

Abstract
It is well known that average levels of population structure are higher on the X chromosome compared to autosomes in humans. However, there have been surprisingly few analyses on the spatial distribution of population structure along the X chromosome. With publicly available data from the HapMap Project and Perlegen Sciences, we show a strikingly punctuated pattern of X chromosome population structure. Specifically, 87% of X-linked HapMap SNPs within the top 1% of FST values cluster into five distinct loci. The largest of these regions spans 5.4 Mb and contains 66% of the most highly differentiated HapMap SNPs on the X chromosome. We demonstrate that the extreme clustering of highly differentiated SNPs on the X chromosome is not an artifact of ascertainment bias, nor is it specific to the populations genotyped in the HapMap Project. Rather, additional analyses and resequencing data suggest that these five regions have been substrates of recent and strong adaptive evolution. Finally, we discuss the implications that patterns of X-linked population structure have on the evolutionary history of African populations.
Introduction
Estimates of human population structure based on X-linked SNPs tend to be more extreme than those based on autosomal markers.1–4 There are at least two reasons for this observation: (1) the X chromosome has a smaller effective population size than the autosomes, which increases the rate of genetic drift for X-linked loci relative to autosomal loci, and (2) males are hemizygous for the X chromosome, which increases the likelihood that local adaptation will produce higher levels of differentiation between geographically isolated populations at X-linked loci, as compared to loci on the autosomes.5,6 More controversially, differences in migration patterns, reproductive success, or generation times of males and females may contribute to the increased population structure associated with X-linked markers. Such parameters themselves are likely to vary between populations, making it difficult to reach definitive conclusions about the relative importance of various forces on the population genetics of the human X chromosome. One reflection of this difficulty is from two recent studies of X-linked variation.7,8 The studies reach opposite conclusions about a relatively simple question: is the X-to-autosome ratio of effective population sizes higher or lower than the value of 0.75 that is predicted by the simplest models? This ratio determines the relative rate of genetic drift for neutral X-linked loci as compared to autosomal loci, and thus provides an important baseline for demographic models of population structure on the X chromosome. Presumably, estimates of the ratio are sensitive to the markers and population samples in a particular study, as well as the method used to correct for site-specific differences in mutation rates.9
We describe analyses of publicly available and newly generated data that highlight the propensity of X-linked markers to display unusual patterns of population structure. Specifically, we found that in a genome-wide scan for markers with large allele-frequency differences between continental-scale populations, the most differentiated loci reside on the X chromosome. Furthermore, these highly differentiated markers cluster into a small number of X-linked regions, individually spanning hundreds of kilobase pairs to a few megabase pairs. We present detailed analyses for all five major clusters, which collectively span 9.6 Mb or approximately 6% of the length of the X chromosome. Finally, our results suggest that detailed analyses of the X chromosome offer a promising framework to constrain the parameter space of human migratory history, particularly those that led to the continental-scale population structure observed in current data sets. To this end, we discuss the implications of X-linked population structure for the genetic history of African populations.
Material and Methods
Public Data Sets
We used the following publicly available data sets in this study: HapMap Release 24, NCBI build 36,10 Perlegen Release 1,11 NCBI build 34, and Stanford University's HGDP-CEPH data.12,13 We also used primate sequences from the March 2006 assembly of the UCSC genome browser:14,15 UCSC versions hg18,16 panTro2,17 and rheMac2.18
The HapMap data consist of 210 unrelated individuals from four populations: (1) 60 Yoruba (YRI) individuals from Ibadan, Nigeria, (2) 60 CEPH (CEU) individuals with ancestry from northern and western Europe, (3) 45 Japanese (JPT) individuals from Tokyo, Japan, and (4) 45 Han Chinese (CHN) individuals from Beijing, China. In all analyses, we combined the JPT and CHB individuals into a single East Asian (ASN) sample. Our inferences are based on HapMap SNPs that were genotyped in all three populations and variable in at least one of them. The Perlegen data consist of 71 unrelated individuals from three populations: 23 African-Americans, 24 European-Americans, and 24 Han Chinese from Los Angeles, CA. The HGDP-CEPH data consist of 940 unrelated individuals from 52 worldwide populations.
We inferred ancestral states by comparing the human alleles to orthologous SNPs in chimpanzee and rhesus macaque sequences, available in the March 2006 assembly of the UCSC genome browser. We considered an allele to be ancestral if it matched the chimpanzee allele, or if that was missing, the rhesus macaque allele. We deemed a SNP to have an ambiguous ancestral state if the primate data conflicted or were missing altogether. For both the HapMap and Perlegen data sets, 11%–14% of the genotyped SNPs on a given chromosome that are variable in at least one population could not be assigned ancestral states, mainly because of conflicting primate alleles.
DNA Sequencing
In total, we sequenced DNA from 101 individuals representing nine geographically diverse populations with ancestry from Africa, the Middle East, Europe, Asia, and South America (see Table S1 available online). DNA samples were obtained from the Coriell Institute for Medical Research Cell Repositories in Camden, NJ. We resequenced approximately 7.2 kb in three loci spanning 221 kb of the gene IL1RAPL2. Primers were designed from the human reference genome16 via the Primer3 Web interface.19 Primer sequences are available upon request. We used standard PCR-based sequencing reactions from Applied Biosystem's Big Dye sequencing protocol on an ABI 3130xl machine. All sequences were assembled with Phred and Phrap20,21 and alignments were inspected for accuracy with Consed 14.0.22,23 Polymorphisms were identified with PolyPhred 4.22.24 All polymorphic sites were manually verified and confirmed by sequence on the opposite strand. To phase sequences from female samples, we used PHASE 2.1.1.25,26
Statistical Analysis
To quantify the magnitude of allele-frequency differences of SNPs genotyped in the HapMap and Perlegen data sets, we calculated a three-population, single-locus FST statistic for each SNP, as described previously.4 To examine levels of genetic variation in the HapMap and Perlegen data, we calculated normalized heterozygosity in nonoverlapping intervals of 100 kb.27 Suppose there are m SNPs in a given interval, where k = 1,2,…,m, and let pAi(k) be the frequency of allele A in population i at SNP k. The heterozygosity statistic for population i in that interval is equal to . For the resequencing data, we treated insertions/deletions in the same way as biallelic SNPs. We calculated standard neutrality tests of the site frequency spectrum for each resequenced locus, including Watterson's estimate of θ,28 nucleotide diversity,29 Tajima's D statistic,30 and Fay and Wu's H statistic.31 In addition, we also calculated a single window-based estimate of the two-population FST statistic to compare allele frequencies between African and non-African samples. To compute this version of FST, we summed the numerator and denominator across all SNPs in a given interval. We assessed statistical significance of the five clusters of highly differentiated SNPs by using coalescent simulations with the software ms.32 Simulations were performed with previously described demographic parameters, adjusted for the smaller effective population size of the X chromosome, that take into account major features of human history.33 Note, simply scaling the effective population by 0.75 in the X chromosome simulations failed to recapitulate average levels of X-linked FST, which is consistent with previous results that suggest a more extensive reduction in the effective population size of the X chromosome, perhaps because of male-biased migrations.8 Therefore, we increased the magnitude of the out-of-Africa bottleneck such that the simulated average FST matched that of the observed data. The command line argument for ms implementing this demographic model is available upon request. Finally, in evaluating the significance of the five highly differentiated clusters in the HapMap data, we approximated ascertainment bias by probabilistically selecting SNPs from the entire set of simulated data to match the minor allele frequency distribution observed in the empirical data.
