

Abstract
The need for newer compounds to treat depression is an ever-growing concern due to the enormous societal and financial ramifications of this disorder. Here, we review some of the candidate systems that could potentially be involved in depression, or an inherent resistance to depression termed resilience, and the numerous protein targets for these systems. A substantial body of literature provides strong evidence that neurotrophic factors, glutamate receptors, hypothalamic feeding peptides, nuclear hormone receptors, and epigenetic mechanisms, among others, will make for interesting targets when examining depressive behavior or resilience in preclinical models, and eventually clinical trials. Although some of these targets for depression already appear promising, new waves of more selective compounds for any molecular system should promote a better understanding of this complex disease and perhaps improved treatments.
1. Introduction
In addition to traditional agonists and antagonists that act predominantly at cellular membrane receptors, a surge in the number of specific compounds affecting intracellular proteins has recently been observed in experimental reports, furthering our understanding of, and putative treatments for, psychiatric and neurological diseases. While much remains to be learned concerning the roles of neurotransmitter systems and neuromodulatory peptides that act directly at the cell surface of neurons and glia, we postulate that the steady rise in compounds aimed at targeting intracellular mechanics reflects remarkable advances in neuroscience. Probing deeper into the mechanisms of cellular function reveals increasingly complex interactions between enzymes and structural proteins that mediate processes ranging from genomic regulation to cellular morphology. Unraveling the roles of particular intracellular signaling molecules, transcription factors, and chromatin modifying enzymes has elucidated cell-type specific events that may be dysregulated during disease states. Further advancements will undoubtedly lead to the generation of newer classes of compounds that are capable of targeting such cellular events, with the added potential of cell-type specificity. The fundamental goal of this strategy is to better treat psychiatric and neurological diseases in a timely manner without undesired negative side effects.
The need for improved antidepressant treatments is long-standing. Despite a steady increase in the number of people treated for depression over the past thirty years, the prevalence of the disorder remains stable. This, along with other factors, demonstrates the inadequacy of current treatments and a lack of improvement over the conventional monoaminergic-based therapies discovered by serendipity decades ago (Healy, 1999; Kessler and Wang, 2008). In addition, available medications are slow to produce effects: mood elevation is seen only after several weeks of treatment, and severely depressed patients reach remission after an average of four successive treatments over 38 weeks (Rush et al., 2006). A need for maintenance therapies after a remission of symptoms is also typically warranted in patients that have a lifetime history of depressive episodes, comorbid anxiety disorder, substance abuse disorders, as well as those who are likely to experience frequent bouts of stress (Blier et al., 2007; Davis et al., 2007). A high lifetime prevalence rate (16.6%), with high rates of recurrence and morbidity, contribute substantially to the global burden of the disease which supports a need for newer medications with greater efficiency, faster onset of action, and improved tolerability (Berton and Nestler, 2006; Kessler and Wang, 2008; Rush, 2007).
A key obstacle in developing newer medications has been a limited knowledge of the pathophysiology of depression (Krishnan and Nestler, 2008). Indeed, there is a profound heterogeneity in the phenotype of depression (i.e., its clinical presentation, age of onset, course of illness, and treatment response), signifying that depression encompasses many different disease states with distinct etiologies (Berton and Nestler, 2006; Rush, 2007). Epidemiologic studies have found that depression is roughly 40% heritable, yet no specific genes have yet been identified definitively. In light of these problems, studies examining the role of gene-environment interactions on the emergence of particular symptoms or treatment responses have indicated several ‘susceptibility’ genes that may indicate a higher risk for major depressive disorder or resistance to treatment (Aan Het Rot et al., 2009; Lekman et al., 2008). Recent neuroimaging and post-mortem studies of human brain have advanced our knowledge of neural systems involved in depression, particularly brain areas that might underlie cognitive impairments and dysregulation of emotional processing (Drevets et al., 2008). As a complement to these findings, deep brain stimulation of the subgenual cingulate cortex or nucleus accumbens has been shown to alleviate some depressive symptoms in treatment-refractory patients (Mayberg et al., 2005; Schlaepfer et al., 2008).
Functional and morphological differences between brains of depressed and non-depressed subjects are likely to be derived from distinct molecular and cellular correlates (Manji et al., 2001). In clinical depression, and through the use of depression models in animals, numerous anomalies at the cellular level in distinct brain areas have indeed been revealed, which has in effect “opened the flood gates” for examining newer antidepressant targets (Berton and Nestler, 2006; Mathew et al., 2008). Also contributing to this wealth of potential new targets is the realization that the search for new antidepressants should not focus solely on mechanisms that prevent or reverse the deleterious effects of stress, but should also include mechanisms that promote resilience—continued health and well-being despite the onslaught of severe stress (Feder et al., 2009). Amongst such newer targets are amino acid neurotransmitter systems that are known for their dominant role in regulating neural activity and synaptic plasticity (e.g., glutamate and GABA), neurotrophic factors, and molecules that are readily induced by episodes of stress (e.g., CRF [corticotropin release factor] and glucocorticoids), neuropeptides that maintain homeostatic energy balance (e.g., hypothalamic feeding peptides), gonadal hormones that fluctuate over time, particularly in females (e.g., estrogen), as well as the abundance of downstream cellular effectors that affect genome-wide transcriptional outcomes (e.g., ΔFosB, CREB [cAMP response element binding protein], PDEs [phosphodiesterases], GSK-3ß [glycogen synthase kinase 3ß], and HDACs [histone deacetylases]). The majority of these potential targets are broadly expressed throughout the brain and have a diversity of functions. Therefore, the overwhelming challenge is to find specific targets that generate predicted outcomes, without producing adverse side effects. The development of allosteric modulators of receptor systems represents a particularly nice example of recent successes in drug discovery. Allosteric binding sites, when targeted, allow for increases or decreases in the efficacy of endogenously available receptor ligand. Consequently, tolerance, dependence, and overdoses are less-frequently reported with allosteric pharmacological modulators. The best example of this principle is the popular GABAA positive allosteric modulators used to treat anxiety. Refining our understanding of the pathophysiology of depression and resilience, and uncovering novel antidepressant mechanisms, will ultimately lead to better therapeutic strategies for treating depressive syndromes.
In this review, we summarize our current understanding of depression and highlight examples of recent neural and molecular mechanisms implicated in this disorder. We focus on their therapeutic potential, and critically discuss their strengths and weaknesses in the light of recent preclinical and translational studies. Novel potential treatments for depression that are already actively in clinical trials (i.e., those that target receptors for CRF, vasopressin, glucocorticoids, and neurokinins) are discussed in other reviews (see Berton and Nestler, 2006; Mathew et al., 2008), and therefore are not covered here.
2. Neurotrophic factors and related signal transduction pathways
Sizeable morphological changes in the hippocampus have been reported in depressed humans and after chronic stress in animal models (Gould et al., 1997; Sapolsky, 1996). These observations have prompted the notion that depression might be associated with neuronal loss in this brain region (Sapolsky, 2000). One mechanism by which hippocampal impairments may correspond with depression is via the loss of neurotrophic factors and related signaling cascades. In addition, conventional antidepressant treatments have been shown to increase patterns of neurogenesis in the adult hippocampus, an effect that appears to be important for their antidepressant-like behavioral effects (Santarelli et al., 2003). However, given a lack of selective molecules that target specific signaling cascades involved in the process of hippocampal neurogenesis has made it difficult to achieve empirical assessments of their effects in preclinical depression models. Nonetheless, BDNF [brain-derived neurotrophic factor] is decreased in the hippocampus of depressed patients (Dwivedi et al., 2003) and by stress in animals, and multiple lines of experimental evidence have supported a role for BDNF in the behavioral effects of antidepressants in animals (Duman and Monteggia, 2006). In addition, BDNF polymorphisms can robustly alter its activity-dependant release, and may be linked to depression-related vulnerabilities in humans (Duncan et al., 2008; Gatt et al., 2009).
However, the effects of BDNF in other brain areas generate different behavioral outcomes, and increases in BDNF in the brain’s reward circuitry may actually be pro-depressant (Berton et al., 2006). For example, infusion of BDNF into the ventral tegmental area (VTA) produces depression-like effects (Eisch et al., 2003). Likewise, an increase in BDNF protein levels in the VTA and its nucleus accumbens (NAc) target is triggered by chronic social defeat stress, and this increase is both necessary and sufficient to produce a depressive-like phenotype, while the selective knockout of BDNF from the VTA promotes resilience to stress (Berton et al., 2006; Krishnan et al., 2007). Thus, it is not surprising that broad forebrain deletions of BDNF or its TrkB receptor do not dramatically influence depression-like behaviors (Monteggia et al., 2007; Zorner et al., 2003) and that modulating BDNF or TrkB activity has proven to be clinically ineffective (Sen and Sanacora, 2008; Tanis et al., 2007). The limitations associated with modulating BDNF globally have prompted a revision of the neurotrophic hypothesis of depression, and highlights the difficulties of targeting such complex systems (Groves, 2007; Krishnan and Nestler, 2008).
Despite such difficulties, the neurotrophic hypothesis of depression has opened new doors for research strategies examining the mechanisms of depression and resilience. One example is the insight obtained from studies on the intracellular signaling pathways that are regulated by BDNF and other neurotrophic factors. BDNF activation of TrkB in turn activates several intracellular cascades, including the Ras-Raf-ERK (extracellular-signal regulated kinase), phosphatidylinositol-3-kinase (PI3K)-AKT, and PLCγ (phospholipase Cγ) pathways. These distinct pathways converge on activating the transcription factor CREB, among many other actions. CREB then activates the transcription of numerous genes, including many of the neurotrophins. Not surprisingly, most of the proteins involved in neurotrophic signaling pathways, including ERK, AKT, PLCγ, and CREB, are regulated in animal models of depression or antidepressant treatments (Bolaños et al., 2003; Carlezon et al., 2005; Duman et al., 2007; Dwivedi et al., 2006; Dwivedi et al., 2008; Krishnan et al., 2008). Another potential target for achieving antidepressant effects may be via upregulation of the cAMP-CREB pathway through inhibition of any of several phosphodiesterases, which catalyze the breakdown of cAMP and increase BDNF expression (Fujimaki et al., 2000). Rolipram, a non-selective inhibitor of PDE4, has been reported to have antidepressant effects in depressed patients (Manji et al., 2003). Although these early trials were discontinued due to intense side effects (e.g., nausea and vomiting), efforts to develop safer drugs that target selective PDE isoforms continue [i.e., mice lacking PDE4D display antidepressant-like behaviors (Zhang et al., 2002)], and new PDE4 agents are in clinical trials for treating inflammatory diseases (Spina, 2008). PDEs 2, 5, 6, 10 and 11 all contain a GAF binding domain, and the GAF domain on PDEs 2, 5 and 6 have a higher affinity for cyclic guanosine monophosphate (cGMP) than for cAMP (Zoraghi et al., 2004), which serves to regulate catalytic activity upon association. By virtue of a large and well-defined structure, GAF domains provide a realistic theoretical target for constructing new ligands that function as agonists, or antagonists, and with a high degree of selectivity for particular PDEs (Martinez et al., 2002). Two compounds, sildenafil (a PDE5 inhibitor) and papaverine (an inhibitor of PDE10 among other subtypes), both appear to have anxiogenic effects in mice, indicating a hypothetical role for these enzymes in stabilizing mood (Hebb et al., 2008; Kurt et al., 2004).
