

Abstract
Background and purpose:
Classically, stimulation of muscarinic cholinoceptors exerts negative inotropic and chronotropic effects in the atrium of mammalian hearts. These effects are crucial to the vagal regulation of the heart beat. This effect is assumed to be mediated via GTP binding (G) proteins, because they can be abolished by Pertussis toxin. However, it is unknown which G proteins are involved.
Experimental approach:
We studied contractility in isolated left or right atrium from genetically manipulated mice with deletion of one of two G proteins, either of the α subunit of Gi2 protein (Gi2α) or of the α subunit of Go protein (Goα). Preparations were stimulated with carbachol alone or after pretreatment with the β-adrenoceptor agonist isoprenaline. For comparison, the effects of carbachol on L-type Ca2+-channels in isolated ventricular cardiomyocytes were studied.
Key results:
The negative inotropic and chronotropic effects of carbachol alone or in the presence of isoprenaline were identical in atria from knockout or wild-type mice. However, the effect of carbachol on isoprenaline-activated L-type Ca2+-channel in isolated ventricular cardiomyocytes was greatly attenuated in both types of knockout mice studied.
Conclusions and implications:
These data imply that there is either redundancy of G proteins for signal transduction or that Pertussis toxin-sensitive proteins other than Gi2α and Goα mediate the vagal stimulation in the atrium. Moreover, different G proteins mediate the effect of carbachol in ventricle compared with atrium.
Keywords: acetylcholine, carbachol, muscarinic cholinoceptors, G proteins, parasympathomimetics, vagal stimulation
Introduction
In the mammalian heart, cardiac function is under the control of the sympathetic and parasympathetic nervous system. Whereas sympathetic stimulation leads to an increase of cardiac function, the effects of the parasympathetic system are the opposite and vagal stimulation exerts negative inotropic, negative chronotropic and negative dromotropic effects in the mammalian heart. These effects can occur either directly or indirectly in the presence of stimulation of β-adrenoceptors (Löffelholz and Pappano, 1985). However, species differences and regional differences of these effects do exist (Boyett et al., 1988; McIvor et al., 1988; McMorn et al., 1993; Yang et al., 1996).
The postganglionic release of acetylcholine from parasympathetic nerve terminals activates postsynaptic muscarinic cholinoceptors in the heart. Among the five known muscarinic receptor subtypes (Caulfield and Birdsall 1998), the M2-muscarinic receptor (nomenclature follows Alexander et al., 2008) is the predominant isoform present in the mammalian heart (Brodde and Michel, 1999; Stengel et al., 2000; Wang et al., 2001; Krejcí and Tucek 2002). All muscarinic receptors belong to the superfamily of G protein-coupled, seven-transmembrane-domain receptors (Dhein et al., 2001). The M1-, M3- and M5-muscarinic receptors preferentially couple to G11/q proteins with subsequent activation of the phospholipase C-diacylglycerol-inositol phosphate system. The M2- and M4-muscarinic receptors are coupled to Pertussis toxin (PTX)-sensitive G proteins. Their stimulation results in an inhibition of adenylyl cyclase activity and a decrease in cAMP and protein phosphorylation (Rockman et al., 2002).
However, other groups noted that the negative inotropic effects of acetylcholine (in ventricle or atrium) can occur without a decrease in cAMP (or increase in cGMP) content and have claimed that PTX-sensitive G proteins couple to additional effectors, such as phosphatases (Böhm et al., 1988; Ahmad et al., 1989; Gupta et al., 1993; 1994; 1999; Neumann et al., 1994; 1995a; b;) or phosphodiesterases (Fischmeister and Hartzell, 1991; Lohmann et al., 1991). Nevertheless, these effects were accompanied by a reduction in the phosphorylation state of cardiac regulatory proteins in the ventricle (Lindemann and Watanabe, 1985; Gupta et al., 1994) and this dephosphorylation was PTX-sensitive (Neumann et al., 1994). In the atrium, besides reduction in cAMP content, other mechanisms are currently deemed to mediate vagal effects. For instance, stimulation of muscarinic receptors in the atrium reduces the L-type Ca2+-channel current and enhances the current through potassium currents (IK,Ach) via G proteins (Nagata et al., 2000) and can inhibit IK1 (Dobrzynski et al., 2002). This can lead to hyperpolarization of the cell, shortening of the action potential and less time for the influx of Ca2+. Overall, this chain of events is assumed to reduce the Ca2+ available for contraction and initiates and maintains the negative inotropic effect of vagal stimulation. It is controversial whether the inotropic effects of acetylcholine in the heart involve nitric oxide (NO; Han et al., 1998; Belevych and Harvey, 2000). Different mechanisms may underlie the negative chronotropic effects of carbachol. In vitro data and those from knockout mice strongly implicate hyperpolarization-activated cation channels as effectors for the negative chronotropic action in the sinus node, but IK,Ach and ICa,L are also affected (DiFrancesco and Tromba, 1988; Brown and Denyer, 1989; Honjo et al., 1992; Zhang et al., 2002).
It is agreed that the negative inotropic effects of vagal stimulation in both the atrium and ventricle are PTX-sensitive. This has been shown in intact animals, in isolated cardiac preparations from PTX-pretreated animals or in isolated cardiomyocytes of these animals but importantly also in cardiomyocytes treated in vitro with PTX (Nakajima et al., 1990; Osaka et al., 1993; Robishaw and Hansen, 1994; Stengel et al., 2000).
More recently, these findings could be recapitulated in knockout mice. Thus, carbachol failed to reduce the current through isoprenaline-stimulated L-type Ca2+-channels in isolated ventricular myocytes from Gi2α (Nagata et al., 2000; Chen et al., 2001) or Goα knockout mice (Valenzuela et al., 1997). These findings imply that knockout of one G protein is sufficient to render at least the cardiac ventricle insensitive to vagal stimulation and would easily explain the previous results on PTX pretreatment.
