

Abstract
Two configurationally stable carbon-based analogues of pyochelin have been prepared from Boc-pyroglutamic acid-tert-butyl ester in 11 and 13 steps. Introduction of the amino group was achieved by a highly diastereoselective electrophilic azidation reaction to afford novel bis-α-amino acid proline derivatives.
Pyochelin (1) is a phenolate siderophore isolated from Pseudomonas aeruginosa that enhances microbial growth by solublizing ferric iron and accelerating iron transport.1 Studies on P. aeruginosa iron uptake systems have focused largely on the recognition of pyochelin by bacterial outer membrane receptors.2 These efforts have benefited from the synthesis and evaluation of various pyochelin analogues, many of which chelate and transport iron(III) with similar efficiency compared to the natural product.3
Recently, it has been shown that the endogenous protein siderocalin (also lipocalin 2, NGAL) is able to bind and sequester iron as complexes with siderophores, thus starving pathogens of this required nutrient.4 While siderocalin tightly binds a number of small-molecule phenolate siderophores such as carboxymycobactins, enterochelin, and parabactin, it does not bind to pyochelin. We have proposed that the presence of a sterically demanding sulfur-containing thiazoline ring, which replaces the oxazoline ring present in the other phenolate siderophores, interferes with a conserved water-mediated hydrogen bond between these siderophores and siderocalin.4c,d In contrast, pyochelin does bind to chicken Ex-FABP, a protein with high sequence homology to human siderocalin in the binding calyx region.5 The fact that chicken Ex-FABP also shows affinity for a variable spectrum of microbial siderophores raises the possibility that these proteins are part of an antibacterial defense strategy employed across the phylum.
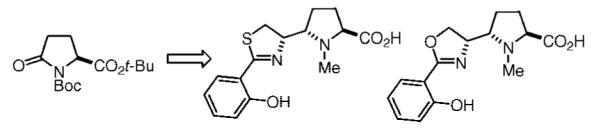
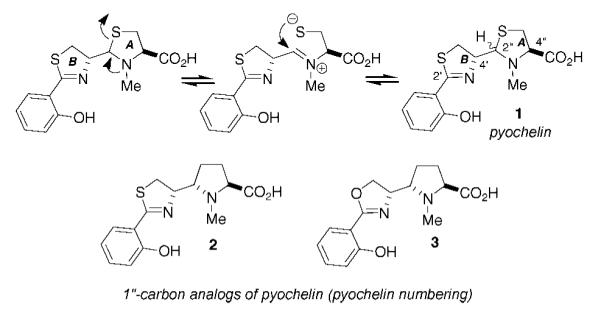
To parse the ligand recognition parameters of both siderocalin and chicken Ex-FABP, we sought to design and synthesize analogues of pyochelin that would demonstrate both protein and iron binding capabilities. Because pyochelin undergoes epimerization at the 2′′ position through the general mechanism shown in Figure 1, efforts to synthesize and isolate 1 in diastereomerically pure form have met with limited success.6 We envisioned this problem could be circumvented by incorporating a methylene unit in place of the ring A sulfur, a position with no established role in iron or siderocalin binding. In addition, we sought to target the oxazoline variant 3 to test our steric clash hypothesis through the design of an oxo analogue. These analogues would potentially provide useful information on the discreet stereochemical requirements for siderophore specific protein–ligand interactions. Here, we describe our efforts toward novel proline-based siderophores, resulting in a diastereoselective synthesis of amino acids 2 and 3.
Figure 1.
Pyochelin and configurationally stable proline-based analogues.
Our initial synthesis plan relied on intramolecular nitrene insertion as the key step for introduction of the ring B nitrogen. Toward this end, we prepared known proline diester 5,7 as an inseparable 16:1 trans:cis mixture via lithium triethylborohydride reduction and Horner–Wadsworth–Emmons olefination of (N-Boc)-tert-butyl pyroglutamate (4).8 Chemoselective ester reduction was followed by carbamate formation to give 6 in 64% yield after purification (Scheme 1). Unfortunately, rhodium(II)-catalyzed C–H insertion9 resulted in poor yields of the desired 5-membered cyclic carbamate. Attempts to employ silver(I)-catalyzed conditions, which have shown enhanced conversion in the case of some secondary C–H bond insertions,10 resulted in slightly improved albeit unsatisfactory yields.
