

Abstract
Genetic variability is considered a key to the evolvability of species. The conversion of an adenosine (A) to inosine (I) in primary RNA transcripts can result in an amino acid change in the encoded protein, a change in secondary structure of the RNA, creation or destruction of a splice consensus site, or otherwise alter RNA fate. Substantial transcriptome and proteome variability is generated by A-to-I RNA editing through site-selective post-transcriptional recoding of single nucleotides. We posit that this epigenetic source of phenotypic variation is an unrecognized mechanism of adaptive evolution. The genetic variation introduced through editing occurs at low evolutionary cost since predominant production of the wild-type protein is retained. This property even allows exploration of sequence space that is inaccessible through mutation, leading to increased phenotypic plasticity and provides an evolutionary advantage for acclimatization as well as long-term adaptation. Furthermore, continuous probing for novel RNA editing sites throughout the transcriptome is an intrinsic property of the editing machinery and represents the molecular basis for increased adaptability. We propose that higher organisms have therefore evolved to systems with increasing RNA editing activity and, as a result, to more complex systems.
Keywords: adaptation, ADAR, evolvability, inosine, phenotypic plasticity, RNA editing
Introduction
Genetic variability and evolvability
Elucidating the molecular mechanisms underlying adaptive phenotypic variation is central to our understanding of the origins and maintenance of biological diversity. This is particularly true in cases where natural selection acting on the genes underlying adaptive traits contributes directly or indirectly to the origins of barriers to gene exchange and, hence, promotes speciation.(1) Identifying the specific genetic changes responsible for phenotypic adaptation, however, is notoriously difficult(2–4) and successful attempts, to date, have been limited to a select group of heavily investigated model systems, such as stickleback fishes,(5) fruit flies,(6,7) and mice.(3) In spite of the challenges involved, research linking genotype and phenotype is beginning to provide answers to some of the most intractable problems in evolutionary biology. In this paper, we focus specifically on our emerging knowledge of novel molecular mechanisms for producing phenotypic variation in response to selection at the genetic and epigenetic level.
As Fisher(8) clearly demonstrated, the potential of organisms to respond to selection (i.e., adapt) is a function of the standing genetic variance in the population. The rate of phenotypic evolution will therefore depend upon the amount of additive genetic variance for a given trait,(9) and the response to selection will also be influenced by factors such as the number of loci underlying the phenotype and whether the fitness of each genotype is linearly related to the trait value.(10,11) The implications are that within species “evolvability” will be facilitated or constrained by the specific patterns of genetic variation, genetic architecture, and developmental constraints operating in any given lineage.(12,13)
Therefore, molecular mechanisms that increase genetic variability and/or flexibility, e.g., may increase the evolvability of species if they increase an organism's ability to express novel phenotypic variation in response to changing environments. If so, then selection may favor systems with high levels of genetic variance and/or significant phenotypic plasticity.(14) This is particularly true for systems that have evolved the ability to switch to increased phenotype variability when needed. For example, Escherichia coli bacteria increase mutation rates under stressful conditions, which allows them to cope better with the altered environment and increases the chance of evading selective pressure.(15)
Types of genetic and epigenetic variation
Traditionally, mutations have been viewed as the major source for genetic variability within populations.(16) For example, it is clear that many single nucleotide polymorphisms (SNPs)(17,18) arise through the relatively simple process of point mutation (i.e., transitions and transversions). Mutations that occur in protein-coding gene transcripts may directly alter an amino acid codon, or be silent. Alternatively (or additionally), they may alter post-transcriptional RNA processing of the primary gene transcripts affecting the splicing, editing, polyadenylation, localization, transport, or stability of the RNA, thereby indirectly creating phenotypic variability. Genetic variability can also arise as a result of differences in the expression of one or several genes, which can be altered by modifications in regulatory sequences within the genomic DNA, e.g., mutations in transcription factor binding sites or in regulatory molecules, such as microRNA (miRNA) sequences.(19) At this moment, it is not clear if differences in gene function or differences in gene regulation are stronger driving forces for adaptation and speciation.
A fundamental property of genomic mutations is that, in diploid organisms, any germline, genomic alteration in one copy of a chromosomal gene will by definition immediately and permanently affect 100% of the gene products generated from that allele and 50% of the overall output for that gene. Exceptions from this general rule exist for imprinted genes, and for mutations of sequences that are not represented in all of the gene's products, e.g., those within alternatively spliced exons or otherwise alternatively processed RNA sequences. The all-or-nothing property of genomic mutations substantially restricts the extent of genetic variation that can be generated within a population since mutations that affect functionally critical residues in proteins are often forbidden to persist within a population due to the detrimental effect on survival. This means that such residues are largely unavailable for mutation-based generation of variability. As a consequence, adaptive evolution based on protein sequence alterations would be strongly limited if mutational events directly affecting genomic, protein-coding sequences were the only available molecular mechanism to generate new variants. However, as discussed below, this limitation does not exist for some epigenetic types of sequence changes.
Adaptation as a result of genomic mutations followed by phenotypic selection of functional variants occurs over the course of generations. In contrast, fast acclimatization of individuals within a species to cope with sudden changes in the environment is typically epigenetic in nature and involves changes that are usually not heritable and thus do not lead to long-term adaptation.(14) For example, recent insights from quantitative gene expression analysis indicate that gene expression exhibits stochastic properties with genomically identical cells varying widely in the expression levels of individual genes.(20,21) Such fluctuations in gene expression profiles have been linked to fast adaptation of organisms to their environment.(22) It has recently been suggested, however, that in some cases the pool of cryptic variation generated through epigenetic events, such as minor RNA splice forms(23) or translational mutations,(24) could be responsible for the fast acclimatization due to the presence of pre-adapted variants, and thus form a basis for long-term adaptation.(23)
Candidate molecular mechanisms that generate epigenetic variability and potentially pre-adapted variants, include allele-specific imprinting as well as changes in RNA processing, translation and stability. It is not known exactly what level of variability is generated using these mechanisms, but, e.g., it has recently been estimated that maybe 4,000 human genes are regulated through allele-specific imprinting.(25) Similarly, most genes in mammals are subject to at least one alternative splicing event.(26) Also, the impact of single epigenetic events, such as those affecting the imprinting of a single transcription unit can be local, or they may have broader effects, e.g., a shift in regulation of one splicing factor that alters the whole alternative splicing program in a cell.