However, in atria, Gi2α and Goα might play different roles in mediating effects of muscarinic receptor stimulation. As reported by Sowell et al. (1997), in cardiocytes derived from embryonic stem cells with targeted inactivation of Gi2α or Gi3α (but not Goα), the muscarinic activation of IK,Ach was disrupted. On the other hand, the chronotropic response remained unaffected in Gi2α- or Gi3α-null cells. Accordingly, Duan et al. (2007) could show that Goα is crucial for the muscarinic regulation of heart rate in isolated working hearts and in addition also for the regulation of heart rate variability in whole animals. In contrast, knockout of Gi2α or Gi3α did not affect regulation of heart rate by muscarinic receptor stimulation.
In order to further investigate the role of Gi2α and Goα in atrium and to complement the above-mentioned data, we performed experiments on isolated, electrically driven, left atria and spontaneously beating right atria from Gi2α and Goα knockout animals. Interestingly, neither Gi2α- nor Goα knockout affected inotropic or chronotropic effects of muscarinic receptor stimulation in isolated atria. Our data indicate that signal transduction of vagal tone uses different G proteins in atrium and in ventricle.
Methods
Animals
Animals were handled and maintained according to protocols approved by the animal welfare committee of the University of Münster, which conform to the NIH Guidelines for the Care and Use of Laboratory Animals. The Gi2α−/− and Goα−/− mice were obtained by crossing the respective heterozygous Gi2α+/− and Goα+/− parents as previously published (Rudolph et al., 1993; Jiang et al., 1998). Wild-type littermates (C57Bl/6-129sv mice) served as controls. The Gi2α and Goα proteins were not detectable in the heart preparations (Rudolph et al., 1993; Jiang et al., 1998).
Measurement of contractile function and pacemaker activity
Mice of either sex, aged between 12 and 14 weeks, weighing 21.3–29.6 g were used. Mice were killed with CO2 inhalation and the hearts removed; right and left atria were dissected from isolated mouse hearts and mounted in an organ bath. Left atrial preparations were continuously electrically stimulated (field stimulation) using a Grass stimulator SD 9 (Quincy, MA, USA) with each impulse consisting of 1 Hz, with a voltage of 10–15% above threshold and 5 ms duration. Right atrial preparations (auricles) were attached in the same set-up but were not electrically stimulated and allowed to contract spontaneously. The resting tension was set at 1 mN and kept constant throughout the experiment.
The bathing solution contained (in mM) NaCl 119.8, KCl 5.4, CaCl2 1.8, MgCl2 1.05, NaH2PO4 0.42, NaHCO3 22.6, Na2EDTA 0.05, ascorbic acid 0.28 and glucose 5.0, continuously gassed with 95% O2 and 5% CO2 and maintained at 35°C resulting in a pH of 7.4. Contractions were measured isometrically. Atria were attached with fine sutures to a hook in the organ bath and an isometric force transducer. Signals were amplified and continuously fed into a chart recorder (Föhr Medical Instruments, Egelsbach, Germany). Carbachol was cumulatively applied with 10 min for each concentration. Contraction experiments with carbachol were performed after addition of adenosine deaminase (1 µg·mL−1 for 30 min) to avoid interference from endogenous adenosine.
PTX pretreatment
PTX (150 µg·kg−1 body weight in sodium phosphate buffer, consisting of 0.1 M of sodium phosphate and 0.5 M of sodium chloride, pH = 7.5) was administered intraperitoneally 72 h before isolation of atria. Control animals were treated in the same way with the corresponding amount of solvent alone.
Isolation of cardiomyocytes
Ventricular cardiomyocytes were isolated from wild type and Gi2α and Goα knockout mouse hearts using a published protocol (Stein et al., 1993). Animals were pretreated with heparin (5 U·g−1 body weight), and later anesthetized with CO2. Mouse hearts were excised and the cannulated aorta was fixed to a Langendorff apparatus. Hearts were perfused for 5 min at 2 mL·min−1 with a Ca2+-free solution (solution A) composed of (in mM) 140 NaCl, 5.8 KCl, 0.5 KH2PO4, 0.4 NaH2PO4, 0.9 MgSO4, 10 HEPES, 11.1 glucose (pH 7.1), followed by a perfusion for 30 min with solution A supplemented with 0.2 mg·mL−1 collagenase (type D, Boehringer Mannheim, Germany). Ca2+ concentration was gradually increased during digestion to 100 µM. After enzymatic digestion, the hearts were perfused for 10 min with solution A. The ventricles were cut into several pieces and subjected to gentle agitation through a nylon mesh to separate the cardiomyocytes. All subsequent experiments on isolated cardiomyocytes were performed in the presence of adenosine deaminase (10 U·mL−1) to avoid interference from endogenous adenosine on treatment (Gupta et al., 1993).
Whole-cell L-type Ca2+current
Isolated cardiomyocytes were studied using the whole-cell variation of the patch-clamp technique. Recordings were performed under conditions that suppress Na+ and K+ currents (Thierfelder et al., 1994). Briefly, cells were plated in a small dish (2 mL) on the stage of an inverted microscope (Leica, Cologne, Germany). The extracellular solution was composed of (in mM): TEA-Cl 130, MgCl2 1, 4-aminopyridine 4, HEPES 10, dextrose 10, CaCl2 2, adjusted to pH 7.3 with TEA-OH. The micropipette electrodes (resistances 1.5–2.5 MΩ) were filled with (in mM): K-aspartate 80, KCl 50, KH2PO4 10, MgCl2 0.5, MgATP 3, HEPES 10, EGTA 1, adjusted to pH 7.4 with KOH. All experiments were done at room temperature. Currents were elicited by voltage steps from a holding potential of −40 mV to a test potential of +10 mV for 200 ms, applied every 10 s. The currents could be blocked either by Ni2+ or Cd2+, which are known to specifically block the L-type Ca2+ currents (see Mitra and Morad, 1986). In the potential range corresponding to the T-type Ca2+ currents (from −100 mV to −30 mV), no inward currents could be registered (data not shown). Cell capacitance and ICa were recorded with an L/M-PC amplifier (LIST-Electronic, Darmstadt, Germany) according to standard protocols. Data were computed with the ISO2 software (MFK, Niedernhausen, Germany).
