

Abstract
Deregulation of the c-Myc oncogene is tightly associated with human and murine plasma cell (PC) neoplasms. Through the analysis of Ag-specific B cell responses in mice where Myc is targeted to the Igh Cα locus, we show here that c-Myc dramatically impairs the primary and secondary Ab response. This impairment is differentiation stage specific, since germinal center B cell formation, affinity maturation, and class switch recombination were intact. Examination of PC viability revealed that c-Myc triggered apoptosis only upon final maturation when Ab is secreted and is resistant to the survival factor BAFF (B cell-activating factor belonging to the TNF family). In contrast, PC precursors (PCpre) that ultimately give rise to mature PCs survived normally and vigorously expanded with BAFF signaling. We further show that c-Myc also facilitates the apoptosis of memory B cells. Thus, Cα-Myc controls both cellular arms of long-lived B cell immunity than previously anticipated. Only when deregulation of c-Myc was combined with enforced Bcl-xL expression were mature PCs able to survive in response to BAFF. These data indicate that the survival requirements for tumor-susceptible PCpre and PCs are distinct and that tumor progression likely develops as PCpre transition to functional PCs when apoptotic pathways such as members of the Bcl-2 family are disabled.
Plasma cells (PC)3 represent a terminal stage of B cell differentiation whose function is to synthesize and secrete large amounts of Ag-specific Ab that provide the host with protective humoral immunity. Following encounter with the activating Ag, mature B cells expand and initiate the primary Ab response that can be generally divided into short- and long-lived responses (1). In short-lived responses, B cell activation often occurs after exposure to a T cell-independent (TI) Ag that results in rapid terminal differentiation to predominantly low- affinity IgM-secreting PCs and limited IgG-secreting PCs. In contrast, long-lived responses are generated upon activation to T cell-dependent (TD) Ag that results in GC formation, where somatic hypermutation coupled with isotype switching occurs that engenders affinity-matured, Ag-specific B cells. These cells are subsequently selected based on improved Ab affinity toward Ag and those that are competitively successful are subject to terminal differentiation into PCs and memory B cells (Bmem). Long-lived PCs migrate to and persist in the bone marrow (BM) for decades and provide up to 80% of protective Ab titers to viral pathogens (2, 3).
As post-GC B cells commit to a PC fate, transforming events can occur that result in PC malignancies such as multiple myeloma (MM) and are characterized by the accumulation of clonotypic neoplastic Ab-secreting cells within lymphoid organs (4). Translocations that involve the Ig H chain are often present in PC neoplasms with the incidence increasing with the stage of tumorigenesis. It is estimated that up to 90% of advanced MM tumors involve the Igh locus (5-7). During the pathogenesis of PC neoplasms, translocations that juxtapose an oncogene and one of the Ig enhancers are common. Deregulation of the Myc (c-Myc) oncogene occurs in several B cell malignancies, including human MM and murine plasmacytoma. Studies using fluorescence in situ hybridization analysis show that ~88% of human MM cell lines and 45% of advanced primary MM tumors have karyotypic abnormalities that juxtapose Myc and an Ig enhancer (8). The most common translocation in murine plasmacytomas is t(12;15), which juxtaposes Myc to the distal Igh Cα gene (9-11), and is the direct counterpart of the t(8;14)(q24;q32) translocation observed in human Burkitt’s lymphoma (BL). Of the numerous outstanding questions concerning this translocation, a long established but still puzzling observation is that the clinical subtype of BL and murine PC neoplasms is tightly associated with the fine structure of translocation. For example, in the case of endemic BL, the translocation breakpoints in IGH always occur 5′ of Eμ, resulting in a rearrangement in which Myc expression can be driven by the cooperative actions of the intronic IGH enhancer Eμ and the 3′ IGH enhancer Eα. In the case of sporadic BL, the translocation break-points in IGH invariably fall 3′ of Eμ, leading to a molecular structure in which Myc can only be deregulated by Eα. Elucidating the biological cause of the differential enhancer availability in endemic and sporadic BL has been an important goal of modeling the t(8;14)(q24;q32) translocation in transgenic mice. However, previous attempts to that end have been largely unsuccessful (12-18). To create an experimental system in which the transformation-inducing potential of endemic vs sporadic BL translocations can be compared directly, a gene-insertion strategy was used to generate c-Myc-transgenic mice where Myc cDNA was inserted into the Cα gene that models the endemic BL translocation and transgenic strains where Eμ was deleted during gene targeting, thus creating a model of the sporadic BL translocation (19). Furthermore, gene insertion of Myc into the Cα gene recapitulates best the t(12;15) translocation most common to murine plasmacytomas (20), which allows Myc to interact with downstream Igh enhancers, including Eα, which is critical for Myc deregulation in PC tumors (18). Thus, the Cα-Myc-transgenic mice have great relevance to studying the origin of endemic BL and plasmacytomas, yet these transgenic mice exhibit little to no tumorigenesis. Only when deregulated Cα-Myc is combined with enforced expression of the antiapoptotic Bcl-xL gene does fulminate PC transformation occur that reflects many features of human MM (20).
Despite its normal function in controlling cell proliferation (21, 22), c-Myc activation can lead to apoptosis under certain cellular stress (23). What determines c-Myc-induced proliferation vs apoptosis is hypothesized to depend on whether sufficient amounts of survival factors are available to prevent death. The failure to obtain sustainable levels of support for growth would lead to the elimination of overproliferating cells, thus providing a safeguard against uncontrolled oncogene-induced proliferation. Thus, heightened levels of c-Myc expression may limit cellular growth by increasing the threshold of survival factor signaling needed to maintain longevity. One such factor that is critical for normal PC survival is the interaction of B cell-activating factor belonging to the TNF family (BAFF) with its PC-expressed receptor B cell maturation Ag (BCMA) (24). Furthermore, several reports have shown that members of the BAFF family are aberrantly expressed in human MM cell lines as well as primary MM tissue and can modulate growth (25, 26).
Because PCs are quiescent by nature, replication events at this differentiation stage leading to cumulative genetic translocations that ultimately confer a growth and/or survival advantage is unlikely. Rather, the target of genetic transformation is more likely to be the proliferating progenitor of BM PCs, the PC precursor (PCpre). We and others have previously identified post-GC B cells that are direct precursors to PCs in both murine and human experimental systems (27-31). Murine PCpre are generated in secondary lymphoid organs and within 2 wk home to the BM where they persist long term by self-renewal and also give rise to mature Ab-secreting PCs (27). The prevailing theory is that acquisition of the Myc-activating chromosomal t(12;15) translocation generates a putative tumor-initiating cell, but is nonpathologic unless additional genetic and environmental factors further promote its proliferation, neoplasticity, and survival (19). Targeted expression in mice reveals that apoptotic pathways such as members of the Bcl-2 family must be disabled for complete transformation of Myc-harboring B cells (32-34). These studies provide strong evidence that B cells poised for tumorigenesis by c-Myc, alone, sense that something is amiss and are instructed to undergo suicide. Yet, whether this occurs in an Ag-dependent manner and, if so, at what stage during terminal differentiation does c-Myc target cell death are unknown. Answers to these questions will likely reveal how Myc-Igh translocations affect the quality of the humoral immune response leading to the nascent tumor precursor.
In this study, we show that targeting Myc to the Igh Cα locus in mice impairs both primary and secondary humoral immune responses by failing to generate mature Ab-secreting PCs to TI and TD Ags. This impairment is differentiation stage specific, since GC B cell formation, affinity maturation, and class switch recombination were intact. The failure to produce PCs results from c-Myc-triggered apoptosis upon final maturation when Ab is secreted and is resistant to BAFF-mediated survival. PCpre survived normally and were responsive to BAFF signaling. Furthermore, c-Myc also assists in the apoptosis of Bmem, resulting in a significantly diminished Ag recall response. Thus, both cellular arms of long-lived B cell immunity are controlled by Cα-Myc. Only when deregulation of c-Myc was combined with enforced Bcl-xL expression were mature PCs able to vigorously respond to BAFF and survive. These results indicate that the Cα-Myc translocation confers distinct survival requirements for tumor-initiating PCpre, PCs, and Bmem, thereby controlling B cell immunity. As such, tumor progression is curbed by c-Myc-induced apoptosis of mature long-lived effector B cells unless apoptotic pathways such as members of the Bcl-2 family are concomitantly disabled.
Materials and Methods
Mice
All experiments were performed with 8- to 12-wk-old mice that were maintained in the specific pathogen-free animal facility at the University of Virginia (Charlottesville, VA). The generation of c-MycCα which express an inserted His-tagged mouse c-Myc coding region in the Cα locus has been previously described (20). C57BL/6-nontransgenic littermates were used as controls. Transgenic mice that express Flag-tagged mouse Bcl-xL driven by the Ig κ L chain 3′ enhancer were generated as previously described (20). All animal procedures were conducted in compliance with the National Institutes of Health guidelines and are approved by the Institutional Animal and Use Committee of the University of Virginia.