RNA editing
RNA editing, defined as the post-transcriptional modification of RNA molecules and distinct from the events of RNA splicing, capping, polyadenylation, or degradation, is a widespread phenomenon and occurs in species as diverse as protozoa, plants, and mammals (for review see ref.(27–32)). Several unrelated mechanisms of editing exist, including the deletion or insertion of nucleotides found in mitochondria of kinetoplastid protozoa and slime molds,(32) as well as base substitutions found both in plant organelles and the nucleus of complex eukaryotes (for review see ref.(33)). Whereas insertion/deletion editing of mRNAs is a guide RNA-mediated process involving the cutting and ligating of the RNA, the most prominent types of substitution editing proceed through base-modification reactions, such as the deamination of cytosine or adenosine residues.(33)
One type of base-modification RNA editing (termed A-to-I RNA editing) has received particular attention with respect to highly complex eukaryotes, including humans. As outlined below, A-to-I editing may be particularly relevant for generating genetic variability as a basis for adaptive evolution. A-to-I RNA editing involves the specific modification of single adenosine nucleotides in RNA molecules to inosine via hydrolytic deamination.(28,30,31) The resulting inosine is interpreted as guanosine by the translational machinery. A-to-I editing within the protein-coding region of an mRNA can therefore result in codon alterations that lead to an amino acid substitution in the protein product(27) (see Fig. 1A). Usually, the edited and unedited proteins are expressed within the same cell and RNA editing levels at a specific site may range from a few to almost 100% (for review see ref.(27)). For physiological targets of mammalian A-to-I RNA editing, the observed recoding at a single site often results in a protein variant with an altered function. In fact, editing often affects highly conserved amino acid residues that constitute molecular determinants for protein function.(27,34)
Figure 1.
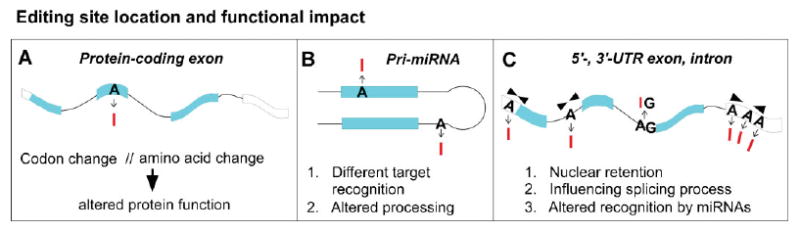
Consequences of A-to-I RNA editing. Location of RNA editing events in pre-mRNA transcripts and known functional implications. A: Editing within the open reading frame of a protein-coding transcript. RNA editing changes a codon, which in turn leads to a protein with amino acid substitution and altered functional properties. Examples: neurotransmitter receptors GluR-2, GluR-5, GluR-6, 5HT2C serotonin receptor, GABA(A) receptor, K(v)1.1 potassium channel, etc. (see ref.(27) for review). B: Editing in pri-miRNA transcripts influencing miRNA biogenesis or target spectrum (see ref.(27) for review). C: RNA editing in mRNA untranslated exons and intronic sequences with impact on RNA fate (transport, splicing) and regulation. The influence on splicing is exemplified through a case where a consensus splice-acceptor site (AG) is destroyed through editing yielding IG (see ref.(27) for review).
Among prominent mammalian editing substrates are various brain-specific ion channels and neurotransmitter receptor genes where single or multiple recoding events generate a spectrum of protein variants with distinct functional properties.(27,35,36) For example, editing within the coding region of the serotonin receptor 5-HT2C transcript occurs at up to five major positions and regulates the sensitivity of the receptor to serotonin.(37) Varying levels of editing at the five positions can result in 25 different gene transcripts, which all arise from a single genomically encoded sequence. In one extreme example of editing-induced functional modulation, a single site-specific editing event within glutamate receptor subunit GluR-2, altering a glutamine (Q) to an arginine (R) codon, determines ion permeability(38) as well as receptor channel transport and assembly(39,40) and, as a result, turned out to be essential for normal brain function.(41) The deficiency or misregulation of A-to-I editing has been implicated in the etiology of neurological diseases, such as epilepsy, amyotrophic lateral sclerosis, as well as depression.(42)
The high specificity of the RNA editing reactions involving physiological substrates is achieved largely through the overall three-dimensional structures of the imperfectly base-paired pre-mRNAs. Double-stranded RNA structures within the substrate molecule are recognized by the adenosine deaminase acting on RNA (ADAR) family of enzymes.(28) Through the characterization of known editing sites, certain 5′ and 3′ base preferences have emerged that increase or decrease the likelihood of a particular adenosine to become edited.(27,28) However, the exact mechanism for the selectivity of ADARs is poorly understood and, despite some progress in computational delineation of editing sites,(43–47) to date it is not possible to predict which adenosine within a given RNA will undergo editing.
Small, single nucleotide changes within an RNA editing structure (caused by heritable point mutations) can drastically alter editing extent or even result in selection of a different editing site.(34) Since such base changes may at the same time also directly alter protein-coding sequences or otherwise influence RNA fate (stability, processing, translation), exonic gene sequences are under multiple constraints. In contrast, the intronic sequence regions that are frequently an essential component of the editing-competent RNA-fold, are usually less restricted.
