

Abstract
Relatively recently, the Atomic Force Microscope (AFM) emerged as a powerful tool for single molecule nanomechanical investigations. Parameters that can be measured by force spectroscopy using AFM, such as the force and total mechanical extension required to break bonds between various proteins can yield valuable insights into the nature of the bond (zippering vs. highly localized binding site), the sequence of its interactions and the energy landscape along the length of the interaction. In this review we discuss the use of AFM in force spectroscopy mode to study intermolecular interactions between the exocytotic proteins of the core SNARE complex. Information gathered by force spectroscopy of protein-protein interactions of this complex supplement previous results acquired with other techniques, and allows a deeper understanding of SNARE protein interactions and their role in exocytosis.
Keywords: single molecule measurements, atomic force microscopy, force spectroscopy, SNARE proteins, exocytosis
Introduction
The structure and function of biological macromolecules, such as proteins and DNA, depend on intermolecular interactions. Hence, an understanding of their interaction forces and related energy landscapes, reaction rates and binding constants greatly improves our knowledge of these molecules. The traditional approach to studying molecular interactions is by performing test tube-type chemical reaction experiments which report an average of numerous molecules being investigated at the same time. To fully understand molecular interactions which involve one or a few interacting molecules, single molecule studies are required. Only recently, however, have researchers managed to manipulate molecules at the single molecule level and to directly measure their properties. Various methods have been employed: (i) pipette suction,1 (ii) magnetic beads,2 (iii) fluorescence resonance energy transfer (FRET),3 (iv) optical traps (laser tweezers),4 and (v) atomic force microscopy (AFM).5–7 Among these techniques, FRET, laser tweezers and AFM have been widely used. AFM has some advantages compared to FRET because of its simplicity of operation and analysis. FRET needs corrective calculations and relatively complicated data analysis. When compared to the laser tweezer technique, which is limited to force applications of up to about hundred pN, AFM has a larger range of measurable forces (0.1 to over 1000 pN). In addition, the AFM technique does not need large »linker« or »handler« molecules to hold the interacting molecules away from the laser focus. In this review, we cover the principals of AFM operation and its use in force spectroscopy to study the mechanical properties of exocytotic proteins at the single molecule level.
Principals of AFM Operation
The AFM was invented in 1986 to study the structure and properties of surfaces of materials.8 The AFM operates like a miniature phonograph stylus, investigating a sample by dragging a probe along its surface. The smallest standard AFM probes have tips with radial curvatures of about 5–10 nm, while a typical radial curvature is ≈50 nm. The tip, located at the end of a cantilever, is used to raster scan over the surface of interest. The cantilever can bend as a consequence of the interaction forces between the tip and the sample surface. The magnitude of bending, or deflection signal, is recorded. With appropriate distance control, the deflection signal of the cantilever can be used to generate topographic images of the sample surface or to study the vertical distance-dependent interactions between the tip and sample. Usually, a piezoactuator is used to control the distance between the tip and substrate. There are different methods to measure the deflection signal: (i) optical lever amplification, (ii) interferometry and (iii) electronic tunneling. Optical lever amplification is used by most commercial AFMs. Since samples can be investigated under physiological conditions in aqueous solutions while offering high temporal (≈1ms), x-, y- (<1 nm) and z- (≈0.1 nm) axes resolution, the AFM has emerged as a technique for imaging submicrometer-sized cellular organelles. The AFM was used to image isolated synaptic vesicles,9–11 secretory granules and their dynamics,12 including pore formation as a result of their fusion with a plasma membrane.13
The AFM setup is presented in Figure 1a. The laser beam from a laser diode is focused to around a 10 micrometer waist radius on the back of the cantilever tip. The beam is reflected off the gold-plated cantilever and monitored, by a photo detector. A piezoactuator is used to provide accurate three-dimensional movement of the sample. Since AFM is able to measure the minute interaction between the tip and sample surface, one can employ it in force spectroscopy mode to probe single molecule interactions. Using various methods (see below), biological molecules are first attached to the tip and a sample plate (substrate), which is usually a glass cover-slip or a mica sheet. As the sample and tip are brought into contact (Figure 1b), there are interactions formed between them due to the attached molecules. The resulting interaction force bends the cantilever when the tip is retracted from the surface. The laser signal reflected off the cantilever is received by the photodetector to record the magnitude of the deflection, which is proportional to the interaction force. Hook's law, F = − k · d, is followed here, where F is the interaction force, k is the spring constant of the cantilever, and d is the deflection magnitude of the cantilever tip. Because of the small spring constant (as low as 10 mN/m) of the cantilever, the force sensitivity of AFM can reach a fraction of a picoNewton, as indicated earlier.
Figure 1.
