

Abstract
Almost all known intracellular fusion reactions are driven by formation of trans-SNARE complexes through pairing of vesicle-associated v-SNAREs with complementary t-SNAREs on target membranes. However, the number of SNARE complexes required for fusion is unknown, and there is controversy about whether additional proteins are required to explain the fast fusion which can occur in cells. Here we show that single vesicles containing the synaptic/exocytic v-SNAREs VAMP/synaptobrevin fuse rapidly with planar, supported bilayers containing the synaptic/exocytic t-SNAREs syntaxin-SNAP25. Fusion rates decreased dramatically when the number of externally oriented v-SNAREs per vesicle was reduced below 5–10, directly establishing this as the minimum number required for rapid fusion. Docking-to-fusion delay time distributions were consistent with a requirement that 5–11 t-SNAREs be recruited to achieve fusion, closely matching the v-SNARE requirement.
Keywords: lipid bilayer, membrane fusion, SNARE mechanisms, supported bilayer
Trafficking of proteins in the cell—as well as secretion of physiological mediators such as hormones and neurotransmitters—depends on intracellular membrane fusion. With few exceptions, intracellular fusion reactions are driven by pairing of vesicle-associated v-SNAREs (soluble N-ethylmaleimide-sensitive factor attachment protein receptors) with cognate t-SNAREs on the target membrane, resulting in a four-helix bundle (SNAREpin) that brings bilayers into close proximity (1 –3). In cells, the action of SNAREs is regulated by auxiliary proteins, some of which, such as the members of the Sec1/Munc18-like (SM) family, are universally required components of the eukaryotic fusion machinery (4). Whether SNAREs alone can catalyze fusion at physiologically meaningful rates in the absence of modulating proteins or peptides (5 –9) remains controversial. In addition, it is unknown how many SNAREpins are required to produce fusion. Here, using an in vitro assay that can resolve single docking and fusion events, we show 5–10 SNAREpins mediate fast fusion in the absence of any auxiliary proteins.
Reconstituted fusion assays have played a key role in elucidating mechanisms of SNARE-mediated membrane fusion (1, 2, 5, 6, 10, 11). SNARE proteins reconstituted into small unilamellar vesicles (SUVs) fused bilayers in a bulk fusion assay, albeit with slow kinetics (1, 2). More recently, single SUVs containing the synaptic/exocytic v-SNAREs VAMP/synaptobrevin were shown to fuse rapidly with planar, supported bilayers (SBLs) containing the synaptic/exocytic t-SNAREs syntaxin 1-SNAP25, with single fusion events occurring in ∼10–100 ms (7, 9) to seconds (8, 12). However, the SNAP25 subunit of the t-SNARE was not required (8, 9), or an artificial peptide was needed (7), raising questions about the physiological relevance of these results. These, and other studies of SNARE-mediated membrane fusion, used lipid bilayers where the active fusion catalysts were the only proteins present. By contrast, natural intracellular membranes are populated by bulk integral membrane proteins at concentrations ranging from 30,000 to 40,000 per μm2 (13), providing a very different environment for fusion. To bring phospholipid bilayers into contact, SNARE proteins in vivo must presumably perform additional work to push aside this repulsive protein layer.
To better mimic the situation realized in cells, we covered bilayer surfaces with a poly(ethylene glycol) (PEG) polymer “brush” of ∼4-nm height using a PEG chain attached to phosphoethanolamine [1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(methoxy[polyethylene glycol]-2000)]. For the conditions of 5 mol % PEG used here, the polymer chain surface density is about 70,000 per μm2 and the conformational flexibility allows each chain to fill out a volume of about 60–70 nm3, corresponding to the cytosolic domain of a folded globular protein of about 50–60 kDa. We find that SNAP25 dependence is restored but rapid fusion remains (mean delay after docking of 130 ms).
Similar PEG brushes have been used previously to provide steric protection to homogeneous bilayers in adhesion studies while allowing specific ligand-receptor interactions to proceed unimpeded (14). PEGylated SUVs are among the most stable known liposomes, and due to their excellent biocompatibility find use in drug delivery applications as “stealth liposomes” (15). The inclusion of lipid-linked PEG chains also confers exceptional stability on supported bilayers. SBLs containing the same PEG brush as used here could be dried, kept in air for >1 day, and then rehydrated to gain their original fluidity without apparent loss of integrity (14). Importantly, the PEG chains also serve to lift the bilayer about 4 nm from the glass coverslip support which has been shown to reduce interactions between the transmembrane proteins and the underlying substrate that otherwise impair lateral diffusion of these proteins (16, 17).
Results
In addition to the use of PEG brushes, we have enhanced the SUV-SBL fusion assay by including a fluorescent lipid [1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl); NBD-PE] in the SBL. This allowed us to quality control the SBL down to the diffraction limit for its continuity and fluidity by a simple fluorescence recovery after photobleaching (FRAP) measurement before every experiment (SI Text).