Immunization and BAFF treatment
To examine primary TD responses, mice were challenged with CFA alone or with 100 μg i.p. of either (4-hydroxy-3-nitrophenyl)acetyl (NP30)-keyhole limpet hemocyanin (KLH) emulsified in CFA or aluminum potassium sulfate (alum), or phosphoryl choline (PhC30)-OVA CFA (Biosearch Technologies). As indicated, some mice also received 200 μg of soluble recombinant BAFF (24) i.p. injections administered twice weekly. To examine TI responses, mice were challenged i.p. with 25 μg of NP30-Ficoll suspended in balanced salt solution (BSS). After days noted, the animals were euthanized and spleen or BM cells were isolated. For secondary TD responses, 42-day primary NP-KLH CFA-immune animals were immunized i.p. with 50 μg of NP-KLH in alum followed by analyses 5 days thereafter.
In vitro LPS/anti-CD40 stimulation
Cells were cultured for 1–5 days in complete RPMI 1640 supplemented with 10% FBS containing IL-4 with or without 40 μg/ml LPS or 10 μg/ml agonistic anti-CD40 mAb FGK115. At the end of the culture, cells were harvested and either enumerated for PCs by ELISPOT or stained for FACS. As indicated, some cells were cultured in the presence of the caspase 3 inhibitor benzylocarbonyl-Val-Ala-Asp(OMe)-fluoromethylketone ZVAD (MP Biomedicals) at a final concentration of 20 μM.
Flow cytometry
Cells were washed and resuspended in 5% bovine calf serum in BSS and stained with a mixture of mAbs, as noted, for the detection of multiple cell surface and intracellular Ags. Transgenic c-Myc protein levels were assessed by intracellular staining of 6-His using an anti-His fluorescein mAb (Miltenyi Biotec) combined with surface B220 and CD138 mAb labeling. For studies that examined NP+ B cell subsets, cells were resuspended in pH 4.0 acetate buffer containing 0.05 M sodium acetate, 0.085 M NaCl, 0.005 M KCl, and 2% FBS in dH2O. Samples were incubated on ice for 1 min, followed by the addition of an equal volume of 0.1 M Tris buffer (pH 8.0) containing 2% FBS, washed, and stained. Purified rat IgG was used as an isotype control. For all studies, nonspecific staining was reduced by the addition of rat serum Ig and the FcR-blocking mAb 2.4G2. Stained cells were incubated for 30 min at 4°C followed by washing in bovine calf serum/BSS. Incubation with streptavidin-PE-cyanin 5.5 was performed for an additional 30 min at 4°C followed by washing. 4′,6-diamidino-2-phenylindole (DAPI) was added to cells at a concentration of 5 μM/ml to discriminate live vs dead cells. After surface staining, samples requiring intracellular staining were stained for histidine (Miltenyi Biotec), caspase 3, cytoplasmic IgM, or IgG using the Cytofix/Cytoperm reagent (BD Biosciences) according to the manufacturer’s instructions. Following staining, cells were washed with BD Permwash and 40 mM of the fluorescent DNA intercalating dye 7-aminoacitnomycin D (7-AAD) was added 5 min before analysis.
Cell acquisition was performed with a DakoCytomation CyAN ADP LX, where a minimum of 300,000 events was collected. Spectral overlap compensation and data analysis were performed using FlowJo software (Tree Star). Profiles are presented as 5% probability contours including outliers with gating based on fluorescence-minus-one and isotype controls. Cell and nuclear morphology of apoptotic cells, as measured by 7-AAD staining of fragmented irregular-shaped nuclei, was performed using the ImageStream 100 cytometer (Amnis) as previously described (35). Briefly, the instrument uses hydrodynamic focusing of cells in a core stream and illuminates cells in flow for fluorescence, dark field, and bright field image detection. All light is collected and separated by wavelength bands using a spectral decomposition optical system, resulting in six subimages being collected for each cell based on spectral properties of bright field, dark field, and four fluorescences. Images of 10,000 cells per sample were analyzed using the IDEAS version 3.0 image analysis software (Amris). Using the brightfield intensity gradient feature that determines image sharpness, images that are considered out of focus are excluded. Spectral overlap among the four fluorescences is removed by creating a compensation matrix using single-color fluorescent control samples that are collected on the instrument without bright field illumination. A matrix coefficient intensity plot is generated for each fluorescence, applied, and validated using control images. After compensation, images of in-focus single cells were selected on the basis of small bright field area, high bright field aspect ratio, and high nuclear contrast.
RT-PCR
After 48 h, in vitro-stimulated cells were harvested and total RNA was isolated using the RNeasy System (Qiagen). cDNA synthesis was performed using the Superscript II System (Life Technologies). PCR amplification was conducted by titrating cDNA samples with Taq (Boehringer Mannheim) and the primers for HPRT to normalize levels of template. Amplification of experimental transcripts was performed using normalized amounts of cDNA. The primers used were as follows: HPRT, Blimp-1, activation-induced cytidine deaminase (AID), germline IgM, germline IgG1, and postswitch IgG1 (36). Conditions used for amplification were 94°C, 55°C/62°C, and 72°C for 35 cycles. For real-time RT-PCR, PCs were positively selected (>90%) using biotinylated anti-CD138 Ab followed by streptavidin-coupled microbeads (Miltenyi Biotec), isolation of total RNA, DNase I treatment, and cDNA synthesis. Gene expression levels were quantified using the SYBR Green PCR Core Kit (Applied Biosystems) on an iCycler iQ instrument (Bio-Rad). Amplification conditions were 95°C for 8 min, followed by 40 cycles of 94°C for 15 s, 63°C for 45 s, and 72°C for 15 s. Primers used were as follows: Bcl-2: forward, 5′-CTTAGAAAATACAGCATTGCGGAG-3′ and reverse, 5′-GGATGTGCTTTGCATTCTTGG-3′; Mcl-1: forward, 5′-GCTCCGGAAACTGGACATTA-3′ and reverse, 5′-CCCAGTTTGTTACGCCATCT-3′; and Bcl-xL: forward, 5′-TTCGGGATGGAGTAAACTGG-3′ and reverse, 5′-TGCAATCCGACTCACCAATA-3′. The relative level of expression for each primer target was calculated by the formula 2−(ΔΔCT) × 1000, where ΔΔCT = (CT gene of interest − CT HPRT in experimental sample) − (CT gene of interest − CT HPRT in a no-template control sample), where CT is cycle threshold. RT-PCR was performed to distinguish wild-type vs transgenic c-Myc by using a common 5′ primer (5′-TCT CCA CTC ACC AGC ACA AC-3′) and 3′ primers specific for MycHis (5′-ATG GTG ATG GTG ATG ATG AC-3′) and wild-type Myc (5′-CCT CGA GTT AGG TCA GTT TA-3′). Conditions used for amplification were 95°C, 62°C, and 72°C for 20 cycles.
Immunoblots
After 48 h, in vitro-stimulated cells were harvested and CD138+ cells were purified using magnetic bead selection (Miltenyi Biotec). Protein samples from whole cell lysates were separated by 10–15% PAGE with SDS. Proteins were transferred to nitrocellulose and membranes were blocked with 1% BSA and/or 5% milk in TBS with 0.05% Tween 20. Primary Ab incubations were done overnight at 4° in blocking buffers that were recommended by the Ab suppliers (Bcl-2 clone 50E3 and Bcl-xL clone 54H6; Cell Signaling Technologies and Mcl-1; Rockland). After washing, secondary Ab incubations were done over 45-min periods in the same blocking buffers using Alexa Fluor 680-conjugated rabbit IgG (H + L; Molecular Probes). Blots were quantified using the Odyssey Infrared Imaging System (Li-Cor Biosciences).
ELISPOT
Spleen and BM Ab-secreting cells were quantified by a total Ig and Ag-specific Ig ELISPOT assay as previously described (37). Briefly, isolated cells were depleted of RBCs by ammonium chloride-Tris lysis, resuspended in complete RPMI 1640, and incubated for 5 h on Multiscreen 96-well plates (Millipore) precoated with either NP4 or NP30 Ag or unlabeled anti-mouse IgM and IgG. Three-fold serial dilutions of cells were made with a start concentration of 150,000 cells/well. After incubation, plates were washed in 0.05% Tween 20 and incubated with secondary HRP-conjugated Abs to detect mouse IgM and IgG (Southern Biotechnology Associates). ELISPOT assays were developed by FAST 5-bromo-4-chlor-3-indolyl phosphate/NBT chromogen substrate (Sigma-Aldrich). The number of Ab-secreting spots was quantified by direct visual counting using a dual-axis light dissecting microscope.
