

Abstract
Onconase® (ONC) is an amphibian member of the bovine pancreatic ribonuclease (RNase A) superfamily that exhibits innate antitumoral activity. ONC has been granted both orphan-drug and fast-track status by the U.S. Food and Drug Administration for the treatment of malignant mesothelioma, and is poised to become the first chemotherapeutic agent based on a ribonuclease. Investigations into the mechanism of ribonuclease-based cytotoxicity have elucidated several important determinants for cytotoxicity, including efficient deliverance of ribonucleolytic activity to the cytosol and preservation of conformation stability. Nevertheless, the most-striking similarity between ONC and bovine seminal ribonuclease, another naturally cytotoxic ribonuclease, is their insensitivity to inhibition by the potent cytosolic ribonuclease inhibitor protein (RI). RI typically binds to its ribonuclease ligands with femtomolar affinity—an extraordinary feat considering the lack of sequence identity among the bound ribonucleases. Mammalian ribonucleases such as RNase A or its human homologue, RNase 1, have the potential to be more desirable chemotherapeutic agents than ONC owing to their higher catalytic activity, low potential for immunogenicity, favorable tissue distribution, and high therapeutic index, but are limited by their sensitivity to RI. These non-toxic mammalian ribonucleases can be transformed into potent cytotoxins by engendering them with RI-evasion using protein engineering strategies such as site-directed mutagenesis, multimerization, fusion to a targeting moiety, and chemical modification. In several instances, these engineered ribonucleases exhibit greater cytotoxicity in vitro than does ONC. Herein, we review the biochemical characteristics of RI·ribonuclease complexes and progress towards the development of mammalian ribonuclease-based chemotherapeutics through the elicitation of RI-evasion.
Keywords: cancer, cytotoxin, pancreatic ribonuclease, ribonuclease A, ribonuclease inhibitor
INTRODUCTION
Since its discovery over eighty years ago [1], bovine pancreatic ribonuclease (RNase A; EC 3.1.27.5) has provided chemical enzymologists, biophysical chemists, structural biologists, and evolutionary biologists with a highly tractable model system. The result has been a myriad of advances in numerous disciplines [2–5]. RNase A catalyzes the cleavage of RNA at a rate approaching the diffusion limit [6] and putatively functions to break down the large amounts of RNA that accumulate in the ruminant gut [7]. In the last several decades, owing to the discovery of homologues of RNase A that exhibit cytotoxic activity at low concentrations both in vitro and in vivo [8–10], interest in this enzyme family has experienced a resurgence directed at exploiting these activities for therapeutic purposes [11–16].
Many of the details of the mechanism of ribonuclease-mediated cytotoxicity are unclear. Nonetheless, several major aspects of the pathway have been elucidated, allowing for the construction of the mechanism depicted in Fig. (1) [11, 14, 17]. The first step in ribonuclease-mediated cytotoxicity is the association of the cationic ribonuclease with the anionic cell membrane [Fig. (1A)] [17, 18]. For many members of the RNase A superfamily, this absorptive process has been shown to be non-saturable and not receptor-mediated [17], but is instead governed by electrostatic association with either heparan sulfate proteoglycans [19–21] or the head groups of phospholipids [22]. The importance of Coulombic interactions in the cellular association and subsequent internalization of ribonucleases has been demonstrated by using both chemical modification [18] and site-directed mutagenesis [23, 24]. Notable exceptions exhibiting more specific cellular interactions include the amphibian sialic acid-binding lectins (SBLs) from Rana catesbeiana and Rana japonica, whose high affinity for sialic acid groups enabled the initial isolation of these ribonucleases [25]. Additionally, the amphibian ribonuclease, Onconase® (ONC; ranpirnase), has been reported to bind to specific sites on cultured 9L glioma cells [26], although these putative receptors have never been identified and could be absent from other cell types [17]. Furthermore, two proteins that bind to angiogenin (ANG; RNase 5) have been identified on the surface of human endothelial cells [27, 28]. Regardless of the exact nature of their cellular association, the subsequent internalization and translocation of ribonucleases into the cytosol [Fig. (1B–C)] has been shown to be an essential aspect of ribonuclease-mediated cytotoxicity [29]. Ribonuclease cytotoxicity has been correlated with the efficiency of endocytic internalization [30], though its route to the cytosol is in dispute [17, 31].
Fig. 1.

Putative mechanism of ribonuclease-based cytotoxicity. A) Ribonucleases bind to the cell surface and B) enter a cell via endocytosis, where C) a fraction escapes from acidifying endosomes and translocates into the cytosol. D) In the cytosol, ribonucleases are either E) bound and inhibited by RI or F) evade RI, allowing them to degrade cellular RNA and ultimately cause cell death.
Many protein toxins derived from bacteria and plants contain a separate domain for the express purpose of translocating across the vesicular membrane to their cytosolic site of action [32, 33]. In contrast, pancreatic ribonucleases are single-domain proteins for which the mechanism of translocation into the cytosol is understood only poorly. For cytotoxic variants of RNase A, escape from the acidifying endosome occurs prior to reaching the trans-Golgi network [17] and is a relatively inefficient process, as <95% of the internalized ribonuclease is degraded in lysosomal compartments without ever reaching the cytosol ([34–36]; Chao, T.-Y.; Lavis, L.D. and Raines, R.T., unpublished results). Ultimately, a few molecules of ribonuclease reach the cytosol, and there encounter the ribonuclease inhibitor protein (RI) [Fig. (1D)]. Tightly bound ribonucleases are rendered harmless to the cell [37] [Fig. (1E)]. In contrast, ribonucleases that are naturally RI-evasive, or have been engineered to be so, degrade cellular RNA and induce apoptosis [Fig. (1F)] [38]. Additional mechanisms unrelated to the disruption of protein synthesis have been proposed for the elicitation of an apoptotic response by ONC [39, 40].
Considering this model of ribonuclease-mediated cytotoxicity, several attributes of a ribonuclease can be identified as determinants of cytotoxicity, and thus represent targets for modification and optimization by the protein engineer. These properties include the ability to bind to the cell surface and be internalized efficiently (which includes the ability to reach the cytosol) [24], the preservation of native three-dimensional structure (i.e., conformational stability) [41], and the ability to catalyze the degradation of RNA [42], specifically in the presence of RI (i.e., RI-evasion) [43–46]. Protein design and engineering efforts aimed at modulating each of these characteristics with the goal of increasing cytotoxic activity have yielded impressive results, while informing our understanding of ribonuclease-mediated cytotoxicity [23, 41, 47, 48]. Much of this work, including tumor cell-targeting [49–51] and sub-cellular localization [36, 52], is beyond the scope of this review but has been described elsewhere [47, 53–55].
Here, we consider the anomalously high affinity of RI for its secretory ribonuclease ligands and the tremendous gains in cytotoxicity that can be made by disruption of this association. We review the strategies nature has used to achieve RI-evasive cytotoxic ribonucleases as well as the state of the art of engendering RI-evasion with the goal of developing ribonuclease-based chemotherapeutics. Before describing the explicit strategies used by nature and protein engineers to achieve RI-evasion, we review RI and its protein complexes so as to provide a detailed understanding of the task at hand and to underscore just how daunting that task can be.
RIBONUCLEASE INHIBITOR
RI is a ~50-kDa protein found exclusively in the cytosol of mammalian cells. (For recent reviews of RI, see [56–58].) Its obligate cytosolic localization maintains its 29 (bovine RI), 30 (porcine RI), or 32 (human RI) cysteine residues in a reduced state, which is required for its function as an inhibitor [59]. Nevertheless, all known ligands of RI are secreted ribonucleases. This apparent paradox, in conjunction with its invariant high concentration of ~4 μM in the cytosol [37, 43], implies a biological role for RI as a intracellular “sentry” to safeguard the cell from the potentially damaging activity of adventitious ribonucleases [37, 60–62]. Further support of this hypothesis comes from experiments in which cytosolic RI is over-produced, diminishing the potency of toxic RNase A variants [37]. Conversely, silencing of RI gene expression by RNA interference increases the sensitivity of cells to exogenous cytotoxic ribonucleases [63]. Other proposed biological roles for RI include the modulation of the biological function of various ribonucleases (e.g., the angiogenic activity of ANG in promoting neovascularization) or as an oxidative sensor to monitor the redox status or age of a cell [64–66]. Despite the uncertainty surrounding the precise physiological role of RI, its potent inhibitory effects have had a strong influence on the natural and artificial evolution of cytotoxic ribonucleases.
RI·RIBONUCLEASE COMPLEXES
RI binds to members of the RNase A superfamily with a 1:1 stoichiometry, inhibiting completely their catalytic activity by steric occlusion of the enzymic active site [67]. The noncovalent complexes formed by RI and its ligands (Kd = 0.3–2.7 × 10−15 M) are among the tightest known in biology [Table 1; Fig. (2)]. The affinity of RI for its ligands is comparable to that of avidin for biotin [68], protein trypsin inhibitor (PTI) for trypsin [69], tissue inhibitor of metalloproteinase (TIMP) for matrix metalloproteases [70], and barstar for barnase [71]. Astonishingly, RI has evolved anomalously high affinity towards protein ligands of low aminoacid sequence identity (≥23%; Table 2). During the last 15 years, many of the atomic details that enable these dichotomous, high affinity/broad specificity interactions have been illuminated by the determination of the three-dimensional structure of free RI [72] as well as that of four RI·ligand complexes [73–76] [Table 3; Fig. (3A,B)].
Table 1.
Comparison of RI·Ribonuclease Complexes to Other Biomolecular Interactions
| Biomolecular complexa | ASA buried (Å2)b | Scc | Kd (M) | Ref. |
|---|---|---|---|---|
| RI·ribonuclease | 2583–3438 | 0.67 ± 0.06 | 0.3–2.7 × 10−15 | 65, 76, 85, 130 |
| Strength of interaction | ||||
| Im9·colicin E9 DNase | 9.3 × 10−17 | 185 | ||
| PTI·trypsin | 1430 | 6 × 10−15 | 69 | |
| biotin·avidin | 0.6 × 10−15 | 68 | ||
| TIMP-2·MMP-2 | 0.6 × 10−15 | 70 | ||
| barstar·barnase | 1590 | 0.70 (0.82)d | 1.3 × 10−14 | 71, 89 |
| Interface area | ||||
| Gtβγ·Phosducin | 4660 | 186, 187 | ||
| EF-Tu·EF-Ts | 3660 | 187, 188 | ||
| inhibitor·enzyme | 2030 ± 630 | 187 | ||
| antibody·antigen | 1680 ± 260 | 0.64–0.68 | 187 | |
| inhibitor·protease | 1530 ± 170 | 0.70–0.76 | 187 | |
| all complexes | 1940 ± 760 | 187 | ||
Im9, immunity protein 9; PTI, protein trypsin inhibitor; TIMP-2, tissue inhibitor of metalloproteinase-2; MMP-2, matrix metalloproteinase-2 (gelatinase A).
Buried accessible surface area (ASA) was calculated with the program NACCESS [189].
The value of Sc reports on geometrical shape complementarity, where Sc = 1.0 for two perfectly complementary surfaces and Sc = 0 for two completely dissimilar surfaces [190].
Value of Sc increases when buried solvent is included in the calculation [89].
Fig. 2.

Typical values of Kd for common biomolecular complexes and precise values of Kd for RI·ribonuclease complexes with known three-dimensional structures (Table 3). The lengths of the vertical lines are proportional to the buried surface areas in the RI·ribonuclease complexes.
Table 2.
RI-Affinity and Amino-Acid Sequence Identity/Similaritya of Bovine Pancreatic Ribonuclease Superfamily Members
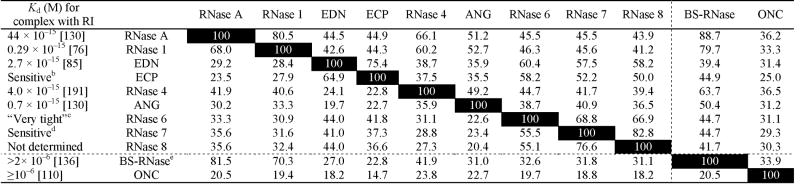 |
The dotted lines separate RI-sensitive and RI-insensitive ribonucleases.
Sequence identities (%) are to the left of the diagonal; sequence similarities (%) are to the right of the diagonal. Both were calculated with the program MacVector v9.5 (MacVector, Inc., Cary, NC). The pairwise matrix was performed with the Gonnet similarity matrix and the default parameters in the “slow” mode. (Open Gap Penalty = 10.0; Extend Gap Penalty = 0.1. Sequences of the human ribonucleases were obtained by using the accession numbers reported in ref. 90. Accession numbers used for RNase A, BS-RNase, and ONC were NM_001014386, NM_181810, and AF332139, respectively.
