

Abstract
Summary: Copy number variants (CNVs) contribute substantially to human genomic diversity, and development of accurate and efficient methods for CNV genotyping is a central problem in exploring human genotype–phenotype associations. SCIMMkit provides a robust, integrated implementation of three previously validated algorithms [SCIMM (SNP-Conditional Mixture Modeling), SCIMM-Search and SCOUT (SNP-Conditional OUTlier detection)] for targeted interrogation of CNVs using Illumina Infinium II and GoldenGate SNP assays. SCIMMkit is applicable to standardized genome-wide SNP arrays and customized multiplexed SNP panels, providing economy, efficiency and flexibility in experimental design.
Availability: Source code and documentation are available for noncommercial use at http://droog.gs.washington.edu/scimmkit.
Contact: troyz@u.washington.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Copy number variation (CNV) in the human genome contributes substantially to genomic diversity and disease etiology (Lupski, 2009; McCarroll, 2008). Use of genome-wide SNP genotype data to perform ab initio discovery of individual CNVs has provided valuable insight into the spectrum of human genomic variation (Itsara et al., 2009; Redon et al., 2006). However, with the development of larger catalogs of common variation (Kidd et al., 2008; McCarroll et al., 2008) and continuing discovery of rare variants with severe phenotypic effects (Sebat et al., 2008; Walsh et al., 2008), it is critical to efficiently genotype specific CNVs in large populations. Targeted detection strategies generally outperform ab initio detection strategies for this task (McCarroll, 2008). Therefore, we have developed SCIMMkit, a toolkit for targeted genotyping of CNVs using Illumina Infinium II and GoldenGate SNP assays.
SNP assays typically generate two measurements per site (‘A’ and ‘B’ allele fluorescence) forming the canonical genotype clusters ‘A/A’, ‘A/B’ and ‘B/B’ when visualized by scatterplot. Deletions of sequence result in decreased signal intensity (i.e. states ‘A/–’, ‘B/–’, ‘–/–’) (Fig. 1), and duplications result in increased signal intensity (i.e. states ‘AAA’ and ‘BBB’) and aberrant allelic ratio (i.e. states ‘AAB’ and ‘ABB’) (Supplementary Fig. S1). States corresponding to individual CNVs often fail to form distinct clusters due to dynamic range limitations; therefore, methods using multiple SNP probes per site are required for robust copy number inference (Cooper et al., 2008; Korn et al., 2008; Mefford et al., 2009).
Fig. 1.
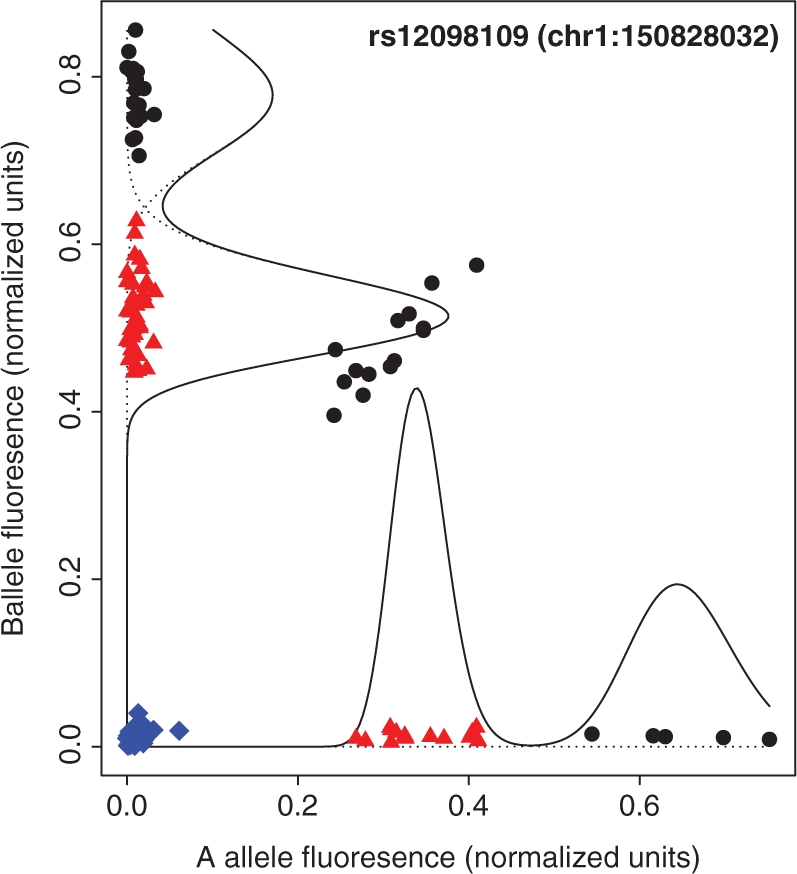
Fluorescence data for 125 HapMap samples (Cooper et al., 2008) at a single SNP probe (rs12098109) within a common deletion polymorphism identified as a susceptibility factor for psoriasis (de Cid et al., 2009). Copy number genotypes (blue diamonds, 0; red triangles, 1; black circles, 2) were computed by SCIMM using three SNP probes; superimposed curves describe components of the estimated mixture distribution.