Results
SNPs with Large Allele-Frequency Differences between Human Populations Are Enriched on the X Chromosome
We performed a scan of human genetic variation with publicly available data from the International HapMap Project.10 In our scan, we searched for SNPs with large allele-frequency differences between populations. To quantify the magnitude of allele-frequency differences among populations, we calculated a three-population, single-site FST statistic for each SNP. Table S2 summarizes the average FST for each chromosome, recapitulating the well-known pattern of higher average levels of structure on the X chromosome.1–4 To test whether SNPs with the most extreme differences in allele frequencies are enriched on the X chromosome, we set a stringent threshold of FST = 0.90. Of the nearly 3.2 million SNPs genotyped in the HapMap data set, there are 100 autosomal and 379 X-linked SNPs with FST ≥ 0.90. Thus, not only does the X chromosome exhibit higher average levels of differentiation, it is also significantly enriched (p < 10−8) for harboring the most extremely structured SNPs in the human genome.
The Spatial Distribution of X-Chromosome Population Structure
To explore the spatial distribution of population structure on the X chromosome, we analyzed FST values in detail for the 91,301 X-linked SNPs in the HapMap data set. The top panel of Figure 1A shows the distribution of FST along the X chromosome. As expected, there is considerable variation in single-locus FST statistics,34 and this obscures any underlying patterns in the data. We therefore counted the number of SNPs that fall in the 99th percentile of the empirical X-linked FST distribution in nonoverlapping 1 Mb bins across the X chromosome.
Figure 1.
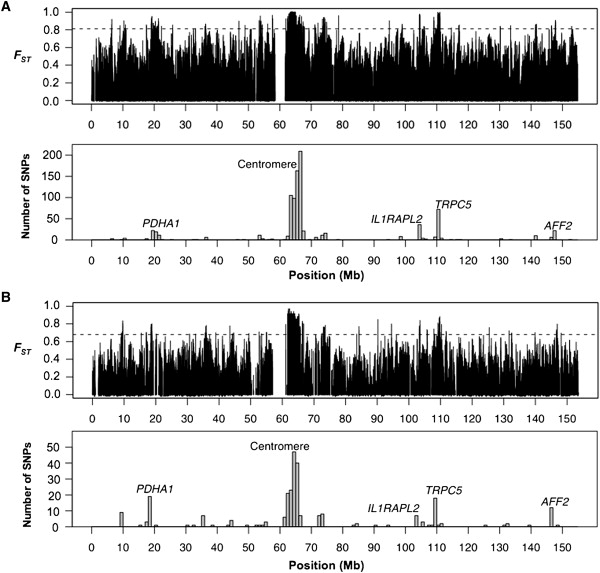
Spatial Distribution of FST on the X Chromosome
The top panels in (A) and (B) show the distribution of single locus estimates of FST in the HapMap and Perlegen data, respectively. The dashed horizontal lines denote the 99th percentile of the empirical FST distribution. The barplots in the bottom panels of (A) and (B) represent counts of high-FST SNPs that fall in nonoverlapping 1 Mb bins in the HapMap and Perlegen data, respectively. The five clusters discussed in the text are labeled.
Figure 1A reveals a striking pattern of FST along the X chromosome, in which the most differentiated HapMap SNPs cluster into several distinct loci. Specifically, 794 of the 914 high-FST SNPs (∼87%) cluster into five distinct regions (Figure 1A and Table 1). The most unusual region spans 5.4 Mb adjacent to the centromere, which contains 66% of the 914 high-FST X-linked SNPs. In addition to the large centromeric cluster, the other four clusters (Table 1) consist of (1) 52 SNPs spanning a 2.3 Mb region encompassing the PDHA1 gene (MIM 300502), (2) 36 SNPs spanning 1.2 Mb and falling in two adjacent introns of the IL1RAPL2 gene (MIM 300277), (3) 79 SNPs spanning a 1.5 Mb region that includes the gene TRPC5 (MIM 300334), and (4) 22 SNPs spanning 137 kb of the AFF2 gene (MIM 300806). For ease of presentation, we refer to these clusters as PDHA1, Centromeric, IL1RAPL2, TRPC5, and AFF2, although it is important to note that each cluster contains multiple genes (Table 1).
Table 1.
Highly Differentiated Loci on the X Chromosome
| Cluster Name | Positiona | Lengtha | Genesb | SNPsc | Percentd | Probability of Clustere |
|---|---|---|---|---|---|---|
| PDHA1 | 19.11–21.41 | 2.30 | 8 | 52 | 5.7 | 0.0139 |
| Centromere | 62.43–67.81 | 5.38 | 14 | 605 | 66.2 | <0.0001 |
| IL1RAPL2 | 104.41–104.64 | 0.23 | 1 | 36 | 3.9 | <0.0001 |
| TRPC5 | 109.56–111.08 | 1.52 | 9 | 79 | 8.6 | 0.0004 |
| AFF2 | 147.71–147.84 | 0.13 | 1 | 22 | 2.4 | 0.0020 |
Positions are expressed in Mb and are from HapMap Release 24 (NCBI build 36).
The number of Refseq genes in a given locus.
The number of high-FST SNPs within the boundaries of a given locus.
The number of high-FST SNPs in the cluster as a percentage of the total number of high-FST SNPs on the X chromosome (914).
The probability of observing as many or more high-FST SNPs under neutrality as determined by 104 coalescent simulations (see Material and Methods).
To determine how unusual the observed clusters of highly differentiated SNPs are under a neutral model, we performed coalescent simulations conditional on the observed number of segregating sites in each region. We performed 104 simulations for each region and asked how many simulation replicates contained as many or more highly differentiated SNPs relative to that observed in the empirical data. For all five regions, the probability of each cluster under a neutral model is small (maximum p = 0.0139 for the PDHA1 cluster; see Table 1).