Data analysis
All data are given as means ± SEM. Statistical significance was estimated using Student's t-test for paired or unpaired observations. A P value of less than 0.05 was considered significant.
Materials
The following compounds were used: adenosine deaminase, isoprenaline (Boehringer Mannheim, Mannheim, Germany), carbachol (Sigma, St. Louis, MO, USA). PTX was purchased from Calbiochem (San Diego, CA, USA). The other chemicals were of best analytical grade. Double-distilled water was used throughout.
Results
In isolated, electrically driven, left atria from wild-type mice, carbachol exerted a concentration-dependent negative inotropic effect. This effect was identical to that observed in atria from Gi2α knockout mice (Figure 1A). From the same animals right atria were isolated and allowed to contract spontaneously. The basal contraction rate was identical in right atrium from Gi2α knockout mice and appropriate controls (Figure 2A). Similar results were obtained in Goα knockout mice (Figures 1B and 2B). We then assessed the efficacy and potency of the β-adrenoceptor agonist isoprenaline in isolated atrium from the knockout mice and found that the inotropic effect of isoprenaline in isolated electrically driven preparations from left atrium was concentration-dependent and identical to that in appropriate controls (Figure 3A). Likewise, isoprenaline exerted a concentration-dependent positive chronotropic effect (Figure 4), which was identical in wild-type mice and Gi2α knockout mice (Figure 4A). Similar results were obtained in left atrium (Figure 3B) and in right atrium of Goα knockout mice (Figure 4B).
Figure 1.
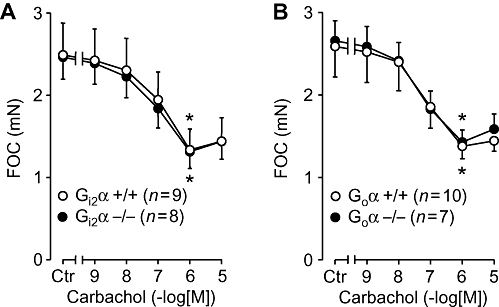
Effects of carbachol alone in isolated left atria on force of contraction (FOC) plotted in mN (A) or in percentage of control values (Ctr, prior to drug addition, B). Littermate wild-type mice (+/+) are compared with Gi2α knockout (Gi2α−/−, A) or Goα knockout mice (Goα−/−, B). Numbers in brackets indicate number of mice studied. * denotes first significant difference versus pre-drug value. No differences were detectable between wild type and knockout mice.
Figure 2.
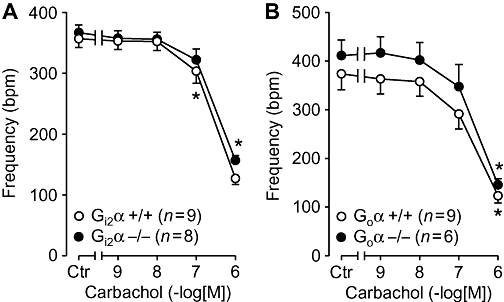
Effects of carbachol alone in isolated right atria on rate of contraction (frequency) plotted in beats per minute (bpm). Ctr indicate control values (prior to drug addition). Littermate wild-type mice (+/+, open circles) are compared with Gi2α knockout (Gi2α−/−, A) or Goα knockout mice (Goα−/−, B). Numbers in brackets indicate number of mice studied. * denotes first significant difference versus pre-drug value. No differences were detectable between wild type and knockout mice.
Figure 3.
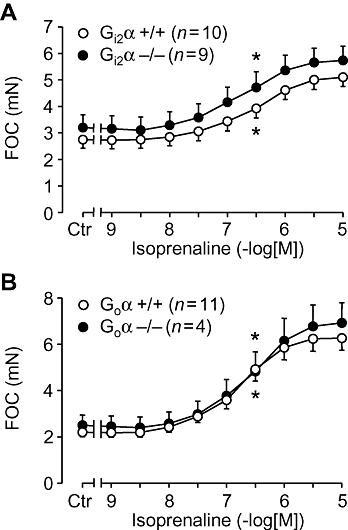
Effects of isoprenaline in isolated left atria on force of contraction (FOC) plotted in mN (B) or in percentage of maximum response to isoprenaline (A). Ctr indicates control values prior to drug addition. Littermate wild-type mice (+/+) are compared with Gi2α knockout (Gi2α−/−, A) or Goα knockout mice (Goα−/−, B). Numbers in brackets indicate number of mice studied. * denotes first significant difference versus pre-drug value. No differences were detectable between wild type and knockout mice.
Figure 4.
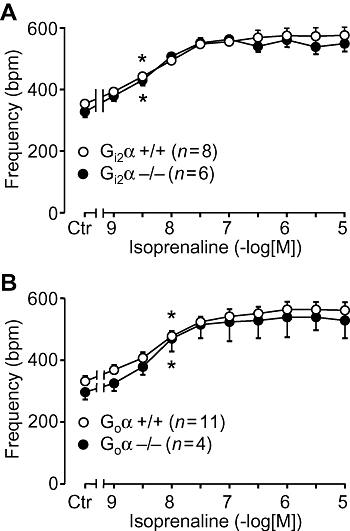
Effect of isoprenaline in isolated right atria on rate of contraction (frequency) plotted in beats per minute (bpm). Ctr indicates control values (prior to drug addition). Littermate wild-type mice (+/+, open circles) are compared with Gi2α knockout (Gi2α−/−, A) or Goα knockout mice (Goα−/−, B). Numbers in brackets indicate number of mice studied. * denotes first significant difference versus pre-drug value (Ctr). No differences were detectable between wild type and knockout mice.
