

Abstract
The transcription factors Notch1 and KLF4 specify epithelial cell fates and confer stem cell properties. suggesting a functional relationship, each gene can act to promote or suppress tumorigenesis in a context-dependent manner, and alteration of KLF4 or Notch pathway genes in mice gives rise to similar phenotypes. Activation of a conditional allele of KLF4 in RK3E epithelial cells rapidly induces expression of Notch1 mRNA and the active, intracellular form of Notch1. KLF4-induced transformation was suppressed by knockdown of endogenous Notch1 using siRNA or an inhibitor of γ-secretase. Chromatin immunoprecipitation assay shows that KLF4 binds to the proximal Notch1 promoter in human mammary epithelial cells, and siRNA-mediated suppression of KLF4 in human mammary cancer cells results in reduced expression of Notch1. Furthermore, KLF4 and Notch1 expression are correlated in primary human breast tumors (N = 89; pearson analysis, r > 0.5, p < 0.0001). Like KLF4, Notch1 was previously shown to induce transformation of rat cells immortalized with adenovirus E1A, similar to RK3E cells. We therefore compared the signaling requirements for Notch1- or KLF4-induced malignant transformation of RK3E. As expected, transformation by Notch1 was suppressed by dominant-negative CSL or MaML1, inhibitors of canonical Notch1 signaling. However, these inhibitors did not suppress transformation by KLF4. Therefore, while KLF4-induced transformation requires Notch1, canonical Notch1 signaling is not required, and Notch1 may signal through a distinct pathway in cells with increased KLF4 activity. These results suggest that KLF4 could contribute to breast tumor progression by activating synthesis of Notch1 and by promoting signaling through a non-canonical Notch1 pathway.
Keywords: KLF4, Notch1, transcription factor, breast cancer, malignant transformation, epithelial cell, carcinoma
Introduction
KLF4 (Krüppel-like factor 4) is expressed in post-mitotic, differentiating epithelial cells. While dispensable for embryonic development, it is required for formation of the permeability barrier in the skin and for differentiation of goblet cells in the gut.1-3 KLF4 was identified as an oncogene in an expression screen that used an adenovirus E1A-immortalized rat kidney cell line, RK3E, as a host to isolate transforming activities present in squamous cell carcinomas (SCCs).4,5 The RK3E line has epithelial features including expression of E-cadherin and formation of junctional complexes.4,6 Oncogene-transformed RK3E cells give rise to dense foci of tumorigenic cells surrounded by a single layer of contact-inhibited parental cells. This model has been used for functional characterization of carcinoma-derived, transforming oncogenes such as KLF4,4 Notch1,7-9 Gli1,5,6,10 β-catenin11,12 and others,13 and observations made in RK3E cells have been well supported by analysis of epithelial tissues.4,6,10,13-15
KLF4 is upregulated in breast cancer, cutaneous SCCs, head and neck cancer (HNSCC) and pancreatic cancer.4,16-19 When KLF4 expression is induced in the basal layer of mouse skin, the animals rapidly develop SCC-like lesions.17,18,20 Consistent with its role as an oncogene, expression of KLF4 along with Myc, Sox2 and Oct4 is sufficient to convert differentiated mammalian cells into stem cells.21 In contrast to its likely role as an oncogene in skin, KLF4 functions as a tumor suppressor in the gut.22-24 The absence or presence of functional p21WAF1 may determine whether KLF4 functions as an oncogene or tumor suppressor.25
Notch1 regulates cell fate and proliferation during development and in adult tissues, such as the skin and gut, where its expression overlaps that of KLF4.26,27 The four mammalian Notch receptors (N1 to N4) are comprised of non-covalently associated extracellular (NEC) and transmembrane (N™) subunits that are generated from the full length protein (NFL) by a proteolytic cleavage in the Golgi. Humans express five membrane-bound Notch ligands [Delta-like (Dll)1, 3, 4 and Jagged (Jag) 1 and 2] that interact with NEC, dissociating it from N™ and triggering a series of proteolytic cleavages in N™. The final cleavage, catalyzed by γ-secretase, releases the intracellular domain, NIC (reviewed in refs. 28–30). NIC translocates to the nucleus where, in the canonical pathway, it forms a transcriptionally active complex with CSL (for CBF-1/RBP-Jκ, Suppressor of Hairless, and Lag-1), histone acetyl-transferases, and co-activators such as Mastermind-like (MAML).28,29,31 In the canonical pathway, the N1IC-CSL-MAML complex activates transcription via CSL binding sites in the promoters of target genes such as Hes1.32 In the absence of Notch signaling, CSL functions as a repressor of its target genes and exists in a complex with Hairless and corepressors.33 Several CSL-independent Notch signaling pathways have been reported.34-36
Constitutively active, N1IC-like proteins are produced in human T-cell acute lymphoblastic leukemia by translocations involving the Notch1 gene (<1%), or more commonly (∼56%), by point mutations.37 Truncated forms of Notch1 and Notch4 are implicated in mouse mammary carcinomas and in the mammary cancer stem cell phenotype.38,39 In vitro, the intracellular domains of Notch1 and 2 were shown to transform E1A-immortalized rodent cells similar to RK3E.5,8 Notch1 is required for maintenance of the Ras-transformed phenotype,40 and may induce transcription of c-Myc.41 Although Notch1 functions as an oncogene in leukemias, conditional knockout of Notch1 in mouse skin leads to BCC,42 indicating that like KLF4, Notch1 can function as a tumor suppressor in some contexts.
Here we report the identification of Notch1 as a direct transcriptional target and a potential mediator of transformation by KLF4. By assessing KLF4 and Notch1 in a context where either gene can induce malignant transformation, we found that N1IC transforms cells in a CSL-dependent manner on its own. However, in response to KLF4 N1IC signals transformation via a CSL-independent pathway. Consistent with this signaling, increased expression of KLF4 or Notch1 was previously shown to identify subsets of aggressive breast, pancreatic, and head and neck cancers.16,19,40,43,44 We now show that KLF4 and Notch1 are frequently co-expressed in human breast cancers. Because Notch1 appears to signal by a different pathway in response to KLF4, tumors co-expressing these proteins may be clinically distinct in their response to Notch inhibitors or to other therapies.
Results
KLF4 upregulates Notch1 expression in RK3E cells
To identify KLF4-regulated transcripts, we used a KLF4-estrogen receptor (KLF4-ER) fusion that is synthesized continuously but is biologically inactive in the absence of 4-hydroxytamoxifen (4OHT).17,45 KLF4-ER levels are not changed by addition of 4OHT.17,20 Instead, when 4OHT binds the ER domain, the fusion protein migrates to the nucleus and can transform RK3E cells, like the wild-type KLF4 protein. 4OHT induction in the presence of a protein synthesis inhibitor, such as cycloheximide (CHX) is expected to permit transcriptional activation by the KLF4-ER already present in the cell, but secondary effects due to de novo protein synthesis would be largely abrogated. Accordingly, we treated RK3E cells stably expressing either KLF4-ER or Vector with 4OHT for 2 h, or with 4OHT in combination with the protein synthesis inhibitor cycloheximide (CHX), or with vehicle alone. In two independent microarray experiments, Notch1 mRNA was upregulated an average of 7.5 fold in KLF4-ER cells treated with 4OHT + CHX, but not in the control.
Increased expression of Notch1 mRNA following induction with 4OHT was observed by northern blot (Fig. 1A, top, lanes 4 and 5). Induction was likewise observed in the presence of 4OHT + CHX, indicating that regulation of Notch1 by KLF4 does not require translation of an intermediary (i.e., the effect appears to be direct). The Notch1 mRNA levels obtained in 4OHT + CHX were actually greater than in 4OHT alone, consistent with loss of a labile regulator of Notch1 transcription or mRNA in the presence of CHX. In contrast, Notch 2, 3, 4 and Dll/Jag ligands were unchanged at 2 h of induction (not shown).
Figure 1.
