

Abstract
Using Strepavidin as a scaffold, we have assembled a composite immunotoxin that consists of recombinant Pseudomonas exotoxin A subunit (PE38) and recombinant 25-D1.16 Fab fragment which recognizes the SIINFEKL (pOV8) peptide from ovalbumin in association with H-2Kb MHC class I protein. The composite immunotoxin exercises cytotoxicity against H-2Kb+ cells sensitized with pOV8 peptide but not with irrelevant peptide. Specific binding of the immunotoxin to H-2Kb+ cells infected with recombinant rabies virus (RV) expressing pOV8 epitope (RV-pOV8) resulted in the suppression of the production of virus particles by the infected cells. This strategy allows readily produce different immunotoxins with desired specificity by combining various targeting and toxin molecules. The results provide a proof of concept that composite immunotoxins can be utilized as novel immunotherapeutics to stop virus spread in the acute phase of the infection allowing winning time for the development of protective immune response.
Keywords: TCR-like antibody, peptide-MHC ligands, Pseudomonas exotoxin, composite immunotoxin, rabies virus
1. INTRODUCTION
Although natural immune mechanisms to control viral infections are available at the host's disposal, they are not always induced in a timely fashion to protect the host. Virus often spreads very rapidly before the host immune system responds. Therefore, it would be beneficial to develop therapeutics that can complement natural immunity. In particular, curbing the spread of virus in the acute phase of the infection could help winning sufficient time to develop strong anti-viral immune responses to control the invaders.
Among various approaches to target unwanted cells immunotoxins have been particularly utilized (Pastan et al., 1992; Sweeney and Murphy, 1995). All strategies that have been used thus far are based on the production of chimeric proteins in which the targeting molecule is fused to a toxin moiety (Kreitman et al., 1990; Williams et al., 1990a; Williams et al., 1990b; Brinkmann et al., 1991; Puri et al., 1991; Reiter et al., 1997; Sweeney et al., 1998; Onda et al., 2008). While some of these strategies have proved to be useful, there are several limitations that preclude therapeutic applications of immunotoxins. The side effects include liver and kidney toxicity and induction of neutralizing antibodies against the toxin (Bera et al., 1998). In addition, because of their toxicity, toxins cannot be expressed in eukaryotic cells and must be expressed in bacterial cells. Meanwhile, the expression of various targeting molecules requires chaperone proteins to facilitate appropriate protein folding that influences their biological activity. This is particularly true for antibodies and their fragments.
Here we describe a novel strategy in which a targeting molecule and a toxin moiety are assembled into a composite immunotoxin on Strepavidin scaffold. This strategy permits expression of the targeting molecule and the toxin molecule in optimal expression systems. We utilized genes encoding heavy and light chains of TCR-like antibody 25-D1.16 recognizing pOV8 peptide from ovalbumin in association with H-2Kb class I MHC (Porgador et al., 1997; Mareeva et al., 2004) to produce recombinant Fab fragment in Drosophila melanogaster cells, which was used as a targeting protein. Pseudomonas exotoxin A subunit PE38 (Pastan et al., 1992; Pastan et al., 2006) expressed in E.coli served as a toxin subunit. We have shown that this composite immunotoxin specifically binds to cells presenting pOV8-Kb molecules on the cell surface. Binding of the composite immunotoxin to cells infected with recombinant RV that expresses pOV8 epitope resulted in significant decrease of the production of virus particles by these cells.
2. MATERIAL AND METHODS
2.1. Cells
The mouse thymoma EL4 (H-2Kb) and TAP-deficient cell line RMA-S were kindly provided by Herman Eisen (Koch Institute for Cancer Research, M.I.T.). The cells were grown in Dulbecco's Modified Eagles medium (DMEM) containing 10% inactivated FCS. BSR hamster kidney cells, which are clonal derivative of BHK-21 cells were grown and infected with rabies virus in DMEM containing 5% inactivated FCS and 1% penicillin-streptomycin as described (Plesa et al., 2006). DH5α (Invitrogen Life Technologies, CA) and JM109 (Promega, WI) competent cells were used for cloning and sequencing. BL21(DE3) cells (Novagen, WI) were utilize for expression of recombinant PE38 toxin subunit. Drosophila S2 cells were obtained from Invitrogen Life Technologies and used for expression of recombinant 25-D1.16 Fab fragments. BRS cells (BKH clone) were grown in DMEM medium supplemented with 10% FBS as described (Plesa et al., 2006).
2.2. Antibodies and Streptavidin
Streptavidin labeled with either Alexa Fluor® 488 or Phycoerythrin (PE) was purchased from Molecular Probes Inc. FITC labeled goat anti-mouse Ig was from BD Biosciences; MTT reagent was from Promega, and anti-rabies virus nuclear protein (anti RV-N) was from FDI FUJIREBIO Diagnostics Inc. AF6-88.5.3 antibody specific for H-2Kb antigen was purchased from AbD Serotec or produced from AF6-88.5.3 hybridoma (American Type Culture Collection).
2.3. Peptides
The peptide from chicken ovalbumin (257–264) SIINFEKL (pOV8) and vesicular stomatitis virus nucleocapsid protein (52–59) RGYVYQGL (VSV) were synthesized by BioSynthesis. Purity of the peptides was confirmed by HPLC and mass spectrometric analysis.
2.4. Construction of expression plasmids
Plasmids containing genes encoding for 3B3-PE38 immunotoxin and enzymatically inactive mutant 3B3-PE38Asp-553 were a kind gift from Dean H. Hamer (NCI, National Institutes of Health) (Root and Hamer, 2003). The PE38 fragment with or without inactive point mutation was amplified by four sequential PCR reactions using the following forward primers: 5′–GCC CAG AAG ATC GAG TGG CAC GAG AAT TCC GGA GGT CCC GAG GGC–3′ (PE38FOR-1); 5′–GGC CTG AAC GAC ATC TTC GAG GCC CAG AAG ATC GAG TGG CAC–3′(PE38FOR-2); 5′–CAT CAC CAT CAC CAT CAC GGC CTG AAC GAC ATC TTC GAG–3′ (PE38FOR-3) and 5′–AAT ATT ATG GCC ATG GAT CAT CAC CAT CAC CAT CAC GGC –3′ (PE38FOR-4). The PCR product of each reaction was used as a template cDNA for the next round of PCR. The same backward primer 5′–AAT ATT AAG CTT TTA CTT CAG GTC CTC GCG CGG C–3′ (PE38BACK) was used for all PCR reactions. Since the PE38 gene is a GC-rich target, Herculase Enchanced DNA Polymerase (Stratagene) in the presence of 4% DMSO was used in PCR reactions to achieve high fidelity amplification and high yield of PCR products. Because REDLK C-terminus of PE38 is critical for the toxin translocation to the cytoplasm and should be preserved (Kreitman and Pastan, 1995), we fused hexahistidine sequence in-line with the sequence (GLNDIFEAQKIEWHE) for site-specific biotinylation by the BirA biotin ligase to the N terminus of PE38. A short linker (NS) containing site for EcoRI restriction endonuclease (underlined) was introduced between the site of biotinylation and PE38 fragment (Fig. S1A). The PE38 cDNA was subcloned into the bacterial expression vector pET22b(+) downstream of PelB leader sequence at the NcoI and HindIII restriction sites under control of T7 promoter. The vector contains the ampicillin resistance gene to provide a selectable marker for clone selection. The final plasmid was verified by DNA sequencing and transformed into Escherichia coli BL21(DE3) (Novagen).