Our experimental design is based on a purpose-designed microfluidic flow system in which the fluorescently labeled v-SUVs flow at closely controlled rates and concentrations over the SBL, which is situated in a standard epifluorescence microscope. This enables quantitative determination of fusion rates under steady-state conditions. At low vesicle concentrations, we can readily observe individual vesicles attaching and then fusing with the SBL without need for total internal reflection fluorescence microscopy (TIRFM) technology to distinguish bulk vesicles from those that have docked. This allows us to measure both the absolute efficiency of fusion and the delay between docking [by the first SNAREpin (8)] and fusion (marked by the commencement of lateral diffusion of fluorescent lipid from the SUV in the plane of the SBL at the docking site). By using two microfluidic flow channels in parallel over a common coverslip, we can reliably compare two different reaction conditions side by side. The small volume of the microfluidic channels (∼1 μL) allows us to run the entire experiment under constant flow with minimal consumption of SUVs. The flow also serves to carry away weakly and presumably nonspecifically bound (8) v-SUVs and maintains a constant v-SUV concentration, facilitating analysis.
Supported bilayers are made by incubation of t-SNARE-containing SUVs over a hydrophilic glass substrate in microfluidic flow channels (Fig. 1 A–C and SI Text). After extensive rinsing with buffer and quality checks of the SBL (SI Text), a solution of 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (LR-PE) -labeled v-SUVs is introduced into the channel at a typical concentration of 40−60 nM lipid and constant flow rate of 2–3 μL/min. Given that the mean v-SUV diameter is ∼50 nm (SI Text) and assuming every lipid occupies 0.7 nm2, this corresponds to 2−3 pM v-SUVs, a sufficiently small concentration that individual docking and fusion events can be detected without interference from the SUVs in bulk, which appear as streaks due to flow.
Fig. 1.

The SUV-SBL fusion assay and detection of single fusion events. (A) Schematic of a v-SUV reconstituted with Syb and a t-SBL reconstituted with the t-SNARE complex Syx·SNAP25. The v-SUV is doped with 2 mol % LR-PE (red) to detect single-vesicle docking and fusion events. The SBL is labeled with 2 mol % NBD-PE (green) to check its homogeneity and fluidity before every fusion test with v-SUVs (Materials and Methods). (B) Schematic view of the setup. A PDMS block containing microfabricated grooves is attached to a glass coverslip to form the microfluidic channels. A solution of SUVs (or buffer) is aspirated into the channel using a syringe pump. (C) Photograph of two flow channels in parallel, in the same PDMS block. (D) A single fusion event. The vesicle docked in frame 2 (marked D) remained docked for two more frames, then fused in frame 5 (marked F). The signature of fusion is the radial spread of fluorescence as LR-PE is transferred from the v-SUV into the t-SBL. Frames are 100 ms apart. Each square is 40 × 40 pixels (11 μm by 11 μm). (E) Two-dimensional Gaussian fits to frames 4–7 in D. (F) Variance, 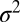 , peak amplitude (ampl.), and total integrated intensity (int.) versus time for Gaussian fits shown in E. Time 0 corresponds to frame 5 in D. Linear fit to the postfusion portion of
, peak amplitude (ampl.), and total integrated intensity (int.) versus time for Gaussian fits shown in E. Time 0 corresponds to frame 5 in D. Linear fit to the postfusion portion of 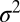 (t) (red) was used to extract the diffusion coefficient of LR-PE in the SBL (see text).
(t) (red) was used to extract the diffusion coefficient of LR-PE in the SBL (see text).
A single docking event followed by fusion is shown in Fig. 1D for a t-SBL with lipid-to-protein ratio (L:P) 104 and a v-SUV with L:P = 667, at 32 °C. The vesicle docked in the frame marked D and fused in the frame marked F, 3 frames (100 ms each) after docking. The signature of fusion is the radial spread of the fluorescent lipids in the SBL after transfer from the v-SUV upon fusion. This spread is quantified in Fig. 1E, where we fitted two-dimensional Gaussians to the fluorescence profiles in the image sequence (D). The variance of the Gaussian fits, 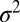 , increases linearly with time after fusion, as expected for simple diffusion from a point source, until signals drop to noise levels (Fig. 1F). Because
, increases linearly with time after fusion, as expected for simple diffusion from a point source, until signals drop to noise levels (Fig. 1F). Because 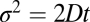 , a fit to the linear, postfusion portion of data such as in Fig. 1F yielded the lipid diffusion coefficient
, a fit to the linear, postfusion portion of data such as in Fig. 1F yielded the lipid diffusion coefficient 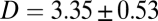 μm2/s (±SEM, 62 vesicles from 6 SBLs), in good agreement with FRAP measurements (SI Text) and the large lipid diffusivities found in high-quality, PEG-cushioned SBLs (14, 16). By comparison, lipid diffusion is 3–4 times slower in SBLs devoid of PEG-lipids (9). The peak amplitude and total intensity of the Gaussian fits as a function of time are also shown in Fig. 1F. The slight increase in the intensity indicates slight dequenching of the LR-PE lipids as they dilute after fusion.
