

Abstract
The mechanism of retinol isomerization in the vertebrate retina visual cycle remains controversial. Does the isomerase enzyme RPE65 operate via nucleophilic addition at C11 of the all-trans substrate, or via a carbocation mechanism? To determine this, we modeled the RPE65 substrate cleft to identify residues interacting with substrate and/or intermediate. We find that wild-type RPE65 in vitro produces 13-cis and 11-cis isomers equally robustly. All Tyr-239 mutations abolish activity. Trp-331 mutations reduce activity (W331Y to ∼75% of wild type, W331F to ∼50%, and W331L and W331Q to 0%) establishing a requirement for aromaticity, consistent with cation-π carbocation stabilization. Two cleft residues modulate isomerization specificity: Thr-147 is important, because replacement by Ser increases 11-cis relative to 13-cis by 40% compared with wild type. Phe-103 mutations are opposite in action: F103L and F103I dramatically reduce 11-cis synthesis relative to 13-cis synthesis compared with wild type. Thr-147 and Phe-103 thus may be pivotal in controlling RPE65 specificity. Also, mutations affecting RPE65 activity coordinately depress 11-cis and 13-cis isomer production but diverge as 11-cis decreases to zero, whereas 13-cis reaches a plateau consistent with thermal isomerization. Lastly, experiments using labeled retinol showed exchange at 13-cis-retinol C15 oxygen, thus confirming enzymatic isomerization for both isomers. Thus, RPE65 is not inherently 11-cis-specific and can produce both 11- and 13-cis isomers, supporting a carbocation (or radical cation) mechanism for isomerization. Specific visual cycle selectivity for 11-cis isomers instead resides downstream, attributable to mass action by CRALBP, retinol dehydrogenase 5, and high affinity of opsin apoproteins for 11-cis-retinal.
Keywords: Metabolism/Retinoid, Methods/Site-directed Mutagenesis, Protein/Isomerase, Tissue/Organ Systems/Retina, Vision, Vitamins and Cofactors/Vitamin A
Introduction
A sequence of metabolic events, termed the visual cycle (1, 2), keeps retinal visual pigments, such as rhodopsin, in a state capable of responding to light. In brief, 11-cis-retinal bound to rhodopsin is photo-isomerized to all-trans-retinal, activating rhodopsin. To regenerate rhodopsin, all-trans-retinal is released, reduced to all-trans-retinol that is transported to the retinal pigment epithelium (RPE),3 and esterified to all-trans-retinyl esters, the substrate for the retinol isomerase (3). All-trans-retinyl esters are enzymatically isomerized to yield 11-cis-retinol that is oxidized to 11-cis-retinal and returned to the photoreceptors (3, 4). Recently, the RPE protein RPE65 (5) has been identified as the isomerase central to this cycle (6–8). The importance of RPE65 in chromophore regeneration had been well established by Rpe65 knock-out mice, which display extreme chromophore starvation (no rhodopsin) in the photoreceptors concurrent with overaccumulation of the all-trans-retinyl ester substrate of RPE65 in the RPE (9). Consequently, Rpe65−/− mice are extremely insensitive to light. Mutations in the human RPE65 gene cause Leber congenital amaurosis 2, a condition of severe early onset blindness (10–13), which has been the subject of phase one clinical trials by somatic gene therapy (14, 15, 16).
RPE65 belongs to a family of carotenoid oxygenases in plant, bacterial, and animal systems, which typically oxidatively cleave conjugated double bonds in the polyene backbone of isoprenoids (carotenoids or lignostilbenes). The other representatives in mammals are β-carotene monooxygenases 1 (17, 18) and 2 (19) (BCMO1 and BCMO2). In insects, NinaB (20) is a combined carotenoid oxygenase and retinoid isomerase (21). All are non-heme iron proteins in which ferrous iron is bound in an unusual four-histidine coordination scheme lacking a negatively charged ligand, as seen in the solved structure for Synechocystis apocarotenal oxygenase (ACO) (22). Three of these histidines are fixed by hydrogen bonds to conserved glutamate residues (22). Alteration of any of these leads to total loss of activity in BCMO1 (23) and RPE65 (8).
By virtue of its conjugated double bonds, retinol can exist in several isomeric forms, of which the all-trans, 9-cis, 11-cis, and 13-cis isomers are relevant in animals. While 11-cis-retinal is the only physiological ligand for opsins, 9-cis-retinal can make a pigment, isorhodopsin, with rod opsin (24, 25). Its only known physiological utility is in the Rpe65−/− mouse, where 9-cis-retinal, probably arising by thermal isomerization, forms the isorhodopsin responsible for its tiny level of light sensitivity (26). The 13-cis retinoids, formed readily by thermal isomerization, are also found in the retina of certain knock-out models (27, 28) but cannot form pigment with opsins.
The mechanism of isomerization of retinol in the vertebrate visual cycle is controversial. In principle, specificity of isomerization may be due entirely to the activity of isomerase itself (29, 30), or the isomerase in conjunction with binding proteins (31–34), such as cellular retinal-binding protein (CRALBP), and opsins. Two alternative mechanisms of retinol isomerization have been proposed: one involving addition of a nucleophile to C11 (29) and the other a carbocation-mediated mechanism (35). A retinyl carbocation acquires a delocalized bond order upon protonation (Fig. 1A), e.g. via loss of palmitate anion from retinyl palmitate. A mechanism involving a radical cation intermediate (Fig. 1B) is an alternative possibility. Although isomerization to 11-cis-retinol only might be the most elegant scenario, we have found (unpublished observations) that, in the in vitro visual cycle system we employed (8), 11-cis- and 13-cis-retinols are produced equally robustly. Also, analysis of retinoids in Rdh5−/− mice suggests a “leaky” isomerase capable of producing both 11-cis- and 13-cis-retinols (27, 28). Is this a legitimate characteristic of RPE65? We analyzed RPE65 isomerase activity to provide answers to these important questions. In this report we show that RPE65 is indeed a leaky retinol isomerase and that this feature is consistent with a carbocation mechanism of action in RPE65 retinol isomerase activity.
FIGURE 1.

Retinoid structures. A, depiction of a generalized retinyl carbocation. Upon protonation, indicated by “+,” charge is delocalized and bond order is lost, as indicated by dotted lines. The extent of charge delocalization and hence of loss of bond order is not necessarily over the entire polyene chain and may be restricted to a few double bonds, depending on the site of binding. B, depiction of a generalized retinyl ester radical cation formed by one-electron oxidation of substrate (positive charge indicated by “+”; radical nature is indicated by “.”). Bond delocalization may include bonds other than those depicted. C, structure of [15-2H,18O]all-trans-retinol showing positions of heavy isotope labels. If enzymatic isomerization occurs via retinol isomerase the 18O label is replaced by 16O; 18O label is retained if thermal isomerization occurs. The deuterium label is not lost in isomerization and thus provides a marker for the labeled retinol if the 18O is lost due to enzymatic isomerization. The carbons in the polyene chain are numbered in italics (7–15), defining the positions of the double bonds that may be in trans (as shown) or cis.
EXPERIMENTAL PROCEDURES
Site-directed Mutagenesis of RPE65
QuikChange XL site-directed mutagenesis kit (Stratagene, La Jolla, CA) was used for mutagenesis of the RPE65 open reading frame cloned in pVitro2 (Invivogen, San Diego, CA). Primer sequences for mutagenesis are given in supplemental Table S1. Mutants were verified by sequence analysis of DNA minipreps (Northwoods DNA, Solway, MN). Validated mutant and wild-type plasmids were purified by Qiagen purification kits (Qiagen, Valencia, CA).