Genes encoding N-terminal half (VH and CH1 domains) of the heavy chain (Fd fragment) and the light chain of 25-D1.16 (Mareeva et al., 2004) were derived by PCR using Taq DNA Polymerase (Eppendorf). The Fd fragment was amplified by four sequential PCR reactions. The PCR product of each reaction was used as a template cDNA for the next PCR reaction. The same forward primer 5′–AAT ATT TGA ATT CAC TTA GTG ATT ATG GGA TGG AGC TGG ATC TTT CTC–3′(FdFOR) was used for each PCR reaction. The backward primers were: 5′–GTC GTT CAG GCC GGA CAG GCC ACA ATC CCT GGG CAC–3′ (FdBACK1), 5′–CTT CTG GGC CTC GAA GAT GTC GTT CAG GCC GGA CAG GCC–3′ (FdBACK2), 5′–GAT GGT GAG ATC TCT CGT GCC ACT CGA TCT TCT GGG CCT CGA AG–3′ (FdBACK3), 5′–AAT ATT ACC GGT CTA ATG GTG ATG GTG ATG GTG AGA TC–3′ (FdBACK4). Each PCR step was used to introduce cloning sites (Sfl I and Blg II), the site for biotinylation, and His6 tag (see Fig. S1B). The light chain was amplified by 2 sequential PCR reactions with a single forward primer 5′–AAT ATT TGA ATT CAC TTA GTG ATT ATG AAG TTT CCT TCT CAA CTT CTG CTC–3′ (LFOR) and two backward primers 5′–CCT CTT CTG AGA TGA GTT TTT GTT CTG CGG CCG CAC ACT CAT TCC TGT TGA AGC TCT TG–3′ (L-BACK1) and 5′–AAT ATT ACC GGT CTA TGC GGC CCC ATT CAG ATC CTC TTC TGA GAT GAG TTT TTG TTC TGC–3′ (L-BACK-2). This enabled to fuse C-terminus of the light chain with the c-myc epitope (Fig. S1C). Natural signal sequence was introduced at the 5′ end of both Fd fragment and the light chain which were subcloned as a EcoR I-Age I fragments into the pMT/V5-His expression vector (Invitrogen) for expression in Schneider cells (S2). Expression of both chains was controlled by a methallothionein promoter as previously described (Anikeeva et al., 2003a; Anikeeva et al., 2003b).
2.5. Expression and purification of recombinant PE38 toxin
5 ml of overnight culture of E.coli BL21 (DE3) transformed with the pET22b recombinant plasmid containing PE38 and was inoculated in 500 ml LB medium containing 100 μg/ml ampicillin. The culture was incubated at 37°C with shaking at 200 rpm until OD600 reached 0.6–1.0. The protein expression was induced with 1mM IPTG and the culture further incubated for 3–4 h at 30°C and 180 rpm. The culture was harvested and centrifuged at 10,000 rpm for 10 min at 4°C and the pellet was used to isolate cytoplasmic soluble protein according to the manufacturer protocol (Novagen Inc). Briefly, the pellet was resuspended in 40 ml of lysis buffer (50 mM Tris-HCl, pH 8.0, 200 mM NaCl, 1 mM PMSF, aprotinin 0.1 μg/ml, leupeptin 0.5 μg/ml, pepstatin 0.07 μg/ml, iodoacetamide 55 μg/ml, 0.02% sodium azide) containing 0.25 mg/ml lysozyme and incubated for 30 min at room temperature. The suspension was homogenized by sonication (10 × 30 msec and 10 × 50 msec) and centrifuged at 12,000 rpm for 20 min at 4°C. The supernatant was harvested as the cytoplasmic fraction containing soluble protein.
The protein was purified from the cell lysate by metal-chelate affinity chromatography on Ni-NTA Agarose (Qiagen). Analysis of material eluted at 250 mM of imidazole shows that ~90% of the material represents PE38 (data not shown). The material was further purified by size-exclusion chromatography on Sephacryl S-200 gel filtration column to remove aggregated material (Fig. S2A). The protein eluted in the last peak revealed a single band SDS PAGE at approximately 45 kD (Fig S2A). The same strategy was utilized to purify the enzymatically inactive point mutant PEAsp-553.
2.6. Expression and purification of recombinant 25-D1.16 Fab
Plasmids containing the cDNA of Fd fragment and the light chain of 25-D1.16 as well as the plasmid containing the neomycin–resistance gene were co-transfected into Schneider cells (S2) and the transfected cells were cultured in the presence of G418 at 500 μg/ml for about 3 weeks to select stable polyclonal transfectants essentially as previously described (Anikeeva et al., 2003a; Anikeeva et al., 2003b; Mareeva et al., 2004; Mareeva et al., 2008). Selected cells were grown in Schneider's medium supplemented with 200 mM L-glutamine and 10% inactivated FCS at the density 1–10×106 per ml and 27°C. For large-scale expression (5–10 liters) the cells were grown in an Erlenmeyer shake flask in a serum free medium (either INSECT EXPRESS medium (Bio Whittaker) or Sf-900 II SFM medium (Gibco)) at room temperature (23–25°C). Protein expression was induced by 0.7 mM CuSO4 for about 56 hours at a cell density 5–10×106 per ml. The culture supernatant was cleared by centrifugation (10 min at 800 g) and concentrated to about 100–200 ml and then dialyzed against 50 mM PB, 0.140 M NaCl, pH 8.0 (PBS) containing 100 μM PMSF (Sigma) using a tangential flow concentrator (Pall Filtron). Recombinant His6-tagged protein was purified by anionexchange chromatography on MonoQ column (Amersham) and was eluted at 250 mM of imidazole. The material was further subjected to gel-filtration on Sephacryl S200 (Amersham) to remove aggregates. A majority of the protein was eluted from the Sephacryl S-200 column as a single peak after a peak of aggregates (Fig. S2B). SDS PAGE analysis in non-reducing conditions showed a homogeneous band with molecular weight corresponding single Fab fragment, i.e. 50 kD (Fig. S2B). The specificity of the recombinant 25-D1.16 Fab was confirmed by SPR measurements of the Fab interaction with soluble pOV8-Kb (see Fig. S3). The parameters of the interaction were virtually identical to those previously described for the interactions of native 25-D1.16 with pOV8-Kb protein (Fig. S3)(Mareeva et al., 2004).
