinox]Cl2, 10 mol% NaHCO3.
30 °C, 2 eq. of NaN3.
er of minor diastereomer is 92:8.
1 eq. NaHCO3
 |
(1) |
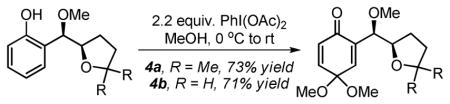
While the incorporation of an o-phenol in the substrate is a mechanistic necessity at this stage, it can be used as a synthetic handle for further functionalization. To demonstrate this, the phenol was oxidized using PhI(OAc)2 to access p-benzoquinone ketals 3 and 4 with no loss of diastereomeric purity (eq. 1).10
To extend the scope of this process to carbon nucleophiles, we submitted an enol ether, a classic inverse electron demand Diels-Alder partner with quinone methides,11 to the reaction conditions. To our delight, the Diels-Alder products 5a and 5b are isolated in good diastereoselectivity and excellent enantioselectivity.12 This complexity generating reaction sets four contiguous stereocenters and allows access to novel functionalized chroman derivatives, an important pharmacophore.13
 |
(2) |
In conclusion, we have developed a highly enantioselective Pd-catalyzed alkene difunctionalization reaction involving the addition of two distinct nucleophiles, a process which allows for the formation of complex chiral molecules from relatively simple starting materials. Future work will focus on expansion of the scope to include other types of nucleophiles and Diels Alder partners, improvement of reaction conditions to reduce the required amount of the second nucleophile, and development of a deeper understanding of the mechanistic details of the reaction.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (NIGMS RO1 GM3540). Crystal structure analysis was performed by Atta Arif. We thank Gary Keck and Ryan Looper for insightful discussions.
Footnotes
Supporting Information Available: Experimental procedures and full spectroscopic data for new compounds are available free of charge via the internet at http://pubs.acs.org.
References
- 1.Kolb HC, VanNieuwenhze MS, Sharpless KB. Chem Rev. 1994;94:2483. [Google Scholar]
- 2.For recent examples, see: Zabawa TP, Kasi D, Chemler SR. J Am Chem Soc. 2005;127:11250. doi: 10.1021/ja053335v.Zeng W, Chemler SR. J Am Chem Soc. 2007;129:12948. doi: 10.1021/ja0762240.Fuller PH, Chemler SR. Org Lett. 2007;9:5477. doi: 10.1021/ol702401w.Michaelis DJ, Shaffer CJ, Yoon TP. J Am Chem Soc. 2007;129:1866. doi: 10.1021/ja067894t.Fuller PH, Kim JW, Chemler SR. J Am Chem Soc. 2008;130:17638. doi: 10.1021/ja806585m.Michaelis DJ, Ischay MA, Yoon TP. J Am Chem Soc. 2008;130:6610. doi: 10.1021/ja800495r.Paderes MC, Chemler SR. Org Lett. 2009;11:1915. doi: 10.1021/ol9003492.Donohoe TJ, Callens CKA, Thompson AL. Org Lett. 2009;11:2305. doi: 10.1021/ol900631y.Benkovics T, Du J, Guzei IA, Yoon TP. J Org Chem. 2009;74:5545. doi: 10.1021/jo900902k.For a review of alkene 1,2-diamination see: Cardona F, Goti A. Nature Chem. 2009;1:269. doi: 10.1038/nchem.256.
- 3.For a review of pioneering work in this area, see: Bäckvall J-E. Metal-Catalyzed Cross-Coupling Reactions. 2. Vol. 2. 2004. p. 479.Bäckvall JE, Nordberg RE. J Am Chem Soc. 1981;103:4959.For a leading reference on alkoxycarbonylation, see: Semmelhack MF, Bodurow C. J Am Chem Soc. 1984;106:1496.For a review of advances in Pd-catalyzed alkene difunctionalization, see: Jensen KH, Sigman MS. Org Biomol Chem. 2008;6:4083. doi: 10.1039/b813246a.Kalyani D, Sanford MS. J Am Chem Soc. 2008;130:2150. doi: 10.1021/ja0782798.Wang A, Jiang H, Chen H. J Am Chem Soc. 2009;131:3846. doi: 10.1021/ja900213d.Rosewall CF, Sibbald PA, Liskin DV, Michael FE. J Am Chem Soc. 2009;131:9488. doi: 10.1021/ja9031659.Rodriguez A, Moran WJ. Eur J Org Chem. 2009:1313.Urkalan KB, Sigman MS. Angew Chem, Int Ed. 2009;48:3146. doi: 10.1002/anie.200900218.Tsujihara T, Takenaka K, Onitsuka K, Hatanaka M, Sasai H. J Am Chem Soc. 2009;131:3452. doi: 10.1021/ja809965e.
- 4.Beccalli EM, Broggini G, Martinelli M, Sottocornola S. Chem Rev. 2007;107:5318. doi: 10.1021/cr068006f. [DOI] [PubMed] [Google Scholar]
- 5.Schultz MJ, Sigman MS. J Am Chem Soc. 2006;128:1460. doi: 10.1021/ja0579053.Zhang Y, Sigman MS. J Am Chem Soc. 2007;129:3076. doi: 10.1021/ja070263u.For other examples of Pd-catalyzed dioxygenation of o-vinyl phenols, see: Chevrin C, Le Bras J, Henin F, Muzart J. Synthesis. 2005:2615.Thiery E, Chevrin C, Le Bras J, Harakat D, Muzart J. J Org Chem. 2007;72:1859. doi: 10.1021/jo062491x.
- 6.E- and Z-alkene isomers are observed to isomerize rapidly under the reaction conditions, and thus a mixture of isomers is allowed.
- 7.This experiment results in a complex mixture of uncharacterized byproducts beleived to be a result of oligomerization.
- 8.(a) Bäckvall JE, Akermark B, Ljunggren SO. J Am Chem Soc. 1979;101:2411. [Google Scholar]; (b) Henry PM. J Am Chem Soc. 1966;88:1595. [Google Scholar]
- 9.See supporting information for details.
- 10.For a leading reference, see: Yu M, Danishefsky SJ. J Am Chem Soc. 2008;130:2783. doi: 10.1021/ja7113757.
- 11.Arduini A, Bosi A, Pochini A, Ungaro R. Tetrahedron. 1985;41:3095. [Google Scholar]
- 12.Relative stereochemistry determined by NMR spectroscopy and derivatization followed by x-ray crystal structural analysis.9
- 13.Shen HC. Tetrahedron. 2009;65:3931. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.