Transient Transfection and Cell Culture
Cell culture methods and transient transfection protocols were previously published (8). In a typical experiment, 3 × 107 293-F (Invitrogen) cells were transfected with 30 μg of pVitro2 plasmid (containing RPE65 (wild type or mutant) and CRALBP open reading frames) and 30 μg of pVitro3 (Invivogen) plasmid (containing lecithin-retinol acyl transferase and retinol dehydrogenase 5) open reading frames in the presence of 40 μl of 293fectin transfection reagent (Invitrogen), all in a total volume of 30 ml. 24 h after transfection, all-trans-retinol was added to a final concentration of 2.5 μm, and the cells were cultured for a further 7 h and then harvested for analysis.
Retinoid Extractions and HPLC
Culture fractions of 20-ml volumes of transfected 293-F cells were centrifuged, and cells were harvested and retinoids extracted and saponified as previously described (8). Isomeric retinols were analyzed on 5-μm particle Lichrospher (Alltech, Deerfield, IL) normal phase columns (2 × 250 mm) on an isocratic HPLC system equipped with a diode-array UV-visible detector (Agilent 1100/1200 series, Agilent Technologies, New Castle, DE), following Landers and Olson (36) as modified by us (8). Data were analyzed using ChemStation32 software (Agilent).
Preparation of [15-2H,18O]All-trans-retinol and MALDI-TOF Mass Spectrometry
The method of McBee et al. (35) was followed, with modifications, to synthesize [15-2H,18O]all-trans-retinol (Fig. 1C). To 200 μl of 3 mg/ml all-trans-retinal in acetonitrile was added 250 μl of acetonitrile, 150 μl of H218O (Cambridge Isotope Laboratories, Andover, MA) and 4 mg of p-toluenesulfonic acid, with stirring overnight at room temperature. The next day, 10 mg of solid NaBD4 (Cambridge Isotope Laboratories) was added, while stirring briefly, and the vial was put on ice for 20 min. The reaction was extracted with 2 × 1 ml of hexane, and the extracts were pooled, dried, redissolved in hexane, and fractionated by normal phase HPLC as described above with n-octanol replaced by chloroform (85.4% n-hexane, 11.2% ethyl acetate, 2% 1,4-dioxane, 1.4% chloroform), and the all-trans-retinol peak was collected and quantified. The labeled all-trans-retinol was dried under vacuum and redissolved in 10 μl of 2,5-dihydroxybenzoic acid in acetone (0.5 m) and analyzed by a matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) method (Voyager-DE STR, Applied Biosystems, Foster City, CA) in positive ion reflective mode, modified from Wingerath et al. (37). A pulsed nitrogen laser with a wavelength of 337 nm was used. Ions were accelerated to a kinetic energy of 20 keV. The following instrument parameters were used: guide wire, 0.002%; grid voltage, 80%; and delay time, 250 ns. Exchange was invariably complete (supplemental Fig. S1) as seen by the presence only of major all-trans-retinol species of molecular mass 289.45, and a minor m+1 species of 290.45 (ascribed to stochastic occurrence of 13C) on MALDI-TOF analysis (supplemental Fig. S1B). The purified [15-2H,18O]all-trans-retinol was added in ethanol to cultures to a final concentration of 2.5 μm, transfected as above with constructs containing either wild-type or mutant RPE65 for 7 h. Retinol isomers were extracted from cells as above, purified by normal phase HPLC as for labeled retinol standard, dried under vacuum for 10 min, mixed with 2,5-dihydroxybenzoic acid (0.5 m), and analyzed by MALDI-TOF, as above.
Immunoblot Analysis
Expression levels of RPE65 were quantitated by fluorescent Western blot. Cell pellets (∼2 × 106 cells) from 1-ml culture aliquots were lysed in 150 μl of CytoBuster detergent (Novagen) containing Complete protease inhibitor (Roche Applied Science), incubated on ice for 10 min and centrifuged at 16,000 × g for 10 min, and the supernatant was harvested for SDS-PAGE analysis. Denatured samples were separated on 12% BisTris NuPage (Invitrogen) gels along with ECL-plex Rainbow protein standard markers (Amersham Biosciences) and electrotransferred to Hybond-ECL nitrocellulose membranes (Amersham Biosciences). Primary antibodies were rabbit anti-RPE65 (1:4,000) and mouse monoclonal antibody anti-CRALBP (1:20,000, gift of Dr. John Saari). Secondary antibodies used were Cy5-conjugated goat anti-rabbit and Cy3-conjugated goat anti-mouse ECLplex fluorescent antibodies (both 1:2,500, Amersham Biosciences). Processed blots, following manufacturer's protocols, were scanned in a Typhoon 9410 scanner (Amersham Biosciences) and quantitated using ImageQuant TL image analysis software. Wild-type and mutant RPE65 levels were normalized to co-expressed CRALBP levels, and mutant levels were calculated relative to wild-type RPE65 expression (set at 100%).
Molecular Modeling
A three-dimensional model of all-trans-retinyl palmitate was obtained using the PRODRG program (38). The model was aligned at the ester bond with the 15,15′ bond of the apocarotenal substrate modeled in the ACO structure (22) and transferred to the RPE65 structure modeled from ACO using the Swiss-Pdb viewer as previously described (39). The position of all-trans-retinyl palmitate was then manually adjusted to minimize clashes with the predicted RPE65 structure. Orientation of the retinyl moiety in the substrate cleft was based on the experimental findings of a requirement for aromaticity at aa 331 commensurate with cation-π stabilization of the retinoid intermediate.
RESULTS
Modeling of the Substrate-binding Cleft: Consideration of the Lining Residues
An assemblage of aromatic and non-aromatic residues lines the substrate-binding cleft of ACO and may interact with substrate (22). We modeled RPE65 on the ACO template (Fig. 2A and supplemental Fig. S2A). Paralogous residues found in RPE65 that are mostly identical or conserved include (RPE65 residues in parentheses): Phe-69 (Phe-61), Phe-113 (Phe-103), Trp-149 (Thr-147), Phe-236 (Tyr-239), and Tyr-322 (Trp-331). (For alignment see supplemental Fig. S2B.) We also identified Tyr-275 of RPE65 as being in the predicted RPE65 tunnel adjacent to Phe-61 and Phe-103, although there is no aromatic paralog in ACO (supplemental Fig. S2B). β-Carotene cleavage by BCMO1 requires the binding of both β-ionone rings of β-carotene in the substrate cleft, unlike ACO where the β-ionone ring is postulated not to enter due to a bottleneck in the opening (22). Because RPE65 is more homologous to BCMO1 (∼40% identity) than ACO (∼26% identity), the β-ionone ring of the retinyl ester could conceivably lie at either end of the substrate cleft. Furthermore, NinaB is capable of accepting not only β-ionone rings but epsilon rings and hydroxylated versions of both (20). We predict that the β-ionone ring of the substrate of RPE65 also enters the cleft and that the retinoid polyene backbone interacts with Trp-331. This proposed orientation is based on the mutagenesis data for Trp-331 (see below) and places the 11–12 bond (Fig. 2B, arrow) of the retinyl ester adjacent to Trp-331 to accommodate the proposed cation-π interaction of the retinoid polyene backbone with this residue, while the ester C–O bond is in register with the iron ligand (Fig. 2B, arrowhead). The residues Thr-147, Phe-103, and Tyr-239 would be predicted to interact closer toward the ester bond of the retinyl ester substrate. We undertook mutagenesis experiments on these residues to determine the role they play in the mechanism of RPE65.
FIGURE 2.