Results
PC development is impaired in transgenic c-Myc mice
To determine whether deregulation of c-Myc affected Ag-specific humoral immune responses, we assessed the development of NP-specific PCs in spleen and BM of mice that were immunized with the TD Ag NP-KLH. Results from ELISPOT assays, which quantify the frequency of PCs based upon Ab secretion, showed that immune nontransgenic littermates (control) developed high numbers of spleen anti-NP IgM PCs by day 3 after challenge and diminished to naive levels after 50 days (Fig. 1A, middle column). Coinciding with the decline in spleen IgM PCs, anti-NP PCs that had class switched to produce IgG were measured by day 7 after challenge and significantly decreased in number after 50 days. From day 14 postimmunization onward, the majority of anti-NP IgG PCs were of high affinity as measured by Ab binding to a low hapten ratio (NP3), indicating that affinity maturation had occurred. The reduction in spleen NP-specific IgM and IgG PCs by day 14 illustrates the short-lived nature of most of the splenic PCs quantified. Because it is known that long-lived PCs originate in the spleen, but subsequently migrate to the BM where they persist for weeks to months (38, 39), anti-NP IgG PCs within the BM of immune mice were also quantified. High numbers of anti-NP IgG PCs, the majority being high affinity, were observed by day 50 after challenge (Fig. 1B). Comparison of total IgM and IgG PCs from spleen and BM of immune control animals showed a similar pattern relative to naive groups. In contrast, approximately one-half the number of spleen anti-NP IgM PCs was found in immune transgenic c-Myc heterozygous (c-Myc+/−) mice that decreased to naive levels by day 14 after challenge, and a small yet detectable number of IgG-producing PCs were measured after 14 days. Decreased numbers of splenic IgG PCs did not result from increased migration to BM, since very few NP-specific as well as total IgG PCs were found in BM of immune c-Myc+/− mice. More strikingly was virtually the complete absence of NP-specific IgM and IgG PCs in spleen and BM of immune transgenic c-Myc homozygous (c-Myc+/+) mice. Thus, enforced expression of c-Myc dramatically inhibits the development of NP-specific PC differentiation. These results correlate with higher transgenic c-Myc protein expression in PCs compared with naive B cells in c-Myc+/− and c-Myc+/+ mice (Fig. 2). However, PCs from c-Myc+/− animals expressed comparable c-Myc protein levels to B cells from c-Myc+/+ mice, suggesting that high levels of c-Myc is not generally toxic to peripheral B cells. We further examined the frequency and tissue distribution of B cell subsets of transgenic c-Myc and control mice. Using a previously established flow cytometric strategy (40-42), three transitional B cell subsets, T1, T2, and T3, in addition to follicular (FO) and marginal zone (MZ) B cells can be identified in spleen based on expression of B220, AA4.1, IgM, CD23, and CD21. The number of FO B cells was slightly decreased in spleens from c-Myc+/+ mice compared with controls (Fig. 3A). In contrast, we did not find decreased numbers of MZ, T1, T2, and T3 B cells, suggesting that transgenic c-Myc expression may be important in the development or maintenance of FO B cells. The percentage of CD5+ peritoneal B cells was significantly increased in c-Myc+/+ animals (Fig. 3B). Further analysis of B cell subsets in the peritoneal cavity demonstrated that the percentage of both the B220lowCD19highCD5+ B1a and B220lowCD19highCD5− B1b B cells were increased in c-Myc+/+ mice compared with controls. Thus, c-Myc appears to be important for the development and/or maintenance of B1 B cells. Finally, B cell development was analyzed in BM from c-Myc+/+ and control mice by flow cytometry (Fig. 3C). We detected a decrease in the number of pre-B cells, but no significant difference in the distribution of pro-B, immature, and mature B cells. The reduced number of pre-B cells suggests that increased c-Myc expression may inhibit their differentiation and/or maintenance.
FIGURE 1.

Single transgenic c-Myc mice are impaired in generating Ag-specific PCs. A, Spleen cells prepared at the indicated days from naive and NP-KLH-immune mice were analyzed by ELISPOT for total NP-specific (NP30 binding), high-affinity NP-specific (NP3 binding), and total IgM- or IgG-secreting PCs. B, On days 14 and 50 after challenge, BM cells from mice were analyzed by ELISPOT for NP-specific and total IgG-secreting PCs. Results are expressed as the mean ± SEM for nine mice per group.
FIGURE 2.
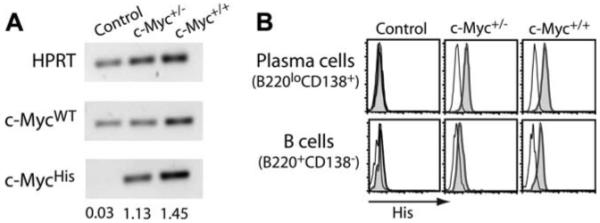
c-Myc transgene expression. A, RT-PCR for endogenous (c-MycWT) and transgenic c-Myc (c-MycHis) was performed using RNA isolated from spleen B cells of control and single transgenic c-Myc+/− or c-Myc+/+ mice. Densitometry values of c-Myc expression relative to HPRT levels are shown. B, Protein levels of transgenic c-Myc were measured on PCs and B cells by intracellular staining of 6-His tag (gray histograms). Rat Ig staining is shown in the black-lined histograms.
FIGURE 3.
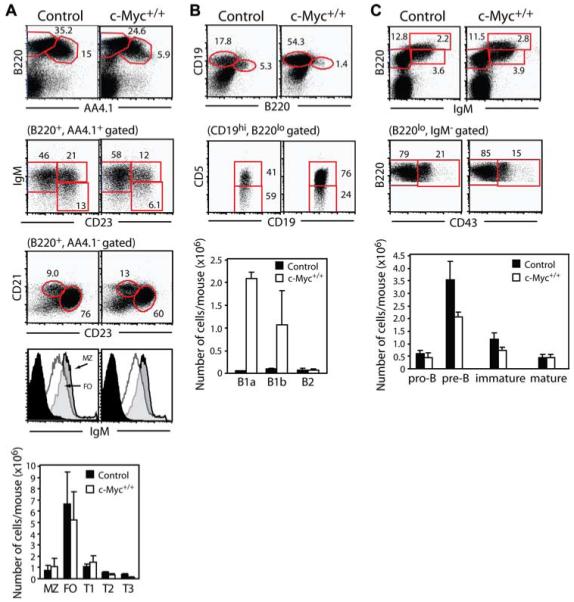
c-Myc transgene expression results in fewer FO B cells, but increased numbers of B1 B cells. A, Freshly isolated splenocytes from control and c-Myc+/+ mice were analyzed using a live cell gate to identify B220+AA4.1− mature B cells and B220+AA4.1+ immature B cells. Immature B cells can be further segregated into IgM+CD23− T1, IgM+CD23+ T2, and IgMlowCD23+ T3 transitional B cell subsets. Mature B cells can be further segregated into CD21highCD23− MZ B cells and CD21lowCD23+ FO B cells. Additional gating on mature B cell subsets shows higher IgM surface expression on MZ B cells (gray histograms) relative to FO B cells (open histograms). Rat Ig staining for nonspecific binding is shown in black histograms. Numbers represent the percentage of total live lymphocytes in each gate or of cells in the gate indicated in parentheses above the plots. Absolute mean number ± SEM of each splenic B cell subset was calculated by multiplying the percentage of total live cells times total cells harvested from spleen of six mice. B, Peritoneal cells were analyzed to identify B220highCD19low B2 cells or B220lowCD19high B1 cells. B1a cells are CD5+ while B1b cells are CD5low/−. Numbers represent the percentage of total live lymphocytes in each gate or of cells in the gate indicated in parentheses above the plots. Absolute numbers of B1a, B1b, and B2 B cells were calculated from the same six mice as described in A. C, BM cells were analyzed to identify B220lowIgM− pro/pre-B cells, B220lowIgM+ immature B cells, and B220highIgM+ mature B cells. Pro-B cells are CD43+ while pre-B cells are CD43−. Numbers are the percentage of total BM cells in each gate or of cells in the gate indicated in parentheses above the plots. Absolute numbers of BM B cell subsets were calculated as described in A.
To evaluate whether poor Ag-specific PC development in transgenic c-Myc mice was limited to either the TD Ag or adjuvant, mice were challenged with various combinations of Ag and adjuvant for 14 days, followed by spleen and BM ELISPOT assays (Fig. 4). Compared with mice immunized with NP-KLH emulsified in CFA, control animals that received NP-KLH precipitated in alum and PhC-OVA in CFA developed a weaker yet measurable IgG PC response. In contrast, significantly reduced numbers of Ag-specific PCs were measured in immune transgenic c-Myc animals. These data indicate that the impaired generation of Ag-specific PCs in c-Myc mice is independent of TD Ag and adjuvant.