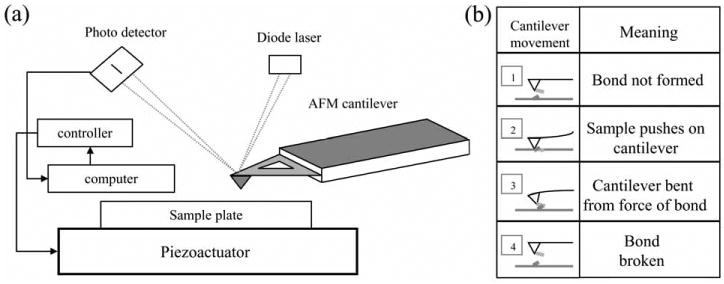
Schematic of AFM operation. (a) A laser beam from a laser diode is reflected off the back of the cantilever and collected by a photodetector which is used by a computer to generate a topographical representation of the scanned sample. The computer controls a piezoactuator which provides accurate three-dimensional movement of the sample. (b) In force spectroscopy mode, the AFM measures the interaction between molecules attached to the tip and substrate. (1) An AFM tip functionalized with a molecule of interest is brought into contact with the sample functionalized by a different molecule that can interact with the molecule on the AFM tip (2). As the tip is retracted from the sample it bends due to the adhesion force as a result of the intermolecular bond (3). At a finite force and distance the intermolecular bond breaks (4). Drawings are not to scale.
A typical example of force spectroscopy using AFM is measuring the unbinding force of a biotin-avidin interaction as a ligand-receptor model.5,6 Biotin molecules were deposited onto agarose beads that served as a substrate, while AFM tips were functionalized with avidin molecules. Under conditions that allow only a small number of interacting pairs to occur, the average force required to take apart a single pair of molecules was (160 ± 20) pN. Such force spectroscopy measurements on biotin-avidin interactions were in good agreement with thermodynamic calculations.
Besides measuring bond strength, AFM can also be used to study intramolecular interactions such as protein folding/unfolding, e.g. unfolding of the modular protein, titin.14 Recombinant titin was tagged at its C-terminus with two consecutive cysteine molecules and was adsorbed onto a freshly evaporated gold surface. The AFM tip was then brought into contact with the titin-decorated gold surface for several seconds to allow titin to adhere to the tip. The protein was stretched by separating the cantilever and sample surface. This caused the unfolding of the titin domains. A saw-tooth pattern force curve was obtained, and the force curves fit well to the worm like chain (WLC) model.4 This model is a widely used to describe the dependence of force and distance, F(x) = kBT/b [1/4(1 − x/L)−2 − 1/4 + x/L], where F is the interaction force, x is the distance, kB is the Boltzmann constant, T is temperature, b and L are the persistence and contour lengths, respectively. By fitting the force curve with the formula, one can extract the persistence and contour lengths. Based on these parameters, one can then draw inferences about the elastic properties of the molecule.
Taken together, these two examples of force spectroscopy experiments carried out by AFM clearly demonstrate the power of the AFM for studies of mechanical properties of single molecules or molecular pairs.
Force Spectroscopy as a Tool for Studying Snare Proteins at the Single Molecule Level
Recently, two different groups employed the AFM in force spectroscopy mode to study intermolecular interactions between the proteins of the core (ternary) soluble N-ethyl-maleimide-sensitive fusion protein (NSF) attachment protein receptor (SNARE) complex, which is known to have coiled-coil interactions.15,16 We briefly discuss the function of this complex and its molecular anatomy, followed by the description of procedures for attachment of individual proteins to the AFM tips and substrates. Information gathered by force spectroscopy of protein-protein interactions within the complex supplement previous findings and bring similar conclusions to those drawn from other techniques.
The SNARE complex is involved in exocytosis, where the secretory vesicle releases its cargo, transmitter molecules, into extracellular space after it fuses with the plasma membrane (Figure 2). The core SNARE complex is comprised of synaptobrevin 2 (Sb2), also known as vesicle-associated membrane protein 2 (VAMP 2), located on the vesicular membrane; syntaxin (Sx) and synaptosome-associated protein of 25 kDa (SNAP25), both located on the plasma membrane. These proteins are sensitive to Clostridial toxins, which are peptidases with specificity to exocytotic proteins (reviewed in Refs. 17, 18); Sb2 is cleaved by tetanus neurotoxin (TeNT) and botulinum neurotoxin (BoNT) type B, D, F and G, Sx by BoNT-C, while SNAP25 is targeted by BoNT-A, -C and -E.
Figure 2.

Exocytotic release of transmitter utilizes SNARE proteins. (a) The ternary SNARE complex consists of synaptobrevin 2 (red), also known as vesicle-associated membrane protein 2 (VAMP 2), located on the vesicular membrane; syntaxin (green) and synaptosome-associated protein of 25 kDa (SNAP25; blue), both located on the plasma membrane. Arrows and numbers indicate truncation sites of recombinant proteins used in Ref. 16. Proteins are oriented in parallel fashion. N, N-terminus. Drawing is not to scale. (b) Protein sequences: synaptobrevin 2, GeneBank accession number BC074003; syntaxin 1A, GeneBank accession number AF217191; and SNAP25B, GeneBank accession number AB003992. Shaded areas indicate SNARE domains. Numbers denote the position of amino acids in the sequence. Arrows indicate truncation sites. Lysine residues (K) are lettered in red and represent possible glutaraldehyde cross-linking sites. Histidine residues (H) are lettered in blue; note the absence of consecutive histidines in the protein sequences of SNARE proteins, which eases the purification and directed deposition of these proteins when they are tagged with a stretch of six histidines (not shown).