Scheme 1.
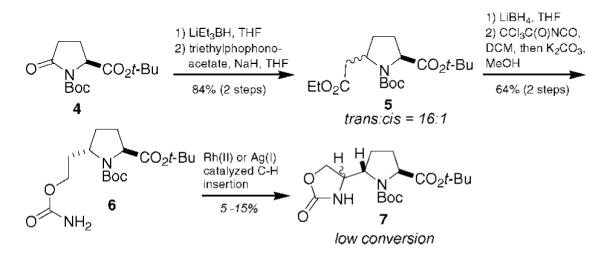
Attempted C–H Insertion Route
Given these results, we decided to explore potentially regio- and diastereoselective electrophilic azidation11 as an alternative strategy (Scheme 2). When 5 was treated with LHMDS and trisyl azide at −78 °C, followed by AcOH quench, we obtained a mixture of starting material, ring-opened starting material (9), and low yields of desired product 8, along with unidentified byproducts. We reasoned that the electron-withdrawing and sterically demanding Boc -group prevents reaction of the desired carbanion with the azide source. Anticipating the necessary introduction of an N-methyl group, we opted to replace the N-Boc group prior to azide transfer. Thus, acidic cleavage of the Boc group followed by treatment with anion exchange resin in MeOH and filtration gave the crude amine, which was then subjected to reductive alkylation to afford 10.
Scheme 2.
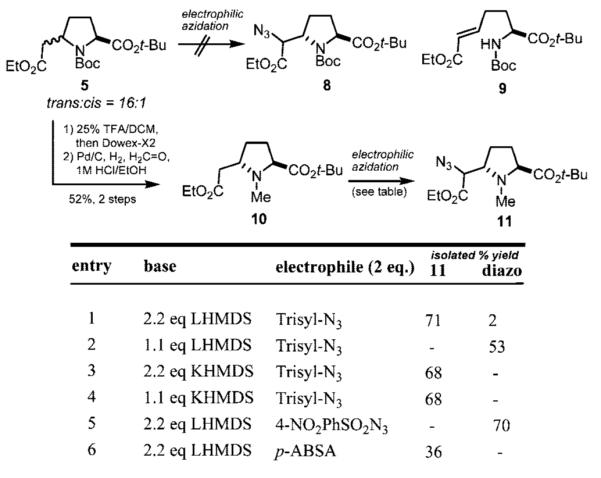
Electrophillic Azidation Reactions
When N-methyl proline 10 was subjected to the same azide transfer condition used for substrate 5, we observed a marked increase in conversion and yield of the desired azidoester. Optimal conditions utilized 2.2 equiv of LHMDS and 2.0 equiv of trisyl azide in THF with AcOH quench (entry 1). Reducing the amount of LHMDS to 1.1 equiv (entry 2) resulted in exclusive formation of the α-diazoester, as did the use of 4-nitrophenylsulfonyl azide as an electrophile (entry 5). The potassium enolate of 10 reacted to give only the desired azide product, irrespective of the amount of base used (entries 3 and 4).
Analysis of the 1H NMR of 11 indicated the presence of only one diastereomer (under optimized azidation conditions). Although the configuration of the newly formed stereocenter could not be determined at this stage, we considered the two possible transition states shown in Figure 2. Path A depicts the minimization of 1,3-allylic strain, followed by approach of trisyl azide from the Re-face of the enolate. Alternatively, a sixmembered chelation transition state may lead to axial approach of trisyl azide onto a low-energy half-chair conformer (Fürst–Plattner principle). Both of these models result in the formation of the anti product. On the basis of this, we anticipated that the newly formed α-amino acid center would possess an S configuration to match that of the 4′ position in pyochelin.12 This tentative assignment was later confirmed by X-ray diffraction of an amide derivative (vide infra).
Figure 2.