FIGURE 4.
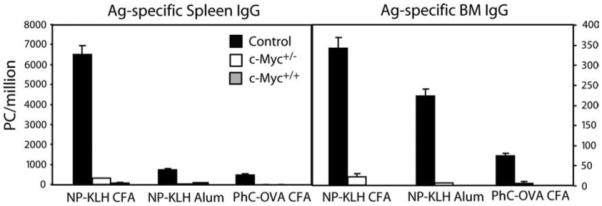
Transgenic c-Myc mice have defective development of Ag-specific PCs regardless of TD Ag and adjuvant. Spleen and BM cells were prepared from mice immunized for 14 days with the indicated TD Ag and adjuvant. NP30- or PC30-specific IgG-secreting PCs were quantified by ELISPOT. Results are expressed as the mean ± SEM for six mice per group.
Previous reports have demonstrated that the humoral immune responses to the TI and TD forms of the hapten NP are dramatically different. In contrast to TD forms, TI Ags such as NP-Ficoll primarily induce a rapid, local IgM response and to a lesser extent IgG (43, 44). To test whether c-Myc-transgenic B cells were impaired in their ability to generate PCs in the absence of T cell help, mice were immunized with NP-Ficoll for 7 days. Control animals showed a normal increase in spleen anti-NP IgM and IgG PCs, but c-Myc+/− and c-Myc+/+ mice had a greatly diminished number of PCs (Fig. 5).
FIGURE 5.
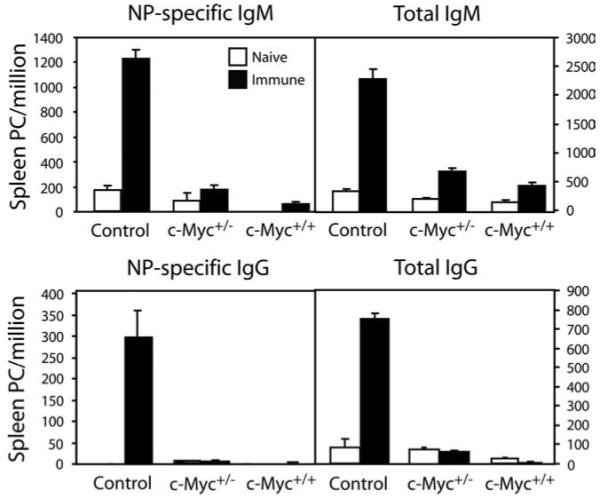
Generation of Ag-specific short-lived PCs is impaired in transgenic c-Myc mice. Spleen cells were prepared from naive mice or animals immunized with the TI Ag NP-Ficoll for 7 days and analyzed by ELISPOT for NP-specific and total IgM- or IgG-secreting PCs. Results are expressed as the mean ± SEM for six mice per group.
Therefore, although transgenic animals can generate Ag-specific PCs, enforced expression of c-Myc to the IgH Cα locus significantly limits the magnitude of the Ag-specific PC response. This is regardless of the form of Ag, adjuvant, and short- or long- lived nature of PCs.
Transgenic c-Myc B cells undergo class switch recombination
The limited IgM- and IgG-secreting PCs in c-Myc mice could result from an inability of B cells to turn on the transcriptional machinery required for PC commitment and/or Ig class switch recombination. Thus, we examined whether transcription of the germline transcripts for IgM and IgG class-specific mRNAs was intact in transgenic c-Myc B cells. Splenic B cells from control and c-Myc animals were cultured in vitro with the polyclonal B cell stimuli, LPS, and agonistic anti-CD40 mAb. After 48 h, RNA was isolated from equivalent numbers of purified B cells. Using primers specific for CH genes (36, 45), germline transcripts as well as postswitch transcripts, in which the Iμ exon is spliced onto the 5′ exon of another CH gene (46), were identified by RT-PCR (Fig. 6). Transcription of germline IgM and IgG1 was intact in c-Myc B cells compared with control groups. In the presence of IL-4, a cofactor that promotes IgG class switching, both LPS and anti-CD40 successfully produced postswitch Iμ-Cγ1 transcripts in all B cells. Furthermore, activation of the transcriptional repressor Blimp-1, which is required for PC differentiation (47, 48), and AID, which is critical for class-switching and somatic hypermutation (49), were equivalent between transgenic c-Myc and control groups. Together, these data indicated that the attenuation of PC development in transgenic c-Myc mice was not due to an inability to activate transcription factors that drive class switching and the terminal differentiation of B cells.
FIGURE 6.
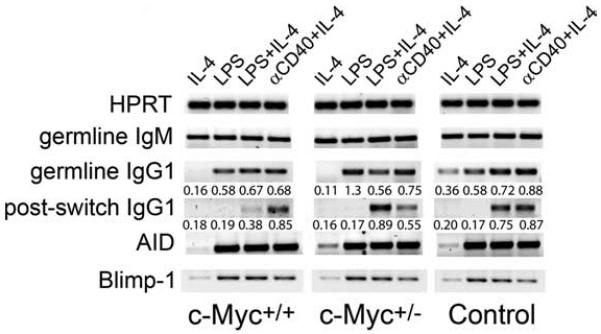
Class switch recombination is intact in transgenic c-Myc B cells. Spleen B cells from transgenic c-Myc and control mice were cultured with the indicated stimuli for 48 h. Cells were tested by RT-PCR for their ability to produce AID, Blimp-1, germline IgM, and IgG1 transcripts as well as postswitch transcripts for IgG1. Data shown are representative of four independent experiments.
Transgenic c-Myc mice have defective Ag-specific PCs and early Bmem despite intact GC responses
To investigate the mechanism responsible for decreased numbers of persisting PCs in c-Myc mice, the expansion and differentiation of Ag-activated B cells into NP-specific subsets were measured following immunization with NP-KLH in CFA. Using a previously established six-color flow cytometric strategy that combines fluorescent-labeled Ag binding with cell surface phenotype (47, 50), NP-specific GC B cells, PCs, and their immediate precursors and Bmem can be identified. To avoid possible cytophilic Ig binding to cells that would erroneously appear as NP+, cells from mice were acid treated to remove any nonmembrane-expressed Ab (51). Staining of spleen B cells with a rat IgG isotype control demonstrated <0.01% nonspecific binding (Fig. 7A, first column). Spleen cells from mice that received adjuvant alone (naive) also demonstrated negligible frequencies of NP+ B cells (Fig. 7A, second column). Fourteen days following challenge, the frequency of NP+IgDlow B cells in spleens of immune control animals significantly expanded to 0.7 ± 0.2% of total live cells (Fig. 7A, third column). In contrast, the percent NP+IgDlow B cells was reduced ~1.4-fold in c-Myc+/− mice and 7-fold in c-Myc+/+ mice relative to control mice. To determine whether the decreased frequencies of NP+IgDlow B cells in immune c-Myc animals segregated with a particular subset, the NP+IgDlow B cell population was gated upon and the expression of CD138 and B220 was examined. This strategy identifies Ag-specific GC B cells (B220+CD138−GL7+), Bmem (B220−CD138−CD79b+), and PCs and their precursors (B220lowCD138+). In control mice, all three NP+ B cell subsets were distinguished (Fig. 7A, fourth column). Expression of the GC marker GL7 (52, 53) further confirmed that NP-specific B cells within the B220+CD138− subset of immune control animals were undergoing a GC response (Fig. 7A, fifth column). In contrast, immune c-Myc-transgenic animals generated reduced numbers of NP-specific CD138+B220low PCs and CD138−B220− Bmem, with the majority of NP+ B cells comprising the CD138−B220+ subset that contained GL7+ GC B cells. Absolute numbers of each NP+ B cell subset were quantified and are proportional to the frequencies measured by FACS (Fig. 7B). Thus, upon enforced Cα-Myc expression, a deficiency of Ag-specific B cells terminally differentiating to long-lived PCs does not result from impaired GC B cell responses. These findings also suggest that c-Myc expression diminishes the development of Bmem, which is further evidenced by a significant decrease in the frequency of NP+IgDlowCD138−B220− B cells in the BM of immune c-Myc+/− and c-Myc+/+ mice relative to control animals (Fig. 7, C and D) where memory can persist (47).
FIGURE 7.