The precise molecular structure of the ternary SNARE complex has been recently resolved using X-ray crystallography.19 Crystal structure has revealed that the complex is formed by four alpha-helices; Sb2 and Sx1 each contribute one alpha-helix, while SNAP25 contributes two. These helices are physiologically oriented in parallel fashion, as denoted by N-termini alignment in Figure 2. They appear bundled through their entire regions of interaction, also known as SNARE domains. Alignment of the complex appears at its ionic pocket or »0« layer where arginine from Sb2 is stabilized by three glutamine residues from other alpha-helices.19 Once formed, the complex is very stable, resistant to denaturation with sodium dodecyl sulfate20 and temperatures up to 90 °C.21 Although enormously informative, these studies could not offer information on the mechanical characteristics of the protein interactions, a necessary component for detailed understanding of exocytosis. Such information has been recently provided by force spectroscopy using AFM. A prerequisite to study SNARE proteins using AFM in force spectroscopy mode is to specifically attach proteins of interest onto the AFM tips and substrates without affecting their ability to mechanically interact.
Non-directional Cross-linking of Exocytotic/SNARE Proteins Using Glutaraldehyde
Yersin et al.15 were first to measure unbinding forces among SNARE proteins using AFM. They used recombinant SNARE proteins with glutathione S-transferase (GST) or six consecutive histidines (His6) tags that were expressed in bacteria. The purified chimera contained full length Sb2 and SNAP25, while Sx1 was truncated and contained only its cytoplasmic tail. These proteins were covalently attached to mica sheets using glutaraldehyde (Figure 3a). To achieve this cross-linking freshly cleaved mica sheets were treated with aminopropyltri-etoxysilane (ATEPS).22 After silanization, mica sheets were exposed to proteins in solution containing glutaraldehyde, which cross linked amines in ATEPS and lysines in the proteins of interest23,24 (consult protein sequences for lysine residue positions; Figure 2b). Thus, this would allow the covalent binding of proteins to mica. However, this homobifunctional amine cross-linker would also cross-link lysines within a single protein molecule and between protein molecules, tending to yield high molecular weight aggregates (Figure 3a, right), leading to reduced reproducibility and batch-to-batch inconsistency. Moreover, the aqueous solution of glutaraldehyde contains variable amounts of polyglutaraldehyde, which also forms during monoglutaraldehyde reaction with proteins. This polymer, having relatively long chains, facilitates reactions with proteins.25 Nonetheless, Yersin et al. deposited proteins onto mica and AFM tips (usually Si3N4) surfaces using glutaraldehyde cross-linking. Thus, this procedure tethers proteins to the surface while reducing the proteins' ability to freely interact. Therefore the interactions measured are severely restricted and/or random, with the molecules possibly forming non-aligned parallel and anti-parallel configurations.
Figure 3.
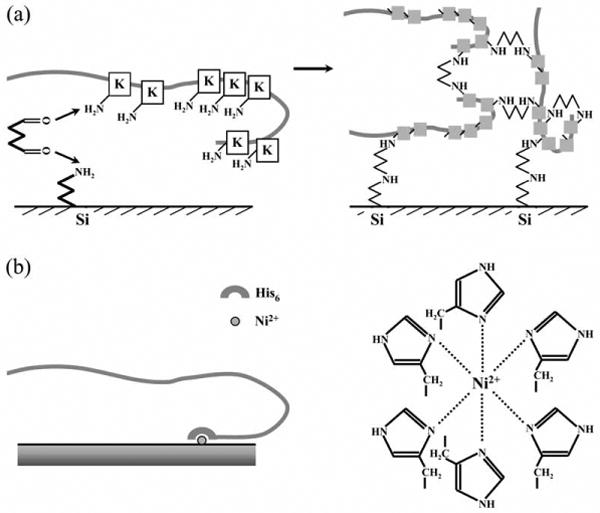
Schematic presentation of methods for the deposition of exocytotic proteins to surfaces of interest, the AFM tips and substrates. (a) Cross-linking: After silanization of mica/tips, proteins can be covalently cross-linked with glutaraldehyde via lysine residues (K). This procedure tends to yield high molecular weight aggregates (right) by cross-linking proteins (squares, lysine residues). (b) Steric coordination: Recombinant proteins containing a His6 tag at, e.g., C-terminus can be deposited onto Ni2+-coated glass coverslips/tips through steric coordination (right). Drawings are not to scale.
Following protein deposition onto the tips and mica, various combinations of SNARE proteins were probed, and the interaction forces measured. The piezoactuator was used to move the mica plate away from the tip at a velocity of 355 nm/s, corresponding to an ≈ 21 nN/s force loading rate. A larger value of the force needed to rupture the interaction was taken to mean stronger binding. First the interaction between any two of the three SNARE proteins was measured and the rupture forces were found to be different. The strongest interaction was observed for the Sx1-SNAP25 pair at (265 ± 4) pN. When studying the ternary SNARE complex containing Sx1, SNAP25 and Sb2, a functionalized tip containing one of the proteins was used to probe the two remaining proteins/binary complex deposited on mica. Here the binary complexes on mica were prepared in two different ways: (i) the mica surface was first functionalized with one protein followed by the addition of the other protein, or (ii) two different proteins of interest were pre-incubated, allowing their interaction prior to cross-linking them onto the mica surface. In the presence of all three SNARE proteins (ternary complex) the strongest interaction of (279 ± 3) pN was measured when Sx1 and SNAP25 were premixed, cross-linked to mica and then probed with the Sb2 functionalized tip.