Modeling of RPE65 substrate binding cleft based on crystal structure of ACO. A, superposition of the RPE65 model on the ACO template. The catalytic center is shown (at 50% opacity for clarity) with histidines coordinating the Fe2+ (magenta sphere). Silver, ACO carbon; green, RPE65 carbon; red, oxygen; blue, nitrogen; yellow, apocarotenol; and orange, retinyl ester. B, modeled cleft of RPE65 showing lining residues. RPE65 residues studied are: carbon (green), oxygen (red), nitrogen (blue), and retinyl ester (orange). Histidines coordinating the Fe2+ (magenta sphere) are depicted in silver/blue. The red arrow indicates the 11–12 double bond of the retinyl moiety, and the red arrowhead indicates the ester oxygen of the substrate.
RPE65 Substrate Binding Cleft Residues Requiring Conservation of Aromaticity
Phe-69 (RPE65 paralog: Phe-61) is located in the substrate cleft of ACO (22) and is part of the highly conserved FDG motif in the family (F and D are invariant, G almost so). We made F61Y, F61W, and F61L mutants. Phe appears to be an essential residue at aa 61, because activity of both F61L and F61W was abolished, while normalized activity of F61Y was reduced to 13.7% of wild type (Table 1). Despite these severe reductions in activity, protein expression, although reduced, was not abolished (Table 1). We also interrogated Tyr-275, predicted to be nearby. Y275F retained 44% of wild-type activity, normalized to protein expression, whereas Y275W and Y275I were totally inactive, although all three were expressed at roughly 30% of wild type (Table 1). These data suggest that aromaticity at both 61 and 275 is required, but that steric considerations may disqualify tryptophan.
TABLE 1.
Effects of mutations on RPE65 isomerase activity and protein expression
| Mutant | 11-cis-Retinol production | Expression | Activity normalized to expression | na |
|---|---|---|---|---|
| %WTb | %WTc | %WTd | ||
| F61L | 1.02 ± 1.18 | 38.65 ± 14.73 | <0.1 | 4 |
| F61Y | 7.57 ± 0.57 | 56.03 ± 7.66 | 13.7 ± 2.6 | 4 |
| F61W | 1.28 ± 1.47 | 22.5 | <0.1 | 4 |
| Y275F | 14.44 ± 0.37 | 33.77 ± 6.6 | 44 ± 8.3 | 4 |
| Y275W | 0 | 27.65 ± 8.0 | 0 | 4 |
| Y275I | 0 | 29.12 ± 7.8 | 0 | 4 |
| Y239W | 0 | 21.6 ± 9.64 | 0 | 3 |
| Y239F | 0 | 17.86 | 0 | 3 |
| Y239L | 0 | 14.74 | 0 | 3 |
| Y239S | 0 | 17.65 ± 10.24 | 0 | 3 |
| Y239T | 0 | 27.98 ± 8.24 | 0 | 3 |
| Y239C | 0 | 17.32 ± 5.46 | 0 | 3 |
| Y239D | 0 | 0 | 0 | 3 |
| W331Y | 26.72 ± 2.26 | 33.74 ± 10.47 | 74 ± 17.1 | 3 |
| W331F | 8.34 ± 2.64 | 17.86 ± 3.4 | 53 ± 6.5 | 3 |
| W331L | 0 | 21.1 ± 3.53 | 0 | 3 |
| W331Q | 0 | NDe | 0 | 3 |
| T147W | 0 | 26.05 ± 6.64 | 0 | 6 |
| T147Y | 0 | 10.55 ± 5.55 | 0 | 6 |
| T147V | 0 | 10.95 ± 4.83 | 0 | 6 |
| T147A | 12.63 ± 0.24 | 34.00 ± 12.46 | 12.6 ± 0.2 | 3 |
| T147C | 20.18 ± 3.69 | 43.22 ± 9.66 | 20.2 ± 3.7 | 3 |
| T147G | 30.02 ± 2.77 | 97.8 ± 20.2 | 29.9 ± 2.8 | 3 |
| T147S | 84.00 ± 5.79 | 98.7 ± 11.2 | 84 ± 5.8 | 6 |
a Number of replicates.
b Mutant activity is normalized to and represented as % of wild-type RPE65 isomerase activity ± S.D.
c Mutant expression is normalized to and represented as % of wild type RPE65 expression ± S.D.
d Mutant isomer production is normalized to protein expression and expressed as % of wild type activity ± S.D.
e ND, not determined.
Trp-331 is conserved in RPE65, and to address its potential role, we replaced it with other aromatic residues: W331Y (its paralog in BCMO1 and ACO) and W331F. We also made W331L (conserving hydrophobicity) and W331Q (its paralog in BCMO2) and measured isomerase activity. Interestingly, W331Y activity was reduced to ∼75% when normalized to wild-type expression, whereas W331F was reduced to ∼50% of wild type when normalized, but W331L and W331Q were completely inactive (Table 1 and Fig. 3). These data suggest that aromaticity at this residue is of importance in the catalytic activity of RPE65. It is interesting to note at this point that W > Y > F in the pi-binding energy series of the aromatic residues, consistent with observed activity of the mutants compared with wild type.
FIGURE 3.

Requirement for aromaticity at residue 331 of RPE65. Mutation of Trp-331 to any other aromatic residue reduces but does not abolish isomerase activity of RPE65, but mutation to leucine or to glutamine (paralogous residue in BCMO2) abolishes activity. The 11-cis-retinol production in 293-F cells transfected with constructs expressing wild type and mutants of dog RPE65 Trp-331 was determined. Mutant activities are expressed as percentage of wild-type RPE65 activity normalized to the expression of the respective RPE65 mutant ± S.D. (n = 4).
Tyrosine Is Essential at aa 239 of RPE65
Tyr-239 in RPE65 is paralogous to the partially conserved Phe-236 of ACO (22) located in the latter's carotenoid-binding cleft, and a tyrosine is the paralog in all metazoan examples. To discern the role of Tyr-239 in the mechanism of RPE65, we made substitutions with aromatic or hydrophobic residues. All mutations, Y239W, Y239F, and Y239L, led to complete loss of activity (Table 1). Similarly, replacement with hydroxyl/thiol-containing residues, Y239S, Y239T, and Y239C, abolished activity, as did Y239D, a human Leber congenital amaurosis 2 mutation (Table 1). Although all of these mutants (except Y239D) exhibited a reduction in expression, protein was still produced. Only with Y239D was expression absent. Taken together, these data point to an absolute and unique requirement for a tyrosine at residue 239 of RPE65. In contrast, Tyr-235, the paralogous residue in BCMO1 can be changed to other aromatics without major loss of activity.4
Site-directed Mutagenesis of Thr-147 and Phe-103 Leads to Opposite Effects on Isomerase Specificity
In the ACO structure (22), Trp-149, adjacent to the fixing Glu-150, is predicted to be within 4 Å of the β-ionone ring of the apocarotenal substrate. Its paralog in RPE65 is Thr-147, adjacent to the paralogous glutamate (Glu-148), which forms a hydrogen bond with the iron-coordinating His-241. The isomerase activity of the T147W mutant (paralogous to ACO) was abolished, as was that of T147Y (Table 1 and Fig. 4A). Expression levels of both mutations were reduced (T147Y > T147W (Table 1)). To further determine the role of Thr-147 we made serine, cysteine, alanine, glycine, and valine mutants. Only T147V was completely inactive, whereas the others showed some degree of activity (Fig. 4A and Table 1). T147G had a marked reduction in isomerase activity (∼30%, Table 1), but its expression level remained comparable to wild type (Table 1), suggesting a significant shift in catalytic efficiency (Fig. 4A). Of the others, T147S had the most interesting effect: overall 11-cis-retinol production was similar to wild type (∼86%), however 13-cis-retinol production was reduced to ∼61% (Tables 1 and 2 and Fig. 4B), a consistent effect unlike that of any other mutant tested, indicating more specific synthesis of 11-cis-retinol by T147S. The threonine is replaceable by serine, but with a significant shift in specificity. Taken together, these data suggest that Thr-147 is involved in RPE65 catalytic activity.