2.7. 25-D1.16 Fab tetramer production and staining of live cells
Purified 25-D1.16 Fab was biotinylated using the BirA ligase enzyme according to the manufacturer's protocol (Avidity). To assemble the Fab tetramer, nonlabeled streptavidin or streptavidin labeled with either Alexa Fluor 488 (Str-Alexa) or Phycoerythrin (Str-PE) were used. The labeled or non-labeled Streptavidin (1 mg/ml) was added drop-wise to biotinylated recombinant Fab fragment (1.3 mg/ml) to achieve the final molar ratio of 1:4. The tetramer formation was controlled by SDS PAGE (not shown). The tetramer concentration was measured in terms of molar concentration of Streptavidin.
RMA-S or EL4 cells were pulsed with either cognate (pOV8) or irrelevant (VSV) peptide at various concentrations for 4 h in complete medium. Cells were then washed with staining buffer (Dilbecco's PBS containing 10% FCS) and resuspended in 100 μl of the staining buffer. 1 μg of 25 -D1.16 Fab tetramer (1 mg/ml) was added to the cell suspension for 10–30 min on ice. The mixture was then washed 3 times with 150 μl of the staining buffer and the cells were analyzed by flow cytometry and confocal microscopy.
2.8. Assembly of composite immunotoxin
A single biotin was introduced at the N-terminus of the recombinant PE38 with BirA as above and the biotinylated PE38 (1 mg/ml) was combined with the biotinylated 25-D1.16 Fab at 1:3, 1:1 or 3:1 molar ratios. Unlabeled or Alexa Fluor 488-labeled streptavidin (1 mg/ml) was added drop-wise to the Fab/PE38 mixture over a period of 10 minutes at room temperature to a final streptavidin : (Fab/PE38) ratio of 1:4 (v/v). The formation of composite immunotoxin (ImTx) was controlled by SDS PAGE (Fig. 1). To produce PE-labeled ImTx, the ratio of streptavidin-PE added to the Fab/PE38 mixture was empirically determined using SDS-PAGE. The same procedure was used to assemble ImTx containing PE38Asp-553. Molar concentration of the streptavidin was used to measure the composite ImTx concentration.
Figure 1. Assembly of ImTx.
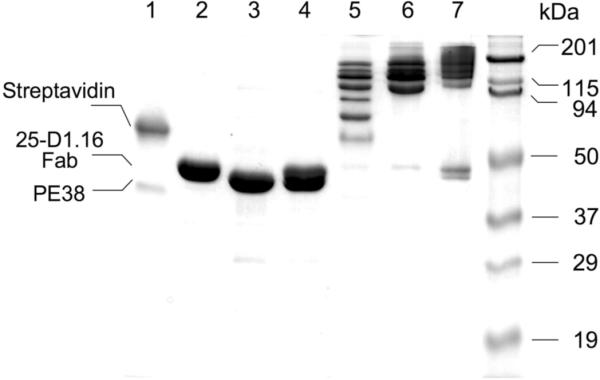
SDS PAGE at non-reducing condition shows that all biotinylated 25-D1.16 Fab and PE38 molecules taken at the equimolar (1:1) ratio were bound to accessible biotin binding sites of Streptavidin. Lane 5 shows that multiple Streptavidin/Fab/PE38 species are formed at (Fab/PE38):Streptavidin ratio 1:1. At the 3:1 ratio, mostly high molecular weight species were seen indicating that majority of Streptavidin binding sites were occupied (Lane 6). When the ratio was increased to 5:1, free Fab and PE38 species were present in the mixture inactive of complete saturation of the Streptavidin binding sites (Lane 7). Lane 1, 2,3 and 4 show Streptavidin, 25-D1.16 Fab, PE38 and a mixture of the Fab and PE38 proteins, respectively.
2.9. The cytotoxicity assay
5000 RMA-S cells in 50 μl of complete medium were aliquoted in a round-bottom 96-well plates and were sensitized with 3 μM of cognate (pOV8) or irrelevant (VSV) peptide for 4 h at 37°C, 5% CO2. Then equal volume of a complete medium containing ImTx at various concentrations (from 10 pM to 1 μM) was added to each well. Complete medium without ImTx was added to some wells, which were used as a negative control. The plates were incubated for 42 h at 37°C in atmosphere of 5% CO2. The cell viability was evaluated by the MTS assay. The assay is based on the ability of viable cells to convert 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS, inner salt) into the aqueous soluble formazan product by dehydrogenase enzymes found in metabolically active cells in the presence of an electron coupling reagent phenazine methosulfate (PMS). 1 ml of MTS solution (2 mg/ml in DPBS) were mixed with 50 μl of PMS solution (0.92 mg/ml in DPBS) and with 4 ml of growth medium. 50 μl of this reagent was added into each well and cells were incubated at 37°C, 5% CO2 for another 3 h. The quantity of formazan product as measured by the level of absorbance at 490 nm is directly proportional to the number of living cells in culture. Optical density of the samples at 490 nm was determined by Emax plate reader (Molecular Devices).
2.10. Infection of EL4 cells with recombinant RV expressing pOV8 eptitope
We produced recombinant virus expressing pOV8 as previously described (Plesa et al., 2006). Recovered rabies virus expressing pOV8 was used to grow a new lot of virus. Briefly, 70 – 80% confluent BSR cells were infected at a low multiplicity of infection (MOI) with Rabies virus expressing OVA257–264 (pOV8) epitope (RV-pOV8). 2 hours post infection, the media was aspirated and the cells were washed two times with Dulbecco's PBS devoid of Ca2+ and Mg2+. Serum free media was then added to the cells and virus was harvested from the culture supernatant on day 3 and day 6. Viral titers were determined on BSR cells as described (Plesa et al., 2006). 2.5×105 EL4 cells were infected with RV-pOV8 at MOI of 5 or 10 and harvested at 48 and 72 hours post infection.
