

Abstract
 An efficient Cu-catalyzed protocol for enantioselective addition of a dimethylphenylsilanyl group to a wide range of cyclic and acyclic unsaturated ketones, esters, acrylonitriles and α,β,γ,δ-dienones is disclosed. Reactions are performed in the presence of 1–2 mol % of commercially available and inexpensive CuCl, a readily accessible monodentate imidazolinium salt as well as commercially available (dimethylphenylsilyl)pinacolatoboron. Cu-catalyzed enantioselective conjugate additions proceed to completion within only two hours to afford the desired silanes in 87–97% yield and 90:10–99:1 enantiomeric ratio (er). Use of a proton source (e.g., MeOH) is not required; accordingly, synthetically versatile α-silyl boron enolates can be obtained. The special utility of the present protocol, in comparison with the related catalytic enantioselective aldol and boronate conjugate additions, are discussed and illustrated through various functionalizations of the enantiomerically enriched β-silylcarbonyls.
An efficient Cu-catalyzed protocol for enantioselective addition of a dimethylphenylsilanyl group to a wide range of cyclic and acyclic unsaturated ketones, esters, acrylonitriles and α,β,γ,δ-dienones is disclosed. Reactions are performed in the presence of 1–2 mol % of commercially available and inexpensive CuCl, a readily accessible monodentate imidazolinium salt as well as commercially available (dimethylphenylsilyl)pinacolatoboron. Cu-catalyzed enantioselective conjugate additions proceed to completion within only two hours to afford the desired silanes in 87–97% yield and 90:10–99:1 enantiomeric ratio (er). Use of a proton source (e.g., MeOH) is not required; accordingly, synthetically versatile α-silyl boron enolates can be obtained. The special utility of the present protocol, in comparison with the related catalytic enantioselective aldol and boronate conjugate additions, are discussed and illustrated through various functionalizations of the enantiomerically enriched β-silylcarbonyls.
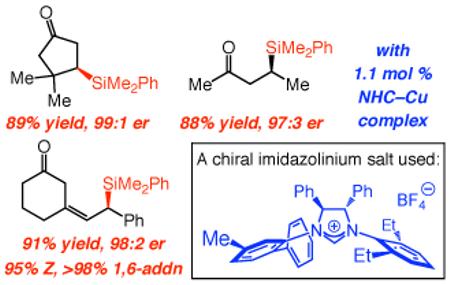
Development of practical and efficient methods for catalytic enantioselective formation of C–Si bonds is an important and challenging goal of research in chemical synthesis;1 transformations delivering β-silylcarbonyls are particularly attractive. A Si-based substituent, among other functions, serves as a masked hydroxyl group; it is sufficiently robust to allow for a range of functionalization processes that involve the carbonyl unit without causing decomposition or side reactions (e.g., retro-aldol).2 If silyl conjugate addition is used,3,4,5 the resulting enol resides adjacent to a sizeable silyl group and an electron-donating C–Si bond (e.g., iii, Scheme 1) and can thus react efficiently and stereoselectively with electrophiles. A number of catalytic enantioselective silyl conjugate additions to α,β-unsaturated carbonyls have been disclosed.6,7 Such protocols, promoted by Pd- and Rh-based phosphine complexes, are noteworthy but operate within a relatively narrow substrate range (e.g., require cis alkenes7b), at times proceed with low to moderate enantioselectivity6 or moderate efficiency,7 or demand reagents (e.g., Cl2PhSi–SiMe3), which afford products that must be alkylated (MeLi) prior to efficient product isolation. Herein, we outline a Cu-catalyzed protocol for enantioselective addition of a dimethylphenylsilanyl group to unsaturated cyclic and acyclic ketones, lactones, esters, as well as acrylonitriles and cyclic α,β,γ,δ-dienones. Reactions proceed in 87–97% yield and 90:10–99:1 er with 1–2 mol % of an inexpensive commercially available Cu salt and silylborane reagent, as well as easily accessible monodentate chiral imidazolinium salts (3–4 steps from diphenylethylenediamine in ~50% overall yield).8
Scheme 1.

