

Abstract
Protein-RNA interactions are important for many cellular processes. The Nut-utilization site (N)-protein of bacteriophages contains an N-terminal arginine-rich motif that undergoes a folding transition upon binding to the boxB RNA hairpin loop target structure. Molecular dynamics simulations were used to investigate the dynamics of the P22 N-peptide-boxB complex and to elucidate the energetic contributions to binding. In addition, the free-energy changes of RNA and peptide conformational adaptation to the bound forms, as well as the role of strongly bound water molecules at the peptide-RNA interface, were studied. The influence of peptide amino acid substitutions and the salt dependence of interaction were investigated and showed good agreement with available experimental results. Several tightly bound water molecules were found at the RNA-binding interface in both the presence and absence of N-peptide. Explicit consideration of the waters resulted in shifts of calculated contributions during the energetic analysis, but overall similar binding energy contributions were found. Of interest, it was found that the electrostatic field of the RNA has a favorable influence on the coil-to-α-helix transition of the N-peptide already outside of the peptide-binding site. This result may have important implications for understanding peptide-RNA complex formation, which often involves coupled folding and association processes. It indicates that electrostatic interactions near RNA molecules can lead to a shift in the equilibrium toward the bound form of an interacting partner before it enters the binding pocket.
Introduction
Protein-RNA interactions are crucially important for numerous cellular processes, such as gene expression and its regulation, and for many catalytic activities required for protein synthesis. To understand these processes at a molecular level, it is important to know how a RNA molecule specifically recognizes its partner protein. Recent advancements in the field of structure determination have provided three-dimensional (3D) atomic details for a number of protein-RNA complexes and allowed analysis of the binding interface (1–10). However, the binding process and the associated energetic and conformational changes are still not well understood.
It is recognized that the conformation of interacting partners can change dramatically upon protein-RNA complex formation (11,12). For example, the N-proteins (Nut-utilization site proteins) of bacteriophages contain arginine-rich conserved RNA recognition motifs of ∼20 amino acids that undergo a folding transition to form a bent α-helix upon binding to the RNA target structure (13). The function of N proteins (from bacteriophages P22, λ, and ϕ21) is to prevent transcription termination upon binding to the boxB RNA of the N utilization site (nut site) in the bacteriophage transcript. This protein-RNA complex then associates stably with RNA polymerase during transcription elongation and inhibits transcription termination (14). It plays an important role in the transcription antitermination process in bacteriophages (14), and has been well studied in many biophysical and biochemical experiments. The N-terminal arginine-rich domain and even a truncated fragment alone (termed N-peptide) bind specifically to the boxB RNA hairpin target (13,15–17).
Structures of several N-peptide-RNA complexes have been determined and indicate overall similar complex geometries (13). In the case of the phage P22, the boxB RNA forms a hairpin with a five-basepair stem, and a loop of five nucleotides (GACAA) that adopts a GNRA tetraloop fold (13,15,16). Although all five nucleotides at the loop region are essential for the function of antitermination, gel mobility shift assays indicate that only nucleotides 1, 3, and 5 are important for the peptide binding, and the other two (nucleotides 2 and 4) are required for the interaction of the N protein/nut complex with additional elongation factors (18–20). The bent α-helix recognizes primarily the shape and negatively charged surface of the RNA through multiple hydrophobic and electrostatic interactions. Structural studies have shown that the hydrophobic interactions between the peptide and the C11 loop nucleotide also play a critical role in stabilizing the N-peptide-boxB RNA complex (13,15).
Investigators have thoroughly examined the involvement of electrostatic interactions in N-peptide boxB binding by studying the salt dependence of peptide-RNA binding (5,21). Biochemical and mutational data show that three of the seven amino acid residues that are conserved in the N-terminal region of the peptide are important for antitermination function and critical for boxB recognition (22,23). Because of its small size, the availability of experimentally determined 3D structures, and the large number of biochemical and biophysical studies that have focused on it, the N-peptide-boxB system is an excellent model system in which to study the dynamics and energetics of peptide-RNA complexes.
Molecular dynamics (MD) simulations can be used to characterize molecular motions in biomolecules on the nanosecond timescale at atomic resolution. The MD approach has already been applied to many RNA-containing molecules (24–28), and has been used to characterize the flexibility of RNA, as well as hydration and ion binding (27,28). The dynamics of structures as large as a complete ribosome have been investigated in the nanosecond time regime (29). In combination with advanced sampling methods, it has also been used to study folding of RNA (30,31) and DNA (32) hairpins, structural transitions in RNA motifs (33), and peptide-RNA interactions (34).
In this study, MD simulations were employed to investigate the dynamics of the P22 N-peptide-boxB complex and to elucidate the energetic contributions to binding. In addition, the free-energy changes associated with adaptation of RNA and peptide conformation toward the bound forms, and the role of strongly bound water molecules at the peptide-RNA interface were studied. The influence of peptide amino acid substitutions and the salt dependence of interaction were investigated and showed good agreement with available experimental results. Of interest, it was found that the electrostatic field of the RNA has a favorable influence on the coil-to-α-helix transition of the N-peptide outside of the peptide-binding site. This result may have important implications for understanding peptide-RNA complex formation, which very frequently involves structural changes or folding transitions in the peptide (14). It indicates that electrostatic interactions near RNA molecules can lead to a shift in the equilibrium toward the bound form of an interacting partner before direct contacts with the binding pocket are formed.
Materials and Methods
MD simulations in explicit solvent
The initial structure of the complex between the N-peptide and boxB RNA of bacteriophage P22 corresponded to the experimental structure (13) (first entry Protein Data Bank ID: 1a4t). The N-peptide contained 19 residues (sequence NAKTRRHERRRKLAIERDT) and the boxB RNA included 15 nucleotides (sequence 5′-GCGCUGACAAAGCGC). MD simulations were performed on the whole complex structure, as well as on the N-peptide and RNA alone (starting from the experimental structures taken from the complex).
The Amber9 program package (35) in combination with the parm99 force field (36) was used for all simulations. Each starting structure's complex, receptor (RNA hairpin), and ligand (peptide) were solvated in an octahedral water box using the TIP3P water model (37) and a minimum distance from the solute and the box boundaries of 10 Å, resulting in 6392 water molecules for the complex, 3841 for the RNA, and 4302 for the peptide. Standard ionization states were used for ionizable side chains at pH 7. The pH for the NMR structure determination was 5.7 (13). It is expected that low experimental pH further stabilizes the protonated forms of the basic residues in the peptide such that the ionization states are also valid at pH = 5.7. Each system was neutralized by the addition of counterions (eight K+ ions for the complex, 14 K+ for RNA, and six Cl− to neutralize the isolated peptide). Each system was energy-minimized (1000 steps) using the sander module. During MD simulations, the systems were initially harmonically restrained (25 kcal · mol−1 · Å−2) to the energy-minimized start coordinates and heated up to 300 K in steps of 100 K followed by gradual removal of positional restraints over a period of 0.1 ns, and a final 1 ns unrestrained equilibration at 300 K. The respective resulting systems were used as starting structures for bound and free MD simulations. Each simulation was extended to an additional 20 ns at constant pressure (1 bar) and constant temperature (300 K). The long-range electrostatic interactions were treated with the particle-mesh Ewald method (38) using a real-space cutoff distance of 9 Å. The SETTLE algorithm (39) was used to constrain bond vibrations involving hydrogen atoms, allowing a time step of 2 fs. Coordinates were recorded every 4 ps.