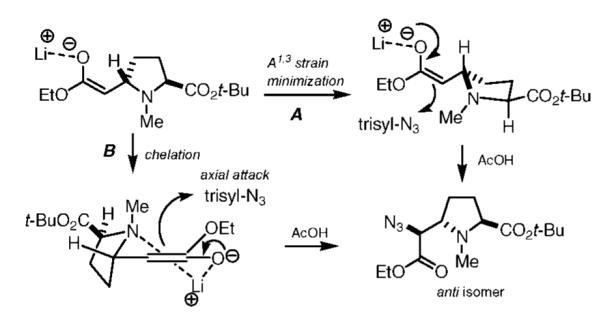
Proposed transition state models for the electrophilic azidation of 10.
Completion of the synthesis of pyochelin analogues 2 and 3 is depicted in Scheme 3. Thus, azide reduction of 11 and EDC-mediated coupling of the crude amine to 2-benzyloxybenzoic acid gave amide 12 in 93% yield. Reduction of the ethyl ester was followed by hydrogenolysis of the benzyl ether and dehydrative cyclization with the Burgess reagent13 to give oxazoline 15. The thiazoline ring was formed over two steps by thiolysis of the oxazoline ring with hydrogen sulfide in MeOH/NEt3, and subsequent dehydration again with the Burgess reagent.14
Scheme 3.
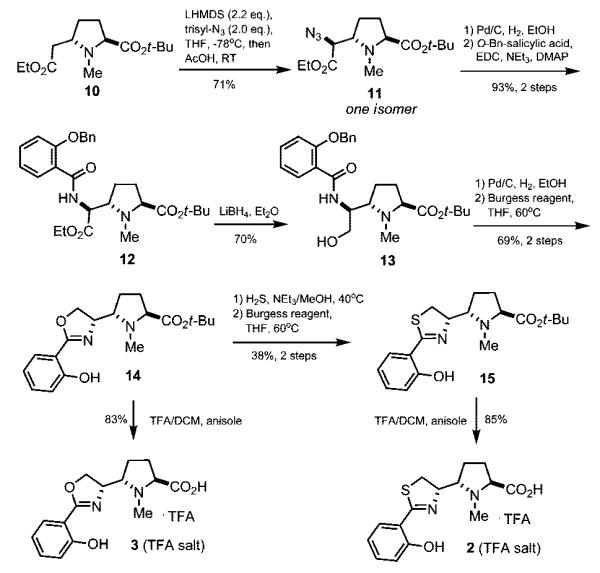
Synthesis of Carbapyochelins 2 and 3
Our efforts to carry out TFA-mediated deprotection of tert-butyl esters 14 and 15 (with anisole scavenger) were initially met with difficulties. The apparent sensitivity of the oxazoline and thiazoline rings in the presence of water and excess TFA resulted in appreciable amounts (>20%) of hydrated ring-opened products. This side reaction could be minimized by a careful workup procedure involving dilution of the reaction mixture with ethyl acetate, concentration in vacuo, and addition of a precooled diethyl ether/water mixture. Separation of the water layer and lyophilization then gave 2 and 3 in high yields without appreciable hydrolysis. RP-HPLC (C18) analysis of the crude amino acids showed each to have >95% purity following deprotection (see the Supporting Information).
To determine the configuration of the newly formed stereocenter of 11 we relied primarily on the synthesis of crystalline derivatives, since detailed 2D NMR studies gave ambiguous results. While some intermediates in Scheme 3 were solids, none of these compounds provided crystals of sufficient quality for X-ray diffraction. We were pleased to find that reduction of azide 11 followed by coupling to 2-methoxymethyloxybenzoic acid15 gave 84% yield of amide 16, which readily crystallized out of CH2Cl2/hexane. Single-crystal X-ray diffraction revealed an S configuration at the 1′ center (IUPAC numbering), thus confirming our earlier prediction.
In summary, we have described a route toward two configurationally stable analogues of pyochelin. We are currently investigating the ability of ferric complexes of 2 and 3 to bind siderocalin and chicken Ex-FABP. Studies are also underway to explore the scope of the highly diastereoselective electrophilic azidation for the synthesis of other functionalized proline chimeras with orthogonally protected N- and C-termini. To the best of our knowledge, this work represents the first stereoselective synthesis of bis-proline structures related to 11 (Scheme 4). This class of residues should find utility in a variety of peptidomimetic applications.