Two recently characterized proteins, named SPROUTY (SPRY) and SPRED, have been shown to repress the action of fibroblast growth factor (FGF) and associated receptor tyrosine kinase-dependent signaling pathways (Bundschu et al., 2007; Cabrita and Christofori, 2008). Antagonism of these proteins has been postulated to be a potential therapeutic treatment for depression. SPRY2 is downregulated in the prefrontal cortex after chronic antidepressant treatment (Ongur et al., 2007), and disruption of this protein in the dorsal hippocampus has long-lasting effects on neurogenensis and depressive-like behavior (Dow et al., 2008). However, this protein, like many others, is enriched across several brain areas and its action may lead to different behavioral outcomes depending on its location. SPRY3, on the other hand, may be a more promising target in this regard (Minowada et al., 1999; Sanchez et al., 2008). As more inhibitors of SPRY (i.e., other modulators of FGF signaling) become increasingly available, their potential effects on depressive-like behavior, through their presumable effects on neurogensis and cellular plasticity, can begin to be fully addressed in preclinical models (Bachis et al., 2008).
The SPRED family of proteins specifically inhibits the Ras-ERK pathway in response to several growth factors, including vascular endothelial growth factor (VEGF) (Bundschu et al., 2007). Studies with Spred1 knockout mice have demonstrated that this protein plays an important role in hippocampal-dependant learning and synaptic plasticity, making this an interesting target for antidepressant treatment (Denayer et al., 2008). Future studies should determine whether or not other family members of these proteins are selectively enriched in certain brain areas. It is promising that some of these proteins, such as SPRED3, are expressed predominantly in brain (Kato et al., 2003). However, until pharmacological agents are made available that alter their activity in vivo, it will remain challenging to unravel the promise in modulating SPRED in depressive-like behavioral responses.
The precursor of BDNF, proBDNF, is widely and abundantly expressed in adult brain and binds to the pan-neurotrophin receptor p75NTR. Upon binding to p75NTR, proBDNF can elicit long-term depression, reduce spine density in hippocampal neurons, and induce apopotosis in basal forebrain neurons (Martinowich et al., 2007; Volosin et al., 2006; Woo et al., 2005; Zagrebelsky et al., 2005). Control over the cleavage of proBDNF may thus represent a potentially relevant therapeutic target. Indeed, several recent studies have shown that the enzymes responsible for converting proBDNF to mature BDNF (i.e., tissue plasminogen activator [tPA], as well as several regulators of tPA like plasminogen activator inhibitor type 1 [PAI1], and p11, an activator of tPA) are implicated in depression (Martinowich et al., 2007; Tsai et al., 2008). Interestingly, the use of STATINs to treat hypercholesterolemia is reported to be associated with a reduction in the incidence of depression (Tsai, 2007). The mechanisms underlying an antidepressive effect of STATINs are currently unknown, but their inhibitory role over PAI1 might increase tPA activity, thereby increasing the cleavage of proBDNF into BDNF. Since tPA is expressed in the blood and the tPA-plasmin proteolytic cascade is involved in cardiovascular function, this approach may target a subset of depressed patients suffering from cardiovascular disease (Hou et al., 2009). Due to a lack of empirical data that support a role for STATINs, and their regulation of associated pathways, in depressive-like behavioral responses, more studies are necessary to build upon these claims. Additional neurotrophic factors, such as VEGF, FGF , and VGF (non-acronymic) have been implicated in the etiology and treatment of depression (Tsai et al., 2009; Evans et al. 2004; Warner-Schmidt and Duman, 2007). Such neurotrophic factors are induced by chronic antidepressant treatments in the hippocampus, while chronic stress reduces the expression of vegf (as well as its receptor fllk-1) and fgf2 (as well as its receptor fgfr1) in this brain region (Heine et al., 2005; Turner et al., 2008). There are more than 20 endogenous ligands for FGF receptors alone (Reuss et al. 2003). Assessments of neurotrophic factor manipulations during ongoing conventional antidepressant treatments may be particularly valuable, given their prominent role in various forms of neuronal plasticity. All in all, a careful elucidation of the networks that rely on transcriptional regulation by these and other neurotrophic factors has tremendous potential for providing new therapeutic targets for the treatment of depression.
3. Glutamate acting drugs
Ever since the discovery in 1959 that D-cycloserine, a partial NMDA glutamate receptor agonist, has antidepressant effects, it has been generally accepted that the glutamatergic system contributes to the pathophysiology of depression (Crane, 1959; Pittenger et al., 2007). Many reports have highlighted alterations in glutamate signaling as well as changes in the expression of AMPA or NMDA receptor subunits in depression, although there are significant variations across brain areas, and the functional significance of these changes remains unclear (Feyissa et al., 2009; Karolewicz et al., 2009; Sanacora et al., 2008). No approved antidepressant treatment is currently based solely on targeting the glutamatergic system, although the glutamatergic agent riluzole is sometimes administered for its antidepressant effects. Originally developed as an anticonvulsant and subsequently granted approval by the FDA for treating amyotrophic lateral sclerosis, riluzole has recently been demonstrated to be a successful augmentation strategy in subjects with treatment resistant depression (Zarate et al., 2004).
In parallel, there is growing interest in the non-competitive NMDA receptor antagonist, ketamine, which produces a rapid and sustained antidepressant response in patients with treatment-resistant depression (Berman et al., 2000; Zarate et al., 2006; Aan Het Rot et al., 2009). Importantly, such effects of ketamine are seen at sub-psychomimetic doses of the drug. Moreover, ketamine produces a profound reduction in suicidality (Price et al., 2009). Based on findings with ketamine, there is interest in developing subtype-selective NMDA antagonists, particularly those that act through allosteric mechanisms. Allosteric modulators of glutamate receptors might have higher selectivity, retain the spatial and temporal aspects of endogenous receptor activity, and avoid many of the drawbacks associated with more conventional ligands (Conn et al., 2009).
NMDA receptors are complex ion channels formed by the combination of two NR1 subunits that contain the glycine/D-serine co-agonist site, and two NR2 subunits, which contain the glutamate-binding site. Among the four NR2 subunits, NR2A and NR2B are expressed in forebrain. These subunits have different pharmacological properties and localization, and they play an important role in adjusting a cell’s excitability threshold for synaptic modification (Yashiro and Philpot, 2008). Therefore, specific targeting of these distinct subunits may reveal useful antidepressant treatments. In this regard, CP-101,606 (an NR2B selective antagonist) is reported to produce rapid and robust antidepressant effects in patients with treatment-refractory depression with good tolerability and without producing dissociative reactions (Preskorn et al., 2008; Pittenger et al., 2007). Contrary to ketamine, which blocks the receptor-gated ion channel, CP-101,606 inhibits NMDA receptors through an allosteric mechanism (Mott et al., 1998), which may account for its fewer adverse side effects. Recent studies have indicated that activation of synaptic (or extrasynaptic) NMDA receptors can have apparently opposite effects on the function and survival of neurons (Hardingham et al., 2002). Specifically, it appears that activation of synaptic NMDA receptors promotes cell survival, in part through activation of CREB and BDNF, while extrasynaptic NMDA receptors initiate cell death. Pre-clinical studies have also revealed antidepressant-like effects of NMDA receptor partial agonists at the glycine/D-serine site (Pittenger et al., 2007).
AMPA and kainate ionotropic glutamate receptors represent additional potential drug targets (Lodge, 2009). AMPA receptors mediate fast excitatory postsynaptic currents in most neurons, however, the time course and amplitude of these effects depend on the subunit composition of the receptors, which vary across brain regions, neurons, and even synapses, which influences their role in synaptic plasticity and behavior (Kessels and Malinow, 2009). Allosteric modulators fail to rapidly desensitize AMPA receptors like full agonists do, and several classes of positive allosteric modulators of AMPA receptors increase BDNF expression levels, which can stimulate neurogenesis as well as neuronal sprouting in hippocampal neurons (O’Neill and Witkin, 2007). Such potentiation of AMPA receptors promotes antidepressant-like effects in rodents (Bleakman et al., 2007). In line with these findings, mice lacking the AMPA subunit GluR1 show depression-like behavior (Chourbaji et al., 2008). It is also possible that the antidepressant effects of ketamine are mediated in part through the activation of AMPA receptors (Maeng et al., 2008).
Despite these promising results, no clinical trial with AMPA receptor potentiators has yet succeeded as a result of their risk of toxicity (Mathew et al., 2008). Nonetheless, large-scale gene candidate studies have revealed an association between the GluR3 AMPA receptor and the K2 kainate receptor and suicidal ideation (Laje et al., 2007). Associations between the K4 kainate subunit and the outcome of antidepressant treatment have been identified as well (Paddock et al., 2007). Such pharmacogenomic approaches have further established a prominent role for the glutamatergic system in the neural effects of antidepressant treatment (Lekman et al., 2008).
AMPA receptors are highly regulated at the synapse via phosphorylation and their direct interaction with a multitude of proteins, particularly, the transmembrane AMPA receptor regulatory proteins (TARPs) and the recently discovered cornichon protein family, among many others like GRIP/ABP and PICK1 (Fig. 1). Targeting these proteins may be a feasible way to influence AMPA receptor function. TARPs are expressed exclusively in excitatory post-synaptic densities, which make them relatively selective for AMPA receptors (Tomita et al., 2003). TARPs mediate AMPA receptor surface expression and synaptic clustering (Tomita et al., 2003) and modulate the receptor’s electrophysiological properties by slowing its desensitization and deactivation (Korber et al., 2007; Priel et al., 2005). TARPs also control the pharmacological effects of AMPA receptor potentiators (Tomita et al., 2006) and antagonists (Cokic and Stein, 2008; Kott et al., 2007). Each TARP isoform displays a specific pattern of expression in brain. For instance, g3 and g8 are almost exclusively expressed in the cerebral cortex and hippocampus, respectively (Tomita et al., 2003). Recent studies implicate dysregulation of TARPγ2 mRNA in prefrontal cortex in bipolar disorder, further suggesting an involvement of these proteins in mood disorders (Silberberg et al., 2008). Cornichon homolog 2 and 3 (CNIH-2, -3) proteins are the main auxiliary subunits integrated into AMPA receptors complexes, at least in rodents (Schwenk et al., 2009). Similar to TARPs, cornichon proteins increase the expression of AMPA receptors and slow the kinetics of their deactivation and desensitization (Schwenk et al., 2009). The specific binding domains for enzymes, such as TARPs, remain rather elusive (Milstein and Nicoll, 2008). Thus, as information regarding the physical properties of these proteins becomes available, so may a future for compounds that have the ability to regulate their cellular functions.