Binding free-energy calculations
Binding free-energy calculations based on the molecular mechanics Poisson-Boltzmann surface area (MM/PBSA) method were carried out over the last 10 ns of the MD trajectory of the respective simulations (11 ns equilibration phase). In one set of calculations, all of the solvent molecules and counterions were removed during the MM/PBSA calculations. To study the effect of several tightly bound water molecules in a second set of calculations, eight water molecules located at the interface between the peptide and RNA were explicitly included during the MM/PBSA calculations. These eight water molecules were present at approximately the same locations in both simulations of the RNA in the presence and absence of the N-peptide. The binding free energy was calculated from the following equation:
The free energies of the complex (ΔGcomplex), RNA (ΔGrna), and peptide (ΔGpeptide) were calculated as averages over 2500 snapshots form the MD trajectories. The calculated free-energy changes were split into several components, including molecular mechanics force-field terms and solvation terms as explained in detail in the Supporting Material.
The energetic contributions to the binding of each amino acid residue in the N-peptide were estimated by calculating the change in free energy of binding while the residues were mutated (in silico) to alanine. The alanine mutant structures were generated from snapshots of the wild-type using the method developed by Massova and Kollman (40). The last 10 ns of the whole MD trajectory were taken to calculate the change in free energy of binding due to the mutation.
Results and Discussion
Dynamics of the RNA and N-peptide in complex and in the absence of partner structures
An MD simulation at 300 K for 20 ns was performed using the experimental boxB-N-peptide complex from bacteriophage p22 as a starting structure (13). In addition, MD simulations of the same length were also used to study the N-peptide and boxB RNA alone. The root mean-square deviation (RMSD) of the simulated complex structure from the starting structure reached a stable level of ∼2.5 Å after a few nanoseconds of simulation time (Fig. 1 A, black). Significantly larger RMSD levels were reached transiently during an interval of 7–9 ns simulation time. In the complex, all N-peptide residues except for the last four C-terminal residues (Glu-16, Arg-17, Asp-18, and Thr-19) and the Glu-7 on the opposite face of the helix contacted the 15 nucleotide boxB-RNA. These contacts were largely preserved throughout the whole simulation of the complex. During the 7–9 ns interval, reversible conformational transitions in the C-terminal region (residues 16–19) of the bound peptide were observed that resulted in increased RMSDs for both the whole complex and the bound peptide (Fig. 1 A, black and green). Indeed, multiple conformations were reported for this apparently flexible segment in an NMR structure determination study (13). The RNA hairpin in the complex that forms a GNRA tetraloop was remarkably stable (Fig. 1 A, red). The average RMSD from the starting structure was ∼1.6 Å (Fig. 1 A, red), which is close to the 1.3 Å obtained by comparing the ensemble of available NMR structures (13).
Figure 1.
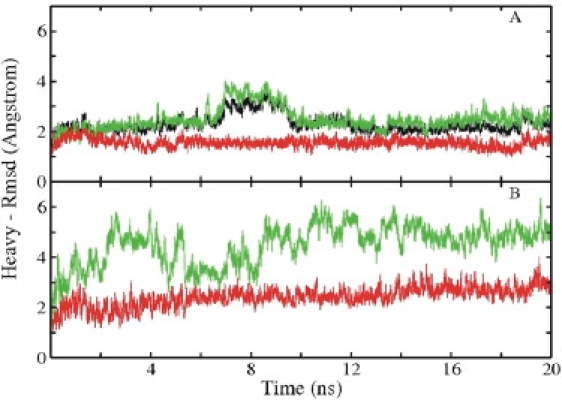
RMSD (heavy atoms) of sampled conformations of complex, peptide, and RNA from the native structure (Protein Data Bank: 1A4T) versus simulation time. (A) RMSD of the N-peptide-boxB RNA complex (black), RNA in complex (red), and peptide in complex (green/light gray) starting from the experimental structure. (B) RMSD of isolated RNA (red/dark gray) and isolated peptide (green/light gray) starting from the experimental structure.
A superposition of the simulated complex structure taken from the final stage of the 20 ns MD trajectory with the experimental starting structure also indicates close agreement with the starting conformation (Fig. 2 A). The overall complex is stable, with a large contact surface area (1410 Å2) buried at the interface between the hairpin RNA and peptide; ∼1/3 of the surface area of the RNA and half of that of the peptide buried at the interface are contributed by the hydrophobic groups. On average, nine hydrogen (H)-bonds were found at the RNA-peptide interface that covers 340 Å2 (24%) of the total buried surface area at the interface. Six of these bonds were formed by the four arginine residues (Arg-5, Arg-6, Arg-9, and Arg-10) that interact with the major groove nucleotides. Moreover, the side-chain nitrogen atoms from Lys-3 and His-7 were also involved in the H-bonds with hairpin nucleotides.
Figure 2.
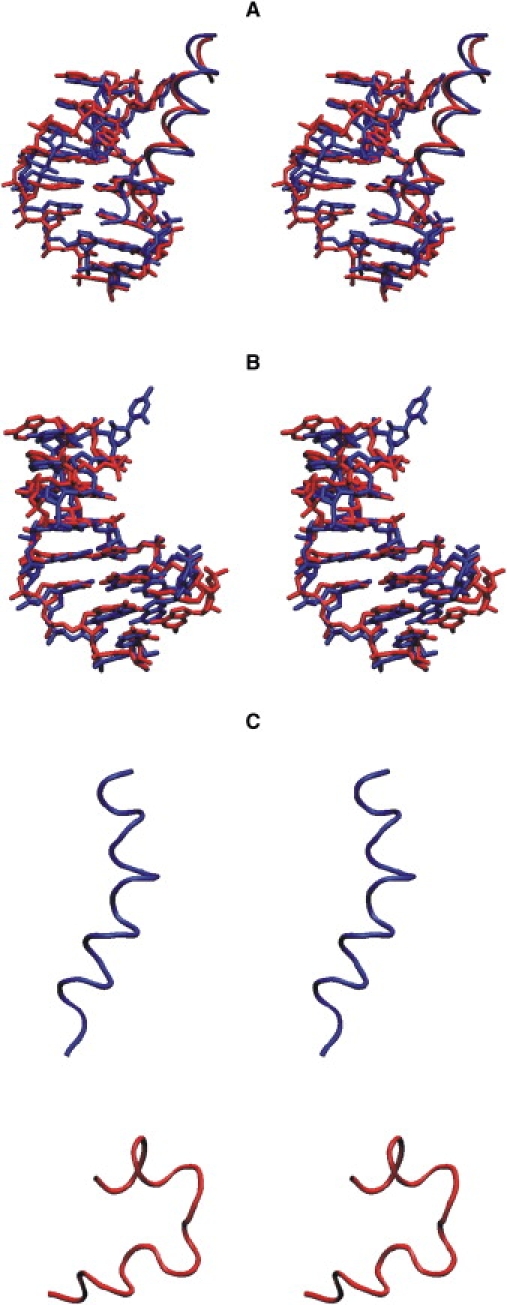
Superposition (in stereo) of the final structures (red/light gray) of complex (A) and isolated RNA hairpin (B) on the experimental starting structures (blue/dark gray). The RNA hairpin is shown in stick representation and the N-peptide is in backbone tube representation. (C) Stereo view of the N-peptide (tube representation) taken from the final stage of simulation of the complex (blue/dark gray) and the final structure of the MD simulation of the isolated peptide (red/light gray).