Scheme 4.
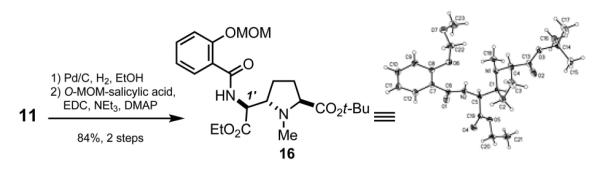
Synthesis and X-ray Structure of 16
Experimental Section
(2S,5S)-5-Ethoxycarbonylmethyl-1-methylpyrrolidine-2-carboxylic Acid tert-Butyl Ester (10)
A solution of 5 (9.20 g, 25.7 mmol) in 50 mL of 25% TFA/DCM was stirred at rt for 30 min, diluted with 100 mL of EtOAc, then evaporated under reduced pressure and dried under high vacuum to remove excess trifluoroacetic acid. The crude amine trifluoroacetate salt was taken up in 100 mL of EtOH and treated with 38 g of Dowex-X2 anion exchange resin (OH- form). The mixture was stirred gently until the pH of the solution reached 8–9. The resin was then removed by filtration and washed repeatedly with EtOH. Evaporation of filtrate gave the crude amine as a yellow oil (attempts to isolate the free amine by aqueous extraction were hampered by its water solubility). The amine was treated with formaldehyde (30% solution in EtOH, 41.8 mL, 515 mmol) and 1 M aq. HCl until a pH of 2 was obtained. A catalytic amount of 10% Pd/C was added and the flask was then purged and filled with an H2 atmosphere (balloon). The reaction mixture was stirred 18 h at rt and purged with a stream of Ar, then the catalyst was removed by filtration over celite with MeOH rinsing. The filtrate was concentrated, diluted with sat. aq. NaHCO3, and extracted with EtOAc. Drying over Na2SO4, filtration, and concentration under reduced pressure gave a crude 16:1 diastereomeric mixture of N-methylated amines which were separated by chromatography over silica gel (33% Et2O/hexanes as eluant). The trans isomer 10 was obtained as a pale yellow oil (3.63 g, 52% from 5). 1H NMR (300 MHz, CDCl3) δ 4.13 (q, J = 7.0 Hz, 2H), 3.53 (dd, J = 8.2, 2.3 Hz, 1H), 3.42 (septet, J = 4.4 Hz, 1H), 2.59 (dd, J = 14.4, 4.1 Hz, 1H), 2.43 (s, 3H), 2.30–2.04 (m, 3H), 1.86–1.76 (m, 1H), 1.70–1.60 (m, 1H), 1.47 (s, 9H), 1.22 (t, J = 7.0 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 173.3, 172.2, 80.7, 67.0, 60.2, 59.7, 38.8, 35.3, 29.9, 28.2, 27.3, 14.2. HRMS (ESI-TOF) (m/z) [MH]+ calcd for C14H25NO4 272.1856, found 272.1859.
(2S,5S,1′S)-5-(Azidoethoxycarbonylmethyl)-1-methylpyrrolidine-2-carboxylic Acid tert-Butyl Ester (11)
A solution of 10 (900 mg, 3.32 mmol) in dry THF (7 mL) was cooled to −78 °C and treated with LHMDS (1 M solution in THF, 7.30 mL, 7.30 mmol) dropwise. The enolate was allowed to form over 1 h at the same temperature and treated with a precooled (−78 °C) solution of 2,4,6-triisopropylbenzenesulfonyl azide (2.05 g, 6.63 mmol) in THF (12 mL). After 2 min the reaction was quenched by adding glacial AcOH (750 μL, 13.3 mmol) and then allowed to stir 16 h at rt. The reaction was concentrated, taken up in 10% aq. Na2CO3, and extracted with EtOAc, then the organic extracts were dried over Na2SO4, filtered, and concentrated in vacuo. The crude residue was purified by chromatography over silica gel (10–20% EtOAc/hexanes gradient as eluant). The desired azide 11 was obtained as a yellow oil (735 mg, 71%). 1H NMR (300 MHz, CDCl3) δ 4.27 (d, J = 2.6 Hz, 1H), 4.25 (q, J = 7.0 Hz, 2H), 3.71 (d, J = 7.6 Hz, 1H), 3.58 (dt, J = 8.8, 3.5 Hz, 1H), 2.53 (s, 3H), 2.10 (m, 2H), 1.87 (m, 2H), 1.47 (s, 9H), 1.29 (t, J = 7.0 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 173.3, 168.9, 80.9, 67.3, 64.7, 63.0, 61.7, 34.8, 28.2, 28.1, 25.1, 14.1; HRMS (ESI-TOF) (m/z) [MH]+ calcd for C14H24N4O4 313.1876, found 313.1884.