Catalytic Cycle for Silane Conjugate Additions Promoted by a NHC–Cu Complexa
a B(pin) = pinacolatoboron.
Our investigations were initiated partly based on observations by Sadighi and co-workers,9 who demonstrated that NHC–Cu-alkoxides (e.g., i, Scheme 1) react with bis(pinacolato)diboron [B2(pin)2] to afford the derived NHC–Cu-B(pin). Such a process is likely driven by the formation of the B–O bond in pinacolatoboron alkoxide that is generated as a byproduct. As outlined in Scheme 1, we sought to determine whether an NHC–Cu-alkoxide (i) reacts with the sterically more congested (dimethylphenylsilyl)pinacolatoboron to deliver an NHC–Cu-silane (ii), which would undergo reaction with an unsaturated carbonyl to effect formation of a C–Si bond (iii). We surmised that ii would be preferred over NHC–Cu-boronate since formation of a Si–O is energetically less favored than a B–O bond.10 Reaction of the resulting copper enolate with dimethylphenylsilylpinacolatoboron (1, Scheme 1) regenerates ii, affording boron enolate iv. We have illustrated that NHC–Cu-enolates (e.g., iii) react readily with B2(pin)2 to release the catalytically active NHC–Cu-B(pin).11 If the same process proceeds with 1, the catalytic process would not require an alcohol additive (MeOH), which is used to induce turnover in catalytic Cu–B(pin) additions to alkenes.12 The versatile boron enolate (vs protonated ketone) would thus be obtained as a result of the projected conjugate silane addition.
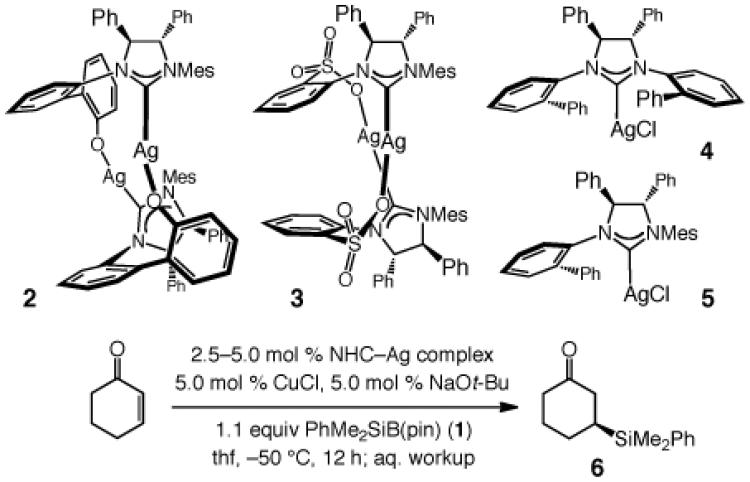
We began by probing the ability of a number of chiral NHC–Cu complexes in promoting the addition of 1 to cyclohexenone to afford β-silylketone 6; key results are summarized in Table 1. All NHC–Cu complexes, generated in situ from reaction of bidentate Ag-based carbenes 213 and 314 as well as monodentate variants 415 and 58 with CuCl, promote conjugate addition of the silane unit. It should be noted that none of the products derived from the formation of a C–B bond is observed (<2% by 400 MHz 1H NMR), and the presence of a proton source (MeOH) is not required. Moreover, enantioselectivity is significantly higher with monodentate complexes 4 and 5, with the C1-symmetric chiral catalyst (5) delivering the optimal er (96:4, entry 4).
Table 1.
Initial Examination of Various Chiral NHC Complexesa
 | ||||
|---|---|---|---|---|
| entry | NHC–Ag; mol % | conv (%)b | yield (%)c | erd |
| 1 | 2; 2.5 | >98 | 91 | 87:13 |
| 2 | 3; 2.5 | >98 | 94 | 89:11 |
| 3 | 4; 5.0 | >98 | 90 | 92.5:7.5 |
| 4 | 5; 5.0 | >98 | 92 | 96:4 |
Under N2 atm.
Determined by analysis of 400 MHz 1H NMR spectra of unpurified mixtures.