FIGURE 4.

Thr-147 is not essential for activity but is important in determining specificity of RPE65. A, Thr-147 is not essential for activity of RPE65. Thr-147 was mutated to Trp, Tyr, Val, Ala, Cys, Gly, and Ser. Mutation to Trp, Tyr, and Val abolished activity, whereas mutation to Ala, Cys, Gly, and Ser retained 30–84% of wild-type activity. Mutant activities are expressed as percentage of wild-type RPE65 activity normalized to expression of the respective RPE65 mutant ± S.D. (n ≥ 3). B, Thr-147 plays a role in determining specificity of isomerization. The 11-cis/13-cis ratio produced by the T147S mutant is elevated 40% compared with wild type. The 11-cis- and 13-cis-retinol production in 293-F cells transfected with constructs expressing wild type and mutants of dog RPE65 Thr-147 was determined.
TABLE 2.
Effect of mutations at F103 and T147 on RPE65 isomerase activity and protein expression
| Mutant | 11-cis-Retinol production | 13-cis-Retinol production | Protein expression | Normalized 11-cis-retinol production | Normalized 13-cis-retinol production | na |
|---|---|---|---|---|---|---|
| %WTb | %WTb | %WTc | -fold WTd | -fold WTd | ||
| T147S | 86.33 ± 5.05 | 60.85 ± 3.58 | 112.24 ± 9.51 | 0.83 ± 0.12 | 0.58 ± 0.07 | 4 |
| F103Y | 20.06 ± 2.95 | 36.47 ± 6.4 | 32.89 ± 7.36 | 0.68 ± 0.09 | 1.23 ± 0.23 | 4 |
| F103W | 24.37 ± 0.93 | 60.51 ± 3.51 | 57.03 ± 11.16 | 0.4 ± 0.04 | 1.01 ± 0.18 | 4 |
| F103I | 2.51 ± 1.04 | 43.8 ± 3.83 | 26.69 ± 6.51 | 0.1 ± 0.02 | 1.91 ± 0.31 | 3 |
| F103L | 28.02 ± 1.27 | 121.89 ± 10.7 | 60.68 ± 12.4 | 0.48 ± 0.08 | 2.14 ± 0.36 | 8 |
| T147S/F103L | 28.79 ± 2.07 | 80.08 ± 5.05 | 79.04 ± 10.88 | 0.37 ± 0.04 | 1.03 ± 0.16 | 4 |
a Number of replicates.
b Mutant activity is normalized to and represented as % of wild-type RPE65 isomerase activity ± S.D.
c Mutant expression is normalized to and represented as % of wild-type RPE65 expression ± S.D.
d Mutant isomer production is normalized to protein expression and expressed as -fold of wild-type activity ± S.D.
Facing Trp-149 across the ACO substrate cleft is Phe-113 (22), an almost invariant residue in the family, except in plant representatives, and whose paralog in RPE65 is Phe-103. Overall, protein expression of a panel of Phe-103 mutants was only moderately affected, ranging from 32 to 79% of wild type (Table 2). F103Y showed markedly altered isomerase activity, with 11-cis production at 0.68-fold and 13-cis production at 1.2-fold of wild type. F103W had production of 11-cis reduced to 0.4-fold wild type, whereas 13-cis production remained at wild-type level (Table 2 and Fig. 5). However, the alteration in activity of F103L and F103I were quite remarkable. In F103L, 11-cis production is reduced to 0.48-fold wild type and 13-cis production is increased to 2.1-fold of wild type (Table 2 and Fig. 5). In the mutant F103I, 11-cis production was reduced to 0.1-fold of wild type, whereas 13-cis remained relatively high at 1.9-fold of wild type. We asked whether the double mutant T147S/F103L balances the opposite effects of the two separate mutants. However, we found that its 11-cis activity was only 0.37-fold of wild type, whereas 13-cis activity was equal (1-fold) to wild type (Fig. 5), suggesting that the identity of the residue at 103 was dominant over the residue at 147. Taken together, the data on Phe-103 and Thr-147 mutants strongly suggest that these residues play a role in governing the orientation of substrate and/or intermediate in the binding tunnel and so help specify the isomerization state of the product.
FIGURE 5.

Phe-103 of RPE65 is not essential for activity but is crucial and opposite-acting to T147S in determination of isomerization specificity. Substitution of Phe-103 with Trp, Tyr, Ile, or Leu alters RPE65 isomerase activity by elevating 13-cis isomer production relative to 11-cis production. The 11-cis- and 13-cis-retinol production in 293-F cells transfected with constructs expressing wild type and mutants of dog RPE65 Phe-103 and/or Thr-147 was determined. Mutant 11-cis and 13-cis production activities are expressed as -fold activity of wild-type RPE65 activities normalized to expression of the respective RPE65 mutant ± S.D. (n ≥ 4).
Coordinated Production of 11-cis- and 13-cis-Retinols by RPE65 in Vitro
The preceding mutational data put in context our previously unpublished observations using the in vitro visual cycle assay (8), where we found that wild-type dog RPE65 produced 13-cis-retinol at levels close to 11-cis-retinol (Fig. 6A), as did mouse and chicken RPE65 (data not shown), indicating that robust production of 13-cis as well as 11-cis-retinol is a legitimate characteristic of the enzymatic activity of RPE65, not an artifact due to thermal isomerization; significant levels of 9-cis or other isomers were not evident. Using these conditions (Ref. 8 and present work), we determined that wild-type RPE65 isomerizes 15.2 ± 2.17% of total retinols to 11-cis-retinol and 14.7 ± 4.2% of total retinols to 13-cis-retinol, estimated for a large pool of assays (see also supplemental Fig. S3). To determine if this is a coordinated effect, we plotted 11-cis/13-cis-retinol production by wild-type RPE65 and several mutant RPE65 enzymes, co-expressed with CRALBP, lecithin-retinol acyl transferase, and retinol dehydrogenase 5, in the HEK 293-F in vitro visual cycle system (Fig. 6B). These mutants include the variants C329S (a mutation made to test requirement for Cys at aa 329), C330T (paralogous chicken residue at aa 330), and A434V (human variant of uncertain pathogenicity) (8), T457N (human variant of uncertain pathogenicity) (39), and the human mutations L22P, P25L, Y79H, and E95Q (directly associated with Leber congenital amaurosis 2) (40). We found that 13-cis-retinol production closely tracks 11-cis-retinol production until ∼50% of wild-type activity where 13-cis production overtakes 11-cis. In mutants with abolished 11-cis-retinol synthesis, a baseline level of 13-cis-retinol production was observed, consistent with non-enzymatic 13-cis-retinol thermal isomerization. In Fig. 6B this level is shown as ∼20% of the wild-type RPE65 activity conversion of all-trans-retinol to 13-cis-retinol, measured as 14.7%, above. That is, there was ∼3% overall conversion to 13-cis-retinol in the absence of RPE65 activity. As the cells are incubated with all-trans-retinol for 7 h at 37 °C, there is ample scope for nonspecific thermal isomerization to occur. To quantify this, we measured the background level of isomerization in untransfected 293-F cells incubated with 2.5 μm all-trans-retinol for 7 h. We found 13-cis-retinol to be 5.3 ± 0.07% of total retinols (all-trans + 13-cis + 11-cis + 9-cis); interestingly 9-cis-retinol was also evident at 1.6 ± 0.13% of total retinols in these untransfected cells, whereas 11-cis-retinol was estimated at 0.12 ± 0.04% of total retinols. The value for 11-cis-retinol is consistent with chemical isomerization equilibrium mixtures where the 11-cis isomer was determined not to be kinetically favored (41), whereas those of 13-cis and 9-cis were lower than in these equilibrium mixtures because no chemical catalysis was employed. The level of 13-cis-retinol we find for these cultures compares closely with an average of 4.05% of total retinols for a selection of cultures expressing RPE65 mutants with no activity. This amount (3–5%) then is the background of 13-cis-retinol due to thermal isomerization. Even if this thermal level is subtracted, the level of 13-cis-retinol production by active RPE65 remains significant, leading us to conclude that it is a legitimate product of RPE65.