We also wanted to test the antagonism of the effects of β-adrenoceptor stimulation by carbachol. To this end, isolated right atria of Gi2α knockout mice were stimulated by 30 nM isoprenaline in order to elicit a positive chronotropic effect (Figure 5A). Thereafter, increasing concentrations of carbachol were cumulatively applied. No differences between Gi2α knockout mice and controls were noted (Figure 5A). Similarly, the indirect negative chronotropic effect of carbachol in the presence of 30 nM isoprenaline did not differ between wild type and Goα knockout atria (Figure 5B). We studied also indirect negative inotropic effects of carbachol in the additional presence of 30 nM isoprenaline in isolated, electrically driven, left atria. As observed in right atria, genetic deletion of Gi2α or Goα did not change the inotropic effect of carbachol in the presence of isoprenaline (data not shown).
Figure 5.
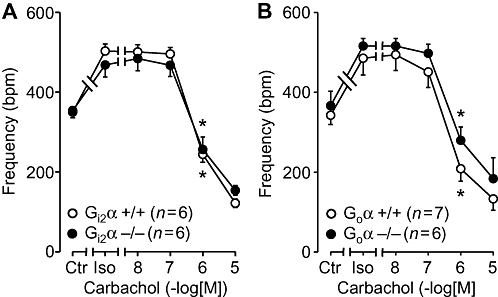
Effect of isoprenaline alone (Iso, 30 nM) or in the additional presence of cumulatively applied carbachol in isolated right atria on rate of contraction (frequency) plotted in beats per minute (bpm). Ctr indicates control values (prior to drug addition). Littermate wild-type mice (+/+, open circles) are compared with Gi2α knockout (Gi2α−/−, A) or Goα knockout mice (Goα−/−, B). Numbers in brackets indicate number of mice studied. * denotes first significant difference of additionally applied carbachol versus Iso (alone) value. No differences were detectable between wild type and knockout mice.
In addition, we wanted to test whether under our experimental conditions we could attenuate or abolish the supraventricular effects of carbachol by pretreatment with PTX. Thus, knockout mice were pretreated with PTX or vehicle (as control). As seen in Figure 6A the negative inotropic effect of carbachol was greatly attenuated in left atria of PTX-pretreated Gi2α knockout mice. The antagonism of the effects of β-adrenoceptor stimulation by carbachol in Gi2α knockout mice were also PTX-sensitive, as shown in Figure 6B.
Figure 6.
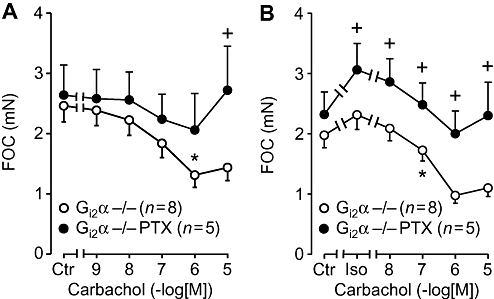
Effects of carbachol alone or in the presence of isoprenaline (30 nM) in isolated left atria on force of contraction (FOC) plotted in mN (A) or in percentage of control values (Ctr, prior to drug addition, B). Solvent-treated Gi2α knockout (Gi2α−/−) mice are compared with PTX-treated mice (PTX). Numbers in brackets indicate number of mice studied.* denotes first significant difference versus pre-drug value (A) or versus Iso alone (B). + denotes significant differences versus solvent pretreatment.
Further, we checked whether under our experimental conditions the effects of carbachol on current through the L-type Ca2+-channel were comparable to previous studies. Indeed, isoprenaline (10 µM) increased current through the L-type Ca2+-channel in isolated ventricular cardiomyocytes from Gi2α knockout mice. Additionally applied carbachol was ineffective (Figure 7B). In cardiomyocytes from wild-type mice, carbachol reduced isoprenaline-stimulated currents, as expected (Figure 7A). Similar findings were obtained for Goα knockout mice. In isolated ventricular cardiomyocytes from Goα knockout mice, carbachol was ineffective (Figure 8B), whereas in appropriate control cells, carbachol did attenuate the isoprenaline-induced current (Figure 8A).
Figure 7.
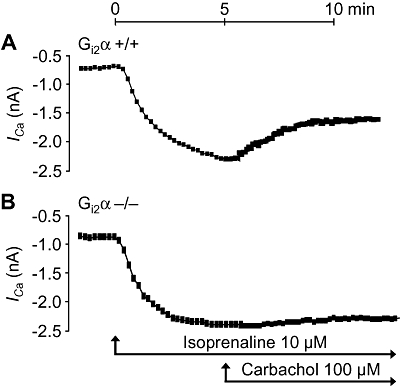
Time-dependent effects of isoprenaline alone (Iso, 10 µM) and in the additional presence of carbachol (Carb) on current through the L-type Ca2+-channels (ICa, nA) in isolated, patch-clamped, ventricular cardiomyocytes from littermate wild-type mice (Gi2α+/+, A) compared with Gi2α knockout mice (Gi2α−/−, B). Representative tracings from five cardiomyocytes from three different Gi2α+/+ and Gi2α−/− hearts are shown.
Figure 8.
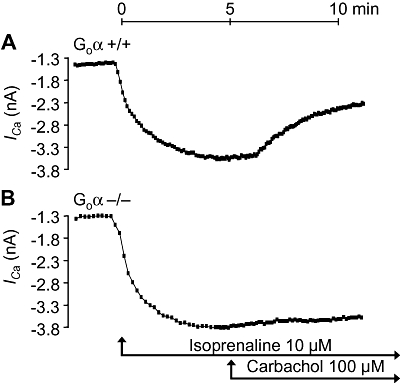
Time-dependent effects of isoprenaline alone (Iso, 10 µM) and in the additional presence of carbachol (Carb) on current through the L-type Ca2+-channels (ICa, nA) in isolated, patch-clamped ventricular cardiomyocytes from littermate wild-type mice (Go+/+, A) compared with Goα knockout mice (Goα−/−, B). Representative tracings from five cardiomyocytes from three different Goα+/+ and Goα−/− hearts are shown.
Discussion
The main new finding of this work is that vagal control of contractility in atrium and ventricle uses different G proteins.