Yields of purified products.
By HPLC analysis;
see the Supporting Information for details. Mes = 2,4,6-trimethylphenyl.
Imidazolinium salts are more robust (less light sensitive) than the derived monodentate NHC–Ag complexes (e.g., 4–5, Table 1). Accordingly, simultaneous with our efforts to identify an optimal catalyst that can be utilized in lower loading (e.g., 1 mol %), we turned to variants of the aforementioned chiral C1-symmetric imidazolinium salts. As shown in Scheme 2, ligands where the symmetric NAr unit bears larger substituents deliver lower enantioselectivity (7–9). Next, we examined candidates containing a 2,4,6-trimethylaniline (NMes; cf. 7, not shown in Scheme 2)16 or a 2,6-diethylphenylamine (cf. 8) along with dissymmetric NAr groups. Optimal enantioselectivities were obtained with 12 (Scheme 2); as before (i.e., with 7–8), the catalyst bearing a smaller meta substituent (Me) furnishes higher selectivity (97.5:2.5 vs 91:9 er with 10).16 Two additional points merit mention: 1) C1-Symmetric NHC complexes17 offer a larger degree of diversity versus C2-symmetric variants (i.e., each NAr unit can be modified independently), and thus are more advantageous in connection with reaction optimization. 2) The ligands corresponding to 10–12, but bearing an NMes unit, promote less selective conjugate additions,16,18 indicating cooperativity between the two NAr units of the chiral ligand. The lower er observed with C2-symmetric 4 versus C1-symmetric 5 (Table 1) further supports the above notion.
Scheme 2.

Enantioselective Synthesis of β-Silylketone 6 with Various Chiral C1-Symmetric NHC Complexesa
a Under conditions in Table 1, except with 1.0 mol % CuCl, 1.1 mol % 7–12, 2.2 mol % NaOt-Bu. All conv >98% by analysis of 400 MHz 1H NMR spectra of unpurified mixtures. Enantiomeric ratios by HPLC analysis (see the SI for details).
Cyclic unsaturated ketones undergo enantioselective silyl conjugate addition in the presence of 1.1 mol % 12 and 1.0 mol % CuCl (Table 2). Reactions proceed to >98% conversion after only one hour at −78 °C, affording the desired β-silylketones in 87–95% yield and 90:10–99:1 er. Five- (entries 1 and 5), six- (entries 2 and 6), as well as seven- (entries 3 and 7) and eight-membered (entry 4) ring enones are effective substrates. Enones that contain sterically congested electrophilic sites readily undergo conjugate silyl addition within one hour (entries 5–6, Table 2). Conjugate addition to an unsaturated lactone is efficient (entry 8, Table 2), but proceeds with lower enantioselectivity (vs the related ketone: 92:8 er vs 97.5:2.5 er in entry 2).
Table 2.
NHC–Cu-Catalyzed Enantioselective Conjugate Additions of Silanes to Cyclic α,β-Unsaturated Carbonylsa
| entry | product | yield (%)b | erc |
|---|---|---|---|
| 1 |  |
87 | 90:10 |
| 2 | 92 | 97.5:2.5 | |
| 3 | 95 | 97:3 | |
| 4 | 95 | 98:2 | |
| 5 |  |
89 | 99:1 |
| 6 | 94 | 99:1 | |
| 7 | 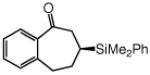 |
91 | 96.5:3.5 |
| 8d | 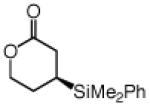 |
95 | 92:8 |
Under N2 atm with 1.1 mol % 12, 1 mol % CuCl and 2.2 mol % NaOt-OBu at −78 °C for 1 h; >98% conv in all cases.
Yields of purified products.
By HPLC analysis; see the SI for details.
Imidazolinium salt 11 (2.2 mol % with 2.0 mol % CuCl and 4.4 mol % NaOt-Bu) used for 12 h.