Figure 1.
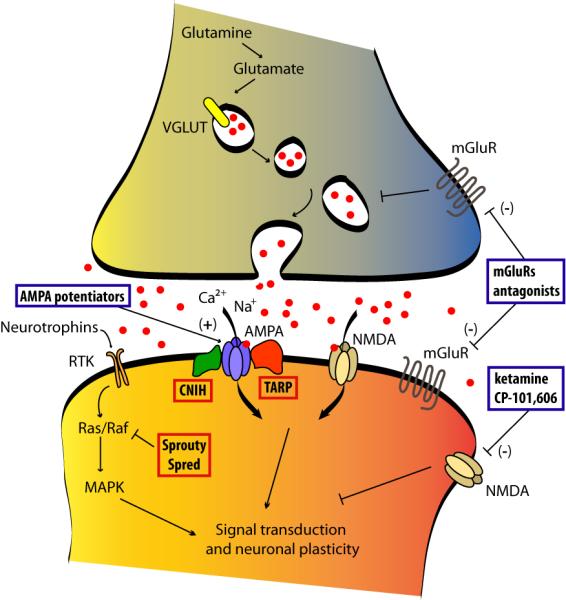
Emerging antidepressant targets from neurotrophic and glutamatergic signaling pathways. Negative modulators of downstream neurotrophic signaling acting at SPROUTY and SPRED protein families represent potential mechanisms for increasing neurotrophic function. AMPA receptor potentiators and NMDA receptor allosteric modulators (with specific subunit selectivity) are now in clinical trials. Allosteric modulators of mGluRs are also being explored in preclinical studies. Modulators of AMPA receptor expression and function, such as TARPs and CNIH, represent potential therapeutic targets as well. Compounds under development for their antidepressants effect are shown in blue boxes. New target proteins are highlighted in red boxes. CNIH, cornichon homolog; MAPK, mitogen-activated protein kinase; RTK, receptor tyrosine kinase; TARP, transmembrane AMPAR regulatory proteins; VGLUT, vesicular glutamate transporter.
These same approaches can be used to examine other components of the glutamatergic system, for instance, by targeting the receptor auxiliary protein NETO2 which functions to modulate kainite receptor channel properties (Zhang et al., 2009). More work is needed to fully explore this possibility (Gallyas et al., 2003; Zhang et al., 2009). Interestingly, blockade of K2 kainate subunits produces anxiolytic-like effects in rats, suggesting that this could be an adjunctive therapy for depression associated with high levels of anxiety (Alt et al., 2007).
Increasing evidence suggests that other aspects of glutamate signaling are regulated by stress or antidepressant treatments, including effects on presynaptic glutamate release and glutamate homeostasis via glial cells. Vesicular transporters for glutamate, SNARE complexes that mediate vesicle exocytosis, and plasma membrane glutamate transporters are under current investigation as potential targets in the treatment of depression (Sanacora et al., 2008).
A final glutamatergic strategy for treating depression may be through modulating metabotropic (G protein-coupled) glutamate receptors (Fig.1). One receptor in particular, mGluR5, increases neuronal excitability and potentiates NMDA-evoked currents, suggesting that antagonism of mGluR5 might dampen NMDA function. Indeed, MPEP and MTEP, selective mGluR5 allosteric antagonists, induce antidepressant effects in rodent models (Pilc et al., 2008). The antidepressant-like effects of MPEP are lost in mGluR5 knockout mice (Li et al., 2006). However, negative allosteric modulators of mGluR5, like MPEP, MTEP or fenobam, function like inverse agonists, and such actions might be the cause of cognitive deficits and psychotomimetic effects observed in some patients with severe anxiety after treatment with fenobam (Porter et al., 2005). The recent development of an mGluR5 partial allosteric antagonist might lead to the generation of improved drugs with fewer side effects (Rodriguez et al., 2005). Contrary to mGluR5, mGluR2 and mGluR3 negatively modulate glutamatergic neurotransmission. Preclinical studies have demonstrated that antagonists of these receptors may possess antidepressant-like effects when administered to rodents (Pilc et al., 2008). Such effects are prevented after blockade of AMPA receptors with NBQX, suggesting an AMPA-dependant antidepressant effect of these mGluRs (Bespalov et al., 2008; Pilc et al., 2008). Similarly, mGluR7 knockout mice generate antidepressant-like behaviors (Cryan et al., 2003).
Targeting glutamate receptors has the appeal of relieving depressive symptoms much faster than conventional monoaminergic strategies. In addition, the number of administrations or daily doses may be reduced based on the persistence of antidepressant effects after a single treatment, as observed with ketamine (Aan Het Rot et al., 2009). Problematic side effects of targeting ionotropic glutamate receptors arise from the acute disruptive effects (e.g., sedation and cognitive impairments) of strongly regulating glutamatergic synapses. Metabatropic glutamate receptor acting drugs, however, are less likely to produce undesirable side effects due to their weaker, modulatory effects on excitatory synapses, and are currently in clinical trials for a number of neurological disorders (Marek, 2004; Moldrich et al., 2003).
4. Hypothalamic feeding peptides
Hypothalamic peptides are best known for their prominent role in the regulation of feeding behavior (Grossman, 1975; Hoebel and Teitelbaum, 1966). Recent studies have demonstrated that these peptides also contribute to emotional behavior (Nestler and Carlezon, 2006). The anhedonic and lethargy symptoms and significant changes in body weight that occur in many depressed patients suggest the involvement of hypothalamic mechanisms in a subtype of depression. For instance, orexin (hypocretin) stimulates feeding in response to energy deficiencies (Sakurai et al., 1998), and is critical for the antidepressant-like effects produced by calorie restriction in animals (Lutter et al., 2008a). Likewise, experimental treatments with this peptide promote antidepressant-like effects (Lutter et al., 2008a). Stimulation of hypothalamic orexin neurons appears to be mediated partly by the feeding-promoting gut hormone, grhelin, and its activation of growth hormone secretagogue receptor (GHSR, also known as the ghrelin receptor) on orexin neurons (Lutter et al., 2008b). Interestingly, social stress increases ghrelin secretion, indicating that this peptide may promote resilience (Lutter et al., 2008b). Mice lacking GSHRs display significant increases in stress-induced depressive-like behaviors (Lutter et al., 2008b). Targeting GHSRs to stimulate receptor function may therefore be a promising strategy for relieving symptoms of major depression, particularly when weight loss is a prominent symptom, as in anorexia nervosa (Lutter and Nestler, 2009).
Melanin-concentrating hormone (MCH) is another major orexigenic (pro-appetite) peptide. Like orexin, its expression is limited to a subset of lateral hypothalamic neurons. MCH activates the MCH1 receptor, which is remarkably enriched in the NAc. (Humans also express an MCH2 receptor, about which much less is known.) Targeted administration of MCH into the NAc, hypothalamus, or lateral ventricles robustly increases feeding behavior; whereas MCH1 receptor antagonists produce the opposite effect (Georgescu et al., 2005). Interestingly, antagonism of MCH1 receptors in the NAc promotes antidepressant-like effects, and similar actions are found in mice lacking these receptors (Georgescu et al., 2005). Over-expressing MCH is correspondingly pro-depressant (Shimazaki et al., 2006). Inhibiting MCH may therefore serve as an effective treatment in the subset of depressed patients that demonstrate weight gain.
Several other hypothalamic feeding peptides may also be targeted for treating depression. Of note are the anorexigenic peptides, including melanocortin (αMSH) and cocaine- and amphetamine-regulated transcript (CART), and the orexigenic peptides, ARP (agouti-related peptide) and NPY (neuropeptide Y). Preliminary evidence indicates that these peptides not only participate in the control of feeding behavior, but also regulate behavioral responses to emotional stimuli (Nestler and Carlezon, 2006). NPY, for example, is an attractive antidepressant target because of its expression in limbic circuits, as well as the hypothalamus (Karl and Herzog, 2007). Well-known for having a role in stress responses, NPY has been intensively examined in animal models and in clinical studies. Indeed, lower levels of NPY in CSF, plasma, and prefrontal cortex have been reported for depressed subjects as well as suicide victims (Caberlotto and Hurd, 2001; Widdowson et al., 1992). In support of these clinical findings, experimental decreases in NPY levels promote depressive-like behavioral responses in animals. Perhaps most encouraging are reports in mice and rats where stimulation of NPY neurotransmission produces antidepressant-like and anxiolytic-like effects. Two of the six known NPY receptors have received attention as potential mediators of the NPY-mediated antidepressant-like effects. For instance, activation of the primarily postsynaptic Y1 receptor (that is highly expressed in the hippocampus and cerebral cortex) via intra-cranial administration of the Y1 selective agonist [Leu31;Pro34]PYY promotes antidepressant effects. Further confirming a role for this receptor are findings that direct hippocampal delivery of the Y1 non-peptidic antagonists BIBP3226 or BIBO3304 prevents the antidepressant-like effects produced by NPY administration (Ishida et al., 2007). NPY Y2 receptors could also be an interesting target for treating depressive symptoms due to its function as an inhibitory presynaptic autoreceptor, and by virtue of it being a heteroreceptor (Tschenett et al., 2003). Significant increases in the release of NPY are accomplished by deletion or blockade of Y2 receptors. Consequently, Y2 knockout mice or administration of the selective Y2 receptor antagonist BIIE0246 reveals a promising anxiolytic- and antidepressant-like phenotype (Bacchi et al., 2006). Clinical trials for NPY-based compounds have already been initiated for the treatment of obesity.
5. Estrogen receptors
The odds of being diagnosed with a depressive or anxiety disorder is at least twice as high for women than for men (Earls, 1987). Fluctuations in endogenous levels of gonadal hormones during premenstrual, postpartum, and perimenopausal times often occur concomitant with changes in mood, which is exemplified by the two-to-five fold increase in incidence of depressive illnesses that occurs during the onset of menopause (Cohen et al., 2006; Freeman et al., 2006). As reviewed below, such hormonal events highlight a critical role for gonadal hormones in the regulation of affective behavioral responses. Indeed, strong evidence for a lack of estrogen receptor beta (ERß) activation in brain during depressive-like behavioral responses has been identified. Positive treatment outcomes in females with depressive illnesses can be deduced from reports on the robust antidepressant effects of estrogen-derived treatments when endogenous estrogen levels are low. Although the effects of estrogen levels during the course of depression in males is remains uncertain, data indicate that gonadectomy-related changes in mood within this population can be alleviated by estrogen treatments (Hughes et al., 2008).
