

Abstract
Cationic peptides known as protein transduction domains (PTDs) provide a means to deliver molecules into mammalian cells. Here, nonaarginine (R9), the most efficacious of known PTDs, is used to elucidate the pathway for PTD internalization. Although R9 is found in the cytosol as well as the nucleolus when cells are fixed, this peptide is observed only in the endocytic vesicles of live cells. Co-localization studies with vesicular markers confirm that PTDs are internalized by endocytosis rather than by crossing the plasma membrane. The inability of R9 to enter living cells deficient in heparan sulfate (HS) suggests that binding to HS is necessary for PTD internalization. This finding is consistent with the high affinity of R9 for heparin (Kd = 109 nM). Finally, R9 is shown to promote the leakage of liposomes, but only at high peptide:lipid ratios. These and other data indicate that the PTD-mediated delivery of molecules into live mammalian cells involves: (1) binding to cell-surface HS, (2) uptake by endocytosis, (3) release upon HS degradation, and (4) leakage from endocytic vesicles.
Facilitating the delivery of molecules into mammalian cells is of substantial interest (1–6). Developing the means to do so can generate both insights into cellular function and potential applications in biomedicine. As early as 1965, Ryser noted that mixing polylysine or other cationic macromolecules with proteins greatly increases the uptake of those proteins by cells (7). In 1988, Frankel and Green independently discovered that the RNA-binding HIV1 TAT protein was capable of crossing lipid bilayers (8, 9). The dissection of HIV TAT function has revealed that a small cationic domain, residues 47–57, is responsible for transduction (10). Other cationic, nucleic acid-binding sequences are likewise capable of entering cells (11, 12). Peptide-like molecules and even non-peptidyl polycations are capable of crossing lipid bilayers (11, 13). These cationic “protein transduction domains” (PTDs) are not only capable of entering cells but also can be used to deliver molecular cargo (14–16), such as proteins, oligonucleotides, and small molecules (1, 17–20).
What are the physicochemical characteristics of cationic PTDs? A natural backbone is not necessary for transduction, as D-amino acid (21), peptoid (18), oligocarbamate (22), β-peptide (23, 24), and 6-aminocaproic acid (25) analogs are capable of entering cells. In contrast, the positive charge of cationic PTD is essential for transduction, and arginine is preferred over lysine or histidine (13). Nonaarginine (R9) is the most efficacious known PTD that is composed of natural L-amino acid residues (18).
Although the use of PTDs as macromolecular delivery devices has received much attention, the mechanism of PTD internalization remains controversial (4). To date, most mechanistic studies have been performed with fixed cells (11, 13, 22–28). Recently, Johansson, Lebleu, and their coworkers showed that cell fixation alters the transduction of cationic peptides (29, 30). Internalization was found to be energy-dependent, and the internalized peptide was observed in the endocytic vesicles of living cells (30). Dervan, Johansson, and their coworkers have reported similar cell-fixation artifacts for the transduction of N-methylpyrrole/N-methylimidazole polyamides (31) and PTD–green fluorescent protein fusions (32).
There have also been conflicting reports on whether PTDs bind to cell-surface glycosaminoglycans (GAGs) (11, 27, 33, 34). The cell surface is coated with polysaccharides such as HS and sialic acid, and these molecules are known to mediate the binding of many proteins to cells (34). GAGs are actively internalized from the cell surface in vesicles (35), and this turnover could allow for the passive uptake of interacting macromolecules. Tyagi has shown that the internalization of HIV TAT protein is dependent on the presence of heparan sulfate proteoglycans (HSPGs) (33). Subsequently, however, Sugiura reported that this constraint does not exist for polyarginine internalization into fixed cells (11).
Here, we examine the internalization of R9 into both fixed and living cells. We also analyze R9 internalization into living GAG-deficient Chinese hamster ovary (CHO) cells, and we extend those analyses with both qualitative and quantitative experiments on the binding of R9 to heparin. Finally, we determine whether R9 is able to promote the leakage of liposomes. Together, the data reveal provisions for the entry of a cationic PTD into mammalian cells.
EXPERIMENTAL PROCEDURES
Materials
Amino acids for peptide synthesis were from Novabiochem (Darmstadt, Germany). 5-(and 6-)Carboxytetramethylrhodamine (TAMRA), Hoescht 33342, and FM 1-43 were from Molecular Probes (Eugene, OR). Solvents were from Aldrich Chemical (Milwaukee, WI). Egg phosphatidylcholine (PC) and low-molecular weight heparin (Mr 3000) were from Sigma Chemical (St. Louis, MO). All other chemicals were from Fisher Scientific (Pittsburgh, PA) or Aldrich Chemical (Milwaukee, WI), and were used without further purification.
CHO-K1 cells were from the American Type Culture Collection (Manassas, VA). CHO–pgsD-677 cells (36) and CHO–pgsA-745 cells (37) were gifts of R. I. Montgomery. Cell culture medium and supplements were from Life Technologies (Gaithersburg, MD).
Peptide Synthesis
R9 was synthesized by normal Fmoc-based solid-phase peptide synthesis with an Applied Biosystems Model 432A peptide synthesizer. R9 was cleaved from the Rink acid resin by incubation for 4 h in trifluoroacetic acid/ethanedithiol/phenol/thioanisole (32:4:3:1) and precipitated in diethylether. R9 was purified by reversed-phase HPLC on a C4 column using a gradient of water:acetonitrile containing trifluoroacetic acid (0.1% v/v), and analyzed by matrix-assisted laser desorption ionization–time-of-flight (MALDI–TOF) mass spectrometry using a Bruker Biflex III instrument (m/z 1424.97; expected: 1423.69).