Reactions of acyclic α,β-unsaturated ketones proceed with equally high efficiency and enantioselectivity (Table 3). Substrates bearing alkyl (entries 1–3, Table 3) as well as aryl (entries 4–7) substituents undergo reaction to afford the desired products in 87–97% yield and up to 98.5:1.5 er. Neither the efficiency nor the enantioselectivity is affected by the electronic attributes (entries 4–6) or the presence of an ortho substituent (entry 7).
Table 3.
NHC–Cu-Catalyzed Enantioselective Conjugate Additions of Silanes to Acyclic α,β-Unsaturated Enonesa

The Cu-catalyzed protocol extends beyond reactions of alkyl ketones, as illustrated by the formation of 13 (Scheme 3) in 92% yield and 95.5:4.5 er. As the additional cases in Scheme 3 indicate, acrylonitriles (e.g., 14), a class of substrates that is inert to Rh-catalyzed silyl conjugate additions,7b and unsaturated esters (15 and 16) are effective substrates.
Scheme 3.

a
a See Table 2 for reaction conditions.
Our preliminary investigations indicate that the present catalytic protocol can be readily carried out with α,β,γ,δ-dienones, affording the 1,6-addition products exclusively (>98% site-selectivity), with effective control of olefin geometry and in high enantioselectivity; the examples in Scheme 4 are illustrative. Several points regarding the observations in Scheme 4 are worthy of note: 1) The optimal catalyst for this class of transformations is derived from C2-symmetric imidazolinium salt 19 (Scheme 4), since NHC–Cu complex 12 delivers substantially lower E/Z ratios and levels of enantiomeric purity (20 in 2:1 Z:E and 94.5:5.5 and 54:46 er, respectively; 21 in >98% E and 69.5:30.5 er). 2) The high alkene stereoselectivity and enantioselectivity with which 21 is formed indicates that reaction likely occurs through an s-cis diene. 3) The enantiomerically enriched allylsilanes can be used in reactions with various electrophiles; the diastereoselective oxidation affording 22 is a case in point. Further development of this class of conjugate additions is ongoing in these laboratories.
Scheme 4.

Cu-Catalyzed Enantioselective Additions to Cyclic Dienones
The Cu-catalyzed enantioselective silane conjugate additions described herein are of substantial utility and complement the related processes involving boronates,19 which, upon oxidation, can also furnish the corresponding β-hydroxy carbonyls. The present set of protocols, however, offers distinct advantages; two examples are illustrated in Scheme 5. First, whereas the boron enolate derived from catalytic boronate conjugate addition to six-membered ring enones undergoes facile aldol addition,11,19d the corresponding enolates from reactions of cyclopentenone or cycloheptenone are unreactive.20 In contrast, as depicted in Scheme 5, the boron enolates of all ring sizes (only five- and seven-membered rings shown) obtained through catalytic silyl additions react readily with aldehydes to afford the desired β-silyl,β-hydroxyketones 23 and 24. The neighboring donor C–Si bonds likely enhances the nucleophilicity of the boron enolates through hyperconjugative effects;21 in contrast, the low-lying C–B σ* in the related boronate addition products can diminish enolate nucleophilicity.
Scheme 5.

β-Silylketones versus the Corresponding Boronates
Second, pinacolatoboronates are sensitive to common organometallics such as aryl- or alkyllithiums as well as the derived Grignard reagents. In contrast, β-silylketones can be easily functionalized, often with high diastereoselectivity, through reaction with such reagents. The example in Scheme 5 (→26) is representative; the β-boronate ketones are converted to unidentifiable products.22 As also presented in Scheme 5, subsequent oxidation23 (→27) furnishes the enantiomerically enriched syn-1,3-diol. A similar procedure with a boronate product would require prior oxidation and protection of the resulting carbinol (to avoid retro-aldol upon treatment with arylmetal).
The Cu-catalyzed additions should prove to be of utility in complex molecule synthesis. For example, ketoester 28 (eq 1), accessed through a racemic synthesis followed by HPLC separation of the enantiomers, was recently utilized in an approach to biologically active natural product (+)-erysotramidine.24 As shown in eq 1, the desired intermediate can now be easily synthesized by a one-pot procedure in 92% yield, as a single diastereomer and in 97.5:2.5 er. The stability of the silyl group towards n-BuLi (see above) allows for conversion of the boron enolate to its more nucleophilic Li-based derivative.