Estrogen receptor alpha (ERα) and ERß are differentially expressed in brain, and have divergent roles regarding depressive-like behaviors. Agonist activity at ERα can positively regulate libido in females (Mazzucco et al., 2008; Walf and Frye, 2005). However, targeting ERß is well documented for its effect on a number of mood-related behaviors. Total knockout of ERß, and not ERα, significantly increases depressive- and anxiety-like behaviors (Imwalle et al., 2005; Krezel et al., 2001). Systemic administration of trilostane, a 3ß-hydroxysteroid dehydrogenase inhibitor, produces antidepressant-like effects in the forced swim test, an effect eliminated in mice lacking ERß (Koonce et al., 2009). ERß is highly expressed in serotonergic neurons of the dorsal raphe nucleus, and within the hippocampus and amygdala where antidepressant actions are considered to be important (Hu et al., 2005; Savitz et al., 2009). In the dorsal raphe, estrogen acts to increase tryptophan hydroxlase (the rate limiting enzyme in serotonin synthesis), and serotonin levels are decreased in the hippocampus of ERß knockout mice (Hiroi et al., 2006; Imwalle et al., 2005). Direct hippocampal activation of estrogen receptors promotes antidepressant- and antianxiety-like effects, and the hippocampus expresses more ERß than ERα (Shughrue et al., 1997; Walf and Frye, 2007). Recent clinical data support the promise of combining estrogen and serotonin modulators as an approach to treating depression in postmenopausal women (Wise et al., 2008). In fact, studies in women with severe postpartum depression have revealed surprising beneficial effects of estradiol treatments when administered alone that far exceed recovery rates after conventional antidepressant treatments (i.e., SSRIs). Gregoire et al. (1999) and Ahokas et al. (2001) both observed a significant reduction in depression scores in at least 80% of their patients treated with estradiol within 3 months or 2 weeks, respectively, after starting treatment. More clinical studies are required to elucidate the parameters of safe and successful estrogen treatment strategies; administration of estrogens that effectively push circulating levels above the “normal” physiological range have no effects, or produce negative outcomes, on mood and cognition in both males and females (Galea et al., 2002; Patisaul et al., 2009).
An intriguing component of gonadal hormone-mediated effects on mood may be derived from their interaction with the hypothalamic-pituitary-adrenal “stress” axis. Estrogen dysregulation can lead to exacerbated stress responses (Walf and Frye, 2007; Young et al., 2000). Affective vulnerabilities to chronic environmental and social stressors may be reduced by estrogen treatments, and potentiated by low levels of endogenous estrogen (Gerrits et al., 2006; Young et al., 2000). Overall, it would appear that physiologically “normal” levels of estrogen positively regulate mood and increase resiliency to stress.
6. Epigenetic mechanisms
While all available antidepressant medications rapidly increase the activity of monoaminergic systems in brain, the mood-enhancing effects of these compounds require weeks of administration. Thus, the nature of drug-induced neural plasticity underlying the clinical actions of classical antidepressants has recently highlighted chromatin remodeling mechanisms as an essential process in these drugs’ progressive therapeutic effects (Lee et al., 2006; Tsankova et al., 2006). Such epigenetic modifications can alter gene transcription in neurons in several ways, including covalent changes to DNA (e.g., DNA methylation) and to histone N-terminal tails (e.g., acetylation, methylation, phosphorylation, among many others). Environmental experiences that modify gene function through epigenetic mechanisms do so in the absence of altering the sequence of DNA, thereby providing a strong rationale for studying epigenetic changes in depression, which is particularly evident when considering the large number of inconsistent genetic association studies. In addition, chronic exposure to stress or antidepressant drugs influences histone acetylation and methylation in brain areas important for emotional processing (Tsankova et al., 2006).
At least two lines of evidence indicate how DNA methylation may play an important role in the emergence and alleviation of depression. In rodents, adult levels of hippocampal DNA methylation (of cytosine) are reported to be under the control of early rearing styles by the mother, with deficiencies in mother-pup interactions between post-natal days 1-10 effectively increasing DNA methylation of the glucocorticoid receptor gene, as well as increasing anxiety-like behaviors throughout the lifespan (Szyf et al., 2007; Weaver et al., 2004). By this mechanism, low amounts of maternal care reduce the expression of hippocampal glucocorticoid receptors. Such maternally induced increases in hippocampal DNA methylation in the pup can be attenuated by direct infusion of trichostatin A, an HDAC inhibitor (Weaver et al., 2004). In contrast, early life stress reduces the methylation of other gene promoters, such as that of the arginine vasopressin gene (Avp) in hypothalamus, which leads to life-long increases in AVP expression and perhaps to hypercortisolism (Murgatroyd et al., 2009).
A second line of evidence supports a prominent role for increasing global DNA methylation as a potential treatment for depression. S-Adenosyl-L-methionine (SAMe) functions as a donor of methyl groups for many cellular functions, including DNA methylation (Lieber and Packer, 2002). Clinical trials have examined the antidepressant effects of administering SAMe with promising results (Mischoulon and Fava, 2002). Because SAMe serves a number of important cellular functions (including a prominent role in the synthesis of monoamines), it is difficult to attribute any particular molecular effects of SAMe to one behavioral phenotype. Nonetheless, experimental work is beginning to provide critical insight regarding the brain areas, and the particular gene promoters, where altering levels of DNA methylation might be therapeutic for treating depression. For instance, chronic stress-induced increases in DNA methylation, within the NAc, occurs via a selective upregulation of certain DNA methyltransferases (LaPlant et al. 2009). Inhibiting DNA methyltransferase activity via RG-108 infusion into this brain area has antidepressant-like effects. Data such as these indicate packaging and unpackaging of heterochromatic domains of the genome may be a stress-sensitive, and reversible, phenomenon.
In neuronal tissue, HDAC inhibitors increase histone acetylation through the inactivation of class I or class II HDACs, and thereby alter patterns of gene expression (Tsankova et al., 2007). In addition, histone acetylation, which is most often associated with activating transcription through its ability to relax ‘condensed’ areas of chromatin, appears sufficient to induce antidepressant effects in animals (Schroeder et al., 2007; Tsankova et al., 2006). One of these reports observed an increase in histone acetylation at certain BDNF gene promoters in the hippocampus after chronic imipramine treatment, and this increase in acetylation appears to be necessary for reversing a depressive-like phenotype induced by chronic stress (Tsankova et al., 2006). More recently, the direct infusion of more specific HDAC inhibitors into the NAc has been shown to induce potent antidepressant-like actions in several rodent models, and alter stress-induced patterns of gene expression in a similar manner to that of the conventional antidepressant fluoxetine (Covington et al., 2009). When examining particular genes in the NAc that are regulated by stress, and oppositely regulated by HDAC inhibitor, numerous potentially interesting gene targets for future scientific studies become apparent (Fig. 2). It should be noted, however, that inhibition of HDACs via systemic routes of administration would be expected to be accompanied by intolerable side effects, hence developmental efforts for more potent agents that are selective for specific HDACs may provide more promising results (Haggarty, 2005; Tsankova et al., 2007).
Figure 2.

Portrayed here is a molecular pathway analysis of genes regulated in the mouse nucleus accumbens by a direct infusion of the HDAC inhibitor MS-275 after chronic social defeat stress. Infusion of MS-275 promotes antidepressant-like behavioral responses and significantly regulates genes as revealed by microarray analysis. Examples of highly regulated molecular pathways that may provide novel targets for treating depression include genes that encode presynaptic vesicular proteins, plasma membrane receptors, intracellular signaling molecules, proteins that regulate the actin cytoskeleton, and the transcriptional regulatory machinery. Reprinted with permission from The Journal of Neuroscience (Covington et al., 2009).
7. Conclusion
A considerable amount of knowledge has been gained since the original discovery of monoamines as a target for antidepressant treatments (Schildkraut, 1965). A multitude of diverse neurobiological systems have now been identified that could potentially be implicated in the pathophysiology of depression and its treatment. This diversity highlights the overwhelming heterogeneity of this complex disease and might represent a first step toward the development of novel drug targets for specific subtypes of depression. Unfortunately, animal models used to screen for newer compounds must first be validated according to the traditional effects produced by classical antidepressants, and thus the potential for false negative findings still remains a source of contention (Berton and Nestler, 2006). The incorporation of new behavioral approaches with a strong focus on neuroadaptive responses to stress has shown some success (Berton et al., 2006; Tsankova et al., 2006).
It is encouraging that there are many drugs in active clinical trials aimed at targeting a several different systems (e.g., corticotrophin-releasing factor, vasopressin, glucocorticoids, and neurokinin) (Mathew et al., 2008). Such trials incorporating mediators of glutamatergic activation appear to be particularly promising (Preskorn et al., 2008). Hypothalamic feeding peptides or hormones like estrogens may also soon find their way in the treatment of subtype-selective depression. The ability to regulate mood and the hypothalamic feeding system in both directions may provide the necessary tools for treating depressive symptoms that are also associated with dietary imbalances. With regard to the long-term stability of depressive symptoms, epigenetic modifications may also play a prominent role. Taken together, as scientific advances uncover the basic neurobiological mechanisms in diverse limbic brain regions that underlie the behavioral disturbances associated with distinct depression syndromes, it is anticipated that major advances in treatment will be achieved.