Peptide Labeling
R9 was labeled on resin by incubation for 4 h with TAMRA (4 equivalents) in dimethylformamide containing benzotriazole-1-yl-oxy-tris-pyrolidino-phosphonium hexafluorophosphate (4 equivalents) and diisopropylethylamine (8 equivalents). The resin was washed with dimethylformamide and dichloromethane before deprotection as described above. TAMRA–R9 was purified by reversed-phase HPLC and analyzed by MALDI–TOF mass spectrometry (m/z 1837.92; expected: 1837.13).
Liposomal Leakage
Egg phosphatidyl choline liposomes containing carboxyfluorescein (100 mM) were formed as described previously (38). Liposomes of 100-μm diameter were generated by passage (≥10 times) through a mini-extruder (Avanti Polar Lipids). Promotion of vesicular leakage by R9 was monitored by an increase in fluorescence intensity at 515 nm (excitation: 495 nm) using a QuantaMaster 1 photon-counting fluorometer from Photon Technology International (South Brunswick, NJ). Experiments were performed in 20 mM HEPES–NaOH buffer, pH 7.2. Percent leakage (%L) was calculated as 100 × (I − Io)/(Idetergent − Io) where Io is the initial fluorescence intensity and Idetergent is the intensity following the addition of Triton X-100 (to 0.1% v/v). Following each experiment, exact phospholipid concentrations were measured by using the method of Ames (39).
Cell Culture
CHO cell lines were cultured in Ham’s F-12 medium supplemented with fetal calf serum (to 10% v/v) and antibiotics. Cells were grown in a CO2(g) (5% v/v) atmosphere at 37 °C.
Peptide Internalization
CHO cells were seeded in 4-well Lab-Tek-II chamber slides (Nalge Nunc International, Naperville, IL) in Ham’s F-12 medium. Labeled peptide was added to each slide, which was then incubated at 37 °C. After 60 min, cells were washed (6 times) with PBS containing Ca2+ (0.1 g/L) and Mg2+ (0.1 g/L). Fixed cells were prepared according to the procedure that we described previously (24). Cellular localization was visualized using an Eclipse E800 fluorescence microscope from Nikon (Tokyo, Japan) equipped with an × 60 or × 100 lens.
Heparin-Affinity Chromatography
The interaction of R9 and heparin was assessed qualitatively by affinity chromatography. A1.0-mL HiTrap Heparin HP column from Amersham Pharmacia Biotech (Piscataway, NJ) was equilibrated with 50 mM sodium acetate buffer (pH 5.0). Labeled peptide (~2 mg) was loaded onto the column, and eluted with a linear gradient of NaCl (0–2 M) in the same buffer. Peptide elution was monitored by absorbance at 280 and 535 nm.
Binding of TAMRA–R9 to Heparin
An increase in the fluoresence of TAMRA–R9 at 590 nm (excitation: 531 nm) accompanies its binding to soluble heparin. This increase was used to quantitate the affinity of TAMRA–R9 (1.0 nM) for heparin (Mr 3000) in 20 mM HEPES–NaOH buffer, pH 7.5, using an EnVision multilabel plate reader from PerkinElmer (Wellesley, MA).
RESULTS
R9 Internalization in CHO Cells
Earlier this year, the intracellular localization of HIV TAT peptide in fixed cells was reported to differ from that in living cells (30). To explore whether this dichotomy is a general feature of cationic PTDs, TAMRA–R9 internalization was examined in living and fixed CHO-K1 cells. In living cells, TAMRA–R9 was found in what appeared to be vesicles within the cell (Figure 1A). Upon fixing with paraformaldehyde (4% w/v), TAMRA–R9 migrated to the nucleolus and diffused throughout the cytoplasm (Figure 1B). Similar results were observed when cells were fixed with acetone/methanol (1:1) (data not shown).
Figure 1.
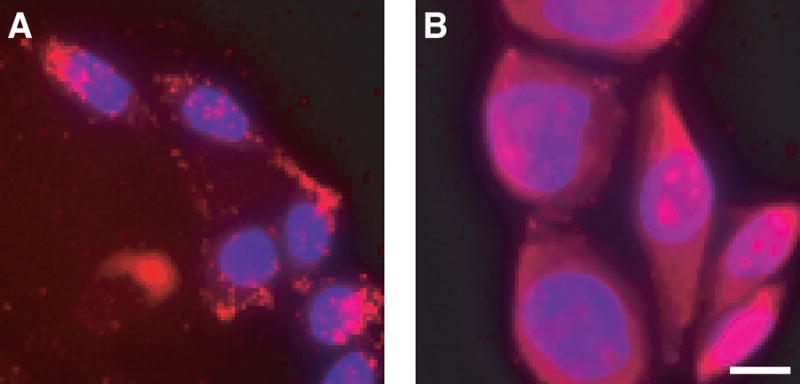
Localization of TAMRA–R9 in living and fixed cells. The internalization of TAMRA–R9 (1.0 μM) was analyzed by fluorescence microscopy. Cells were counterstained with Hoescht 33342. (A) living CHO-K1 cells. (B) CHO-K1 cells fixed with paraformaldehyde. Bar, 10 μm.