The ribonucleolytic activity of ECP was shown to be sensitive to a commercial preparation of RI [192].
The interaction of hRI with RNase 6 was described as being “very tight” [74].
A pyrimidine-specific, ~13-kDa enzyme with endoribonucleolytic activity isolated from human skin was sensitive to hRI [193]. RNase 7 is expressed primarily in keratinocytes [194].
Only the dimeric form of BS-RNase is insensitive to RI. hRI has a Ki value of 9.3 × 10−12 M for monomeric C31K/C32S BS-RNase [136].
Table 3.
Characteristics of RI·Ribonuclease Complexes with Known Three-dimensional Structures
| No. of contact residuesd | Character of interface residuese [No. (%)] | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Complex | Kda (fM) | Buried ASAb (Å2) | Scc | From RI | From RNase | Non-polar | Uncharged polar | Charged | H-bondsd | Non-bonded contactsd | PDB code |
| pRI·RNase A | 67 | 2583 | 0.58 | 26 | 23 | 14 (29%) | 13 (27%) | 22 (45%) | 8 (3.01) | 90 | 1djf [73] |
| hRI·RNase 1 | 0.29 | 2802 | 0.70 | 28 | 23 | 17 (33%) | 14 (27%) | 20 (39%) | 19 (2.79) | 177 | 1z7xf [76] |
| hRI·EDN | 2.7 | 3438 | 0.69 | 42 | 31 | 23 (32%) | 26 (36%) | 24 (33%) | 27 (2.87) | 256 | 2bexg [75] |
| hRI·ANG | 0.7 | 2659 | 0.70 | 30 | 28 | 16 (28%) | 17 (29%) | 25 (43%) | 14 (2.92) | 141 | 1a4yh [74] |
Buried accessible surface area (ASA) was calculated with the program NACCESS [189].
Shape complementarity values (Sc) for pRI·RNase A, hRI·EDN, and hRI·ANG were from refs 67, 75, and 74, respectively. Sc for hRI·RNase 1 was calculated with SC v5.0 [190] and the default parameters: dot density = 15.00/Å2; interface separation = 8.00 Å; trim width = 1.50 Å; probe radius = 1.70 Å; weight factor = 0.50/Å2.
Number of contact residues, H-bonds, and non-bonded contacts were determined with the program PDBsum [195]. These values differ from those reported previously [67, 74, 75], but this uniform analysis enables more meaningful comparisons between the complexes.
Contact residues were identified by PDBsum as non-polar (A,F,G,I,L,M,P,V,W,Y), uncharged polar (C,N,Q,S,T), or charged (D,E,H,K,R).
Calculations were performed with chain Y (hRI) and chain Z (RNase 1) due to the presence of bound citrate in the active site of RNase 1 in the other complex in the asymmetric unit.
Calculations were performed with chain A (hRI) and chain C (EDN).
Calculations were performed with chain A (hRI) and chain B (ANG).
Fig. 3.

A) Overlay of the structures of six ribonucleases bound to hRI. Each ribonuclease was aligned with RNase 1 in the hRI(gray)·RNase 1(yellow) complex (chains Y and Z, respectively, of PDB entry 1z7x [76]). EDN (blue): chain C of 2bex [75]; ECP (green): 1qmt [202]; RNase 4 (teal): 1rnf [203]; ANG (pink): chain B of 1a4y [74]; RNase A (orange): 7rsa [204]. B) Aminoacid sequence alignment of the four ribonucleases whose structure has been solved in a complex with RI (RNase A, RNase 1, EDN, and ANG) and two RI-evasive homologues (BS-RNase and ONC) [205]. Residue numbers correspond to RNase A. The assigned secondary structure is above the sequence (α = α-helix; β = β-sheet) [67]. Residues that contact RI are in the colors of panel A. Completely conserved residues are boxed. Enzymic subsites of RNase A are indicated in italics typeface above the relevant residues [4, 206]. The sequences corresponding to the nuclear localization signal and receptor-binding loop of ANG are underlined [207, 208]. C) Overlay of the structures of cytotoxic amphibian ribonucleases (ONC: olive green, 1ONC [108]; Amphinase: forest green, 2p7s [99]; RC-RNase 3: pea green, 1z5f [104]) aligned with RNase A (orange, 7rsa [204]). D) Structural alignment of RNase A (black) and ONC (gray). The most prominent surface loops of RNase A are labeled explicitly. The side chains of the nine residues in these loops for which ONC possesses no analogous residues are shown explicitly. E) Structure of the M×M form of BS-RNase [209]. F) Structure of a monomeric subunit of the M=M form of BS-RNase (gold: 1r3m [210]) aligned with RNase 1 (not shown) in the hRI·RNase 1 complex [76]. Alignments were performed and images created with the program PyMOL (DeLano Scientific, South San Francisco, CA).
The highly symmetrical but nonglobular horseshoe-shape of RI provides a broad platform for extensive protein–protein interactions, which is a hallmark of leucine-rich repeat (LRR)-containing proteins [77]. Few heterologous protein complexes bury more solvent accessible surface area (ASA) than do RI·ribonuclease complexes, which bury 27–60% more surface area than do the average inhibitor·enzyme complex and 54–105% more surface area than is observed in typical antibody·antigen complexes (Table 1). Despite the expansive size of the interfaces formed between RI and its ligands, much of the free energy of complex formation is contributed by small regions or “hot spots” [78–80], with the majority of the interface contributing little to complex stability [46, 81–83]. For example, truncated variants of RI that lack residues 144–257 or 315–371 retain picomolar affinity for RNase A and ANG [84].
Cursory inspection of the four known RI·ribonuclease structures suggests that RI binds to each of its ligands in a similar manner. Approximately one-third of the kidney-shaped ligand protrudes into the central cavity of RI, which is lined exclusively with parallel β-strands contributed by 15 LRR domains. The active-site cleft of the bilobal ribonuclease is nestled against the C-terminal portion of RI, with the remainder of the ribonuclease spilling outside of the inner circumference of the horseshoe and onto the face of the inhibitor molecule [Fig. (3A)].
Actually, RI interacts distinctly with its ligands [Table 3; Fig. (3B)], binding each with extraordinary avidity despite their low sequence conservation. RI achieves this feat by recognizing features that are unique to each of its ribonuclease ligands. Although the 23–31 ribonuclease residues that mediate interactions with RI appear to be grouped in similar regions along the enzymic amino-acid sequences [Fig. (3B)], this apparent similarity of interaction is misleading because either the cognate contact residue on RI or the nature of the interaction usually differs among the ribonuclease ligands [75]. Further evidence for the unique means by which RI recognizes divergent ribonucleases has come from alanine-scanning mutagenesis studies, which demonstrate that both the location of the energetic “hot-spots” and the nature of the cooperativity between these regions vary greatly from one ligand to another [75, 81, 82, 85]. Another illustration of the divergence of interactions within the various RI·ribonuclease interfaces is provided by the ability to endow RI with selectivity for different ribonuclease ligands. For example, subtle remodeling of a single loop provided >109-fold selectivity for ANG over RNase A [86]. Finally, the lack of conservation among the modes of ligand-recognition is evidenced most succinctly by the conservation of only two hydrogen-bond interactions in the four known structures of RI·ribonuclease complexes [75, 76] [Fig. (3B); Table 4].
Table 4.
Equivalent Interactions in RI·Ribonuclease Complexes with Known Three-dimensional Structures
| RI | RNase A | RNase 1 | EDN | ANG |
|---|---|---|---|---|
| Hydrogen bonds | ||||
| Asp435 | Lys41a | Lys41 | Lys38 | Lys40 |
| Ser460b | Gln11 | Gln11 | Trp10 | Gln12 |
| van der Waals contacts | ||||
| Tyr434 | Lys41 | Lys41 | Lys38 | Lys40 |
| Tyr437 | His119 | His119 | His129 | His114 |
This contact was not observed in the crystal structure [67] due to the presence of a bound sulfate molecule, but its existence has been inferred from site-directed mutagenesis experiments [81, 196].
Although the ribonuclease residues do not correspond in position in the sequence alignment [Fig. (3B)], these residues occupy similar positions in the three-dimensional structure. This hydrogen bond is mediated by the C-terminal carboxyl group of RI with RNase A and RNase 1, but by the side-chain hydroxyl group with ANG and EDN.
The problem of preserving the integrity of intracellular RNA is not unique to vertebrates. The prokaryote Bacillus amyloliquifaciens produces the intracellular ribonuclease inhibitor barstar for protection from its cognate extracellular ribonuclease, barnase [87]. Thus, across two domains of life, nature has chosen to solve a fundamentally identical problem by using the same strategy—a cytosolic proteinaceous inhibitor with extraordinary affinity for its potentially lethal ligand. Though the bacterial barstar–barnase system bears no evolutionary or structural relatedness to the RI–ribonuclease system, it is both interesting and informative to compare and contrast these two solutions to an ancient problem.
Barnase and barstar have a high affinity for one another (Kd ~1014 M [71]), but the means by which this affinity is achieved differ significantly from that of RI and its ribonuclease ligands. Electrostatic interactions play a large role in the formation of both complexes, particularly in the use of electrostatic steering to enhance the association rate [76, 88]. The surface area buried at the interface of the barstar·barnase complex (1590 Å2) is, however, dramatically less than that buried upon ligand binding by RI (2600–3400 Å2). Also, buried solvent molecules play a much larger role in the barstar·barnase complex, filling otherwise deleterious voids at the interface and also increasing shape complementarity from Sc = 0.70 to Sc = 0.82. The latter value is significantly greater than the shape complementarity of, for example, the complex between porcine RI (pRI) and RNase A (Sc = 0.58). In addition, barstar comes much closer to mimicking the interactions with an RNA substrate than does RI [89].
Compared to barstar, RI has the greater challenge of inactivating numerous divergent ligands and has evolved to do so with femtomolar affinity for each. In humans, RI has as many as 13 potential ribonuclease ligands. There are genes encoding 20 and 17 ribonucleases in the mouse and rat genomes, respectively [90]. Exposure of human tumor cells to exogenous but homologous ribonucleases that are naturally RI-evasive, such as amphibian ribonucleases and BS-RNase has demonstrated their potential as cancer chemotherapeutic agents. Next, we review briefly the structural basis for the natural evasion of the amphibian ribonucleases and BS-RNase. Finally, inspired by these naturally RI-evasive ribonucleases, we describe efforts to engineer otherwise non-toxic mammalian ribonucleases to evade RI, thereby unleashing their cytotoxicity and enabling the development of new chemotherapeutic agents.
EVASION OF RI BY NATURAL CYTOTOXIC RIBONUCLEASES
The first observation of ribonuclease-mediated cytotoxicity occurred in the 1950’s, when RNase A was shown to be toxic to tumor cells both in vitro [91] and in vivo [92–94]. Effects were observed only after milligram quantities of enzyme were injected into solid tumors—smaller doses of RNase A had no effect. Thirty years ago, BS-RNase, was discovered in bull seminal fluid and found to be cytotoxic at low concentrations [9, 95]. In the last two decades, more cytotoxic homologues were isolated from the eggs of the bullfrog Rana catesbeiana [96, 97], the Japanese rice paddy frog Rana japonica [98], and the Northern leopard frog Rana pipiens [10, 99]. These amphibian ribonucleases are toxic to tumor cells in vitro with IC50 values <1 μM [26, 99–101]. The ability of both BS-RNase and the amphibian ribonucleases to evade RI is a primary determinant of their cytotoxicity toward cancer cells. The aspects of their structures that enable them to evade RI are described below.
The Rana pipiens ribonuclease, ONC, deserves special attention as it is currently in a Phase IIIb confirmatory clinical trial as a second-line chemotherapic agent for malignant mesothelioma [102], and has been granted both orphan-drug and fast-track status by the U.S. Food and Drug Administration. In this trial ONC is being administered to patients by intravenous injection along with doxorubicin, and the life-extension is being compared to that from doxorubicin alone. ONC has also completed Phase II human clinical trials for the treatment of refractory breast cancer, prostate cancer, and metastatic kidney cancer [103]. The enzyme is administered intravenously, and is able to destroy cancer cells selectively. If the Phase IIIb trial is successful, ONC would be approved in 2008 for use as the first ribonuclease chemotherapeutic agent.