Acknowledgements
Preparation of this review was supported by grants from the National Institute of Mental Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aan Het Rot M, Mathew SJ, Charney DS. Neurobiological mechanisms in major depressive disorder. CMAJ. 2009;180:305–313. doi: 10.1503/cmaj.080697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aan Het Rot M, Collins KA, Murrough JW, Perez AM, Reich DL, Charney DS, Mathew SJ. Safety and efficacy of repeated-dose intravenous ketamine for treatment-resistant depression. Biol Psychiatry. 2009 doi: 10.1016/j.biopsych.2009.08.038. in press. [DOI] [PubMed] [Google Scholar]
- Ahokas A, Kaukoranta J, Wahlbeck K, Aito M. Estrogen deficiency in severe postpartum depression: successful treatment with sublingual physiologic 17beta-estradiol: a preliminary study. J Clin Psychiatry. 2001;62:332–336. doi: 10.4088/jcp.v62n0504. [DOI] [PubMed] [Google Scholar]
- Alt A, Weiss B, Ornstein PL, Gleason SD, Bleakman D, Stratford RE, Jr., Witkin JM. Anxiolytic-like effects through a GLUK5 kainate receptor mechanism. Neuropharmacology. 2007;52:1482–1487. doi: 10.1016/j.neuropharm.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Bacchi F, Mathe AA, Jimenez P, Stasi L, Arban R, Gerrard P, Caberlotto L. Anxiolytic-like effect of the selective neuropeptide Y Y2 receptor antagonist BIIE0246 in the elevated plus-maze. Peptides. 2006;27:3202–3207. doi: 10.1016/j.peptides.2006.07.020. [DOI] [PubMed] [Google Scholar]
- Bachis A, Mallei A, Cruz MI, Wellstein A, Mocchetti I. Chronic antidepressant treatments increase basic fibroblast growth factor and fibroblast growth factor-binding protein in neurons. Neuropharmacology. 2008;55:1114–1120. doi: 10.1016/j.neuropharm.2008.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, Krystal JH. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47:351–354. doi: 10.1016/s0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- Berton O, McClung CA, Dileone RJ, Krishnan V, Renthal W, Russo SJ, Graham D, Tsankova NM, Bolanos CA, Rios M, Monteggia LM, Self DW, Nestler EJ. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 2006;311:864–868. doi: 10.1126/science.1120972. [DOI] [PubMed] [Google Scholar]
- Berton O, Nestler EJ. New approaches to antidepressant drug discovery: beyond monoamines. Nature Reviews Neuroscience. 2006;7:137–151. doi: 10.1038/nrn1846. [DOI] [PubMed] [Google Scholar]
- Bespalov AY, van Gaalen MM, Sukhotina IA, Wicke K, Mezler M, Schoemaker H, Gross G. Behavioral characterization of the mGlu group II/III receptor antagonist, LY-341495, in animal models of anxiety and depression. Eur J Pharmacol. 2008;592:96–102. doi: 10.1016/j.ejphar.2008.06.089. [DOI] [PubMed] [Google Scholar]
- Bleakman D, Alt A, Witkin JM. AMPA receptors in the therapeutic management of depression. CNS & Neurological Disorders. 2007;6:117–126. doi: 10.2174/187152707780363258. [DOI] [PubMed] [Google Scholar]
- Blier P, Keller MB, Pollack MH, Thase ME, Zajecka JM, Dunner DL. Preventing recurrent depression: long-term treatment for major depressive disorder. J Clin Psychiatry. 2007;68:e06. [PubMed] [Google Scholar]
- Bolaños CA, Perrotti LI, Edwards S, Eisch AJ, Barrot M, Olson VG, Russell DS, Neve RL, Nestler EJ. Viral-mediated expression of phospholipase Cγ in distinct regions of the ventral tegmental area differentially modulates mood-related behaviors. J Neurosci. 2003;23:7569–7576. doi: 10.1523/JNEUROSCI.23-20-07569.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bundschu K, Walter U, Schuh K. Getting a first clue about SPRED functions. Bioessays. 2007;29:897–907. doi: 10.1002/bies.20632. [DOI] [PubMed] [Google Scholar]
- Caberlotto L, Hurd YL. Neuropeptide Y Y(1) and Y(2) receptor mRNA expression in the prefrontal cortex of psychiatric subjects. Relationship of Y(2) subtype to suicidal behavior. Neuropsychopharmacology. 2001;25:91–97. doi: 10.1016/S0893-133X(00)00231-1. [DOI] [PubMed] [Google Scholar]
- Cabrita MA, Christofori G. Sprouty proteins, masterminds of receptor tyrosine kinase signaling. Angiogenesis. 2008;11:53–62. doi: 10.1007/s10456-008-9089-1. [DOI] [PubMed] [Google Scholar]
- Carlezon WA, Jr., Duman RS, Nestler EJ. The many faces of CREB. Trends Neurosci. 2005;28:436–445. doi: 10.1016/j.tins.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Chourbaji S, Vogt MA, Fumagalli F, Sohr R, Frasca A, Brandwein C, Hortnagl H, Riva MA, Sprengel R, Gass P. AMPA receptor subunit 1 (GluR-A) knockout mice model the glutamate hypothesis of depression. FASEB J. 2008;22:3129–3134. doi: 10.1096/fj.08-106450. [DOI] [PubMed] [Google Scholar]
- Cohen LS, Soares CN, Vitonis AF, Otto MW, Harlow BL. Risk for new onset of depression during the menopausal transition: the Harvard study of moods and cycles. Arch Gen Psychiatry. 2006;63:385–390. doi: 10.1001/archpsyc.63.4.385. [DOI] [PubMed] [Google Scholar]
- Cokic B, Stein V. Stargazin modulates AMPA receptor antagonism. Neuropharmacology. 2008;54:1062–1070. doi: 10.1016/j.neuropharm.2008.02.012. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covington HE, 3rd, Maze I, LaPlant QC, Vialou VF, Ohnishi YN, Berton O, Fass DM, Renthal W, Rush AJ, 3rd, Wu EY, Ghose S, Krishnan V, Russo SJ, Tamminga C, Haggarty SJ, Nestler EJ. Antidepressant actions of histone deacetylase inhibitors. J Neurosci. 2009;29:11451–11460. doi: 10.1523/JNEUROSCI.1758-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane GE. Cyloserine as an antidepressant agent. Am J Psychiatry. 1959;115:1025–1026. doi: 10.1176/ajp.115.11.1025. [DOI] [PubMed] [Google Scholar]
- Cryan JF, Kelly PH, Neijt HC, Sansig G, Flor PJ, van Der Putten H. Antidepressant and anxiolytic-like effects in mice lacking the group III metabotropic glutamate receptor mGluR7. Eur J Neurosci. 2003;17:2409–2417. doi: 10.1046/j.1460-9568.2003.02667.x. [DOI] [PubMed] [Google Scholar]
- Davis LL, Frazier EC, Gaynes BN, Trivedi MH, Wisniewski SR, Fava M, Barkin J, Kashner TM, Shelton RC, Alpert JE, Rush AJ. Are depressed outpatients with and without a family history of substance use disorder different? A baseline analysis of the STAR*D cohort. J Clin Psychiatry. 2007;68:1931–1938. doi: 10.4088/jcp.v68n1214. [DOI] [PubMed] [Google Scholar]
- Denayer E, Ahmed T, Brems H, Van Woerden G, Borgesius NZ, Callaerts-Vegh Z, Yoshimura A, Hartmann D, Elgersma Y, D’Hooge R, Legius E, Balschun D. Spred1 is required for synaptic plasticity and hippocampus-dependent learning. J Neurosci. 2008;28:14443–14449. doi: 10.1523/JNEUROSCI.4698-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dow AL, Lin T, Patel T, Neve RL, Ongur D, Carlezon WA., Jr. Society for Neuroscience, list of abstracts. Washington, DC: 2008. Disruption of Sprouty2 function in the dorsal hippocampus produces antidepressant-like effects in rats. [Google Scholar]
- Drevets WC, Price JL, Furey ML. Brain structural and functional abnormalities in mood disorders: implications for neurocircuitry models of depression. Brain Struct Funct. 2008;213:93–118. doi: 10.1007/s00429-008-0189-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duman CH, Schlesinger L, Kodama M, Russell DS, Duman RS. A role for MAP kinase signaling in behavioral models of depression and antidepressant treatment. Biological Psychiatry. 2007;61:661–670. doi: 10.1016/j.biopsych.2006.05.047. [DOI] [PubMed] [Google Scholar]
- Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psychiatry. 2006;59:1116–1127. doi: 10.1016/j.biopsych.2006.02.013. [DOI] [PubMed] [Google Scholar]
- Duncan LE, Hutchison KE, Carey G, Craighead WE. Variation in brain-derived neurotrophic factor (BDNF) gene is associated with symptoms of depression. J Affect Disord. 2008;115:215–219. doi: 10.1016/j.jad.2008.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwivedi Y, Rizavi HS, Conley RR, Pandey GN. ERK MAP kinase signaling in post-mortem brain of suicide subjects: differential regulation of upstream Raf kinases Raf-1 and B-Raf. Mol Psychiatry. 2006;11:86–98. doi: 10.1038/sj.mp.4001744. [DOI] [PubMed] [Google Scholar]
- Dwivedi Y, Rizavi HS, Conley RR, Roberts RC, Tamminga CA, Pandey GN. Altered gene expression of brain-derived neurotrophic factor and receptor tyrosine kinase B in postmortem brain of suicide subjects. Arch Gen Psychiatry. 2003;60:804–815. doi: 10.1001/archpsyc.60.8.804. [DOI] [PubMed] [Google Scholar]
- Dwivedi Y, Rizavi HS, Teppen T, Zhang H, Mondal A, Roberts RC, Conley RR, Pandey GN. Lower phosphoinositide 3-kinase (PI 3-kinase) activity and differential expression levels of selective catalytic and regulatory PI 3-kinase subunit isoforms in prefrontal cortex and hippocampus of suicide subjects. Neuropsychopharmacology. 2008;33:2324–2340. doi: 10.1038/sj.npp.1301641. [DOI] [PubMed] [Google Scholar]
- Earls F. Sex differences in psychiatric disorders: origins and developmental influences. Psychiatr Dev. 1987;5:1–23. [PubMed] [Google Scholar]
- Eisch AJ, Bolanos CA, de Wit J, Simonak RD, Pudiak CM, Barrot M, Verhaagen J, Nestler EJ. Brain-derived neurotrophic factor in the ventral midbrain-nucleus accumbens pathway: a role in depression. Biological Psychiatry. 2003;54:994–1005. doi: 10.1016/j.biopsych.2003.08.003. [DOI] [PubMed] [Google Scholar]
- Evans SJ, Choudary PV, Neal CR, Li JZ, Vawter MP, Tomita H, Lopez JF, Thompson RC, Meng F, Stead JD, Walsh DM, Myers RM, Bunney WE, Watson SJ, Jones EG, Akil H. Dysregulation of the fibroblast growth factor system in major depression. Proc Natl Acad Sci U S A. 2004;101:15506–15511. doi: 10.1073/pnas.0406788101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feyissa AM, Chandran A, Stockmeier CA, Karolewicz B. Reduced levels of NR2A and NR2B subunits of NMDA receptor and PSD-95 in the prefrontal cortex in major depression. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33:70–75. doi: 10.1016/j.pnpbp.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman EW, Sammel MD, Lin H, Nelson DB. Associations of hormones and menopausal status with depressed mood in women with no history of depression. Arch Gen Psychiatry. 2006;63:375–382. doi: 10.1001/archpsyc.63.4.375. [DOI] [PubMed] [Google Scholar]