To confirm that TAMRA–R9 did indeed localize to the vesicles of living cells, the localization of TAMRA–R9 was compared to that of the dyes FM 1-43 (which is a marker for endocytotic vesicles) and Hoescht 33342 (which is a cell-permeable nuclear stain). Both TAMRA–R9 and FM 1-43 appeared in punctate vesicular structures (Figure 2). No TAMRA–R9 was visible in the nucleus of living cells.
Figure 2.
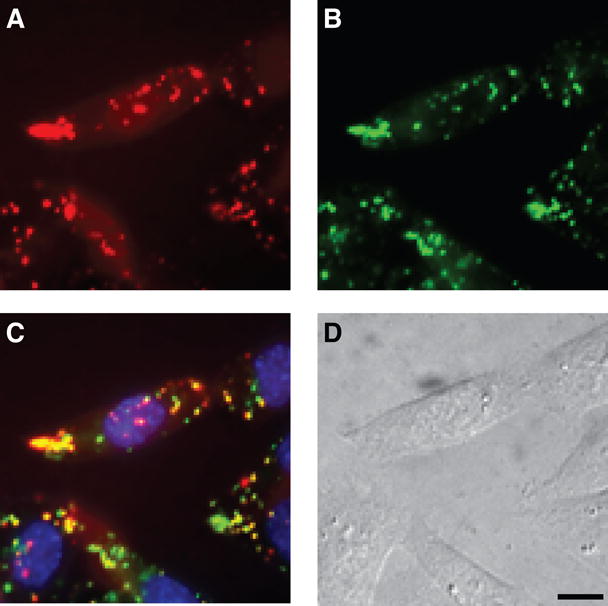
Co-localization of TAMRA–R9 in living cells. The internalization of TAMRA–R9 (1.0 μM) and marker dyes into living CHO-K1 cells was analyzed by fluorescence microscopy. (A) TAMRA–R9. (B) FM 1-43. (C) TAMRA–R9, FM 1-43, and Hoescht 33342. (D) Bright-field image of CHO-K1 cells. Bar, 10 μm.
R9 Uptake by Glycosaminoglycan-Deficient CHO Cells
The CHO–pgsD-677 and CHO–pgsA-745 cell lines have mutations in the glycosaminoglycan (GAG) biosynthetic pathway. Specifically, CHO–pgsD-677 cells do not produce HS (36), and CHO–pgsA-745 cells produce neither HS nor chondroitin sulfate (CS) (37). TAMRA–R9 was incubated with these mutant cells and wild-type CHO-K1 cells (positive control experiment), and its uptake was examined by fluorescence microscopy. Although TAMRA–R9 was clearly visible in CHO-K1 cells, there was almost no detectable fluorescence from TAMRA–R9 in GAG-deficient cell lines (Figure 3). These data indicate that interaction with GAGs on the cell surface is critical for TAMRA–R9 internalization. Adding exogenous heparin to CHO-K1 cells prior to the addition of TAMRA–R9 leads to results similar to that with the GAG-deficient cell lines (data not shown), presumably because the exogenous heparin competes with cell-surface GAGs for TAMRA–R9.
Figure 3.
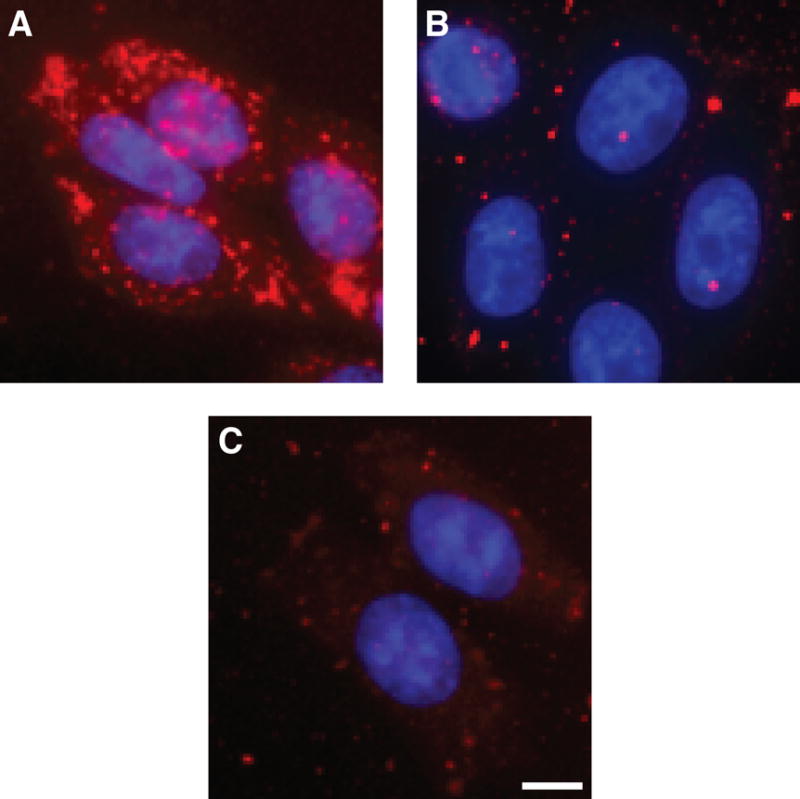
Dependence of TAMRA–R9 internalization into living cells on glycosaminoglycans. Internalization of TAMRA–R9 (1.0 μM) into living wild-type and mutant CHO cells. (A) CHO-K1 cells (wild-type). (B) CHO–pgsD-677 cells (which lack HS). C, CHO–pgsA-745 cells (which lack HS and CS). Bar, 10 μm.