μm2/s (±SEM, 62 vesicles from 6 SBLs), in good agreement with FRAP measurements (SI Text) and the large lipid diffusivities found in high-quality, PEG-cushioned SBLs (14, 16). By comparison, lipid diffusion is 3–4 times slower in SBLs devoid of PEG-lipids (9). The peak amplitude and total intensity of the Gaussian fits as a function of time are also shown in Fig. 1F. The slight increase in the intensity indicates slight dequenching of the LR-PE lipids as they dilute after fusion.
All detectable individual events were tabulated from every experiment and represented as plots of the cumulative number of fusion events versus time. Several examples are shown in Fig. 2A. Because the v-SUV density is low, the SBL t-SNAREs are slowly consumed over hours, so the rate of fusion does not diminish noticeably during the first 0.5 h of reaction (SI Text). The slopes of the lines in Fig. 2A yield the unnormalized fusion rates, 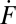 (fusions/s), which are converted to normalized fusion rates,
(fusions/s), which are converted to normalized fusion rates, 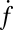 [(s·μm2·pM SUV)−1], after dividing by the detection area and the molar v-SUV concentration (SI Text). The background fusion rate of identically prepared v-SUVs with protein-free SBLs was extremely low (Fig. 2A, red). Normalized fusion rates averaged over all experiments are shown in Fig. 2B. Fusion did not occur when either v- or t-SNAREs were omitted; was efficiently blocked when tetanus neurotoxin was used to cleave Syb on v-SUVs; and was also blocked by soluble v-SNAREs (Syb residues 1−92). Thus, SNARE complex assembly is responsible for driving fusion in this system.
[(s·μm2·pM SUV)−1], after dividing by the detection area and the molar v-SUV concentration (SI Text). The background fusion rate of identically prepared v-SUVs with protein-free SBLs was extremely low (Fig. 2A, red). Normalized fusion rates averaged over all experiments are shown in Fig. 2B. Fusion did not occur when either v- or t-SNAREs were omitted; was efficiently blocked when tetanus neurotoxin was used to cleave Syb on v-SUVs; and was also blocked by soluble v-SNAREs (Syb residues 1−92). Thus, SNARE complex assembly is responsible for driving fusion in this system.
Fig. 2.

Fusion rates. (A) Cumulative number of fusions as a function of time for t-SBLs (t-L:P = 10K) and v-SUVs (4.9 pM, v-L:P = 120) shown in blue for various individual acquisitions. Data for identically prepared v-SUVs over protein-free SBLs are shown in red. (B) Mean fusion rates normalized by detection area and SUV concentration for various conditions as indicated. TeNT, 50 nM tetanus neurotoxin; Syb1–92, cytoplasmic domain of Syb. Numbers of experiments and total numbers of detected fusion events are indicated. All data were obtained at 27 °C.
Individual docking events were recorded, cumulated, and normalized in the same fashion as for fusion events. The normalized total docking rate, 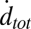 , was about twice the fusion rate
, was about twice the fusion rate 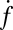 (SI Text), indicating that the efficiency of fusion after docking is about 50%. Most of the docked vesicles that did not end up fusing within 20–30 s gradually photobleached during this period and appeared to remain at the docking site. Only a small fraction (∼4%) visibly dissociated from the SBL to rejoin the flow (SI Text).
(SI Text), indicating that the efficiency of fusion after docking is about 50%. Most of the docked vesicles that did not end up fusing within 20–30 s gradually photobleached during this period and appeared to remain at the docking site. Only a small fraction (∼4%) visibly dissociated from the SBL to rejoin the flow (SI Text).
Both 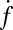 and
and 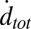 eventually decreased as a function of time with a half-time of 2000–3000 s, ultimately dropping to background levels for t > 5000 s for v-SUV L:P = 150–200 (SI Text). This is presumably due to consumption of SBL t-SNAREs by free v-SNAREs delivered by fusing v-SUVs. This is corroborated by the observation that
eventually decreased as a function of time with a half-time of 2000–3000 s, ultimately dropping to background levels for t > 5000 s for v-SUV L:P = 150–200 (SI Text). This is presumably due to consumption of SBL t-SNAREs by free v-SNAREs delivered by fusing v-SUVs. This is corroborated by the observation that 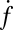 decreased more rapidly for v-SUVs containing higher copy numbers of Syb (SI Text), as more Syb were released into the SBL per fusion event. This further suggests that either t- or v-SNAREs or both are mobile in the SBL (17) and are able to form inactive cis-SNARE complexes upon encounter.
decreased more rapidly for v-SUVs containing higher copy numbers of Syb (SI Text), as more Syb were released into the SBL per fusion event. This further suggests that either t- or v-SNAREs or both are mobile in the SBL (17) and are able to form inactive cis-SNARE complexes upon encounter.