Data for the α-diazoester byproduct: 1H NMR (300 MHz, CDCl3) δ 4.21 (q, J = 6.9 Hz, 2H), 4.05 (dd, J = 8.4, 4.9 Hz, 1H), 3.56 (d, J = 8.2 Hz, 1H), 2.43 (s, 3H), 2.14–2.03 (m, 1H), 1.90–1.72 (m, 3H), 1.26 (t, J = 7.1 Hz, 3H), 1.48 (s, 9H); ESI-MS (m/z) [MH]+ calcd for C14H23N3O4 298.17, found 298.26.
(2S,5S,4’S)-5-[2-(2-Hydroxyphenyl)-4,5-dihydrooxazol-4-yl]-1-methylpyrrolidine-2-carboxylic Acid (3)
Oxazoline ester 14 (40 mg, 116 μmol) was treated with anisole (126 μL, 1.16 mmol) followed by 1.5 mL of a 90% TFA/DCM solution. The reaction was stirred at rt for 6 h, then diluted with 5 mL of EtOAc, and concentrated in vacuo (this step is necessary to remove excess TFA and avoid hydrolytic opening of the oxazoline ring). The crude residue was then partitioned between water and Et2O (precooled to 0 °C) and separated, then the aqueous layer was washed with an additional portion of Et2O. The aqueous layer was then frozen and lyophilized to yield pure 3 TFA as a hygroscopic white solid (38 mg, 83%). 1H NMR (400 MHz, DMSO-d6) δ 7.63 (dd, J = 7.6, 1.6 Hz, 1H), 7.45 (ddd, J = 8.6, 7.4, 2.0 Hz, 1H), 6.99 (dd, J = 8.6, 0.8 Hz, 1H), 6.92 (dt, J = 7.4, 1.2 Hz, 1H), 4.91 (ddd, J = 10.2, 7.8, 3.9 Hz, 1H), 4.61 (t, J = 9.4 Hz, 1H), 4.27 (t, J = 8.4 Hz, 1H), 3.92 (m, 1H), 2.93 (s, 3H), 2.16 (m, 1H), 2.01 (m, 1H), 1.72 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ 170.1, 166.3, 159.1, 158.0 (q, TFA), 134.2, 128.2, 119.1, 116.7, 109.6, 69.3, 66.7, 63.3, 51.9, 36.4, 26.7, 23.1; HPLC C18 retention time (0–90% MeCN in H2O with 0.1% formic acid, linear gradient, 20 min) = 9.78 min; HRMS (ESI-TOF) (m/z) [MH]+ calcd for C15H18N2O4 291.1339, found 291.1349.