Next, by varying the mica plate retraction velocity, loading rate experiments were performed for the binary interaction between Sx1-SNAP25, and for the ternary complex. Such experiments allowed measurements of potential barrier widths, and by extrapolating loading rate at zero force, one can estimate spontaneous lifetime, or dissociation rate for the given potential barrier. Here, a dissociation rate of ≈ 3 · 10−7 s−1 for Sx1-SNAP25 and ≈ 2· 10−10 s−1 for the ternary complex were obtained. Furthermore, based on force measurements Yersin et al. estimated that 4–5 ternary complexes would be necessary to hold a single vesicle in the vicinity of the plasma membrane.
This initial study of SNARE proteins by AFM in force spectroscopy mode used only the rupture force as a representation of the binding energy for understanding single molecule interactions between SNARE proteins. However, the work done, which is a vector product of the applied force and the corresponding extension, is accounted for, in part, (i) by the energy for breaking intermolecular bonds, (ii) by the energy required to compensate the thermal entropy of the free sections of the stretched proteins and (iii) by dissipation. Thus the final force required to rupture all the bonds will not necessarily correspond to the total interaction energy of the bound proteins, as assumed in the study above, due to: (i) the different extensions for each system, (ii) unknown angle of the applied force with respect to the axis of the protein system, (iii) entropy contributions and (iv) dissipation.
Liu et al.(16) extended the use of AFM in force spectroscopy measurements to show that both the total extension and the rupture force provide critical information on the binding mechanism of SNARE proteins.
Directional Deposition of SNARE Proteins Using Sterical Coordination of His6 Tags by Ni2+
Liu et al.,16 studying mechanical interaction between molecules of the SNARE complex, deposited recombinant proteins based on the principle of metal ion affinity chromatography,29 a method used widely for the purification30,31 and immobilization of proteins.32 Metal ions having a coordination number of six, e.g., nickel(II) ions (Ni2+), selectively bind proteins containing stretches of consecutive histidine residues33 (Figure 3b). Since proteins containing isolated histidines do not form stable complexes, recombinant proteins and peptides containing a His6 tag added at either of their termini have been commonly engineered to facilitate purification steps.34,35
The limitation of this method is that only protein interactions whose strength is less than the strength of binding between Ni2+ and His6 can be investigated. Therefore, the Ni2+-His6 interaction was studied first.16 AFM tips and glass coverslips were coated with nickel films which were partially oxidized by exposure to air.36 Nickel-coated tips and coverslips were then functionalized with a His6 peptide. His6 functionalized coverslips were subsequently decorated with Ni2+-nitrilotriacetate (NTA) agarose beads that were then probed with His6 functionalized tips (Figure 4a). The mean value of the single molecule unbinding force between His6 and Ni2+ was found to be (525 ± 41) pN by measuring the force required to rupture the interaction of His6 functionalized AFM tips with the Ni2+-NTA agarose beads (Figure 4b, c). It should be noted that NTA is a quadridentate chelating adsorbent, which occupies four positions in the Ni2+ octahedral coordination sphere, leaving two remaining positions available for selective interaction.33 Therefore, even though these force measurements are in good agreement with previously reported mechanical strengths of the coordination bond between a His6 tag and Ni2+-NTA,37 they are an underestimate for unbinding in the absence of NTA, when direct Ni2+-His6 interaction occurs as used in SNARE proteins interactions below. Nonetheless, the measured forces are still much greater than the forces required for taking apart recombinant SNARE proteins. Therefore, a Ni2+-His6 bond can be effectively used to attach SNARE proteins to the nickel-coated tips and coverslips as outlined below.37
Figure 4.
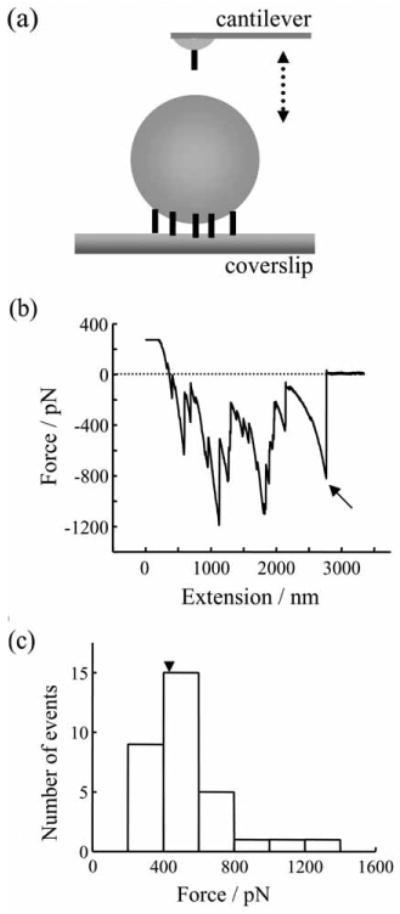
Strength of the single molecule binding force between six consecutive histidine molecules (His6) tag and Ni2+. (a) His6 coated tip was used to probe Ni2+/NTA agarose beads immobilized on a His6 functionalized glass coverslip. Tip and bead were brought in contact (approach; arrow pointing down) by means of the piezoelectric element and then taken apart (retract, arrow pointing up). The drawing in (a) is not to scale. (b) The retraction part of a typical force-distance (extension) curve using an experimental design in (a). There are several events indicating unbinding/rupture of His6 -Ni2+/NTA. The very last events (arrow) were used in analysis of a single unbinding event, since it is possible that the other steps are a nonlinear convolution of multiple unbinding events. The dashed line indicates zero force. (c) Distributions of the rupture forces for His6 -Ni2+/NTA single bonds. Retraction velocity, 1.6 μm/s. Arrowhead indicates the mean value.