All amphibian ribonucleases characterized to date have been found to be toxic to mammalian tumor cells [99, 104], which is in stark contrast to mammalian ribonucleases. For this reason the structural and biochemical differences between amphibian and mammalian ribonucleases are of particular interest. Amphibian ribonucleases, which are isolated from oocytes and early embryos, preferentially cleave RNA between pyrimidine and guanine residues instead of the pyrimidine–adenine substrates preferred by RNase A [101]. Like human RNase 4 and ANG, most amphibian ribonucleases (with the exception of the recently described “Amphinase” variants from Rana pipiens [99]) contain an N-terminal pyroglutamate residue that, unlike in RNase 4 and ANG [105], plays an important role in catalysis by preorganizing active-site residues for binding to the rate-limiting transition state [104, 106]. Despite ribonucleolytic activity being required for cytotoxicity, amphibian ribonucleases typically exhibit markedly less ribonucleolytic activity than does RNase A [99, 101].
The divergent, compact structure of the amphibian ribonucleases enables them to evade RI. The amphibian ribonucleases share three of the four conserved disulfide bonds of RNase A, lacking the cystine residue corresponding to Cys65–Cys72 and possessing instead a fourth disulfide bond near the C-terminus [107]. Although amphibian ribonucleases and RNase A share a similar overall fold [104, 108, 109], they have low sequence identity. Moreover, the amphibian ribonucleases are typically shorter (104–111 amino-acid residues), giving rise to a more compact structure. The amino-acid deletions occur predominantly in surface loops, and decimate favorable interactions with RI [26, 67, 110] [Fig. (3B,C)]. For example, of the 24 residues of RNase A that contact RI within the pRI·RNase A complex, only three are conserved in ONC, three are replaced with similar residues, nine with dissimilar residues, and the remaining nine contact residues are found in the surface loops of RNase A and have no structural counterparts on ONC [67]. These nine unique residues in the surface loops of RNase A are shown explicitly in Fig. (3D).
BS-RNase is another member of the bovine pancreatic ribonuclease family that exerts selective toxicity toward cancer cells. The naturally dimeric nature of BS-RNase is unique among the members of the RNase A superfamily [111] and uses adverse steric interactions to preclude binding by RI [112]. The constituent RI-sensitive monomers (M) [Fig. (3F)] of the BS-RNase homodimer are linked by two intermolecular disulfide bonds between Cys31 of one subunit and Cys32 of the other. At equilibrium, ⅔ of the dimeric molecules are stabilized further by the swapping of N-terminal domains (denoted as “M×M”) [Fig. (3E)]. This additional degree of quaternary association affords greater protection against dissociation upon internalization into the reducing environment of the cytosol as compared to the non-domain swapped form (“M=M”) [113, 114]. The insensitivity of dimeric BS-RNase to inhibition by RI allows it to exert unique biological effects, including toxicity towards tumor cells as well as aspermatogenic, embryotoxic, and immunosuppressive activities [115, 116]. Thus, despite being >80% identical in sequence to RNase A, which is bound by RI with femtomolar affinity, the unique quaternary structure of BS-RNase renders it completely insensitive to RI.
Despite the discovery and characterization of these naturally-occurring cytotoxic ribonucleases, and the success of ONC in the clinic, the amphibian ribonucleases and BS-RNase exhibit at least five undesirable properties that could be circumvented by using related monomeric mammalian ribonucleases or their variants. (1) Greater potential immunogenicity. In general, exogenous proteins with a low degree of similarity to native homologues are immunogenic [117]. For example, RNase A shares 70% sequence identity with murine pancreatic ribonuclease (Rib1), whereas the amphibian ONC shares only 18% identity with Rib1, and mice produce neutralizing antibodies against ONC but not against RNase A [118]. In a clinical study investigating the use of RNase A for the treatment of tick-borne encephalitis, 1 gram of RNase A was administered to 246 patients over a 6-day period and produced no evident toxic or allergic effects [119]. It should be noted, however, that numerous clinical studies have found ONC to possess an acceptable safety profile, being generally well-tolerated by most patients [103]. (2) Unfavorable tissue distribution following administration. A practical limitation of ONC as a potential chemotherapeutic agent stems from its dose-limiting renal toxicity [103, 120]. Pharmacokinetic and biodistribution studies have revealed the renal retention of ONC to be 50- to 100-fold greater than that of mammalian members of the RNase A superfamily [121]. (3) Rapid clearance from circulation. Proteins having a low molecular mass (≤40 kDa) are cleared rapidly from circulation by the kidneys via renal filtration [122]. RNase A administered intravenously to mice and rats was observed to possess a half-life in circulation of 3.5 and 5 minutes, respectively [35, 123]. ONC is cleared at a similar rate [121]. Considering that ONC is administered intravenously on a weekly basis in its current clinical trial [103], modification of a ribonuclease to possess an enhanced time in circulation could permit less frequent or lower dosing, which would translate to improved tolerance, reduced cost, and greater patient compliance. (4) Modest ribonucleolytic activity (amphibian ribonucleases). As mentioned above, ribonucleolytic activity is essential for cytotoxicity [26, 42]. The catalytic efficiency of RNase A, BS-RNase, and human pancreatic ribonuclease (RNase 1) against known substrates is 104–105-fold greater than that of ONC [8, 106, 110], suggesting a much greater cytotoxic potential. (5) Dissociation of the BS-RNase dimer in the cytosol and subsequent inhibition by RI. The unique quaternary structure of BS-RNase, though a powerful deterrent of RI, is metastable, and thus diminishes its potential as a cytotoxic ribonuclease [45]. Moreover, BS-RNase is <10-fold less cytotoxic than ONC in a variety of assays [118].
Several microbial ribonucleases, including the prokaryotic RNase Sa3 from Streptomyces aureofaciens and a ribonuclease from Bacillus intermedius, have been found to be cytotoxic to human tumor cells [124, 125]. Additionally, ribotoxins from the fungal genus Aspergillus (such as α-sarcin, restrictocin, and mitogillin) exert cytotoxic activity toward mammalian cells by cleaving the 28 S RNA of the large ribosomal subunit and thereby inhibiting protein synthesis [126]. Not surprisingly, these microbial ribonucleases are not inhibited by RI [125, 127]. The lack of cancer-cell selectivity of fungal ribonucleases [25], along with the high immunogenicity and systemic toxicity of microbial enzymes [128, 129], precludes their use as human chemotherapeutics agents.
These limitations of naturally cytotoxic ribonucleases could be overcome by the development of new chemotherapeutic agents based on mammalian (especially human) ribonucleases. A major hurdle in realizing the full cytotoxic potential of mammalian ribonucleases is the potent inhibition of these enzymes by RI. The remainder of this review will focus on the various protein engineering strategies that have been applied towards thwarting the undesirable influence of RI on cytotoxicity.
ENGINEERING EVASION OF RI
A. SITE-DIRECTED MUTAGENESIS
One especially successful strategy for attenuating the affinity of RI for mammalian ribonucleases has been the use of site-directed mutagenesis to install steric or electrostatic incompatibilities in the interface of the complex. Although site-directed mutagenesis has elucidated the means by which RI recognizes its divergent ligands, this work has primarily involved making single or multiple alanine substitutions in RI. The RI variants have been characterized to reveal effects on the affinity of RI for native ribonuclease ligands [75, 81–83, 85]. The focus of this review is to describe progress that has been made through protein engineering toward the development of chemotherapeutic ribonucleases and thus is concerned only with the disruption of RI–ribonuclease interactions by the modification of ribonucleases.
Guided by the (then) recent report of the structure of the pRI·RNase A complex [67], our group first demonstrated the feasibility of transforming a non-toxic ribonuclease into a potent cytotoxin. In the pRI·RNase A complex, Gly88 was observed to reside in a hydrophobic pocket of pRI defined by Trp257, Trp259, and Trp314. By replacing Gly88 in the β4–β5 loop of RNase A with an arginine or aspartate residue, the affinity of RI was reduced by 104- and103-fold, respectively. Because of the high sequence identity between pRI and hRI (23 of the 28 contact residues in pRI are identical in hRI, and 3 are replaced conservatively), these contacts were expected to be preserved in the hRI·RNase A complex, which has a Kd value nearly identical to that of the pRI·RNase A complex [130, 131]. The specific amino acids used for the substitution were chosen to introduce a bulky protrusion or “knob” that would not be well-accommodated by any “hole” on the surface of RI. In essence, this strategy is the converse of alanine-scanning mutagenesis, which seeks to truncate the side chain so as to discern its contribution to the binding interaction [132]. Arginine and aspartate possess the two most polar side chains [133], whose hydration imparts yet additional bulk. Furthermore, arginine is the second largest amino acid. Importantly, both G88R RNase A and G88D RNase A were found to inhibit the proliferation of a continuous human erythroleukemia line (K-562) with IC50 values of 7 and 30 μM, respectively [43]. Though G88R RNase A is ~25-fold less toxic than ONC, it has inspired numerous subsequent efforts to disrupt the RI–RNase A interface further (See Table 5). These efforts ultimately resulted in variants such as K31A/D38R/R39D/N67R/G88R RNase A, whose multiple amino-acid substitutions disrupt simultaneously several regions within the expansive interface. The five substitutions in this variant decrease affinity for RI by 60 million-fold, and endow RNase A with a greater ability than ONC to inhibit the proliferation of human erythroleukemia cells in vitro. A related variant, D38R/R39D/N67R/G88R RNase A, displays cytotoxicity equal to or greater than that of ONC for four different human tumor cell lines [46]. Similar efforts to reduce the sensitivity of RNase 1 [44, 134], RNase 1-immunotoxins [135], and monomeric BS-RNase [45, 136] to inhibition by RI have proven the broad utility of this engineering strategy (Table 5).
Table 5.
Effect of Site-directed Mutagenesis on Affinity of Ribonucleases for hRI
| Ribonuclease | Variant | Ribonucleolytic Activity (%)a | Ki or Kd (nM) | IC50b (μM) | Ref. |
|---|---|---|---|---|---|
| ONC | Wild-type | 0.01–0.1c | >1000d | 0.3 | 46 |
| RNase A | Wild-type | 100 | 44 × 10−6e | >25 | 43, 46, 138, 196 |
| G88D | 56 | 0.052 | 30 | 43 | |
| G88R | 51 | 0.41 | 7 | 43 | |
| K41R/G88R | 1.4 | 3.0 | 2 | 138 | |
| K7A/G88R | 20 | 7.2 | 1 | 196 | |
| D38R/R39D/N67R/G88R | 73 | 510 | 0.2 | 46 | |
| K31A/D38R/R39D/N67R/G88R | 92 | 2500 | 0.2 | 46 | |
| RNase 1 | Wild-type | 100 | 0.3 × 10−6f | >10 | 44 |
| K7A/N88R/G89D/R91D | 115g | —h | 6.7 | 135 | |
| K41R/Q69A/N88R/G89D/R91D | 2.2g | —h | 7.4 | 135 | |
| L86E/N88R/G89D/R91D | 193 | 0.21 | 7 | 44 | |
| K7A/N71A/E111A | 115 | 0.5 | 5.55 | 134 | |
| R4C/L86E/N88R/G89D/R91D/V118C | 71 | 2.6 | 3 | 44 | |
| BS-RNase | C31A/C32A | 100 | 9.3 × 10−3i | >50 | 45 |
| C31K/C32S/G88R/S89E | 103 | 2.5 | 1.5j | 136 | |
| C31K/C32S/T87WG88R/S89E/S90W | 100 | 3.1 | 1.2j | 136 | |
| C31A/C32A/G88R | 121 | 2.3 | 0.11 | 45 | |
| C31A/C32A/G38K/K39G/G88R | 89 | 100 | 0.046 | 45 | |
The ribonucleolytic activity of each variant relative to the wild-type enzyme within each study.
Values of IC50 for variants of RNase A are for human erythroleukemia cells (K-562).
ONC is a 104–105-fold less active than RNase A against typical substrates [110].
Value of Kd for ONC is from ref. 110.
Value of Kd for wild-type RNase A is from ref. 130.
Values of Kd for RNase 1 are from ref. 76.
UpA was used as the substrate for enzymatic activity determinations.
Affinity for RI was determined only qualitatively (Kd <250 nM) but varied inversely with cytotoxicity.
Value of Kd is for C31K/C32S BS-RNase and is from ref. 136.
Values of IC50 are SV40-transformed mouse cells (SVT2).
Important considerations for selecting amino-acid substitutions to generate RI-evasive, and thus cytotoxic, ribonuclease variants are evident from the work summarized in Table 5 and reported elsewhere [24, 76]. Substitutions should be chosen to minimize deleterious effects on either catalytic activity or conformational stability (which is related to proteolytic susceptibility [41, 137]), as these attributes are important determinants of cytotoxicity [41, 138]. In addition, positive charge, manifested either as a high net molecular charge (Z) or in discrete regions of cationicity, is crucial for the favorable Coulombic interactions with anionic components of the cell surface [22–24, 48, 139].