Next, to assess whether the highly punctuated spatial distribution of high-FST SNPs is an artifact of ascertainment bias in the HapMap data, we performed a similar analysis with data from Perlegen BioSciences.11 Although the number of X-linked SNPs genotyped in the Perlegen data (26,937) is considerably smaller than the number genotyped in the HapMap data, the Perlegen SNPs were discovered in a more uniform manner, which attenuates potential artifacts induced by ascertainment bias.35 We restricted our analysis to class A Perlegen SNPs, which were ascertained by array-based genomic resequencing, to further minimize any biasing effects. Figure 1B summarizes results from this analysis. The Perlegen data recapitulates the major features we observed in the HapMap data, with the five labeled clusters accounting for 193 of the 271 high-FST Perlegen SNPs on the X chromosome (71%). We therefore conclude that the punctuated spatial distribution of X-linked population structure is a consequence of evolutionary history and not a statistical artifact of SNP ascertainment bias.
Genetic Variation in the Clusters of Highly Differentiated SNPs
Figure 2 displays male haplotypes in the HapMap data for representative regions spanning 1–2 Mb from each of the five clusters of high-FST SNPs. In all five loci, the non-African haplotypes exhibit dramatically reduced levels of genetic variation relative to the African haplotypes. Low levels of variation in the CEU samples extends for approximately 400 kb in the PDHA1 cluster, 2.8 Mb near the centromere, 300 kb in the IL1RAPL2 gene, 1.3 Mb in the TRPC5 cluster, and 500 kb at the AFF2 gene. For all of the loci, the pattern extends even farther in the ASN samples: 1.3 Mb in the PDHA1 cluster, 4.7 Mb near the centromere, 400 kb in the IL1RAPL2 gene, 2.2 Mb in the TRPC5 cluster, and 700 kb encompassing the AFF2 gene. Immediately outside these regions, levels of variation return to background levels, as quantified by the profiles of normalized heterozygosity shown in Figure 3. The pattern among the five loci suggests that the non-African haplotypes originally shared common signals of low variation, which were then partially recombined away in the CEU samples.
Figure 2.
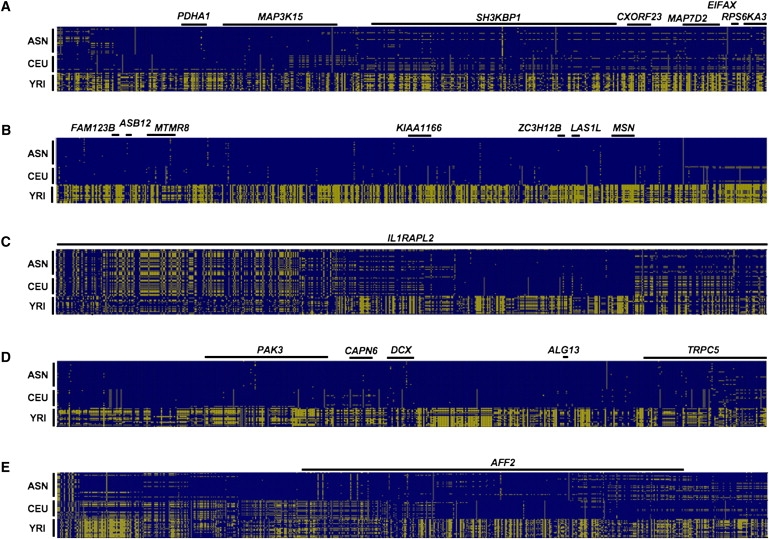
Visual Haplotypes of the Highly Differentiated Loci
Each panel displays haplotypes for all 105 unrelated males in the HapMap data. SNPs are listed in order of coordinate position across the top of each panel, and each row depicts the haplotype for a single HapMap sample. A yellow rectangle indicates the minor allele for a given SNP across the 105 samples, while a blue box indicates the common allele.
(A) A 1 Mb interval (coordinates 19–20 Mb) in the PDHA1 cluster.
(B) A 2 Mb interval (coordinates 63–65 Mb) in the centromeric cluster.
(C) A 1.2 Mb interval (coordinates 103.7–104.9 Mb) in the IL1RAPL2 cluster. Note, the 1.4 kb gene TEX13A is not shown.
(D) A 1 Mb interval (coordinates 110–111 Mb) in the TRPC5 cluster.
(E) A 1 Mb interval (coordinates 147–148 Mb) centered on the 500 kb gene AFF2. All genes in the interval are denoted by black rectangles at the top of each visual haplotype.
Figure 3.
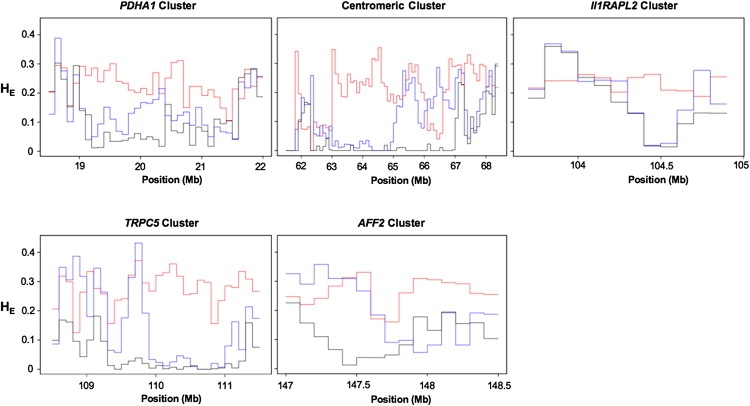
Levels of Genetic Variation in the Highly Differentiated Loci
Each panel displays profiles of normalized heterozygosity that were calculated with all HapMap SNPs in nonoverlapping, 100 kb bins. Red, blue, and black lines denote heterozygosity for the YRI, CEU, and ASN samples, respectively.
The extreme levels of population structure and reduced levels of genetic variation are consistent with the action of recent positive selection for each of the five clusters. Indeed, previous studies have found evidence of adaptive evolution in the PDHA1,36–39 TRPC5,40 AFF2,40 and Centromeric41 clusters (see Discussion). Below we describe more detailed population genetics analyses of the IL1RAPL2 and centromeric clusters that yield new insights into the evolutionary history of these loci.
The IL1RAPL2 Cluster
Within the IL1RAPL2 gene, low levels of variation in non-African haplotypes extends for 300–400 kb, encompassing the 230 kb cluster of high-FST SNPs and spanning two adjacent introns (Figures 2C and 3). To our knowledge, there have been no prior studies examining patterns of genetic variation at IL1RAPL2. We therefore sequenced three loci across 221 kb in this gene to better understand its evolutionary history. In total, we resequenced 7.2 kb in 101 individuals from nine geographically diverse populations, four from Africa and five from outside of Africa (Table S1). For each resequenced locus, we calculated standard neutrality tests of the site frequency spectrum for three sets of individuals: all 101 samples, only the 29 African samples, and only the 72 non-African samples. The resequencing data indicate a clear skew in the site frequency spectrum, as summarized in Table 2. One of the loci we sequenced has a Tajima's D value of −2.1 and a Fay & Wu's H of 7.8 across seven segregating sites in non-African samples; both of these test are significant (p < 0.05). Together, these values of D and H indicate an overall lack of variation and an excess of high-frequency derived alleles in the non-African samples, consistent with the action of recent positive selection.