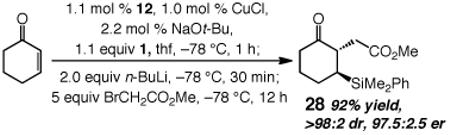 |
(1) |
Supplementary Material
Acknowledgment
Financial support was provided by the NSF (CHE-0715138) and the NIH (GM-57212). K-s. L. is grateful for a Schering-Plough Graduate Fellowship. We thank Ms. Jamie O'Brien for helpful suggestions. Mass spectrometry facilities at Boston College are supported by the NSF (DBI-0619576).
Footnotes
Supporting Information Available: Experimental procedures and spectral, analytical data for all products (PDF). This material is available on the web: http://www.pubs.acs.org
References
- 1.For reviews on the use of organosilanes in organic synthesis, see: Chan TH, Wang D. Chem. Rev. 1992;92:995. Jones GR, Landais Y. Tetrahedron. 1996;52:7599. Fleming I, Barbero A, Walter D. Chem. Rev. 1997;97:2063. doi: 10.1021/cr941074u. Suginome M, Ito Y. Chem. Rev. 2000;100:3221. doi: 10.1021/cr9902805.
- 2.For a review on the utility of β-silylcarbonyls in synthesis, see: Fleming I. Science of Synthesis. Vol. 4. Thieme; Stuttgart, Germany: 2002. p. 927.
- 3.Enantiomerically enriched β-silylcarbonyls have been prepared by catalytic conjugate hydride additions to trisubstituted Si-substituted enones. See: Lipshutz BH, Tanaka N, Taft BR, Lee C.-t. Org. Lett. 2006;8:1963. doi: 10.1021/ol0529593.
- 4.Enantiomerically enriched β-silylcarbonyls can be accessed by catalytic conjugate additions of alkyl or aryl groups to silyl-substituted enones. See: Shintani R, Okamoto K, Hayashi T. Org. Lett. 2005;7:4757. doi: 10.1021/ol051978+. Balskus EP, Jacobsen EN. J. Am. Chem. Soc. 2006;128:6810. doi: 10.1021/ja061970a. Kacprzynski MA, Kazane SA, May TL, Hoveyda AH. Org. Lett. 2007;9:3187. doi: 10.1021/ol071331k.
- 5.Catalytic non-enantioselective methods that afford β-silylcarbonyls have been disclosed. For example, see: Conjugate silane additions: Lipshutz BH, Sclafani JA, Takanami T. J. Am. Chem. Soc. 1998;120:4021. Auer G, Weiner B, Oestreich M. Synthesis. 2006:2113. Conjugate disilane additions: Tamao K, Okazaki S, Kumada M. J. Organomet. Chem. 1978;146:87. Ito H, Ishizuka T, Tateiwa J.-i., Sonoda M, Hosomi A. J. Am. Chem. Soc. 1998;120:11196. Ogoshi S, Tomiyasu S, Morita M, Kurosawa H. J. Am. Chem. Soc. 2002;124:11598. doi: 10.1021/ja0272944. Clark CT, Lake JF, Scheidt KA. J. Am. Chem. Soc. 2004;126:84. doi: 10.1021/ja038530t.