Thus, the evolution of RNA editing sites involves heritable genetic variation in the form of genetic changes in the genome, which is a critical prerequisite for this process to play a role in adaptation. However, any phenotypic variation is expressed only indirectly on the epigenetic level in form of RNA modification events that alter transcriptome and proteome composition.
Prevalence of RNA editing
To support our hypothesis that RNA editing serves a general evolutionary role, it should also be of widespread prevalence within the system. It is currently not known how many protein-coding sequences in mammals are subject to A-to-I RNA editing. Although only about two dozen recoding sites are known,(27) it is estimated that many more exist.
Besides editing of protein-coding sequences (see Fig. 1A), A-to-I alterations also modify other regions of pre-mRNAs and noncoding RNAs, modulating RNA fate (see Fig. 1B and C). For example, editing of intronic adenosines can create novel splice consensus sites or destroy existing ones,(48–50) and editing in untranslated exons can cause retention of the edited substrate within the nucleus(51,52) (Fig. 1C). In addition, several groups have recently shown that a large number of miRNA transcripts are also subject to A-to-I editing (for review see ref.(27)), which may cause changes in miRNA biogenesis(53) or in the retargeting of mature miRNAs(54) (see Fig. 1B). The partially double-stranded nature of miRNA precursor molecules makes them especially prone to modification by ADARs. Currently, up to 16% of miRNA transcripts are believed to undergo A-to-I editing.(53)
Recently, we and others discovered widespread, high-level A-to-I RNA editing in Alu-type repeat elements (for review see ref.(55)), suggesting that based on this group of prevalent transposon-derived sequences alone, a minimum of 85% of all pre-mRNAs in primates undergo A-to-I editing in at least one position.(48) Interestingly, in transcriptomes of other mammals the intensity of adenosine deamination editing is an order of magnitude smaller compared to that in primates due to the lack of Alu repeats.(55) The mechanistic explanation for the massive base modification in primate-specific Alu sequences springs from the basic properties of the editing machinery to target double-stranded RNA structures for modification. Alu-repeat elements are present in either forward or reverse orientation within the genomes of primates with a frequency of about 12 repeats per average human gene.(48,56) If two oppositely orientated Alu elements are both transcribed within the same RNA molecule, they can fold back onto each other forming an excellent substrate for ADAR editing enzymes.
RNA editing is a highly regulated process and editing levels have been shown to change during development.(46,57) Furthermore, editing is often under cell-type or tissue-specific control.(58,59) Intriguingly, the editing extent of a particular substrate can also be influenced by environmental alterations. For example, RNA editing at multiple sites within the mammalian 5-HT2C serotonin receptor gene changed when rodents were fed with a serotonin uptake inhibitor (fluoxetine).(60,61) It is believed that this change was brought about by the increased effective levels of serotonin in the brain of these rodents. Since it is known that 5-HT2C editing reduces the responsiveness of the serotonin receptor to the neurotransmitter, the alteration of editing levels in fluoxetine-treated rodents can be interpreted as a compensatory mechanism to dynamically adapt to alterations in synaptic serotonin levels. Intriguingly, in human patients suffering from suicidal depression, which is believed to be accompanied by a reduction in serotonin levels, editing of the 5HT2C receptor was also changed. Taken together, these findings indicate that regulation of RNA editing levels can be part of a short-term adaptive response.(60)
When it comes to long-term adaptation based on heritable genetic variation, evolutionary models to assess positive and negative selection of alleles usually include only non-synonymous base changes within the protein-coding sequences of a given gene.(62) This means that amino acid changes brought about by RNA editing will be overlooked, because novel editing events may be induced by synonymous sequence changes in translated exons or non-coding sequences. Therefore, current efforts to detect patterns of selection at the molecular level based on these traditional evolutionary models may be incomplete or inaccurate.
Although adaptive variation can arise as a novel mutation that is strongly favored immediately, most mutations will initially be neutral, and thus likely lost due to drift, or selectively disfavored and removed from the population via purifying selection. Some subset of these mutations, however, may become beneficial under certain environmental conditions (e.g., in novel habitats resulting from range expansion).(63) In the latter case, the mutations will have been present in the ancestral population before they became adaptive (see discussion of cryptic variation above).
In contrast to variation introduced through mutation, new variants created by RNA editing come at low evolutionary cost, because initially the wild-type product is still present at a high level. Therefore, the new variant can be tested out, often with little cost to organismal fitness. It also can persist longer because selection pressure is small to eliminate it. Therefore, there is a higher chance that a variant produced by RNA editing, and beneficial in a changed environment is present when that change occurs and new selection pressures arise. This model parallels that postulated for alternatively spliced exons(23) and may be mirrored by other epigenetic mechanisms of variation, such an alternative gene promoters and alternative poly(A) processing.
Variability through RNA editing
As outlined above, the post-transcriptional nature of A-to-I RNA editing allows for the variation of amino acid residues that are not accessible for direct mutational changes in the genomic sequence because of their functional importance (Fig. 2A).
Figure 2.