Binding of TAMRA–R9 to Heparin
The interaction of R9 with heparin (Figure 4A), which mimics the GAGs on the surface of a mammalian cell,2 was probed in both qualitative and quantitative assays. The qualitative assay used affinity chromatography with immobilized heparin. TAMRA–R9 was found to bind extremely tightly to immobilized heparin, eluting at approximately 1.5 M NaCl in 50 mM sodium acetate buffer, pH 5.0 (Figure 4B). This affinity is comparable to that of the HIV TAT peptide under similar conditions (40).
Figure 4.

Affinity of TAMRA–R9 for heparin. (A) Prevalent structural motif in heparin.2 (B) Elution profile from affinity chromatography of TAMRA–R9 on immobilized heparin. TAMRA–R9 was eluted with a linear gradient of NaCl (0–2 M) (conductivity, thin line), and monitored by its absorbance at 280 nm (thick line). (C) Fluorescence titration of TAMRA–R9 (1.0 nM) with heparin (Mr 3000) in 20 mM HEPES–NaOH buffer, pH 7.5. The equilibrium dissociation constant is Kd = 109 ± 13 nM.
The affinity of TAMRA–R9 for soluble heparin was quantitated with a direct titration monitored by fluorescence spectroscopy. To determine the value of the equilibrium dissociation constant (Kd), binding data were fitted by nonlinear regression analysis to the equation: Kdn = [heparin][TAMRA–R9]n/[heparin·nTAMRA–R9] with the program Prism 4 from GraphPad (San Diego, CA). Because heparin (Mr 3000) and TAMRA–R9 (Mr 1836) are essentially homopolymers of similar mass, the value of n is likely to be near unity. For n = 1, Kd = 109 ± 13 nM (Figure 4C). Allowing the value of n to vary gave n = 0.87 ± 0.05 and Kd = 119 ± 16 nM, which is not distinguishable from the value of Kd calculated with n = 1.
Liposomal Leakage
Some cell-permeating peptides enter mammalian cells by interacting directly with the lipid bilayer (41). The ability of peptides to disrupt membrane integrity can be measured with a liposomal leakage assay (38). Carboxyfluorescein was encapsulated in liposomes at a high concentration (>100 mM), which quenches its intrinsic fluorescence. Upon vesicle disruption, leakage occurs, resulting in an increase in fluorescence. In a negative control experiment, no leakage leakage of carboxyfluorescein from egg PC vesicles was observed in buffer alone (data not shown). R9-Induced leakage in a peptide concentration-dependent manner (Figure 5A). This result indicates that R9 is capable of disrupting the lipid bilayer. Yet, a peptide:lipid molar ratio of nearly 1:1 was required to induce leakage by R9 (Figure 5B). In a positive control experiment, melittin (which forms pores in lipid bilayers (42)) caused more rapid leakage at 30 nM than did R9 at 260 nM (data not shown).
Figure 5.

R9-Induced leakage of egg PC liposomes. (A) Time course for the leakage of carboxyfluorescein from egg PC liposomes (100-μm diameter) upon exposure to various concentrations of R9. (B) Leakage of carboxyfluorescein from egg PC liposomes (100-μm diameter) as a function of peptide:lipid molar ratio. Data in panel B are derived from data in panel A at 180 s.
DISCUSSION
The efficacy of nascent chemotherapeutics can be limited by their ability to enter mammalian cells (43, 15). PTDs can expedite delivery (1–6). Determining the requirements for PTD entry into cells could enable the creation of even better molecular devices to effect internalization, as well as reveal new aspects of cellular biochemistry and biophysics.
Previous work has led to conflicting conclusions regarding the interaction of PTDs with cells. The mechanism by which peptide toxins, such as mellitin and magainin, interact with cells has been studied extensively, and these peptides are known to form pores in the plasma membrane (42). Penetratin, which is a cationic peptide corresponding to the third α-helix of the Antennapedia homeodomain, is capable of crossing membranes without causing vesicle disruption. In the presence of a lipid bilayer, penetratin appears to form an amphipathic α-helix, which facilitates its insertion into the bilayer (44, 45). HIV TAT peptide has likewise be found in a helical structure by NMR spectroscopy, suggesting that HIV TAT and perhaps other PTDs could act in a manner similar to penetratin (46).
Early mechanistic studies on the cellular entry of PTDs were performed with fixed cells (11, 13, 22–28). Fixation is believed to permeate membranes (30), and allows vesicle-entrapped peptides and proteins to travel to new locations (29, 30, 32). Likewise, we find that TAMRA–R9 is found in different cellular locations in fixed and living cells. Specifically, TAMRA–R9 localizes in the nucleolus and cytoplasm of fixed cells (Figure 1B). In living cells, however, TAMRA–R9 is found primarily in vesicles (Figures 1A and 2). As a result, our efforts to elucidate the requirements for PTD internalization have focused on living cells.