Apparently, the site-directed mutagenesis approach has reached a limit [46]. The value of Kd for the complex of RI with highly evasive variants of RNase A is close to the cytosolic concentration of RI (~4 μM [37, 43]). Additional evasion of RI does not lead to a substantial increase in the concentration of free ribonuclease in the cytosol and hence does not increase cytotoxicity. Further increases in cytotoxicity are likely to be achievable, but will require modulating other parameters, such as cellular binding, uptake, and translocation.
B. MULTIMERIZATION
The naturally dimeric nature of BS-RNase is unique in the RNase A superfamily. Pro19, Leu28, Cys31, and Cys32 were identified as the residues that enable BS-RNase to form its unique M=M ⇄ M×M structure [140–142]. Replacement of the corresponding residues in RNase 1 (where necessary) with the aforementioned residues resulted in a variant that adopted a BS-RNase-like structure spontaneously [143]. This dimeric RNase 1, also composed of a mixture of M=M and M×M forms, was selectivity toxic towards human tumor cells, albeit with a reduced potency compared to BS-RNase. Adding the E111G substitution does, however, lead to a dimeric RNase 1 that is more cytotoxic than BS-RNase to a SV40-transformed 3T3 fibroblast cell line. The quaternary structure of these RNase 1 dimers was essential not only for cytotoxicity, but also to reduce their sensitivity to RI [144].
Subsequent to the discovery of the antitumoral activity of BS-RNase, interest grew in generating chemically-linked dimers of RNase A in an attempt to reproduce by semisynthesis the unique biological activities of BS-RNase [145, 146]. In contrast to BS-RNase or the noncovalent, domain-swapped oligomers of RNase A, which are metastable, dimers produced by chemical coupling are not able to dissociate [146]. Dimers of RNase A that were cross-linked with bifunctional amine-reactive imido esters, inhibited tumor cell proliferation in vitro [35], exhibited antitumoral activity in mice [147, 148], possessed both a newly acquired enzymatic activity against dsRNA, and had an enhanced persistence in the circulation of rats [123]. Furthermore, these cross-linked dimers have a reduced affinity for RI [123]. More recently, cross-linked trimers of RNase A generated with a dimethylsuberimidate linker displayed higher cytotoxic activity against a cervical carcinoma cell line than did cross-linked dimers of RNase A. The cross-linked trimers also possessed enhanced activity against dsRNA, compared to that of cross-linked dimers [149], conforming to the trend observed with the noncovalent domain-swapped oligomers of RNase A [150].
A marked disadvantage of utilizing amine-reactive linkers such as dimethylsuberimidate for the chemical cross-linking of RNase A is the heterogeneity of the products that derives from its eleven amino groups. This heterogeneity confounds biochemical characterization of the conjugates, making this methodology undesirable for the development of chemotherapeutic agents. Furthermore, modification of active-site lysine residues, such as Lys41, can reduce catalytic efficiency by up to 105-fold [151].
Homogeneous homodimers of both RNase 1 and eosinophil-derived neurotoxin (EDN; RNase 2) have been produced by introducing a cysteine residue via site-directed mutagenesis and reacting its thiol with 1,6-bis(maleimido)hexane to generate a thioether-linked dimer [152]. The cysteine residues were introduced at positions that were predicted to be the most disruptive to RI-binding. This site-specific tethering resulted in dimeric conjugates that were ≥104-fold less sensitive to inhibition by RI, and effectively transformed these two non-toxic, RI-sensitive, monomeric ribonucleases into cytotoxic dimers [152].
Recently, the in vacuo incubation of lyophilized RNase A at 85 °C was shown to promote the condensation of the side chains of Glu9 of one monomer and Lys66 of another to form a dimer linked by an amide bond [153]. This dimer was distinct from the noncovalent dimers prepared by lyophilization of RNase A from dilute solutions of acetic acid [150]. The covalent dimer exhibited a slightly increased catalytic activity but was insensitive to RI at near-physiological concentrations. No data have been reported on its cytotoxicity.
C. FUSION TO A TARGETING PROTEIN
Mammalian ribonuclease-based immunotoxins have been engineered by linkage of the N- or C-terminus of the enzyme to a targeting moiety, either by chemical coupling [53] or by fusion to another protein [154, 155]. Targeting moieties have included transferrin, growth factors, or antibody/antibody fragments directed against cell-surface antigens or receptors abundant only on cancer cells. The use of mammalian ribonucleases (especially human) as the effector portion of the immunotoxin was intended to supplant more traditional plant or bacterial toxins (e.g., ricin and diphtheria toxin), which can elicit strong immunogenic responses and promote vascular leak syndrome in patients [156]. The resulting conjugates had impressive toxicity for specific cell types but remained sensitive to RI [53, 154, 155], limiting their potency [152].
Semisynthesis has been used to link a targeting moiety to a ribonuclease residue in a manner that enables evasion of RI. For example, a free cysteine residue was introduced into the β4–β5 loop of both human RNase 1 (G89C) and EDN (T87C) to allow for linkage via a thioether bond to either transferrin or an anti-human transferrin receptor monoclonal antibody. Unlike other less-specific chemical coupling methods (e.g., via multiple amino groups), these homogeneous conjugates preserved enzymatic activity and displayed a 104-fold reduction in affinity for RI. The data suggest that gains in cytotoxicity achieved through RI-evasion and cell-targeting were additive [152], consistent with RI-evasion being an important consideration when designing mammalian ribonuclease-based immunotoxins.
Two immunotoxins have been prepared in which the C-terminus of the Δ1–7 fragment of RNase 1 is fused to the N-terminus of either hEGF or hFGF [49, 157]. This truncated variant of RNase 1 possesses an attenuated affinity for RI, which is accompanied by a substantial decrease in ribonucleolytic activity [158]. The fusion containing the Δ1–7 RNase 1 was more effective at inhibiting the growth of A431 cells than was the fusion containing the full-length protein, despite a 250-fold reduction in enzymatic activity [49]. Apparently, the reduced ability to degrade RNA is more than compensated by the decreased affinity for RI—similar to what was observed for monomeric variants of RNase A [138].
In RI·ribonuclease complexes, neither terminus of ribonuclease is in contact with RI. Accordingly, the tandem fusion of a ribonuclease to another protein is unlikely to enable the evasion of RI. A new gene fusion technique—“insertional fusion”—overcomes this limitation [159]. With this technique, human basic fibroblast growth factor has been inserted between Gly89 and Ser90 of RNase 1 (RNF89) to achieve cell-targeting and RI-evasion simultaneously. CL-RFN89, which contains an additional disulfide bond between Cys4 and Cys118, retained >85% of its enzymatic activity in the presence of a 200-fold molar excess of RI, and its interaction with cells was specific for the FGF receptor [160]. In contrast, constructs in which the insertional fusion was made outside of the RI ribonuclease interface (RNF19, CL-RNF19; bFGF inserted between Pro19 and Ser20) retained sensitivity to RI and were >10-fold less cytotoxic than CL-RNF89 ([161]; Tada, H., personal communication).
D. CHEMICAL MODIFICATION
The influence of random chemical cationization on the internalization of RNase A and RNase 1 has been studied in detail. These two ribonucleases have net charges of Z = +4 and +6, respectively. The basicity of these two proteins was enhanced by the amidation of the carboxyl groups (RNase A has 5 aspartate and 5 glutamate residues) with either ethylenediamine [18] or polyethylenimine (PEI) [158], and a positive correlation was observed between the value of Z and cytotoxic activity. The increase in cytotoxicity was due not only to enhanced internalization but also to diminished affinity for RI. Modification with either reagent was found to have a deleterious effect on enzymatic activity making the opimization of derivatization problematic [48]. The most cytotoxic preparation of RNase A (Z = +15.6) exhibited a >4×104-fold reduced affinity for RI (Kd = 19 nM; Table 8).
Table 8.
Effect of Chemical Modification on Affinity of Ribonucleases for RI
| Ribonuclease | Modification | Ribonucleolytic activity | Ki or Kd | IC50 or phenotype | Ref. |
|---|---|---|---|---|---|
| C31S BS-RNase or C32S BS-RNase | Sulfhydryl group carboxymethylation | Not quantitated | Reduced affinity for RI | Specifically toxic to spermatogenic layers | 200 |
| RNase A | Carboxyl group amidation with ethylenediamine | 1.6% of RNase A | Ribonucleolytic activity reduced by 10-fold molar excess of RI | 0.17 μM (3T3-SV40) | 18 |
| RNase 1 | Carboxyl group amidation with ethylenediamine | 0.38% of RNase A | Ribonucleolytic activity reduced by 10-fold molar excess of RI | 0.13 μM for (3T3-SV40) | 18 |
| RNase A | Carboxyl group amidation with ethylenediamine to Z = +15.6 | 5% of RNase A | 19 nM | 0.085 μM (MCF-7) 0.075 μM (3T3-SV40) | 48 |
| RNase A | Carboxyl group amidation with polyethylenimine (250, 600, 1000, 1800 Da) | Not determined | “Markedly decreased” affinity for RI” | 0.33–3.3 μM (3T3-SV40) | 201 |
| RNase A | Amino group amidation with PEG (5, 22 kDa) | Not determined | Not determined | Aspermatogenic and anti-tumoral activity | 168 |
| RNase A | Amino group amidation with PHPMA (classic and star-like) | Not determined | Not determined | Tumor growth inhibited in CD-1 nude mice bearing human tumors | 170 |
The efficacy of biological chemotherapeutic agents is limited largely by their bioavailability and circulating half-life, which is in turn affected by rate of plasma clearance, degradation, and immunogenicity [162]. A common strategy to remedy these problems is to conjugate inert polymers such as poly(ethylene glycol) (PEG), which under normal dosing circumstances does not elicit an immune response [163]. Instead, PEG acts to shield potentially immunogenic epitopes on the surface of the protein from the host immune system and also acts as a sheath to prevent proteolytic degradation [164]. PEGylation serves by increasing dramatically the hydrodynamic radius of the species, thereby increasing serum half-life by reducing the rate of renal filtration. Indeed, plasma clearance time has been shown to be directly proportional to PEG chain length [165]. Other water-soluble polymers such as poly(alkylene oxide), poly(oxyethylated polyols), and poly(vinyl alcohols) have also been shown to achieve similar benefits [166].
First-generation PEG conjugates were produced through the modification of protein amino groups. This strategy endowed RNase A with greater conformational stability and resistance to proteolytic degradation [167] as well as greater antitumoral activity in vivo [168, 169]. RNase A has also been modified with poly[N-(2-hydroxypropyl)methylacrylamide] (PHPMA), producing a conjugate that inhibited tumor growth in nude mice [170]. Considering the cloaking effect that PEG imparts to a conjugated protein, it is tempting to speculate that the enhanced biological activities of PEGylated RNase A are the result, at least in part, of reduced affinity for RI [168, 171].
A disadvantage of amino-group PEGylation is its creation of a heterogeneous population of conjugates. Moreover, the PEGylation of lysine residues can be deleterious to the biological function of a protein. In addition, the PEGylation of amino groups destroys positive charges, which has an adverse effect on the cellular internalization of ribonucleases [172]. Therefore, recent generations of PEGylated conjugates have had large PEG groups at one or two specific sites on the protein [163]. For example, our group has prepared site-specifically PEGylated RNase A by using maleimide-derivatized PEG and free-thiol containing variants of RNase A. We have found that the affinity of RNase A for RI could be reduced by 106-fold through such PEGylation and that the chain length, branching, and site of the PEG all influence the evasion of RI and, consequently, the cytotoxicity (Rutkoski, T.J.; Kink, J.A.; Strong, L.E. and Raines, R.T., unpublished results).
Finally, we note that mammalian ribonucleases exhibit diverse natural chemical modification in the form of glycosylation [173, 174]. The pendant carbohydrates could enhance bioavailability and circulating half-life [175], as well as engender evasion of RI. For example, RNase 1 is known to be glycosylated at an RI-contact residue, which is part of a consensus N-glycosylation sequence: Asn88–Gly89–Ser90 [176]. Likewise, a putative RI-contact residue in Rib1, residue 38 [46], is part of a consensus N-glycosylation sequence: Asn38–Gly39–Ser40. Nothing is known about its glycosylation. In addition, Trp7 of EDN, which is an RI-contact residue [75], undergoes an unusual post-translationsal modification with an α-mannopyranose moiety [177]. Finally, it is noteworthy that the glycosylation of ONC by its heterologous production in Pichia pastoris increases its toxicity for human erythroleukemia cells by 50-fold—presumably by increasing its conformational stability and resistance to proteolysis—without compromising its cellular internalization [178]. Its amphibian homologue, Amphinase, is glycosylated naturally and is toxic for submaxillary gland carcinoma cells [99]. Thus, the glycosylation of ribonucleases could provide mammals with an endogenous toxin for cancer cells.