2.11. Detection of pOVA epitope on the surface of infected cells
Expression of pOV8-Kb complexes on the surface of infected was tested by flow cytometry using 25-D1.16 Fab/tetramer and antibodies specific for RV nucleoprotein (anti-RV-N). Briefly, infected EL4 cells were washed with staining buffer. Cells were then incubated in staining buffer containing 3.3 μg streptadivin per 100μl of staining buffer for 1 hour, washed 3 times with staining buffer, and incubated on ice with 25-D1.16 Fab tetramer (10 μg /ml) for 30 minutes. Cells were then washed two times in staining buffer and fixed in 100 μl of BD cytofix buffer for 10 minutes on ice. After two washes, the cells were incubated for 30 minutes at room temperature with labeled anti-RV-N diluted in BD cytoperm buffer containing saponin. Following incubation, the cells were washed and resuspended in staining buffer for analysis by flow cytometry.
2.12. One-step growth curve
EL4 cells were plated in 12-well plates at 250,000 cells/well. The cells were infected with recombinant rabies virus expressing pOV8 at a multiplicity of infection of 3 and incubated at 37°C for 1 h. Since EL4 cells are suspension cells, they were aliquoted into 15 ml falcon tubes and washed two times with PBS. The cells were then resuspended in 2 ml DMEM. 5 μl of immunotoxin containing either active or inactive PE38 subunit was added to assigned wells at time zero. 100 μl of supernatant was collected from cells at time 0, 24, 48 and 72 hours post infection. At each time point, the cells were split at 1:2 ratio to avoid cell overgrowth. 5 μl of the active or the inactive immunotoxin was added to the respective wells at 24 and 48 hours post infection. The viral titers in the supernatants were determined in duplicate by infecting BSR cells as previously described (Plesa et al., 2006).
3. RESULTS
3.1. Composite immunotoxin: assembly and analysis of the binding specificity
To assemble fluorescent-tagged immunotoxin, fluorescent-labeled streptavidin was combined with a mixture of 25-D1.16 Fab and PE38 where the two components were present at various molar ratios. The Fab/PE38 ratios used were 1:3, 1:1 or 3:1. The formation of composite immunotoxin was evident from SDS PAGE analysis (Fig. 1). Addition of the Strepavidin to equimolar Fab/PE38 mixture at 1:1 ratio led to formation of multiple ImTx species containing variable numbers of the Fab and the PE38 arms bound to the Streptavidin scaffold (Lane 5). As the (Fab/PE38) : Strepavidin ratio increased to 3:1, majority of the biotin binding sites of the Strepavidin were occupied by the Fab and the PE38 biotinylated proteins resulting in the formation of high-molecular weight ImTx species (Lane 6). At the ratio 5:1, all accessible binding sites of the Strepavidin scaffold were engaged leaving some Fab and PE38 molecules unbound (Lane 7). In our experiments we always used composite ImTx produced at the (Fab/PE38) : Strepavidin 4:1 ratio. Keeping with this ratio, we utilized different ratios of biotinylated Fab and PE38 in the mixture to vary the number of targeting 25-D1.16 moiety and of PE38 toxin subunit within the ImTx preparations.
To test the ability of fluorescent-labeled ImTx to recognize pOV8-Kb proteins on the surface of live cells, we sensitized RMA-S cells with either cognate (pOV8) or irrelevant (VSV) peptides. The presence of pOV8-Kb molecules of the cell surface was evident from staining of these cells with 25-D1.16 Fab tetramer (Fig. S4A). We then let fluorescent-labeled ImTx interact with the cells. Fig. 2 shows that the ImTx specifically bound to pOV8-Kb proteins on RMA-S cells as established by flow cytometry (A) and fluorescent microscopy (B) analyses. The specificity of the binding was also evident from the titration of the ImTx concentration and the titration of pOV8 peptide added to the extracellular medium (Fig. S5).
Figure 2. Binding specificity of composed immunotoxin.

(A) RMA-S cells sensitized with peptide pOV8 but not with VSV peptide were specifically stained with ImTx containing Alexa Fluor 488 Streptavidin (Str-Alexa) as established by flow cytometry analysis on Beckman Coulter FACS analyser. (B). Fluorescent microscopy of the same cells showed that fluorescent-labeled ImTx was specifically bound to pOV8-sensitized RMA-S cells. Representative images from 2 independent experiments are shown; interference reflection microscopy images are on the top and fluorescent images on the bottom. The fluorescence intensity was adjusted by subtracting cell autofluorescence (green).
3.2. Composite immunotoxin exercises cytotoxicity against H-2Kb+ cells sensitized with pOV8
The ImTx specifically bound to pOV8-Kb bearing RMA-S cells effectively killed these cells (Fig. 3). ImTx concentration required to achieve half-maximal killing was about 4.5 nM (Fig. 3). The killing with ImTx was specific since RMA-S cells presenting VSV-Kb were not killed (Fig. 3). The cytotoxicity was mediated by PE38 subunit of ImTx as evident from complete lack of the cytolytic activity with ImTx containing inactive PE38Asp-553 subunit (Fig. 3). Incubation of pOV8-sensitized RMA-S cells with PE38 tetramer in the absence of 25-D1.16 Fab did not resulted in the specific interaction with the cell surface and the cell killing (data not shown) indicating the toxin PE38 subunit must be specifically targeted to the cell surface protein to utilize PE38 biological activity. The engagement of pOV8-Kb molecules on the surface of target cells with either ImTx or 25-D1.16 tetramer at 37°C initiated rapid uptake of pOV8-Kb-bound ImTx and 25-D1.16 tetramer (see Fig. 2 and Fig. S4B), a necessary prerequisite for the pE38 subunit to exercise its effector function (FitzGerald, 1999; Pastan et al., 2006).
Figure 3. The cytotoxicity of composite immunotoxin.
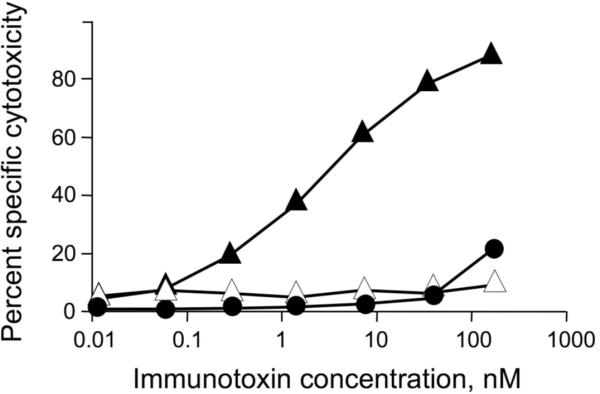
RMA-S cells sensitized with either pOV8 (▲) or VSV (●) peptide were incubated with ImTx containing active PE38 subunit at indicated concentrations for 4 h at 37° C in atmosphere of 5% CO2; pOV8-sensitized cells were also incubated with ImTx containing inactive PE38Asp-553 (▵). The cells viability was evaluated by the MTS assay and percent specific cytotoxicity was determined (see Material and Methods) for each concentration of ImTx.