Larger deviations from the starting structures were observed in the simulations of isolated RNA and N-peptide (Fig. 1 B). However, in the case of boxB RNA, the structure remained overall close to the starting structure (Fig. 1 B, red), with an average RMSD of all heavy atoms of ∼2.5 Å from the starting structure (Fig. 2 B). In contrast, the RMSD of the peptide from the α-helical starting configuration increased rapidly during the first few nanoseconds of the simulation and remained at a level of 5–6 Å (Fig. 1 B, green), representing an unfolded structure with only a partial residual helical structure (Fig. 2 C). This agrees with the experimental observation that the N-peptide is unstructured in free solution and forms an α-helix only upon binding to boxB RNA (5,13,21). Fig. S1 in the Supporting Material shows a comparison of the average B-factor of each RNA nucleotide during the MD simulations in the absence and presence of the N-peptide. It demonstrates overall lower atomic fluctuations of the RNA in the bound form compared to the form with no binding partner.
Free energy of the peptide and RNA to adopt the structure in the complex
After the explicit water molecules and ions were removed, the generated MD trajectories were subjected to an MM/PBSA analysis. To calculate the electrostatic solvation contributions, both the finite-difference PB and (for comparison) generalized Born (GB) methods were used. In this approach, the solute is presented as a low dielectric cavity (ɛ = 1) embedded in a high-dielectric aqueous continuum (ɛ = 80). The MM/PBSA analysis was performed for the complex as well as for the isolated RNA and N-peptide partners using conformations extracted from the trajectory of the complex (bound forms) or from MD simulations of the isolated partners (free forms). The calculations were performed over the last 10 ns of each simulation (2500 frames).
The convergence of the results was monitored by computing the cumulative average over the trajectories (Fig. S2). In all cases the cumulative average showed a final drift of the calculated free energies of <1 kcal · mol−1 ns−1. This was the case even for the isolated peptide, which samples a variety of conformational states. It is likely that the simulation time was too short to exhaustively sample all relevant unfolded peptide states. However, most of the conformational energy differences between these unfolded or partially folded states are small and therefore result in only a small final drift of the average calculated peptide free energy (Fig. S2).
By comparing the average energies of the RNA and N-peptide trajectories in the bound versus free forms, we were able to estimate the free-energy change associated with the process of transforming the structures from the unbound to the bound form (termed free energy of adaptation; Tables 1 and 2). The calculations predict that the free-energy change of peptide folding toward the bound helical structure is favorable (−11.7 kcal · mol−1 for MM/PBSA and −11.4 kcal · mol−1 for MM/GBSA; Table 1). In contrast, the calculated free energy change associated with the formation of the bound form compared to the unbound from of the boxB RNA was positive (19.8 kcal · mol−1 for MM/PBSA and 13.6 kcal·mol−1 for MM/GBSA; Table 1). Note that the calculations do not include contributions due to changes in the conformational entropy of the peptide and RNA. Normal-mode or quasiharmonic analysis is frequently used to estimate the conformational entropy of a molecule (41). However, it is unlikely that this approach could provide realistic results in the case presented here because the peptide in the unfolded form can visit multiple conformational substates, and hence calculations based on a quadratic approximation around a single energy minimum (as used in normal-mode analysis) do not give an accurate estimate of the entropy of the peptide. Therefore, the data in Table 1 represent mainly the energetic part of the RNA and peptide states (including possible entropic solvent contributions).
Table 1.
Adaptation energy for the peptide and hairpin RNA
| Energy | N-peptide |
boxB RNA |
||||
|---|---|---|---|---|---|---|
| Bound | Free | Adaptation energy∗ | Bound | Free | Adaptation energy∗ | |
| BOND† | 460.1 (± 2) | 456.0 (± 2) | 4.1 (± 2) | 757.8 (± 2) | 763.7 (± 1) | −5.9 (± 1) |
| VDW‡ | −49.5 (± 1) | −24.0 (± 1) | -25.5 (± 1) | -128.1 (± 1) | -134.3 (± 2) | 6.2 (± 1) |
| ELE§ | −22.3 (± 5) | −418.2 (± 3) | 395.9 (± 4) | −404.7 (± 12) | −538.4 (± 15) | 133.7 (± 10) |
| SURNP¶ | 17.5 (± 0.7) | 18.1 (± 0.01) | −0.6 (± 0.1) | 22.5 (± 0.02) | 21.9 (± 0.03) | 0.6 (± 0.01) |
| PB‖ | −1557.2 (± 6) | −1171.5 (± 4) | −385.7 (± 5) | −3426.0 (± 13) | −3311.3 (± 15) | −114.7 (± 10) |
| GB∗∗ | −1559.5 (± 6) | −1174.0 (± 3) | −385.4 (± 5) | −3344.2 (± 12) | −3223.3 (± 8) | −121.0 (± 9) |
| ΔG††PB | −1151.3 (± 3) | −1139.6 (± 1) | −11.7 (± 2) | −3178.5 (± 2) | −3198.4 (± 2) | 19.8 (± 2) |
| ΔG††GB | −1153.6 (± 3) | −1142.2 (± 1) | −11.4 (± 1) | −3096.7 (± 2) | −3110.3 (± 1) | 13.6 (± 2) |
MM/PBSA free energies are averages over the last 10 ns of the trajectories (2500 frames).
Adaptation energy (kcal/mol) is calculated by subtracting the energy of the free state from that of the bound state.
Bond, angle, dihedral energy.
van der Waals energy.
Electrostatic energy.
Contribution of the nonpolar surface area to the solvation free energy.
Electrostatic solvation free energy with the PB model.
Electrostatic solvation free energy with the GB model.
Total energy, sum of all the above energy terms.
Table 2.