In living cells, cationic proteins such as HIV TAT protein and some analogues of ribonuclease A (which is a small cationic protein (47)) are thought to bind to anionic carbohydrates on the cell surface (33, 48). Moreover, polysaccharides such as heparan sulfate are believed to play important roles in cellular signaling by initiating the binding of certain proteins to their cellular receptors (34, 49, 50). On the cell surface, heparan sulfate is attached to either transmembrane proteins to form syndecans or GPI-anchored proteins to form glypicans (51, 52). The HS side chains of these heparan sulfate proteoglycans (HSPGs) undergo hydrolytic degradation by heparanases en route to the lysosome (53).
A number of proteins are known to bind to specific HSPGs. For some of these proteins, HS is believed to act as the receptor for cellular entry (54). We find that the entry of a cationic PTD into living mammalian cells relies on the presence of HSPGs. Specifically, the entry of TAMRA–R9 into CHO cells that are deficient in HS is decreased greatly relative to that into wild-type cells (Figure 3). Similar results have been observed with the intact HIV-1 TAT protein (54).
HS and heparin are analogous glycosaminoglycans (51, 55).2 Accordingly, heparin has been used as a surrogate for HS in a wide variety of biochemical experiments (56–58). We reasoned that if HSPGs mediate the cellular entry of PTDs, then cationic peptides such as TAMRA–R9 should have a measurable affinity for heparin. Indeed, we find that TAMRA–R9 binds strongly to immobilized heparin (Figure 4B), which mimics the HS side chains of cell-surface HSPGs. Moreover, the soluble heparin·TAMRA–R9 complex has a value of Kd near 0.1 μM (Figure 4C), which is similar to that of a typical receptor–ligand interaction.
How does R9 release from HS and escape from endocytic vesicles? HS is cleaved by heparanases, first in vesicles of neutral pH and then in acidic endosomes (34, 53, 59). HS cleavage would diminish its anion valency and should thus diminish its affinity for R9. We find that free R9 is capable of disrupting a lipid bilayer and causing a liposome to leak (Figure 5). This leakage is maximal between pH 7.5 and 9.5 (data not shown), suggesting that R9 could escape from endosomes prior to their acidification. Regardless, R9-induced liposomal leakage occurs at a peptide:lipid molar ratio that is 10- to 100-fold greater than that for known pore-forming peptides (38). Thus, the disruption of lipid bilayers is likely to play a limited role in transduction at low concentrations of R9, and a relatively high concentration could be necessary for efficient transduction.
Our findings are consistent with a four-step pathway for the entry of cationic PTDs into the cytoplasm of mammalian cells (Figure 6). First, peptides bind to HSPGs on the cell surface. These peptides are then taken up a cell by heparan sulfate-mediated endocytosis. Once in vesicles, HS is degraded by heparanases, releasing the PTD. Finally, unbound PTD escapes from the vesicles after achieving a concentration high enough to promote vesicular leakage. This pathway bears some analogy to those proposed for the cellular entry of other HS-binding proteins, such as bFGF (60) or follistatin (61). The identity of the rate-limiting step for PTD entry (Figure 6) is not known. Identifying that step could lead to more efficacious PTDs or to transduction agonists.
Figure 6.

Pathway for the transduction of R9 into cells. Cationic PTDs such as R9 bind to HSPGs on the cell surface. PTDs are internalization by endocytosis. HS is degraded by heparanases. Free PTDs leak from endocytic vesicles and enter the cytosol.
Acknowledgments
We are grateful to R. I. Montgomery for mutant CHO cell lines, C. Wiese for use of her fluorescence microscope, the Keck Center for Chemical Genomics at the University of Wisconsin Madison for use of their multilabel plate reader, and L. L. Kiessling, S. H. Gellman, and A. Menon for contributive discussions.
Footnotes
Abbreviations: CHO, Chinese hamster ovary; CS, condroitin sulfate; GAG, glycosaminoglycan; HIV, human immunodeficiency virus; HS, heparan sulfate; HSPG, heparan sulfate proteoglycan; MALDI–TOF, matrix-assisted laser desorption ionization–time-of-flight; PC, phosphatidyl choline; PTD, protein transduction domain; R9, nonaarginine; TAT, transactivator of transcription; TAMRA–R9, 5-(and 6-)carboxytetramethylrhodamine conjugated to nonaarginine.
Both HS and heparin are acidic glycosaminoglycans characterized by a repeating unit of α-D-glucosamine linked 1→4 to a uronic (α-L-iduronic or β-D-glucuronic) acid and modified to a variable extent by O- and N-sulfation, and N-acetylation.
This work was supported by National Institutes of Health Grant GM44783.