The dependence of 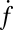 on t-SNARE density in the SBLs was nonlinear.
on t-SNARE density in the SBLs was nonlinear. 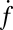 increased only slightly as the t-SNARE density was increased from t-L:P = 30,000 (∼48 t-SNAREs/μm2) to 10,000 (∼140 t-SNAREs/μm2) and then dropped rapidly at higher densities (SI Text), consistent with an earlier report (9) that inactive t-SNARE aggregates may develop at high concentrations. By contrast,
increased only slightly as the t-SNARE density was increased from t-L:P = 30,000 (∼48 t-SNAREs/μm2) to 10,000 (∼140 t-SNAREs/μm2) and then dropped rapidly at higher densities (SI Text), consistent with an earlier report (9) that inactive t-SNARE aggregates may develop at high concentrations. By contrast, 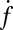 depended linearly on v-SUV concentration in the range we studied.
depended linearly on v-SUV concentration in the range we studied.
Most fusions occurred extremely rapidly after docking. The mean delay before fusion occurred was about 160 ms for the data shown in Fig. 3 A and B. A small fraction (<15–25%) of fusions entailed far longer delays (Fig. 3B). The long delays followed the same delay time distribution as that for docking and fusion of v-SUVs to protein-free SBLs (SI Text), suggesting a small, nonspecific component was present in the overall fusion rate.
Fig. 3.

Delays between individual docking and fusion events and SNAP25 dependence of the overall fusion rate. (A) Distribution of delay times, normalized to integrate to unity (t-LP = 10K, v-LP = 150, 32 °C, 175 delays from 8 acquisitions; bin width = 100 ms). (B) The same delays as in A, presented as a survivor function (SI Text). Inset shows the full span of the distribution, including a small fraction of delays (<15–20%) which occur on >1 s timescales. (C) Effect of the limited time resolution on the sampling of the true delays. The mean delay for fast fusions (delays 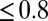 s) versus acquisition period Tacq. The mean delay extrapolated to Tacq = 0 ms is
s) versus acquisition period Tacq. The mean delay extrapolated to Tacq = 0 ms is 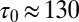 ms. (D) Comparison of fusion rates between SBLs reconstituted with Syx·SNAP25 (190 fusions, 7 acquisitions, t-LP = 10K) and with Syx alone (60 fusions in 19 acquisitions, t-LP = 10K). SBLs were formed from t-SUVs reconstituted side by side. Identical v-SUV preparations were used. Omission of SNAP25 resulted in a 12-fold reduction in the normalized fusion rate (v-LP = 200, T = 30–32 °C).
ms. (D) Comparison of fusion rates between SBLs reconstituted with Syx·SNAP25 (190 fusions, 7 acquisitions, t-LP = 10K) and with Syx alone (60 fusions in 19 acquisitions, t-LP = 10K). SBLs were formed from t-SUVs reconstituted side by side. Identical v-SUV preparations were used. Omission of SNAP25 resulted in a 12-fold reduction in the normalized fusion rate (v-LP = 200, T = 30–32 °C).
Owing to limits on our time resolution the measured fast delays may be overestimated, especially at higher t-SNARE densities, where delays are shorter (see below). To quantify this, in one series of experiments (t-L:P = 10K) we varied the acquisition period, 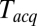 , obtained the apparent mean delay for delays
, obtained the apparent mean delay for delays 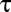
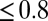 s as in Fig. 3 A and B for every
s as in Fig. 3 A and B for every 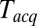 , and plotted these mean delays versus
, and plotted these mean delays versus 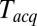 , as shown in Fig. 3C. Apparent mean delay values decreased as a function of decreasing
, as shown in Fig. 3C. Apparent mean delay values decreased as a function of decreasing 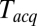 , extrapolating to
, extrapolating to 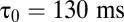 at
at 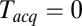 . At large
. At large 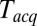 the signal-to-noise ratio is better, facilitating detection, but the large bin width leads to overestimation of delay times. As a good compromise under our experimental conditions, we used
the signal-to-noise ratio is better, facilitating detection, but the large bin width leads to overestimation of delay times. As a good compromise under our experimental conditions, we used 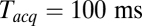 for all data reported here, unless noted otherwise. The mean delay values we report elsewhere in this article are not extrapolated values, as implementation of this procedure for every experimental condition is impractical; consequently, they should be viewed as upper bounds of the true mean delays.
for all data reported here, unless noted otherwise. The mean delay values we report elsewhere in this article are not extrapolated values, as implementation of this procedure for every experimental condition is impractical; consequently, they should be viewed as upper bounds of the true mean delays.
Notably, this system is sufficiently robust that relatively weak interactions between syntaxin (Syx) and VAMP/synaptobrevin (Syb) that are well-documented (18) are unable to drive fusion (Fig. 3D), in contrast to earlier reports using SBLs (8, 9). Adding back SNAP25 in our assay does not reconstitute functional t-SNAREs, consistent with the long incubation times required for proper assembly in vitro (8).