Adaptation energy for the peptide and hairpin RNA including eight explicit interface water molecules
| Energy | N-peptide |
boxB RNA |
||||
|---|---|---|---|---|---|---|
| Bound | Free | Adaptation energy∗ | Bound | Free | Adaptation energy∗ | |
| BOND† | 460.1 (± 2) | 456.0 (± 2) | 4.1 (± 1) | 757.8 (± 2) | 763.2 (± 1) | −5.9 (± 1) |
| VDW‡ | −49.5 (± 1) | −24.1 (± 1) | −25.4 (± 1) | −118.2 (± 1) | −123.8 (± 2) | 5.7 (± 0.4) |
| ELE§ | −22.3 (± 5) | −418.2 (± 3) | 395.9 (± 4) | −535.4 (± 6) | −664.2 (± 5) | 128.8 (± 5) |
| SURNP¶ | 17.5 (± 0.1) | 18.1 (± 0.01) | −0.6 (± 0.1) | 23.1 (± 0.01) | 22.3 (± 0.03) | 0.1 (± 0.01) |
| PB‖ | −1557.2 (± 6) | −1171.5 (± 3) | −385.7 (± 5) | −3407.4 (± 9) | −3289.8 (± 6) | −117.6 (± 8) |
| GB∗∗ | −1559.5 (± 6) | −1174.0 (± 3) | −385.4 (± 5) | −3322.9 (± 4) | −3202.7 (± 2) | −120.1 (± 3) |
| ΔG††PB | −1151.3 (± 3) | −1139.6 (± 1) | −11.7 (± 2) | −3280.4 (± 1) | −3291.6 (± 2) | 11.2 (± 2) |
| ΔG††GB | −1153.6 (± 3) | −1142.2 (± 1) | −11.4 (± 1) | −3195.9 (± 1) | −3204.6 (± 2) | 8.7 (± 2) |
MM/PBSA free energies are averages over the last 10 ns of the trajectories (2500 frames).
Adaptation energy (kcal/mol) is calculated by subtracting the energy of the free state from that of the bound state.
Bond, angle, dihedral energy.
van der Waals energy.
Electrostatic energy.
Contribution of the nonpolar surface area to the solvation free energy.
Electrostatic solvation free energy with the PB model.
Electrostatic solvation free energy with the GB model.
Total energy, sum of all the above energy terms.
The predicted favorable structural adaptation free-energy change in the case of the peptide disagrees with the experimental result for the related λ N-peptide of +3.04 kcal · mol−1 (21) (extrapolated to zero salt concentration). If one assumes that the MM/PBSA results are dominated by the energetic changes associated with binding (although the continuum solvent calculations may include possible solvent entropy effects), it is useful to compare it with experimental data on the enthalpy change for a coil-helix transition. Garcia-Garcia and Draper (21) reported a ΔHcoil->helix = −12.6 kcal · mol−1, which is close to our predicted adaptation MM/PBSA energy change (−11.7 kcal · mol−1) for the coil-helix transition of the P22 N-peptide. Note, however, that the λ N-peptide in their work consisted of 22 residues, whereas our calculations were performed on a 19-residue P22 N-peptide, which may also contribute to the difference between the calculation and experimental results.
We also checked the consistency of the calculation results by estimating the entropy change for a helix-coil transition and comparing it with the experimental results. We assumed that the MM/PBSA energy for the coil-helix transition is dominated by energetic contributions, and that the experimental free energy of the coil-helix transition for the P22 N-peptide is similar to that of the λ N-peptide (+3 kcal · mol−1; see above). This allowed us to estimate the conformational entropy contribution to the conformational adaptation (−TΔSconf) to be on the order of ∼15 kcal · mol−1 (ΔG − ΔH = 3 + 11.7 kcal · mol−1). The entropy change per residue for coil-helix transitions has been reported to be ∼−3 cal · mol−1 (42). This translates to an entropy contribution at 300 K (19 residues) of 17.1 kcal · mol−1, which is in very reasonable agreement with the above estimate based on the difference between the experimental free energy and the calculated MM/PBSA energetic contributions. Presumably, because the conformational entropy is neglected, the calculated absolute free energies of N-peptide binding to boxB are much more favorable than the corresponding experimental data.
The calculation of MM/PBSA energetic contributions allowed us to identify the main driving forces for binding. In the case of the peptide, the van der Waals interactions and the electrostatic solvation terms make the most favorable contributions to the transition from the unfolded peptide structure to the folded helical structure. The Coulomb interaction strongly opposes helix formation, presumably because of the many positive charges of the N-peptide that come closer together in the helix versus the unfolded conformations (see Fig. S3, ligand). In the case of the RNA, van der Waals and Coulomb interactions oppose the formation of the bound form, whereas the bonded contributions and the electrostatic solvation terms favor the transition to the bound structure (Fig. S3, receptor). The formation of the bound RNA structure results in a slightly more compact form that overall decreases the average distance between charged phosphate groups, which results in an increase of Coulomb repulsion that is partially outbalanced by a more negative Born solvation free energy. The various energetic contributions associated with the transition from the unbound to the bound conformation of RNA (receptor) and peptide (ligand) are illustrated in Fig. S3.
A conceptual weakness of the MM/PBSA approach is the representation of all water molecules as a continuum. This may be an oversimplified description of the aqueous environment, especially for water molecules at the interface between the peptide and RNA. We therefore went a step beyond the standard MM/PBSA method by including several interface water molecules explicitly during the MM/PBSA calculations.
Previous studies have included explicit water molecules in MMPBSA calculations (43,44). In the system presented here, we were able to identify eight water molecules that occupied nearly the same positions (including some exchanges with the bulk) during the entire last 10 ns of the simulation of the complex (Fig. S4). Fortunately, the same positions were also occupied during the simulation of the free RNA (at very similar positions and also including exchanges with bulk water molecules).
This observation indicates that at least a significant fraction of the interface water molecules in the RNA partner that may stabilize the complex (for example, as H-bond bridging waters) are already present in the absence of the binding partner (prehydration). This is not the case for the peptide, since it undergoes large conformational changes in the absence of the RNA. The MM/PBSA calculations for complex and isolated RNA were repeated with the eight water molecules included (for both bound and unbound RNA). It must be emphasized that we assume here that the water molecules belong to a strongly bound hydration shell of the RNA that is present in the free and bound forms (not considering possible differences in the water-binding free energy in the absence or presence of the peptide partner). The addition of explicit interface water molecules lowers the total calculated adaptation free energy of the RNA by ∼6–8 kcal · mol−1 (compare Tables 1 and 2). However, the influence of explicitly accounting for the interface water molecules on the different contributions to conformational adaptation is relatively modest (Table 2). Hence, the qualitative conclusions regarding the energetic contributions are similar to the results from the analysis without explicit interface waters.
Free energy of binding
The MM/PBSA free energy of binding was calculated as the difference between the MM/PBSA analysis of the trajectory of the complex minus the results for the N-peptide and boxB RNA (in either the bound or free form). The difference between taking either the bound or the free state of the binding partners as a reference corresponds to the overall structural adaptation contribution (the sum of the adaptation of peptide and RNA). Both the GB and PB models gave qualitatively very similar results. The calculated total free energy of binding is strongly favorable (< −110 kcal · mol−1; Table 3). Note that the calculated binding free energy does not account for changes in the conformational entropy of the binding partners (see discussion in the previous paragraph). Since for both partners binding is associated with a considerable loss of conformational flexibility, it is expected that accounting for this contribution would shift the predicted free energy to more positive (and hence more realistic) values. Indeed, an estimate of conformational entropy changes based on normal-mode calculations averaged over 10 snapshots of complex and partners resulted in an entropy difference of 46 kcal · mol−1 opposing complex formation. Accounting for this contribution brings the calculations closer to experiment, but still significantly overestimates the binding free energy. One should keep in mind that the normal-mode approach results only in approximate entropies within selected snapshots (not accounting for the full conformational space accessible for the molecules). However, the neglect of a discussion of the conformational entropy contribution does not affect any conclusions based on the calculated energetic contributions.