Variation of the Fab/PE38 ratio within the ImTx led to different efficiency the target cell killing (Fig. S6). At the Fab/PE38 ratio 1:3, the immunotoxin showed maximal potential to destroy target cells sensitized with pOV8 at saturated concentration (3×10−6 M), while the immunotoxin containing on average 3 Fab and 1 PE38 molecules displayed the least potential to kill. These data suggest that the amount of the toxin delivered into the target cell, but not the number of engaged targeted molecules (pOV8-Kb) on the cell surface limit the effectiveness of target cell lysis.
3.3. Processed pOV8 epitope is presented on the surface of target cells infected with RV-pOV8
To evaluate the ability of ImTx to target virally infected cells, we utilized recombinant rabies virus that express pOV8 peptide epitope, RV-pOV8 (Plesa et al., 2006). H-2Kb+ EL4 cells were infected with RV-pOV8 or control RV virus that does not express pOV8 epitope. The infected cells were stained with 25-D1.26 tetramer to detect the presence of pOV8-Kb complexes on the cells surface. At the same time, the infected cells were stained intracellularly with anti-RV-N antibody to reveal the presence of RV nucleoprotein in the cytoplasm of these cells. Flow cytometry analysis showed that virtually all RV-N-positive cells displayed pOV8-Kb ligand on their surface (Fig. 4). Fluorescence-tagged ImTx also specifically bound to the infected cells, while PE38 tetramer did not (Fig. S7).
Figure 4. Detection of pOV8-Kb proteins on the surface of RV-pOV8 infected EL4 cells.

A. Staining with 25-D1.16 tetramer showed presence of pOV8-Kb molecules on the surface of EL4 cells infected with RV-pOV8 but not with control RV virus. B. RV nuclear protein is detected in the cytoplasm of EL4 cells infected with both RV-pOV8 and control RV virus. C. The double staining of RV-pOV8- and RV-infected cells demonstrated that practically all RV-pOV8-infected IL4 cells were pOV8-Kb positive.
3.4. Composite immunotoxin inhibits viral replication in the infected cells
We next tested the ability of ImTx to suppress replication of the recombinant virus RV-pOV8 in the infected EL4 cells by measuring the number of viral particles released into the supernatant using one-step growth curve approach (Plesa et al., 2006). Figure 5 shows that presence of ImTx containing active PE38 resulted in a significant decrease of virus release by the infected cells. In contrast, the presence of ImTx containing inactive PE38Asp-553 subunit did not decrease RV-pOV8 virus production which was similar to that observed for RV virus production in the absence of either active or inactive ImTx (Fig. 5). Since RV-pOV8 recombinant virus showed even slightly greater ability to replicate in the infected cells as compared to a control RV virus expressing Gag epitope (data not shown), the data presented clearly indicate that a composite ImTx interfered with virus production indicative of the suppression of the virus replication in the infected cells.
Figure 5. ImTx suppresses RV-pOV8 production by EL4 infected cells.
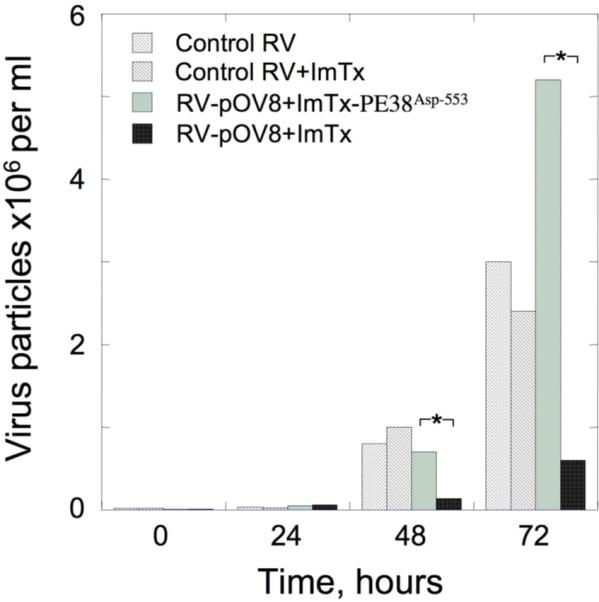
ImTx containing active PE38 subunit as opposed to that with inactive PE38Asp-553 subunit significantly suppressed production of recombinant RV-pOV8 virus by the infected cells. The difference in RV-pOV8 virus particles measured in the supernatant was statistically significant as established by Student's t-tests for paired data. *: P<0.02. Presence of ImTx in the supernatant practically did not affect production of a control RV virus by the infected cells. The recombinant RV that has Gag gene cloned into the backbone similar to that of RV-pOV8 was used as a control virus. The control RV-Gag virus showed somewhat impaired ability to replicate in EL4 cells as compared to RV-pOV8 most likely due to a toxic effect of Gag protein on the infected cells.
That ImTx blocks protein synthesis in RV-pOV8 infected cells is evident from a significantly lower amount of pOV8 peptide generated in the infected cells treated with the ImTx. Consequently, the level of pOV8-Kb expression on the surface of RV-pOV8 infected cells in the presence of the ImTx decreased by about a hundred-fold (Fig. 6A). The decrease of the density of pOV8-Kb complexes on RV-pOV8 infected cells was not due to the loss of the cell surface MHC since the total level of H-2Kb class I, which restricts presentation the pOV8 and many other endogenous peptides, on the infected cells was very similar in the presence or absence of the ImTx (Fig. 6B).
Figure 6. Exposure of RV-pOV8-infected cells to ImTx results in substantial decrease of the cell surface level of pOV8-Kb proteins while the total level of MHC class I expression remains the same.

A. Left panel. EL4 cells were infected with RV-pOV8 in the presence (green) or absence (red) of ImTx and stained with fluorescent-labeled 25-D1.16 Fab/tetramer (see Materialand Methods for details). The level of pOV8-Kb expression of the infected cells was determined by mean fluorescent intensity. Right panel. EL4 cells were sensitized with either pOV8 or VSV peptides at various concentrations and the cells were stained with fluorescent-labeled 25-D1.16 Fab/tetramer. The level of pOV8-Kb expression was evaluated by measuring MFI of the staining. The difference in peptide concentration required to achieve levels of pOV8-Kb expression similar to that found on RV-pOV8-infected cells in the presence or absence of ImTx (see left panel) amounted to approximately 100 fold (indicated by errors).
B. Uninfected cells EL4 cells as well as those infected with either RV-pOV8 or control RV viruses were incubated in the presence (green) or absence (red) of ImTx or in the presence of ImTx containing inactive PE38Asp-553 subunit (blue) and were stained with H-2Kb-specific AF6-88.5.3 antibodies followed by FITC-labeled anti-mouse antibody. In control experiment the cells were incubated with FITC-labeled anti-mouse antibody only (grey). The level of H-2Kb expression was found to be similar in all cases.