An essential parameter that would help elucidate molecular details of the action of SNAREs is the number of SNAREpins required for fusion, 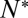 . Although a few estimates have been made from live-cell studies (19), these span a wide range (between 3 and 16) and are inferential rather than direct estimates. To estimate
. Although a few estimates have been made from live-cell studies (19), these span a wide range (between 3 and 16) and are inferential rather than direct estimates. To estimate 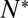 , we measured the normalized fusion rate,
, we measured the normalized fusion rate, 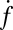 , while varying the SUV L:P, or equivalently the number of Syb per SUV,
, while varying the SUV L:P, or equivalently the number of Syb per SUV, 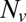 . When
. When 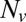 was lowered between 20 and 10,
was lowered between 20 and 10, 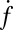 dropped precipitously by approximately two orders of magnitude to background levels, as shown in Fig. 4. Because about half the v-SNAREs face the lumen of the SUV at these low densities (11), we conclude that about 5–10 v-SNAREs are required on the outside of the vesicle for efficient fusion. Assuming all v-SNAREs are active, this suggests fusion requires
dropped precipitously by approximately two orders of magnitude to background levels, as shown in Fig. 4. Because about half the v-SNAREs face the lumen of the SUV at these low densities (11), we conclude that about 5–10 v-SNAREs are required on the outside of the vesicle for efficient fusion. Assuming all v-SNAREs are active, this suggests fusion requires 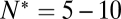 SNAREpins.
SNAREpins.
Fig. 4.

The normalized fusion rate, 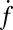 , as a function of the number of Sybs per v-SUV,
, as a function of the number of Sybs per v-SUV, 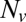 , plotted on a semilogarithmic scale. Note the precipitous drop in
, plotted on a semilogarithmic scale. Note the precipitous drop in 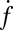 spanning approximately two orders of magnitude as
spanning approximately two orders of magnitude as 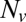 is decreased from 20 to 10. (Inset) Blow-up of small
is decreased from 20 to 10. (Inset) Blow-up of small 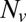 region, linear scale (72 experiments, 1609 fusion events total, 32 °C). At least 6 experiments per data point for
region, linear scale (72 experiments, 1609 fusion events total, 32 °C). At least 6 experiments per data point for 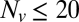 , 2–14 experiments per data point for
, 2–14 experiments per data point for 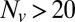 . Error bars are ±SEM.
. Error bars are ±SEM.
We found that delays between docking and fusion became greater as the t-SNARE density was lowered, whereas they were insensitive to variations in 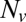 provided
provided 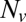 was sufficiently high to sustain fusion (SI Text), suggesting that delays are limited by how fast t-SNAREs can be recruited to fusion sites. Motivated by these observations, we adopted an indirect but independent approach to confirm our direct measurements of
was sufficiently high to sustain fusion (SI Text), suggesting that delays are limited by how fast t-SNAREs can be recruited to fusion sites. Motivated by these observations, we adopted an indirect but independent approach to confirm our direct measurements of 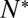 by predicting the distribution of delay times on the sole assumption that lateral diffusion of t-SNAREs to the docked vesicle should be rate-limiting for fusion after docking. In this model, the distribution of delay times reflects the variability of time required for p number of individual t-SNAREs to cumulatively diffuse from surrounding regions of the SBL to the docking site at which one SNAREpin is already engaged (SI Text). The only input parameters are the diffusion constant, the concentration of actively reconstituted t-SNAREs in the SBL, and a molecular size, all of which are either known or can be estimated independently. We found the model describes the observed delays well for p = 4–10, or
by predicting the distribution of delay times on the sole assumption that lateral diffusion of t-SNAREs to the docked vesicle should be rate-limiting for fusion after docking. In this model, the distribution of delay times reflects the variability of time required for p number of individual t-SNAREs to cumulatively diffuse from surrounding regions of the SBL to the docking site at which one SNAREpin is already engaged (SI Text). The only input parameters are the diffusion constant, the concentration of actively reconstituted t-SNAREs in the SBL, and a molecular size, all of which are either known or can be estimated independently. We found the model describes the observed delays well for p = 4–10, or 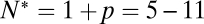 SNAREpins (SI Text), closely matching the directly measured v-SNARE requirements.
SNAREpins (SI Text), closely matching the directly measured v-SNARE requirements.
Using a similarly indirect modeling approach, a recent in vitro study suggested 6–9 SNAREpins might be required (7). However, because SNARE densities were not varied, different models of delay distributions could not be distinguished. In addition, the delays might have been affected by the osmotic gradient employed and by the artificial peptide (Syb49–96) used to assemble the t-SNARE complex that needed to be displaced by the full-length Syb for fusion to occur. Our finding that delays increase when the t-SNAREs are depleted precludes a model in which t-SNAREs are preassembled into clusters that act as functional docking/fusion sites in this assay (SI Text).