Liu et al. directionally attached the cytoplasmic domains of recombinant Sb2 and Sx1A with a His6 tag on their C-termini that was coordinated by nickel ions on the AFM tip and glass coverslip surfaces, thus allowing these proteins to freely interact in a physiologically more prevalent parallel fashion3,20,38 (Figures 3b and 5). Rather than relying on force measurements alone, Liu et al. reported on both the interaction force and the total mechanical extension for individual Sx1A-Sb2 pairs (Figure 5a). The mean rupture force necessary to take apart the interaction between single Sx1A-Sb2 pairs was (237 ± 4) pN, when using a retraction velocity of 1.6 μm/s, corresponding to an ≈ 20 nN/s force loading rate. Such a force would be sufficient to hold an ≈ 40 μm diameter Ni2+-NTA agarose bead off the cantilever tip (see development of a BoNT-B sensor based on this finding in Ref. 36), given the buoyancy of the bead in the fluid, indicating that a single Sx1A-Sb2 pair would be more than sufficient to hold a vesicle an order of magnitude smaller (≈ 50 nm in diameter) in close proximity to the plasma membrane. Extension values of (23.0 ± 0.6) nm were measured. Similar force and extension measurements were obtained when the cantilever tips were functionalized using a truncated form of syntaxin, Sx1A178–266−His6, lacking a part of the molecule towards the N-terminal from its SNARE domain, and used to probe Sb2-His6 functionalized glass coverslips. This finding favors the notion that Sb2 directly interacts with the SNARE domain of Sx1 A in closed form without inducing a large conformation change of Sx1A from its closed to open state as recently suggested.3,39
Figure 5.
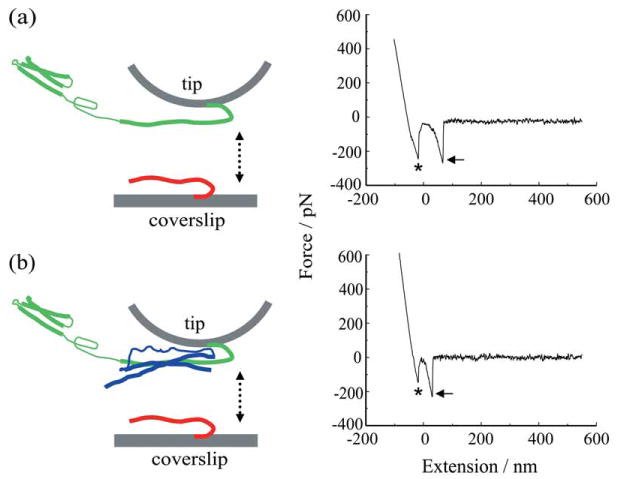
Force spectroscopy of SNARE proteins. (a) Recombinant synaptobrevin 2 (Sb2-His6; red) is attached to the nickel coated coverslip surface through histidine residue tags (His6) at its C terminus leaving its cytoplasmic domain free to interact with recombinant syntaxin 1A (Sx1A-His6; green) which is similarly attached by means of a C-terminus His6 tag to the nickel-coated cantilever tip. These two proteins are brought into close proximity (approach; arrow pointing down) by means of the piezoelectric element and then taken apart (retract, arrow pointing up). The retraction part of a typical force-distance (extension) curve using a Sx1A-His6 functionalized tip and a Sb2-H6 functionalized coverslip (right). Asterisk indicates the segment of the curve while the coverslip and the cantilever tip are still in contact. The arrow indicates the rupture of the Sx1A-Sb2 intermolecular »bond.« (b) SNAP25B (blue) reduces the extension of Sx1A-Sb2. Co-functionalized tips with equimolar ratio of Sx1A-His6/His6-SNAP25B were used to probe Sb2-His6 functionalized coverslips as shown in the force-distance curve (right). Asterisk as in (a); arrow, the rupture of the ternary complex intermolecular interaction. Retraction velocity, 1.6 μm/s. The drawings are not to scale. Modified from Ref. 16.
Following the study of the mechanical properties of Sx1A-Sb2 intermolecular interactions, Liu et al. then measured the single intermolecular interaction events between all three core proteins of the SNARE complex, Sb2, Sx1A and SNAP25B (Figure 5b). Here, the AFM cantilevers were functionalized with Sx1A-His6 and then pre-incubated with full length SNAP25B with GST at its N-terminus to form a binary complex, or Sx1A-His6 and His6-SNAP25B were pre-incubated in an equimolar ratio in a tube to form binary complexes, which were then used to co-functionalize AFM tips. These tips were used to probe Sb2-His6 functionalized coverslips. Upon contact of the two surfaces, a binary Sx1A-SNAP25B complex at the tip binds Sb2 on the coverslip to form a ternary Sb2-Sx1A-SNAP25B core SNARE complex. Retracting the coverslip dissociates this complex and the extension and rupture forces for this type of single intermolecular interaction were measured (Figure 5b). Here the presence of SNAP25B on the tip did not cause significant changes in force measurements ((243 ± 5) pN; at ≈ 20 nN/s force loading rate, but see below for different rates) when compared to the Sx1A-Sb2 interaction, while the extension measurements exhibited a significant shortening to (12.5 ± 0.4) nm.