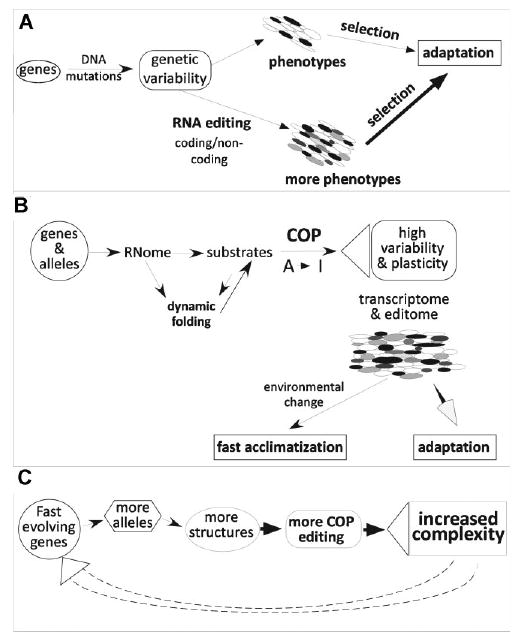
RNA editing and adaptation. A: Schematic describing the basic mechanism for increased genetic variability through post-transcriptional RNA editing. B: Continuous probing (COP) hypothesis. Dynamic folding of RNome causes complex pattern of RNA editing in individual cells through continued probing of RNA structures by the editing machinery. The high variability and plasticity within the phenome allows for fast acclimation after environmental change and facilitates adaptive evolution. C: Feed forward regulation of complexity-generating mechanisms. Genes that evolve faster than others generate a larger number of alleles in the population, which leads to an increased diversity in COP editing due to the larger number of alternative RNA-folds. The resulting increased complexity of the system further feeds into the generation of novel alleles in the population.
A key element of our model implicating RNA editing in adaptive evolution is that novel RNA editing sites emerge continuously. The basis for this assumption is the current understanding regarding the dynamics of RNA folding and the known properties of the RNA editing machinery. The key feature for an adenosine to become targeted for editing is a partially double-stranded foldback structure that provides a high-affinity binding site for the editing enzyme and positions the enzyme to edit a nearby adenosine.(31) No specific primary RNA sequence is required, but the flanking nucleotides to an editing site may modulate the extent of editing.
RNA folding is a highly dynamic process and under physiological conditions a single pre-mRNA molecule (with an average size of ∼30kb in human) will assume many alternative secondary structures, all in equilibrium with the other possible folds, and further modulated by RNA–protein interactions.(64,65) We have shown previously that, in vivo, a single RNA with several computer-predicted secondary structures, dominated by pairs of Alu-repeat elements, all exist and that the level of editing within each of the Alu sequences can be used to estimate the relative prevalence of each alternative structure within the cell.(48)
We expect that many of the transient RNA secondary structures will sometimes persist long enough to result in an editing event. With thousands of alternative structures, this means that through continuous probing (COP) of pre-mRNAs by the editing machinery there will probably be low-level editing at many sites. Only sites that are part of a dominant structure will give rise to prominent, high-level editing, such as in Alu repeats or one of the well-characterized mammalian recoding editing sites.
The low-level background editing due to targeting of minor structures within the dynamic equilibrium of pre-mRNA-folds will then produce an extensive array of novel transcript variants, albeit at a low level (see Fig. 2B). These edited RNA molecules can result in a low fraction of proteins with an altered function. The vast majority of these editing events are not expected to have any beneficial effects under normal conditions, but at the same time, they are produced with a low cost on fitness as outlined above. However, when the environment changes such spurious amounts of edited transcripts might become important. Proteins translated from edited RNAs could fulfill a function that is critical in the changed environment, resulting in an instant acclimatization and survival advantage for the organism, and lead to adaptation. Therefore, the cryptic variation generated by editing represents a feature that enhances evolvability. Although driven by small genomic changes, the phenotypic variability results indirectly from an epigenetic mechanism (A-to-I editing). In contrast, genomic mutations can directly produce new alleles only in an all-or-nothing fashion and have therefore a much smaller likelihood of producing a pre-adapted (cryptic) property that could be used during fast acclimatization (see Fig. 2B).
A change introduced through COP background editing can be limited to the alteration of the function of a single protein. However, if this protein has an important role, e.g., in the function or expression of other proteins, a single editing event can have a wide impact. Furthermore, background editing is not expected to be limited to the alteration of the coding sequence, but will also occur in regulatory RNAs, such as miRNAs. Editing within regulatory RNAs can consequently cause a broad effect on the expression of proteins; miRNAs, for example generally regulate the expression of multiple genes. In these instances, the effect of a single editing event can substantially alter proteome composition.
How can the COP hypothesis be tested?
The majority of the edited RNA molecules that arise through COP are most likely not detected due to the limitations of the currently employed techniques for the analysis of editing. A-to-I editing is predominantly investigated by gene-specific RT-PCR of the RNA substrate followed by sequencing, in which the edited position appears as guanosine in the sequencing reaction, while at the same genomic position an adenosine is specified. At editing levels below 100% efficiency, overlapping A and G signals can be observed at that position. However, if the editing occurs in only about 5% or less of all the transcriptsof a gene, this methodology is not sensitive enough. The COP background editing by definition leads to modification rates lower than a few percent and will therefore not be detectable using these conventional techniques.
A potential solution for the analysis of low-level editing is the subcloning of gene-specific amplicons followed by isolation and sequencing of a high number of individual templates that each correspond to a single cDNA molecule (after amplification). One drawback of this approach is that it is very laborious and time consuming. Furthermore, this strategy introduces additional amplification steps (via secondary PCR and within bacteria during cloning) that increase the overall error rate of the analysis and therefore reduces the signal-to-noise ratio.
An accurate and high-throughput solution to these technical hurdles may be provided by next-generation sequencing technologies. For example, the 454 KS GLS platform (Roche) with average read lengths of ∼250 bp is well suited for this purpose.(66,67) With a single sequencing run, several hundred gene-specific PCR amplicons together with several hundred genomic control fragments can be analyzed obtaining coverage of about 1,000 reads per cDNA fragment.(66,67) This would allow for the detection of editing events with a sub-percentage penetrance. The 454 direct sequencing approach is also characterized by a smaller intrinsic error rate than conventional Sanger-based sequencing technology. (66,67)
Two studies that apply next-generation sequencing technology to the investigation of RNA editing have recently been published.(68,69) However, neither study was designed with something like the COP hypothesis in mind and, therefore, did not analyze with sufficient resolution potential background editing. Rather, they either focused on several known high-level editing sites,(69) or single A/G discrepancy positions without flanking sequence information.(68) Therefore, it is not possible to prove or disprove the COP hypothesis based on currently available data. Nevertheless, it is notable that, based on the so far most comprehensive sampling of adenosines through high-throughput sequencing,(68) it can be concluded that most RNA editing sites show in vivo editing levels of below 2%, whereas only few additional bona fide editing sites with a higher editing rate were identified.(68) This trend may indicate that, as predicted by the COP hypothesis, modification sites with low levels of editing far outnumber high percentage editing events.