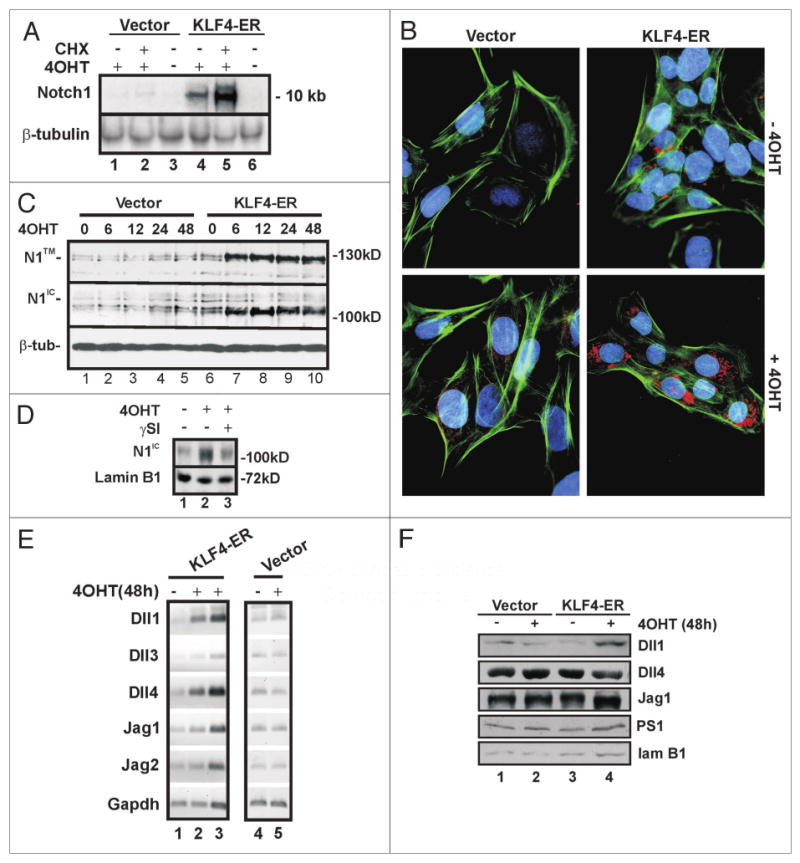
KLF4 induces Notch1 expression in RK3E cells. (A) Notch1 is induced by KLF4 in RK3E cells without de novo protein synthesis. KLF4-ER cells or vector cells were treated with 4OHT +/- CHX for 2 h. Notch1 and -tubulin (loading control) mRNAs were analyzed by northern blot. (B) KLF4-ER cells or vector cells were induced with 4OHT for 10 h. Notch1 expression (red) was analyzed by immunoflouresence using N1-C20 antibody, which primarily detects cytoplasmic Notch1. DAPI (blue) and phalloidin (green) stain the nucleus and cytoskeleton, respectively. (C) Vector or KLF4-ER cells were treated with 4OHT for the number of hours indicated above the lanes. Notch1 expression was analyzed by immunoblot using antibodies N1-C20 (top) and N1-Nter (center) which predominantly recognize N1™ and N1IC respectively. -tubulin (bottom) served as a loading control. Molecular weight markers are indicated on the right. (D) KLF4-ER cells were treated with 4OHT or vehicle for 48 h. To test for a role of -secretase, cells were treated with SI or vehicle for 3 h prior to preparation of extract. Immunoblots were probed with N1IC-Nter and lamin B1 (loading control) antibodies. (E) Semi-quantitative RT-PCR analysis was performed using total RNA isolated from Vector or KLF4-ER-cells treated with 4OHT for 48 h. To demonstrate that PCR reactions were not saturated, lane 3 contained twice as much input RNA as lane 2. Gapdh served as control. (F) Immunoblot analysis of Notch pathway genes. Lamin B1 served as a loading control. Cells were treated as indicated for 48 h.
Antibody N1-C20, directed against the C-terminus of Notch1, showed strong staining of KLF4-ER cells treated with 4OHT for 10 h (Fig. 1B). By immunoblot, the same antibody detected an increase in the membrane-bound form of Notch1, N1™, at 6 h of induction (Fig. 1C, upper). N1-C20 does not recognize the active form of Notch1, N1IC, efficiently by immunoblot. In contrast, antibody N1IC-Nter, specific for the N-terminus of N1IC generated by γ-secretase cleavage, showed accumulation of N1IC with increasing time in 4OHT (Fig. 1C, middle). As expected, this accumulation was diminished by inclusion of a γ-secretase inhibitor (γSI) in the medium (Fig. 1D). As cleavage of N1FL to N1IC is dependent upon the expression of Notch ligands and components of γ-secretase, we examined these for regulation by KLF4. Semi-quantitative RT-PCR showed that mRNAs for the Notch ligands Dll1, Dll4 and Jag1 are present in RK3E cells and are increased similarly to 1.5–2.0 fold by 48 h of induction (Fig. 1E, lanes 1–2). To demonstrate that PCR reactions were not saturated, lane 3 contained twice as much input RNA as lane 2. As these increases occurred between 12 and 24 h (Suppl. Fig. 1), it is possible that the ligands are induced indirectly. Immunoblotting indicated that Dll1, Dll4 and Jag1 proteins are present in RK3E cells and were unchanged or else slightly upregulated in response to KLF4 (Fig. 1F). The γ-secretase component Presenillin1 (PS1) likewise showed little change (Fig. 1F). These results indicate that Notch1 is a transcriptional target of KLF4, and that it is processed to the active form, N1IC, in response to activation of KLF4 and in the presence of several Notch1 ligands and γ-secretase.
Expression of KLF4 and Notch1 in human breast carcinomas
Compared with adjacent uninvolved mammary epithelium, KLF4 mRNA and protein are upregulated in the majority of breast cancers, and KLF4 immunostaining identifies tumors with an aggressive phenotype.16,43 To correlate KLF4 and Notch1 in human tumors we immunostained paraffin sections of primary breast cancers. For Notch1, we first characterized N1-C20 and N1IC-Nter antibodies by staining subjacent sections representing 12 cases of infiltrating ductal carcinoma and adjacent uninvolved epithelium. Eight cases showed intense, uniform staining of tumor cells with each Notch1 antibody, while the other four cases showed only low-level staining (Fig. 2A and Suppl. Fig. 2). Concordant staining of these 12 cases was statistically significant as indicated by 2 × 2 contingency analysis (p = 0.002; Fisher's exact test, two-tailed).
Figure 2.
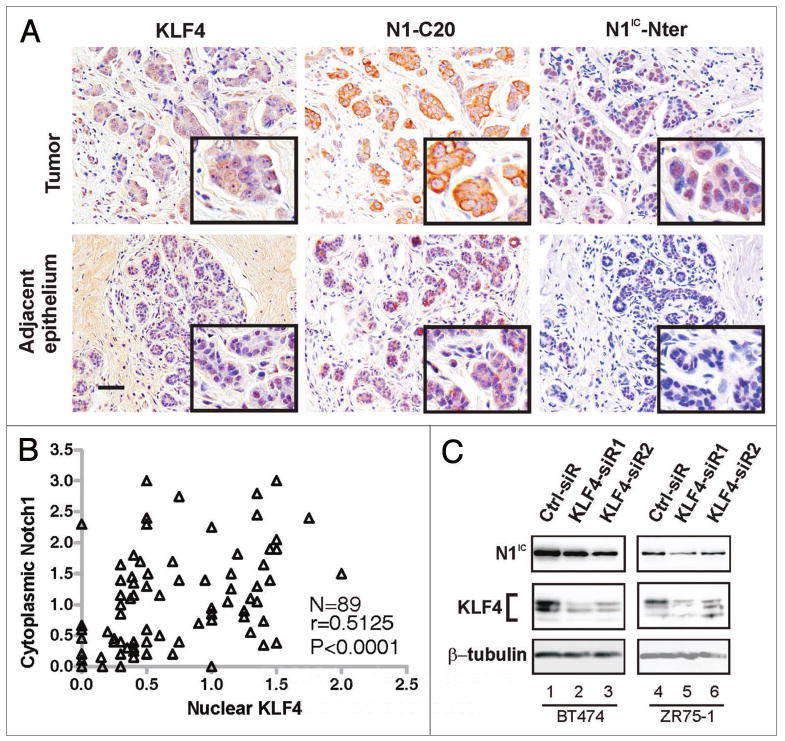
Co-expression of KLF4 and Notch1 in human breast tumors. (A) Subjacent sections of a primary human breast tumor were stained with the indicated antibodies. Within the section were areas representing either infiltrating ductal carcinoma (Tumor, upper row) or adjacent, uninvolved epithelium (lower row). Scale bar, 50 μm. (B) Pearson correlation analysis of KLF4 and N1-C20 immunostaining in 89 cases of breast cancer. Tumors with the same scores appear as one data point. (C) Immunoblot analysis of N1IC following siRNA-mediated suppression of KLF4 in BT474 and ZR75-1 human breast cancer cell lines. The upper and lower portions of a filter representing a single SDS-PAGE gel were queried in parallel with N1IC-Nter or KLF4 (H180) antibodies, respectively. The lower portion was reprobed with β-tubulin antibody (loading control).
Although staining by the two Notch antibodies within and between sections was similar, N1-C20 predominantly stained the cytoplasm while N1IC-Nter mainly stained the nucleus, consistent with our observation that N1-C20 recognizes N1IC less efficiently than other forms of Notch1, such as N1™ (Fig. 1C). The reason for this differential detection is currently unknown.