Ag-specific PCs and early developing Bmem are impaired in c-Myc mice. A, The frequency of NP-specific IgDlow B cells was determined from DAPI−CD4−CD8− gated spleen cells of naive and 14-day NP-immune mice. Insets represent the region used for calculating mean ± SEM cell frequencies from four mice per group. Nonspecific binding of rat Ig is shown in the first column. The fourth column shows expression of CD138 and B220 on NP+IgDlow gated cells, with the CD138+B220low population to include PCs and PCpre, the CD138− B220− population to include Bmem, and the CD138−B220+ population to include GC B cells. Representative percentages of each population are shown. Expression of the GC marker GL7 on CD138−B220+ gated cells is shown in the fifth column and is represented by the open black-lined histograms. Rat Ig staining on CD138−B220+ cells is shown in the light gray histograms. B, Total numbers were calculated for the indicated NP-specific cell population from immune animals shown in A. C, The frequency of NP-specific IgDlow B cells was determined from DAPI−CD4−CD8− gated BM cells of 14-day NP-immune mice shown in A. Rat Ig isotype controls for nonspecific binding are shown in the first column. The expression of CD138 and B220 of NP+IgDlow gated cells is shown in the third column. D, Total NP+IgDlow B cells from immune BM of mice in C are shown. Data represent one of three independent experiments.
Enforced c-Myc expression impairs Bmem responses
Bmem function to support the long-lived BM PC pool by providing the host with a rapid burst of Ag-specific PCs at a pace and quality manyfold greater than that observed during the primary encounter with Ag (54). Because the signals that control PC differentiation and survival from post-GC B cells vs Bmem may differ, we determined the functional impact of c-Myc on Bmem during a secondary challenge. Mice were immunized with NP-KLH in CFA for 42 days, followed by secondary challenge to NP-KLH in alum for an additional 5 days (Fig. 8A, denoted by day 47). Recall responses were compared with baseline levels of NP-specific spleen B cells in mice that received adjuvant alone (naive), mice that were immunized with NP-KLH CFA for 42 days only (day 42), and in mice that received NP-KLH alum only for 5 days (day 5). NP+ B cells were absent in naive animals (Fig. 8A, first column), expanded to 1.6 ± 0.1% on day 42 before rechallenge in control animals (Fig. 8A, second column), but were virtually absent in c-Myc mice. Persisting NP+ B cells in day 42 control mice contained Bmem since 5 days after secondary challenge, both the percentage (Fig. 8A, fourth and fifth columns) and absolute number of NP+ B cells (Fig. 8C) showed a significant expansion with 80% representing the CD138−B220− Bmem compartment. This is further supported by the unimodal up-regulated expression of the Igβ BCR coreceptor CD79b (Fig. 8A, sixth column) previously demonstrated to be expressed by this Bmem population (47, 50). In contrast, both the percentage and number of total NP+ B cells as well as CD138−B220− cells were reduced in c-Myc transgenic mice following secondary challenge. Furthermore, NP+IgDlowCD138−B220− Bmem within the spleen of day 47 c-Myc+/− mice bimodally expressed CD79b, which was completely absent in the memory fraction of c-Myc+/+ animals (Fig. 8A, sixth column). Together, the nominal expansion of NP+ spleen B cells, the graded reduction in CD79b expression, and the significantly diminished numbers of total Bmem indicates that increasing levels of Cα-Myc expression impairs the persistence of Bmem. This is further supported by a graded reduction of NP+CD138−B220− Bmem in BM of c-Myc mice (Fig. 8, B and C).
FIGURE 8.

Transgenic heterozygous and homozygous c-Myc mice show a graded defect in the expansion and differentiation of NP-specific Bmem upon secondary Ag exposure. A, The frequency of NP+IgDlow B cells was determined from DAPI−CD4−CD8− gated spleen cells of mice administered adjuvant only (naive), mice that received a primary challenge for 42 days (day 42), mice that received only the secondary challenge over 5 days (day 5), and from 42-day NP-immune animals that received a secondary NP-KLH challenge for an additional 5 days (day 47). Insets represent the region used for calculating mean ± SEM cell frequencies from four mice per group. The fifth column shows expression of CD138 and B220 on NP+IgDlow gated cells as described in Fig. 5. Expression of the Bmem marker CD79b on the CD138−B220− Bmem population is shown in the sixth column and is represented by the black-lined histograms. The gray histograms represent CD79b expression on naive B cells. B, The frequency of NP-specific IgDlow B cells was determined from DAPI−CD4−CD8− gated BM cells of mice shown in A. C, Total numbers were calculated for the indicated NP-specific cell population from spleen (left column) and BM (right column) of immune animals shown in A. D, Spleen and BM cells were prepared from mice examined in A–C. The numbers of PCs secreting high-affinity NP-specific (NP3 binding) IgG relative to total NP-specific (NP30 binding) IgG were quantified by ELISPOT. Data are expressed as the mean ± SEM for eight mice per group.
The functional capacity of Ag-responding Bmem to generate PCs was investigated by performing ELISPOT analyses of spleen and BM cells from immunized mice (Fig. 8D). Control mice that received a secondary challenge generated high numbers of spleen and BM NP-specific IgG PCs, of which the majority were high affinity. In contrast, c-Myc+/− and c-Myc+/+ mice showed approximately a 2- and 8-fold reduction in anti-NP IgG PC numbers. These results are consistent with a graded decrease in Bmem. Thus, although c-Myc does not affect GC responses, affinity maturation, and commitment to PCs or Bmem, increasing c-Myc levels severely diminished the long-term survival of these effector cell populations.
Impaired PC development in transgenic c-Myc mice results from apoptosis and is restricted to mature PCs, but not PCpre
It has been previously shown that naive Cα-Myc-harboring B cells within spleen and lymph nodes have a 2- to 3- fold higher spontaneous proliferative rate and can undergo spontaneous apoptosis or upon in vitro LPS stimulation, compared with control B cells (20). The propensity of transgenic Myc B cells to undergo apoptosis in these studies did not distinguish whether dying B cells were directly linked to a specific activation state. Whether enforced c-Myc expression in B cells led to apoptosis in the context of terminal differentiation was investigated in vitro after stimulation with LPS and agonistic anti-CD40 mAb (Fig. 9A). Purified spleen B cells from control and transgenic c-Myc mice were cultured 48 h with stimuli, followed by FACS to measure apoptosis via intracellular caspase 3 activation in B cells (B220+CD138−) and terminally differentiating PCs (B220lowCD138+). Neither B cells nor PCs of control mice showed positive staining for the cleaved, active form of caspase 3 (Fig. 9, red histograms). Confirmation that caspase 3 staining was indeed at baseline levels was provided by staining of cells from parallel cultures that contained the caspase 3 inhibitor ZVAD (Fig. 9, light gray histograms). Cultured B cells from c-Myc mice also showed negative staining for caspase 3. In contrast, terminally differentiating CD138+ PCs from c-Myc+/− and c-Myc+/+ mice showed positive staining for caspase 3, indicating that a portion of PCs were undergoing apoptosis. This was abrogated with the addition of ZVAD. In vivo examination of NP-specific PCs and Bmem from day 7-challenged mice confirmed that both of these effector B cell populations were undergoing apoptosis in transgenic c-Myc mice (Fig. 9B). Together, these results indicate that c-Myc induces apoptosis of B cells terminally differentiating to a PC and Bmem fate.
FIGURE 9.
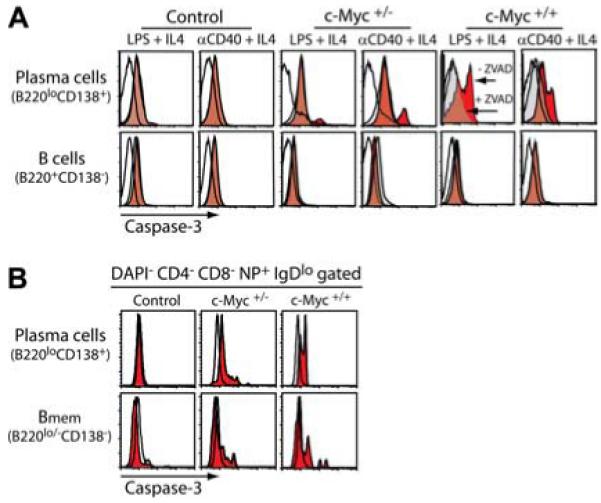
B cells differentiating to a PC fate undergo apoptosis in transgenic c-Myc mice. A, Spleen cells prepared from the indicated mice were depleted of T cells and cultured in vitro with IL-4 and either LPS or agonistic anti-CD40 mAb for 48 h. Cells were collected and apoptosis was measured by intracellular caspase 3 staining of B cells (B220+CD138−) and PCs (B220lowCD138+) by multicolor FACS. Caspase 3 staining of cells cultured with stimuli is represented by the red histograms. As a specificity control, stimulated cells were also cultured in the presence of the caspase 3 inhibitor ZVAD throughout the 48 h and subjected to caspase 3 staining as shown by the light gray histograms. Nonspecific intracellular binding of rat Ig is represented by the open black-lined histograms. Results are representative of four independent experiments. B, Apoptosis of spleen NP+ (DAPI−CD4−CD8−NP+IgDlow gated) PCs and early Bmem from 7-day NP-immune mice was measured by caspase 3 staining as shown by the red histograms. An irrelevant isotype control is represented by the open black-lined histograms. Data shown are representative of six mice per group.