In humans, more than 6.5 million validated SNPs are currently annotated in the SNP database(17) and many more sites with low penetrance exist. This means that no two genomes are identical with respect to the nature of all SNP positions. As a result, the secondary RNA structures of most primary transcripts will also be slightly different between individuals including those double-stranded structures that give rise to RNA editing. Therefore, RNA editing patterns and levels will also vary between individuals. This intriguing conclusion can be tested experimentally by comparing the background editing patterns between different family members and individuals with larger genetic distance. Closely related family members will share a large number of SNPs, making it more likely to have a similar set of background-edited RNAs. Due to this variation in SNP positions, it is expected that between individuals of the same species a different set of RNA transcripts or different positions within the same RNA will be edited leading to a transcriptome-wide editing signature that differs between individuals and may even be unique in every cell within an organism at a particular time point. Interestingly, it has been shown that RNA editing, even of prominent recoding targets, differ between mice of distinct genetic background.(70)
How can RNA editing be beneficial for long-term adaptation over generations, like DNA mutations? Organisms with a certain set of SNPs may exhibit an editing event that leads to a survival advantage compared to other individuals of the same species. If this beneficial effect persists over an extended period of time, this SNP becomes more prevalent (positive selection). Subsequent genomic mutations can result in a further increase in editing efficiency, without losing the un-edited version (which still might serve a function as well) up to an equilibrium that is optimal for fitness.
RNA editing and brain complexity
The notion of a person-specific or even cell-specific pattern of RNA editing is particularly interesting regarding the mammalian central nervous system, where the activity of the RNA editing machinery is especially high.(71) The brain is considered the most complex organ and also accomplishes such complex tasks as learning and memory. The molecular mechanisms of memory formation and long-term potentiation are not well understood.(72) We may speculate that the extraordinary diversity of the transcriptome in the brain generated through RNA editing could constitute a memory-forming process if dynamic changes in the microenvironment of single cells alter intracellular editing levels. Synaptic signals represent such changes in local environment and the ensuing change in editing pattern may constitute a form of cellular memory mirroring its input and activation history.
Most of the known recoding editing events in mammals occur in brain-specific genes that have gone through multiple gene duplication events generating sequence-related sub-units and whose primary transcripts are also subject to a high degree of alternative splicing.(73) Intriguingly, our COP hypothesis predicts such a behavior. Genes that evolve faster, which means that more distinct alleles exist in the population, will also display an increased complexity of editing variants due to the higher number of polymorphisms in the population. As a result, they may evolve even faster (see Fig. 2C).
Comparison of the human and chimpanzee genomes revealed that the primate brain did not evolve through addition of novel genes, but rather was associated with changes in gene expression.(74) The human lineage is characterized by accelerated gene expression changes in the brain compared with the chimpanzee lineage.(75) A recent study that investigated 200 brain-expressed genes analyzing the KA/KS ratios between mammalian species concluded that genes involved in brain development have a higher tendency to be under positive selection between macaques and humans than between mice and rats.(76,77) Thus, genetic plus epigenetic mechanisms create extraordinary variability allowing the organism to explore a maximum of new variations.
Concluding remarks
RNA editing may be a principal contributor to the evolution of phenotypic complexity in mammals and particularly to the complexity of the brain. It has yet to be shown exactly how much variability is generated by editing in vivo. If recent insights from the analysis of alternative splicing are any indication, then the increased sensitivity of transcriptome analysis through the use of deep sequencing technology, as suggested above, will likely reveal large numbers of low-level editing sites. For example, deep sequencing of human cells showed that at least 95% of human multiexon genes undergo at least one alternative splicing event, up from previous estimates of 60–70%.(26) Furthermore, a substantial fraction of alternative splicing events displayed individual-specific variation (between 10 and 30% of all events).(26) The latter is believed to be due to polymorphisms that alter splicing dynamics and outcome. We would speculate that the variation due to RNA editing strongly contributes to these observed person-to-person differences.
With the widespread prevalence of A-to-I editing, the functional variation generated through editing and the fact that editing position and extents evolve through small inheritable changes in genomic sequences, A-to-I editing exhibits key properties to support a general evolutionary role. The limitation of genetic variability introduced by RNA editing lies in the overall balance between the beneficial impact of editing events and the accumulation of non-beneficial variants. It has been shown that organisms with higher complexity tend to be more robust due to increased functional redundancy. Furthermore, experimental and theoretical evidence suggests that in complex systems, genes act as capacitators that buffer, or canalize genotypic variation under normal conditions.(12,78) Therefore, as long as the editing-induced variability is still buffered sufficiently by the inherent robustness of the complex, dynamic system, additional editing sites would be allowable. This could suggest that, in principal, more complex organisms could allow a higher prevalence of editing and still remain stable. At the same time, RNA editing further enhances the complexity of the system. This means that overall, RNA editing may be subject to a feed-forward regulation (Fig. 2C) where more editing within an organism opens up the sequence space for more variability, which may be achieved through additional editing sites further increasing complexity.
Acknowledgments
Research in the laboratory of S. M. is supported by the National Institutes of Health (grant number NS057739).