For the cases above, KLF4 antibody gave uniform staining of the eight Notch1-high cases but showed little staining in four Notch1-low cases (Suppl. Fig. 2). To further test whether KLF4 and Notch are correlated in breast tumors we stained 89 cases with KLF4 and N1-C20 antibodies (Fig. 2A and B). KLF4-stained slides and Notch1-stained slides were scored independently. As described previously, intensity scores were determined using a 0.0–4.0 scale, and weighted according to the percentage of cells exhibiting that intensity.43 Immunoscores for each of the 89 cases are provided as Supplementary Data. Median values were: KLF4 (nuclear), 0.5, Notch1 (cytoplasmic), 0.75. Pearson analysis revealed that KLF4 and Notch1 are correlated in these samples (r > 0.5, p < 0.0001; Fig. 2B). Using the median scores as cutoffs, 65 of 89 cases (73%) exhibited concordant expression of KLF4 and Notch1 (Fig. 2B and Table 1, p < 0.001). Thus, KLF4 and Notch1 are expressed concordantly in most breast cancers.
Table 1.
2 × 2 contingency analysis of Notch1 and KLF4
| Notch1 > median | Notch1 < median | Total | |
|---|---|---|---|
| KLF4 > median | 33 | 12 | 45 |
| KLF4 < median | 12 | 32 | 44 |
| Total | 45 | 44 | 89 |
Immunostaining scores corresponded to nuclear KLF4 and cytoplasmic Notch1. p < 0.001 (Fisher's exact test, two-tailed).
We next examined the expression of Notch and KLF4 in human breast cancer cell lines. In BT474 and ZR75-1 cells, siRNA-mediated knockdown of KLF4 resulted in a corresponding decrease in endogenous N1IC (Fig. 2C). In these lines, and several others that we tested, there was no effect on cell proliferation in response to KLF4 knockdown as determined by cell count (not shown).
KLF4 binds to the Notch1 promoter and activates transcription
Chromatin immunoprecipitation (ChIP) was used to determine if KLF4 could bind to the human Notch1 promoter (Fig. 3A). We transduced MCF10A cells with pBpuro-HA-KLF4 retrovirus and selected a puromycin-resistant pool. From these cells, the Notch1 promoter fragment P was specifically immunoprecipitated by HA antibody but not by control IgG (Fig. 3B, lanes 1–4). In contrast, a GAPDH promoter fragment was brought down by TFIIB antibody, but not HA or control IgG (Fig. 3B, lanes 5–8). This is expected as the GAPDH promoter is not regulated by KLF4 although it contains several consensus KLF4 binding sites (unpublished data). Further scanning of the Notch1 promoter by ChIP showed that fragments L, M, N and O, within a ∼2.1 kb region encompassing the start of the Notch1 open reading frame, were bound by KLF4. In contrast, fragment K is not bound (Fig. 3B, lanes 9–13). Thus KLF4 binds the Notch1 promoter in human mammary epithelial cells.
Figure 3.
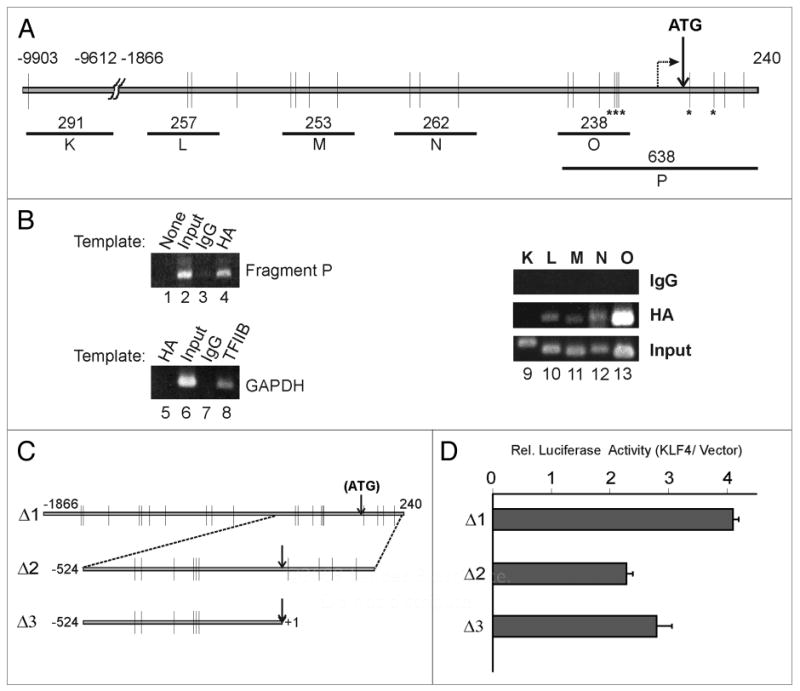
KLF4 binds to the human Notch1 promoter and activates transcription. (A) Schematic of the human Notch1 promoter (-9903 to +240 bp relative to the ATG initiating codon). Consensus KLF4 binding sites (RRGGYGY) are indicated by vertical lines, and sites conserved in mouse and rat are indicated (*). A broken arrow indicates the putative start site of transcription. Chromatin immunoprecipitation (ChIP) fragments K to P and their sizes in bp are indicated. (B) ChIP analysis was performed on MCF10A/HA-KLF4 cells using the indicated antibodies. (C) Fragments 1 to 3 of the human Notch1 promoter were inserted into the luciferase reporter pGL3-Pro. (D) Relative transcription of Notch1 promoter constructs co-transfected into HEK293 cells with either KLF4 expression vector or control. Three experiments were performed, each in triplicate. standard deviation (SD) bars are shown.
To determine if the Notch1 promoter is responsive to KLF4, we performed promoter-reporter assays in HEK293 cells, which express little endogenous KLF4. The 2.1 kb fragment Δ1 (-1866/+240) and two truncations, Δ2 and Δ3, were cloned into the luciferase reporter vector, pGL3-Pro (Fig. 3C). Each construct was co-transfected with either KLF4 or vector, and a control for transfection efficiency. Relative to the vector, Δ1 was activated four-fold by KLF4 (Fig. 3D). Δ2 which contains 524 nt upstream and 240 nt downstream of the translation start site, showed ∼2.3 fold activation (Fig. 3D). Δ3, which removes 240 nt downstream of the initiation codon, showed similar activation (2.7 fold). These results are consistent with interaction data obtained by ChIP (Fig. 3B).
Notch1 mediates transformation by KLF4
To determine if N1IC is required for transformation by KLF4 we used two strategies to suppress N1IC in RK3E cells (Fig. 4). To test for an essential role for γ-secretase we performed transformation assays in the presence or absence of the γSI L685, 458 (Fig. 4A). As a control we utilized a vector that expresses N1IC. As N1IC is the product of γ-secretase, transformation by N1IC should not be inhibited by γSI. As previously reported,17 KLF4-ER retrovirus induced foci in the presence of 4OHT (Fig. 4A). The frequency of focus formation by KLF4-ER, but not by N1IC retrovirus, was reduced in the presence of γSI (Fig. 4A, bottom right). Although γ-secretase has multiple cellular substrates in addition to Notch1, our results indicate that none of these is required for transformation by Notch1, and are consistent with a role for Notch1 in transformation by KLF4.
Figure 4.
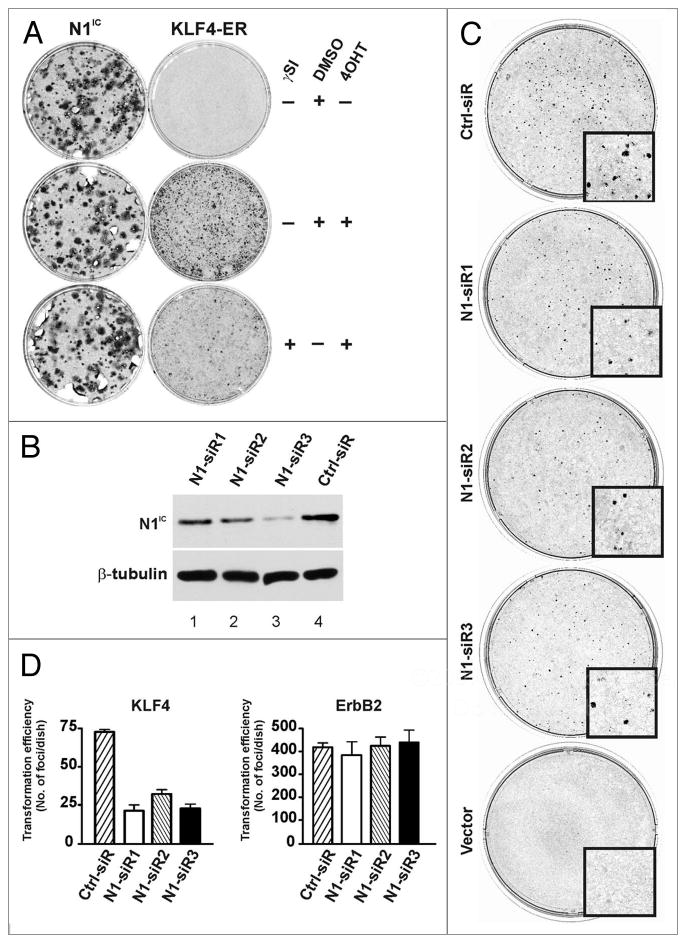
Transformation by KLF4 requires active Notch1. (A) To determine whether processing of Notch1 to the active form (N1IC) contributes to malignant transformation by KLF4, RK3E cells were transduced with N1IC or KLF4-ER retroviruses and cultured in the presence/absence of γsI. The experiment was performed twice in triplicate. (B) KLF4-ER cells were transfected with Notch1-specific siRNAs, and N1IC expression was determined by immunoblot 72 h later. A portion of the filter was queried in parallel with β-tubulin antibody (loading control). (C) RK3E cells were transfected with Notch1-specific siRNAs and then transduced with KLF4-ER retrovirus 24 h later. Transformed foci were scored at 20 d. ErbB2 served as a control oncogene and showed no inhibition by siRNas. Three independent experiments were performed in duplicate. (D) Quantification of transformed foci from the experiment shown in panel C and not shown (ErbB2). Bars, SD.