References
- 1.Hallbrink M, Floren A, Elmquist A, Pooga M, Bartfai T, Langel U. Cargo delivery kinetics of cell-penetrating peptides. Biochem Biophys Acta. 2001;1515:101–109. doi: 10.1016/s0005-2736(01)00398-4. [DOI] [PubMed] [Google Scholar]
- 2.Denicourt C, Dowdy SF. Protein transduction technology offers novel therapeutic approach for brain ischemia. Trends Pharmacol Sci. 2003;24:216–218. doi: 10.1016/S0165-6147(03)00074-9. [DOI] [PubMed] [Google Scholar]
- 3.Green I, Christison R, Voyce CJ, Bundell KR, Lindsay MA. Protein transduction domains: Are they delivering? Trends Pharmacol Sci. 2003;24:213–215. doi: 10.1016/S0165-6147(03)00076-2. [DOI] [PubMed] [Google Scholar]
- 4.Leifert JA, Lindsay Whitton J. “Translocatory proteins” and “protein transduction domains”: A critical analysis of their biological effects and underlying mechanisms. Mol Ther. 2003;8:13–20. doi: 10.1016/s1525-0016(03)00151-5. [DOI] [PubMed] [Google Scholar]
- 5.Thierry AR, Vives E, Richard JP, Prevot P, Martinand-Mari C, Robbins I, Lebleu B. Cellular uptake and intracellular fate of antisense oligonucleotides. Curr Opin Mol Ther. 2003;5:133–138. [PubMed] [Google Scholar]
- 6.Vives E, Richard JP, Rispal C, Lebleu B. TAT peptide internalization: Seeking the mechanism of entry. Curr Protein Pept Sci. 2003;4:125–132. doi: 10.2174/1389203033487306. [DOI] [PubMed] [Google Scholar]
- 7.Ryser HJ, Hancock R. Histones and basic polyamino acids stimulate the uptake of albumin by tumor cells in culture. Science. 1965;150:501–503. doi: 10.1126/science.150.3695.501. [DOI] [PubMed] [Google Scholar]
- 8.Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 1988;55:1189–1193. doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- 9.Green M, Loewenstein PM. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell. 1988;55:1179–1788. doi: 10.1016/0092-8674(88)90262-0. [DOI] [PubMed] [Google Scholar]
- 10.Vives E, Brodin P, Lebleu B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J Biol Chem. 1997;272:16010–16017. doi: 10.1074/jbc.272.25.16010. [DOI] [PubMed] [Google Scholar]
- 11.Suzuki T, Futaki S, Niwa M, Tanaka S, Ueda K, Sugiura Y. Possible existence of common internalization mechanisms among arginine-rich peptides. J Biol Chem. 2002;277:2437–2443. doi: 10.1074/jbc.M110017200. [DOI] [PubMed] [Google Scholar]
- 12.Coeytaux E, Coulaud D, Le Cam E, Danos O, Kichler A. The cationic amphipathic α-helix of HIV-1 viral protein R (Vpr) binds to nucleic acids, permeablizes membranes, and efficiently transfects cells. J Biol Chem. 2003;278:18110–18116. doi: 10.1074/jbc.M300248200. [DOI] [PubMed] [Google Scholar]
- 13.Mitchell DJ, Kim DT, Steinman L, Fathman CG, Rothbard JB. Polyarginine enters cells more efficiently than other polycationic homopolymers. J Pept Res. 2000;56:318–325. doi: 10.1034/j.1399-3011.2000.00723.x. [DOI] [PubMed] [Google Scholar]
- 14.Fawell S, Seery J, Daikh Y, Moore C, Chen LL, Pepinsky B, Barsoum J. Tat-mediated delivery of heterologous proteins into cells. Proc Natl Acad Sci USA. 1994;91:664–668. doi: 10.1073/pnas.91.2.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: Delivery of a biologically active protein into the mouse. Science. 1999;285:1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- 16.Eguchi A, Akuta T, Okuyama H, Senda T, Yokoi H, Inokuchi H, Fujita S, Hayakawa T, Takeda K, Hasegawa M, Nakanishi M. Protein transduction domain of HIV-1 Tat protein promotes efficient delivery of DNA into mammalian cells. J Biol Chem. 2001;276:26204–26210. doi: 10.1074/jbc.M010625200. [DOI] [PubMed] [Google Scholar]
- 17.Schwarze SR, Dowdy SF. In vivo protein transduction: Intracellular delivery of biologically active proteins, compounds and DNA. Trends Pharmacol Sci. 2000;21:45–48. doi: 10.1016/s0165-6147(99)01429-7. [DOI] [PubMed] [Google Scholar]
- 18.Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, Rothbard JB. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: Peptoid molecular transporters. Proc Natl Acad Sci USA. 2000;97:13003–13008. doi: 10.1073/pnas.97.24.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pooga M, Kut C, Kihlmark M, Hallbrink M, Fernaeus S, Raid R, Land T, Hallberg E, Bartfai T, Langel U. Cellular translocation of proteins by transportan. FASEB J. 2001;15:1451–1453. doi: 10.1096/fj.00-0780fje. [DOI] [PubMed] [Google Scholar]
- 20.Backer MV, Aloise R, Przekop K, Stoletov K, Backer JM. Molecular vehicles for targeted drug delivery. Bioconjug Chem. 2002;13:462–467. doi: 10.1021/bc0155770. [DOI] [PubMed] [Google Scholar]
- 21.Derossi D, Calvet S, Trembleau A, Brunissen A, Chassaing G, Prochiantz A. Cell internalization of the third helix of the Antennapedia homeodomain is receptor-independent. J Biol Chem. 1996;271:18188–18193. doi: 10.1074/jbc.271.30.18188. [DOI] [PubMed] [Google Scholar]