Table 2.
Summary Statistics for the IL1RAPL2 Resequencing Data
|
Region |
|||
|---|---|---|---|
| I | II | III | |
| Positiona | 104,421,433–104,425,062 | 104,633,434–104,635,208 | 104,641,039–104,642,851 |
| Length (bp) | 3630 | 1776 | 1815 |
| FSTb | 0.674 | 0.809 | 0.679 |
| Segregating sitesc | |||
| All | 21 (4) | 13 (4) | 11 (4) |
| African | 16 | 7 | 10 |
| Non-African | 11 | 7 | 1 |
| θ per site × 10−3 | |||
| All | 1.03 | 1.31 | 1.08 |
| African | 1.03 | 0.92 | 1.29 |
| Non-African | 0.57 | 0.75 | 0.1 |
| π per site × 10−3 | |||
| All | 0.66 | 0.81 | 0.75 |
| African | 0.71 | 0.96 | 1.52 |
| Non-African | 0.25 | 0.07 | 0.01 |
| Tajima's D | |||
| All | −0.996 | −0.976 | −0.765 |
| African | −0.992 | 0.102 | 0.538 |
| Non-African | −1.469 | −2.086∗ | −1.015 |
| Fay and Wu's H | |||
| All | 0.084 | −7.285∗ | −0.147 |
| African | −0.738 | −1.105 | 0.322 |
| Non-African | −2.563 | −7.804∗ | 0.018 |
Statistical significance: ∗ indicates p < 0.05 determined from 104 coalescent simulations.
Positions are from the March 2006 assembly of the UCSC genome browser (NCBI Build 36.1).
Window-based estimate of the two-population FST statistic between African and non-African populations.
The number in parentheses is the number of segregating sites where the common allele (i.e., the allele having a frequency greater than 0.5) is different in the African samples versus the non-African samples.
The Centromeric Region
The centromeric cluster of high-FST SNPs is located adjacent to the centromere on the q-arm of the X chromosome. The clustering of highly differentiated SNPs near the X chromosome centromere does not appear to be a characteristic of centromeres in general, because analyses of loci near autosomal centromeres did not reveal similar clusters (data not shown). In the HapMap YRI sample, heterozygosity is at background levels across the entire cluster with the exception of two regions (Figure 3): (1) a 1 Mb region spanning 61.95–62.95 Mb and (2) a 600 kb region spanning 66.05–66.65 Mb. The 1 Mb region is closest to the centromere and, as such, contains few SNPs that are both genotyped in the HapMap data and variable in at least one population. In this region, an average of 14 such SNPs comprise the 100 kb windows shown in Figure 3. The 600 kb region is much more densely genotyped, containing an average of 45 genotyped SNPs per 100 kb window.
To investigate the 600 kb region in more detail, we analyzed the spatial distribution of SNPs with derived alleles at high frequencies in the YRI. Figure 4 shows the number of high-FST HapMap SNPs that fall in nonoverlapping, 100 kb bins across the centromeric locus. In the YRI sample, 111 highly differentiated SNPs have derived alleles at or near fixation in the YRI and are concentrated in a single location: the large red peak in the figure corresponds to 60 SNPs spanning 544 kb that coincide exactly with the region of low YRI heterozygosity (coordinates 66,036,841-66,580,944). This concentration of high-frequency derived alleles in the YRI is extremely rare in the HapMap data. In total, there are only 65 highly differentiated autosomal SNPs for which the derived alleles are at high frequencies in the YRI samples. Moreover, these 65 SNPs tend to be randomly distributed among autosomes, with only four regions containing two or more such SNPs. Therefore, the 544 kb centromeric region is characterized not only by low levels of variation in the YRI samples, but also an unusually high concentration of derived alleles nearly fixed in the YRI. To our knowledge, the only SNP previously known to have a similar geographic distribution, where the derived allele is at high frequency in Africa but low frequency outside of Africa, is the single-base mutation responsible for the Duffy-O blood type42,43 (MIM 110700).
Figure 4.
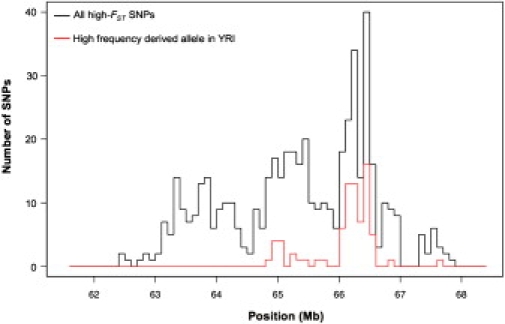
Distribution of YRI High-Frequency Derived Alleles in the Centromeric Cluster
The black lines depict the distribution of HapMap SNPs with (1) FST values in the 99th percentile on the X chromosome and (2) ancestral states that were unambiguously assigned. The red lines depict the subset of those SNPs with derived alleles at high frequencies in the YRI samples. The difference between the black and red curves thus represents all highly differentiated SNPs with ancestral alleles at high frequencies in the YRI.
Although it is possible that recurrent mutation obscures the ancestral states of some SNPs in the 544 kb region, it is highly unlikely to have affected all of them. Of the 60 high-FST HapMap SNPs that have high YRI-derived allele frequencies in this region, nearly all (51) are such that the chimpanzee and rhesus macaque alleles match the allele at high frequency in non-African samples; the remaining nine SNPs have missing data in the chimpanzee reference genome. Furthermore, only five high-FST SNPs in this region are located in the hypermutable cytosine position of CpG dinucleotides in the YRI samples.
To better understand worldwide patterns of genetic variation at the centromere cluster, we obtained genotype data from the HGDP-CEPH,12,13 which consists of 940 unrelated individuals44 from 52 worldwide populations. The HGDP-CEPH data set contains 16,400 SNPs on the X chromosome. Within the centromeric cluster of high-FST SNPs, 80 of the 605 highly differentiated HapMap SNPs were also genotyped in HGDP-CEPH. Notably, 19 of the 80 are located within the 600 kb region of low YRI heterozygosity and have derived alleles at high frequencies in all populations from sub-Saharan Africa, not simply the Yoruba. Figure 5 shows the global distribution of allele frequencies in HGDP-CEPH populations for 1 of the 19 SNPs. The pattern is striking: the derived allele is nearly fixed in populations across sub-Saharan Africa but exists only at low frequencies outside of Africa, and nearly identical patterns are observed with the other 18 SNPs (data not shown). Thus, the pattern of genetic variation in this region is not specific to the populations genotyped in the HapMap Project. Moreover, the pattern cannot be explained by gene flow from recent African migrations, such as the expansion of Bantu agriculturalists from western Africa into eastern and southern Africa within the past 3000–4000 years.45–47 The HGDP-CEPH populations from sub-Saharan Africa represent both Bantu and non-Bantu cultures, and three are highly diverse hunter-gather populations.46,47 These data, combined with previous analyses of this region (see Discussion), strongly suggest that the centromeric cluster has been subject to independent selective events in African and non-African populations.