To determine if Notch1 is a specific downstream effector of transformation by KLF4 we sought to knock down endogenous Notch1. Attempts to generate stable knockdown lines using lentiviral shRNA vectors were unsuccessful. For oncogene-transduced cells to successfully establish transformed foci, they must continue to divide as the surrounding cells become confluent during the first 2–3 d following retroviral transduction. We reasoned that suppression of KLF4 transforming activity during the immediate post-transduction period would suffice to reduce the overall size and/or number of transformed foci. For example, we previously showed that transient expression of a Snail dominant-negative allele could block transformation by another oncogene, Gli1.6 We therefore used a transient approach to knock down endogenous Notch1 by directly transfecting RK3E cells with siRNAs.
As shown in Figure 4B, Notch1 siRNAs reduced the expression of N1IC relative to control (compare lanes 1–3 with lane 4). To assess the effect of siRNAs on transformation by KLF4, cells were first transfected with siRNAs, infected with KLF4-ER or ErbB2 retroviruses 24 h later, and then allowed to form foci (Fig. 4C and not shown). Following fixation and staining, foci larger than 0.5 mm were counted. Three independent experiments were performed in duplicate (Fig. 4C and D). Knockdown of Notch1 diminished the frequency of focus formation by KLF4 but not by the control, ErbB2, indicating that Notch1 promotes transformation by KLF4.
Canonical Notch1 signaling is not required for transformation by KLF4
To determine if canonical Notch1 signaling is required for transformation by KLF4, we made use of dominant-negative inhibitors of this pathway. A point mutation in CSL (CSLDN) and truncated versions of MAML1 (MAMLDN1 and MAMLDN2) were previously shown to dominantly interfere with activation of the Hes1 promoter by the N1IC-CSL-MAML complex.46-48 We used retroviral constructs and antibiotic selection to generate pools of RK3E cells that stably expressed vector, MAMLDN1, MAMLDN2 or CSLDN. MAMLDN1, which lacks the epitope recognized by the MAML-specific antibody, was expressed as a fusion protein detectable by GFP antibody (Fig. 5A, left). MAMLDN2 was detected by immunoblotting with a MAML-specific antibody (Fig. 5A, left). RT-PCR with species-specific primers was used to detect expression of CSLDN (Fig. 5A, right). We next examined the activity of a Notch1 reporter construct, Hes1-luc, in the presence of exogenous N1IC in each of these pools of cells (Fig. 5B). Compared with vector, stable expression of MAMLDN1, MAMLDN2 or CSLDN inhibited activation of the Hes1 promoter by N1IC, indicating suppression of canonical Notch1 signaling (Fig. 5B).
Figure 5.
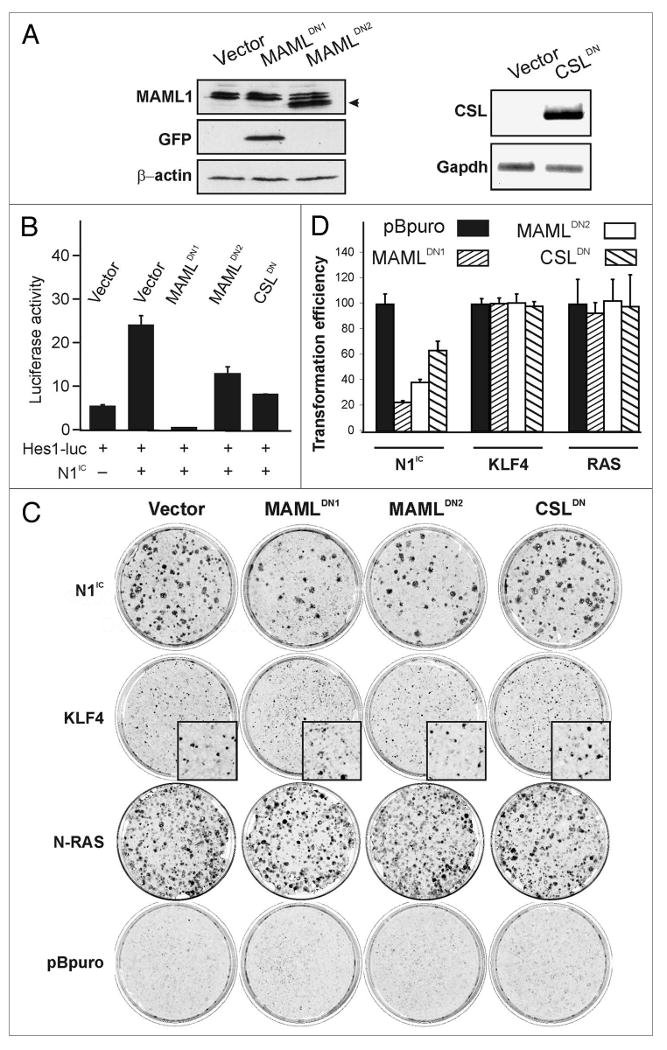
Dominant-negative (DN) MAML and CSL block transformation by N1IC but not by KLF4. (A) RK3E cells were transduced with retroviral vectors encoding CSLDN, MAMLDN1, MAMLDN2, or empty vector and stably transduced cells were selected using antibiotic or FACS (for MAMLDN1-GFP)(left). Immunoblotting with MAML1 antibody was used to detect MAMLDN2 (arrow). As MAMLDN1 is not recognized by this antibody, GFP antibody was used to detect MAMLDN1-GFP fusion protein (middle). β-actin served as a loading control (bottom)(right): A DN allele of mouse CSL was detected by semi-quantitative RT-PCR using species-specific primers. GAPDH served as a loading control. (B) The ability of DN alleles to suppress canonical Notch1 signaling was assessed by transfection of established cell lines with a Hes1-luc reporter in combination with N1IC expression vector or control. Luciferase activity was measured 48 h post-transfection. One experiment was performed in triplicate (bars, SD). Similar results were obtained in an independent experiment also performed in triplicate. (C) Cells stably expressing vector, MAMLDN1, MAMLDN2 or CSLDN (as indicated across the top of the panel) were superinfected with transforming retroviruses encoding N1IC, KLF4, RAS or the control, pBpuro (left). Plates are representative of three independent experiments performed in triplicate for N1IC and KLF4 and one experiment performed in triplicate for N-RAS. (D) Quantitation of foci from experiments shown in (C). The histogram indicates the transformation efficiency of the DN cell line relative to the Vector control line superinfected with the same retroviral supernatant. Bars, SD.
To test for a role of canonical Notch1 signaling we assayed the transforming activities of retroviruses encoding KLF4, N1IC, N-RAS, Gli1 or c-MYC. Transformation by N1IC was suppressed in each of the three cell lines expressing attenuators of the canonical pathway. In contrast, transformation by KLF4, N-RAS, Gli1 or c-MYC was unaffected (Fig. 5C and D, and not shown). These results indicate that the canonical Notch1 signaling pathway is dispensable for transformation by KLF4. In contrast, in the absence of KLF4, N1IC induces transformation via the canonical pathway.
Discussion
A productive approach to the study of cancer biology is the use of analytic (top-down) strategies, where the malignant phenotype of established tumor cells is altered by genetic or pharmacologic means. The complementary approach, utilized in the current study, relies upon a synthetic (bottom-up) strategy, where more normal cells are converted into malignant cells by introduction of an oncogene or by inhibition of a tumor suppressor. Because introduction of oncogenes into primary cells typically leads to responses other than malignant transformation (e.g., no response, growth arrest or cell death), most investigators have studied malignant transformation in the context of an intermediate model such as an established cell line. Available human epithelial models include various spontaneously immortalized cells49 as well as primary human epithelial cells immortalized using SV40 Large T and hTERT.50 For analysis of transformed phenotypes, potential limitations of current cell culture models, including the available human models, include the genetically-undefined nature of spontaneously immortalized cells and the use of immortalizing proteins from DNA tumor viruses such as SV40 or adenovirus, as these are not present in human tumors.