Table 3.
Average binding free energy
| Energy | Bound form | Free form | Adaptation |
|---|---|---|---|
| BOND | 0.0 (± 0.0) | −1.8 (± 1) | 1.8 (± 1) |
| VDW | −49.1 (± 0.3) | −68.3 (±1) | 19.2 (± 1) |
| ELE | −3074.5 (± 2) | −2544.9 (± 4) | −529.6 (± 2) |
| SURNP | −9.1 (± 0.1) | −9.2 (± 0.3) | 0.1 (± 0.1) |
| PB | 3011.1 (± 3) | 2510.4 (± 4) | 500.7 (± 3) |
| GB | 3011.7 (± 3) | 2505.3 (± 4) | 506.4 (± 2) |
| ΔG∗PB | −121.6 (± 1) | −113.7 (± 1) | −7.9 (± 1) |
| ΔG∗GB | −121.0 (± 1) | −118.8 (± 1) | −2.2 (± 1) |
Total energy, sum of all the above energy terms.
Taking into account that the adaptation free energy lowers the calculated binding free energy by 2–8 kcal · mol−1, depending on whether the PB or GB electrostatic model is used (Table 3), in the case of including eight interface water molecules explicitly, the calculated contribution of the structural adaptation is close to zero (−2.7 kcal · mol−1 for the GB model). This indicates, surprisingly, that energetically the structural adaptation of the binding partners is predicted to make only a small overall contribution relative to the total gain in binding free energy. This is mainly because the adaptation free energy is negative for the N-peptide but is predicted to be positive for the RNA, and overall they nearly compensate for each other (Table 1). Of interest, the change in the Coulomb interaction strongly favored the complex formation (although it strongly opposed the process of structural adaptation toward the bound forms of both peptide and RNA). In addition, the van der Waals interactions and the surface area reduction significantly favor complex formation, whereas the electrostatic solvation part opposes binding. Qualitatively very similar results were obtained when the interface waters were treated explicitly (Table 4).
Table 4.
Average binding free energy including eight explicit interface water molecules
| Energy | Bound form | Free form | Adaptation |
|---|---|---|---|
| BOND | 0.0 (± 0) | −2.0 (± 1) | 2.0 (± 1) |
| VDW | −52.1 (± 1) | −71.0 (± 1) | 18.9 (± 1) |
| ELE | −3108.5 (± 3) | −2584.5 (± 3) | −524 (± 3) |
| SURNP | −10.1 (± 0.2) | −10.5 (± 0.3) | 0.4 (± 0.1) |
| PB | 3041.7 (± 3) | 2538.5 (± 2) | 503.2 (± 4) |
| GB | 3036.1 (± 3) | 2530.9 (± 2) | 506.2 (± 4) |
| ΔG∗PB | −128.9 (± 1) | −129.6 (± 1) | 0.7 (± 1) |
| ΔG∗GB | −134.6 (± 1) | −137.3 (± 1) | −2.7 (± 1) |
Binding free energy (kcal/mol) calculated over the last 10 ns from the bound and free trajectories. The adaptation energy is calculated by subtracting the total energy of the free state from that of the bound state.
Total energy, sum of all the above energy terms.
RNA electrostatic field stabilizes the helical peptide structure outside of the binding region
The analysis of the N-peptide adaptation free energy (i.e., the comparison of peptide conformations compatible with the bound state versus conformations obtained in the absence of RNA) indicates that the helical bound structure is energetically more favorable by ∼−11.7 kcal · mol−1 (−11.4 kcal · mol−1 for the GB model; Table 1). However, the fact that unfolding was observed during the MD simulation starting from the initial helical (bound) structure indicates that this favorable energetic contribution is insufficient to fully compensate for the increase in conformational entropy upon unfolding (otherwise it should have stayed in a folded structure).
It would be of significant interest if, during the process of peptide binding, the helical (bound) structure is further stabilized by the RNA electrostatic field outside of the binding region. This would mean that a nearby RNA electrostatic field creates a bias toward the bound form (or induces the bound form) before the structure enters the binding site. To investigate this possibility, the trajectory of the N-peptide in the bound state was shifted relative to the bound position by 15 Å outside of the binding pocket. The trajectory of the free peptide was also shifted to the same position (by optimal superposition onto the bound structures; Fig. 3). We found that 15 Å was the minimum distance that guaranteed that no overlap of peptide atoms with any RNA atom occurred for any trajectory frame. Indeed, the calculated total free-energy difference between conformations representing the bound ensemble versus the unbound ensemble was 6.9 kcal · mol−1, which is more favorable at a distance of 15 Å from the binding pocket compared to the free-energy difference in the absence of boxB RNA (mainly due to a favorable electrostatic contribution; Fig. 3). Although the effect decreased with salt concentration, the stabilization even at 0.2 M salt amounts to ∼3.2 kcal · mol−1, which is still significantly above the thermal energy at room temperature per degree of freedom (RT = 0.6 kcal · mol−1, where R is the gas constant, and T is the temperature). The result confirms that for the example presented here, the RNA electrostatic field can shift the equilibrium toward the helical peptide structure outside of the binding site, and contributes to an induced folding of the peptide during the association process.
Figure 3.
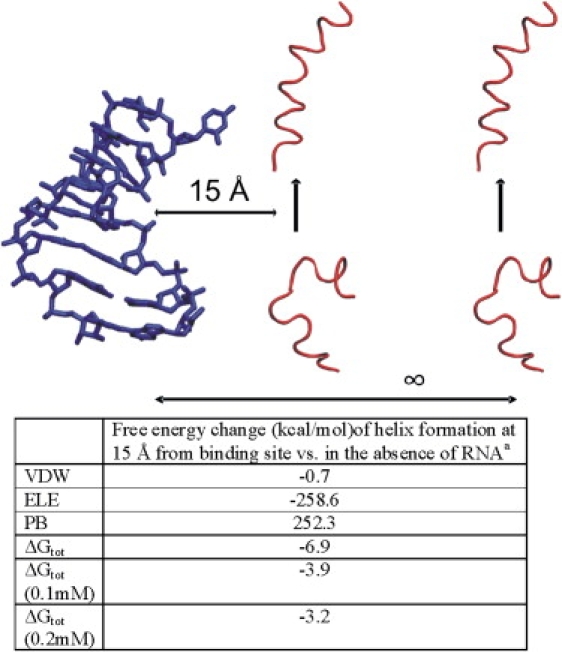
Stabilization of the coil-to-helix transition near the RNA binding site. (Upper panel) Schematic illustration of the process. (Lower panel) MM/PBSA free-energy contributions averaged over the last 10 ns of the MD simulations (2500 snapshots). Structures representing the helical bound form were taken from the simulation of the complex (helix shifted by 15 Å from the binding site). The structures of the isolated N-peptide were taken from the simulation of the isolated peptide shifted to the same position 15 Å from the binding site. The placement was sufficiently far from the RNA to avoid any sterical overlap.