To provide additional evidence that terminally differentiating B cells were selectively undergoing apoptosis, we used multispectral imaging flow cytometry, a novel technology that integrates cell microscopy with flow cytometry in real time. This flow-imaging platform creates digital images of each cell as it passes through the detector, discriminating specific cell types within a heterogeneous population based on fluorescence combined with morphology. The application of this technology to measure apoptosis has been previously established (35, 55). In this study, we take advantage of this platform to determine whether apoptosis of terminally differentiating B cells is restricted to mature Ab-secreting CD138+ PCs, as measured by cytoplasmic Ig production, or their immediate precursors that do not secrete Ab yet also express CD138+.
Spleen cells from control and c-Myc+/+ mice were stimulated with LPS for 48 h, followed by staining for surface B220 and CD138 expression and intracellular Ig. The cationic nucleotide-binding dye 7-AAD was added to samples before analysis. Although 7-AAD binds to both necrotic and apoptotic cells, each type of dying cell is morphologically distinguishable. Necrotic cells harbor intact nuclei, whereas apoptotic cells have fragmented nuclei with condensed chromatin. Apoptotic cells were identified using two standard image-based features from the IDEAS software analysis program: Area and Spot Small Total. Condensed apoptotic nuclei have lower nuclear area values compared with live cells. Also, images of fragmented apoptotic nuclei exhibit small, bright regions with higher Spot Small Total values compared with the uniform images of normal nuclei. Representative digital images of mature Ab-secreting PCs (B220low/− intracellular Ig+CD138+), PCpre (B220+ intracellular Iglow/−CD138+), and B cells (B220+ intracellular Ig−CD138−) from control and c-Myc+/+ B cell cultures shown in Fig. 10A. Results showed that although mature Ab-secreting control PCs had largely intact nuclei, as demonstrated by singular nuclear 7-AAD staining, a significantly greater percentage of c-Myc PCs harbored a condensed, fragmented 7-AAD nuclear-staining pattern. In contrast, neither control nor c-Myc PCpre and B cells showed a discernable 7-AAD-staining pattern for apoptosis. The total percentages of apoptotic mature PCs, PCpre, and B cells are quantified in Fig. 10B. These results confirm that enforced c-Myc expression induces apoptosis of B cells terminally differentiating to PCs and suggests that death is initiated in mature end-stage PCs, whereas PCpre remain intact.
FIGURE 10.
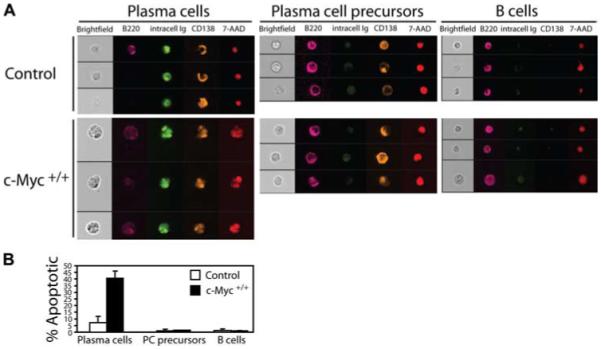
c-Myc- induced apoptosis of terminally differentiating B cells is restricted to mature Ab-secreting PCs. A, Spleen cells from control and c-Myc+/+ mice were depleted of T cells and cultured in vitro with LPS for 48 h. Cells were then collected and surface labeled for expression of B220 and CD138, followed by intracellular staining for Ig. 7-AAD was added to samples immediately before data acquisition on the ImageStream100 cytometer. Bright field (white), B220 (magenta), IgM and IgG (green), CD138 (yellow), and 7-AAD (red) composite images for three representative cells are shown for B220low/− intracellular Ig+CD138+ end-stage PCs, B220+ intracellular Iglow/−CD138+ PCpre, and B220+ intracellular Ig−CD138− B cells. B, The percentage of apoptotic PCs, PCpre, and B cells were quantified using two standard image-based software analysis features: Area and Spot Small Total. Data shown are based on these algorithms calculated from 10,000 images/group and are representative of three independent experiments.
Mature PCs from transgenic c-Myc mice fail to undergo BAFF-mediated survival
We have previously demonstrated that normal long-lived BM PCs express heightened levels of the BCMA receptor, but not transmembrane activator and calcium-modulator and cytophilin ligand interactor (TACI) or BAFF-R relative to mature naive B cells (24). Furthermore, the interaction between BCMA and BAFF or APRIL is critical for long-term PC survival since BCMA−/− mice sustain significantly reduced numbers of BM PCs compared with wild-type control animals. We therefore asked whether BAFF could mediate survival of transgenic c-Myc PCs. To this end, the surface expression of the BAFF receptors TACI, BCMA, and BAFF-R on Ag-responding B cell subsets was examined (Fig. 11A). Because so few PCs were found in the BM of c-Myc animals (Fig. 1B), we chose to examine mature B cells, GC B cells, PCpre, and PCs from spleen of mice 14 days after NP-KLH immunization. We and others have previously demonstrated that 2 wk following immunization is sufficient time for the spleen to generate post-GC PCs destined to become long-lived (38, 56). No significant differences in BAFF-R expression levels were observed in the various B cell subsets among the mice. BCMA was expressed at slightly higher levels on GC B cells and PCpre from c-Myc animals, but was up-regulated on PCs at levels equivalent to control PCs. In contrast, TACI expression levels were significantly up-regulated on PCs from c-Myc animals compared with control PCs.
FIGURE 11.
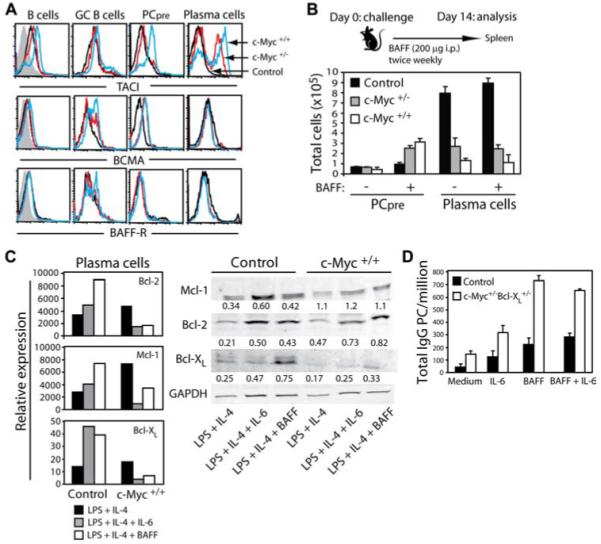
PCs from transgenic c-Myc mice fail to respond to BAFF-mediated survival. A, Expression of the BAFF receptors TACI, BCMA, and BAFF-R on the indicated population is shown from spleen cells of 14-day NP-immune mice. Receptor expression levels on gated cells are represented by the black-lined histograms for control mice, red-lined histograms for c-Myc+/− mice, and blue-lined histograms for c-Myc+/+ mice. The gray histograms in the first column represent nonspecific rat Ig staining. Data are representative of eight mice per group. B, Mice were immunized with NP-KLH and administered BAFF i.p. twice weekly for 2 wk. Total numbers of spleen PC and PCpre were quantified on day 14, based on their respective phenotype. Results are expressed as the mean ± SEM for eight mice per group. C, Spleen B cells from control and c-Myc+/+ mice were cultured in vitro for 48 h with LPS plus IL-4 with or without IL-6 or BAFF. Cells were then collected and RNA was isolated from purified CD138+ PCs. Real-time PCR analysis was performed and the relative expression of antiapoptotic genes was determined compared with HPRT. Protein levels of each gene were determined from CD138+ PCs by Western blot analysis. Densitometry values of protein expression relative to GAPDH are shown. D, BM cells containing spontaneous-developing PCs were prepared from c-Myc+/−Bcl-xL +/− mice and cultured in vitro for 5 days with the indicated stimuli. BM cells from 42-day NP-immune control mice were cultured in the same manner as a source of normal long-lived PCs. The survival of total IgG-secreting PCs was quantified by ELISPOT. Data are expressed as the mean ± SEM for three independent experiments.