References
- 1.Nosil P, Mooers AO. Testing hypotheses about ecological specialization using phylogenetic trees. Evolution. 2005;59:2256–2263. [PubMed] [Google Scholar]
- 2.Hoekstra HE, Coyne JA. The locus of evolution: evo devo and the genetics of adaptation. Evolution. 2007;61:995–1016. doi: 10.1111/j.1558-5646.2007.00105.x. [DOI] [PubMed] [Google Scholar]
- 3.Hoekstra HE, Hirschmann RJ, Bundey RA, Insel PA, Crossland JP. A single amino acid mutation contributes to adaptive beach mouse color pattern. Science. 2006;313:101–104. doi: 10.1126/science.1126121. [DOI] [PubMed] [Google Scholar]
- 4.Stinchcombe JR, Hoekstra HE. Combining population genomics and quantitative genetics: finding the genes underlying ecologically important traits. Heredity. 2008;100:158–170. doi: 10.1038/sj.hdy.6800937. [DOI] [PubMed] [Google Scholar]
- 5.Colosimo PF, Hosemann KE, Balabhadra S, Villarreal G, Jr, Dickson M, et al. Widespread parallel evolution in sticklebacks by repeated fixation of ectodysplasin alleles. Science. 2005;307:1928–1933. doi: 10.1126/science.1107239. [DOI] [PubMed] [Google Scholar]
- 6.Greenberg AJ, Moran JR, Coyne JA, Wu CI. Ecological adaptation during incipient speciation revealed by precise gene replacement. Science. 2003;302:1754–1757. doi: 10.1126/science.1090432. [DOI] [PubMed] [Google Scholar]
- 7.Presgraves DC, Balagopalan L, Abmayr SM, Orr HA. Adaptive evolution drives divergence of a hybrid inviability gene between two species of Drosophila. Nature. 2003;423:715–719. doi: 10.1038/nature01679. [DOI] [PubMed] [Google Scholar]
- 8.Fisher RA. The genetical theory of natural selection. Oxford: Clarendon; 1930. [Google Scholar]
- 9.Lynch M, Walsh B. Genetics and analysis of quantitative traits. Sunderland, MA: Sinauer Associates; 1998. [Google Scholar]
- 10.Barton NH, Turelli M. Evolutionary quantitative genetics: how little do we know? Annu Rev Genet. 1989;23:337–370. doi: 10.1146/annurev.ge.23.120189.002005. [DOI] [PubMed] [Google Scholar]
- 11.Futuyma DJ. Evolutionary biology. Sunderland, MA: Sinauer Associates; 1998. [Google Scholar]
- 12.Pigliucci M. Is evolvability evolvable? Nat Rev Genet. 2008;9:75–82. doi: 10.1038/nrg2278. [DOI] [PubMed] [Google Scholar]
- 13.Wagner GP, Alternberg L. Complex adaptations and the evolution of evolvability. Evolution. 1996;50:967–976. doi: 10.1111/j.1558-5646.1996.tb02339.x. [DOI] [PubMed] [Google Scholar]
- 14.West-Eberhard MJ. Developmental plasticity and evolution. New York: Oxford University Press; 2003. [Google Scholar]
- 15.Rosenberg SM, Hastings PJ. Microbiology and evolution. Modulating mutation rates in the wild. Science. 2003;300:1382–1383. doi: 10.1126/science.1085691. [DOI] [PubMed] [Google Scholar]
- 16.Dobzhansky T. Genetics and the origin of species. New York: Columbio University Press; 1937. [Google Scholar]
- 17.Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taylor JG, Choi EH, Foster CB, Chanock SJ. Using genetic variation to study human disease. Trends Mol Med. 2001;7:507–512. doi: 10.1016/s1471-4914(01)02183-9. [DOI] [PubMed] [Google Scholar]
- 19.Hunter P. The great leap forward. Major evolutionary jumps might be caused by changes in gene regulation rather than the emergence of new genes. EMBO Rep. 2008;9:608–611. doi: 10.1038/embor.2008.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Golding I, Paulsson J, Zawilski SM, Cox EC. Real-time kinetics of gene activity in individual bacteria. Cell. 2005;123:1025–1036. doi: 10.1016/j.cell.2005.09.031. [DOI] [PubMed] [Google Scholar]
- 21.Yu J, Xiao J, Ren X, Lao K, Xie XS. Probing gene expression in live cells, one protein molecule at a time. Science. 2006;311:1600–1603. doi: 10.1126/science.1119623. [DOI] [PubMed] [Google Scholar]
- 22.Lopez-Maury L, Marguerat S, Bahler J. Tuning gene expression to changing environments: from rapid responses to evolutionary adaptation. Nat Rev Genet. 2008;9:583–593. doi: 10.1038/nrg2398. [DOI] [PubMed] [Google Scholar]
- 23.Modrek B, Lee CJ. Alternative splicing in the human, mouse and rat genomes is associated with an increased frequency of exon creation and/or loss. Nat Genet. 2003;34:177–180. doi: 10.1038/ng1159. [DOI] [PubMed] [Google Scholar]
- 24.Goldsmith M, Tawfik DS. Potential role of phenotypic mutations in the evolution of protein expression and stability. Proc Natl Acad Sci U S A. 2009;106:6197–6202. doi: 10.1073/pnas.0809506106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gimelbrant A, Hutchinson JN, Thompson BR, Chess A. Widespread monoallelic expression on human autosomes. Science. 2007;318:1136–1140. doi: 10.1126/science.1148910. [DOI] [PubMed] [Google Scholar]
- 26.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gommans WM, Dupuis DE, McCane JE, Tatalias NE, Maas S. Diversifying exon code through A-to-I RNA editing. In: Smith H, editor. DNA RNA editing. Wiley & Sons, Inc.; Hoboken, NJ: 2008. pp. 3–30. [Google Scholar]