To study the nature of the binding between SNARE proteins, i.e., zippering versus a highly localized binding site, and to deduce the life times necessary to spontaneously dismantle protein-protein interactions, the authors measured force and extension at the point of rupture of the single intermolecular bond as a function of the force loading rate (Figure 6a). As in Yersin et al., by extrapolating the loading force rate to zero force using an obtained exponential relationship (Figure 6a), Liu et al. estimated dissociation rates; the Sx1A-Sb2 interaction displayed a spontaneous dissociation lifetime of 0.16 s, while the ternary SNARE complex containing Sx1A, Sb2 and SNAP25B showed a spontaneous lifetime of 2.1 s, indicating that the ternary SNARE complex is substantially more stable than the Sx1A-Sb2 interaction. Note that these measurements do not agree with Yersin et al.
Figure 6.
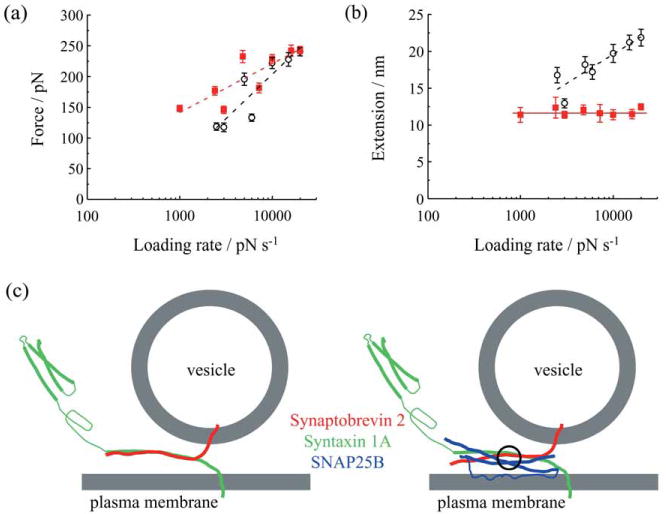
Force and extension values for dissociation of SNARE proteins as a function of the force loading rate. (a) Force necessary to take apart Sx1A-Sb2 complex in the absence (open circles) or presence of SNAP25B (red squares) increases exponentially with an increase in the loading rate. (b) Extension changes exponentially with loading rate only when Sx1A-Sb2 interactions are ruptured, but not when SNAP25B is present with Sx1A-Sb2. Dashed lines indicate exponential fits to the data, while solid red line indicates that the extension value is constant. (c) A model describing interactions between SNARE proteins. Sx1A and Sb2 are zippered through their entire SNARE domains (left). When SNAP25B is present within the complex (right), the interaction is localized C-terminally from a Sx1A-Sb2 cuffing position at »0« layer (circle). Drawings in (c) are not to scale. Modified from Ref. 16.
The extension measurements, as a function of the force loading rate, not preformed by Yersin et al., provided critical information regarding the nature of the bonding mechanism in the Sx1A-Sb2 intermolecular bond by comparison with the ternary SNARE complex (Figure 6b). Here, the extension as well as the force increased exponentially as a function of the force loading rate, pointing to the relatively high spontaneous dissociation rate of the zipper type non-localized interaction. By contrast, the extension measurements with the ternary SNARE complex remained constant as the loading rate was varied, although the rupture force increased exponentially with an increasing loading rate, pointing to cuffing, a strong intermolecular bond localized at the“0” layer induced by SNAP25B (for detailed discussion consult Ref. 16). This finding supplements static information on bundling of SNARE proteins in the ternary complex obtained from X-ray crystallography,19 and conversely crystallography offers an interpretation of the extension data in respect to the location of the cuffing.
This work introduced intermolecular extension as an important parameter in studying the characteristics of protein-protein interactions and, when combined with measuring rupture force, has provided additional insights into the binding mechanisms of the proteins in the SNARE complex. Additionally, these findings suggest the idea that intracellularly there could be two modes of vesicular positioning with respect to the plasma membrane. At vesicle-plasma membrane distances smaller than ≈12 nm, the ternary SNARE complex would play the major role in vesicular positioning, while at distances of 12-23 nm this role could be accomplished by Sx1A-Sb2 pairs (Figure 6c).
Reconciling Differences and Concluding Remarks
The studies presented above have investigated SNARE proteins using AFM in force spectroscopy mode. Although they have brought new insights to the function of SNARE proteins, they display some differences in their results and interpretation. Since both groups determined the spring constant of the cantilevers by the same method,40 observed differences might be due to the method of protein deposition. Yersin et al.15 cross-linked proteins on the mica/tip surface. This procedure greatly restricts protein-protein interactions. Although the procedure has been successfully used to study antigen-antibody interactions, where the binding pocket does not appear to be affected, it might not be the best choice to study coiled-coil interactions.41,42 To form a bundle (zippering), proteins need three-dimensional flexibility and freedom of movement, which is restricted by cross-linking, as is schematically presented in Figure 3a. Furthermore, in physiological conditions, Sx1A and Sb2 predominantly form parallel interactions,3,26,38 which were enforced in Liu et al. by directionally attaching proteins with His6 tags located at their C-termini. Once His6 tags were coordinated by Ni2+, the N-termini of the proteins could freely move and would predominantly form parallel interactions as the tip and coverslip were vertically brought into close proximity. In Yersin et al., however, cross-linked proteins were tethered to the surface. This non-directional attachment results in restricted protein motility and also random formation of both parallel and anti-parallel interactions.