- 6.(a) Hayashi T, Matsumoto Y, Ito Y. J. Am. Chem. Soc. 1988;110:5579. [Google Scholar]; (b) Matsumoto Y, Hayashi T, Ito Y. Tetrahedron. 1994;50:335. [Google Scholar]
- 7.(a) Walter C, Auer G, Oestreich M. Angew. Chem., Int. Ed. 2006;45:5675. doi: 10.1002/anie.200601747. [DOI] [PubMed] [Google Scholar]; (b) Walter C, Oestreich M. Angew. Chem., Int. Ed. 2008;47:3818. doi: 10.1002/anie.200800361. [DOI] [PubMed] [Google Scholar]; (c) Walter C, Fröhlich R, Oestreich M. Tetrahedron. 2009;65:5513. [Google Scholar]
- 8.Lee K-s., Hoveyda AH. J. Org. Chem. 2009;74:4455. doi: 10.1021/jo900589x. [DOI] [PubMed] [Google Scholar]
- 9.(a) Laitar DS, Müller P, Sadighi JP. J. Am. Chem. Soc. 2005;127:17196. doi: 10.1021/ja0566679. [DOI] [PubMed] [Google Scholar]; (b) Laitar DS, Tsui EY, Sadighi JP. Organometallics. 2006;25:2405. [Google Scholar]
- 10.A value of ~125 kcal/mol−1 is attributed to a B–O bond (vs. ~110 kcal/mol−1 for a Si–O). See: Sanderson RT. Chemical Bonds and Bond Energy. Academic Press; New York: 1976. p. 128. Sanderson RT. Polar Covalence. Academic Press; New York: 1983. p. 82.
- 11.Lee K-s., Zhugralin AR, Hoveyda AH. J. Am. Chem. Soc. 2009;131:7253. doi: 10.1021/ja902889s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Lee Y, Hoveyda AH. J. Am. Chem. Soc. 2009;131:3160. doi: 10.1021/ja809382c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lee Y, Jang H, Hoveyda AH. J. Am. Chem. Soc. 2009;131:18234. doi: 10.1021/ja9089928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Veldhuizen JJ, Campbell JE, Giudici RE, Hoveyda AH. J. Am. Chem. Soc. 2005;127:6877. doi: 10.1021/ja050179j. [DOI] [PubMed] [Google Scholar]
- 14.Brown MK, May TL, Baxter CA, Hoveyda AH. Angew. Chem., Int. Ed. 2007;46:1097. doi: 10.1002/anie.200604511. [DOI] [PubMed] [Google Scholar]
- 15.For representative applications of the corresponding C2-symmetric imidazolinium salt, see: Chaulagain MR, Sormunen GJ, Montgomery J. J. Am. Chem. Soc. 2007;129:9568. doi: 10.1021/ja072992f. Lillo V, Prieto A, Bonet A, Díaz-Requejo MM, Ramírez J, Pérez PJ, Fernández E. Organometallics. 2009;28:659. Ref 12a.
- 16.See the Supporting Information for details regarding these screening studies.
- 17.For design of C1-symmetric monodentate chiral NHC–Cu complexes and comparison of their utility vs C2-symmetric variants, see ref 8.
- 18.For example, the NMes-containing derivative of 12 promotes formation of β-silylketone 6 in >98% conversion and 91:9 er (vs 97.5:2.5 er).
- 19.(a) Lee J-E, Yun J. Angew. Chem., Int. Ed. 2008;47:145. doi: 10.1002/anie.200703699. [DOI] [PubMed] [Google Scholar]; (c) Sim H-S, Feng X, Yun J. Chem. Eur. J. 2009;15:1939. doi: 10.1002/chem.200802150. [DOI] [PubMed] [Google Scholar]; (d) Chen I-H, Yin L, Itano W, Kanai M, Shibasaki M. J. Am. Chem. Soc. 2009;131:11664. doi: 10.1021/ja9045839. [DOI] [PubMed] [Google Scholar]
- 20.Lee K-s., Hoveyda AH. unpublished results. [Google Scholar]
- 21.Houk KN, Moses SR, Wu Y-D, Rondan NG, Jäger V, Schohe R, Fronczek FR. J. Am. Chem. Soc. 1984;106:3880. [Google Scholar]
- 22.Fernández E, Maeda K, Hooper MW, Brown JM. Chem. Eur. J. 2000;6:1840. doi: 10.1002/(sici)1521-3765(20000515)6:10<1840::aid-chem1840>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 23.Fleming I, Sanderson PEJ. Tetrahedron Lett. 1987;28:4229. [Google Scholar]
- 24.Tietze LF, Tölle N, Kratzert D, Stalke D. Org. Lett. 2009;11:5230. doi: 10.1021/ol901980u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.