- Fujimaki K, Morinobu S, Duman RS. Administration of a cAMP phosphodiesterase 4 inhibitor enhances antidepressant-induction of BDNF mRNA in rat hippocampus. Neuropsychopharmacology. 2000;22:42–51. doi: 10.1016/S0893-133X(99)00084-6. [DOI] [PubMed] [Google Scholar]
- Galea LA, Lee TT, Kostaras X, Sidhu JA, Barr AM. High levels of estradiol impair spatial performance in the Morris water maze and increase ‘depressive-like’ behaviors in the female meadow vole. Physiol Behav. 2002;77:217–225. doi: 10.1016/s0031-9384(02)00849-1. [DOI] [PubMed] [Google Scholar]
- Gallyas F, Jr., Ball SM, Molnar E. Assembly and cell surface expression of KA-2 subunit-containing kainate receptors. J Neurochem. 2003;86:1414–1427. doi: 10.1046/j.1471-4159.2003.01945.x. [DOI] [PubMed] [Google Scholar]
- Gatt JM, Nemeroff CB, Dobson-Stone C, Paul RH, Bryant RA, Schofield PR, Gordon E, Kemp AH, Williams LM. Interactions between BDNF Val66Met polymorphism and early life stress predict brain and arousal pathways to syndromal depression and anxiety. Mol Psychiatry. 2009 doi: 10.1038/mp.2008.143. [DOI] [PubMed] [Google Scholar]
- Georgescu D, Sears RM, Hommel JD, Barrot M, Bolanos CA, Marsh DJ, Bednarek MA, Bibb JA, Maratos-Flier E, Nestler EJ, DiLeone RJ. The hypothalamic neuropeptide melanin-concentrating hormone acts in the nucleus accumbens to modulate feeding behavior and forced-swim performance. Journal of Neuroscience. 2005;25:2933–2940. doi: 10.1523/JNEUROSCI.1714-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerrits M, Bakker PL, Koch T, Ter Horst GJ. Stress-induced sensitization of the limbic system in ovariectomized rats is partly restored by cyclic 17beta-estradiol administration. Eur J Neurosci. 2006;23:1747–1756. doi: 10.1111/j.1460-9568.2006.04701.x. [DOI] [PubMed] [Google Scholar]
- Gould E, McEwen BS, Tanapat P, Galea LA, Fuchs E. Neurogenesis in the dentate gyrus of the adult tree shrew is regulated by psychosocial stress and NMDA receptor activation. J Neurosci. 1997;17:2492–2498. doi: 10.1523/JNEUROSCI.17-07-02492.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregoire AJ, Kumar R, Everitt B, Henderson AF, Studd JW. Transdermal oestrogen for treatment of severe postnatal depression. Lancet. 1996;347:930–933. doi: 10.1016/s0140-6736(96)91414-2. [DOI] [PubMed] [Google Scholar]
- Grossman SP. Role of the hypothalamus in the regulation of food and water intake. Psychol Rev. 1975;82:200–224. [PubMed] [Google Scholar]
- Groves JO. Is it time to reassess the BDNF hypothesis of depression? Mol Psychiatry. 2007;12:1079–1088. doi: 10.1038/sj.mp.4002075. [DOI] [PubMed] [Google Scholar]
- Haggarty SJ. The principle of complementarity: chemical versus biological space. Curr Opin Chem Biol. 2005;9:296–303. doi: 10.1016/j.cbpa.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Healy D. The three faces of the antidepressants: a critical commentary on the clinical-economic context of diagnosis. J Nerv Ment Dis. 1999;187:174–180. doi: 10.1097/00005053-199903000-00007. [DOI] [PubMed] [Google Scholar]
- Hebb AL, Robertson HA, Denovan-Wright EM. Phosphodiesterase 10A inhibition is associated with locomotor and cognitive deficits and increased anxiety in mice. Eur Neuropsychopharmacol. 2008;18:339–363. doi: 10.1016/j.euroneuro.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Heine VM, Zareno J, Maslam S, Joels M, Lucassen PJ. Chronic stress in the adult dentate gyrus reduces cell proliferation near the vasculature and VEGF and Flk-1 protein expression. Eur J Neurosci. 2005;21:1304–1314. doi: 10.1111/j.1460-9568.2005.03951.x. [DOI] [PubMed] [Google Scholar]
- Hiroi R, McDevitt RA, Neumaier JF. Estrogen selectively increases tryptophan hydroxylase-2 mRNA expression in distinct subregions of rat midbrain raphe nucleus: association between gene expression and anxiety behavior in the open field. Biol Psychiatry. 2006;60:288–295. doi: 10.1016/j.biopsych.2005.10.019. [DOI] [PubMed] [Google Scholar]
- Hoebel BG, Teitelbaum P. Weight regulation in normal and hypothalamic hyperphagic rats. J Comp Physiol Psychol. 1966;61:189–193. doi: 10.1037/h0023126. [DOI] [PubMed] [Google Scholar]
- Hou SJ, Yen FC, Tsai SJ. Is dysfunction of the tissue plasminogen activator (tPA)-plasmin pathway a link between major depression and cardiovascular disease? Med Hypotheses. 2009;72:166–168. doi: 10.1016/j.mehy.2008.09.009. [DOI] [PubMed] [Google Scholar]
- Hu S, Lu SF, Kaplan JR, Adams MR, Simon NG. ERbeta protein expression in female cynomolgus monkey and CF-1 mouse brain: Western analysis. J Neurobiol. 2005;64:298–309. doi: 10.1002/neu.20139. [DOI] [PubMed] [Google Scholar]
- Hughes ZA, Liu F, Platt BJ, Dwyer JM, Pulicicchio CM, Zhang G, Schechter LE, Rosenzweig-Lipson S, Day M. WAY-200070, a selective agonist of estrogen receptor beta as a potential novel anxiolytic/antidepressant agent. Neuropharmacology. 2008;54:1136–1142. doi: 10.1016/j.neuropharm.2008.03.004. [DOI] [PubMed] [Google Scholar]
- Imwalle DB, Gustafsson JA, Rissman EF. Lack of functional estrogen receptor beta influences anxiety behavior and serotonin content in female mice. Physiol Behav. 2005;84:157–163. doi: 10.1016/j.physbeh.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Ishida H, Shirayama Y, Iwata M, Katayama S, Yamamoto A, Kawahara R, Nakagome K. Infusion of neuropeptide Y into CA3 region of hippocampus produces antidepressant-like effect via Y1 receptor. Hippocampus. 2007;17:271–280. doi: 10.1002/hipo.20264. [DOI] [PubMed] [Google Scholar]
- Karl T, Herzog H. Behavioral profiling of NPY in aggression and neuropsychiatric diseases. Peptides. 2007;28:326–333. doi: 10.1016/j.peptides.2006.07.027. [DOI] [PubMed] [Google Scholar]
- Karolewicz B, Szebeni K, Gilmore T, Maciag D, Stockmeier CA, Ordway GA. Elevated levels of NR2A and PSD-95 in the lateral amygdala in depression. Int J Neuropsychopharmacol. 2009;12:143–153. doi: 10.1017/S1461145708008985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato R, Nonami A, Taketomi T, Wakioka T, Kuroiwa A, Matsuda Y, Yoshimura A. Molecular cloning of mammalian Spred-3 which suppresses tyrosine kinase-mediated Erk activation. Biochem Biophys Res Commun. 2003;302:767–772. doi: 10.1016/s0006-291x(03)00259-6. [DOI] [PubMed] [Google Scholar]
- Kessels HW, Malinow R. Synaptic AMPA receptor plasticity and behavior. Neuron. 2009;61:340–350. doi: 10.1016/j.neuron.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler RC, Wang PS. The descriptive epidemiology of commonly occurring mental disorders in the United States. Annu Rev Public Health. 2008;29:115–129. doi: 10.1146/annurev.publhealth.29.020907.090847. [DOI] [PubMed] [Google Scholar]
- Koonce CJ, Walf AA, Frye CA. Trilostane exerts antidepressive effects among wild-type, but not estrogen receptor [beta] knockout mice. Neuroreport. 2009;20:1047–1050. doi: 10.1097/wnr.0b013e32832e0c44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korber C, Werner M, Kott S, Ma ZL, Hollmann M. The transmembrane AMPA receptor regulatory protein gamma 4 is a more effective modulator of AMPA receptor function than stargazin (gamma 2) J Neurosci. 2007;27:8442–8447. doi: 10.1523/JNEUROSCI.0424-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kott S, Werner M, Korber C, Hollmann M. Electrophysiological properties of AMPA receptors are differentially modulated depending on the associated member of the TARP family. J Neurosci. 2007;27:3780–3789. doi: 10.1523/JNEUROSCI.4185-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krezel W, Dupont S, Krust A, Chambon P, Chapman PF. Increased anxiety and synaptic plasticity in estrogen receptor beta -deficient mice. Proc Natl Acad Sci U S A. 2001;98:12278–12282. doi: 10.1073/pnas.221451898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan V, Han MH, Graham DL, Berton O, Renthal W, Russo SJ, Laplant Q, Graham A, Lutter M, Lagace DC, Ghose S, Reister R, Tannous P, Green TA, Neve RL, Chakravarty S, Kumar A, Eisch AJ, Self DW, Lee FS, Tamminga CA, Cooper DC, Gershenfeld HK, Nestler EJ. Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell. 2007;131:391–404. doi: 10.1016/j.cell.2007.09.018. [DOI] [PubMed] [Google Scholar]
- Krishnan V, Han MH, Mazei-Robison M, Iniguez SD, Ables JL, Vialou V, Berton O, Ghose S, Covington HE, 3rd, Wiley MD, Henderson RP, Neve RL, Eisch AJ, Tamminga CA, Russo SJ, Bolanos CA, Nestler EJ. AKT signaling within the ventral tegmental area regulates cellular and behavioral responses to stressful stimuli. Biol Psychiatry. 2008;64:691–700. doi: 10.1016/j.biopsych.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan V, Nestler EJ. The molecular neurobiology of depression. Nature. 2008;455:894–902. doi: 10.1038/nature07455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurt M, Bilge SS, Aksoz E, Kukula O, Celik S, Kesim Y. Effect of sildenafil on anxiety in the plus-maze test in mice. Pol J Pharmacol. 2004;56:353–357. [PubMed] [Google Scholar]
- Laje G, Paddock S, Manji H, Rush AJ, Wilson AF, Charney D, McMahon FJ. Genetic markers of suicidal ideation emerging during citalopram treatment of major depression. Am J Psychiatry. 2007;164:1530–1538. doi: 10.1176/appi.ajp.2007.06122018. [DOI] [PubMed] [Google Scholar]
- Lee MG, Wynder C, Schmidt DM, McCafferty DG, Shiekhattar R. Histone H3 lysine 4 demethylation is a target of nonselective antidepressive medications. Chem Biol. 2006;13:563–567. doi: 10.1016/j.chembiol.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Lekman M, Paddock S, McMahon FJ. Pharmacogenetics of major depression: insights from level 1 of the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) trial. Mol Diagn Ther. 2008;12:321–330. doi: 10.1007/BF03256297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Need AB, Baez M, Witkin JM. Metabotropic glutamate 5 receptor antagonism is associated with antidepressant-like effects in mice. J Pharmacol Exp Ther. 2006;319:254–259. doi: 10.1124/jpet.106.103143. [DOI] [PubMed] [Google Scholar]