- 28.Bass BL. RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem. 2002;71:817–846. doi: 10.1146/annurev.biochem.71.110601.135501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Conticello SG. The AID/APOBEC family of nucleic acid mutators. Genome Biol. 2008;9:229. doi: 10.1186/gb-2008-9-6-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoopengardner B. Adenosine-to-inosine RNA editing: perspectives and predictions. Mini Rev Med Chem. 2006;6:1213–1216. doi: 10.2174/138955706778742812. [DOI] [PubMed] [Google Scholar]
- 31.Maydanovych O, Beal PA. Breaking the central dogma by RNA editing. Chem Rev. 2006;106:3397–3411. doi: 10.1021/cr050314a. [DOI] [PubMed] [Google Scholar]
- 32.Stuart KD, Schnaufer A, Ernst NL, Panigrahi AK. Complex management: RNA editing in trypanosomes. Trends Biochem Sci. 2005;30:97–105. doi: 10.1016/j.tibs.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 33.Gott JM. Expanding genome capacity via RNA editing. C R Biol. 2003;326:901–908. doi: 10.1016/j.crvi.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 34.Reenan RA. Molecular determinants and guided evolution of species-specific RNA editing. Nature. 2005;434:409–413. doi: 10.1038/nature03364. [DOI] [PubMed] [Google Scholar]
- 35.Jepson JE, Reenan RA. RNA editing in regulating gene expression in the brain. Biochim Biophys Acta. 2008;1779:459–470. doi: 10.1016/j.bbagrm.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 36.Seeburg PH, Hartner J. Regulation of ion channel/neurotransmitter receptor function by RNA editing. Curr Opin Neurobiol. 2003;13:279–283. doi: 10.1016/s0959-4388(03)00062-x. [DOI] [PubMed] [Google Scholar]
- 37.Burns CM, Chu H, Rueter SM, Hutchinson LK, Canton H, et al. Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature. 1997;387:303–308. doi: 10.1038/387303a0. [DOI] [PubMed] [Google Scholar]
- 38.Sommer B, Kohler M, Sprengel R, Seeburg PH. RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell. 1991;67:11–19. doi: 10.1016/0092-8674(91)90568-j. [DOI] [PubMed] [Google Scholar]
- 39.Greger IH, Akamine P, Khatri L, Ziff EB. Developmentally regulated, combinatorial RNA processing modulates AMPA receptor biogenesis. Neuron. 2006;51:85–97. doi: 10.1016/j.neuron.2006.05.020. [DOI] [PubMed] [Google Scholar]
- 40.Greger IH, Khatri L, Kong X, Ziff EB. AMPA receptor tetramerization is mediated by Q/R editing. Neuron. 2003;40:763–774. doi: 10.1016/s0896-6273(03)00668-8. [DOI] [PubMed] [Google Scholar]
- 41.Higuchi M, Maas S, Single FN, Hartner J, Rozov A, et al. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature. 2000;406:78–81. doi: 10.1038/35017558. [DOI] [PubMed] [Google Scholar]
- 42.Maas S, Kawahara Y, Tamburro KM, Nishikura K. A-to-I RNA editing and human disease. RNA Biol. 2006:3. doi: 10.4161/rna.3.1.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Clutterbuck DR, Leroy A, O'Connell MA, Semple CA. A bioinformatic screen for novel A-I RNA editing sites reveals re-coding editing in BC10. Bioinformatics. 2005;21:2590–2595. doi: 10.1093/bioinformatics/bti411. [DOI] [PubMed] [Google Scholar]
- 44.Gommans WM, Tatalias NE, Sie C, Dupuis DE, Vendetti NJ, et al. Screening of human SNP database identifies recoding sites of A-to-I RNA editing. RNA. 2008;14:2074–2085. doi: 10.1261/rna.816908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Levanon EY, Hallegger M, Kinar Y, Shemesh R, Djinovic-Carugo K, et al. Evolutionarily conserved human targets of adenosine to inosine RNA editing. Nucleic Acids Res. 2005;33:1162–1168. doi: 10.1093/nar/gki239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ohlson J, Pedersen JS, Haussler D, Ohman M. Editing modifies the GABA(A) receptor subunit alpha3. RNA. 2007;13:698–703. doi: 10.1261/rna.349107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sixsmith J, Reenan RA. Comparative genomic and bioinformatic approaches for the identification of new adenosine-to-inosine substrates. Meth Enzymol. 2007;424:245–264. doi: 10.1016/S0076-6879(07)24011-X. [DOI] [PubMed] [Google Scholar]
- 48.Athanasiadis A, Rich A, Maas S. Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol. 2004;2:e391. doi: 10.1371/journal.pbio.0020391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lev-Maor G, Sorek R, Levanon EY, Paz N, Eisenberg E, et al. RNA-editing-mediated exon evolution. Genome Biol. 2007;8:R29. doi: 10.1186/gb-2007-8-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rueter SM, Dawson TR, Emeson RB. Regulation of alternative splicing by RNA editing. Nature. 1999;399:75–80. doi: 10.1038/19992. [DOI] [PubMed] [Google Scholar]
- 51.Chen LL, DeCerbo JN, Carmichael GG. Alu element-mediated gene silencing. EMBO J. 2008;27:1694–1705. doi: 10.1038/emboj.2008.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Prasanth KV, Prasanth SG, Xuan Z, Hearn S, Freier SM, et al. Regulating gene expression through RNA nuclear retention. Cell. 2005;123:249–263. doi: 10.1016/j.cell.2005.08.033. [DOI] [PubMed] [Google Scholar]
- 53.Kawahara Y, Megraw M, Kreider E, Iizasa H, Valente L, et al. Frequency and fate of microRNA editing in human brain. Nucleic Acids Res. 2008;36:5270–5280. doi: 10.1093/nar/gkn479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kawahara Y, Zinshteyn B, Sethupathy P, Iizasa H, Hatzigeorgiou AG, et al. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science. 2007;315:1137–1140. doi: 10.1126/science.1138050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eisenberg E, Levanon EY. A-To-I editing of ALU repeats. In: Smith H, editor. RNA and DNA editing: molecular mechanisms and their integration into biological systems. Wiley & Sons, Inc.; Hoboken, NJ: 2008. pp. 257–279. [Google Scholar]