- 22.Wender PA, Rothbard JB, Jessop TC, Kreider EL, Wylie BL. Oligocarbamate molecular transporters: Design, synthesis, and biological evaluation of a new class of transporters for drug delivery. J Am Chem Soc. 2002;124:13382–13383. doi: 10.1021/ja0275109. [DOI] [PubMed] [Google Scholar]
- 23.Rueping M, Mahajan Y, Sauer M, Seebach D. Cellular uptake studies with β-peptides. Chembiochem. 2002;3:257–259. doi: 10.1002/1439-7633(20020301)3:2/3<257::AID-CBIC257>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 24.Umezawa N, Gelman MA, Haigis MC, Raines RT, Gellman SH. Translocation of a β-peptide across cell membranes. J Am Chem Soc. 2002;124:368–369. doi: 10.1021/ja017283v. [DOI] [PubMed] [Google Scholar]
- 25.Rothbard JB, Kreider E, VanDeusen CL, Wright L, Wylie BL, Wender PA. Arginine-rich molecular transporters for drug delivery: Role of backbone spacing in cellular uptake. J Med Chem. 2002;45:3612–3618. doi: 10.1021/jm0105676. [DOI] [PubMed] [Google Scholar]
- 26.Futaki S, Nakase I, Suzuki T, Youjun Z, Sugiura Y. Translocation of branched-chain arginine peptides through cell membranes: Flexibility in the spatial disposition of positive charges in membrane-permeable peptides. Biochemistry. 2002;41:7925–7930. doi: 10.1021/bi0256173. [DOI] [PubMed] [Google Scholar]
- 27.Mai JC, Shen H, Watkins SC, Cheng T, Robbins PD. Efficiency of protein transduction is cell type-dependent and is enhanced by dextran sulfate. J Biol Chem. 2002;277:30208–30218. doi: 10.1074/jbc.M204202200. [DOI] [PubMed] [Google Scholar]
- 28.Ragin AD, Morgan RA, Chmielewski J. Cellular import mediated by nuclear localization signal peptide sequences. Chem Biol. 2002;9:943–948. doi: 10.1016/s1074-5521(02)00189-8. [DOI] [PubMed] [Google Scholar]
- 29.Lundberg M, Johansson M. Positively charged DNA-binding proteins cause apparent cell membrane translocation. Biochem Biophys Res Commun. 2002;29:367–371. doi: 10.1006/bbrc.2002.6450. [DOI] [PubMed] [Google Scholar]
- 30.Richard JP, Melikov K, Vives E, Ramos C, Verbeure B, Gait MJ, Chernomordik LV, Lebleu B. Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake. J Biol Chem. 2003;278:585–590. doi: 10.1074/jbc.M209548200. [DOI] [PubMed] [Google Scholar]
- 31.Belitsky JM, Leslie SJ, Arora PS, Beerman TA, Dervan PB. Cellular uptake of N-methylpyrrole/N-methylimidazole polyamide-dye conjugates. Bioorg Med Chem. 2002;10:3313–3318. doi: 10.1016/s0968-0896(02)00204-3. [DOI] [PubMed] [Google Scholar]
- 32.Lundberg M, Wikstrom S, Johansson M. Cell surface adherence and endocytosis of protein transduction domains. Mol Ther. 2003;8:143–150. doi: 10.1016/s1525-0016(03)00135-7. [DOI] [PubMed] [Google Scholar]
- 33.Tyagi M, Rusnati M, Presta M, Giacca M. Internalization of HIV-1 tat requires cell surface heparan sulfate proteoglycans. J Biol Chem. 2001;276:3254–3261. doi: 10.1074/jbc.M006701200. [DOI] [PubMed] [Google Scholar]
- 34.Belting M. Heparan sulfate proteoglycan as a plasma membrane carrier. Trends Biochem Sci. 2003;28:145–151. doi: 10.1016/S0968-0004(03)00031-8. [DOI] [PubMed] [Google Scholar]
- 35.Yanagishita M. Cellular catabolism of heparan sulfate proteoglycans. Trends Glycosci Glycobiol. 1998;10:57–63. [Google Scholar]
- 36.Lidholt K, Weinke JL, Kiser CS, Lugemwa FN, Bame KJ, Cheifetz S, Massague J, Lindahl U, Esko JD. A single mutation affects both N-acetylglucosaminyltransferase and glucuronosyltransferase activities in a Chinese hamster ovary cell mutant defective in heparan sulfate biosynthesis. Proc Natl Acad Sci USA. 1992;89:2267–2271. doi: 10.1073/pnas.89.6.2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Esko JD, Stewart TE, Taylor WH. Animal cell mutants defective in glycosaminoglycan biosynthesis. Proc Natl Acad Sci USA. 1985;82:3197–3201. doi: 10.1073/pnas.82.10.3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Medina ML, Chapman BS, Bolender JP, Plesniak LA. Transient vesicle leakage initiated by a synthetic apoptotic peptide derived from the death domain of neurotrophin receptor, p75NTR. J Pept Res. 2002;59:149–158. doi: 10.1034/j.1399-3011.2002.1o971.x. [DOI] [PubMed] [Google Scholar]
- 39.Ames BN. Assay of inorganic phosphate, total phosphate, and phosphatases. Methods Enzymol. 1966;8:115–118. [Google Scholar]
- 40.Hakansson S, Jacobs A, Caffrey M. Heparin binding by the HIV-1 tat protein transduction domain. Protein Sci. 2001;10:2138–2139. doi: 10.1110/ps.23401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prochiantz A. Messenger proteins: Homeoproteins, TAT and others. Curr Opin Cell Biol. 2000;12:400–406. doi: 10.1016/s0955-0674(00)00108-3. [DOI] [PubMed] [Google Scholar]