PROSPECTUS
Ribonucleases represent a promising new class of chemotherapeutic agents [11–16]. Mammalian ribonucleases, in particular human homologues of RNase A, are especially attractive because of their pronounced ribonucleolytic activity and selective toxicity for cancer cells [46, 76, 143]. Moreover, mammalian ribonucleases are tolerated extremely well by humans. The primary hurdle in the development of mammalian ribonuclease-based chemotherapeutics is their inactivation by RI.
Some workers have reported on engineered ribonucleases that retain sensitivity to RI but demonstrate cytotoxicity, in apparent conflict with the work described above [36, 179, 180]. There are at least three scenarios in which RI-evasion would not be essential for ribonuclease-mediated cytotoxicity. (1) The mechanism of cytotoxicity is independent of enzymatic activity. In these examples, which include bacteriocidal pore-forming mechanism of eosinophil cationic protein (ECP; RNase 3) or other membrane destabilization [22, 181], protein basicity appears to be solely important. (2) Enough ribonuclease can reach the cytosol to overwhelm RI [11, 179, 180, 30, 182]. This scenario was apparent for the human anti-ErbB-2 scFv-RNase 1 immunotoxin (hERB-RNase 1), which is completely sensitive to RI but displays low-nanomolar toxicity toward cells displaying ErbB-2. Quantitative immunoblotting demonstrated that the anti-ErbB targeting moiety effectively delivers the conjugate to the cytosol [182]. (3) The ribonuclease enters a sub-cellular compartment (e.g., the nucleus) that is devoid of RI. The ability to evade RI would not be expected to influence cytotoxicity [36]. All three of these scenarios deviate significantly from the mechanism of ribonuclease-mediated cytotoxicity depicted in Fig. (1) in which the ribonuclease is internalized via non-receptor-mediated endocytosis to deliver ribonucleolytic activity to the cytosol of a target cell. In this mechanism, RI-evasion is essential.
Although its importance is indisputable, RI-evasion is but one of several determinants of cytotoxicity. Equations that correlate multiple biochemical attributes of a ribonuclease with cytotoxic activity have been both useful [138] and informative [46]. Nonetheless, a complete mathematical model describing ribonuclease-mediated cytotoxicity remains elusive. Only by thorough and careful consideration and optimization of all relevant factors can the cytotoxic potential of a ribonuclease be realized fully [48]. As research transitions from the petri dish to whole animals, techniques for instilling RI-evasion along with increased macromolecular size (e.g., by multimerization, fusion to a targeting protein, or PEGylation) could impart the distinct advantage of enhanced persistence in circulation [183, 184]. Further progress is not merely possible, but likely.
Table 6.
Effect of Multimerization on Affinity of Ribonucleases for RI
| Ribonuclease | Preparation | Affinity for RI | Cytotoxicity (IC50) a | Comments | Ref. |
|---|---|---|---|---|---|
| BS-RNase dimer | Natural | Ki >2 μM [136] | 1.3 μM (K-562) [45] | 2:1 M×M/M=M | 113 |
| HHP-RNase | Disulfide linked dimer of Q28L/R31C/R32C/N34K RNase 1 | Reduced sensitivity to RI | 0.15–6.3 μM (six human tumor cell lines); selective for malignant cell lines | 143 | |
| HHP2-RNase | Disulfide linked dimer of Q28L/R31C/R32C/N34K/E111G RNase 1 | Reduced sensitivity to RI | 1.2 μM (SVT2); twofold more cytotoxic than HHP-RNase; more cytotoxic than BS-RNase | 144 | |
| (RNase A)2 | Amino-group cross-linking [146] | Reduced [123] | Cytotoxic in vitro [35, 147, 148] | Cleaves dsRNA; enhanced persistence in circulation of mice and rats | 123 |
| (RNase A)3 | Amino-group cross-linking [146] | Not determined | More cytotoxic than (RNase A)2 [149] | Cleaves dsRNA | 149 |
| (G89C RNase 1)2 | Sulfhydryl-group cross-linking | 104-fold less affinity than wild-type RNase 1 | 80 nM (U251) 150 nM (Wehi 7.1; mouse T lymphoma) |
152 | |
| Lys66–Glu9 RNase A dimers | Lyophilized in vacuo at 85 °C for 96 h | Insensitive to RI at 1.4 μM | Not determined | 153 |
IC50 values reported are based on the concentration of constituent monomeric active sites.
Table 7.
Effect of Fusion to a Cell-targeting Moiety on Affinity of Ribonucleases for RI
| Ribonuclease | Design | Affinity for RI | Cytotoxicity | Comments | Ref. |
|---|---|---|---|---|---|
| Tf–G89C RNase 1 5E9–G89C RNase 1 Tf–T87C EDN |
Site-specific attachment of either transferin (Tf) or anti-human TfR via a thioether bond | 103-fold lower than bismaleimidohexane–G89C RNase 1; 104-fold lower than RNase 1 | IC50 = 1–2 nM (human glioma cells); 5,000-fold more cytotoxic than wild-type RNase 1 | 200-fold increase in cytotoxicity attributed to RI-evasion and 25-fold to cellular targeting moiety | 152 |
| hERB–RNase 1 | C-Terminus of human anti- ErbB-2 receptor scFv fused to N-terminus of RNase 1 with a His6 tag | Very high, but shown to overwhelm cytosolic RI | IC50 = 12.5–60 nM (four cell lines displaying ErbB-2) | 86% tumor growth inhibition in mice bearing TUBO tumors | 197 |
| Ber-H2-scFv–RNase 1 | C-Terminus of Ber-H2- scFv fused to N-terminus of RNase 1; Ber-H2-scFv binds to CD30 | Evasive | Cytotoxic to CD30+ cell lines | Dramatically reduced tumor growth in mice bearing CD30+ TS/A cells | 198 |
| des.1–7 RNase 1–hEGF | C-Terminus of Δ1–7 RNase 1 fused to N-terminus of human EGF | des.1–7 RNase 1 required 3-fold more RI to acheieve equivalent inhibition of RNase 1 [158] | IC50 = 0.35 μM (A431cells) versus 0.55 μM for RNase 1–hEFG (which has 250-fold higher catalytic activity) | Protein was unstable; 0.34% of RNase 1 catalytic activity | 49 |
| des.1–7 RNase 1–hFGF | C-Terminus of Δ1–7 RNase 1 fused to N-terminus of human FGF | Not determined | IC50 ~2 μM (mouse melanoma B16/BL6 cells) | Comparable cytotoxicity to RNase 1–human FGF, which has 20-fold higher catalytic activity | 157 |
| RNase 1–human bFGF | C-Terminus of human bFGF fused to N-terminus of RNase 1 | Sensitive Ki = 2.1 |
IC50 = 1.6 (mouse melanoma B16/BL6 cells) | 157, 161 | |
| CL-RNase 1 | Disulfide bond added at residues 4 and 118 | Ki = 1.8 nM | None detected | Additional disulfide bond reduces affinity for RI by 13-fold [44] | 161 |
| CL-RNF19 | β-trefoil core region (residues 19–146) of bFGF inserted into RNase 1 between Pro19 and Ser20 | Ki = 2.1 | >3 μM | Negative control, as Pro19 and Ser20 are distal from RI-binding site [199] | 161 |
| CL-RFN89 | β-trefoil core region (residues 19–146) of bFGF inserted to RNase 1 between Gly89 and Ser90 | Ki = 110 | 0.32 μM | Tumor growth inhibition in mice bearing human A431 SCC tumors (anti-angiogenic effect) [50] | 161 |
| CL-RFN89-2 | β-trefoil core region (residues 21–144) of bFGF inserted to RNase 1 between Gly89 and Ser90 | Ki = 193 nM | 0.23 μM | Removal of linker residues enabled more constrained attachment of FGF moiety | 161 |
Acknowledgments
We are grateful to E. Butzlaff for performing calculations of buried surface area for RI·ribonuclease complexes, and to J.G. McCoy and J.C. Mitchell for their assistance in determining the shape-complementarity values (Sc) of these complexes. We also thank B.R. Becklund, T.-Y. Chao, G.A. Ellis, R.J. Johnson, V.M. Kung, L.D. Lavis, and R.W. Watkins for many contributive discussions and their critical reading of this manuscript.
References
- 1.Jones W. J Am Physiol. 1920;52:203–207. [Google Scholar]
- 2.Richards FM, Wyckoff HW. The Enzymes. 1971;IV:647–806. [Google Scholar]
- 3.Blackburn P, Moore S. The Enzymes. 1982;XV:317–433. [Google Scholar]
- 4.Raines RT. Chem Rev. 1998;98:1045–1065. doi: 10.1021/cr960427h. [DOI] [PubMed] [Google Scholar]
- 5.Marshall GR, Feng JA, Kustler DJ. Biopolymers. 2007 In Press. [Google Scholar]
- 6.Park C, Raines RT. Biochemistry. 2003;42:3509–3518. doi: 10.1021/bi026076k. [DOI] [PubMed] [Google Scholar]
- 7.Barnard EA. Nature. 1969;221:340–344. doi: 10.1038/221340a0. [DOI] [PubMed] [Google Scholar]
- 8.Floridi A, D’Alessio G, Leone E. Eur J Biochem. 1972;26:162–167. doi: 10.1111/j.1432-1033.1972.tb01752.x. [DOI] [PubMed] [Google Scholar]
- 9.Dostál J, Matoušek J. J Reprod Fertil. 1973;33:263–274. doi: 10.1530/jrf.0.0330263. [DOI] [PubMed] [Google Scholar]
- 10.Ardelt W, Mikulski SM, Shogen K. J Biol Chem. 1991;266:245–251. [PubMed] [Google Scholar]
- 11.Leland PA, Raines RT. Chem Biol. 2001;8:405–413. doi: 10.1016/s1074-5521(01)00030-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matoušek J. Comp Biochem Physiol. 2001;129C:175–191. [Google Scholar]
- 13.Makarov AA, Ilinskaya ON. FEBS Lett. 2003;540:15–20. doi: 10.1016/s0014-5793(03)00225-4. [DOI] [PubMed] [Google Scholar]
- 14.Benito A, Ribó M, Vilanova M. Mol Biosyst. 2005;1:294–302. doi: 10.1039/b502847g. [DOI] [PubMed] [Google Scholar]
- 15.Arnold U, Ulbrich-Hofmann R. Biotechnol Lett. 2006;28:1615–1622. doi: 10.1007/s10529-006-9145-0. [DOI] [PubMed] [Google Scholar]
- 16.Lee JE, Raines RT. Biodrugs. 2008;22 doi: 10.2165/00063030-200822010-00006. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haigis MC, Raines RT. J Cell Sci. 2003;116:313–324. doi: 10.1242/jcs.00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Futami J, Maeda T, Kitazoe M, Nukui E, Tada H, Seno M, Kosaka M, Yamada H. Biochemistry. 2001;26:7518–7524. doi: 10.1021/bi010248g. [DOI] [PubMed] [Google Scholar]