In the current study we used the RK3E cell model, derived from primary rat cells by immortalization with adenovirus E1A,5 to identify Notch1 as an effector of KLF4-induced transformation and to show that an alternative or non-canonical pathway mediates this effect. Unlike other culture models that we have tested, these cells undergo malignant transformation in response to either KLF4 or Notch1, facilitating a comparative analysis of oncogene signaling. E1A induces the epithelial phenotype in a wide variety of cells, including even human tumor cells, a useful property for modeling carcinoma.4,51 In addition, E1A inhibits the p105Rb pathway that is known to be inactivated by genetic alteration in many human cancers. At subconfluence RK3E cells actually divide more rapidly than transformed derivatives (e.g., using c-Myc, Gli1 or KLF4 to induce transformation).4 Therefore, rather than indicating mere increases in cell growth rate, the RK3E focus assay detects other characteristics of the malignant phenotype, such as insensitivity to growth inhibitory signals. Notably, these foci are consistently composed of cells that are tumorigenic in athymic mice.4,5 Therefore, focus formation in vitro is a reasonable surrogate endpoint that enables detailed mechanistic studies.
Although few regulators of Notch transcription have been characterized, many human carcinomas show increased expression of Notch1 mRNA and protein. By microarray analysis we identified upregulation of Notch1 mRNA as an early response to activation of KLF4. The human Notch1 promoter is bound by KLF4, and suppression of KLF4 in human breast cancer cells resulted in attenuation of Notch1 expression. By immunostaining, KLF4 and Notch1 were concordant in 73% of human primary breast tumors. Our results identify KLF4 as an activator of Notch1 transcription and a potential effector of Notch1 upregulation in human tumors such as breast cancer, cutaneous SCC, HNSCC or pancreatic cancer.17-19,43,44,52,53
Although Notch1 and KLF4 have well-established roles as oncogenes in breast cancer,25,44 in other KLF4-positive tumor types the role of Notch1 is less clear. For example, the Notch pathway is widely regarded to suppress tumorigenesis in the skin, a tissue in which KLF4 promotes SCC tumorigenesis.17 It is clear from conditional gene deletion that Notch1 functions to suppress cutaneous BCC, probably through regulation of Hedgehog pathway signaling, and Notch1 expression is very low in human BCCs.42,53 Others have introduced exogenous alleles of truncated MAML to inhibit canonical Notch pathway signaling.54,55 These studies provide compelling evidence that canonical Notch signaling (i.e., through CSL-MAML) suppresses SCC tumorigenesis, but do not directly address the role of Notch1. In other settings such as the mammary gland, it appears that non-canonical, or alternate Notch pathway signaling can lead to tumorigenesis independently of CSL/MAML.34,56 Furthermore, Notch1 expression is upregulated in many SCCs, including most oral cancers and the cutaneous SCCs that develop on sun-protected areas of the skin.52,53 In these ectodermally-derived tissues, which are developmentally related to mammary tissue, Notch1 may function in an alternate pathway to promote tumorigenesis. In summary, CSL/MAML (canonical Notch1) signaling appears to suppress SCC, while Notch1 itself suppresses BCC. However the role of Notch1 in SCC requires further study.
Our data indicate that induction by KLF4 of the active signaling form of Notch1, N1IC, is important for transformation in vitro. However, when DN mutants of MAML1 or CSL were used to suppress canonical Notch signaling, KLF4 transformation was permitted. As a control, these same alleles abrogated transformation by exogenous N1IC. Therefore, on its own N1IC may signal transformation by activation of the canonical CSL-MAML pathway. In contrast, when induced by KLF4, Notch1 signals transformation through a CSL-independent pathway. Possibly, KLF4 may switch Notch signaling to favor an alternate pathway. Further insight will require identification of the relevant alternate pathways and determining their roles in tumorigenesis. As additional functional assays for KLF4 are identified in human cells or in transgenic mice, it will be possible to design experiments that test a role for Notch1 signaling in these other contexts.
KLF4 may regulate Notch1 in settings other than cancer. KLF4 was shown to be required for embryonic stem cell renewal,57 and was isolated in a screen as one of only four genes that together confer stem-cell like properties on adult and embryonic fibroblasts.21 Notch1 signaling is also required for maintenance of the undifferentiated state and is implicated in the mammary cancer stem cell phenotype.39,58,59 It will be interesting to determine whether KLF4 signals via Notch1 to perpetuate pluripotency and self-renewal of stem cells in both normal tissues and in tumors.
Materials and Methods
Constructs
pRK5 HA-KLF4 pBpuro-myc-KLF4-ER pCTV3K-KLF4, pCTV4-N-RAS and pCTV3K-c-MYC were described earlier.17,43 pBpuro N1IC was derived from pcDNA3.1 N1IC, (provided by C.J. McGlade). Mouse CSL cDNA (NCBI accession #BC051387) was purchased from Invitrogen and mouse CSLDN, containing a single mutation,46 was generated by site-directed mutagenesis (Quick-change Kit; Stratagene). Oligonucleotides used for mutagenesis are shown in Table 2. MAMLDN2 (MAML1 1-305) was provided by A.J. Capobianco and shuttled into pQCXIN (Clontech). MAMLDN1 (MSCV-MAML1 13-74 GFP) was a gift of J.C. Aster, andHes1-luc was a gift from R. Kageyama.
Table 2.
Sequences of DNA oligonucleotides used in this study
| Name | Olignucleotides used for cloning |
|---|---|
| Rn Notch1 promoter- BamHI (S) | 5′ AGAGATCTTCGCCAATGGAGGCACACAGGCAG 3′ |
| Rn Notch1 promoter- REcoRI (AS) | 5′ GGAATTCGAAGGTCGGTCCTCCCTGAT 3′ |
| hsnotchATG (S) | 5′ CAGCCGGTGGGGAGGCTAACCGCCGCTCCTGGC3′ |
| hsnotchATG (AS) | 5′ GCCAGGAGCGGCGGTTAGCCTCCCCACCGGCTG 3′ |
| hsnotch740ATG (S) | 5′ GCGGGGAGGCCAGCATCTAGAGGGAAAAGCG 3′ |
| hsnotch740ATG (AS) | 5′ CGCTTTTCCCTCTAGATGCTGGCCTCCCCGC 3′ |
| Oligonucleotides used for ChIP analysis | |
| K (S) | 5′ GGCCCTGACACAGGCTGGCTTCC 3′ |
| K (AS) | 5′ GGTTCGTCTGGGTCACAAACGTCCC 3′ |
| L (S) | 5′ AGCCCCGCGCTTCCTTCTATGGA 3′ |
| L (AS) | 5′ CCTGGC CACCTCTTGCCAAATGC 3′ |
| M (S) | 5′ CCTGGCACACCTCTTGCCAAATGC 3′ |
| M (AS) | 5′ TTCTCCACCACGATGCCAGGCACG 3′ |
| N (S) | 5′ GGCACTGGTCCCGGCTGCTCCCT 3′ |
| N (AS) | 5′ GAGCAGCTGAGGCCACGTTGGGG 3′ |
| O (S) | 5′ TGGGCTCGGGACGCGCGGCTCAG 3′ |
| O (AS) | 5′ GCTCCGCGCCCGGCTCGTTCCTT 3′ |
| P (S) | 5 ′ GCTCAGCTCGGGGAGGCGCAAAG 3 ′ |
| P (AS) | 5 ′ GAAGGTCGGTCCTCCCTGAT 3 ′ |
| Oligonucleotides used for semiquantitative RT-PCR | |
| Rn DLL1 (S) | 5 ′ TGACAAGAGCAGCTTTAAGGCCCG 3 ′ |
| Rn DLL1 (AS) | 5 ′ CTGTTTCTCAGCAGCAGTCCCTGG 3 ′ |
| Rn DLL3 (S) | 5 ′ GCTGACTCACAGCGCTTCCTTCT 3 ′ |
| Rn DLL3 (AS) | 5 ′ GTCGGTGTGGGGCAGGGATTGGA 3 ′ |
| Rn DLL4 (S) | 5 ′ TGTGGGCAGCCGCTGCGAGTTTC 3 ′ |
| Rn DLL4 (AS) | 5 ′ GTAACCGAAGTGGCACCTTCTCTCC 3 ′ |
| Rn Jagged1 (S) | 5 ′ CAGACAGTGCGTGCCGCCATAGGT 3 ′ |
| Rn Jagged1 (AS) | 5 ′ CAGCAACTGCTGACATCAAATCCCC 3 ′ |
| Rn Jagged2 (S) | 5 ′ TCTGCTCTGGAATCCGAGCCCTG 3 ′ |
| Rn Jagged2 (AS) | 5 ′ TTGTTGGCGCTCTCGTCCCGTGG 3 ′ |
| Rn GAPDH (S) | 5 ′ TACTAGCGGTTTTACGGGCG 3 ′ |
| Rn GAPDH (AS) | 5 ′ TCGAACAGGAGGAGCAGAGAGAGCGA 3 ′ |
A ∼2.1 kb human Notch1 promoter fragment (-1866/+240 relative to the A of the initiating codon) was amplified from human genomic DNA using a GC-RICH PCR System (Roche), cloned and sequenced. For efficient translation of luciferase, the initiating codon of Notch1 and an upstream ATG were mutated (Quick-change Kit; Stratagene) and verified by sequencing. Plasmid maps and constructs are available upon request.