Figure 5.
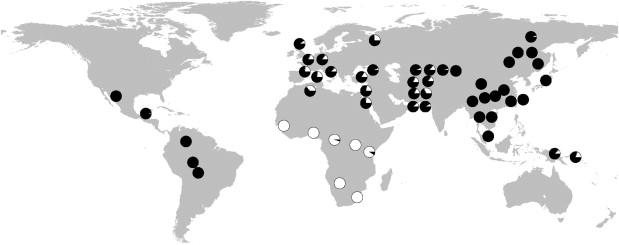
Allele Frequencies for SNP rs1511061 in the HGDP-CEPH Populations
Allele frequencies are indicated by pie charts. Derived and ancestral alleles are shown in white and black, respectively.
Discussion
Patterns of Population Structure on the X Chromosome
Highly differentiated X-linked SNPs cluster dramatically into discrete blocks that individually span hundreds of kilobases and collectively encompass nearly 6% of the X chromosome. The punctuated pattern of X-linked population structure is not simply a reflection of the bias toward higher overall FST values on the X chromosome as compared to the autosomes. Because the X chromosome has a smaller effective population size relative to the autosomes, genetic drift is expected to be stronger, resulting in higher overall levels of population structure.1–4 As shown in Figure 6, average FST on the X is larger than average autosomal FST even in the absence of the most differentiated X-linked loci. The distribution of FST values for all X-linked SNPs is virtually identical to the distribution obtained when SNPs from highly differentiated regions are excluded (Figure 6). Similar results were obtained with class A SNPs from the Perlegen data set (data not shown). Thus, because recent attempts to infer human demographic history from X-linked SNPs7,8 depend on the shift in mass of the FST distribution between X-linked and autosomal SNPs, they should be relatively robust to the clusters of highly differentiated SNPs that we describe here.
Figure 6.
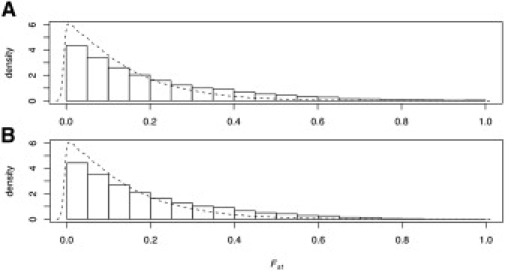
FST Probability Densities for X-Linked HapMap SNPs
Each histogram is a probability density for X-linked SNPs that are variable in at least one HapMap population. The corresponding probability density for autosomal SNPs, displayed in both panels by the dotted line, is provided for comparison.
(A) Empirical density for all 91,301 X-linked HapMap SNPs.
(B) Empirical density for 85,883 X-linked SNPs that are not located within highly differentiated regions of the X chromosome.
Positive Selection Results in Clustering of Highly Differentiated X-Linked SNPs
Although current data sets involve far too sparse a sampling of genetic variation to identify specific targets of selection, there is corroborating evidence that positive selection has acted on genes within four of the five clusters we identified: the PDHA136–39 and TRPC540 clusters, the AFF2 gene,40 and the centromeric region.41 Here, we will focus on discussing the novel insights our analysis has revealed for the evolutionary history of IL1RAPL2, AFF2, and the centromeric region.
Patterns of genetic variation between populations and resequencing data within the IL1RAPL2 gene suggest that it underwent a selective sweep in non-African populations. The IL1RAPL2 gene is a member of the interleukin-1 family of genes,48–50 although its exact function in the context of the interleukin-1 signaling pathways remains largely uncharacterized. The cluster of high-FST SNPs localizes to the sixth exon and noncoding sequence from its two adjacent introns. According to the InterPro database,51 the IL1RAPL2 protein contains three consecutive immunoglobulin domains, and the link between the second and third immunoglobulin domain corresponds exactly to exon 6. Thus, exon 6 is a strong candidate for being the target of selection, although without additional resequencing of the surrounding introns, it is not possible to rule out selection on other nearby sequences.
Although natural selection appears to have affected AFF2 as well, the patterns of variation in this gene are slightly more complex. AFF2 is an RNA-binding protein that causes FRAXE syndrome (MIM 309548), a rare form of mental retardation, when transcription is silenced.52–54 In an analysis of ten X-linked genes implicated in mental retardation, Kitano et al.55 found the number of nonsynonymous polymorphisms in AFF2 to be twice the number of synonymous polymorphisms; the results are suggestive of adaptive evolution but are not statistically significant in the study. Based on extended haplotype homozygosity, however, Sabeti et al.40 identified a region within the AFF2 gene that has significant evidence for selection in the HapMap ASN samples. The region is contained within the first and largest of the gene's 20 introns and overlaps the signal of low variation we found in the ASN sample (Figure 3). Interestingly, however, the region harboring the LD-based signature of selection does not coincide with the cluster of high-FST SNPs, which spans the fourth through ninth exons of the gene along with flanking intronic sequence. It is also not coincident with the signal of low variation in the CEU samples, which extends for 500 kb and encompasses the last 18 exons of AFF2, but not the first 3. Whereas the cluster of high-FST SNPs overlaps the region of low variation shared by both the CEU and ASN samples, the LD-based signal is located upstream of this region and occurs only in the ASN. To explain these observations, we hypothesize that LD-based statistics may imprecisely localize signatures of selection in cases where a sweep is at or nearing completion as it loses power in regions of low nucleotide variation,56 which is expected to be most pronounced around the specific adaptive allele.