- 19.Fredens K, Dahl R, Venge P. Allergy. 1991;46:27–29. doi: 10.1111/j.1398-9995.1991.tb00538.x. [DOI] [PubMed] [Google Scholar]
- 20.Soncin F, Strydom DJ, Shapiro R. J Biol Chem. 1997;272:9818–9824. doi: 10.1074/jbc.272.15.9818. [DOI] [PubMed] [Google Scholar]
- 21.Fuchs SM, Raines RT. Cell Mol Life Sci. 2006;63:1819–1822. doi: 10.1007/s00018-006-6170-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Notomista E, Mancheño JM, Crescenzi O, Di Donato A, Gavilanes J, D’Alessio G. FEBS J. 2006;273:3687–3697. doi: 10.1111/j.1742-4658.2006.05373.x. [DOI] [PubMed] [Google Scholar]
- 23.Fuchs SM, Rutkoski TJ, Kung VM, Groeschl RT, Raines RT. Protein Eng Des Sel. 2007;20:505–509. doi: 10.1093/protein/gzm051. [DOI] [PubMed] [Google Scholar]
- 24.Johnson RJ, Chao TY, Lavis LD, Raines RT. Biochemistry. 2007;46:10308–10316. doi: 10.1021/bi700857u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Youle RJ, D’Alessio G. In: Ribonucleases: Structures and Functions. D’Alessio G, Riordan JF, editors. Academic Press; New York: 1997. pp. 491–514. [Google Scholar]
- 26.Wu Y, Mikulski SM, Ardelt W, Rybak SM, Youle RJ. J Biol Chem. 1993;268:10686–10693. [PubMed] [Google Scholar]
- 27.Hu GF, Strydom DJ, Fett JW, Riordan JF, Vallee BL. Proc Natl Acad Sci USA. 1993;90:1217–1221. doi: 10.1073/pnas.90.4.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu GF, Riordan JF, Vallee BL. Proc Natl Acad Sci USA. 1997;94:2204–2209. doi: 10.1073/pnas.94.6.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saxena SK, Rybak SM, Winkler G, Meade HM, McGray P, Youle RJ, Ackerman EJ. J Biol Chem. 1991;266:21208–21214. [PubMed] [Google Scholar]
- 30.Leich F, Stohr N, Rietz A, Ulbrich-Hofmann R, Arnold U. J Biol Chem. 2007;282:27640–27646. doi: 10.1074/jbc.M702240200. [DOI] [PubMed] [Google Scholar]
- 31.Rodriguez M, Torrent G, Bosch M, Rayne F, Dubremetz JF, Ribó M, Benito A, Vilanova M, Beaumelle B. J Cell Sci. 2007;120:1405–1411. doi: 10.1242/jcs.03427. [DOI] [PubMed] [Google Scholar]
- 32.Olsnes S, Kozlov JV. Toxicon. 2001;39:1723–1728. doi: 10.1016/s0041-0101(01)00158-1. [DOI] [PubMed] [Google Scholar]
- 33.Collier RJ. Toxicon. 2001;39:1793–1803. doi: 10.1016/s0041-0101(01)00165-9. [DOI] [PubMed] [Google Scholar]
- 34.Davidson SJ, Hughes WL, Barnwell A. Exp Cell Res. 1971;67:171–187. doi: 10.1016/0014-4827(71)90633-1. [DOI] [PubMed] [Google Scholar]
- 35.Bartholeyns J, Baudhuin P. Proc Natl Acad Sci USA. 1976;73:573–576. doi: 10.1073/pnas.73.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bosch M, Benito A, Ribó M, Puig T, Beaumelle B, Vilanova M. Biochemistry. 2004;43:2167–2177. doi: 10.1021/bi035729+. [DOI] [PubMed] [Google Scholar]
- 37.Haigis MC, Kurten EL, Raines RT. Nucleic Acids Res. 2003;31:1024–1032. doi: 10.1093/nar/gkg163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ilinskaya ON, Makarov AA. Mol Biol (Moscow) 2005;39:1–10. [Google Scholar]
- 39.Iordanov MS, Ryabinina OP, Wong J, Dinh TH, Newton DL, Rybak SM, Magun BE. Cancer Res. 2000;60:1983–1994. [PubMed] [Google Scholar]
- 40.Ardelt B, Ardelt W, Darzynkiewicz Z. Cell Cycle. 2003;2:22–24. doi: 10.4161/cc.2.1.232. [DOI] [PubMed] [Google Scholar]
- 41.Klink TA, Raines RT. J Biol Chem. 2000;275:17463–17467. doi: 10.1074/jbc.M001132200. [DOI] [PubMed] [Google Scholar]
- 42.Kim JS, Souček J, Matoušek J, Raines RT. Biochem J. 1995;308:547–550. doi: 10.1042/bj3080547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leland PA, Schultz LW, Kim BM, Raines RT. Proc Natl Acad Sci USA. 1998;98:10407–10412. doi: 10.1073/pnas.95.18.10407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leland PA, Staniszewski KE, Kim BM, Raines RT. J Biol Chem. 2001;276:43095–43102. doi: 10.1074/jbc.M106636200. [DOI] [PubMed] [Google Scholar]
- 45.Lee JE, Raines RT. Biochemistry. 2005;44:15760–15767. doi: 10.1021/bi051668z. [DOI] [PubMed] [Google Scholar]
- 46.Rutkoski TJ, Kurten EL, Mitchell JC, Raines RT. J Mol Biol. 2005;354:41–54. doi: 10.1016/j.jmb.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 47.Rybak SM, Newton DL. Exp Cell Res. 1999;253:325–335. doi: 10.1006/excr.1999.4718. [DOI] [PubMed] [Google Scholar]
- 48.Futami J, Nukui K, Maeda T, Kosaka M, Tada H, Seno M, Yamada H. J Biochem (Tokyo) 2002;132:223–228. doi: 10.1093/oxfordjournals.jbchem.a003214. [DOI] [PubMed] [Google Scholar]
- 49.Hoshimoto S, Ueda M, Jinno H, Kitajima M, Futami J, Seno M. Anticancer Res. 2006;26:857–863. [PubMed] [Google Scholar]
- 50.Yagi H, Ueda M, Jinno H, Aiura K, Mikami S, Tada H, Seno M, Yamada H, Kitajima M. Cancer Sci. 2006;97:1315–1320. doi: 10.1111/j.1349-7006.2006.00336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Daniels TR, Delgado T, Helguera G, Penichet ML. Clin Immunol. 2006;121:159–176. doi: 10.1016/j.clim.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 52.Rodriguez M, Benito A, Tubert P, Castro J, Ribó M, Beaumelle B, Vilanova M. J Mol Biol. 2006;360:548–557. doi: 10.1016/j.jmb.2006.05.048. [DOI] [PubMed] [Google Scholar]
- 53.Rybak SM, Saxena SK, Ackerman EJ, Youle RJ. J Biol Chem. 1991;266:21202–21207. [PubMed] [Google Scholar]
- 54.Youle RJ, Newton D, Wu YN, Gadina M, Rybak SM. Crit Rev Ther Drug Carrier Syst. 1993;10:1–28. [PubMed] [Google Scholar]
- 55.Schein CH. Nat Biotechnol. 1997;15:529–536. doi: 10.1038/nbt0697-529. [DOI] [PubMed] [Google Scholar]
- 56.Hofsteenge J. In: Ribonucleases: Structures and Functions. D’Alessio G, Riordan JF, editors. Academic Press; New York: 1997. pp. 621–658. [Google Scholar]
- 57.Shapiro R. Methods Enzymol. 2001;341:611–628. doi: 10.1016/s0076-6879(01)41180-3. [DOI] [PubMed] [Google Scholar]
- 58.Dickson KA, Haigis MC, Raines RT. Prog Nucleic Acid Res Mol Biol. 2005;80:349–374. doi: 10.1016/S0079-6603(05)80009-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Blázquez M, Fominaya JM, Hofsteenge J. J Biol Chem. 1996;271:18638–18642. doi: 10.1074/jbc.271.31.18638. [DOI] [PubMed] [Google Scholar]
- 60.Roth JS. J Biol Chem. 1958;231:1085–1095. [PubMed] [Google Scholar]
- 61.Beintema JJ, Schüller C, Irie M, Carsana A. Prog Biophys Mol Biol. 1988;51:165–192. doi: 10.1016/0079-6107(88)90001-6. [DOI] [PubMed] [Google Scholar]
- 62.Lee FS, Vallee BL. Prog Nucleic Acid Res Mol Biol. 1993;44:1–30. doi: 10.1016/s0079-6603(08)60215-9. [DOI] [PubMed] [Google Scholar]
- 63.Monti DM, D’Alessio G. J Biol Chem. 2004;279:39195–39198. doi: 10.1074/jbc.C400311200. [DOI] [PubMed] [Google Scholar]
- 64.Moenner M, Vosoghi M, Ryazantsev S, Glitz D. Blood Cells Mol Dis. 1998;24:149–164. doi: 10.1006/bcmd.1998.0182. [DOI] [PubMed] [Google Scholar]
- 65.Johnson RJ, Lavis LD, Raines RT. Biochemistry. 2007;46:13131–13140. doi: 10.1021/bi701521q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Monti DM, Montesano Gesualdi N, Matoušek J, Esposito F, D’Alessio G. FEBS Lett. 2007;581:930–934. doi: 10.1016/j.febslet.2007.01.072. [DOI] [PubMed] [Google Scholar]
- 67.Kobe B, Deisenhofer J. J Mol Biol. 1996;264:1028–1043. doi: 10.1006/jmbi.1996.0694. [DOI] [PubMed] [Google Scholar]
- 68.Green NM. Adv Protein Chem. 1975;29:85–133. doi: 10.1016/s0065-3233(08)60411-8. [DOI] [PubMed] [Google Scholar]
- 69.Vincent JP, Lazdunski M. Biochemistry. 1972;11:2967–2977. doi: 10.1021/bi00766a007. [DOI] [PubMed] [Google Scholar]
- 70.Hutton M, Willenbrock F, Brocklehurst K, Murphy G. Biochemistry. 1998;37:10094–10098. doi: 10.1021/bi980616p. [DOI] [PubMed] [Google Scholar]
- 71.Schreiber G, Fersht AR. Biochemistry. 1993;32:5145–5150. doi: 10.1021/bi00070a025. [DOI] [PubMed] [Google Scholar]
- 72.Kobe B, Deisenhofer J. Nature. 1993;366:751–756. doi: 10.1038/366751a0. [DOI] [PubMed] [Google Scholar]
- 73.Kobe B, Deisenhofer J. Nature. 1995;374:183–186. doi: 10.1038/374183a0. [DOI] [PubMed] [Google Scholar]
- 74.Papageorgiou A, Shapiro R, Acharya K. EMBO J. 1997;16:5162–5177. doi: 10.1093/emboj/16.17.5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Iyer S, Holloway DE, Kumar K, Shapiro R, Acharya KR. J Mol Biol. 2005;347:637–655. doi: 10.1016/j.jmb.2005.01.035. [DOI] [PubMed] [Google Scholar]
- 76.Johnson RJ, McCoy JG, Bingman CA, Phillips GN, Jr, Raines RT. J Mol Biol. 2007;368:434–449. doi: 10.1016/j.jmb.2007.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kobe B, Deisenhofer J. Trends Biochem Sci. 1994;19:415–421. doi: 10.1016/0968-0004(94)90090-6. [DOI] [PubMed] [Google Scholar]
- 78.Clackson T, Wells JA. Science. 1995;267:383–386. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- 79.DeLano WL. Curr Opin Struct Biol. 2002;12:14–20. doi: 10.1016/s0959-440x(02)00283-x. [DOI] [PubMed] [Google Scholar]
- 80.Moreira IS, Fernandes PA, Ramos MJ. Proteins. 2007;68:803–812. doi: 10.1002/prot.21396. [DOI] [PubMed] [Google Scholar]
- 81.Chen CZ, Shapiro R. Proc Natl Acad Sci USA. 1997;94:1761–1766. doi: 10.1073/pnas.94.5.1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chen CZ, Shapiro R. Biochemistry. 1999;38:9273–8285. doi: 10.1021/bi990762a. [DOI] [PubMed] [Google Scholar]
- 83.Shapiro R, Ruiz-Gutierrez M, Chen CZ. J Mol Biol. 2000;302:497–519. doi: 10.1006/jmbi.2000.4075. [DOI] [PubMed] [Google Scholar]
- 84.Lee FS, Vallee BL. Biochemistry. 1990;29:6633–6638. doi: 10.1021/bi00480a012. [DOI] [PubMed] [Google Scholar]
- 85.Teufel DP, Kao RY, Acharya KR, Shapiro R. Biochemistry. 2003;42:1451–1459. doi: 10.1021/bi026852o. [DOI] [PubMed] [Google Scholar]