Antibodies
Polyclonal antibody to human KLF4 was used for immunostaining17 and H-180 (Santa Cruz Biotechnology) was used for immunoblot. N1-C20 (sc6014-R), Jag1 and Dll1 were from Santa Cruz; Dll4 (ab7280), GFP (ab290), lamin B1 (ab16048) and N1IC-Nter (ab8925) were from Abcam. Other antibodies used were β-actin (AC74) and β-tubulin (D66) from Sigma, TFIIB (Active Motif), MAML1 (C1237, Bethyl laboratories), Presenillin1 (AB5757, Millipore) and HA (12CA5, Roche).
Immunoblotting and RNA analysis
Preparation of cell extracts, isolation of RNA, RT-PCR, immunoblotting, microarray analysis and northern blotting were as described.4,14 Oligonucleotide sequences are shown in Table 2.
Cell lines and tissue culture
RK3E cells were grown in DMEM with 10% fetal bovine serum. RK3E/pBpuro, RK3E/KLF4-ER, RK3E/HA-KLF4, RK3E/pQCXIN, RK3E/MAMLDN1, RK3E/MAMLDN2 and RK3E/CSLDN were derived by retroviral transduction of RK3E cells as described.4,14 MCF10A cells were cultured and transduced as described.49 Virus transduced pools were grown in the presence of 0.4 μg/ml puromycin (Sigma) or 400 μg/ml G418 (Mediatech). Transformation assays utilized retroviral transduction, as described previously.4 The γSI L685, 458 (Sigma) was used at 10 μM in DMSO, and media were changed daily.
siRNAs (Table 3) were synthesized by Thermo Fisher/Dharmacon, and transfected into RK3E or human tumor cell lines using Lipofectamine™RNAiMAX reagent (Invitrogen). Trypsinized cells were plated directly into wells containing lipid-siRNA complexes. Cells were then incubated in serum-free medium (Optimem) at 37°C for 2 h before adding complete medium. For analysis of Notch1 expression in KLF4 knockdown cells (Fig. 2C), the siRNA-transfected breast cancer cells were analyzed at 96 h posttransfection.
Table 3.
Sequences of siRNas used in this study
| Name | RNA sequence |
|---|---|
| Hs KLF4-siR1 (S) | 5′ACCUCGCCUUACACAUGAAUU3′ |
| Hs KLF4-siR1 (AS) | 5′UUCAUGUGUAAGGCGAGGUUU3′ |
| Hs KLF4-siR2 (S) | 5′ GAGAGACCGAGGAGUUCAAUU3′ |
| Hs KLF4-siR2 (AS) | 5′UUGAACUCUCGGUCUCUCUU3′ |
| Rn N1-siR1 (S) | 5′CUACAAGAUCGAAGCCGUAUU3′ |
| Rn N1-siR1 (AS) | 5′UACGGCUUCGAUCUUGUAGUU3′ |
| Rn N1-siR2 (S) | 5′CCAUGGAGCUUGCCGGGAUUU3′ |
| Rn N1-siR2 (AS) | 5′AUCCCGGCAAGCUCCAUGGUU3′ |
| Rn N1-siR3 (S) | 5′GCGAGGAAGAGCUACGCAAUU3′ |
| Rn N1-siR3 (AS) | 5′UUGCGUAGCUCUUCCUCGCUU3′ |
| Control (S) | 5′GAAUAUGGUUGUUUGAAGAUU3′ |
| Control (AS) | 5′UCUUCAAACAACCAUAUUCUU3′ |
Immunostaining
Breast tumors were obtained from the University of Alabama Hospital. Tissues were fixed and processed as described.43 Adjacent sections from 89 tumors were stained with KLF4 and N1-C20 antibodies separately and statistical analysis was performed as described.43 twelve of these 89 cases were also stained with N1IC-Nter using heat-induced antigen retrieval. All experiments used normal rabbit immunoglobulin as a negative control (not shown). Human studies conformed to NIH guidelines and were approved by the Institutional Review Board for Human use at the University of Alabama at Birmingham.
For detection of Notch1 in cultured cells (Fig. 1B), KLF4-ER cells were plated on poly-L-lysine coated coverslips at 30% confluence. 24 h later, 4OHT or DMSO was added to the medium. 10 h later the cells were fixed and stained with N1-C-20 antibody. Indirect immunofluorescence and digital imaging were performed as described.43
Digital imaging
For immunofluorescence studies, black and white images were collected using an Axio-Cam HRc digital camera (Zeiss), and the pseudo-colored images were merged using Axiovision software (version 3.1). Color images of stained slides were acquired similarly using brightfield microscopy. All images were exported from Axiovision as tiff files, and these were imported into CorelDraw (version 10). Within each Figure, minor adjustments were made to gamma, brightness, contrast and intensity so as to more accurately portray the appearance of the actual samples. For the experimental and control panels within each figure or subfigure, all manipulations and adjustments were performed identically and in parallel. Within CorelDraw, color bitmaps were changed from RGB to CMYK and then the entire figure was exported from CorelDraw as a tiff file for submission.
Chromatin immunoprecipitation
MCF10A cells at 95% confluence were washed twice with PBS. Formaldehyde (37% solution; Fisher Scientific) was added to a final concentration of 1% in PBS, and the plates were incubated at room temperature for 10 min. Cells were rinsed with ice-cold PBS and scraped into 6 ml of TD buffer (100 mM Tris-HCl, pH 9.4, 10 mM DTT), transferred to a 15 ml tube and incubated at 30°C for 15 min. The cells were centrifuged for 5 min at 3,000 rpm, washed and centrifuged sequentially with 1.0 ml of buffer I (0.25% Triton X-100, 10 mM EDTA, 0.5 mM EGTA, 10 mM HEPES-KOH, pH 6.5) and 1.0 ml of buffer II (200 mM NaCl, 1.0 mM EDTA, 0.5 mM EGTA, 10 mM HEPES-KOH, pH 6.5). Cells were re-suspended in 0.6 ml of lysis buffer with freshly added protease inhibitors (1% SDS, 10 mM EDTA, 50 mM Tris-HCl, pH 8.1, 1.0 mM PMSF, 1.0 mM benzamidine, 5.0 μg/ml leupeptin, 5.0 μg/ml aprotinin) and incubated on ice for 10 min. Samples were then sonicated with an Ultrasonic processor at 25% amplitude (twenty pulses for 10 s each) on ice, then centrifuged at 4°C at 10,000 rpm for 10 min. Supernatants were collected and 4.5 ml of ChIP Dilution Buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl, pH 8.1, 167 mM NaCl, with protease inhibitors as in lysis buffer above) was added. The chromatin was pre-cleared with 12 μg of sheared salmon sperm DNA, 10 μl normal mouse IgG (0.5 mg/ml, Santa Cruz) and protein A-Sepharose (100 μl of 50% slurry in 10 mM Tris-HCl pH 8.0, 1.0 mM EDTA) with agitation for 2 h at 4°C.
Pre-cleared chromatin was incubated with 4.0 μg HA antibody, 9.0 μl of TFIIB antibody, or 9.0 μl normal IgG (Chip-IT Kit, Active Motif, Cat# 53001), and rocked at 4°C overnight. 100 μl of protein A-Sepharose (50% slurry) and 20 μg salmon sperm DNA were added to each sample, followed by agitation for a further 2.5 h at 4°C. Beads were pelleted by centrifugation at 2,000 xg for 3 min at 4°C and washed sequentially [10 min, in 1.0 ml with low salt buffer (0.1% SDS, 1% Triton X-100, 2.0 mM EDTA, 20 mM Tris-HCl pH 8.1, 150 mM NaCl), TSE2 (0.1% SDS, 1% Triton X-100, 2.0 mM EDTA, 20 mM Tris-HCl pH 8.1, 500 mM NaCl) and Buffer 3 (0.25% LiCl, 1% NP40, 1% sodium deoxycholate, 1.0 mM EDTA, 10 mM Tris-HCl pH 8.0)], followed by three washes in TE (10 mM Tris-HCl pH 9.0, 0.1 mM EDTA). The complexes were eluted in 250 μl of elution buffer (1% SDS, 0.1 M NaHCO3) with agitation for 15 min. The beads were pelleted by centrifugation and the supernatants were transferred to clean tubes. The elution was repeated and the two eluates were combined. Crosslinks were removed by adding NaCl to 0.3 M with incubation at 65°C overnight. To reverse crosslinks in the input DNA, 100 μl inputs were heated without addition of NaCl. DNA was purified using Qiaquick columns (Qiagen) and eluted in 50 μl of 10 mM Tris-HCl, pH 8.0. 2 μl of DNA was used in semi-quantitative PCR reactions. Oligonucleotides are listed in Table 2.