Finally, the region near the X chromosome centromere is the largest and most complex of the five loci we analyzed. Indeed, our analysis of this locus points to a selective history that is more complicated than is typically assumed.57 The patterns of genetic variation near the centromere suggest that there may have been multiple selective events at this locus: one within Africa and one outside of Africa. The 2.8 Mb region of low variation shared by the CEU and ASN samples (Figure 3) does not overlap the 544 kb region containing the high density of SNPs with derived alleles nearly fixed in the YRI samples. The hypothesis of natural selection in both African and non-African populations is supported by previous studies of the centromeric locus. Specifically, Nachman et al.41 resequenced 4.6 kb from the MSN (MIM 309845) gene and 4.7 kb from the ALAS2 (MIM 301300) gene in a global sampling of 41 males. The two genes are situated nearly 10 Mb apart on either side of the centromere; MSN is located in the cluster of high-FST SNPs. Within their African samples, the authors observed extremely low levels of variation and significant LD across the centromere. Within the non-African samples, they observed low nucleotide diversity, long stretches of LD, and a skew in the site frequency spectrum toward rare variants. In both populations, the patterns of variation could not be explained by simple demographic models and were consistent with natural selection. The signatures of selection clearly differed between the two populations, however.
The 2.8 Mb region overlapped by both the cluster of high-FST SNPs and low levels of genetic variation in the CEU and ASN samples contains nine genes. In contrast, the 544 kb region (characterized by both low heterozygosity and a high concentration of derived alleles nearly fixed in YRI samples) contains no genes. It is situated 47 kb upstream of the nearest gene, the androgen receptor (AR), and nearly 300 kb downstream of EDA2R (MIM 300276), which has a role in NF-κB signaling. Although there are plausible selective candidates throughout the centromeric locus, current data implicate only the region and not specific genes within it.
Implications for Human Population History
Our most dramatic finding is the existence of a large number of derived alleles within a 544 kb region that are nearly fixed in populations across sub-Saharan Africa but virtually absent outside of Africa. Many loci show the reciprocal pattern. For example, a derived nonsynonymous SNP in the SLC24A5 (MIM 609802) gene that contributes to light skin color in individuals of European ancestry exists at high frequencies outside of Africa but low frequencies within African populations.58 The cluster of high-frequency African-specific derived alleles that we describe here may place new constraints on human population history.
The modern Recent African Origin model for human evolution explains the high genetic variation in contemporary African populations, relative to genomic regions with sharply reduced variation in non-Africans, by presupposing that human migrations out of Africa involved strong founder effects.59 Hence, a combination of genetic drift and local adaptation can readily account for the existence of derived alleles at high frequencies in non-African populations but low frequencies within Africa. Much less is known about African population history, particularly in the past 50,000–100,000 years during which founders of contemporary non-African populations emigrated into Europe and Asia.46 Our results suggest that a single African population, ancestral to contemporary Africans, may have remained a relatively coherent and local entity long enough for natural selection to sweep the cluster of derived alleles we describe to near fixation. This process would have occurred either after the initial out-of-Africa migrations or, equally as plausible based on current data, in an African population different than the one from which these out-of-Africa migrations occurred. Under this model, the ancestral African population would necessarily have been large to account for both the levels of variation and substructure evident in contemporary African populations.47,60
The most compelling prior evidence for this kind of selective event in African populations is the single-base mutation associated with the Duffy-O blood type,43 which may reflect, at least in part, this same historical trajectory. The Duffy-O mutation exists at high frequencies in many populations from sub-Saharan Africa but low frequencies outside of Africa, and confers complete resistance to malaria caused by the parasite Plasmodium vivax. The Duffy-O mutation has been shown to be recurrent,61 however, so the patterns of genetic variation within African populations may not have been caused by a simple selective sweep on a single variant.42 In contrast, the X-linked cluster of derived alleles that we analyzed can only be the result of a single historical process.
An alternative model involves both natural selection and gene flow between structured ancestral populations in sub-Saharan Africa. We consider this model to be less plausible, however, because it requires the cluster of derived alleles to have arisen locally and then spread continentally under sustained selection across Africa's extraordinarily diverse topographic, biological, and social environments. Clearly, dense sampling of African populations and full resequencing of the 544 kb region will be needed to completely understand the evolutionary history of this locus and its implications on human prehistory.
Conclusions
By analyzing genotype data from both the International HapMap Project and Perlegen Biosciences, we discovered a punctuated distribution of population differentiation on the human X chromosome, and additional analyses suggest that patterns of X-linked variation are mediated in bulk by neutral forces such as genetic drift, but in the extremes by natural selection. As the deluge of next-generation sequencing data begins to accumulate, the analysis of completely resequenced X chromosomes will provide greater power and resolution for fine-scale mapping targets of adaptive evolution, and the comparison of X and autosomal patterns of genetic variation in the context of whole-genome resequencing data will be a powerful framework for testing hypotheses of human demographic and cultural history.7–9
Supplemental Data
Supplemental Data include two tables and can be found with this article online at http://www.cell.com/AJHG.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Coriell Institute for Medical Research Cell Repositories, http://coriell.undmj.edu
The Human Genome Diversity Panel cell line, http://www.cephb.fr/HGDP-CEPH-Panel
The International HapMap Project, http://www.hapmap.org
The InterPro Database, http://www.ebi.ac.uk/interpro
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Perlegen Sciences, Inc., http://genome.perlegen.com
SeattleSNPs software programs, http://pga.gs.washington.edu/software.html
Stanford University's HGDP-CEPH data set, http://shgc.stanford.edu/hgdp
The UCSC Genome Browser, http://genome.ucsc.edu
Acknowledgments
The authors would like to thank Shameek Biswas and Laura Scheinfeldt for assistance with the HGDP-CEPH analysis, as well as Joseph Felsenstein and Sarah Tishkoff for helpful comments and discussions. This work was supported by NIH grants 1R01GM076036-01 (J.M.A.) and P50HG02351 (M.V.O.) and a Sloan Fellowship in Computational Biology (J.M.A.).