- 86.Kumar K, Brady M, Shapiro R. Proc Natl Acad Sci USA. 2004;101:53–58. doi: 10.1073/pnas.0307268101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Smeaton JR, Elliott WH, Coleman G. Biochem Biophys Res Commun. 1965;18:36–42. doi: 10.1016/0006-291x(65)90878-8. [DOI] [PubMed] [Google Scholar]
- 88.Buckle AM, Fersht AR. Biochemistry. 1994;33:1644–1653. doi: 10.1021/bi00173a005. [DOI] [PubMed] [Google Scholar]
- 89.Buckle AM, Schreiber G, Fersht AR. Biochemistry. 1994;33:8878–8889. doi: 10.1021/bi00196a004. [DOI] [PubMed] [Google Scholar]
- 90.Cho S, Beintema JJ, Zhang J. Genomics. 2005;85:208–220. doi: 10.1016/j.ygeno.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 91.Ledoux L, Baltus E. Experientia. 1954;10:500–501. doi: 10.1007/BF02166182. [DOI] [PubMed] [Google Scholar]
- 92.Ledoux L. Nature. 1955;175:258–259. doi: 10.1038/175258b0. [DOI] [PubMed] [Google Scholar]
- 93.Ledoux L. Nature. 1955;176:36–37. doi: 10.1038/176036a0. [DOI] [PubMed] [Google Scholar]
- 94.Aleksandrowicz J. Lancet. 1958;2:420. [Google Scholar]
- 95.Floridi A, D’Alessio G. Bull Soc Ital Biol Sper. 1967;43:32–36. [PubMed] [Google Scholar]
- 96.Lewis M, Hunt L, Barker W. Protein Sequences Data Anal. 1989;2:101–105. [PubMed] [Google Scholar]
- 97.Liao YD. Nucleic Acids Res. 1992;20:1371–1377. doi: 10.1093/nar/20.6.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kamiya Y, Oyama F, Oyama R, Sakakibara F, Nitta K, Kawauchi H, Takayanagi Y, Titani K. J Biochem (Tokyo) 1990;108:139–143. doi: 10.1093/oxfordjournals.jbchem.a123153. [DOI] [PubMed] [Google Scholar]
- 99.Singh UP, Ardelt W, Saxena SK, Holloway DE, Vidunas E, Lee HS, Saxena A, Shogen K, Acharya KR. J Mol Biol. 2007;371:93–111. doi: 10.1016/j.jmb.2007.04.071. [DOI] [PubMed] [Google Scholar]
- 100.Nitta K, Ozaki K, Ishikawa M, Furusawa S, Hosono M, Kawauchi H, Sasaki K, Takayanagi Y, Tsuiki S, Hakomori S. Cancer Res. 1994;54:920–927. [PubMed] [Google Scholar]
- 101.Liao YD, Huang HC, Leu YJ, Wei CW, Tang PC, Wang SC. Nucleic Acids Res. 2000;28:4097–4104. doi: 10.1093/nar/28.21.4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mikulski SM, Costanzi JJ, Vogelzang NJ, McCachren S, Taub RN, Chun H, Mittelman A, Panella T, Puccio C, Fine R, Shogen K. J Clin Oncol. 2002;20:274–281. doi: 10.1200/JCO.2002.20.1.274. [DOI] [PubMed] [Google Scholar]
- 103.Pavlakis N, Vogelzang NJ. Expert Opin Biol Ther. 2006;6:391–399. doi: 10.1517/14712598.6.4.391. [DOI] [PubMed] [Google Scholar]
- 104.Lou YC, Huang YC, Pan YR, Chen C, Liao YD. J Mol Biol. 2006;355:409–421. doi: 10.1016/j.jmb.2005.10.069. [DOI] [PubMed] [Google Scholar]
- 105.Shapiro R, Harper JW, Fox EA, Jansen HW, Hein F, Uhlmann E. Anal Biochem. 1988;175:450–461. doi: 10.1016/0003-2697(88)90569-6. [DOI] [PubMed] [Google Scholar]
- 106.Lee JE, Raines RT. Biochemistry. 2003;42:11443–11450. doi: 10.1021/bi035147s. [DOI] [PubMed] [Google Scholar]
- 107.Rosenberg HF, Zhang J, Liao YD, Dyer KD. J Mol Evol. 2001;53:31–38. doi: 10.1007/s002390010188. [DOI] [PubMed] [Google Scholar]
- 108.Mosimann SC, Ardelt W, James MNG. J Mol Biol. 1994;236:1141–1153. doi: 10.1016/0022-2836(94)90017-5. [DOI] [PubMed] [Google Scholar]
- 109.Hsu CH, Liao YD, Pan YR, Chen LW, Wu SH, Leu YJ, Chen C. J Mol Biol. 2003;326:1189–1201. doi: 10.1016/s0022-2836(02)01472-9. [DOI] [PubMed] [Google Scholar]
- 110.Boix E, Wu Y, Vasandani VM, Saxena SK, Ardelt W, Ladner J, Youle RJ. J Mol Biol. 1996;257:992–1007. doi: 10.1006/jmbi.1996.0218. [DOI] [PubMed] [Google Scholar]
- 111.D’Alessio G, Di Donato A, Mazzarella L, Piccoli R. In: Ribonucleases: Structures and Functions. D’Alessio G, Riordan JF, editors. Academic Press; New York: 1997. pp. 383–423. [Google Scholar]
- 112.Murthy BS, Sirdeshmukh R. Biochem J. 1992;281:343–348. doi: 10.1042/bj2810343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Piccoli R, Tamburrini M, Piccialli G, Di Donato A, Parente A, D’Alessio G. Proc Natl Acad Sci USA. 1992;89:1870–1874. doi: 10.1073/pnas.89.5.1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kim JS, Souček J, Matoušek J, Raines RT. J Biol Chem. 1995;270:10525–10530. doi: 10.1074/jbc.270.18.10525. [DOI] [PubMed] [Google Scholar]
- 115.Vescia S, Tramontano D, Augusti-Tocco G, D’Alessio G. Cancer Res. 1980;40:3740–3744. [PubMed] [Google Scholar]
- 116.Murthy BS, De Lorenzo C, Piccoli R, D’Alessio G, Sirdeshmukh R. Biochemistry. 1996;35:3880–3885. doi: 10.1021/bi952429m. [DOI] [PubMed] [Google Scholar]
- 117.De Groot AS, Scott DW. Trends Immunol. 2007;28:482–490. doi: 10.1016/j.it.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 118.Matoušek J, Souček J, Slavík T, Tománek M, Lee JE, Raines RT. Comp Biochem Physiol C: Toxicol Pharmacol. 2003;136:343–356. doi: 10.1016/j.cca.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 119.Glukhov BN, Jerusalimsky AP, Canter VM, Salganik RI. Arch Neurol. 1976;33:598–603. doi: 10.1001/archneur.1976.00500090004002. [DOI] [PubMed] [Google Scholar]
- 120.Mikulski SM, Grossman AM, Carter PW, Shogen K, Costanzi JJ. Int J Oncol. 1993;3:57–64. doi: 10.3892/ijo.3.1.57. [DOI] [PubMed] [Google Scholar]
- 121.Vasandani VM, Wu YN, Mikulski SM, Youle RJ, Sung C. Cancer Res. 1996;56:4180–4186. [PubMed] [Google Scholar]
- 122.Maack T, Johnson V, Kau ST, Figueiredo J, Sigulem D. Kidney Int. 1979;16:251–270. doi: 10.1038/ki.1979.128. [DOI] [PubMed] [Google Scholar]
- 123.Bartholeyns J, Moore S. Science. 1974;186:444–455. doi: 10.1126/science.186.4162.444. [DOI] [PubMed] [Google Scholar]
- 124.Ilinskaya O, Decker K, Koschinski A, Dreyer F, Repp H. Toxicology. 2001;156:101–107. doi: 10.1016/s0300-483x(00)00335-8. [DOI] [PubMed] [Google Scholar]
- 125.Sevcik J, Urbanikova L, Leland PA, Raines RT. J Biol Chem. 2002;277:47325–47330. doi: 10.1074/jbc.M208425200. [DOI] [PubMed] [Google Scholar]
- 126.Olmo N, Turnay J, Gonzalez de Buitrago G, Lopez de Silanes I, Gavilanes JG, Lizarbe MA. Eur J Biochem. 2001;268:2113–2123. doi: 10.1046/j.1432-1327.2001.02086.x. [DOI] [PubMed] [Google Scholar]
- 127.Cho SW, Joshi JG. Anal Biochem. 1989;176:175–179. doi: 10.1016/0003-2697(89)90289-3. [DOI] [PubMed] [Google Scholar]
- 128.Bugelski PJ, Treacy G. Curr Opin Mol Ther. 2004;6:10–16. [PubMed] [Google Scholar]
- 129.Fu CH, Sakamoto KM. Expert Opin Pharmacother. 2007;8:1977–1984. doi: 10.1517/14656566.8.12.1977. [DOI] [PubMed] [Google Scholar]
- 130.Lee FS, Shapiro R, Vallee BL. Biochemistry. 1989;28:225–230. doi: 10.1021/bi00427a031. [DOI] [PubMed] [Google Scholar]
- 131.Vicentini AM, Kieffer B, Mathies R, Meyhack B, Hemmings BA, Stone SR, Hofsteenge J. Biochemistry. 1990;29:8827–8834. doi: 10.1021/bi00489a046. [DOI] [PubMed] [Google Scholar]
- 132.Wells JA. Methods Enzymol. 1991;202:390–411. doi: 10.1016/0076-6879(91)02020-a. [DOI] [PubMed] [Google Scholar]
- 133.Radzicka A, Wolfenden R. Biochemistry. 1988;27:1664–1670. doi: 10.1021/bi00412a047. [DOI] [PubMed] [Google Scholar]
- 134.Gaur D, Swaminathan S, Batra JK. J Biol Chem. 2001;276:24978–24984. doi: 10.1074/jbc.M102440200. [DOI] [PubMed] [Google Scholar]
- 135.Erickson HA, Jund MD, Pennell CA. Protein Eng Des Sel. 2006;19:37–45. doi: 10.1093/protein/gzi073. [DOI] [PubMed] [Google Scholar]
- 136.Antignani A, Naddeo M, Cubellis MV, Russo A, D’Alessio G. Biochemistry. 2001;40:3492–3496. doi: 10.1021/bi002781m. [DOI] [PubMed] [Google Scholar]
- 137.McLendon G. Biochem Biophys Res Commun. 1977;77:959–966. doi: 10.1016/s0006-291x(77)80071-5. [DOI] [PubMed] [Google Scholar]
- 138.Bretscher LE, Abel RL, Raines RT. J Biol Chem. 2000;275:9893–9896. doi: 10.1074/jbc.275.14.9893. [DOI] [PubMed] [Google Scholar]
- 139.Huang YC, Lin YM, Chang TW, Wu SJ, Lee YS, Chang MDT, Chen C, Wu SH, Liao YD. J Biol Chem. 2007;282:4626–4633. doi: 10.1074/jbc.M607321200. [DOI] [PubMed] [Google Scholar]
- 140.Di Donato A, Cafaro V, D’Alessio G. J Biol Chem. 1994;269:17394–17396. [PubMed] [Google Scholar]
- 141.Di Donato A, Cafaro V, Romeo I, D’Alessio G. Protein Sci. 1995;4:1470–1477. doi: 10.1002/pro.5560040804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ciglic MI, Jackson PJ, Raillard SA, Haugg M, Jermann TM, Opitz JG, Trabesinger-Ruf N, Benner SA. Biochemistry. 1998;37:4008–4022. doi: 10.1021/bi972203e. [DOI] [PubMed] [Google Scholar]
- 143.Piccoli R, Di Gaetano S, De Lorenzo C, Grauso M, Monaco C, Spalletti-Cernia D, Laccetti P, Cinatl J, Matoušek J, D’Alessio G. Proc Natl Acad Sci USA. 1999;96:7768–7773. doi: 10.1073/pnas.96.14.7768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Di Gaetano S, D’Alessio G, Piccoli R. Biochem J. 2001;358:241–247. doi: 10.1042/0264-6021:3580241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hartman FC, Wold F. Biochemistry. 1967;6:2439–2448. doi: 10.1021/bi00860a021. [DOI] [PubMed] [Google Scholar]
- 146.Wang D, Wilson G, Moore S. Biochemistry. 1976;15:660–665. doi: 10.1021/bi00648a033. [DOI] [PubMed] [Google Scholar]
- 147.Tarnowski GS, Kassel RL, Mountain IM, Blackburn P, Wilson G, Wang D. Cancer Res. 1976;36:4074–4078. [PubMed] [Google Scholar]
- 148.Bartholeyns J, Zenebergh A. Eur J Cancer. 1979;15:85–91. doi: 10.1016/0014-2964(79)90209-3. [DOI] [PubMed] [Google Scholar]
- 149.Gotte G, Testolin L, Costanzo C, Sorrentino S, Armato U, Libonati M. FEBS Lett. 1997;415:308–312. doi: 10.1016/s0014-5793(97)01147-2. [DOI] [PubMed] [Google Scholar]