- 56.Grover D, Majumder PP, Rao CB, Brahmachari SK, Mukerji M. Nonrandom distribution of alu elements in genes of various functional categories: Insight from analysis of human chromosomes 21 and 22. Mol Biol Evol. 2003;20:1420–1424. doi: 10.1093/molbev/msg153. [DOI] [PubMed] [Google Scholar]
- 57.Lomeli H, Mosbacher J, Melcher T, Hoger T, Geiger JR, et al. Control of kinetic properties of AMPA receptor channels by nuclear RNA editing. Science. 1994;266:1709–1713. doi: 10.1126/science.7992055. [DOI] [PubMed] [Google Scholar]
- 58.Barbon A, Vallini I, La Via L, Marchina E, Barlati S. Glutamate receptor RNA editing: a molecular analysis of GluR2, GluR5 and GluR6 in human brain tissues and in NT2 cells following in vitro neural differentiation. Brain Res Mol Brain Res. 2003;117:168–178. doi: 10.1016/s0169-328x(03)00317-6. [DOI] [PubMed] [Google Scholar]
- 59.Geiger JR, Melcher T, Koh DS, Sakmann B, Seeburg PH, et al. Relative abundance of subunit mRNAs determines gating and Ca2+ permeability of AMPA receptors in principal neurons and interneurons in rat CNS. Neuron. 1995;15:193–204. doi: 10.1016/0896-6273(95)90076-4. [DOI] [PubMed] [Google Scholar]
- 60.Gurevich I, Tamir H, Arango V, Dwork AJ, Mann JJ, et al. Altered editing of serotonin 2C receptor pre-mRNA in the prefrontal cortex of depressed suicide victims. Neuron. 2002;34:349–356. doi: 10.1016/s0896-6273(02)00660-8. [DOI] [PubMed] [Google Scholar]
- 61.Iwamoto K, Nakatani N, Bundo M, Yoshikawa T, Kato T. Altered RNA editing of serotonin 2C receptor in a rat model of depression. Neurosci Res. 2005;53:69–76. doi: 10.1016/j.neures.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 62.Goldman N, Yang Z. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol Biol Evol. 1994;11:725–736. doi: 10.1093/oxfordjournals.molbev.a040153. [DOI] [PubMed] [Google Scholar]
- 63.Gonzalez J, Lenkov K, Lipatov M, Macpherson JM, Petrov DA. High rate of recent transposable element-induced adaptation in Drosophila melanogaster. PLoS Biol. 2008;6:e251. doi: 10.1371/journal.pbio.0060251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schroeder R, Barta A, Semrad K. Strategies for RNA folding and assembly. Nat Rev Mol Cell Biol. 2004;5:908–919. doi: 10.1038/nrm1497. [DOI] [PubMed] [Google Scholar]
- 65.Shcherbakova I, Mitra S, Laederach A, Brenowitz M. Energy barriers, pathways, and dynamics during folding of large, multidomain RNAs. Curr Opin Chem Biol. 2008;12:655–666. doi: 10.1016/j.cbpa.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 67.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li JB, Levanon EY, Yoon JK, Aach J, Xie B, et al. Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science. 2009;324:1210–1213. doi: 10.1126/science.1170995. [DOI] [PubMed] [Google Scholar]
- 69.Wahlstedt H, Daniel C, Enstero M, Ohman M. Large-scale mRNA sequencing determines global regulation of RNA editing during brain development. Genome Res. 2009;19:978–986. doi: 10.1101/gr.089409.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Du Y, Davisson MT, Kafadar K, Gardiner K. A-to-I pre-mRNA editing of the serotonin 2C receptor: comparisons among inbred mouse strains. Gene. 2006;382:39–46. doi: 10.1016/j.gene.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 71.Paul MS, Bass BL. Inosine exists in mRNA at tissue-specific levels and is most abundant in brain mRNA. EMBO J. 1998;17:1120–1127. doi: 10.1093/emboj/17.4.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Neves G, Cooke SF, Bliss TV. Synaptic plasticity, memory and the hippocampus: a neural network approach to causality. Nat Rev Neurosci. 2008;9:65–75. doi: 10.1038/nrn2303. [DOI] [PubMed] [Google Scholar]
- 73.Burnashev N, Rozov A. Genomic control of receptor function. Cell Mol Life Sci. 2000;57:1499–1507. doi: 10.1007/PL00000634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hill RS, Walsh CA. Molecular insights into human brain evolution. Nature. 2005;437:64–67. doi: 10.1038/nature04103. [DOI] [PubMed] [Google Scholar]
- 75.Enard W, Khaitovich P, Klose J, Zollner S, Heissig F, et al. Intra- and interspecific variation in primate gene expression patterns. Science. 2002;296:340–343. doi: 10.1126/science.1068996. [DOI] [PubMed] [Google Scholar]
- 76.Consortium CSaA. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature. 2005;437:69–87. doi: 10.1038/nature04072. [DOI] [PubMed] [Google Scholar]
- 77.Dorus S, Vallender EJ, Evans PD, Anderson JR, Gilbert SL, et al. Accelerated evolution of nervous system genes in the origin of Homo sapiens. Cell. 2004;119:1027–1040. doi: 10.1016/j.cell.2004.11.040. [DOI] [PubMed] [Google Scholar]
- 78.Bergman A, Siegal ML. Evolutionary capacitance as a general feature of complex gene networks. Nature. 2003;424:549–552. doi: 10.1038/nature01765. [DOI] [PubMed] [Google Scholar]