Genetic studies in mice have demonstrated TACI to negatively regulate B cell proliferation, Ig secretion, and survival (57-59), and under certain conditions direct engagement of TACI using agonistic mAbs results in apoptosis (57, 59). To determine whether high TACI expression on c-Myc PCs influenced apoptosis, ex vivo PCs and those generated in vitro upon LPS stimulation were cultured 48 h with graded doses of recombinant BAFF and APRIL ligand or agonistic anti-TACI mAb. Results demonstrated that there was no difference in the percentage of caspase 3-positive PCs from cultures containing ligand and anti-TACI mAb compared with control groups (data not shown). These findings implied that within the context of c-Myc-induced apoptosis of PCs, signaling through TACI did not initiate death. An alternative hypothesis is that heightened TACI expression on c-Myc PCs increased the threshold for BAFF signaling required for their continued survival. To test this possibility, mice were challenged with NP-KLH and administered exogenous BAFF twice weekly for 2 wk. The absolute numbers of spleen PCs and PCpre were quantified after 14 days (Fig. 11B). Results demonstrated that increasing BAFF levels did not improve the persistence of mature PCs beyond numbers observed in mice expressing endogenous BAFF only. Consistent with the aforementioned in vitro studies, these findings indicate that BCMA/TACI-BAFF interactions fail to support survival in c-Myc-harboring PCs. In contrast, heightened levels of BAFF resulted in a 3-fold greater number of PCpre in c-Myc mice, indicating that exogenous BAFF was biologically active in vivo and that PCpre but not end-stage PCs are sensitive to BAFF-mediated growth and/or survival.
Studies in both human and mouse PC malignancies have demonstrated that up-regulation of antiapoptotic genes of the Bcl-2 family contributes to tumor survival. In human MM, increased expression of Bcl-xL and Mcl-1 control survival (60-62), whereas mouse plasmacytomas and healthy PCs appear to rely more on Bcl-xL and Bcl-2 for their survival (63-65). To determine whether enforced c-Myc expression altered expression of Bcl-2 family members, in vitro-generated control and c-Myc spleen CD138+ PCs were purified and transcription of Bcl-2, Mcl-1, and Bcl-xL were tested by real-time PCR (Fig. 11C). Results demonstrated that expression of these genes was equivalent, if not higher, in c-Myc-harboring PCs compared with control PCs generated in response to LPS plus IL-4. This indicates that c-Myc-induced apoptosis of PCs does not result from an inability to transcribe these genes. Of interest, however, is that in contrast to control PCs, the survival factors IL-6 and BAFF significantly diminished the expression of all three genes in c-Myc PCs. Further examination at the protein level demonstrated that Mcl-1, Bcl-2. and Bcl-xL expression levels increased in control PCs with IL-6 and BAFF. PCs from c-Myc+/+ mice had a similar pattern of Mcl-1 and Bcl-2 protein expression in response to IL-6 and BAFF. In contrast, basal levels of Bcl-xL protein expression was low in c-Myc+/+ PCs and was not significantly increased after IL-6 and BAFF stimulation. These findings suggest that although targeted expression of c-Myc to the IgH Cα locus prevents BAFF-induced expression of anti-apoptotic genes in mature PCs, c-Myc impairs PC survival by inhibiting Bcl-xL protein expression.
Because Bcl-xL plays a critical role in the survival of human and mouse PC malignancies, we crossed single transgenic c-Myc mice with mice that express constitutive Bcl-xL within the B cell compartment to test whether enforced expression of this Bcl-2 family member might permit PC sensitivity to IL-6 and BAFF, thus restoring survival. It has been previously demonstrated that Myc/Bcl-xL double-transgenic mice spontaneously generate PCs and develop PC tumors that mimic human MM (20). Spleen cells containing spontaneous-developing PCs from c-Myc+/−Bcl-xL +/− double-transgenic mice were compared with normal long-lived PCs from control animals for survival in response to IL-6 and BAFF. After 5 days, cell cultures were harvested and ELISPOT analysis was performed to quantify the number of mature Absecreting IgG PCs (Fig. 11D). Results demonstrated that relative to cultures containing medium only, the addition of IL-6, BAFF, or the combination of the two stimuli significantly increased the survival of control PCs. Survival of PCs from double-transgenic mice was supported by IL-6, but was dramatically increased with the addition of BAFF. These data indicate that although enforced expression of Bcl-xL promotes neoplastic transformation of c-Myc-harboring PCs, these mature Ab-secreting cells evade death by now being responsive to BAFF.
Discussion
Deregulation of c-Myc is tightly associated with many B cell neoplasms, including those that involve the transformation of terminally differentiated PCs. Yet, how c-Myc controls the differentiation fate of post-GC B cells resulting in a tumor-initiating effector cell is unknown. In this study, we have demonstrated that targeting Myc to the Igh Cα locus controls the development of short- and long-lived effector cells. The findings establish that enforced c-Myc expression dramatically impairs the primary Ab response to TI and TD Ags, as measured by reduced numbers of PCs (Figs. 1 and 5). These data indicated that regardless of T cell help, there was an inherent defect in the magnitude of the short- and long-lived PC response. Reduced PC frequencies were not attributable to a particular Ag and/or adjuvant specificity (Fig. 4) and, furthermore, were not caused by an inability to form GC responses, class switch, or to turn on the transcriptional machinery required for PC commitment (Figs. 6 and 7). Thus, Ag-activated B cells harboring a T (12, 15) translocation where Myc is inserted into the Cα gene that preserves Eμ,Eα, and all other regulatory elements within the Igh locus progress normally through the molecular and cellular processes leading up to the final maturation of a PC. Further examination of PC viability revealed that c-Myc triggered apoptosis of PCs (Figs. 9 and 10), thus accounting for reduced PC numbers generated in vitro and in vivo. Of particular interest is that apoptosis mediated by c-Myc is specific to the final stage of PC maturation when Ab is secreted while PCpre that express membrane Ig, but do not actively secrete Ab yet, give rise to end-stage PCs survived normally. These data indicate that the survival requirements for PCpre and PCs are distinct and that transitioning from a PCpre to a mature PC renders them susceptible to c-Myc-induced apoptosis. The same c-Myc pathway that provokes PC apoptosis also facilitates the apoptosis of Bmem, resulting in a significantly diminished Ag recall response (Figs. 8 and 9). Thus, both cellular arms of long-lived B cell immunity are controlled by Myc juxtaposed to the Cα locus. These results pose at least two major questions. First, why is c-Myc-induced suicide restricted to Bmem and mature Ab-secreting PCs? And, second, what additional elements relieve cell death and allow c-Myc tumor-initiating PCpre to become persisting malignant PCs?
To address the first question it appears that where the t(12;15) translocation occurs within the IgH locus determines which B cell developmental stage is targeted for tumorigenesis by c-Myc. Targeting Myc to the Igh Cα locus has vastly different consequences than expressing Myc under the control of the Ig H chain enhancer Eμ. Transgenic Eμ-Myc mice develop clonal early B cell lymphomas and leukemias as opposed to plasmacytomas (12). These clinical features are consistent with the timing of the translocation in that VDJ recombination-mediated chromosomal breakage in a pre-B cell underlies the endemic BL-typical t(8;14) exchange, whereas class switch recombination-mediated breakage in a GC B cell underlies the sporadic BL-typical t(8;14) exchange. Recent experiments using mice deficient in AID support this view (66-69) even though definitive evidence in humans is still lacking. c-Myc-mediated apoptosis is also observed in Eμ-Myc mice, but in contrast to Cα-Myc-transgenic mice takes place in immature and mature B cells. Interestingly, Eμ-Myc B cell apoptosis is blocked if Bcl-xL and Bcl-2 expression is enforced (70), which are both found to be highly expressed in spontaneously occurring lymphomas from Eμ-Myc mice (32). Therefore, it seems that the positioning of Myc within the Igh locus determines the B cell developmental stage for tumor susceptibility, but regardless of position requires disabling of Bcl-xL and/or Bcl-2 suppression to birth a fully malignant cell. A point of note, however, is that although c-Myc and Bcl-xL may drive 100% tumor incidence in mice, there are likely other events that must occur in addition to attenuation of apoptosis for full penetrance since most animals get single or limited clonal malignancies (21).