Reporter assays
In Figure 3D, 0.2 μg of pTK-β-gal (internal reference), 0.5 μg Notch1 promoter-reporter, and 0.2 μg pRK5-HA-KLF4 or control vector were transfected into HEK293 cells at 50% confluence in six-well dishes, using 1.8 μl TransIT-LT1 (Mirus) according to the manufacturer's protocol. Cells were extracted 48 h later and assayed for luciferase and β-gal activity with the Dual Light System (Applied Biosystems). Luciferase activity was normalized to β-gal, and the fold activation (KLF4/Vector) was determined. Using the same protocol in Figure 5B, 0.5 μg of the Hes1-luc reporter and 0.2 μg of pTK-β-gal were transiently transfected into RK3E cells with (+) or without 1.0 μg of pcDNA3.1-N1IC. The total DNA was kept constant by the inclusion of vector as necessary. Luciferase activity was normalized to β-gal, and the fold activation (N1IC/vector) was determined.
Supplementary Material
Acknowledgments
We thank Drs. Jon Aster, Tony Capobianco, Ryoichiro Kageyama and C. Jane McGlade for constructs, and Jeffrey Kudlow for the ChIP protocol. This work was funded by NIH grants R01 CA127405, RO1 CA094030, P50 CA89019 and P30 AR050948.
Abbreviations
- BCC
basal cell carcinoma
- ChIP
chromatin immunoprecipitation
- CHX
cycloheximide
- SI
secretase inhibitor
- SCC
squamous cell carcinoma
- HNSCC
head and neck squamous cell carcinoma
- NEC
notch extracellular domain
- NFL
notch full length protein
- NIC
notch intracellular domain
- N™
notch transmembrane domain
- 4OHT
4-hydroxytamoxifen
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/9440
References
- 1.Segre JA, Bauer C, Fuchs E. Klf4 is a transcription factor required for establishing the barrier function of the skin. Nat Genet. 1999;22:356–60. doi: 10.1038/11926. [DOI] [PubMed] [Google Scholar]
- 2.Katz JP, Perreault N, Goldstein BG, Lee CS, Labosky PA, Yang VW, et al. The zinc-finger transcription factor Klf4 is required for terminal differentiation of goblet cells in the colon. Development. 2002;129:2619–28. doi: 10.1242/dev.129.11.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Swamynathan SK, Katz JP, Kaestner KH, shery-Padan R, Crawford MA, Piatigorsky J. Conditional deletion of the mouse Klf4 gene results in corneal epithelial fragility, stromal edema and loss of conjunctival goblet cells. Mol Cell Biol. 2007;27:182–94. doi: 10.1128/MCB.00846-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Foster KW, Ren S, Louro ID, Lobo-Ruppert SM, McKie-Bell P, Grizzle W, et al. Oncogene expression cloning by retroviral transduction of adenovirus E1A-immortalized rat kidney RK3E cells: transformation of a host with epithelial features by c-MYC and the zinc finger protein GKLF. Cell Growth Differ. 1999;10:423–34. [PubMed] [Google Scholar]
- 5.Ruppert JM, Vogelstein B, Kinzler KW. The zinc finger protein GLI transforms primary cells in cooperation with adenovirus E1A. Mol Cell Biol. 1991;11:1724–8. doi: 10.1128/mcb.11.3.1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li X, Deng W, Lobo-Ruppert SM, Ruppert JM. Gli1 acts through Snail and E-cadherin to promote nuclear signaling by β-catenin. Oncogene. 2007;26:4489–98. doi: 10.1038/sj.onc.1210241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ascano JM, Beverly LJ, Capobianco AJ. The C-terminal PDZ-ligand of JAGGED1 is essential for cellular transformation. J Biol Chem. 2003;278:8771–9. doi: 10.1074/jbc.M211427200. [DOI] [PubMed] [Google Scholar]
- 8.Capobianco AJ, Zagouras P, Blaumueller CM, Artavanis-Tsakonas S, Bishop JM. Neoplastic transformation by truncated alleles of human NOTCH1/TAN1 and NOTCH2. Mol Cell Biol. 1997;17:6265–73. doi: 10.1128/mcb.17.11.6265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tonon G, Modi S, Wu L, Kubo A, Coxon AB, Komiya T, et al. t(11;19)(q21;p13) translocation in mucoepidermoid carcinoma creates a novel fusion product that disrupts a Notch signaling pathway. Nat Genet. 2003;33:208–13. doi: 10.1038/ng1083. [DOI] [PubMed] [Google Scholar]
- 10.Li X, Deng W, Nail CD, Bailey SK, Kraus MH, Ruppert JM, et al. Snail induction is an early response to Gli1 that determines the efficiency of epithelial transformation. Oncogene. 2006;25:609–21. doi: 10.1038/sj.onc.1209077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kolligs FT, Hu G, Dang CV, Fearon ER. Neoplastic transformation of RK3E by mutant β-catenin requires deregulation of Tcf/Lef transcription but not activation of c-myc expression. Mol Cell Biol. 1999;19:5696–706. doi: 10.1128/mcb.19.8.5696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chamorro MN, Schwartz DR, Vonica A, Brivanlou AH, Cho KR, Varmus HE. FGF-20 and DKK1 are transcriptional targets of β-catenin and FGF-20 is implicated in cancer and development. EMBO J. 2005;24:73–84. doi: 10.1038/sj.emboj.7600460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rahman L, Voeller D, Rahman M, Lipkowitz S, Allegra C, Barrett JC, et al. Thymidylate synthase as an oncogene: a novel role for an essential DNA synthesis enzyme. Cancer Cell. 2004;5:341–51. doi: 10.1016/s1535-6108(04)00080-7. [DOI] [PubMed] [Google Scholar]
- 14.Louro ID, Bailey EC, Li X, South LS, McKie-Bell PR, Yoder BK, et al. Comparative gene expression profile analysis of GLI and c-MYC in an epithelial model of malignant transformation. Cancer Res. 2002;62:5867–73. [PubMed] [Google Scholar]
- 15.Hoseong YS, Andl T, Grachtchouk V, Wang A, Liu J, Syu LJ, et al. Pathological responses to oncogenic Hedgehog signaling in skin are dependent on canonical Wnt/β-catenin signaling. Nat Genet. 2008;40:1130–5. doi: 10.1038/ng.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Foster KW, Frost AR, McKie-Bell P, Lin CY, Engler JA, Grizzle WE, et al. Increase of GKLF messenger RNA and protein expression during progression of breast cancer. Cancer Res. 2000;60:6488–95. [PubMed] [Google Scholar]
- 17.Foster KW, Liu Z, Nail CD, Li X, Fitzgerald TJ, Bailey SK, et al. Induction of KLF4 in basal keratinocytes blocks the proliferation-differentiation switch and initiates squamous epithelial dysplasia. Oncogene. 2005;24:1491–500. doi: 10.1038/sj.onc.1208307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang CC, Liu Z, Li X, Bailey SK, Nail CD, Foster KW, et al. KLF4 and PCNA identify stages of tumor initiation in a conditional model of cutaneous squamous cell epithelial neoplasia. Cancer Biol Ther. 2005;4:1401–8. doi: 10.4161/cbt.4.12.2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prasad NB, Biankin AV, Fukushima N, Maitra A, Dhara S, Elkahloun AG, et al. Gene expression profiles in pancreatic intraepithelial neoplasia reflect the effects of Hedgehog signaling on pancreatic ductal epithelial cells. Cancer Res. 2005;65:1619–26. doi: 10.1158/0008-5472.CAN-04-1413. [DOI] [PubMed] [Google Scholar]
- 20.Jiang W, Deng W, Bailey SK, Nail CD, Frost AR, Brouillette WJ, et al. Prevention of KLF4-mediated tumor initiation and malignant transformation by UAB30 rexinoid. Cancer Biol Ther. 2009;8:287–96. doi: 10.4161/cbt.8.3.7486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–76. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 22.Katz JP, Perreault N, Goldstein BG, Actman L, McNally SR, Silberg DG, et al. Loss of Klf4 in mice causes altered proliferation and differentiation and precancerous changes in the adult stomach. Gastroenterology. 2005;128:935–45. doi: 10.1053/j.gastro.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 23.Zhao W, Hisamuddin IM, Nandan MO, Babbin BA, Lamb NE. Yang VW Identification of Kruppel-like factor 4 as a potential tumor suppressor gene in colorectal cancer. Oncogene. 2004;23:395–402. doi: 10.1038/sj.onc.1207067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghaleb AM, McConnell BB, Nandan MO, Katz JP, Kaestner KH, Yang VW. Haploinsufficiency of Kruppel-like factor 4 promotes adenomatous polyposis coli dependent intestinal tumorigenesis. Cancer Res. 2007;67:7147–54. doi: 10.1158/0008-5472.CAN-07-1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rowland BD, Bernards R, Peeper DS. The KLF4 tumour suppressor is a transcriptional repressor of p53 that acts as a context-dependent oncogene. Nat Cell Biol. 2005 doi: 10.1038/ncb1314. [DOI] [PubMed] [Google Scholar]