Another reason that could contribute to the observed differences in the results could possibly arise from differences in the isoforms of SNAP25 and Sx1 that were used. While Liu et al. specified that they used SNAP25B and Sx1A, Yersin et al. did not disclose that information. Furthermore, Yersin et al. used the full length of Sb2, while Liu et al. used only cytoplasmic domains; it is then possible that the presence of the transmembrane and intravesicular moieties of Sb2, which normally would not be available to interact with the cytoplasmic domain of Sx1 could contribute to discrepancies in the measurements.
Interestingly, while force measurements with ≈ 20 nN/s loading rate yielded similar measurement in both studies, there was a divergent interpretation of the data. Yersin et al. estimated that 4–5 ternary SNARE complexes are necessary to hold a vesicle in close proximity to the plasma membrane. Liu et al., however, implied that a single ternary complex or even a single Sx1A-Sb2 pair would be sufficient to hold a vesicle at the plasma membrane, albeit for different durations. An independent study that used FRET demonstrated that as few as one complex per liposome was sufficient for its docking to a supported lipid bilayer,3 thus favoring the Liu et al. interpretation of force measurements.
Intermolecular extension is an important parameter in studying single molecule interactions between proteins, particularly when those proteins are involved in exocytosis, where vesicle-plasma membrane distance is of critical importance. Introduction of extension as a parameter in studying the dynamics of the SNARE proteins allowed correlation of AFM data originating from different methods, especially X-ray crystallography. This resulted in the assignment of a cuffing/stabilizing role for SNAP25B in the ternary complex that occurs at the ionic layer.
Atomic force mi-croscopy proves to be a powerful tool when used in force spectroscopy mode to study macromolecular interactions at the single molecule level. Combining data gathered using AFM with those obtained using other complementary techniques, allows us to comprehensively understand how the protein machinery operates in the vital cellular process of exocytosis.
Acknowledgments
The authors' work is supported by a grant from the National Institute of Mental Health (MH 069791), and a grant from Department of Defense/Defense Microelectronics Activity under Award No. DOD/DMEA-H94003-06-2-0608. We thank Dr Erik B. Malarkey for comments on the previous versions of this manuscript.
References
- 1.Evans E, Ritchie K. Biophys J. 1997;72:1541–1555. doi: 10.1016/S0006-3495(97)78802-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith SB, Cui Y, Bustamante C. Science. 1996;271:795–799. doi: 10.1126/science.271.5250.795. [DOI] [PubMed] [Google Scholar]
- 3.Bowen ME, Weninger K, Brunger AT, Chu S. Biophys J. 2004;87:3569–3584. doi: 10.1529/biophysj.104.048637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bustamante C, Marko JF, Siggia ED, Smith S. Science. 1994;265:1599–1600. doi: 10.1126/science.8079175. [DOI] [PubMed] [Google Scholar]
- 5.Lee GU, Chrisey LA, Colton RJ. Science. 1994;266:771–773. doi: 10.1126/science.7973628. [DOI] [PubMed] [Google Scholar]
- 6.Florin EL, Moy VT, Gaub HE. Science. 1994;264:415–417. doi: 10.1126/science.8153628. [DOI] [PubMed] [Google Scholar]
- 7.Merkel R, Nassoy P, Leung A, Ritchie K, Evans E. Nature. 1999;397:50–53. doi: 10.1038/16219. [DOI] [PubMed] [Google Scholar]
- 8.Binnig G, Quate CF, Gerber C. Phys Rev Lett. 1986;56:930–933. doi: 10.1103/PhysRevLett.56.930. [DOI] [PubMed] [Google Scholar]
- 9.Garcia RA, Laney DE, Parsons SM, Hansma HG. J Neurosci Res. 1998;52:350–355. doi: 10.1002/(SICI)1097-4547(19980501)52:3<350::AID-JNR11>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 10.Laney DE, Garcia RA, Parsons SM, Hansma HG. Biophys J. 1997;72:806–813. doi: 10.1016/s0006-3495(97)78714-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parpura V, Doyle RT, Basarsky TA, Henderson E, Haydon PG. Neuroimage. 1995;2:3–7. doi: 10.1006/nimg.1995.1003. [DOI] [PubMed] [Google Scholar]
- 12.Parpura V, Fernandez JF. Biophys J. 1996;71:2356–2366. doi: 10.1016/S0006-3495(96)79483-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jena BP, Schneider SW, Geibel JP, Webster P, Oberleithner H, Sritharan KC. Proc Natl Acad Sci USA. 1997;94:13317–13322. doi: 10.1073/pnas.94.24.13317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rief M, Gautel M, Oesterhelt F, Fernandez JF, Gaub HE. Science. 1997;276:1109–1112. doi: 10.1126/science.276.5315.1109. [DOI] [PubMed] [Google Scholar]