Discussion
In previous studies using SBLs lacking polymer brushes, rapid fusion of SUVs mediated by synaptic/exocytic SNARE proteins has been observed, but SNAP25 has not been required (8, 9). These observations suggest that even within the minimal fusion machinery there is a catalytic core consisting of Syb and Syx. This view is directly supported by the long-known affinity between Syb and Syx (18) and by the recent x-ray crystal structure of the postfusion state of the complete SNARE complex, including the membrane anchors of Syx and Syb (20). Both Syx and Syb form continuous interacting helices from the beginning of their N-terminal (membrane-distal) SNARE motif, through the linker regions, and into and across the lipid bilayer. SNAP25 does not extend into the lipid bilayer, and according to our results presumably imparts additional binding energy to stabilize Syb and Syx and to pay for the free-energy cost of clearing bulk membrane proteins out of the vicinity required for fusion. Taken together, these and earlier results (1, 3) provide clear physical chemical proof that SNARE proteins are indeed engines of membrane fusion. The energy supplied by 5–10 SNAREpins, ∼35 kT each (21), is more than sufficient to overcome known activation energies for fusion (40–140 kT) both in vivo (22, 23) and in model systems (24). Other proteins position or regulate this engine; in particular, it is likely that the universally important SM proteins play a key role in organizing SNAREpins (4). It will be interesting to see whether the same number of SNAREpins is required in this SM-organized structure as with the SNAREs alone.
It is widely recognized that SNARE proteins constitute the core of many different membrane fusion machineries required for intracellular trafficking. However, it has been a matter of recent debate as to whether SNAREs alone can drive fusion efficiently in the absence of modulating proteins or peptides. A recent report (6) showed that Rabs together with tethers and associated proteins resulted in multiple rounds of robust fusion when endosomal SNARE proteins were present, whereas little fusion appeared to occur with SNAREs alone. This raised the possibility that Rab GTPases and associated tethering might contribute physically to bilayer fusion. However, this conclusion cannot be reached because the conditions used to load the vesicles with reporter molecules filled only about 15% and 44% of the donor (t-SUV) and acceptor (v-SUV) vesicles, respectively, so only 6.6% of the fusion events in the first round could be recorded, even if this occurred with 100% efficiency. The assay therefore primarily measured subsequent rounds of fusion (i.e., SNARE recycling) rather than the initial events, which presumably did not require the recycling machinery. It can be concluded that Rabs and tethers, in addition to NSF and SNAP, play an important role in recycling SNAREs. This would be consistent with the need to favor trans-SNARE interactions over cis interactions, especially in homotypic fusion processes, in which Rabs and tethers have previously been implicated (5).
The mean time of 130 ms for fusion after docking is more than ample to explain the speed of virtually all fusion events in the cell (25), and indeed closely matches the speed at which single synaptic vesicles undergo exocytosis upon stimulation in retinal bipolar neurons, where they are held in reserve ∼20 nm from the presynaptic membrane by ribbon structures, just outside the reach of t- and v-SNAREs (26). However, in many other neurons, 2–10 synaptic vesicles are predocked at the active zone and neurotransmitter release occurs 100 times faster (27). The SNAREs in this readily releasable pool of vesicles are preassembled (28, 29) and the fusion process at this point is clamped by complexin (30). Thus, only the very final step in the fusion process remains to be accomplished when calcium enters to trigger synaptotagmin (27, 30), which may very well contribute directly to the speed of fusion at this late stage (10, 11, 31).
Materials and Methods
Recombinant Protein Expression and Purification.
Details are given in SI Materials and Methods.
Preparation of SUVs and SBLs.
SNARE proteins were reconstituted into liposomes essentially as described (32) with small differences. All lipids were from Avanti Polar Lipids, and were dissolved in a 2:1 (vol:vol) mixture of CHCl3:methanol. Typically, 1 μM total lipid was used, with a composition that was 78 mol % 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 15 mol % 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (DOPS), 5 mol % 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (PEG2000-PE), and 2 mol % fluorescently labeled lipids, either 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl) (NBD-PE) or 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (LR-PE).
SBLs were formed by incubating protein-free or t-SUVs over very hydrophilic #1.5 glass coverslips (Waldemar Knittel Glasbearbeitungs), which were prepared by, in sequence, cleaning in a hot Hellmanex II (Hellma) solution, extensive rinsing with Milli-Q (Millipore)-purified water (MQ water), Piranha cleaning (a 2:1 mixture of sulfuric acid and hydrogen peroxide), extensive rinsing with MQ water, drying, and plasma cleaning (Harrick PDC-32G plasma cleaner/sterilizer; Harrick Plasma). A clean coverslip was bonded with an elastomer block made of poly(dimethyl siloxane) (PDMS) containing microfabricated grooves which formed flow channels (Fig. 1). After extensive rinsing with buffer and for every SBL we formed, we checked the homogeneity of the SBL down to the diffraction limit using the NBD-labeled lipids included in the bilayers. Then the fluidity of the SBL was verified by fluorescence recovery after photobleaching (FRAP; a sample trace is shown in SI Materials and Methods). Only if an SBL passed these quality checks did we introduce a solution of v-SUVs into the channel, at a typical concentration of 40–60 nM lipid. Given that the mean vesicle diameter is ∼50 nm (see below), and assuming 0.7 nm2 per lipid (33), this corresponds to 2–3 pM SUV. Typically, protein-free or v-SUVs were diluted 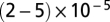 times before use. See SI Materials and Methods for further details.
times before use. See SI Materials and Methods for further details.