As the study was focussed on the role of Gi2α or Goα for the muscarinic responses in atria, the experiments were performed in isolated electrically driven left atria and in isolated spontaneously beating right atria. Moreover, adenosine deaminase was used throughout the experiments to avoid interference from endogenous adenosine, which has the same effects as carbachol in atria. Compared with the more physiological working heart model used by Duan et al. (2007) our experimental approach enables the analysis of atrial function without possible interferences from ventricles (release of various humoral factors, e. g. adenosine; influence from the sympathetic nervous system).
As mentioned in the Introduction, vagal stimulation is a powerful regulator of the heart beat. In the isolated atrium stimulation of muscarinic cholinoceptors, for instance via carbachol alone, decreases force of contraction (left atrium) or frequency (right atrium) in man and animals. In the ventricle of man and mouse, carbachol has little effect on basal cardiac function but greatly attenuates the contractile response to β-adrenoceptor stimulation, termed indirect inhibitory action (Endoh, 1999). A similar pattern is also known for the cardiac actions of adenosine, which our group has extensively studied (Böhm et al., 1986; Gupta et al., 1993; Neumann et al., 1999; Kirchhof et al., 2003; Gergs et al., 2009). In atrium and ventricle of mouse the negative chronotropic and inotropic effects of carbachol are probably mediated by M2-muscarinic receptors, on the basis of studies with antagonists and appropriate knockout mice (Gupta et al., 1994; Neumann et al., 1994; Fisher et al., 2004). This M2-receptor couples to effectors via G proteins, in broken cell preparations and also in isolated cardiac cells. In the heart, nearly all known G proteins are detectable; however, there is some evidence that their expression is cell type- and region-specific (Eschenhagen, 1993). Hence, we hypothesized that Gi2α and/or Goα should be necessary for the inotropic and chronotropic effects of carbachol in the atrium. This hypothesis gained further plausibility from the fact that these G proteins were reported to be essential for the ventricular effects of carbachol (Valenzuela et al., 1997; Nagata et al., 2000; Chen et al., 2001).
Disruption of any of these G proteins impaired the action of carbachol on contractility or on the ventricular L-type Ca2+ current in the ventricle. Our results however, were the opposite of those predicted by our starting hypothesis. The inotropic and chronotropic effects of carbachol alone were still present in atrium from knockout mice. Even the antagonism of the effects of β-adrenoceptor stimulation by carbachol in atrium was independent of the presence of the investigated G proteins. However, pretreatment with PTX was still able to attenuate the direct and indirect effects of carbachol in knockout and wild-type mice, as reported before (Nakajima et al., 1990; Neumann et al., 2003). As another control, we could show that ablation of either G protein abolished the electrophysiological consequences of carbachol's antagonism of the effects of β-adrenoceptor stimulation in the ventricle, as described before (Valenzuela et al., 1997; Chen et al., 2001). Hence, PTX can block vagal effects both in the atrium and in the ventricle, but these G proteins are only essential for the effects in the ventricle and not in the atrium. One could argue that in knockout animals, the expression of muscarinic receptors is altered (for instance increased) and that under these conditions additional effector systems might be recruited. However, the density and affinity of M2-muscarinic receptors in our Gi2α knockout mice have been shown to be unaltered in comparison with wild-type controls (Chen et al., 2001). Furthermore, in Gi2α knockout mice the expression of other G proteins, notably of Goα, was found to be unaltered (Rudolph et al., 1996). Hence, non-specific effects of the knockout of one G protein on the protein levels of other G proteins, which would compensate for this knockout, are an unlikely explanation for our findings. Likewise, in Goα knockout mice the expression of Gi2α and Gi3α was unchanged (Valenzuela et al., 1997). Interestingly, while in these mice the effects of carbachol on isoprenaline stimulated L-type Ca2+ currents in ventricular myocytes were abolished, the effects of carbachol on the IK,Ach in isolated atrial cells from knockout and wild-type mice were identical (Valenzuela et al., 1997). These electrophysiological findings are in line with our contractile studies in isolated atrial preparations. We extended these electrophysiological recordings to force measurements in left atrium and beating rate measurements in right atrium. Hence, our findings support the assumption that Goα does not affect IK,Ach channel function and thus contractility in the atrium (Valenzuela et al., 1997). It is tempting to speculate that, in a similar manner, Gi2α in the atrium does not open IK,Ach after carbachol stimulation. The activation of IK,Ach is only one putative mediator of inotropic and chronotropic effects of muscarinic receptor stimulation in atria. A most straightforward explanation for indirect (in the presence of isoprenaline) effects would be the reversal of cAMP increase by isoprenaline. Indeed, the inhibition of adenylyl cyclase seems to play a more dominant role in mediating the indirect effect of carbachol. As shown by Ray and MacLeod (1993), this is not the only mechanism and a cAMP-independent component, which can be blocked by the K+-channel blocker, 4-aminopyridine, does exist. Thus, the indirect negative inotropic effect of carbachol is most probably a combination of two mechanisms. In addition, the inhibition of L-type Ca2+ currents by carbachol might involve an activation of NO synthases via Goα (Ye et al., 1999). This NO-mediated mechanism of inhibition of L-type Ca2+ currents would be independent of any effects on adenylyl cyclase activity but at the same time dependent on intracellular cAMP concentrations. The proposed mechanism involves NO-dependent activation of a cGMP-activated phosphodiesterase with resulting degradation of cAMP.
Our data on isolated atria of Gi2α knock out mice are also in agreement with the findings of Duan et al. (2007). Inactivation of the Gαi2 gene had no effect on negative chronotropic responses of carbachol alone or in the presence of isoprenaline in working heart preparations. In contrast to our data, however, sensitivity to muscarinic stimulation by carbachol was decreased in mouse hearts lacking Goα (Duan et al., 2007). There are several possible explanations for this apparent discrepancy between the findings by Duan et al. (2007) and the data of the present study. First of all, our Goα knockout model is not identical with that used by Duan et al. (2007); for instance, different embryonic stem cells were used for the generation of knockout and genetic backgrounds of mice were likewise different. Apparently, this has also an important effect on phenotypic manifestations. Our mice show various disturbances during postnatal development, especially multiple neurological abnormalities leading to higher mortality, compared with the model used by Duan et al. (2007). Thus, one should be cautious in interpreting and comparing data from different animal models, even when they lack the same gene. Under this assumption, findings from one knockout model cannot be simply generalized. On the other hand, different experimental approaches might also explain the discrepancy between our data and findings by Duan et al. (2007). As already discussed above, both experimental procedures have their advantages and disadvantages. We suggest that interference from ventricles in the working heart configuration should also be taken into account as a possible reason for the different findings obtained in isolated atria, as in the present study.