2 DESCRIPTION OF FUNCTIONALITY
SCIMMkit provides three tools for targeted interrogation of CNVs, each of which assumes prior knowledge of the approximate location of each interrogated variant: SCIMM (SNP-Conditional Mixture Modeling), for genotyping polymorphic deletions (frequency exceeding 1%); SCIMM-Search, for automatically generating informative probe sets to be used by SCIMM; and SCOUT (SNP-Conditional OUTlier detection), for detecting rare deletion and duplication variants (frequency <1%). Each of these tools uses a statistical model of observed fluorescence data which contains, for each SNP probe, separate location parameters for each homozygous allelic state (i.e. ‘A/A’, ‘A/–’, ‘B/B’, ‘B/–’) and a single dispersion parameter shared by all homozygous allelic states.
SCIMM assigns diallelic insertion/deletion genotypes (i.e. copy number ‘0’, ‘1’ and ‘2’), using SNP calls and normalized fluorescence measurements for a set of n SNP probes hybridizing specifically to sequence spanning the deleted region (Cooper et al., 2008). Two rounds of mixture likelihood-based clustering are used: the first round uses intensity data to call samples near the origin as ‘0’, and the second round uses intensity data and supplied SNP genotypes to call remaining samples as ‘1’ or ‘2’, using a two-component, 2n-variate lognormal mixture model. Copy number for each sample is assumed to be constant for all probes in a set; accordingly, samples that are SNP heterozygous at any probe are assumed to have copy number 2 for the purposes of model fitting and copy number assignment. These statistical assumptions do not hold for SNP probes that hybridize non-specifically (Supplementary Fig. S2); such probes are rejected during probe set generation, below. SCIMM also generates a score for the probe set, defined as the difference of the Bayesian information criterion (BIC) value for the two-component model and the BIC value for the corresponding one-component model. Genotypes are reported only for sites with positive scores.
SCIMM-Search can be used to automatically generate informative probe sets in circumstances where the specificity assumptions of SCIMM may not be satisfied for all probes in the putatively deleted region. SCIMM-Search uses the BIC to select between alternate probe sets, and allows the investigator to specify constraints on consistency with reference genotypes, internal consistency of the probe set, probe spacing and dynamic range (Cooper et al., 2008).
SCOUT detects rare deletions and duplications at each targeted site by initially calculating per-probe scores for each sample, using a one-component SCIMM model extended to describe fluorescence data for SNP heterozygotes (Mefford et al., 2009). For SNP homozygotes, per-probe score is determined solely by intensity; for SNP heterozygotes, per-probe score is determined by intensity and deviation from 1:1 allelic ratio (specifically, by distance of the observed datum from the line connecting the origin to the center of the heterozygote cluster). Per-probe scores are approximately normally distributed, with samples at the center of each canonical SNP genotype cluster receiving a score of zero. The per-probe scores are combined additively to obtain per-site scores, which are then compared with an empirically determined threshold to generate a list of putative deletion and duplication events. Hemizygous and duplicated haplotypes for strongly scoring events are also reported, allowing inference of complex allelic states (e.g. ‘AAB’, Supplementary Fig. S1) and parental chromosome of origin (in cases where parental data are available). SCIMMkit also implements an initial SCOUT quality-control pass which rejects samples with a genome-wide excess of extreme per-probe scores, improving the positive predictive value of the per-site SCOUT scores generated for the remaining samples (Supplementary Fig. S1).
SCIMMkit requires as input a target file and one or more data files. Each line of the target file specifies a set of probes (with probe ID and coordinates) and an action associated with the probe set (i.e. SCIMM genotyping, SCIMM-Search probe set generation or SCOUT scoring). Input is supplied in Illumina BeadStudio genotype report format (or similar tabular format).
SCIMMkit generates two primary output files: a comma-delimited matrix with scores and numeric genotype codes (one row per sample and one column per target site), and a comma-delimited table with per-site summary information including genotype counts, probe set scores and SCIMM-Search generated probe sets. SCIMMkit can optionally generate scatterplots with superimposed SCIMM genotypes and mixture distribution curves in postscript format. SCIMMkit is implemented in PERL (used for command-line interpretation, input parsing and data consolidation) and R (used for numerically intensive tasks), and has been tested on Apple Macintosh OS X, Linux and Microsoft Windows platforms.
3 DISCUSSION
SCIMM and SCOUT use a common statistical model to facilitate distinct applications. SCIMM genotypes polymorphic deletions by estimating the location of each genotype cluster (‘–/–’, ‘A/–’, ‘B/–’, ‘A/A’, ‘A/B’, ‘B/B’). SCOUT detects rare deletion and duplication variants by analyzing the location of each sample relative to the canonical SNP genotype clusters (‘A/A’, ‘A/B’, ‘B/B’), avoiding estimation of location parameters for rare allelic states (e.g. deletion states ‘A/–’, ‘B/–’ and duplication states ‘AAB’, ‘ABB’).