References
- 1.Schaffner S.F. The X chromosome in population genetics. Nat. Rev. Genet. 2004;5:43–51. doi: 10.1038/nrg1247. [DOI] [PubMed] [Google Scholar]
- 2.Payseur B.A., Nachman M.W. Natural selection at linked sites in humans. Gene. 2002;300:31–42. doi: 10.1016/s0378-1119(02)00849-1. [DOI] [PubMed] [Google Scholar]
- 3.Vicoso B., Charlesworth B. Evolution on the X chromosome: Unusual patterns and processes. Nat. Rev. Genet. 2006;7:645–653. doi: 10.1038/nrg1914. [DOI] [PubMed] [Google Scholar]
- 4.Akey J.M., Zhang G., Zhang K., Jin L., Shriver M.D. Interrogating a high-density SNP map for signatures of natural selection. Genome Res. 2002;12:1805–1814. doi: 10.1101/gr.631202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu J., Wu C.-I. Weak selection revealed by the whole-genome comparison of the X chromosome and autosomes of human and chimpanzee. Proc. Natl. Acad. Sci. USA. 2005;102:4063–4067. doi: 10.1073/pnas.0500436102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nielsen R., Bustamante C., Clark A.G., Glanowski S., Sackton T.B., Hubisz M.J., Fledel-Alon A., Tanenbaum D.M., Civello D., White T.J. A scan for positively selected genes in the genomes of humans and chimpanzees. PLoS Biol. 2005;3:e170. doi: 10.1371/journal.pbio.0030170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hammer M.F., Mendez F.L., Cox M.P., Woerner A.E., Wall J.D. Sex-biased evolutionary forces shape genomic patterns of human diversity. PLoS Biol. 2008;4:e10000202. doi: 10.1371/journal.pgen.1000202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keinan A., Mullikin J.C., Patterson N., Reich D. Accelerated genetic drift on chromosome X during the human dispersal out of Africa. Nat. Genet. 2009;41:66–70. doi: 10.1038/ng.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bustamante C.D., Ramachandran S. Evaluating signatures of sex-specific processes in the human genome. Nat. Genet. 2009;41:66–70. doi: 10.1038/ng0109-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frazer K.A., Ballinger D.G., Cox D.R., Hinds D.A., Stuve L.L., Gibbs R.A., Belmont J.W., Boudreau A., Hardenbol P., Leal S.M., International HapMap Consortium A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hinds D.A., Stuve L.L., Nilsen G.B., Halperin E., Eskin E., Ballinger D.G., Frazer K.A., Cox D.R. Whole-genome patterns of common DNA variation in three human populations. Science. 2005;307:1072–1079. doi: 10.1126/science.1105436. [DOI] [PubMed] [Google Scholar]
- 12.Cann H.M., de Toma C., Cazes L., Legrand M.-F., Morel V., Piouffre L., Bodmer J., Bodmer W.F., Bonne-Tamir B., Cambon-Thomsen A. A human genome diversity cell line panel. Science. 2002;296:261–262. doi: 10.1126/science.296.5566.261b. [DOI] [PubMed] [Google Scholar]
- 13.Li J.Z., Absher D.M., Tang H., Southwick A.M., Casto A.M., Ramachandran S., Cann H.M., Barsh G.S., Feldman M., Cavalli-Sforza L.L., Myers R.M. Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008;319:1100–1104. doi: 10.1126/science.1153717. [DOI] [PubMed] [Google Scholar]
- 14.Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karolchik D., Hinrichs A.S., Furey T.S., Roskin K.M., Sugnet C.W., Haussler D., Kent W.J. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004;32(Database issue):D493–D496. doi: 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W., International Human Genome Sequencing Consortium Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 17.Chimpanzee Sequencing and Analysis Consortium Initial sequence of the chimpanzee genome and comparison with the human genome. Nature. 2005;437:69–87. doi: 10.1038/nature04072. [DOI] [PubMed] [Google Scholar]
- 18.Gibbs R.A., Rogers J., Katze M.G., Bumgarner R., Weinstock G.M., Mardis E.R., Remington K.A., Strausberg R.L., Venter J.C., Wilson R.K., Rhesus Macaque Genome Sequencing and Analysis Consortium Evolutionary and biomedical insights from the rhesus macaque genome. Science. 2007;316:222–234. doi: 10.1126/science.1139247. [DOI] [PubMed] [Google Scholar]
- 19.Rozen S., Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. In: Misener S., Krawetz S.A., editors. Bioinformatics Methods and Protocols: Methods in Molecular Biology. Humana Press; Totowa, NJ: 2000. pp. 365–386. [DOI] [PubMed] [Google Scholar]
- 20.Ewing B., Hillier L., Wendl M.C., Green P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998;8:175–185. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- 21.Ewing B., Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998;8:186–194. [PubMed] [Google Scholar]
- 22.Gordon D., Abajian C., Green P. Consed: A graphical tool for sequence finishing. Genome Res. 1998;8:195–202. doi: 10.1101/gr.8.3.195. [DOI] [PubMed] [Google Scholar]
- 23.Gordon D. Viewing and editing assembled sequences using Consed. In: Baxevanis A.D., editor. Current Protocols in Bioinformatics. John Wiley & Sons, Inc.; New York: 2003. unit 11.2. [DOI] [PubMed] [Google Scholar]
- 24.Stephens M., Sloan J.S., Robertson P.D., Scheet P., Nickerson D.A. Automating sequence-based detection and genotyping of SNPs from diploid samples. Nat. Genet. 2006;38:375–381. doi: 10.1038/ng1746. [DOI] [PubMed] [Google Scholar]
- 25.Stephens M., Smith N.J., Donnelly P. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stephens M., Scheet P. Accounting for decay of linkage disequilibrium in haplotype inference and missing-data imputation. Am. J. Hum. Genet. 2005;76:449–462. doi: 10.1086/428594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Watterson G.A. The homozygosity test of neutrality. Genetics. 1978;88:405–417. doi: 10.1093/genetics/88.2.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Watterson G.A. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 1975;7:256–276. doi: 10.1016/0040-5809(75)90020-9. [DOI] [PubMed] [Google Scholar]
- 29.Tajima F. Evolutionary relationship of DNA sequences in finite populations. Genetics. 1983;105:437–460. doi: 10.1093/genetics/105.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fay J.C., Wu C.-I. Hitchhiking under positive Darwinian selection. Genetics. 2000;155:1405–1413. doi: 10.1093/genetics/155.3.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hudson R.R. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics. 2002;18:337–338. doi: 10.1093/bioinformatics/18.2.337. [DOI] [PubMed] [Google Scholar]