4. DISCUSSION
Normally, virus-infected cells can be destroyed by cytotoxic T lymphocytes (CTL) that recognize viral peptides presented by MHC proteins on the surface of infected cells. The recognition of the peptide-MHC molecules (pMHC) is mediated by antigen-specific T-cell receptor (TCR) that is able to discriminate between pMHC containing peptides from the virus and those carrying peptides from indigenous (self) proteins. Success in containment of virus infection is often determined by the outcome of the race between the development of effective immune response and viral spread. When virus spreads faster than a strong anti-virus immune response can be developed, it may overwhelm the host immune system leading to death in the acute phase of the infection. At such circumstances, it seems essential to develop immunotherapeutic interventions to decrease production of virus particles in the infected cells and to curb the spread of the virus.
Since small peptide fragments from viral proteins displayed on the cell surface by MHC proteins uniquely mark infected cells for recognition by TCR on T cells, we sought to utilize TCR-like reagent, which specifically recognize a particular pMHC complexes on infected cells as a tool to target a toxin moiety to the infected cells. We exploited 25-D1.16 monoclonal antibody specific for pOV8-Kb complex as a targeting protein (Porgador et al., 1997). Sequences of the heavy and light chains of this antibody have been determined and the crystal structure of the 25-D1.16 Fab/pOV8-Kb complex has been solved (Mareeva et al., 2004; Mareeva et al., 2008). In addition, we have recently evaluated the ability of both recombinant 25-D1.16 Fab tetramer and the monovalent Fab fragment specifically detect pOV8-Kb proteins on the cell surface and have found that both reagents can detect the pMHC proteins at relatively low densities (Anikeeva et al., 2009).
The pOV8 peptide epitope has been successfully used to produce various recombinant viruses to investigate the induction of virus-specific T cell responses and the development of protective anti-viral immunity (Brown et al., 2000; McAllister et al., 2000; Wherry et al., 2002) or autoimmunity (Turner et al., 2008). In addition, the pOV8 epitope has also been utilized in a model system to investigate immunization strategies in anti-cancer vaccine design (Sherritt et al., 2001). In this study, we exploited recently developed noncytopathic recombinant rabies virus that express pOV8; the latter is presented by H-2Kb MHC class I molecules on the surface of the infected cells (Plesa et al., 2006). Thus, the RV-pOV8 virus is very well suited to evaluate the virus replication over a long period of time (up to 72 hours) in the presence or absence of the composite ImTx.
To produce a recombinant toxin moiety, we chose PE38 toxin consisting of the translocation and ADP-ribosylation domains of Pseudomonas exotoxin A (Pastan et al., 1992). When delivered into a cell, the toxin is translocated by retrograde transport to the cytosole and catalyzes the irreversible ADP-ribosylation and subsequent inactivation of elongation factor 2 shutting down protein synthesis. Blocking the host protein synthesis machinery is expected to halt the production of viral proteins resulting in a decrease of newly assembled viral particles and spread of the virus. Indeed, we have shown that presence of the composite ImTx containing PE38 moiety effectively suppress production of RV-pOV8 in the infected cells (Fig. 5). Our result is consistent with previous findings showing that a recombinant chimeric immunotoxin containing Fv fragment of an antibody specific for HIV gp120 kills HIV-infected cells (Bera et al., 1998) and stop spreading the virus in primary macrophages (Kennedy et al., 2006).
Instead of producing the chimeric immunotoxin, we decided to assemble targeting and toxin subunits on Streptavidin scaffold. This strategy has several advantages. Assembly of immunotoxin on Streptavidin scaffold does not require refolding of the fusion protein that can result in a low yield of properly folded chimeric protein comprising of functionally active targeting and toxin subunits. Moreover, because of its toxicity, toxin moiety should be express in E.coli, but not in eukaryotic cells. The latter, however, may be more suitable for expression of targeting protein. Even production of antibody fragment in E.coli, which are usually refolded from inclusion bodies fairly well, could result in a lost of their fine specificity. In fact, our initial attempts to produce a fusion protein containing single chain 25-D1.16 Fv fragment and PE38 led to a loss of the ability of the Fv to discriminate pOV8 peptide bound to Kb protein (Mareeva and Sykulev, unpublished). In addition, assembly of composite ImTx permits to vary the ratio of targeting and toxin subunits to establish an optimal ratio required for successful delivery of necessary amount of toxin molecules into a target cells. This may be especially useful for targeting proteins whose intrinsic affinity for a cell surface receptor is low. For instance, soluble peptide-MHC molecules that could be utilized to target unwanted T cells with known specificity have typically low intrinsic affinity for TCR, and inclusion of 2 or 3 peptide-MHC proteins along with 1–2 toxin subunits into the composite immunotoxin assembled on Streptavidin scaffold would produce an effective tool for targeting T cells. Thus, the proposed strategy represents a versatile system that allows combining various targeting and toxin molecules to generate various immunotoxins with desired specificity instead of developing a chimera protein containing both target protein and toxin subunit for every viral peptide epitope. If necessary, multivalency of a composite ImTx can be further increased using nanoparticles as a scaffold (Anikeeva et al., 2006; Anikeeva et al., 2008).
While immunotoxins have been previously implicated for treatment of viral infections, HIV in particular (Bera et al., 1998), targeting a small number of infected cells that present viral peptide epitopes at low density during chronic phase of the infection would very unlikely to be an effective strategy leading to virus eradication. The ability to control virus production in the acute phase of the infection using the strategy described here appears to be more promising. The number of infected cells and the density of viral peptides on the cell surface in the acute phase are significantly higher that increases the probability of productive ImTx encounters with the infected cells. The proposed strategy is expected to be useful for treatment of viral infections caused by rapidly spreading viruses such as Ebola, influenza or Variola viruses (Sullivan et al., 2000; Feldmann et al., 2003; Amanna et al., 2006; Taubenberger and Morens, 2008) that cause death during acute phase of the infection. In the case of the highly pathogenic avian influenza virus H5H1, there is an indication that viral pathology in humans is caused at least in part by high level of viral replication (Taubenberger and Morens, 2008), therefore any reduction of virus-infected cells would probably result in better outcome of the infection. It seems, therefore, essential to develop novel immunotherapeutic interventions to curb virus production cells infected with rapidly spread viruses. This could permit winning the time for the immune system to develop effective anti-virus immune response. In this context, we propose to utilize composite ImTx not to eradicate virus-infected cells, but as an effective tool to stop virus spread in the acute phase of the infection. Since several peptide epitopes from Variola and Ebola viruses recognizable by T cells have been identified (Drexler et al., 2003; Sundar et al., 2007; Tang et al., 2008), it is feasible to produce TCR-like antibodies (Biddison et al., 2003; Cohen et al., 2003) or soluble TCR with enhanced affinity (Holler et al., 2000; Chlewicki et al., 2005) specific for these peptide epitopes in context with respected MHC proteins paving the way to the development of composite immunotoxins to target the infected cells.