Influence of amino acid substitutions in the N-peptide on RNA binding and dependence on salt concentration
To elucidate the importance of individual peptide side chains for the RNA binding, we substituted the 11 amino acid residues (forming the binding contact region) of the peptide by alanine and recalculated the MM/PBSA energies (using the trajectories obtained from the simulations involving the wild-type peptide). This results in sterically possible conformations because the alanine is smaller and represents a substructure of the substituted side chains. Some of the substitutions have a very modest influence on the calculated binding free energy, whereas others result in a large drop of the calculated binding free energy (Fig. 4). It is important to note that since the calculations were simply performed on the trajectory obtained from the wild-type simulations, any conformational adaptation (due to the substitution) that would reduce the magnitude of the influence on binding was not accounted for. Basically, all of the substituted residues make contacts with the nucleotides of the RNA hairpin (except for one, Glu-8, which is a conserved residue) along with six other residues (Ala-2, Thr-4, Arg-5, Arg-6, Arg-9, and Arg-10) at the N-terminal region of the N-proteins of bacteriophages (45).
Figure 4.
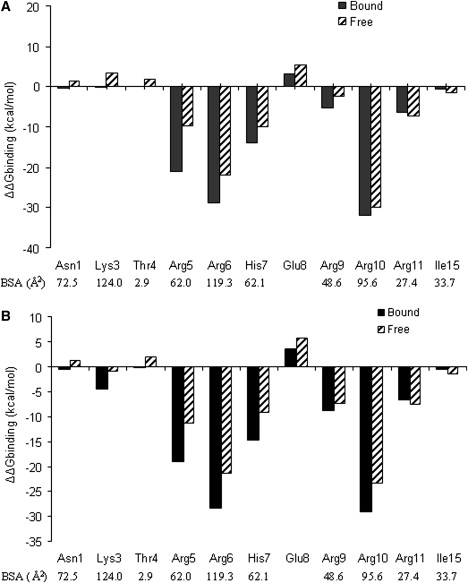
Change in the calculated binding free energy upon substitution by alanine. Negative ΔΔG values correspond to unfavorable substitutions, and positive values indicate improved binding due to alanine mutation. The change in the solvent-accessible surface area that is buried at the protein-RNA interface for each amino acid residues is also indicated. Results are given for the PB (A) and GB (B) approaches.
Experimental mutagenesis studies have shown that three of the conserved residues—Ala-2, Arg-6, and Arg-10—are important for antitermination functions in λ-phage (23). In agreement with experiment, the calculations indicate that the substitution of Arg-6 and Arg-10 to alanine causes the largest drop in calculated binding free energy (Fig. 4). These two residues contribute a large surface area to the binding of the hairpin RNA, and also participate in several H-bonds with the hairpin nucleotides to stabilize the complex. Experimental data for the effect of the same mutations in the P22 N-peptide are not available. However, in general, the effect of substituting Arg residues at the center of related N-peptides (e.g., N-peptide of phage λ (22)) by other residues results in smaller binding free-energy changes (on the order of a few kcal · mol−1) than predicted by our calculations. This is at least in part due to the conformational relaxation upon substitution that is not accounted for in our calculations, which should reduce the effect of amino acid substitutions on the calculated change in binding free energy. However, the calculations can give valuable insight into the relative importance of peptide residues for RNA binding.
Other substitutions that are energetically unfavorable are Arg-5, His-7, Arg-9, and Arg-11 to Ala. Moreover, Arg-5 is conserved in bacteriophages, and along with the other arginine residues it also plays an important role in the binding of arginine-rich peptide motifs that recognize an RNA structural element (19). In general, Arg residues play an important role in both protein-RNA and protein-protein interfaces by participating in a large number of H-bonds through their guanidinium group: at protein-protein interfaces, the guanidinium group is involved with other polar residues (46), and in protein-RNA interfaces it recognizes a specific base sequence in the RNA chain (10). In this particular case, the side chain of arginine is important to recognize a G:C basepair (part of a GNRA tetraloop) located at the major groove of the hairpin RNA.
Mutation of Asn-1, Lys-3, Thr-4, Glu-8, and Ile-15 to Ala resulted in smaller changes in ΔΔGbinding because these residues are either outside of the core-binding region or are on the opposite face of the N-peptide helix. Glu-8, for example, does not form any contact with the nucleotides; however, substitution by Ala resulted in a decrease of the calculated binding free energy due to the reduced electrostatic repulsion. The results from the GB model were very similar to those obtained with the more demanding finite-difference PB calculations in almost all cases (Fig. 4).
The influence of the salt concentration in the framework of the nonlinear PB approach on the N-peptide-boxB binding was also investigated. The calculations did not only involve one structure that was representative of each state of the system; instead, in each case, averages over 250 snapshots taken from the final 10 ns of each simulation were used. The slope of the binding free energy versus logarithm of the salt concentration ranged from 4.8 to 5.6, taking the free or bound RNA and peptide structures as references, respectively (Table 5). The calculated slope for the wild-type N-peptide boxB binding is in very good agreement with experimental data reported by Austin et al. (22), and similar to the salt dependence obtained for the related λ N-peptide boxB system (5,21). The difference between the calculated salt dependence taking the bound versus unbound RNA and N-peptide structures as references (representing the free states) can be mainly attributed to the salt dependence of the coil-helix transition of the N-peptide, because the calculated salt dependence of the transition of the RNA from the bound to free form is negligible (data not shown), presumably because of the small conformational difference between unbound and bound RNA structures. The difference corresponds to a calculated salt dependence of ∼−0.8 kcal · mol−1 for the P22 N-peptide coil-helix transition. Remarkably, Draper and Garcia-Garcia (21) experimentally determined the salt dependence of the coil-helix transition for the related but slightly longer λ N-peptide (22 residues, as opposed to 19 in our system) and obtained −0.63(+/−0.08) kcal · mol−1, which is in very close agreement with our calculation.
Table 5.
Effect of salt concentration on the calculated binding free energy
| Slope of the change in binding free energy ΔΔGbinding (kcal/mol) |
||
|---|---|---|
| Bound reference | Free reference | |
| Wild-type | 5.62 | 4.79 |
| Asn-1 | 5.63 | 4.80 |
| Lys-3 | 4.74 | 4.00 |
| Thr-4 | 5.61 | 4.80 |
| Arg-5 | 4.88 | 4.10 |
| Arg-6 | 4.69 | 4.00 |
| His-7 | 4.83 | 4.10 |
| Glu-8 | 6.22 | 5.42 |
| Arg-9 | 4.81 | 4.11 |
| Arg-10 | 4.72 | 4.00 |
| Arg-11 | 4.99 | 4.17 |
| Ile-15 | 5.60 | 4.79 |
Slope was calculated over the last 10 ns of the MD trajectories. Residues were substituted by alanine. Bound and free reference indicates that the trajectories from the complex or the isolated RNA and peptide, respectively, were used to represent the individual partner structures for calculating the binding free energies.