(2S,5S,4’S)-5-[2-(2-Hydroxyphenyl)-4,5-dihydrothiazol-4-yl]-1-methylpyrrolidine-2-carboxylic Acid (2)
Thiazoline ester 15 (35 mg, 110 μmol) was deprotected by using the same procedure described for 14. Lyophilization of the final aqueous extracts yielded pure 2 ·TFA as a white solid (40 mg, 86%). 1H NMR (400 MHz, DMSO-d6) δ 7.48 (m, 2H), 6.99 (m, 2H), 5.17 (dt, J = 9.3, 3.1 Hz, 1H), 4.18 (m, 1H), 3.89 (m, 1H), 3.61 (dd, J = 11.7, 9.4 Hz, 1H), 3.22 (dd, J = 10.9, 9.4 Hz, 1H), 2.85 (s, 3H), 2.15 (m, 2H), 2.03 (m, 1H), 1.81 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ 170.5, 168.1, 158.0 (q, TFA), 156.0, 132.3, 129.2, 118.4, 115.7, 114.9, 73.8, 67.1, 52.0, 46.0, 36.9, 34.8, 27.2; HPLC C18 retention time (0–90% MeCN in H2O with 0.1% formic acid, linear gradient, 20 min) = 10.38 min; mp 68–70 °C; HRMS (ESI-TOF) (m/z) [M + H]+ calcd for C15H18N2O3S 307.1111, found 307.1115.
Supplementary Material
Acknowledgment
This research was supported by a grant from the National Cancer Institute (NIH U54CA132383) and by New Mexico State University. We thank Dr. Eileen Duesler (University of New Mexico) for the X-ray structure of compound 16.
Footnotes
Supporting Information Available: Detailed experimental procedures for compounds 5 and 12–16, and spectral data for all new compounds, as well as crystal structure data (CIF) for 16. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1)(a).Cox CD, Graham R. J. Bacteriol. 1979;137:357. doi: 10.1128/jb.137.1.357-364.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cox CD, Rinehart KL, Jr., Moore ML, Cook JC., Jr Proc. Natl. Acad. Sci. U.S.A. 1981;78:4256. doi: 10.1073/pnas.78.7.4256. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Raymond KN, Müller G, Matzanke BF. In: Topics in Current Chemistry. Boschke FL, editor. Vol. 123. Springer-Verlag; Berlin, Germany: 1984. p. 50. [Google Scholar]
- (2)(a).Pattus F, Abdallah MA. J. Chin. Chem. Soc. 2000;47:1. [Google Scholar]; (b) Poole K, McKay GA. Front. Biosci. 2003;8:D661. doi: 10.2741/1051. [DOI] [PubMed] [Google Scholar]
- (3)(a).Ankenbauer RG, Staley AL, Rinehart KL, Cox CD. Proc. Nat. Acad. Sci. U.S.A. 1991;88:1878. doi: 10.1073/pnas.88.5.1878. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mislin GL, Burger A, Abdallah MA. Tetrahedron. 2004;60:12139. [Google Scholar]; (c) Rivault F, Liebert C, Burger A, Hoegy F, Abdallah MA, Schalk IJ, Mislin GLA. Bioorg. Med. Chem. Lett. 2007;17:640. doi: 10.1016/j.bmcl.2006.11.005. [DOI] [PubMed] [Google Scholar]; (d) Rivault F, Liebert C, Burger A, Hoegy F, Mohamed A. Abdallah, Isabelle J. Schalk, Gaetan L. A. Mislin. Bioorg. Med. Chem. Lett. 2007;17:640. doi: 10.1016/j.bmcl.2006.11.005. [DOI] [PubMed] [Google Scholar]; (e) Rivault F, Schons V, Liebert C, Burger A, Sakr E, Abdallah MA, Schalk IJ, Mislin GLA. Tetrahedron. 2006;62:2247. [Google Scholar]; (f) Zamri A, Schalk IJ, Pattus F, Abdallah MA. Bioorg. Med. Chem. Lett. 2003;13:1147. doi: 10.1016/s0960-894x(03)00010-6. [DOI] [PubMed] [Google Scholar]