Supplementary Material
AKNOWLEDGEMENT
This work was supported by SAP #4100026302 Grant from Commonwealth of PA - Dept. of Health to Y.S. and M.S. T.M. was supported in part by research grant CA56036-08 to the Kimmel Cancer Center. We thank Dean H. Hamer for providing plasmids containing genes encoding for 3B3-PE38 immunotoxin and enzymatically inactive mutant 3B3-PE38Asp-553.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Amanna IJ, Slifka MK, Crotty S. Immunity and immunological memory following smallpox vaccination. Immunol Rev. 2006;211:320–37. doi: 10.1111/j.0105-2896.2006.00392.x. [DOI] [PubMed] [Google Scholar]
- Anikeeva N, Lebedeva T, Clapp AR, Goldman ER, Dustin ML, Mattoussi H, Sykulev Y. Quantum dot/peptide-MHC biosensors reveal strong CD8-dependent cooperation between self and viral antigens that augment the T cell response. Proc Natl Acad Sci U S A. 2006;103:16846–51. doi: 10.1073/pnas.0607771103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anikeeva N, Lebedeva T, Krogsgaard M, Tetin SY, Martinez-Hackert E, Kalams SA, Davis MM, Sykulev Y. Distinct molecular mechanisms account for the specificity of two different T-cell receptors. Biochemistry. 2003a;42:4709–4716. doi: 10.1021/bi026864+. [DOI] [PubMed] [Google Scholar]
- Anikeeva N, Lebedeva T, Sumaroka M, Kalams SA, Sykulev Y. Soluble HIV-specific T-cell receptor: expression, purification and analysis of the specificity. J. Immunol. Meth. 2003b;277:75–86. doi: 10.1016/s0022-1759(03)00179-0. [DOI] [PubMed] [Google Scholar]
- Anikeeva N, Mareeva T, Liu W, Sykulev Y. Can oligomeric T-cell receptor be used as a tool to detect viral peptide epitopes on infected cells? Clin Immunol. 2008 doi: 10.1016/j.clim.2008.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anikeeva N, Mareeva T, Liu W, Sykulev Y. Can oligomeric T-cell receptor be used as a tool to detect viral peptide epitopes on infected cells? Clin Immunol. 2009;130:98–109. doi: 10.1016/j.clim.2008.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bera TK, Kennedy PE, Berger EA, Barbas CF, 3rd, Pastan I. Specific killing of HIV-infected lymphocytes by a recombinant immunotoxin directed against the HIV-1 envelope glycoprotein. Mol Med. 1998;4:384–91. [PMC free article] [PubMed] [Google Scholar]
- Biddison WE, Turner RV, Gagnon SJ, Lev A, Cohen CJ, Reiter Y. Tax and M1 peptide/HLA-A2-specific Fabs and T cell receptors recognize nonidentical structural features on peptide/HLA-A2 complexes. J Immunol. 2003;171:3064–74. doi: 10.4049/jimmunol.171.6.3064. [DOI] [PubMed] [Google Scholar]
- Brinkmann U, Pai LH, FitzGerald DJ, Willingham M, Pastan I. B3(Fv)-PE38KDEL, a single-chain immunotoxin that causes complete regression of a human carcinoma in mice. Proc Natl Acad Sci U S A. 1991;88:8616–20. doi: 10.1073/pnas.88.19.8616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M, Zhang Y, Dermine S, de Wynter EA, Hart C, Kitchener H, Stern PL, Skinner MA, Stacey SN. Dendritic cells infected with recombinant fowlpox virus vectors are potent and long-acting stimulators of transgene-specific class I restricted T lymphocyte activity. Gene Ther. 2000;7:1680–9. doi: 10.1038/sj.gt.3301288. [DOI] [PubMed] [Google Scholar]
- Chlewicki LK, Holler PD, Monti BC, Clutter MR, Kranz DM. High-affinity, peptide-specific T cell receptors can be generated by mutations in CDR1, CDR2 or CDR3. J Mol Biol. 2005;346:223–39. doi: 10.1016/j.jmb.2004.11.057. [DOI] [PubMed] [Google Scholar]
- Cohen CJ, Denkberg G, Lev A, Epel M, Reiter Y. Recombinant antibodies with MHC-restricted, peptide-specific, T-cell receptor-like specificity: new tools to study antigen presentation and TCR-peptide-MHC interactions. J Mol Recognit. 2003;16:324–32. doi: 10.1002/jmr.640. [DOI] [PubMed] [Google Scholar]
- Drexler I, Staib C, Kastenmuller W, Stevanovic S, Schmidt B, Lemonnier FA, Rammensee HG, Busch DH, Bernhard H, Erfle V, Sutter G. Identification of vaccinia virus epitope-specific HLA-A*0201-restricted T cells and comparative analysis of smallpox vaccines. Proc Natl Acad Sci U S A. 2003;100:217–22. doi: 10.1073/pnas.262668999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann H, Jones S, Klenk HD, Schnittler HJ. Ebola virus: from discovery to vaccine. Nat Rev Immunol. 2003;3:677–85. doi: 10.1038/nri1154. [DOI] [PubMed] [Google Scholar]
- FitzGerald DJ. Recombinant immunotoxins. In: Chamow AASM, editor. Antibody Fusion Proteins. Wiley-Liss, Inc; 1999. pp. 111–126. [Google Scholar]
- Holler PD, Holman PO, Shusta EV, O'Herrin S, Wittrup KD, Kranz DM. In vitro evolution of a T cell receptor with high affinity for peptide/MHC. Proc Natl Acad Sci U S A. 2000;97:5387–92. doi: 10.1073/pnas.080078297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy PE, Bera TK, Wang QC, Gallo M, Wagner W, Lewis MG, Berger EA, Pastan I. Anti-HIV-1 immunotoxin 3B3(Fv)-PE38: enhanced potency against clinical isolates in human PBMCs and macrophages, and negligible hepatotoxicity in macaques. J Leukoc Biol. 2006;80:1175–82. doi: 10.1189/jlb.0306139. [DOI] [PubMed] [Google Scholar]
- Kreitman RJ, Chaudhary VK, Waldmann T, Willingham MC, FitzGerald DJ, Pastan I. The recombinant immunotoxin anti-Tac(Fv)-Pseudomonas exotoxin 40 is cytotoxic toward peripheral blood malignant cells from patients with adult T-cell leukemia. Proc Natl Acad Sci U S A. 1990;87:8291–5. doi: 10.1073/pnas.87.21.8291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitman RJ, Pastan I. Importance of the glutamate residue of KDEL in increasing the cytotoxicity of Pseudomonas exotoxin derivatives and for increased binding to the KDEL receptor. Biochem J. 1995;307(Pt 1):29–37. doi: 10.1042/bj3070029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mareeva T, Lebedeva T, Anikeeva N, Manser T, Sykulev Y. Antibody specific for the peptide-MHC complex: is it TCR-like? J Biol Chem. 2004;279:44243–49. doi: 10.1074/jbc.M407021200. [DOI] [PubMed] [Google Scholar]