These findings and assumptions are consistent with the view that carbachol acts in the atrium on force of contraction and beating rate by means of PTX-sensitive proteins other than Gi2α and Goα. These proteins would be expected to couple directly to IK,Ach and/or If in order to mediate the atrial functional responses of vagal stimulation. Interestingly, Gi2α has been shown to mediate muscarinic inhibition of adenylyl cyclase activity (Rudolph et al., 1996). As knockout of Gi2α did not affect the contractile function of carbachol in the atrium (this work), it is unlikely that the negative inotropic effect of carbachol is related (in the atrium) to a decrease in cAMP content, as assumed sometimes in the literature. While one cannot totally exclude the possibility that PTX acts in ventricle and atrium on proteins other than G proteins, this is an unlikely explanation for the present findings.
In summary, our data deepen our knowledge of the signal transduction pathway of vagal stimulation in the mammalian atrium and indicate that different G proteins mediate the effect of carbachol in ventricle compared with atrium.
Acknowledgments
This work was supported in part by the Intramural Research Program of the NIH to LB (Z01-ES-101643) and by the NIH grant to MJ (DK069771).
Glossary
Abbreviations:
- Gi2α
α subunit of Gi2 protein
- Goα
α subunit of Go protein
- PTX
Pertussis toxin
Conflict of interest
None.
References
- Ahmad Z, Green FJ, Subuhi HS, Watanabe AM. Autonomic regulation of type 1 protein phosphatase in cardiac muscle. J Biol Chem. 1989;264:3859–3863. [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 3rd Edition (2008 Edition) Br J Pharmacol. 2008;153(Suppl. 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belevych AE, Harvey RD. Muscarinic inhibitory and stimulatory regulation of the L-type Ca2+ current is not altered in cardiac ventricular myocytes from mice lacking endothelial nitric oxide synthase. J Physiol. 2000;528:279–289. doi: 10.1111/j.1469-7793.2000.00279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böhm M, Brückner R, Neumann J, Schmitz W, Scholz H, Starbatty J. Role of guanine nucleotide-binding protein in the regulation by adenosine of cardiac potassium conductance and force of contraction. Evaluation with pertussis toxin. Naunyn-Schmiedeberg's Arch Pharmacol. 1986;332:403–405. doi: 10.1007/BF00500095. [DOI] [PubMed] [Google Scholar]
- Böhm M, Brückner R, Neumann J, Nose M, Schmitz W, Scholz H. Adenosine inhibits the positive inotropic effect of 3-isobutyl-1-methylxanthine in papillary muscles without effect on cyclic AMP or cyclic GMP. Br J Pharmacol. 1988;93:729–738. doi: 10.1111/j.1476-5381.1988.tb11456.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyett MR, Kirby MS, Orchard CH, Roberts A. The negative inotropic effect of acetylcholine on ferret ventricular myocardium. J Physiol. 1988;404:613–635. doi: 10.1113/jphysiol.1988.sp017309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodde OE, Michel MC. Adrenergic and muscarinic receptors in the human heart. Pharmacol Rev. 1999;51:651–690. [PubMed] [Google Scholar]
- Brown HF, Denyer JC. Low-dose acetylcholine reduces calcium current in isolated sino-atrial node cells of rabbit. J Physiol. 1989;410:65P. [Google Scholar]
- Caulfield MP, Birdsall NJ. International Union of Pharmacology. XVII. Classification of muscarinic acetylcholine receptors. Pharmacol Rev. 1998;50:279–290. [PubMed] [Google Scholar]
- Chen F, Spicher K, Jiang M, Birnbaumer L, Wetzel GT. Lack of muscarinic regulation of Ca2+ channels in Gi2α gene knockout mouse hearts. Am J Physiol. 2001;280:H1989–H1995. doi: 10.1152/ajpheart.2001.280.5.H1989. [DOI] [PubMed] [Google Scholar]
- Dhein S, van Koppen CJ, Brodde OE. Muscarinic receptors in the mammalian heart. Pharmacol Res. 2001;44:161–182. doi: 10.1006/phrs.2001.0835. [DOI] [PubMed] [Google Scholar]
- DiFrancesco D, Tromba C. Muscarinic control of the hyperpolarization-activated current If in rabbit sino-atrial node myocytes. J Physiol. 1988;405:493–510. doi: 10.1113/jphysiol.1988.sp017344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrzynski H, Janvier NC, Leach R, Findlay JB, Boyett MR. Effects of ACh and adenosine mediated by Kir3.1 and Kir3.4 on ferret ventricular cells. Am J Physiol. 2002;283:H615–H630. doi: 10.1152/ajpheart.00130.2002. [DOI] [PubMed] [Google Scholar]
- Duan SZ, Christe M, Milstone DS, Mortensen RM. Go but not Gi2 or Gi3 is required for muscarinic regulation of heart rate and heart rate variability in mice. Biochem Biophys Res Comm. 2007;357:139–149. doi: 10.1016/j.bbrc.2007.03.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endoh M. Muscarinic regulation of Ca2+ signaling in mammalian atrial and ventricular myocardium. Eur J Pharmacol. 1999;375:177–196. doi: 10.1016/s0014-2999(99)00231-9. [DOI] [PubMed] [Google Scholar]
- Eschenhagen T. G proteins and the heart. Cell Biol Int. 1993;17:723–749. doi: 10.1006/cbir.1993.1135. [DOI] [PubMed] [Google Scholar]