FIGURE 6.
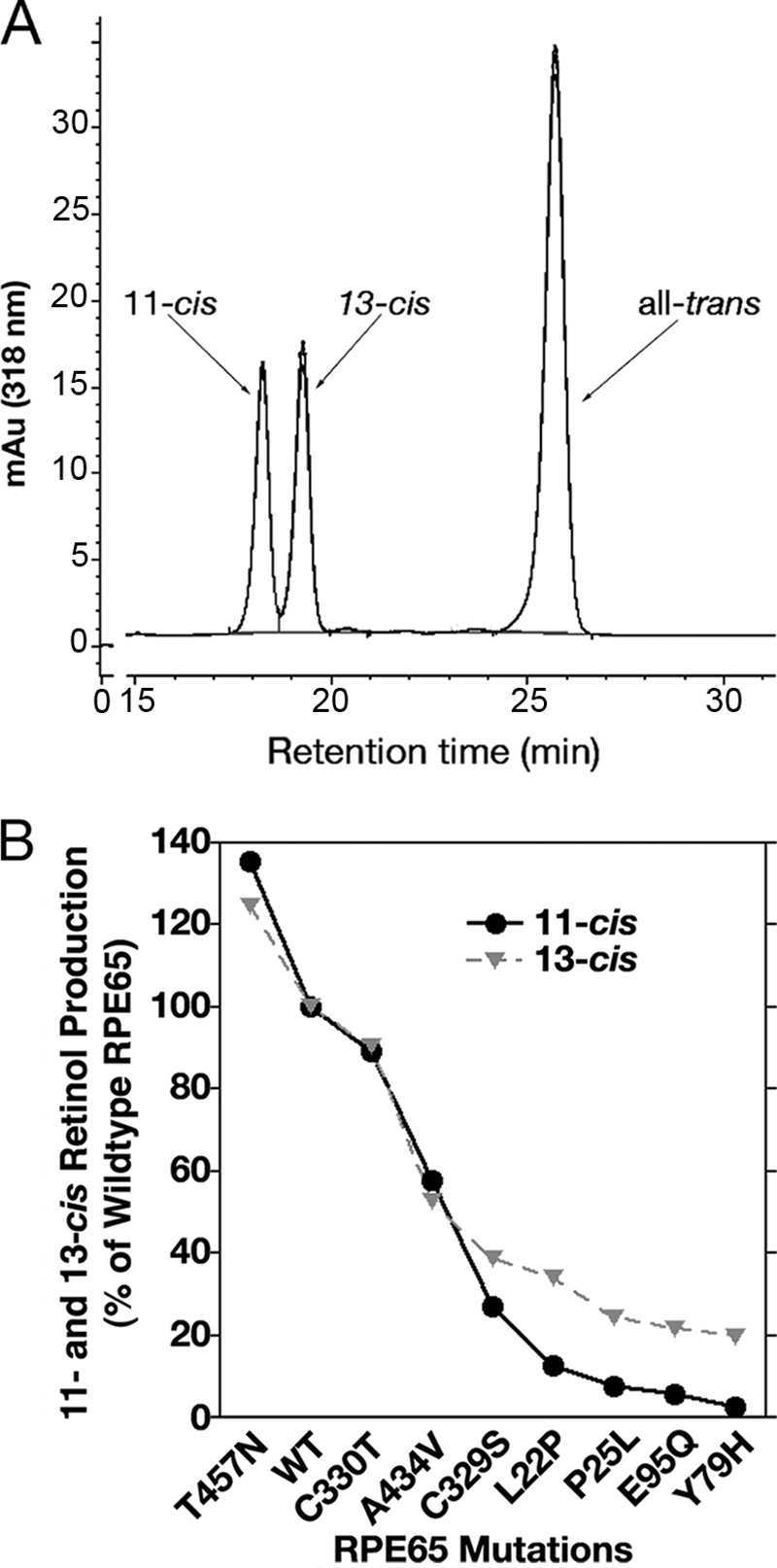
Robust co-production of 11-cis- and 13-cis-retinols by RPE65 in vitro. A, normal-phase HPLC of retinol isomers from saponified retinyl esters isolated from HEK 293-F cells heterologously expressing wild-type dog RPE65, showing locations of 11-cis-, 13-cis-, and all-trans-retinol peaks. B, RPE65 mutations affecting isomerase activity display a coordinated decline in both 11-cis- and 13-cis-retinol production. Whereas 11-cis-retinol production of mutants of progressively decreasing activity declined to zero, 13-cis-retinol production reached a plateau due to 13-cis-retinol derived from thermal isomerization of retinol. Wild-type, pathogenic mutants and variants of RPE65 with activities ranging from 100% to 0% of wild-type level of 11-cis-retinol production were co-expressed with CRALBP, lecithin-retinol acyl transferase, and retinol dehydrogenase 5, and following incubation with all-trans-retinol, retinyl esters were extracted and saponified, and isomeric retinols were measured to compare 11-cis production and 13-cis production by the same mutant. The data were not corrected for RPE65 expression. These include the variants C329S, C330T, and A434V (8), T457N (39), and the pathogenic mutants L22P, P25L, Y79H, and E95Q (40).
Exchange of C15 Oxygen: 13-cis Isomer Is Produced by RPE65-mediated Isomerization
Enzymatic retinol isomerization occurs with inversion of stereochemistry and loss of oxygen at C15 of both 11-cis (29, 35) and 13-cis-retinols (35). To establish that 13-cis-retinol production in our experiments is a result of enzymatic RPE65-mediated isomerization, we supplied cultures expressing wild-type and F103L (where 13-cis production is enhanced) RPE65 with [15-2H,18O]all-trans-retinol (Fig. 1C). Following extraction and separation on HPLC, the 11-cis, 13-cis, and all-trans isomer peaks were isolated and purified, and were then subjected to MALDI-TOF mass spectrometry. We detected a species of 287 mass units showing clear loss of the 2-mass unit increment due to 18O in both 11-cis and 13-cis peaks from cultures expressing either wild-type RPE65 or F103L RPE65 (Fig. 7, A, B, and E) establishing that not only 11-cis but also 13-cis arises from enzymatic isomerization. A small proportion of 13-cis-retinol of 289 mass units was detected (Fig. 7E, arrow) due to the entirely expected occurrence of thermal all-trans to 13-cis isomerization. The all-trans-retinol peak from either showed only small amounts of the 287 species (Fig. 7, C and F, arrows), probably due to cis to trans isomerization of enzymatically isomerized 11-cis and 13-cis products, being mainly the 289-mass unit precursor. These data establish that the 13-cis-retinol is an enzymatically isomerized product of RPE65.
FIGURE 7.