SNP-based genome-wide association studies have generated a wealth of resources for retrospective analysis of CNV (Itsara et al. 2009). The first step in analyzing polymorphic variation in such data is identification of CNVs that can be accurately genotyped. To generate a database of polymorphic deletion sites and validated copy number-informative probe sets, we used SCIMM-Search to analyze data generated by the Illumina 1M-DuoV3 array for 269 HapMap samples. We compared the resulting SCIMM-generated diallelic deletion genotypes with previously published genotypes generated by BirdSuite software using the Affymetrix SNP 6.0 array (McCarroll et al., 2008). SCIMM produced diallelic deletion genotypes for 113 common (sample allele frequency at least 5%) autosomal deletions (84% of which have per-site concordance to BirdSuite genotypes exceeding 99%), 392 autosomal deletions of lower frequency (88.5% of which have genotype concordance exceeding 99% and positive predictive value for deletion status exceeding 80%) and 6 X-linked diallelic deletions (all of which have concordance exceeding 98.5%). These concordance rates are consistent with earlier analyses using independently generated reference genotypes (Cooper et al., 2008). The resulting list of highly concordant sites and corresponding Illumina 1M-DuoV3 probe sets produced by SCIMM-Search are provided on the SCIMMkit web site for genotyping polymorphic deletions in other genome-wide datasets. (See Supplementary Material for details).
Detection of highly pathogenic CNVs presents a distinct challenge: individually, such variants tend to be rare (frequency <1%) in affected individuals and very rare or completely absent in control populations; thus, definitively establishing a difference in allele frequency between cases and controls requires analysis of a large number (many thousands) of samples (International Schizophrenia Consortium, 2009). To assess the feasibility of large-scale targeted detection studies, SCOUT was recently used in conjunction with a customized Illumina BeadXpress assay to genotype deletions and duplications at 69 non-allelic homologous recombination hotspots in 1005 individuals with unexplained intellectual disability (ID). SCOUT correctly detected 48 rare deletion and duplication events, including 22 events known to be pathogenic, with only seven false positives (score threshold |6|, events validated by oligo-array CGH) (Mefford et al., 2009). Although, SCOUT does not explicitly include batch effects in its statistical model, the robustness of its model-fitting procedure at small sample sizes allows known batch effects to be remedied by independent scoring of batches. In the ID study above, each 96-well plate was analyzed independently to provide robustness against plate-to-plate variation in signal intensity and dynamic range.
We anticipate that future studies of association between CNV and phenotype will follow a model similar to SNP-based studies: an ab initio discovery stage (often in a population enriched for the phenotype of interest), an initial phenotypic association testing stage and a validation stage where the strongest associations are tested in a much larger population. SCIMMkit allows efficient and accurate detection of CNVs in the latter two stages of this model, facilitating further exploration of the link between CNV and human phenotypic variation.
Funding: T.Z. and D.A.N. acknowledge support from NIH grants HL66682 and HL66642. G.M.C. is supported by a Merck, Jane Coffin Childs Memorial Fund Fellowship. E.E.E. acknowledges support from NIH grant HG004120 and HD065285. E.E.E. is an investigator of the Howard Hughes Medical Institute.
Conflict of Interest: EEE is a SAB member of Pacific Biosciences.
Supplementary Material
REFERENCES
- Cooper GM, et al. Systematic assessment of copy number variant detection via genome-wide SNP genotyping. Nat. Genet. 2008;40:1199–1203. doi: 10.1038/ng.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Cid R, et al. Deletion of the late cornified envelope LCE3B and LCE3C genes as a susceptibility factor for psoriasis. Nat. Genet. 2009;41:211–215. doi: 10.1038/ng.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Schizophrenia Consortium. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2009;455:237–241. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itsara A, et al. Population analysis of large copy number variants and hotspots of human genetic disease. Am. J. Hum. Genet. 2009;84:148–161. doi: 10.1016/j.ajhg.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd JM, et al. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;453:56–64. doi: 10.1038/nature06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn JM, et al. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat. Genet. 2008;40:1253–1260. doi: 10.1038/ng.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR. Genomic disorders ten years on. Genome Med. 2009;1:42. doi: 10.1186/gm42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarroll SA. Extending genome-wide association studies to copy-number variation. Hum. Mol. Genet. 2008;17:R135–R142. doi: 10.1093/hmg/ddn282. [DOI] [PubMed] [Google Scholar]
- McCarroll SA, et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat. Genet. 2008;40:1166–1174. doi: 10.1038/ng.238. [DOI] [PubMed] [Google Scholar]
- Mefford HC, et al. A method for rapid, targeted CNV genotyping identifies rare variants associated with neurocognitive disease. Genome Res. 2009;19:1579–1585. doi: 10.1101/gr.094987.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redon R, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, et al. Strong association of de novo copy number mutations with autism. Science. 2008;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh T, et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320:539–543. doi: 10.1126/science.1155174. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.