- 42.Yang L, Harroun TA, Weiss TM, Ding L, Huang HW. Barrel-stave model or toroidal model? A case study on melittin pores. Biophys J. 2001;81:1475–1485. doi: 10.1016/S0006-3495(01)75802-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rojas M, Donahue JP, Tan Z, Lin YZ. Genetic engineering of proteins with cell membrane permeability. Nat Biotechnol. 1998;16:370–375. doi: 10.1038/nbt0498-370. [DOI] [PubMed] [Google Scholar]
- 44.Fischer PM, Zhelev NZ, Wang S, Melville JE, Fahraeus R, Lane DP. Structure activity relationship of truncated and substituted analogues of the intracellular delivery vector Penetratin. J Pept Res. 2000;55:163–172. doi: 10.1034/j.1399-3011.2000.00163.x. [DOI] [PubMed] [Google Scholar]
- 45.Thoren PE, Persson D, Karlsson M, Norden B. The Antennapedia peptide penetratin translocates across lipid bilayers—the first direct observation. FEBS Lett. 2000;482:265–268. doi: 10.1016/s0014-5793(00)02072-x. [DOI] [PubMed] [Google Scholar]
- 46.Silhol M, Tyagi M, Giacca M, Lebleu B, Vives E. Different mechanisms for cellular internalization of the HIV-1 Tat-derived cell penetrating peptide and recombinant proteins fused to Tat. Eur J Biochem. 2002;269:494–501. doi: 10.1046/j.0014-2956.2001.02671.x. [DOI] [PubMed] [Google Scholar]
- 47.Raines RT. Ribonuclease A. Chem Rev. 1998;98:1045–1065. doi: 10.1021/cr960427h. [DOI] [PubMed] [Google Scholar]
- 48.Ogawa Y, Iwama M, Ohgi K, Tsuji T, Irie M, Itagaki T, Kobayashi H, Inokuchi N. Effect of replacing the aspartic acid/glutamic acid residues of bullfrog sialic acid binding lectin with asparagine/glutamine and arginine on the inhibition of cell proliferation in murine leukemia P388 cells. Biol Pharm Bull. 2002;25:722–727. doi: 10.1248/bpb.25.722. [DOI] [PubMed] [Google Scholar]
- 49.Bernfield M, Gotte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, Zako M. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem. 1999;68:729–777. doi: 10.1146/annurev.biochem.68.1.729. [DOI] [PubMed] [Google Scholar]
- 50.Renato V. Heparan sulfate proteoglycans: Intricate molecules with intriguing functions. J Clin Invest. 2001;108:165–167. doi: 10.1172/JCI13560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Varki A, Cummings R, Esko J, Freeze H, Hart G, Marth J. Essentials of Glycobiology. Cold Spring Harbor Laboratory Press; New York: 1999. [PubMed] [Google Scholar]
- 52.Sasisekharan R, Myette JR. The sweet science of glycobiology. Am Sci. 2003;91:432–441. [Google Scholar]
- 53.Yanagishitia M, Hascall VC. Metabolism of proteoglycans in rat ovarian granulosa cell culture. J Biol Chem. 1984;259:10207–10283. [PubMed] [Google Scholar]
- 54.Hashimoto O, Nakamura T, Shoji H, Shimasaki S, Hayashi Y, Sugino H. A novel role of follistatin, an activin-binding protein, in the inhibition of activin action in rat pituitary cells. Endocytotic degradation of activin and its acceleration by follistatin associated with cell-surface heparan sulfate. J Biol Chem. 1997;272:13835–13842. doi: 10.1074/jbc.272.21.13835. [DOI] [PubMed] [Google Scholar]
- 55.Sasisekharan R, Venkataraman G. Heparin and heparan sulfate: Biosynthesis, structure and function. Curr Opin Chem Biol. 2000;4:626–631. doi: 10.1016/s1367-5931(00)00145-9. [DOI] [PubMed] [Google Scholar]
- 56.Bilozur ME, Biswas C. Identification and characterization of heparan sulfate-binding proteins from human lung carcinoma cells. J Biol Chem. 1990;265:19697–19703. [PubMed] [Google Scholar]
- 57.San Antonio JD, Lander AD, Karnovsky MJ, Slayter HS. Mapping the heparin-binding sites on type I collagen monomers and fibrils. J Cell Biol. 1994;125:1179–1188. doi: 10.1083/jcb.125.5.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou H, Casas-Finet JR, Heath Coats R, Kaufman JD, Stahl SJ, Wingfield PT, Rubin JS, Bottaro DP, Andrew Byrd R. Identification and dynamics of a heparin-binding site in hepatocyte growth factor. Biochemistry. 1999;38:14793–14802. doi: 10.1021/bi9908641. [DOI] [PubMed] [Google Scholar]
- 59.McKenzie E, Young K, Hircock M, Bennett J, Bhaman M, Felix R, Turner P, Stamps A, McMillan D, Saville G, Ng S, Mason S, Snell D, Schofield D, Gong H, Townsend R, Gallagher J, Page M, Parekh R, Stubberfield C. Biochemical characterization of the active heterodimer form of human heparanase (Hpa1) protein expressed in insect cells. Biochem J. 2003;373:423–435. doi: 10.1042/BJ20030318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sperinde GV, Nugent MA. Mechanisms of fibroblast growth factor 2 intracellular processing: A kinetic analysis of the role of heparan sulfate proteoglycans. Biochemistry. 2000;39:3788–3796. doi: 10.1021/bi992243d. [DOI] [PubMed] [Google Scholar]
- 61.Sugino H, Ugiro K, Hashimoto O, Shoji H, Nakamura T. Follistatin and its role as an activin-binding protein. J Med Invest. 1997;44:1–14. [PubMed] [Google Scholar]