Quantification of Actual L:P Ratios.
Actual lipid-to-protein ratios were obtained using a combination of densitometry for quantifying protein concentrations and fluorescence for lipids, as described in SI Materials and Methods. The protein and lipid yields coincided to within measurement error. Therefore, the nominal and the actual L:P ratios are the same.
Characterization of SUVs by Dynamic Light Scattering.
Details are given in SI Materials and Methods. We found that the number-average bare SUV diameter is ∼50 nm. This value, which is in close agreement with previous independent measurements by electron microscopy (32), was used to convert lipid concentrations to SUV concentrations and to calculate the number of proteins per vesicle from the lipid:protein ratios.
Microfluidic Flow Channels, Microscopy, and Analysis of Fusion Events.
Details are given in SI Materials and Methods.
Supplementary Material
Acknowledgments
This work was supported by an NIH grant to J.E.R. E.K. would like to thank M. Schöffel for help with some initial experiments, R. Attia and J.-L. Viovy (Institut Curie, Paris) for supplying templates for microfluidic cells at the early stages of this study, and P. Thévenaz (Lausanne) for updating his ImageJ plugin PointPicker. We are grateful to the CNRS and the Laboratoire de Dynamique Membranaire (CNRS FRE 3146, Paris) for granting a leave of absence to E.K. We thank all members of Laboratoire de Dynamique Membranaire and the Rothman lab, T. Melia (Yale University), F. Pincet (Ecole Normale Supérieure, Paris), F. Brochard-Wyart (Institut Curie, Paris), and J. Warner (Columbia University) for fruitful discussions and suggestions. We are grateful to J. Shen (University of Colorado) for the SUMO-syntaxin plasmid, G. Warren (Max F. Perutz Laboratories, Vienna), T. Melia, F. Pincet, and D. Tareste (Inserm, Paris) for reading and commenting on the manuscript. E.K. would like to thank E. Folta-Stogniew (Keck Foundation Biotechnology Resource Laboratory at Yale) for help with dynamic light scattering, M. Powers at the μELM clean-room facility at Yale for help with photolithography, and M. Perez del Rio for drawing the photolithographic masks.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0914723107/DCSupplemental.
References
- 1.Weber T, et al. SNAREpins: Minimal machinery for membrane fusion. Cell. 1998;92:759–772. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- 2.McNew JA, et al. Compartmental specificity of cellular membrane fusion encoded in SNARE proteins. Nature. 2000;407:153–159. doi: 10.1038/35025000. [DOI] [PubMed] [Google Scholar]
- 3.Hu C, et al. Fusion of cells by flipped SNAREs. Science. 2003;300:1745–1749. doi: 10.1126/science.1084909. [DOI] [PubMed] [Google Scholar]
- 4.Südhof TC, Rothman JE. Membrane fusion: Grappling with SNARE and SM proteins. Science. 2009;323:474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mima J, Hickey CM, Xu H, Jun Y, Wickner W. Reconstituted membrane fusion requires regulatory lipids, SNAREs and synergistic SNARE chaperones. EMBO J. 2008;27:2031–2042. doi: 10.1038/emboj.2008.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ohya T, et al. Reconstitution of Rab- and SNARE-dependent membrane fusion by synthetic endosomes. Nature. 2009;459:1091–1097. doi: 10.1038/nature08107. [DOI] [PubMed] [Google Scholar]
- 7.Domanska MK, Kiessling V, Stein A, Fasshauer D, Tamm LK. Single vesicle millisecond fusion kinetics reveals number of SNARE complexes optimal for fast SNARE-mediated membrane fusion. J Biol Chem. 2009;284:32158–32166. doi: 10.1074/jbc.M109.047381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bowen ME, Weninger K, Brunger AT, Chu S. Single molecule observation of liposome-bilayer fusion thermally induced by soluble N-ethyl maleimide sensitive-factor attachment protein receptors (SNAREs) Biophys J. 2004;87:3569–3584. doi: 10.1529/biophysj.104.048637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu T, Tucker WC, Bhalla A, Chapman ER, Weisshaar JC. SNARE-driven, 25-millisecond vesicle fusion in vitro. Biophys J. 2005;89:2458–2472. doi: 10.1529/biophysj.105.062539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hui E, Johnson CP, Yao J, Dunning FM, Chapman ER. Synaptotagmin-mediated bending of the target membrane is a critical step in (Ca2+)-regulated fusion. Cell. 2009;138:709–721. doi: 10.1016/j.cell.2009.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martens S, Kozlov MM, McMahon HT. How synaptotagmin promotes membrane fusion. Science. 2007;316:1205–1208. doi: 10.1126/science.1142614. [DOI] [PubMed] [Google Scholar]