- 26.Lin MH, Kopan R. Long-range, nonautonomous effects of activated Notch1 on tissue homeostasis in the nail. Dev Biol. 2003;263:343–59. doi: 10.1016/j.ydbio.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 27.Sancho E, Batlle E, Clevers H. Live and let die in the intestinal epithelium. Curr Opin Cell Biol. 2003;15:763–70. doi: 10.1016/j.ceb.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 28.Fiuza UM, Arias AM. Cell and molecular biology of Notch. J Endocrinol. 2007;194:459–74. doi: 10.1677/JOE-07-0242. [DOI] [PubMed] [Google Scholar]
- 29.Koch U, Radtke F. Notch and cancer: a double-edged sword. Cell Mol Life Sci. 2007;64:2746–62. doi: 10.1007/s00018-007-7164-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Watt FM, Estrach S, Ambler CA. Epidermal Notch signalling: differentiation, cancer and adhesion. Curr Opin Cell Biol. 2008;20:171–9. doi: 10.1016/j.ceb.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lai EC. Notch signaling: control of cell communication and cell fate. Development. 2004;131:965–73. doi: 10.1242/dev.01074. [DOI] [PubMed] [Google Scholar]
- 32.Jarriault S, Brou C, Logeat F, Schroeter EH, Kopan R, Israel A. Signalling downstream of activated mammalian Notch. Nature. 1995;377:355–8. doi: 10.1038/377355a0. [DOI] [PubMed] [Google Scholar]
- 33.Lai EC. Keeping a good pathway down: transcriptional repression of Notch pathway target genes by CSL proteins. EMBO Rep. 2002;3:840–5. doi: 10.1093/embo-reports/kvf170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brennan K, Brown AM. Is there a role for Notch signalling in human breast cancer? Breast Cancer Res. 2003;5:69–75. doi: 10.1186/bcr559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramain P, Khechumian K, Seugnet L, Arbogast N, Ackermann C, Heitzler P. Novel Notch alleles reveal a Deltex-dependent pathway repressing neural fate. Curr Biol. 2001;11:1729–38. doi: 10.1016/s0960-9822(01)00562-0. [DOI] [PubMed] [Google Scholar]
- 36.Talora C, Campese AF, Bellavia D, Felli MP, Vacca A, Gulino A, et al. Notch signaling and diseases: an evolutionary journey from a simple beginning to complex outcomes. Biochim Biophys Acta. 2008;1782:489–97. doi: 10.1016/j.bbadis.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 37.Weng AP, Ferrando AA, Lee W, Morris JP, Silverman LB, Sanchez-Irizarry C, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–71. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- 38.Callahan R, Egan SE. Notch signaling in mammary development and oncogenesis. J Mammary Gland Biol Neoplasia. 2004;9:145–63. doi: 10.1023/B:JOMG.0000037159.63644.81. [DOI] [PubMed] [Google Scholar]
- 39.Dontu G, Liu S, Wicha MS. Stem cells in mammary development and carcinogenesis: implications for prevention and treatment. Stem Cell Rev. 2005;1:207–13. doi: 10.1385/SCR:1:3:207. [DOI] [PubMed] [Google Scholar]
- 40.Weijzen S, Rizzo P, Braid M, Vaishnav R, Jonkheer SM, Zlobin A, et al. Activation of Notch-1 signaling maintains the neoplastic phenotype in human Ras-transformed cells. Nat Med. 2002;8:979–86. doi: 10.1038/nm754. [DOI] [PubMed] [Google Scholar]
- 41.Sharma VM, Draheim KM, Kelliher MA. The Notch1/c-Myc pathway in T cell leukemia. Cell Cycle. 2007;6:927–30. doi: 10.4161/cc.6.8.4134. [DOI] [PubMed] [Google Scholar]
- 42.Nicolas M, Wolfer A, Raj K, Kummer JA, Mill P, van Noort M, et al. Notch1 functions as a tumor suppressor in mouse skin. Nat Genet. 2003;33:416–21. doi: 10.1038/ng1099. [DOI] [PubMed] [Google Scholar]
- 43.Pandya AY, Talley LI, Frost AR, Fitzgerald TJ, Trivedi V, Chakravarthy M, et al. Nuclear localization of KLF4 is associated with an aggressive phenotype in early-stage breast cancer. Clin Cancer Res. 2004;10:2709–19. doi: 10.1158/1078-0432.ccr-03-0484. [DOI] [PubMed] [Google Scholar]
- 44.Weng AP, Aster JC. Multiple niches for Notch in cancer: context is everything. Curr Opin Genet Dev. 2004;14:48–54. doi: 10.1016/j.gde.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 45.Littlewood TD, Hancock DC, Danielian PS, Parker MG, Evan GI. A modified oestrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic Acids Res. 1995;23:1686–90. doi: 10.1093/nar/23.10.1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kato H, Taniguchi Y, Kurooka H, Minoguchi S, Sakai T, Nomura-Okazaki S, et al. Involvement of RBP-J in biological functions of mouse Notch1 and its derivatives. Development. 1997;124:4133–41. doi: 10.1242/dev.124.20.4133. [DOI] [PubMed] [Google Scholar]
- 47.Jeffries S, Robbins DJ, Capobianco AJ. Characterization of a high-molecular-weight Notch complex in the nucleus of Notch(ic)-transformed RKE cells and in a human T-cell leukemia cell line. Mol Cell Biol. 2002;22:3927–41. doi: 10.1128/MCB.22.11.3927-3941.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maillard I, Weng AP, Carpenter AC, Rodriguez CG, Sai H, Xu L, et al. Mastermind critically regulates Notch-mediated lymphoid cell fate decisions. Blood. 2004;104:1696–702. doi: 10.1182/blood-2004-02-0514. [DOI] [PubMed] [Google Scholar]
- 49.Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30:256–68. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- 50.Elenbaas B, Spirio L, Koerner F, Fleming MD, Zimonjic DB, Donaher JL, et al. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev. 2001;15:50–65. doi: 10.1101/gad.828901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frisch SM. E1a induces the expression of epithelial characteristics. Journal of Cell Biology. 1994;127:1085–96. doi: 10.1083/jcb.127.4.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gillespie J, Pallente M, Ensley JF, Koontongkaew S, et al. Distinct pattern of expression of differentiation and growth-related genes in squamous cell carcinomas of the head and neck revealed by the use of laser capture microdissection and cDNA arrays. Oncogene. 2000;19:3220–4. doi: 10.1038/sj.onc.1203703. [DOI] [PubMed] [Google Scholar]
- 53.Panelos J, Tarantini F, Paglierani M, Di SC, Maio V, Pellerito S, et al. Photoexposition discriminates Notch 1 expression in human cutaneous squamous cell carcinoma. Mod Pathol. 2008;21:316–25. doi: 10.1038/modpathol.3801007. [DOI] [PubMed] [Google Scholar]
- 54.Proweller A, Tu L, Lepore JJ, Cheng L, Lu MM, Seykora J, et al. Impaired notch signaling promotes de novo squamous cell carcinoma formation. Cancer Res. 2006;66:7438–44. doi: 10.1158/0008-5472.CAN-06-0793. [DOI] [PubMed] [Google Scholar]
- 55.LeFort K, Dotto GP. Notch signaling in the integrated control of keratinocyte growth/differentiation and tumor suppression. Semin Cancer Biol. 2004;14:374–86. doi: 10.1016/j.semcancer.2004.04.017. [DOI] [PubMed] [Google Scholar]
- 56.Raafat A, Lawson S, Bargo S, Klauzinska M, Strizzi L, Goldhar AS, et al. Rbpj conditional knockout reveals distinct functions of Notch4/Int3 in mammary gland development and tumorigenesis. Oncogene. 2009;28:219–30. doi: 10.1038/onc.2008.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li Y, McClintick J, Zhong L, Edenberg HJ, Yoder MC, Chan RJ. Murine embryonic stem cell differentiation is promoted by SOCS-3 and inhibited by the zinc finger transcription factor Klf4. Blood. 2005;105:635–7. doi: 10.1182/blood-2004-07-2681. [DOI] [PubMed] [Google Scholar]
- 58.Milner LA, Bigas A, Kopan R, Brashem-Stein C, Bernstein ID, Martin DI. Inhibition of granulocytic differentiation by mNotch1. Proc Natl Acad Sci USA. 1996;93:13014–9. doi: 10.1073/pnas.93.23.13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shawber C, Nofziger D, Hsieh JJ, Lindsell C, Bogler O, Hayward D, et al. Notch signaling inhibits muscle cell differentiation through a CBF1-independent pathway. Development. 1996;122:3765–73. doi: 10.1242/dev.122.12.3765. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.