- 15.Yersin A, Hirling H, Steiner P, Magnin S, Regazzi R, Huni B, Huguenot P, De los Rios P, Dietler G, Catsicas S, Kasas S. Proc Natl Acad Sci USA. 2003;100:8736–8741. doi: 10.1073/pnas.1533137100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu W, Montana V, Bai J, Chapman ER, Mohideen U, Parpura V. Biophys J. 2006;91:744–758. doi: 10.1529/biophysj.105.073312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schiavo G, Matteoli M, Montecucco C. Physiol Rev. 2000;80:717–766. doi: 10.1152/physrev.2000.80.2.717. [DOI] [PubMed] [Google Scholar]
- 18.Parpura V, Chapman ER. Croat Med J. 2005;46:491–497. [PubMed] [Google Scholar]
- 19.Sutton RB, Fasshauer D, Jahn R, Brunger AT. Nature. 1998;395:347–353. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- 20.Otto H, Hanson PI, Jahn R. Proc Natl Acad Sci USA. 1997;94:6197–6201. doi: 10.1073/pnas.94.12.6197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang B, Gonzalez L, Jr, Prekeris R, Steegmaier M, Advani RJ, Scheller RH. J Biol Chem. 1999;274:5649–5653. doi: 10.1074/jbc.274.9.5649. [DOI] [PubMed] [Google Scholar]
- 22.Karrasch S, Dolder M, Schabert F, Ramsden J, Engel A. Biophys J. 1993;65:2437–2446. doi: 10.1016/S0006-3495(93)81327-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Engvall E, Perlmann P. J Immunol. 1972;109:129–135. [PubMed] [Google Scholar]
- 24.Baumert HG, Fasold H. Methods Enzymol. 1989;172:584–609. doi: 10.1016/s0076-6879(89)72035-8. [DOI] [PubMed] [Google Scholar]
- 25.Rembaum A, Margel S, Levy J. J Immunol Meth. 1978;24:239–250. doi: 10.1016/0022-1759(78)90128-x. [DOI] [PubMed] [Google Scholar]
- 26.Schwesinger F, Ros R, Strunz T, Anselmetti D, Guntherodt HJ, Honegger A, Jermutus L, Tiefenauer L, Pluckthun A. Proc Natl Acad Sci USA. 2000;97:9972–9977. doi: 10.1073/pnas.97.18.9972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dettmann W, Grandbois M, Andre S, Benoit M, Wehle AK, Kaltner H, Gabius HJ, Gaub HE. Arch Biochem Biophys. 2000;383:157–170. doi: 10.1006/abbi.2000.1993. [DOI] [PubMed] [Google Scholar]
- 28.Fritz J, Katopodis AG, Kolbinger F, Anselmetti D. Proc Natl Acad Sci USA. 1998;95:12283–12288. doi: 10.1073/pnas.95.21.12283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Porath J, Carlsson J, Olsson I, Belfrage G. Nature. 1975;258:598–599. doi: 10.1038/258598a0. [DOI] [PubMed] [Google Scholar]
- 30.Belew M, Yip TT, Andersson L, Ehrnstrom R. Anal Biochem. 1987;164:457–465. doi: 10.1016/0003-2697(87)90519-7. [DOI] [PubMed] [Google Scholar]
- 31.Lonnerdal B, Carlsson J, Porath J. FEBS Lett. 1977;75:89–92. doi: 10.1016/0014-5793(77)80059-8. [DOI] [PubMed] [Google Scholar]
- 32.Coulet PR, Carlsson J, Porath J. Biotech Bioeng. 1981;23:665–668. [Google Scholar]
- 33.Hochuli E, Dobeli H, Schacher A. J Chromatagr. 1987;411:177–184. doi: 10.1016/s0021-9673(00)93969-4. [DOI] [PubMed] [Google Scholar]
- 34.Nieba L, Krebber A, Pluckthun A. Anal Biochem. 1996;234:155–165. doi: 10.1006/abio.1996.0067. [DOI] [PubMed] [Google Scholar]
- 35.Birko Z, Schauwecker F, Pfennig F, Szeszak F, Vitalis S, Keller U, Biro S. FEMS Microbiol Lett. 2001;196:223–227. doi: 10.1111/j.1574-6968.2001.tb10568.x. [DOI] [PubMed] [Google Scholar]
- 36.Liu W, Montana V, Chapman ER, Mohideen U, Parpura V. Proc Natl Acad Sci USA. 2003;100:13621–13625. doi: 10.1073/pnas.2233819100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Conti M, Falini G, Samori B. Angew Chem, Int Ed Engl. 2000;39:215–218. [PubMed] [Google Scholar]
- 38.Lin RC, Scheller RH. Neuron. 1997;19:1087–1094. doi: 10.1016/s0896-6273(00)80399-2. [DOI] [PubMed] [Google Scholar]
- 39.Munson M, Chen X, Cocina AE, Schultz SM, Hughson FM. Nat Struct Biol. 2000;7:894–902. doi: 10.1038/79659. [DOI] [PubMed] [Google Scholar]
- 40.Hutter JL, Bechhoefer J. Rev Sci Instrum. 1993;64:1868–1873. [Google Scholar]
- 41.Allen S, Chen X, Davies J, Davies MC, Dawkes AC, Edwards JC, Roberts CJ, Sefton J, Tendler SJ, Williams PM. Biochemistry. 1997;36:7457–7463. doi: 10.1021/bi962531z. [DOI] [PubMed] [Google Scholar]
- 42.Allen S, Davies J, Davies MC, Dawkes AC, Roberts CJ, Tendler SJ, Williams PM. Biochem J. 1999;341:173–178. [PMC free article] [PubMed] [Google Scholar]
- 43.Hayashi T, McMahon H, Yamasaki S, Binz T, Hata Y, Sudhof TC, Niemann H. EMBO J. 1994;13:5051–5061. doi: 10.1002/j.1460-2075.1994.tb06834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]