- Mareeva T, Martinez-Hackert E, Sykulev Y. How a T cell receptor-like antibody recognizes major histocompatibility complex-bound peptide. J Biol Chem. 2008;283:29053–9. doi: 10.1074/jbc.M804996200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister A, Arbetman AE, Mandl S, Pena-Rossi C, Andino R. Recombinant yellow fever viruses are effective therapeutic vaccines for treatment of murine experimental solid tumors and pulmonary metastases. J Virol. 2000;74:9197–205. doi: 10.1128/jvi.74.19.9197-9205.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onda M, Beers R, Xiang L, Nagata S, Wang QC, Pastan I. An immunotoxin with greatly reduced immunogenicity by identification and removal of B cell epitopes. Proc Natl Acad Sci U S A. 2008;105:11311–6. doi: 10.1073/pnas.0804851105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastan I, Chaudhary V, FitzGerald DJ. Recombinant toxins as novel therapeutic agents. Annu Rev Biochem. 1992;61:331–54. doi: 10.1146/annurev.bi.61.070192.001555. [DOI] [PubMed] [Google Scholar]
- Pastan I, Hassan R, Fitzgerald DJ, Kreitman RJ. Immunotoxin therapy of cancer. Nat Rev Cancer. 2006;6:559–65. doi: 10.1038/nrc1891. [DOI] [PubMed] [Google Scholar]
- Plesa G, McKenna PM, Schnell MJ, Eisenlohr LC. Immunogenicity of cytopathic and noncytopathic viral vectors. J Virol. 2006;80:6259–66. doi: 10.1128/JVI.00084-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porgador A, Yewdell JW, Deng Y, Bennink JR, Germain RN. Localization, quantitation, and in situ detection of specific peptide-MHC class I complexes using a monoclonal antibody. Immunity. 1997;6:715–26. doi: 10.1016/s1074-7613(00)80447-1. [DOI] [PubMed] [Google Scholar]
- Puri RK, Ogata M, Leland P, Feldman GM, FitzGerald D, Pastan I. Expression of high-affinity interleukin 4 receptors on murine sarcoma cells and receptor-mediated cytotoxicity of tumor cells to chimeric protein between interleukin 4 and Pseudomonas exotoxin. Cancer Res. 1991;51:3011–7. [PubMed] [Google Scholar]
- Reiter Y, Di Carlo A, Fugger L, Engberg J, Pastan I. Peptide-specific killing of antigen-presenting cells by a recombinant antibody-toxin fusion protein targeted to major histocompatibility complex/peptide class I complexes with T cell receptor-like specificity. Proc Natl Acad Sci U S A. 1997;94:4631–6. doi: 10.1073/pnas.94.9.4631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Root MJ, Hamer DH. Targeting therapeutics to an exposed and conserved binding element of the HIV-1 fusion protein. Proc Natl Acad Sci U S A. 2003;100:5016–21. doi: 10.1073/pnas.0936926100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherritt M, Cooper L, Moss DJ, Kienzle N, Altman J, Khanna R. Immunization with tumor-associated epitopes fused to an endoplasmic reticulum translocation signal sequence affords protection against tumors with down-regulated expression of MHC and peptide transporters. Int Immunol. 2001;13:265–71. doi: 10.1093/intimm/13.3.265. [DOI] [PubMed] [Google Scholar]
- Sullivan NJ, Sanchez A, Rollin PE, Yang ZY, Nabel GJ. Development of a preventive vaccine for Ebola virus infection in primates. Nature. 2000;408:605–9. doi: 10.1038/35046108. [DOI] [PubMed] [Google Scholar]
- Sundar K, Boesen A, Coico R. Computational prediction and identification of HLA-A2.1-specific Ebola virus CTL epitopes. Virology. 2007;360:257–63. doi: 10.1016/j.virol.2006.09.042. [DOI] [PubMed] [Google Scholar]
- Sweeney EB, Foss FM, Murphy JR, vanderSpek JC. Interleukin 7 (IL-7) receptor-specific cell killing by DAB389 IL-7: a novel agent for the elimination of IL-7 receptor positive cells. Bioconjug Chem. 1998;9:201–7. doi: 10.1021/bc9701757. [DOI] [PubMed] [Google Scholar]
- Sweeney EB, Murphy JR. Diphtheria toxin-based receptor-specific chimaeric toxins as targeted therapies. Essays Biochem. 1995;30:119–31. [PubMed] [Google Scholar]
- Tang ST, Wang M, Lamberth K, Harndahl M, Dziegiel MH, Claesson MH, Buus S, Lund O. MHC-I-restricted epitopes conserved among variola and other related orthopoxviruses are recognized by T cells 30 years after vaccination. Arch Virol. 2008;153:1833–44. doi: 10.1007/s00705-008-0194-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubenberger JK, Morens DM. The pathology of influenza virus infections. Annu Rev Pathol. 2008;3:499–522. doi: 10.1146/annurev.pathmechdis.3.121806.154316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner MJ, Jellison ER, Lingenheld EG, Puddington L, Lefrancois L. Avidity maturation of memory CD8 T cells is limited by self-antigen expression. J Exp Med. 2008;205:1859–68. doi: 10.1084/jem.20072390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, McElhaugh MJ, Eisenlohr LC. Generation of CD8(+) T cell memory in response to low, high, and excessive levels of epitope. J Immunol. 2002;168:4455–61. doi: 10.4049/jimmunol.168.9.4455. [DOI] [PubMed] [Google Scholar]
- Williams DP, Snider CE, Strom TB, Murphy JR. Structure/function analysis of interleukin-2-toxin (DAB486-IL-2). Fragment B sequences required for the delivery of fragment A to the cytosol of target cells. J Biol Chem. 1990a;265:11885–9. [PubMed] [Google Scholar]
- Williams DP, Wen Z, Watson RS, Boyd J, Strom TB, Murphy JR. Cellular processing of the interleukin-2 fusion toxin DAB486-IL-2 and efficient delivery of diphtheria fragment A to the cytosol of target cells requires Arg194. J Biol Chem. 1990b;265:20673–7. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.