- 150.Libonati M, Gotte G. Biochem J. 2004;380:311–327. doi: 10.1042/BJ20031922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Messmore JM, Fuchs DN, Raines RT. J Am Chem Soc. 1995;117:8057–8060. doi: 10.1021/ja00136a001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Suzuki M, Saxena SK, Boix E, Prill RJ, Vasandani VM, Ladner JE, Sung C, Youle RJ. Nat Biotechnol. 1999;17:265–270. doi: 10.1038/7010. [DOI] [PubMed] [Google Scholar]
- 153.Simons BL, Kaplan H, Fournier SM, Cyr T, Hefford MA. Proteins. 2007;66:183–195. doi: 10.1002/prot.21144. [DOI] [PubMed] [Google Scholar]
- 154.Rybak SM, Hoogenboom HR, Meade HM, Raus JC, Schwartz D, Youle RJ. Proc Natl Acad Sci USA. 1992;89:3165–3169. doi: 10.1073/pnas.89.8.3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Newton DL, Nicholls PJ, Rybak SM, Youle RJ. J Biol Chem. 1994;269:26739–26745. [PubMed] [Google Scholar]
- 156.Pennell CA, Erickson HA. Immunol Res. 2002;25:177–191. doi: 10.1385/IR:25:2:177. [DOI] [PubMed] [Google Scholar]
- 157.Futami J, Seno M, Ueda M, Tada H, Yamada H. Protein Eng. 1999;12:1013–1019. doi: 10.1093/protein/12.11.1013. [DOI] [PubMed] [Google Scholar]
- 158.Futami J, Seno M, Kosaka M, Tada H, Seno S, Yamada H. Biochem Biophys Res Commun. 1995;216:406–413. doi: 10.1006/bbrc.1995.2638. [DOI] [PubMed] [Google Scholar]
- 159.Russell RB. Protein Eng. 1994;7:1407–1410. doi: 10.1093/protein/7.12.1407. [DOI] [PubMed] [Google Scholar]
- 160.Hayashida T, Ueda M, Aiura K, Tada H, Onizuka M, Seno M, Yamada H, Kitajima M. Protein Eng Des Sel. 2005;18:321–327. doi: 10.1093/protein/gzi040. [DOI] [PubMed] [Google Scholar]
- 161.Tada H, Onizuka M, Muraki K, Masuzawa W, Futami J, Kosaka M, Seno M, Yamada H. FEBS Lett. 2004;568:39–43. doi: 10.1016/j.febslet.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 162.Harris JM, Martin NE, Modi M. Clin Pharmacokinet. 2001;40:539–551. doi: 10.2165/00003088-200140070-00005. [DOI] [PubMed] [Google Scholar]
- 163.Harris JM, Chess RB. Nat Rev Drug Discov. 2003;2:214–221. doi: 10.1038/nrd1033. [DOI] [PubMed] [Google Scholar]
- 164.Greenwald RB, Choe YH, McGuire J, Conover CD. Adv Drug Delivery Rev. 2003;55:217–250. doi: 10.1016/s0169-409x(02)00180-1. [DOI] [PubMed] [Google Scholar]
- 165.Yamaoka T, Tabata Y, Ikada Y. J Pharm Sci. 1994;83:601–606. doi: 10.1002/jps.2600830432. [DOI] [PubMed] [Google Scholar]
- 166.Harris JM, Zalipsky S. Poly(ethylene glycol) Chemistry and Biological Applications. Washington, D.C: American Chemical Society; 1997. p. 489. [Google Scholar]
- 167.Monfardini C, Schiavon O, Caliceti P, Morpurgo M, Harris JM, Veronese FM. Bioconjug Chem. 1995;6:62–69. doi: 10.1021/bc00031a006. [DOI] [PubMed] [Google Scholar]
- 168.Matoušek J, Poučková P, Souček J, Škvor J. J Control Release. 2002;82:29–37. doi: 10.1016/s0168-3659(02)00082-2. [DOI] [PubMed] [Google Scholar]
- 169.Matoušek J, Poučková P, Hloušková D, Zadinová M, Souček J, Škvor J. J Control Release. 2004;94:401–410. doi: 10.1016/j.jconrel.2003.10.025. [DOI] [PubMed] [Google Scholar]
- 170.Poučková P, Zadinová M, Hloušková D, Strohalm J, Plocová D, Špunda M, Olejár T, Zitko M, Matoušek J, Ulbrich K, Souček J. J Control Release. 2004;95:83–92. doi: 10.1016/j.jconrel.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 171.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. J Control Release. 2000;65:271–284. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 172.Chapman AP. Adv Drug Deliv Rev. 2002;54:531–545. doi: 10.1016/s0169-409x(02)00026-1. [DOI] [PubMed] [Google Scholar]
- 173.Plummer TH, Jr, Hirs CH. J Biol Chem. 1963;238:1396–1401. [PubMed] [Google Scholar]
- 174.Plummer TH., Jr J Biol Chem. 1968;243:5961–5966. [PubMed] [Google Scholar]
- 175.Veronese FM, Pasut G. Drug Discov Today. 2005;10:1451–1458. doi: 10.1016/S1359-6446(05)03575-0. [DOI] [PubMed] [Google Scholar]
- 176.Beintema JJ, Blank A, Schieven GL, Dekker CA, Sorrentino S, Libonati M. Biochem J. 1988;255:501–505. [PMC free article] [PubMed] [Google Scholar]
- 177.de Beer T, Vliengenthart JF, Loffler A, Hofsteenge J. Biochemistry. 1995;34:11785–11789. doi: 10.1021/bi00037a016. [DOI] [PubMed] [Google Scholar]
- 178.Kim BM, Kim H, Raines RT, Lee Y. Biochem Biophys Res Commun. 2004;315:976–983. doi: 10.1016/j.bbrc.2004.01.153. [DOI] [PubMed] [Google Scholar]
- 179.Naddeo M, Vitagliano L, Russo A, Gotte G, D’Alessio G, Sorrentino S. FEBS Lett. 2005;579:2663–2668. doi: 10.1016/j.febslet.2005.03.087. [DOI] [PubMed] [Google Scholar]
- 180.Leich F, Koditz J, Ulbrich-Hofman R, Arnold U. J Mol Biol. 2006;358:1305–1313. doi: 10.1016/j.jmb.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 181.Navarro S, Aleu J, Jiménez M, Boix E, Cuchillo CM, Nogués MV. Cell Mol Life Sci. 2007 doi: 10.1007/s00018-007-7499-7. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.De Lorenzo C, Di Malta C, Cali G, Troise F, Nitsch L, D’Alessio G. FEBS Lett. 2007;581:296–300. doi: 10.1016/j.febslet.2006.12.034. [DOI] [PubMed] [Google Scholar]
- 183.Noguchi Y, Wu J, Duncan R, Strohalm J, Ulbrich K, Akaike T, Maeda H. Jpn J Cancer Res. 1998;89:307–314. doi: 10.1111/j.1349-7006.1998.tb00563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Dreher MR, Liu W, Michelich CR, Dewhirst MW, Yuan F, Chilkoti A. J Natl Cancer Inst. 2006;98:335–344. doi: 10.1093/jnci/djj070. [DOI] [PubMed] [Google Scholar]
- 185.Wallis R, Moore GR, James R, Kleanthous C. Biochemistry. 1995;34:13743–13750. doi: 10.1021/bi00042a004. [DOI] [PubMed] [Google Scholar]
- 186.Gaudet R, Bohm A, Sigler PB. Cell. 1996;87:577–588. doi: 10.1016/s0092-8674(00)81376-8. [DOI] [PubMed] [Google Scholar]
- 187.Lo Conte L, Chothia C, Janin J. J Mol Biol. 1999;285:2177–2198. doi: 10.1006/jmbi.1998.2439. [DOI] [PubMed] [Google Scholar]
- 188.Kawashima T, Berthet-Colominas C, Wulff M, Cusack S, Leberman R. Nature. 1996;379:511–518. doi: 10.1038/379511a0. [DOI] [PubMed] [Google Scholar]
- 189.Hubbard SJ, Thornton JM. NACCESS. Department of Biochemistry and Molecular Biology, University College London; London: 1993. [Google Scholar]
- 190.Lawrence MC, Colman PM. J Mol Biol. 1993;234:946–950. doi: 10.1006/jmbi.1993.1648. [DOI] [PubMed] [Google Scholar]
- 191.Hofsteenge J, Vicentini A, Zelenko O. Cell Mol Life Sci. 1998;54:804–810. doi: 10.1007/s000180050209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Domachowske JB, Dyer KD, Adams AG, Leto TL, Rosenberg HF. Nucleic Acids Res. 1998;26:3358–3363. doi: 10.1093/nar/26.14.3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Probst J, Brechtel S, Scheel B, Hoerr I, Jung G, Rammensee HG, Pascolo S. Genet Vaccines Ther. 2006;4:4. doi: 10.1186/1479-0556-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Harder J, Schroder JM. J Biol Chem. 2002;277:46779–46784. doi: 10.1074/jbc.M207587200. [DOI] [PubMed] [Google Scholar]
- 195.Laskowski RA, Chistyakov VV, Thornton JM. Nucleic Acids Res. 2005;33:D266–268. doi: 10.1093/nar/gki001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Haigis MC, Kurten EL, Abel RL, Raines RT. J Biol Chem. 2002;277:11576–11581. doi: 10.1074/jbc.M112227200. [DOI] [PubMed] [Google Scholar]
- 197.De Lorenzo C, Arciello A, Cozzolino R, Palmer DB, Laccetti P, Piccoli R, D’Alessio G. Cancer Res. 2004;64:4870–4874. doi: 10.1158/0008-5472.CAN-03-3717. [DOI] [PubMed] [Google Scholar]
- 198.Braschoss S, Hirsch B, Dubel S, Stein H, Durkop HM. Leuk Lymphoma. 2007;48:1179–1186. doi: 10.1080/10428190701272264. [DOI] [PubMed] [Google Scholar]
- 199.Abel RL, Haigis MC, Park C, Raines RT. Anal Biochem. 2002;306:100–107. doi: 10.1006/abio.2002.5678. [DOI] [PubMed] [Google Scholar]
- 200.Matoušek J, Kim JS, Souček J, Rihá J, Ribó M, Leland PA, Raines RT. Comp Biochem Physiol. 1997;118B:881–888. doi: 10.1016/S0305-0491(97)00278-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Futami J, Kitazoe M, Maeda T, Nukui E, Sakaguchi M, Kosaka J, Miyazaki M, Kosaka M, Tada H, Seno M, Sasaki J, Huh NH, Namba M, Yamada H. J Biosci Bioeng. 2005;99:95–103. doi: 10.1263/jbb.99.95. [DOI] [PubMed] [Google Scholar]
- 202.Boix E, Leonidas DD, Nikolovski Z, Nogues MV, Cuchillo CM, Acharya KR. Biochemistry. 1999;38:16794–16801. doi: 10.1021/bi9919145. [DOI] [PubMed] [Google Scholar]
- 203.Terzyan SS, Peracaula R, de Llorens R, Tsushima Y, Yamada H, Seno M, Gomis-Ruth FX, Coll M. J Mol Biol. 1999;285:205–214. doi: 10.1006/jmbi.1998.2288. [DOI] [PubMed] [Google Scholar]
- 204.Wlodawer A, Svensson LA, Sjölin L, Gilliland GL. Biochemistry. 1988;27:2705–2717. doi: 10.1021/bi00408a010. [DOI] [PubMed] [Google Scholar]
- 205.Beintema JJ, Breukelman HJ, Carsana A, Furia A. In: Ribonucleases: Structures and Functions. D’Alessio G, Riordan JF, editors. Academic Press; New York: 1997. pp. 245–269. [Google Scholar]
- 206.Fisher BM, Grilley JE, Raines RT. J Biol Chem. 1998;273:34134–34138. doi: 10.1074/jbc.273.51.34134. [DOI] [PubMed] [Google Scholar]
- 207.Hallahan TW, Shapiro R, Vallee BL. Proc Natl Acad Sci USA. 1991;88:2222–2226. doi: 10.1073/pnas.88.6.2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Moroianu J, Riordan JF. Biochem Biophys Res Commun. 1994;203:1765–1772. doi: 10.1006/bbrc.1994.2391. [DOI] [PubMed] [Google Scholar]
- 209.Mazzarella L, Capasso S, Demasi D, Di Lorenzo G, Mattia CA, Zagari A. Acta Crystallogr, Sect D. 1993;49:389–402. doi: 10.1107/S0907444993003403. [DOI] [PubMed] [Google Scholar]
- 210.Berisio R, Sica F, De Lorenzo C, Di Fiore A, Piccoli R, Zagari A, Mazzarella L. FEBS Lett. 2003;554:105–110. doi: 10.1016/s0014-5793(03)01114-1. [DOI] [PubMed] [Google Scholar]