13-cis-retinol produced by RPE65 isomerase activity shows exchange of oxygen at C15. Cultures expressing wild-type RPE65 and F103L mutant RPE65 were supplied with [15-2H,18O]all-trans-retinol (A, B, C, E, and F) or unmodified all-trans-retinol (D). Following extraction and saponification, isomeric retinols were purified by HPLC and collected and subjected to MALDI-TOF. Loss of 18O label was seen in isomeric retinols from wild-type RPE65: A, 11-cis; B, 13-cis-retinols but not C, all-trans-retinol. A small amount of all-trans-retinol lacking 18O label (C, arrow) was sometimes detected and was probably due to cis-trans isomerization of enzymatically produced 11-cis- and/or 13-cis-retinols. For cultures expressing F103L mutant and supplied with unmodified all-trans-retinol as reagent control, the expected mass (∼286.5) was observed (D); however, loss of 18O label was seen in E, 13-cis-retinol but not F, all-trans-retinol from cultures supplied with [15-2H,18O]all-trans-retinol. A minor amount of 13-cis-retinol retaining both labels was detected (E, arrow). This was probably due to thermal isomerization. Nominal mass of retinols = 286.45, nominal mass of [15-2H,18O]all-trans-retinol = 289.45. The peak with observed mass of ∼273 was derived from the matrix (37); the peak with observed mass of ∼305 (F) is likely an epoxidation product (289 + 16 = 305) of retinol (see also supplemental Fig. S1). Data shown are representative of multiple independent analyses.
DISCUSSION
Here we show that RPE65, the visual cycle retinol isomerase, is not inherently 11-cis specific in its activity, addressing a long standing debate in the visual cycle. We show that RPE65 can produce both 11-cis- and 13-cis-retinols, the proportions of which can be modulated by altering key residues. We propose that this is a result of RPE65 having a carbocation/radical cation (Fig. 1, A and B) intermediate-mediated mechanism, and strengthens the notion that 11-cis selectivity in the visual cycle is governed by mass action imposed by the 11-cis-specific binding protein CRALBP (35) and the ultimate chromophore sink, the apoprotein opsins.
How all-trans-retinol is isomerized to 11-cis-retinol has long been a fundamental question in the visual cycle. Two possible mechanisms have received most attention: one an SN2′ mechanism of nucleophile addition to C11 (29) and the other a carbocation-mediated mechanism (35). Although Rando et al. (29, 30, 42) propose an SN2′ mechanism as consistent with their data, they, explicitly, do not rule out a carbonium (i.e. carbocation) mechanism. Conversely, Palczewski et al. (35, 43) favor only the latter. Clearly, the actual mechanism of retinol isomerization will be that employed by RPE65. We asked which one of these possible mechanisms is consistent with RPE65 activity?
An SN2′ mechanism of nucleophilic addition at C11 will only produce 11-cis-retinol. To produce both 11-cis and 13-cis requires nucleophilic addition at either C11 or C13, followed by hydration of either intermediate (a C11 or a C13 intermediate) by the same active site of the one enzyme, a spatially difficult conjecture. On the other hand, a carbocation mechanism obligately predicts that isomers other than 11-cis could be made. In outline, a carbocation mechanism requires an initial protonation of the substrate, followed by bond rearrangement of the carbocation intermediate, and fixation of product by proton elimination and hydration. In a retinoid carbocation, bond order is reduced through electron delocalization along the part of the polyene chain affected by protonation. The most likely site of protonation is the carbonyl oxygen of retinyl ester, which leads to subsequent loss of palmitate. Direct protonation of the double bond at the C13-C14 position without loss of palmitate is much less likely, but a similar event is described for α-tocopherol cyclase activation of chromanol (44). At equilibrium, several isomeric conformations of intermediate are present, subject to local environment (e.g. residue side chains) in the reaction site. The isomer(s) made would depend on the cellular retinoid-binding proteins present to sequester the products. Thus, because addition of cellular retinol-binding protein 1 enhanced formation of 13-cis-retinol when added to bovine RPE microsomes, using 18O/2H-labeled all-trans-retinol, McBee et al. (35) concluded it was not entirely thermal in origin, a finding that we reproduced in our study. Also, the retinoid retinylamine, positively charged at physiological pH, which is expected to disrupt a carbocation-mediated process, selectively inhibits isomerization (43). Physiologically, potential “leakiness” with significant levels of 13-cis retinoids can be seen in Rdh5−/− mice (27). Some isomerization to 13-cis in these mice was thermal, but much was enzymatic in origin (28), as it was inhibited by retinylamine (43). In support of this hypothesis, we found that 11-cis- and 13-cis-retinol production in hypomorphic RPE65 mutants follows a coordinated decline down to “background” thermal level. Such a coordinated decline would not be expected if 13-cis were not a potential product of RPE65. Instead, 13-cis-retinol isomer levels for all mutants would be similar, i.e. at a level consistent with thermal isomerization.
Carbocation intermediates are important in many aspects of isoprenoid biogenesis. Terpenoid cyclases, catalyzing some of the most complex reactions in biology, use carbocation intermediates, many of them highly unfavorable (45), and stabilize these by aromatic residues (46), as do carotenoid cyclases (47). Aromatic residues are critical as they can engage in cation-π interactions between the side-chain π electrons and charged entities (48, 49). Due to their isoprenoid origin, carotenoid or retinoid pathways may involve carbocations. Therefore, we studied the role of aromatic residues, which we identified by modeling RPE65 on the ACO structure (22), in the mechanism of RPE65. Such residues may be involved in stabilization of a putative carbocation intermediate in isomerization. Although many residues are not conserved between ACO and RPE65, aromaticity is mostly retained. We found that mutations to some RPE65 residues alter specificity and illuminate its underlying mechanism. Although our in vitro assay lacks key physiological elements (e.g. apoprotein opsin) that drive specificity of a specifically operating visual cycle, our strategy exposes the complete spectrum of RPE65 enzymatic activity.
Based on three considerations, 1) that RPE65 belongs to a family of monooxygenases with a catalytic iron center capable of activating oxygen, 2) that, concomitant with inversion of stereochemistry, the oxygen bound to C15 is exchanged in the retinol product, and 3) that mutating active site residues modulate the 11-cis/13-cis-retinol ratio, we propose that RPE65 isomerization involves a cationic delocalized intermediate (carbocation or radical cation), which could employ an isomerooxygenase rather than an isomerohydrolase mechanism (29, 30, 42). Because a radical cation intermediate has been postulated for the ACO reaction mechanism (50), we must also consider this avenue. NinaB, a related protein in insects catalyzing oxidative cleavage of β-cryptoxanthin and isomerization of the hydroxylated end, has also been proposed to be an isomerooxygenase (21). Although the Palczewski mechanism (35) is tenable, some issues remain. First, molecular oxygen is not implicated in any way in their model. Second, without an enzyme-substrate intermediate, allowing rotation around C11 or C13, but not C15, is problematic. At this point we cannot distinguish between hydrolysis and oxygenation at C15 position, but we suggest that oxygen could play a role in formation of a radical cation and/or oxygenation of the substrate. Loss of oxygen at C15 is not typical of ester hydrolysis, in which the bond cleaved is that between the acyl carboxyl carbon and the oxygen, and not that between the alcohol (retinol) terminal carbon (C15) and the oxygen, such as is the case in retinol ester “hydrolysis” and isomerization. Besides the possible carbocation mechanism, we cannot exclude the possibility of one electron oxidation by activated oxygen and formation of a radical cation intermediate, as in the cuproenzyme dopamine β-monooxygenase (51, 52) and Fe2+ extradiol ring-cleaving dioxygenase (53). An active peroxy or oxo species, in principle, could oxygenate C15 and release the acyl moiety by in-line displacement with inversion of stereochemistry (SN2). Radical cations also can be directly stabilized by electron-rich π systems of aromatic residues. Also a water molecule, perhaps coordinated by iron, could replace oxygen at C15 of a retinyl ester radical cation by SN2 mechanism with inversion of stereochemistry. These are all potential mechanisms, given the iron ligand and its predicted structure, which will require thorough testing to determine the correct one.