- (4)(a).Fischbach MA, Lin H, Zhou L, Yu Y, Abergel RJ, Liu DR, Raymond KN, Wanner BL, Strong RK, Walsh CT, Aderem A, Smith KD. Proc. Natl. Acad. Sci. U.S.A. 2006;103:16502. doi: 10.1073/pnas.0604636103. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, Akira S, Aderem A. Nature. 2004;432:917. doi: 10.1038/nature03104. [DOI] [PubMed] [Google Scholar]; (c) Goetz DH, Holmes MA, Borregaard N, Bluhm ME, Raymond KN, Strong RK. Mol. Cell. 2002;10:1033. doi: 10.1016/s1097-2765(02)00708-6. [DOI] [PubMed] [Google Scholar]; (d) Holmes MA, Paulsene W, Jide X, Ratledge C, Strong RK. Structure. 2005;13:29. doi: 10.1016/j.str.2004.10.009. [DOI] [PubMed] [Google Scholar]; (e) Yang J, Goetz D, Li JY, Wang W, Mori K, Setlik D, Du T, Erdjument-Bromage H, Tempst P, Strong R, Barasch J. Mol. Cell. 2002;10:1045. doi: 10.1016/s1097-2765(02)00710-4. [DOI] [PubMed] [Google Scholar]
- (5).Cancedda F. Descalzi, Dozin B, Zerega B, Cermelli S, Cancedda R. Biochim. Biophys. Acta. 2000;1482:127. doi: 10.1016/s0167-4838(00)00159-x. [DOI] [PubMed] [Google Scholar]
- (6)(a).Rinehart KL, Staley AL, Wilson SR, Ankenbauer RG, Cox CD. J. Org. Chem. 1995;60:2786. [Google Scholar]; (b) Zamri A, Abdallah MA. Tetrahedron. 2000;56:249. [Google Scholar]; (c) Ankenbauer RG, Toyokuni T, Staley A, Rinehart KL, Cox CD. J. Bacteriol. 1988;170:5344. doi: 10.1128/jb.170.11.5344-5351.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7)(a).Manzoni L, Arosio D, Belvisi L, Bracci A, Colombo M, Invernizzi D, Scolastico C. J. Org. Chem. 2005;70:4124. doi: 10.1021/jo0500683. [DOI] [PubMed] [Google Scholar]; (b) Collado I, Ezquerra J, Vaquero JJ, Pedregal C. Tetrahedron Lett. 1994;35:8037. [Google Scholar]
- (8).Jiang MXW, Jin B, Gage JL, Priour A, Savela G, Miller MJ. J. Org. Chem. 2006;71:4164. doi: 10.1021/jo060224l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Espino CG, Du Bois J. Angew. Chem., Int. Ed. 2001;40:598. doi: 10.1002/1521-3773(20010202)40:3<598::AID-ANIE598>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- (10)(a).Li Z, Capretto DA, Rahaman R, He C. Angew. Chem., Int. Ed. 2007;46:5184. doi: 10.1002/anie.200700760. [DOI] [PubMed] [Google Scholar]; (b) Cui Y, He C. Angew. Chem., Int. Ed. 2004;43:4210. doi: 10.1002/anie.200454243. [DOI] [PubMed] [Google Scholar]
- (11)(a).For seminal publications on electrophilic azidation of enolates, see: Wasserman HH, Hlasta DJ. J. Am. Chem. Soc. 1978;100:6780–6781.Kühlein K, Jensen H. Liebigs Ann. Chem. 1974:369–402.
- (12)(a).A similar stereochemical outcome has been observed in the electrophilic azidation of a β-proline derivative; Hanessian S, Sharma R. Heterocycles. 2000;52:1231.See also:Yi JJ, Hua ZM, Rong TG. Synth. Commun. 2003;33:3913.
- (13)(a).Atkins GM, Burgess EM. J. Am. Chem. Soc. 1968;90:4744. [Google Scholar]; (b) Wipf P, Miller CP. Tetrahedron Lett. 1992;33:907. [Google Scholar]
- (14)(a).Wipf P, Miller CP. Tetrahedron Lett. 1992;33:6267. [Google Scholar]; (b) Wipf P, Fritch PC. Tetrahedron Lett. 1994;35:5397. [Google Scholar]; (c) Wipf P, Miller CP, Venkatraman S, Fritch PC. Tetrahedron Lett. 1995;36:6395. [Google Scholar]
- (15).Somu RV, Wilson DJ, Bennett EM, Boshoff HI, Celia L, Beck BJ, Barry CE, III, Aldrich CC. J. Med. Chem. 2006;49:7623. doi: 10.1021/jm061068d. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.