The calculated salt dependencies of sequence variants of the N-peptide resulted in a decrease by 0.6–1.0 kcal · mol−1 for replacing an Arg in the core-binding region by Ala, no effect from replacing a neutral side chain, and an increase by ∼0.6 kcal · mol−1 in the case of replacing Glu-8 by Ala (Table 5). The calculations agree well with experimental results obtained in previous studies on the related λ N/box B system that used similar calculations but employed single structures of N-peptide/boxB complexes (5,21), as opposed to the ensembles used in this study. The latter result also indicates that the salt dependencies of binding are quite robust with respect to modest conformational changes (as included in our calculations).
Conclusions
The N-protein of bacteriophages (P22, λ, and ϕ21) plays an essential role in transcription antitermination and regulation of phage gene expression (13–23,47). The MM/PBSA approach was used to analyse various contributions to the P22 N-peptide-boxB interaction. The calculated salt dependence of the coil-helix transition, as well as the salt dependence of the wild-type N-peptide-RNA binding and of several peptide sequence variants, was in good agreement with available experimental data and data on the closely related phage λ N system (5,22).
The process of binding was formally split into two subprocesses: the transition of the RNA and peptide from the ensemble obtained from the simulations in the absence of the partner to conformations that represent the structure in the complex. The calculations that did not account for conformational entropy changes predicted a negative change in the free energy of transforming to the bound form for the peptide (−11.7 kcal · mol−1), but a positive change in the case of the RNA (19.8 kcal · mol−1). In both cases, the Coulomb interactions opposed the transition, whereas the van der Waals and solvation contributions favored the transition to the bound structures. The association of peptide and RNA was strongly opposed by electrostatic solvation, but overall was favored electrostatically and by surface area reduction and increased van der Waals interactions.
It was also possible to identify stable water-binding sites that partially bury water molecules at the peptide-RNA interface and were already occupied at the surface of the RNA in the absence of the peptide partner. This indicated a partial preformation of a hydration layer that mediates interactions in the complex between peptide and RNA. The inclusion of these explicit water molecules during MM/PBSA calculations resulted in qualitatively similar results but an overall reduction of the calculated RNA adaptation free energy. In addition, the inclusion of interface water molecules explicitly predicted that the overall adaptation energy to form the bound structures was zero, because the unfavorable energy of forming the bound RNA structure was outbalanced by the favorable adaptation energy of the peptide.
By analyzing the peptide coil-helix transition near the RNA-binding site, we found that the electrostatic field of the RNA not only attracts the positively charged N-peptide to the binding site, it also provides a significant driving force for a transition to the bound form near the binding cleft. The protein-RNA interaction is frequently accompanied by conformational changes that can include partial unfolding-folding transition events (13). It is, therefore, likely that a mechanism that influences the equilibrium of folded bound versus unfolded protein conformations also plays an important role in other systems of peptide-RNA or protein-RNA interactions.
Supporting Material
Molecular mechanics/Poisson-Boltzmann calculations, four figures, and references are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(09)01518-5.
Supporting Material
Acknowledgments
This work was performed using the computational resources of the Computer Laboratories for Animation, Modeling and Visualization at Jacobs University Bremen, and the supercomputer resources of the Environmental Molecular Science Laboratories at Pacific Northwest National Laboratories.
This work was supported by grant Za153-11 from the Deutsche Forschungsgemeinschaft and grant I/80485 from the VolkswagenStiftung to M.Z.
Footnotes
Martin Zacharias's present address is Physics Department, Technical University Munich, 85747 Garching, Germany.
References
- 1.Hall K.B. RNA-protein interactions. Curr. Opin. Struct. Biol. 2002;12:283–288. doi: 10.1016/s0959-440x(02)00323-8. [DOI] [PubMed] [Google Scholar]
- 2.Pérez-Cañadillas J., Varani G. Recent advances in RNA-protein recognition. Curr. Opin. Struct. Biol. 2000;11:53–58. doi: 10.1016/s0959-440x(00)00164-0. [DOI] [PubMed] [Google Scholar]
- 3.Van Duyne G.D., Yang W. Protein-nucleic acid complexes: large, small, old, and new. Curr. Opin. Struct. Biol. 2008;18:67–69. doi: 10.1016/j.sbi.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 4.Nadassy K., Wodak S., Janin J. Structural features of protein-nucleic acid recognition sites. Biochemistry. 1999;38:1999–2017. doi: 10.1021/bi982362d. [DOI] [PubMed] [Google Scholar]
- 5.Draper D.E. Themes in RNA-protein recognition. J. Mol. Biol. 1999;293:255–270. doi: 10.1006/jmbi.1999.2991. [DOI] [PubMed] [Google Scholar]
- 6.Allers J., Shamoo Y. Structure-based analysis of protein-RNA interactions using the program ENTANGLE. J. Mol. Biol. 2001;311:75–86. doi: 10.1006/jmbi.2001.4857. [DOI] [PubMed] [Google Scholar]
- 7.Jones S., Daley D., Luscombe N., Berman H., Thornton J. Protein-RNA interactions: a structural analysis. Nucleic Acids Res. 2001;29:943–954. doi: 10.1093/nar/29.4.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Treger M., Westhof E. Statistical analysis of atomic contacts at RNA protein interfaces. J. Mol. Recognit. 2001;14:199–214. doi: 10.1002/jmr.534. [DOI] [PubMed] [Google Scholar]
- 9.Ellis J.J., Broom M., Jones S. Protein-RNA interactions: structural analysis and functional classes. Proteins. 2007;66:903–911. doi: 10.1002/prot.21211. [DOI] [PubMed] [Google Scholar]
- 10.Bahadur R.P., Zacharias M., Janin J. Dissecting protein-RNA recognition sites. Nucleic Acids Res. 2008;36:2705–2716. doi: 10.1093/nar/gkn102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ellis J.J., Jones S. Evaluating conformational changes in protein structures binding RNA. Proteins. 2008;70:1518–1526. doi: 10.1002/prot.21647. [DOI] [PubMed] [Google Scholar]
- 12.Janin J., Bahadur R.P. Relating macromolecular function and association: the structural basis of protein-DNA and RNA recognition. Cell. Mol. Bioeng. 2008;1:327–338. [Google Scholar]
- 13.Cai Z., Gorin A., Frederick R., Ye X., Hu W. Solution structure of P22 transcriptional antitermination N peptide-boxB RNA complex. Nat. Struct. Biol. 1998;5:203–212. doi: 10.1038/nsb0398-203. [DOI] [PubMed] [Google Scholar]
- 14.Das A. Conrol of transcription termination by RNA-binding proteins. Annu. Rev. Biochem. 1993;62:893–930. doi: 10.1146/annurev.bi.62.070193.004333. [DOI] [PubMed] [Google Scholar]