- Fischmeister R, Hartzell HC. Cyclic AMP phosphodiesterases and Ca2+ current regulation in cardiac cells. Life Sci. 1991;48:2365–2376. doi: 10.1016/0024-3205(91)90369-m. [DOI] [PubMed] [Google Scholar]
- Fisher JT, Vincent SG, Gomeza J, Yamada M, Wess J. Loss of vagally mediated bradycardia and bronchoconstriction in mice lacking M2 or M3 muscarinic acetylcholine receptors. FASEB J. 2004;18:711–713. doi: 10.1096/fj.03-0648fje. [DOI] [PubMed] [Google Scholar]
- Gergs U, Boknik P, Schmitz W, Simm A, Silber RE, Neumann J. A positive inotropic effect of adenosine in the human heart. Naunyn-Schmiedeberg's Arch Pharmacol. 2009;379:533–540. doi: 10.1007/s00210-008-0374-8. [DOI] [PubMed] [Google Scholar]
- Gupta RC, Neumann J, Durant P, Watanabe AM. A1-adenosine-receptor mediated inhibition of isoproterenol-stimulated protein phosphorylation in ventricular myocytes. Evidence against a cyclic AMP-dependent effect. Circ Res. 1993;72:65–74. doi: 10.1161/01.res.72.1.65. [DOI] [PubMed] [Google Scholar]
- Gupta RC, Neumann J, Boknik P, Watanabe AM. M2-specific muscarinic cholinergic receptor-mediated inhibition of phosphorylation of cardiac regulatory proteins. Am J Physiol. 1994;266:H1138–H1144. doi: 10.1152/ajpheart.1994.266.3.H1138. [DOI] [PubMed] [Google Scholar]
- Gupta RC, Neumann J, Watanabe AM, Sabbah HN. Muscarinic-cholinoceptor mediated attenuation of phospholamban phosphorylation induced by inhibition of phosphodiesterase in ventricular cardiomyocytes: evidence against a cAMP-dependent effect. Exp Clin Cardiol. 1999;4:23–28. doi: 10.1023/a:1006899931151. [DOI] [PubMed] [Google Scholar]
- Han X, Kubota I, Feron O, Opel DJ, Arstall MA, Zhao YY, et al. Muscarinic cholinergic regulation of cardiac myocyte ICa-L is absent in mice with targeted disruption of endothelial nitric oxide synthase. Proc Natl Acad Sci USA. 1998;95:6510–6515. doi: 10.1073/pnas.95.11.6510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honjo H, Kodama I, Zang WJ, Boyett MR. Desensitization to acetylcholine in single sinoatrial node cells isolated from rabbit hearts. Am J Physiol. 1992;263:H1779–H1789. doi: 10.1152/ajpheart.1992.263.6.H1779. [DOI] [PubMed] [Google Scholar]
- Jiang M, Gold MS, Boulay G, Spicher K, Peyton M, Brabet P, et al. Multiple neurological abnormalities in mice deficient in the G protein Go. Proc Natl Acad Sci USA. 1998;95:3269–3274. doi: 10.1073/pnas.95.6.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhof P, Fabritz L, Fortmüller L, Matherne G, Lankford A, Baba HA, et al. Altered sinus nodal and atrioventricular nodal function in freely moving mice overexpressing the A1 adenosine receptor. Am J Physiol. 2003;285:H145–H153. doi: 10.1152/ajpheart.01036.2002. [DOI] [PubMed] [Google Scholar]
- Krejcí A, Tucek S. Quantitation of mRNAs for M1 to M5 subtypes of muscarinic receptors in rat heart and brain cortex. Mol Pharmacol. 2002;61:1267–1272. doi: 10.1124/mol.61.6.1267. [DOI] [PubMed] [Google Scholar]
- Lindemann JP, Watanabe AM. Muscarinic cholinergic inhibition of β-adrenergic stimulation of phospholamban phosphorylation and Ca2+ transport in guinea pig ventricles. J Biol Chem. 1985;260:13122–13129. [PubMed] [Google Scholar]
- Löffelholz K, Pappano AJ. The parasympathetic neuroeffector junction of the heart. Pharmacol Rev. 1985;37:1–24. [PubMed] [Google Scholar]
- Lohmann SM, Fischmeister R, Walter U. Signal transduction by cGMP in heart. Basic Res Cardiol. 1991;86:503–514. doi: 10.1007/BF02190700. [DOI] [PubMed] [Google Scholar]
- McIvor ME, Orchard CH, Lakatta EG. Dissociation of changes in apparent myofibrillar Ca2+sensitivity and twitch relaxation induced by adrenergic and cholinergic stimulation in isolated ferret cardiac muscle. J Gen Physiol. 1988;92:509–529. doi: 10.1085/jgp.92.4.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMorn SO, Harrison SM, Zang WJ, Yu XJ, Boyett MR. A direct negative inotropic effect of acetylcholine on rat ventricular myocytes. Am J Physiol. 1993;265:H1393–H1400. doi: 10.1152/ajpheart.1993.265.4.H1393. [DOI] [PubMed] [Google Scholar]
- Mitra R, Morad M. Two types of calcium channels in guinea pig ventricular myocytes. Proc Natl Acad Sci USA. 1986;83:5340–5344. doi: 10.1073/pnas.83.14.5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata K, Ye C, Jain M, Milstone DS, Liao R, Mortensen RM. Gi2α but not Gi3α is required for muscarinic inhibition of contractility and calcium currents in adult cardiomyocytes. Circ Res. 2000;87:903–909. doi: 10.1161/01.res.87.10.903. [DOI] [PubMed] [Google Scholar]
- Nakajima T, Wu S, Irisawa H, Giles W. Mechanism of acetylcholine-induced inhibition of Ca current in bullfrog atrial myocytes. J Gen Physiol. 1990;96:865–885. doi: 10.1085/jgp.96.4.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann J, Boknik P, Bodor GS, Jones LR, Schmitz W, Scholz H. Effects of adenosine receptor and muscarinic cholinergic receptor agonists on cardiac protein phosphorylation. Influence of pertussis toxin. J Pharmacol Exp Ther. 1994;269:1310–1318. [PubMed] [Google Scholar]