We propose that four of the residues identified play crucial roles in the enzymatic mechanism of RPE65, whatever its precise characteristics. First, the graded pattern of activity of mutants of Trp-331, a binding cleft residue we predict to interact with the substrate/intermediate (distance < 5 Å from C11), is consistent with the established π-binding energy series of the aromatic residues: W > Y > F for cation-π interactions. The predominant role for Trp in enzyme catalysis is as a stabilizer of reaction intermediates (54). Trp-331 is not replaceable by non-aromatic residues such as Leu or Gln. Second, because Tyr-239 is not replaceable, a tyrosine-specific requirement at this position is more likely than a role in carbocation stabilization. Tyr-239 is the only tyrosine predicted to be within 4 Å of the substrate, and its location is close to the carboxyl of the acyl moiety of all-trans retinyl ester. As such, its phenolic side chain may be positioned to donate a proton to the palmitate anion product, consistent with a major role for tyrosines in proton shuttling in addition to stabilization of reaction intermediates (54).
The third and most important finding we present here is that we can modulate the ratio of cis isomers produced by RPE65 by single amino acid changes to Thr-147 and Phe-103, thereby affecting catalysis. This provides an ineluctable case for a carbocation/radical cation intermediate by showing that product isomeric conformation is dependent on the binding cleft environment. We predict that, to adopt the 11-cis conformation, the polyene chain is constrained in a particular orientation that has to interact with cleft residues, such as Phe-103 and Thr-147. How this comes about may be analogous in some respects to the generation of the cranked cis-trans-cis conformation of apocarotenal substrate in ACO (22), but the precise details are not known at this point. We show that 11-cis synthesis by Phe-103 mutants is lowered while 13-cis synthesis is not as adversely affected, or even enhanced. Thus, Phe-103 is not essential for the gross isomerase activity of RPE65 but is pivotal for its specificity. We suggest that this is due to specific constraints that the side chain of Phe places on the putative intermediate to enhance an 11-cis isomer outcome, whereas relief of this constraint by mutating Phe reduces the 11-cis outcome but enhances 13-cis outcome. Thr-147 is less pivotal, and opposite in action to Phe-103, with mutation to Ser modestly enhancing 11-cis production relative to 13-cis. It is possible that the T147S effect is secondary to an interaction with another residue. These findings bridge two separate avenues of thought on the mechanism of retinol isomerization. Deigner et al. (29) established C15 oxygen exchange concomitant with inversion of stereochemistry as a hallmark of 11-cis-retinol isomerization. McBee et al. (35), in addition, showed that 13-cis-retinol is produced, and its C15 oxygen is also replaced when a bovine RPE microsome isomerization reaction is supplemented with cellular retinol-binding protein 1, thereby prompting them to suggest a carbocation mechanism for retinol isomerization. Accordingly, we used 18O-substituted retinol to corroborate the enzymatic origin of the 13-cis-retinol produced by RPE65, showing that the C15 oxygen is replaced in 13-cis-retinol as well as 11-cis-retinol in our experiments. These classic findings (29), predating the identification of RPE65 as the retinol isomerase, define endpoints for retinol isomerization that are met for both 11-cis- and 13-cis-retinol products in our experiments with RPE65. At this juncture, the precise geometry as to how Thr-147 and Phe-103 interact with substrate to affect product fidelity is not known and will require a crystal structure for precise clarification.
In conclusion, RPE65 may harness the ability of retinoids to form carbocations (55) or radical cations (56) to produce cis isomers of retinol. Although RPE65 can generate both 11-cis- and 13-cis-retinol, the innate selectivity of the visual cycle for 11-cis retinoids (35) ensures that only 11-cis-retinol is made under normal physiological conditions. As in aberrant physiological states (28), a relaxation in isomer selectivity allows 13-cis-retinol formation to occur. Taken together, our findings specify that: 1) retinoid isomerization in the visual cycle occurs by a carbocation/radical cation mechanism and 2) 11-cis selectivity ultimately depends on visual cycle proteins downstream of RPE65.
Supplementary Material
Acknowledgment
We thank Dr. John Saari for the gift of mouse monoclonal antibody to CRALBP.
This work was supported, in whole or in part, by National Institutes of Health Intramural Research Program of NEI.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1 and Figs. S1–S3.
Poliakov, E., Gentleman, S., Chander, P., Cunningham, F. X., Jr., Grigorenko, B. L., Nemuhin, A. V., and Redmond, T. M. (2009) BMC Biochem. 10, 31.
- RPE
- retinal pigment epithelium
- ACO
- apocarotenal oxygenase
- BCMO1 and BCMO2
- β-carotene monooxygenases 1 and 2
- CRALBP
- cellular retinaldehyde-binding protein
- MALDI-TOF
- matrix-assisted laser desorption ionization-time of flight
- HPLC
- high-performance liquid chromatography
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- aa
- amino acid(s).
REFERENCES
- 1.Saari J. C. (2000) Invest. Ophthalmol. Vis. Sci. 41, 337–348 [PubMed] [Google Scholar]
- 2.Lamb T. D., Pugh E. N., Jr. (2004) Prog. Retin. Eye Res. 23, 307–380 [DOI] [PubMed] [Google Scholar]
- 3.Moiseyev G., Crouch R. K., Goletz P., Oatis J., Jr., Redmond T. M., Ma J. X. (2003) Biochemistry 42, 2229–2238 [DOI] [PubMed] [Google Scholar]
- 4.Gollapalli D. R., Rando R. R. (2003) Biochemistry 42, 5809–5818 [DOI] [PubMed] [Google Scholar]
- 5.Hamel C. P., Tsilou E., Pfeffer B. A., Hooks J. J., Detrick B., Redmond T. M. (1993) J. Biol. Chem. 268, 15751–15757 [PubMed] [Google Scholar]
- 6.Jin M., Li S., Moghrabi W. N., Sun H., Travis G. H. (2005) Cell 122, 449–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moiseyev G., Chen Y., Takahashi Y., Wu B. X., Ma J. X. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 12413–12418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Redmond T. M., Poliakov E., Yu S., Tsai J. Y., Lu Z., Gentleman S. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 13658–13663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Redmond T. M., Yu S., Lee E., Bok D., Hamasaki D., Chen N., Goletz P., Ma J. X., Crouch R. K., Pfeifer K. (1998) Nat. Genet. 20, 344–351 [DOI] [PubMed] [Google Scholar]
- 10.Gu S. M., Thompson D. A., Srikumari C. R., Lorenz B., Finckh U., Nicoletti A., Murthy K. R., Rathmann M., Kumaramanickavel G., Denton M. J., Gal A. (1997) Nat. Genet. 17, 194–197 [DOI] [PubMed] [Google Scholar]
- 11.Marlhens F., Bareil C., Griffoin J. M., Zrenner E., Amalric P., Eliaou C., Liu S. Y., Harris E., Redmond T. M., Arnaud B., Claustres M., Hamel C. P. (1997) Nat. Genet. 17, 139–141 [DOI] [PubMed] [Google Scholar]