- 33.Schaffner S.F., Foo C., Gabriel S., Reich D., Daly M.J., Altshuler D. Calibrating a coalescent simulation of human genome sequence variation. Genome Res. 2005;15:1576–1583. doi: 10.1101/gr.3709305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weir B.S., Cardon L.R., Anderson A.D., Nielsen D.M., Hill W.G. Measures of human population structure show heterogeneity among genomic regions. Genome Res. 2005;15:1468–1476. doi: 10.1101/gr.4398405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clark A.G., Hubisz M.J., Bustamante C.D., Williamson S.H., Nielsen R. Ascertainment bias in studies of human genome-wide polymorphism. Genome Res. 2005;15:1496–1502. doi: 10.1101/gr.4107905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harding R.M. More on the X files. Proc. Natl. Acad. Sci. USA. 1999;96:2582–2584. doi: 10.1073/pnas.96.6.2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harris E.E., Hey J. X chromosome evidence for ancient human histories. Proc. Natl. Acad. Sci. USA. 1999;96:3320–3324. doi: 10.1073/pnas.96.6.3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu N., Li W.-H. No fixed nucleotide difference between Africans and Non-Africans at the pyruvate dehydrogenase E1 α-subunit locus. Genetics. 2000;155:1481–1483. doi: 10.1093/genetics/155.3.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hammer M.F., Garrigan D., Wood E., Wilder J.A., Mobasher Z., Bigham A., Krenz J.G., Nachman M.W. Heterogeneous patterns of variation among multiple human x-linked loci: The possible role of diversity-reducing selection in non-africans. Genetics. 2004;167:1841–1853. doi: 10.1534/genetics.103.025361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sabeti P.C., Varilly P., Fry B., Lohmueller J., Hostetter E., Cotsapas C., Xie X., Byrne E.H., McCarroll S.A., Gaudet R., International HapMap Consortium Genome-wide detection and characterization of positive selection in human populations. Nature. 2007;449:913–918. doi: 10.1038/nature06250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nachman M.W., D'Agostino S.L., Tillquist C.R., Mobasher Z., Hammer M.F. Nucleotide variation at Msn and Alas2, two genes flanking the centromere of the X chromosome in humans. Genetics. 2004;167:423–437. doi: 10.1534/genetics.167.1.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hamblin M.T., Di Rienzo A. Detection of the signature of natural selection in humans: evidence from the Duffy blood group locus. Am. J. Hum. Genet. 2000;66:1669–1679. doi: 10.1086/302879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hamblin M.T., Thompson E.E., Di Rienzo A. Complex signatures of natural selection at the Duffy blood group locus. Am. J. Hum. Genet. 2002;70:369–383. doi: 10.1086/338628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rosenberg N.A. Standardized subsets of the HGDP-CEPH Human Genome Diversity Cell Line Panel, accounting for atypical and duplicated samples and pairs of close relatives. Ann. Hum. Genet. 2006;70:841–847. doi: 10.1111/j.1469-1809.2006.00285.x. [DOI] [PubMed] [Google Scholar]
- 45.Tishkoff S.A., Williams S.M. Genetic analysis of African populations: Human evolution and complex disease. Nat. Rev. Genet. 2002;3:611–621. doi: 10.1038/nrg865. [DOI] [PubMed] [Google Scholar]
- 46.Campbell M.C., Tishkoff S.A. African genetic diversity: Implications for human demographic history, modern human origins, and complex disease mapping. Annu. Rev. Genomics Hum. Genet. 2008;9:403–433. doi: 10.1146/annurev.genom.9.081307.164258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tishkoff S.A., Reed F.A., Friedlaender F.R., Ehret C., Ranciaro A., Froment A., Hirbo J.B., Awomoyi A.A., Bodo J.-M., Doumbo O. The genetic structure and history of Africans and African Americans. Science. 2009;324:1035–1044. doi: 10.1126/science.1172257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jin H., Gardner R.J., Viswesvaraiah R., Muntoni F., Roberts R.G. Two novel members of the interleukin-1 receptor gene family, one deleted in Xp22.1-Xp21.3 mental retardation. Eur. J. Hum. Genet. 2000;8:87–94. doi: 10.1038/sj.ejhg.5200415. [DOI] [PubMed] [Google Scholar]
- 49.Born T.L., Smith D.E., Garka K.E., Renshaw B.R., Bertles J.S., Sims J.E. Identification and characterization of two members of a novel class of the interleukin-1 receptor (IL-1R) family. Delineation of a new class of IL-1R-related proteins based on signaling. J. Biol. Chem. 2000;275:29946–29954. doi: 10.1074/jbc.M004077200. [DOI] [PubMed] [Google Scholar]
- 50.Sana T.R., Debets R., Timans J.C., Bazan J.F., Kastelein R.A. Computational identification, cloning, and characterization of IL-1R9, a novel interleukin-1 receptor-like gene encoded over an unusually large interval of human chromosome Xq22.2-q22.3. Genomics. 2000;69:252–262. doi: 10.1006/geno.2000.6328. [DOI] [PubMed] [Google Scholar]
- 51.Mulder N.J., Apweiler R., Attwood T.K., Bairoch A., Bateman A., Binns D., Bork P., Buillard V., Cerutti L., Copley R. New developments in the InterPro database. Nucleic Acids Res. 2007;35(Database issue):D224–D228. doi: 10.1093/nar/gkl841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gecz J. The FMR2 gene, FRAXE and non-specific X-linked mental retardation: Clinical and molecular aspects. Ann. Hum. Genet. 2000;64:95–106. doi: 10.1017/S0003480000007983. [DOI] [PubMed] [Google Scholar]
- 53.Bensaid M., Melko M., Bechara E.G., Davidovic L., Berretta A., Catania M.V., Gecz J., Lalli E., Bardoni B. FRAXE-associated mental retardation protein (FMR2) is an RNA-binding protein with high affinity for G-quartet RNA forming structure. Nucleic Acids Res. 2009;37:1269–1279. doi: 10.1093/nar/gkn1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hillman M.A., Gecz J. Fragile XE-associated familial mental retardation protein 2 (FMR2) acts as a potent transcription activator. J. Hum. Genet. 2001;46:251–259. doi: 10.1007/s100380170074. [DOI] [PubMed] [Google Scholar]
- 55.Kitano T., Schwarz C., Nickel B., Pääbo S. Gene diversity patterns at 10 X-chromosomal loci in humans and chimpanzees. Mol. Biol. Evol. 2003;20:1281–1289. doi: 10.1093/molbev/msg134. [DOI] [PubMed] [Google Scholar]
- 56.Voight B.F., Kudaravalli S., Wen X., Pritchard J.K. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Akey J.M. Constructing genomic maps of positive selection in humans: Where do we go from here? Genome Res. 2009;19:711–722. doi: 10.1101/gr.086652.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lamason R.L., Mohideen M.-A.P., Mest J.R., Wong A.C., Norton H.L., Aros M.C., Jurynec M.J., Mao X., Humphreville V.R., Humbert J.E. SLC24A5, a putative cation exchanger, affects pigmentation in zebrafish and humans. Science. 2005;310:1782–1786. doi: 10.1126/science.1116238. [DOI] [PubMed] [Google Scholar]
- 59.Reed F.A., Tishkoff S.A. African human diversity, origins and migrations. Curr. Opin. Genet. Dev. 2006;16:597–605. doi: 10.1016/j.gde.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 60.Fagundes N.J.R., Ray N., Beaumont M., Neuenschwander S., Salzano F.M., Bonatto S.L., Excoffier L. Statistical evaluation of alternative models of human evolution. Proc. Natl. Acad. Sci. USA. 2007;104:17614–17619. doi: 10.1073/pnas.0708280104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zimmerman P.A., Woolley I., Masinde G.L., Miller S.M., McNamara D.T., Hazlett F., Mgone C.S., Alpers M.P., Genton B., Boatin B.A., Kazura J.W. Emergence of FY∗A(null) in a Plasmodium vivax-endemic region of Papua New Guinea. Proc. Natl. Acad. Sci. USA. 1999;96:13973–13977. doi: 10.1073/pnas.96.24.13973. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.