- Neumann J, Gupta RC, Jones LR, Bodor GS, Bartel S, Krause EG, et al. Interaction of β-adrenoceptor and adenosine receptor agonists on phosphorylation. Identification of target proteins in mammalian ventricles. J Mol Cell Cardiol. 1995a;27:1655–1667. doi: 10.1016/s0022-2828(95)90689-4. [DOI] [PubMed] [Google Scholar]
- Neumann J, Kaspareit G, Kirchhefer U, Scholz H. Sodium fluoride attenuates the negative inotropic effects of muscarinic M2 and adenosine receptor agonists. Eur J Pharmacol. 1995b;294:451–457. doi: 10.1016/0014-2999(95)00569-2. [DOI] [PubMed] [Google Scholar]
- Neumann J, Vahlensieck U, Boknik P, Linck B, Lüss H, Müller FU, et al. Functional studies in atrium overexpressing A1-adenosine receptors. Br J Pharmacol. 1999;128:1623–1629. doi: 10.1038/sj.bjp.0702963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann J, Boknik P, Matherne GP, Lankford A, Schmitz W. Pertussis toxin sensitive and insensitive effects of adenosine and carbachol in murine atria overexpressing A1-adenosine receptors. Br J Pharmacol. 2003;138:209–217. doi: 10.1038/sj.bjp.0705012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osaka T, Joyner RW, Kumar R. Postnatal decrease in muscarinic cholinergic influence on Ca2+ currents of rabbit ventricular cells. Am J Physiol. 1993;264:H1916–H1925. doi: 10.1152/ajpheart.1993.264.6.H1916. [DOI] [PubMed] [Google Scholar]
- Ray A, MacLeod KM. A pharmacological investigation of the contribution of muscarinic receptor-linked potassium channels to the reversal by carbachol of positive inotropic responses of rabbit left atrium to cyclic AMP-generating agents. J Pharmacol Exp Ther. 1993;266:1594–1601. [PubMed] [Google Scholar]
- Robishaw JD, Hansen CA. Structure and function of G proteins mediating signal transduction pathways in the heart. Alcohol Clin Exp Res. 1994;18:115–120. doi: 10.1111/j.1530-0277.1994.tb00890.x. [DOI] [PubMed] [Google Scholar]
- Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmembrane-spanning receptors and heart function. Nature. 2002;415:206–212. doi: 10.1038/415206a. [DOI] [PubMed] [Google Scholar]
- Rudolph U, Brabet P, Hasty P, Bradley A, Birnbaumer L. Disruption of the Gi2α locus in embryonic stem cells and mice: a modified hit and run strategy with detection by a PCR dependent on gap repair. Transgenic Res. 1993;2:345–355. doi: 10.1007/BF01976176. [DOI] [PubMed] [Google Scholar]
- Rudolph U, Spicher K, Birnbaumer L. Adenylyl cyclase inhibition and altered G protein subunit expression and ADP-ribosylation patterns in tissues and cells from Gi2α−/− mice. Proc Natl Acad Sci USA. 1996;93:3209–3214. doi: 10.1073/pnas.93.8.3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowell MO, Ye C, Ricupero DA, Hansen S, Quinn SJ, Vassilev PM, et al. Targeted inactivation of αi2 or αi3 disrupts activation of the cardiac muscarinic K+ channel, IK+Ach, in intact cells. Proc Natl Acad Sci USA. 1997;94:7921–7926. doi: 10.1073/pnas.94.15.7921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein B, Mende U, Neumann J, Schmitz W, Scholz H. Pertussis toxin unmasks stimulatory A2-adenosine receptors on ventricular cardiomyocytes. J Mol Cell Cardiol. 1993;25:655–659. doi: 10.1006/jmcc.1993.1078. [DOI] [PubMed] [Google Scholar]
- Stengel PW, Gomeza J, Wess J, Cohen ML. M2 and M4 receptor knockout mice: muscarinic receptor function in cardiac and smooth muscle in vitro. J Pharmacol Exp Ther. 2000;292:877–885. [PubMed] [Google Scholar]
- Thierfelder S, Hirche H, Benndorf K. Anoxia decreases the transient K+ outward current in isolated ventricular heart cells of the mouse. Pflugers Arch. 1994;427:547–549. doi: 10.1007/BF00374273. [DOI] [PubMed] [Google Scholar]
- Valenzuela D, Han X, Mende U, Fankhauser C, Mashimo H, Huang P, et al. Gαo is necessary for muscarinic regulation of Ca2+ channels in mouse heart. Proc Natl Acad Sci USA. 1997;94:1727–1732. doi: 10.1073/pnas.94.5.1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Han H, Zhang L, Shi H, Schram G, Nattel S, et al. Expression of multiple subtypes of muscarinic receptors and cellular distribution in the human heart. Mol Pharmacol. 2001;59:1029–1036. doi: 10.1124/mol.59.5.1029. [DOI] [PubMed] [Google Scholar]
- Yang ZK, Boyett MR, Janvier NC, McMorn SO, Shui Z, Karim F. Regional differences in the negative inotropic effect of acetylcholine within the canine ventricle. J Physiol. 1996;492:789–806. doi: 10.1113/jphysiol.1996.sp021346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye C, Sowell MO, Vassilev PM, Milstone DS, Mortensen RM. Gαi2, Gαi3 and Gαo are all required for normal muscarinic inhibition of the cardiac calcium channels in nodal/atrial-like cultured cells. J Mol Cell Cardiol. 1999;31:1771–1781. doi: 10.1006/jmcc.1999.1015. [DOI] [PubMed] [Google Scholar]
- Zhang H, Holden AV, Noble D, Boyett MR. Analysis of the chronotropic effect of acetylcholine on sinoatrial node cells. J Cardiovasc Electrophysiol. 2002;13:465–474. doi: 10.1046/j.1540-8167.2002.00465.x. [DOI] [PubMed] [Google Scholar]