- 12.Fix M, et al. Imaging single membrane fusion events mediated by SNARE proteins. Proc Natl Acad Sci USA. 2004;101:7311–7316. doi: 10.1073/pnas.0401779101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quinn P, Griffiths G, Warren G. Density of newly synthesized plasma membrane proteins in intracellular membranes II. Biochemical studies. J Cell Biol. 1984;98:2142–2147. doi: 10.1083/jcb.98.6.2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Albertorio F, et al. Fluid and air-stable lipopolymer membranes for biosensor applications. Langmuir. 2005;21:7476–7482. doi: 10.1021/la050871s. [DOI] [PubMed] [Google Scholar]
- 15.Lasic DD, Needham D. The “stealth” liposome: A prototypical biomaterial. Chem Rev. 1995;95:2601–2628. [Google Scholar]
- 16.Diaz AJ, Albertorio F, Daniel S, Cremer PS. Double cushions preserve transmembrane protein mobility in supported bilayer systems. Langmuir. 2008;24:6820–6826. doi: 10.1021/la800018d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wagner ML, Tamm LK. Reconstituted syntaxin1a/SNAP25 interacts with negatively charged lipids as measured by lateral diffusion in planar supported bilayers. Biophys J. 2001;81:266–275. doi: 10.1016/S0006-3495(01)75697-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Calakos N, Bennett MK, Peterson KE, Scheller RH. Protein-protein interactions contributing to the specificity of intracellular vesicular trafficking. Science. 1994;263:1146–1149. doi: 10.1126/science.8108733. [DOI] [PubMed] [Google Scholar]
- 19.Montecucco C, Schiavo G, Pantano S. SNARE complexes and neuroexocytosis: How many, how close? Trends Biochem Sci. 2005;30:367–372. doi: 10.1016/j.tibs.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 20.Stein A, Weber G, Wahl MC, Jahn R. Helical extension of the neuronal SNARE complex into the membrane. Nature. 2009;460:525–528. doi: 10.1038/nature08156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li F, et al. Energetics and dynamics of SNAREpin folding across lipid bilayers. Nat Struct Mol Biol. 2007;14:890–896. doi: 10.1038/nsmb1310. [DOI] [PubMed] [Google Scholar]
- 22.Oberhauser AF, Monck JR, Fernandez JM. Events leading to the opening and closing of the exocytotic fusion pore have markedly different temperature dependencies. Kinetic analysis of single fusion events in patch-clamped mouse mast cells. Biophys J. 1992;61:800–809. doi: 10.1016/S0006-3495(92)81884-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Z, Jackson MB. Temperature dependence of fusion kinetics and fusion pores in Ca2+-triggered exocytosis from PC12 cells. J Gen Physiol. 2008;131:117–124. doi: 10.1085/jgp.200709891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee J, Lentz BR. Secretory and viral fusion may share mechanistic events with fusion between curved lipid bilayers. Proc Natl Acad Sci USA. 1998;95:9274–9279. doi: 10.1073/pnas.95.16.9274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kasai H. Comparative biology of Ca2+-dependent exocytosis: Implications of kinetic diversity for secretory function. Trends Neurosci. 1999;22:88–93. doi: 10.1016/s0166-2236(98)01293-4. and erratum (2000) 23:43. [DOI] [PubMed] [Google Scholar]
- 26.Zenisek D, Steyer JA, Almers W. Transport, capture and exocytosis of single synaptic vesicles at active zones. Nature. 2000;406:849–854. doi: 10.1038/35022500. [DOI] [PubMed] [Google Scholar]
- 27.Sudhof TC. The synaptic vesicle cycle. Annu Rev Neurosci. 2004;27:509–547. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- 28.Hua SY, Charlton MP. Activity-dependent changes in partial VAMP complexes during neurotransmitter release. Nat Neurosci. 1999;2:1078–1083. doi: 10.1038/16005. [DOI] [PubMed] [Google Scholar]
- 29.Xu T, et al. Inhibition of SNARE complex assembly differentially affects kinetic components of exocytosis. Cell. 1999;99:713–722. doi: 10.1016/s0092-8674(00)81669-4. [DOI] [PubMed] [Google Scholar]
- 30.Giraudo CG, Eng WS, Melia TJ, Rothman JE. A clamping mechanism involved in SNARE-dependent exocytosis. Science. 2006;313:676–680. doi: 10.1126/science.1129450. [DOI] [PubMed] [Google Scholar]
- 31.Dai H, Shen N, Araç D, Rizo J. A quaternary SNARE-synaptotagmin-Ca2+-phospholipid complex in neurotransmitter release. J Mol Biol. 2007;367:848–863. doi: 10.1016/j.jmb.2007.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scott BL, et al. Liposome fusion assay to monitor intracellular membrane fusion machines. In: Duzgunes N, editor. Methods in Enzymology. 372, Part B. San Diego, CA: Academic; 2003. pp. 274–300. [DOI] [PubMed] [Google Scholar]
- 33.Leckband D, Israelachvili J. Intermolecular forces in biology. Q Rev Biophys. 2001;34:105–267. doi: 10.1017/s0033583501003687. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.