- Lieber CS, Packer L. S-Adenosylmethionine: molecular, biological, and clinical aspects--an introduction. Am J Clin Nutr. 2002;76:1148S–1150S. doi: 10.1093/ajcn/76/5.1148S. [DOI] [PubMed] [Google Scholar]
- Lodge D. The history of the pharmacology and cloning of ionotropic glutamate receptors and the development of idiosyncratic nomenclature. Neuropharmacology. 2009;56:6–21. doi: 10.1016/j.neuropharm.2008.08.006. [DOI] [PubMed] [Google Scholar]
- Lutter M, Krishnan V, Russo SJ, Jung S, McClung CA, Nestler EJ. Orexin signaling mediates the antidepressant-like effect of calorie restriction. J Neurosci. 2008a;28:3071–3075. doi: 10.1523/JNEUROSCI.5584-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutter M, Nestler EJ. Homeostatic and hedonic signals interact in the regulation of food intake. J Nutr. 2009;139:629–632. doi: 10.3945/jn.108.097618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutter M, Sakata I, Osborne-Lawrence S, Rovinsky SA, Anderson JG, Jung S, Birnbaum S, Yanagisawa M, Elmquist JK, Nestler EJ, Zigman JM. The orexigenic hormone ghrelin defends against depressive symptoms of chronic stress. Nat Neurosci. 2008b;11:752–753. doi: 10.1038/nn.2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeng S, Zarate CA, Jr., Du J, Schloesser RJ, McCammon J, Chen G, Manji HK. Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol Psychiatry. 2008;63:349–352. doi: 10.1016/j.biopsych.2007.05.028. [DOI] [PubMed] [Google Scholar]
- Manji HK, Drevets WC, Charney DS. The cellular neurobiology of depression. Nature Medicine. 2001;7:541–547. doi: 10.1038/87865. [DOI] [PubMed] [Google Scholar]
- Manji HK, Quiroz JA, Sporn J, Payne JL, Denicoff K, N AG, Zarate CA, Jr., Charney DS. Enhancing neuronal plasticity and cellular resilience to develop novel, improved therapeutics for difficult-to-treat depression. Biol Psychiatry. 2003;53:707–742. doi: 10.1016/s0006-3223(03)00117-3. [DOI] [PubMed] [Google Scholar]
- Marek GJ. Metabotropic glutamate 2/3 receptors as drug targets. Curr Opin Pharmacol. 2004;4:18–22. doi: 10.1016/j.coph.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Martinez SE, Beavo JA, Hol WG. GAF domains: two-billion-year-old molecular switches that bind cyclic nucleotides. Mol Interv. 2002;2:317–323. doi: 10.1124/mi.2.5.317. [DOI] [PubMed] [Google Scholar]
- Martinowich K, Manji H, Lu B. New insights into BDNF function in depression and anxiety. Nat Neurosci. 2007;10:1089–1093. doi: 10.1038/nn1971. [DOI] [PubMed] [Google Scholar]
- Mathew SJ, Manji HK, Charney DS. Novel drugs and therapeutic targets for severe mood disorders. Neuropsychopharmacology. 2008;33:2080–2092. doi: 10.1038/sj.npp.1301652. [DOI] [PubMed] [Google Scholar]
- Mayberg HS, Lozano AM, Voon V, McNeely HE, Seminowicz D, Hamani C, Schwalb JM, Kennedy SH. Deep brain stimulation for treatment-resistant depression. Neuron. 2005;45:651–660. doi: 10.1016/j.neuron.2005.02.014. [DOI] [PubMed] [Google Scholar]
- Mazzucco CA, Walker HA, Pawluski JL, Lieblich SE, Galea LA. ERalpha, but not ERbeta, mediates the expression of sexual behavior in the female rat. Behav Brain Res. 2008;191:111–117. doi: 10.1016/j.bbr.2008.03.016. [DOI] [PubMed] [Google Scholar]
- Milstein AD, Nicoll RA. Regulation of AMPA receptor gating and pharmacology by TARP auxiliary subunits. Trends Pharmacol Sci. 2008;29:333–339. doi: 10.1016/j.tips.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minowada G, Jarvis LA, Chi CL, Neubuser A, Sun X, Hacohen N, Krasnow MA, Martin GR. Vertebrate Sprouty genes are induced by FGF signaling and can cause chondrodysplasia when overexpressed. Development. 1999;126:4465–4475. doi: 10.1242/dev.126.20.4465. [DOI] [PubMed] [Google Scholar]
- Mischoulon D, Fava M. Role of S-adenosyl-L-methionine in the treatment of depression: a review of the evidence. Am J Clin Nutr. 2002;76:1158S–1161S. doi: 10.1093/ajcn/76/5.1158S. [DOI] [PubMed] [Google Scholar]
- Moldrich RX, Chapman AG, De Sarro G, Meldrum BS. Glutamate metabotropic receptors as targets for drug therapy in epilepsy. Eur J Pharmacol. 2003;476:3–16. doi: 10.1016/s0014-2999(03)02149-6. [DOI] [PubMed] [Google Scholar]
- Monteggia LM, Luikart B, Barrot M, Theobold D, Malkovska I, Nef S, Parada LF, Nestler EJ. Brain-derived neurotrophic factor conditional knockouts show gender differences in depression-related behaviors. Biol Psychiatry. 2007;61:187–197. doi: 10.1016/j.biopsych.2006.03.021. [DOI] [PubMed] [Google Scholar]
- Mott DD, Doherty JJ, Zhang S, Washburn MS, Fendley MJ, Lyuboslavsky P, Traynelis SF, Dingledine R. Phenylethanolamines inhibit NMDA receptors by enhancing proton inhibition. Nat Neurosci. 1998;1:659–667. doi: 10.1038/3661. [DOI] [PubMed] [Google Scholar]
- Murgatroyd C, Patchev AV, Wu Y, Micale V, Bockmühl Y, Fischer D, Holsboer F, Wotjak CT, Almeida OF, Spengler D. Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nat Neurosci. 2009 doi: 10.1038/nn.2436. in press. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Carlezon WA., Jr. The mesolimbic dopamine reward circuit in depression. Biological Psychiatry. 2006;59:1151–1159. doi: 10.1016/j.biopsych.2005.09.018. [DOI] [PubMed] [Google Scholar]
- O’Neill MJ, Witkin JM. AMPA receptor potentiators: application for depression and Parkinson’s disease. Curr Drug Targets. 2007;8:603–620. doi: 10.2174/138945007780618517. [DOI] [PubMed] [Google Scholar]
- Ongur D, Pohlman J, Dow AL, Eisch AJ, Edwin F, Heckers S, Cohen BM, Patel TB, Carlezon WA., Jr. Electroconvulsive seizures stimulate glial proliferation and reduce expression of Sprouty2 within the prefrontal cortex of rats. Biol Psychiatry. 2007;62:505–512. doi: 10.1016/j.biopsych.2006.11.014. [DOI] [PubMed] [Google Scholar]
- Paddock S, Laje G, Charney D, Rush AJ, Wilson AF, Sorant AJ, Lipsky R, Wisniewski SR, Manji H, McMahon FJ. Association of GRIK4 with outcome of antidepressant treatment in the STAR*D cohort. Am J Psychiatry. 2007;164:1181–1188. doi: 10.1176/appi.ajp.2007.06111790. [DOI] [PubMed] [Google Scholar]
- Patisaul HB, Burke KT, Hinkle RE, Adewale HB, Shea D. Systemic administration of diarylpropionitrile (DPN) or phytoestrogens does not affect anxiety-related behaviors in gonadally intact male rats. Horm Behav. 2009;55:319–328. doi: 10.1016/j.yhbeh.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilc A, Chaki S, Nowak G, Witkin JM. Mood disorders: regulation by metabotropic glutamate receptors. Biochem Pharmacol. 2008;75:997–1006. doi: 10.1016/j.bcp.2007.09.021. [DOI] [PubMed] [Google Scholar]
- Pittenger C, Sanacora G, Krystal JH. The NMDA receptor as a therapeutic target in major depressive disorder. CNS & Neurological Disorders. 2007;6:101–115. doi: 10.2174/187152707780363267. [DOI] [PubMed] [Google Scholar]
- Porter RH, Jaeschke G, Spooren W, Ballard TM, Buttelmann B, Kolczewski S, Peters JU, Prinssen E, Wichmann J, Vieira E, Muhlemann A, Gatti S, Mutel V, Malherbe P. Fenobam: a clinically validated nonbenzodiazepine anxiolytic is a potent, selective, and noncompetitive mGlu5 receptor antagonist with inverse agonist activity. J Pharmacol Exp Ther. 2005;315:711–721. doi: 10.1124/jpet.105.089839. [DOI] [PubMed] [Google Scholar]
- Preskorn SH, Baker B, Kolluri S, Menniti FS, Krams M, Landen JW. An innovative design to establish proof of concept of the antidepressant effects of the NR2B subunit selective N-methyl-D-aspartate antagonist, CP-101,606, in patients with treatment-refractory major depressive disorder. J Clin Psychopharmacol. 2008;28:631–637. doi: 10.1097/JCP.0b013e31818a6cea. [DOI] [PubMed] [Google Scholar]
- Price RB, Nock MK, Charney DS, Mathew SJ. Effects of intravenous ketamine on explicit and implicit measures of suicidality in treatment-resistant depression. Biol Psychiatry. 2009;66:522–526. doi: 10.1016/j.biopsych.2009.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priel A, Kolleker A, Ayalon G, Gillor M, Osten P, Stern-Bach Y. Stargazin reduces desensitization and slows deactivation of the AMPA-type glutamate receptors. J Neurosci. 2005;25:2682–2686. doi: 10.1523/JNEUROSCI.4834-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaPlant Q, Vialou V, Covington HE, III, Warren B, Maze I, Dietz DM, Iñiguez SD, Xiao GH, Neve RL, Ren YH, Hurd YL, Oosting RS, Nestler EJ. Dnmt3a regulates emotional behavior and spine plasticity in the nucleus accumbens (Submitted) 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuss B, von Bohlen und Halbach O. Fibroblast growth factors and their receptors in the central nervous system. Cell Tissue Res. 2003;313:139–157. doi: 10.1007/s00441-003-0756-7. [DOI] [PubMed] [Google Scholar]
- Rodriguez AL, Nong Y, Sekaran NK, Alagille D, Tamagnan GD, Conn PJ. A close structural analog of 2-methyl-6-(phenylethynyl)-pyridine acts as a neutral allosteric site ligand on metabotropic glutamate receptor subtype 5 and blocks the effects of multiple allosteric modulators. Mol Pharmacol. 2005;68:1793–1802. doi: 10.1124/mol.105.016139. [DOI] [PubMed] [Google Scholar]
- Rush AJ. The varied clinical presentations of major depressive disorder. J Clin Psychiatry. 2007;68(Suppl 8):4–10. [PubMed] [Google Scholar]
- Rush AJ, Trivedi MH, Wisniewski SR, Nierenberg AA, Stewart JW, Warden D, Niederehe G, Thase ME, Lavori PW, Lebowitz BD, McGrath PJ, Rosenbaum JF, Sackeim HA, Kupfer DJ, Luther J, Fava M. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163:1905–1917. doi: 10.1176/ajp.2006.163.11.1905. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, Williams SC, Richarson JA, Kozlowski GP, Wilson S, Arch JR, Buckingham RE, Haynes AC, Carr SA, Annan RS, McNulty DE, Liu WS, Terrett JA, Elshourbagy NA, Bergsma DJ, Yanagisawa M. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92:1–696. doi: 10.1016/s0092-8674(02)09256-5. [DOI] [PubMed] [Google Scholar]
- Sanacora G, Zarate CA, Krystal JH, Manji HK. Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nat Rev Drug Discov. 2008;7:426–437. doi: 10.1038/nrd2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez A, Setien F, Martinez N, Oliva JL, Herranz M, Fraga MF, Alaminos M, Esteller M, Rojas JM. Epigenetic inactivation of the ERK inhibitor Spry2 in B-cell diffuse lymphomas. Oncogene. 2008;27:4969–4972. doi: 10.1038/onc.2008.129. [DOI] [PubMed] [Google Scholar]
- Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S, Weisstaub N, Lee J, Duman R, Arancio O, Belzung C, Hen R. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science. 2003;301:805–809. doi: 10.1126/science.1083328. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM. Why stress is bad for your brain. Science. 1996;273:749–750. doi: 10.1126/science.273.5276.749. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM. Stress hormones: Good and bad. Neurobiology of Disease. 2000;7:540–542. doi: 10.1006/nbdi.2000.0350. [DOI] [PubMed] [Google Scholar]
- Savitz J, Lucki I, Drevets WC. 5-HT(1A) receptor function in major depressive disorder. Prog Neurobiol. 2009;88:17–31. doi: 10.1016/j.pneurobio.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schildkraut JJ. The catecholamine hypothesis of affective disorders: a review of supporting evidence. Am J Psychiatry. 1965;122:509–522. doi: 10.1176/ajp.122.5.509. [DOI] [PubMed] [Google Scholar]
- Schlaepfer TE, Cohen MX, Frick C, Kosel M, Brodesser D, Axmacher N, Joe AY, Kreft M, Lenartz D, Sturm V. Deep brain stimulation to reward circuitry alleviates anhedonia in refractory major depression. Neuropsychopharmacology. 2008;33:368–377. doi: 10.1038/sj.npp.1301408. [DOI] [PubMed] [Google Scholar]
- Schroeder FA, Lin CL, Crusio WE, Akbarian S. Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biol Psychiatry. 2007;62:55–64. doi: 10.1016/j.biopsych.2006.06.036. [DOI] [PubMed] [Google Scholar]
- Schwenk J, Harmel N, Zolles G, Bildl W, Kulik A, Heimrich B, Chisaka O, Jonas P, Schulte U, Fakler B, Klocker N. Functional proteomics identify cornichon proteins as auxiliary subunits of AMPA receptors. Science. 2009;323:1313–1319. doi: 10.1126/science.1167852. [DOI] [PubMed] [Google Scholar]
- Sen S, Sanacora G. Major depression: emerging therapeutics. Mt Sinai J Med. 2008;75:204–225. doi: 10.1002/msj.20043. [DOI] [PubMed] [Google Scholar]
- Shimazaki T, Yoshimizu T, Chaki S. Melanin-concentrating hormone MCH1 receptor antagonists: a potential new approach to the treatment of depression and anxiety disorders. CNS Drugs. 2006;20:801–811. doi: 10.2165/00023210-200620100-00002. [DOI] [PubMed] [Google Scholar]
- Shughrue PJ, Lane MV, Merchenthaler I. Comparative distribution of estrogen receptor-alpha and -beta mRNA in the rat central nervous system. J Comp Neurol. 1997;388:507–525. doi: 10.1002/(sici)1096-9861(19971201)388:4<507::aid-cne1>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Silberberg G, Levit A, Collier D, St Clair D, Munro J, Kerwin RW, Tondo L, Floris G, Breen G, Navon R. Stargazin involvement with bipolar disorder and response to lithium treatment. Pharmacogenet Genomics. 2008;18:403–412. doi: 10.1097/FPC.0b013e3282f974ca. [DOI] [PubMed] [Google Scholar]
- Spina D. PDE4 inhibitors: current status. Br J Pharmacol. 2008;155:308–315. doi: 10.1038/bjp.2008.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szyf M, Weaver I, Meaney M. Maternal care, the epigenome and phenotypic differences in behavior. Reprod Toxicol. 2007;24:9–19. doi: 10.1016/j.reprotox.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Tanis KQ, Newton SS, Duman RS. Targeting neurotrophic/growth factor expression and signaling for antidepressant drug development. CNS & Neurological Disorders. 2007;6:151–160. doi: 10.2174/187152707780363276. [DOI] [PubMed] [Google Scholar]
- Tomita S, Chen L, Kawasaki Y, Petralia RS, Wenthold RJ, Nicoll RA, Bredt DS. Functional studies and distribution define a family of transmembrane AMPA receptor regulatory proteins. J Cell Biol. 2003;161:805–816. doi: 10.1083/jcb.200212116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita S, Sekiguchi M, Wada K, Nicoll RA, Bredt DS. Stargazin controls the pharmacology of AMPA receptor potentiators. Proc Natl Acad Sci U S A. 2006;103:10064–10067. doi: 10.1073/pnas.0603128103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai SJ. Statins may enhance the proteolytic cleavage of proBDNF: implications for the treatment of depression. Med Hypotheses. 2007;68:1296–1299. doi: 10.1016/j.mehy.2006.09.043. [DOI] [PubMed] [Google Scholar]
- Tsai SJ, Hong CJ, Liou YJ, Yu YW, Chen TJ. Plasminogen activator inhibitor-1 gene is associated with major depression and antidepressant treatment response. Pharmacogenet Genomics. 2008;18:869–875. doi: 10.1097/FPC.0b013e328308bbc0. [DOI] [PubMed] [Google Scholar]
- Tsai SJ, Hong CJ, Liou YJ, Chen TJ, Chen ML, Hou SJ, Yen FC, Yu YW. Haplotype analysis of single nucleotide polymorphisms in the vascular endothelial growth factor (VEGFA) gene and antidepressant treatment response in major depressive disorder. Psychiatry Res. 2009;169:113–117. doi: 10.1016/j.psychres.2008.06.028. [DOI] [PubMed] [Google Scholar]
- Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci. 2007;8:355–367. doi: 10.1038/nrn2132. [DOI] [PubMed] [Google Scholar]
- Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006;9:519–525. doi: 10.1038/nn1659. [DOI] [PubMed] [Google Scholar]
- Tschenett A, Singewald N, Carli M, Balducci C, Salchner P, Vezzani A, Herzog H, Sperk G. Reduced anxiety and improved stress coping ability in mice lacking NPY-Y2 receptors. European Journal of Neuroscience. 2003;18:143–148. doi: 10.1046/j.1460-9568.2003.02725.x. [DOI] [PubMed] [Google Scholar]
- Turner CA, Gula EL, Taylor LP, Watson SJ, Akil H. Antidepressant-like effects of intracerebroventricular FGF2 in rats. Brain Res. 2008;1224:63–68. doi: 10.1016/j.brainres.2008.05.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volosin M, Song W, Almeida RD, Kaplan DR, Hempstead BL, Friedman WJ. Interaction of survival and death signaling in basal forebrain neurons: roles of neurotrophins and proneurotrophins. J Neurosci. 2006;26:7756–7766. doi: 10.1523/JNEUROSCI.1560-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walf AA, Frye CA. ERbeta-selective estrogen receptor modulators produce antianxiety behavior when administered systemically to ovariectomized rats. Neuropsychopharmacology. 2005;30:1598–1609. doi: 10.1038/sj.npp.1300713. [DOI] [PubMed] [Google Scholar]
- Walf AA, Frye CA. Administration of estrogen receptor beta-specific selective estrogen receptor modulators to the hippocampus decrease anxiety and depressive behavior of ovariectomized rats. Pharmacol Biochem Behav. 2007;86:407–414. doi: 10.1016/j.pbb.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Warner-Schmidt JL, Duman RS. VEGF is an essential mediator of the neurogenic and behavioral actions of antidepressants. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:4647–4652. doi: 10.1073/pnas.0610282104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver IC, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR, Dymov S, Szyf M, Meaney MJ. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;7:847–854. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- Widdowson PS, Ordway GA, Halaris AE. Reduced neuropeptide Y concentrations in suicide brain. J Neurochem. 1992;59:73–80. doi: 10.1111/j.1471-4159.1992.tb08877.x. [DOI] [PubMed] [Google Scholar]
- Wise DD, Felker A, Stahl SM. Tailoring treatment of depression for women across the reproductive lifecycle: the importance of pregnancy, vasomotor symptoms, and other estrogen-related events in psychopharmacology. CNS Spectr. 2008;13:647–662. doi: 10.1017/s1092852900013742. [DOI] [PubMed] [Google Scholar]
- Woo NH, Teng HK, Siao CJ, Chiaruttini C, Pang PT, Milner TA, Hempstead BL, Lu B. Activation of p75NTR by proBDNF facilitates hippocampal long-term depression. Nat Neurosci. 2005;8:1069–1077. doi: 10.1038/nn1510. [DOI] [PubMed] [Google Scholar]
- Yashiro K, Philpot BD. Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology. 2008;55:1081–1094. doi: 10.1016/j.neuropharm.2008.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young EA, Midgley AR, Carlson NE, Brown MB. Alteration in the hypothalamic-pituitary-ovarian axis in depressed women. Arch Gen Psychiatry. 2000;57:1157–1162. doi: 10.1001/archpsyc.57.12.1157. [DOI] [PubMed] [Google Scholar]
- Zagrebelsky M, Holz A, Dechant G, Barde YA, Bonhoeffer T, Korte M. The p75 neurotrophin receptor negatively modulates dendrite complexity and spine density in hippocampal neurons. J Neurosci. 2005;25:9989–9999. doi: 10.1523/JNEUROSCI.2492-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate CA, Jr., Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, Charney DS, Manji HK. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Archives of General Psychiatry. 2006;63:856–864. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]
- Zarate CA, Jr., Payne JL, Quiroz J, Sporn J, Denicoff KK, Luckenbaugh D, Charney DS, Manji HK. An open-label trial of riluzole in patients with treatment-resistant major depression. Am J Psychiatry. 2004;161:171–174. doi: 10.1176/appi.ajp.161.1.171. [DOI] [PubMed] [Google Scholar]
- Zhang HT, Huang Y, Jin SL, Frith SA, Suvarna N, Conti M, O’Donnell JM. Antidepressant-like profile and reduced sensitivity to rolipram in mice deficient in the PDE4D phosphodiesterase enzyme. Neuropsychopharmacology. 2002;27:587–595. doi: 10.1016/S0893-133X(02)00344-5. [DOI] [PubMed] [Google Scholar]
- Zhang W, St-Gelais F, Grabner CP, Trinidad JC, Sumioka A, Morimoto-Tomita M, Kim KS, Straub C, Burlingame AL, Howe JR, Tomita S. A transmembrane accessory subunit that modulates kainate-type glutamate receptors. Neuron. 2009;61:385–396. doi: 10.1016/j.neuron.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoraghi R, Corbin JD, Francis SH. Properties and functions of GAF domains in cyclic nucleotide phosphodiesterases and other proteins. Mol Pharmacol. 2004;65:267–278. doi: 10.1124/mol.65.2.267. [DOI] [PubMed] [Google Scholar]
- Zorner B, Wolfer DP, Brandis D, Kretz O, Zacher C, Madani R, Grunwald I, Lipp HP, Klein R, Henn FA, Gass P. Forebrain-specific trkB-receptor knockout mice: behaviorally more hyperactive than “depressive”. Biol Psychiatry. 2003;54:972–982. doi: 10.1016/s0006-3223(03)00418-9. [DOI] [PubMed] [Google Scholar]