- 15.Legault P., Li J., Mogridge J., Kay L.E., Greenblatt J. NMR structure of the bacteriophage λ N peptide/boxB RNA complex: recognition of a GNRA fold by an arginine-rich motif. Cell. 1998;93:289–299. doi: 10.1016/s0092-8674(00)81579-2. [DOI] [PubMed] [Google Scholar]
- 16.Schärpf M., Sticht H., Schweimer K., Boehm M., Hoffmann S. Antitermination in bacteriophage λ. The structure of the N36 peptide-boxB RNA complex. Eur. J. Biochem. 2000;267:2397–2408. doi: 10.1046/j.1432-1327.2000.01251.x. [DOI] [PubMed] [Google Scholar]
- 17.Cilley C.D., Williamson J.R. Structural mimicry in the phage ϕ21 N peptide-boxB RNA complex. RNA. 2003;9:663–676. doi: 10.1261/rna.2189203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chattopadhyay S., Garcia-Mena J., DeVito J., Wolska K., Das A. Bipartite function of a small RNA hairpin in transcription antitermination in bacteriophage λ. Proc. Natl. Acad. Sci. USA. 1995;92:4061–4065. doi: 10.1073/pnas.92.9.4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tan R., Frankel A.D. Structural variety of arginine-rich RNA-binding peptides. Proc. Natl. Acad. Sci. USA. 1995;92:5282–5286. doi: 10.1073/pnas.92.12.5282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cilley C.D., Williamson J.R. Analysis of bacteriophage N protein and peptide binding to boxB RNA using polyacrylamide gel coelectrophoresis (PACE) RNA. 1997;3:57–67. [PMC free article] [PubMed] [Google Scholar]
- 21.Garcia-Garcia C., Draper D.E. Electrostatic interactions in a peptide-RNA complex. J. Mol. Biol. 2003;331:75–88. doi: 10.1016/s0022-2836(03)00615-6. [DOI] [PubMed] [Google Scholar]
- 22.Austin R.J., Xia T., Ren J., Takahashi T.T., Roberts R.W. Differential modes of recognition in N peptide-boxB complexes. Biochemistry. 2003;42:14957–14967. doi: 10.1021/bi0351312. [DOI] [PubMed] [Google Scholar]
- 23.Franklin N.C. Clustered arginine residues of bacteriophage λ N protein are essential to antitermination of transcription, but their locale cannot compensate for boxB loop defects. J. Mol. Biol. 1993;231:343–360. doi: 10.1006/jmbi.1993.1287. [DOI] [PubMed] [Google Scholar]
- 24.McDowell S.E., Spackova N., Sponer J., Walter N.G. Molecular dynamics simulations of RNA: an in silico single molecule approach. Biopolymers. 2007;85:169–184. doi: 10.1002/bip.20620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Orozco M., Noy A., Pérez A. Recent advances in the study of nucleic acid flexibility by molecular dynamics. Curr. Opin. Struct. Biol. 2008;18:185–193. doi: 10.1016/j.sbi.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 26.Hashem Y., Auffinger P. A short guide for molecular dynamics simulations of RNA systems. Methods. 2009;47:187–197. doi: 10.1016/j.ymeth.2008.09.020. [DOI] [PubMed] [Google Scholar]
- 27.Auffinger P., Westhof E. Water and ion binding around RNA and DNA (C,G)-oligomers. J. Mol. Biol. 2000;300:1113–1131. doi: 10.1006/jmbi.2000.3894. [DOI] [PubMed] [Google Scholar]
- 28.Hashem Y., Auffinger P. Nucleic solvation: from outside to insight. Curr. Opin. Struct. Biol. 2007;17:325–333. doi: 10.1016/j.sbi.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 29.Sanbonmatsu K.Y., Joseph S., Tung C.-S. Simulating movement of tRNA into the ribosome during decoding. Proc. Natl. Acad. Sci. USA. 2005;102:15854–15859. doi: 10.1073/pnas.0503456102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sorin E.J., Rhee Y.M., Pande V.S. Does water play a structural role in the folding of small nucleic acids? Biophys. J. 2005;88:2516–2524. doi: 10.1529/biophysj.104.055087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garcia A.E., Paschek D. Simulation of the pressure and temperature folding/unfolding equilibrium of a small RNA hairpin. J. Am. Chem. Soc. 2008;130:815–817. doi: 10.1021/ja074191i. [DOI] [PubMed] [Google Scholar]
- 32.Kannan S., Zacharias M. Folding of a DNA hairpin loop structure in explicit solvent using replica-exchange molecular dynamics simulations. Biophys. J. 2007;93:3218–3231. doi: 10.1529/biophysj.107.108019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barthel A., Zacharias M. Conformational transitions in RNA single uridine and adenosine bulge structures: a molecular dynamics free energy simulation study. Biophys. J. 2007;90:2450–2462. doi: 10.1529/biophysj.105.076158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mu Y., Stock G. Conformational dynamics of RNA-peptide binding: a molecular dynamics simulation study. Biophys. J. 2006;90:391–399. doi: 10.1529/biophysj.105.069559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Case D.A., Cheatham T.E., Darden T., Gohlke H., Luo R. The Amber biomolecular simulation programs. J. Comput. Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang J., Cieplak P., Kollman P.A. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 2000;21:1049–1074. [Google Scholar]
- 37.Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 38.Darden T., York D., Pedersen L. Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 39.Miyamoto S., Kollman P.A. SETTLE: an analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992;13:952–962. [Google Scholar]
- 40.Massova I., Kollman P.A. Computational alanine scanning to probe protein-protein interactions: a novel approach to evaluate binding free energies. J. Am. Chem. Soc. 1999;121:8133–8143. [Google Scholar]
- 41.Srinivasan J., Cheatham T.E., Cieplak P., Kollman P.A., Case D.A. Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate-DNA helices. J. Am. Chem. Soc. 1998;120:9401–9409. [Google Scholar]
- 42.Padmanabhan S., York E.J., Stewart J.M., Baldwin R.L. Helix propensities of basic amino acids increase with the length of the side-chain. J. Mol. Biol. 1996;257:726–734. doi: 10.1006/jmbi.1996.0197. [DOI] [PubMed] [Google Scholar]
- 43.Spacková N., Cheatham T.E., Ryjácek F., Lankas F., Van Meervelt L. Molecular dynamics simulations and thermodynamics analysis of DNA-drug complexes. Minor groove binding between 4′,6-diamidino-2-phenylindole and DNA duplexes in solution. J. Am. Chem. Soc. 2003;125:1759–1769. doi: 10.1021/ja025660d. [DOI] [PubMed] [Google Scholar]
- 44.Wong S., Amaro R.E., McCammon J. MM-PBSA captures key role of intercalating water molecules at a protein-protein interface. J. Chem. Theory Comput. 2009;5:422–429. doi: 10.1021/ct8003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lazinski D., Grzadzielska E., Das A. Sequence-specific recognition of RNA hairpins by bacteriophage antiterminators requires a conserved arginine-rich motif. Cell. 1989;59:207–218. doi: 10.1016/0092-8674(89)90882-9. [DOI] [PubMed] [Google Scholar]
- 46.Janin J., Rodier F., Chakrabarti P., Bahadur R.P. Macromolecular recognition in the Protein Data Bank. J. Acta Crystallogr. 2007;D63:1–8. doi: 10.1107/S090744490603575X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cocozaki A.I., Ghattas I.R., Smith C.A. Bacteriophage P22 antitermination boxB sequence requirements are complex and overlap with those of λ. J. Bacteriol. 2008;190:4263–4271. doi: 10.1128/JB.00059-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.