- 12.Morimura H., Fishman G. A., Grover S. A., Fulton A. B., Berson E. L., Dryja T. P. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 3088–3093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thompson D. A., Gyürüs P., Fleischer L. L., Bingham E. L., McHenry C. L., Apfelstedt-Sylla E., Zrenner E., Lorenz B., Richards J. E., Jacobson S. G., Sieving P. A., Gal A. (2000) Invest. Ophthalmol. Vis. Sci. 41, 4293–4299 [PubMed] [Google Scholar]
- 14.Maguire A. M., Simonelli F., Pierce E. A., Pugh E. N., Jr., Mingozzi F., Bennicelli J., Banfi S., Marshall K. A., Testa F., Surace E. M., Rossi S., Lyubarsky A., Arruda V. R., Konkle B., Stone E., Sun J., Jacobs J., Dell'Osso L., Hertle R., Ma J. X., Redmond T. M., Zhu X., Hauck B., Zelenaia O., Shindler K. S., Maguire M. G., Wright J. F., Volpe N. J., McDonnell J. W., Auricchio A., High K. A., Bennett J. (2008) N. Engl. J. Med. 358, 2240–2248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bainbridge J. W., Smith A. J., Barker S. S., Robbie S., Henderson R., Balaggan K., Viswanathan A., Holder G. E., Stockman A., Tyler N., Petersen-Jones S., Bhattacharya S. S., Thrasher A. J., Fitzke F. W., Carter B. J., Rubin G. S., Moore A. T., Ali R. R. (2008) N. Engl. J. Med. 358, 2231–2239 [DOI] [PubMed] [Google Scholar]
- 16.Cideciyan A. V., Aleman T. S., Boye S. L., Schwartz S. B., Kaushal S., Roman A. J., Pang J. J., Sumaroka A., Windsor E. A., Wilson J. M., Flotte T. R., Fishman G. A., Heon E., Stone E. M., Byrne B. J., Jacobson S. G., Hauswirth W. W. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 15112–15117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paik J., During A., Harrison E. H., Mendelsohn C. L., Lai K., Blaner W. S. (2001) J. Biol. Chem. 276, 32160–32168 [DOI] [PubMed] [Google Scholar]
- 18.Redmond T. M., Gentleman S., Duncan T., Yu S., Wiggert B., Gantt E., Cunningham F. X., Jr. (2001) J. Biol. Chem. 276, 6560–6565 [DOI] [PubMed] [Google Scholar]
- 19.Kiefer C., Hessel S., Lampert J. M., Vogt K., Lederer M. O., Breithaupt D. E., von Lintig J. (2001) J. Biol. Chem. 276, 14110–14116 [DOI] [PubMed] [Google Scholar]
- 20.von Lintig J., Vogt K. (2000) J. Biol. Chem. 275, 11915–11920 [DOI] [PubMed] [Google Scholar]
- 21.Oberhauser V., Voolstra O., Bangert A., von Lintig J., Vogt K. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 19000–19005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kloer D. P., Ruch S., Al-Babili S., Beyer P., Schulz G. E. (2005) Science 308, 267–269 [DOI] [PubMed] [Google Scholar]
- 23.Poliakov E., Gentleman S., Cunningham F. X., Jr., Miller-Ihli N. J., Redmond T. M. (2005) J. Biol. Chem. 280, 29217–29223 [DOI] [PubMed] [Google Scholar]
- 24.Hubbard R., Wald G. (1952) J. Gen. Physiol. 36, 269–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reuter T. (1964) Nature 204, 784–785 [DOI] [PubMed] [Google Scholar]
- 26.Fan J., Rohrer B., Moiseyev G., Ma J. X., Crouch R. K. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 13662–13667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Driessen C. A., Winkens H. J., Hoffmann K., Kuhlmann L. D., Janssen B. P., Van Vugt A. H., Van Hooser J. P., Wieringa B. E., Deutman A. F., Palczewski K., Ruether K., Janssen J. J. (2000) Mol. Cell Biol. 20, 4275–4287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maeda A., Maeda T., Imanishi Y., Golczak M., Moise A. R., Palczewski K. (2006) Biochemistry 45, 4210–4219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deigner P. S., Law W. C., Cañada F. J., Rando R. R. (1989) Science 244, 968–971 [DOI] [PubMed] [Google Scholar]
- 30.Rando R. R. (1991) Biochemistry 30, 595–602 [DOI] [PubMed] [Google Scholar]
- 31.Carlson A., Bok D. (1992) Biochemistry 31, 9056–9062 [DOI] [PubMed] [Google Scholar]
- 32.Stecher H., Gelb M. H., Saari J. C., Palczewski K. (1999) J. Biol. Chem. 274, 8577–8585 [DOI] [PubMed] [Google Scholar]
- 33.Winston A., Rando R. R. (1998) Biochemistry 37, 2044–2050 [DOI] [PubMed] [Google Scholar]
- 34.Kuksa V., Imanishi Y., Batten M., Palczewski K., Moise A. R. (2003) Vision Res. 43, 2959–2981 [DOI] [PubMed] [Google Scholar]
- 35.McBee J. K., Kuksa V., Alvarez R., de Lera A. R., Prezhdo O., Haeseleer F., Sokal I., Palczewski K. (2000) Biochemistry 39, 11370–11380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Landers G. M., Olson J. A. (1988) J. Chromatogr. 438, 383–392 [DOI] [PubMed] [Google Scholar]
- 37.Wingerath T., Kirsch D., Spengler B., Stahl W. (1999) Analyt. Biochem. 272, 232–242 [DOI] [PubMed] [Google Scholar]
- 38.Schüttelkopf A. W., van Aalten D. M. F. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 1355–1363 [DOI] [PubMed] [Google Scholar]
- 39.Redmond T. M., Weber C. H., Poliakov E., Yu S., Gentleman S. (2007) Mol. Vis. 13, 1813–1821 [PubMed] [Google Scholar]
- 40.Lorenz B., Poliakov E., Schambeck M., Friedburg C., Preising M. N., Redmond T. M. (2008) Invest. Ophthalmol. Vis. Sci. 49, 5235–5242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rando R. R., Chang A. (1983) J. Am. Chem. Soc. 105, 2879–2882 [Google Scholar]
- 42.Cañada F. J., Law W. C., Rando R. R., Yamamoto T., Derguini F., Nakanishi K. (1990) Biochemistry 29, 9690–9697 [DOI] [PubMed] [Google Scholar]
- 43.Golczak M., Kuksa V., Maeda T., Moise A. R., Palczewski K. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 8162–8167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grütter C., Alonso E., Chougnet A., Woggon W. D. (2006) Angewandte Chemie 45, 1126–1130 [DOI] [PubMed] [Google Scholar]
- 45.Jenson C., Jorgensen W. L. (1997) J. Am. Chem. Soc. 119, 10846–10854 [Google Scholar]
- 46.Lesburg C. A., Caruthers J. M., Paschall C. M., Christianson D. W. (1998) Curr. Opin. Struct. Biol. 8, 695–703 [DOI] [PubMed] [Google Scholar]
- 47.Bouvier F., d'Harlingue A., Camara B. (1997) Arch. Biochem. Biophys. 346, 53–64 [DOI] [PubMed] [Google Scholar]
- 48.Dougherty D. A. (1996) Science 271, 163–168 [DOI] [PubMed] [Google Scholar]
- 49.Mecozzi S., West A. P., Jr., Dougherty D. A. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 10566–10571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Borowski T., Blomberg M. R., Siegbahn P. E. (2008) Chemistry 14, 2264–2276 [DOI] [PubMed] [Google Scholar]
- 51.Klinman J. P. (2006) J. Biol. Chem. 281, 3013–3016 [DOI] [PubMed] [Google Scholar]
- 52.Prigge S. T., Eipper B. A., Mains R. E., Amzel L. M. (2004) Science 304, 864–867 [DOI] [PubMed] [Google Scholar]
- 53.Kovaleva E. G., Lipscomb J. D. (2007) Science 316, 453–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Holliday G. L., Mitchell J. B., Thornton J. M. (2009) J. Mol. Biol. 390, 560–577 [DOI] [PubMed] [Google Scholar]
- 55.Pienta N. J., Kessler R. J. (1992) J. Am. Chem. Soc. 114, 2419–2428 [Google Scholar]
- 56.Gurzadyan G. G., Reynisson J., Steenken S. (2007) Phys. Chem. Chem. Phys. 9, 288–298 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.