Next, deregulated Myc expression significantly alters the mammalian cell cycle by tipping the balance toward positive regulators, the cyclin-dependent kinases (CDK) and D-type cyclins (71-73), and away from negative regulation by CDK inhibitors (74-77). Thus, the homeostatic regulation of G1 to S transition in normal mammalian cells is hijacked by Myc overexpression, leading to uncontrolled proliferation. We postulate that cell cycle control is a critical determinant in the demise of Myc-harboring terminally differentiated effector B cells. The CDK inhibitor p18INK4c, in particular, has previously been shown to bind to and negatively regulate CDK6 (78, 79), leading to the inhibition of phosphorylated Rb by CDK6 and cycle arrest in G1 (80). Importantly, p18INK4c is required for generating functional Ab-secreting PCs because a deficiency in this gene results in a profound decrease of Ag-specific primary and secondary Ab responses (81). Strikingly similar to our studies, p18INK4c appears to act at a post-GC stage, since GC formation, somatic mutation, class switch recombination, and BM homing of PCs were all intact. Thus, while Ag-responsive p18INK4c-deficient B cells could commit to a PC fate, as demonstrated by their transcriptional profile, they failed to cycle arrest and consequently underwent apoptosis. These data indicate that withdrawal from cell cycle is necessary for PC survival. Therefore, enforced Myc expression likely disallows cycle arrest upon terminal differentiation and because of this opposing nature the mature PC is triggered to die. Results from preliminary studies to investigate cell cycle genes show that PCs isolated from c-Myc mice express dramatically reduced p18INK4c as well as p27Kip1, suggesting that enforced c-Myc expression may mimic the apoptosis of p18INK4c-deficient PCs (S. E. Khuda and L. D. Erickson, unpublished data). Although deletion of p18INK4c has been reported in human MM cell lines (82), this might explain why a deletion of p18 is rarely seen in primary MM cells (83). We believe that the reason PCpre are unaffected by Myc-induced death is due to their inherent proliferative nature, cells at this stage of terminal differentiation require constant cell cycle progression for both their self-renewal and further differentiation to Ab-secreting PCs (27). This is consistent with the finding that B1 B cells, also a self-renewing population, are increased in transgenic c-Myc mice (Fig. 3). Accumulation of additional survival advantage mutations would be required within the tumor-initiating PCpre compartment or upon final maturation to PCs for tumor progression.
Whether Myc induces apoptosis of Bmem and PCs via an imbalance in cell cycle regulation or simply by these cells having too much Myc that it triggers a stress response leading to death needs further study. But the fact that apoptotic pathways need to be disabled for Myc to promote transformation tells us that a tumor-initiating cell can progress only so far before a survival checkpoint within the cell is breached and instructs it to die. This scenario is likely to occur as the tumor-initiating cell attempts to become a tumor-propagating type cell. The most direct connection of Myc and Bcl-2 family proteins has come from studies demonstrating that Myc suppresses Bcl-2 or Bcl-xL, particularly in precancerous B cells of Eμ-Myc-transgenic mice, and therefore are more susceptible to death. Both of these antiapoptotic genes are normally induced by IL-6 to promote survival (84). Myc could in theory disrupt the IL-6 and/or other survival factor antiapoptotic signaling pathways triggering cell death. Some evidence lending support to this hypothesis comes from our data showing that although both IL-6 and BAFF significantly increased Bcl-2, Mcl-1, and Bcl-xL expression in control PCs, these antiapoptotic genes were profoundly suppressed by IL-6 and BAFF in Myc-harboring PCs (Fig. 11C). Normal PCs are dependent on the BM microenvironment for survival, growth, and differentiation. These processes are, in part, mediated by paracrine IL-6 and BAFF. Our results indicate that these natural means for PC survival are themselves an essential checkpoint that, if disrupted by Myc, guards the cell from further transformation by dying but can be resurrected if this checkpoint is overridden by enforced Bcl-xL (Fig. 11D). This checkpoint appears to be selective to the final stage of PC differentiation, despite BCMA and high TACI receptor expression, since tumor-initiating PCpre not only persist in Myc-transgenic mice but proliferate favorably in response to BAFF (Fig. 11B). Furthermore, it is possible that c-Myc does not allow Bmem to be sustained by BAFF signaling either, but a recent report indicates that Bmem survival is independent of BAFF (85). Exactly why Cα-Myc Bmem are dying is an important question that will require more study.
In human primary MM cells and established cell lines, it has been shown that TACI expression is bimodal with TACIhigh MM cells resembling BM PCs and TACIlow MM cells resembling the characteristics of plasmablasts (86, 87). The gene expression signature of TACI-expressing MM cells suggests that TACIhigh tumors represent those that are dependent on the BM microenvironment for survival (86) and, furthermore, are growth arrested by the BAFF/APRIL inhibitor atacicept (88). These studies suggest that TACI expression may be a valuable predictive marker in identifying patients with plasmacytomas who are likely to respond favorably to atacicept. We suggest that high TACI expression may furthermore be a useful diagnostic indicator for more accurately predicting the risk of progression in patients who are asymptomatic, but harbor proliferating monoclonal PCs as in the monoclonal gammopathy of undetermined significance disorder. All in all, our findings point to the notion that PCpre is the target cell population for transformation and that an additional oncogenic event(s), which enables BAFF dependency for survival, must occur as they mature to functional Ab-secreting malignant PCs.
Acknowledgments
We thank Joanne Lannigan and Michael Solga of the University of Virginia Flow Cytometry Core for expert technical assistance. We thank Drs. Timothy Bender and Ulrike Lorenz for helpful discussions. We thank Dr. Brian P. O’Connor for assistance in preparing this manuscript.
Footnotes
This work was supported by grants from the American Cancer Society and the Leukemia Research Foundation (to L.D.E.) and by the University of Virginia Cancer Center for Core support.
- PC
- plasma cell
- TI
- T cell independent
- TD
- T cell dependent
- GC
- germinal center
- BM
- bone marrow
- MM
- multiple myeloma
- BAFF
- B cell-activating factor belonging to the TNF family
- BL
- Burkitt’s lymphoma
- BCMA
- B cell maturation Ag
- PCpre
- PC precursor
- NP
- (4-hydroxy-3-nitrophenyl)acetyl
- KLH
- keyhole limpet hemocyanin
- ZVAD
- benzylocarbonyl-Val-Ala-Asp(OMe)-fluoromethylketone
- DAPI
- 4′,6-diamidino-2-phenylindole
- 7-AAD
- 7-aminoactinomycin D
- CT
- cycle threshold
- FO
- follicular
- CDK
- cyclin-dependent kinase
- MZ
- marginal zone
- AID
- activation-induced cytidine deaminase
- Bmem
- B memory cell
- TACI
- transmembrane activator and calcium-modulator and cytophilin ligand interactor
Disclosures
The authors have no financial conflict of interest.
References
- 1.Shapiro-Shelef M, Calame K. Regulation of plasma-cell development. Nat. Rev. Immunol. 2005;5:230–242. doi: 10.1038/nri1572. [DOI] [PubMed] [Google Scholar]
- 2.Manz RA, Radbruch A. Plasma cells for a lifetime? Eur. J. Immunol. 2002;32:923–927. doi: 10.1002/1521-4141(200204)32:4<923::AID-IMMU923>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 3.Benner R, Hijmans W, Haaijman JJ. The bone marrow: the major source of serum immunoglobulins, but still a neglected site of antibody formation. Clin. Exp. Immunol. 1981;46:1–8. [PMC free article] [PubMed] [Google Scholar]
- 4.Shaffer AL, Yu X, He Y, Boldrick J, Chan EP, Staudt LM. BCL-6 represses genes that function in lymphocyte differentiation, inflammation, and cell cycle control. Immunity. 2000;13:199–212. doi: 10.1016/s1074-7613(00)00020-0. [DOI] [PubMed] [Google Scholar]
- 5.Bergsagel PL, Chesi M, Nardini E, Brents LA, Kirby SL, Kuehl WM. Promiscuous translocations into immunoglobulin heavy chain switch regions in multiple myeloma. Proc. Natl. Acad. Sci. USA. 1996;93:13931–13936. doi: 10.1073/pnas.93.24.13931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bergsagel PL, Kuehl WM. Chromosome translocations in multiple myeloma. Oncogene. 2001;20:5611–5622. doi: 10.1038/sj.onc.1204641. [DOI] [PubMed] [Google Scholar]
- 7.Avet-Louseau H, Daviet A, Sauner S, Bataille R. Chromosome 13 abnormalities in multiple myeloma are mostly monosomy 13. Br. J. Haematol. 2000;111:1116–1117. doi: 10.1046/j.1365-2141.2000.02488.x. [DOI] [PubMed] [Google Scholar]
- 8.Shou Y, Martelli ML, Gabrea A, Qi Y, Brents LA, Roschke A, Dewald G, Kirsch IR, Bergsagel PL, Kuehl WM. Diverse karyotypic abnormalities of the c-myc locus associated with c-myc dysregulation and tumor progression in multiple myeloma. Proc. Natl. Acad. Sci. USA. 2000;97:228–233. doi: 10.1073/pnas.97.1.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ohno S, Babonits M, Wiener F, Spira J, Klein G, Potter M. Non-random chromosome changes involving the Ig gene-carrying chromosomes 12 and 6 in pristane-induced mouse plasmacytomas. Cell. 1979;18:1001–1007. doi: 10.1016/0092-8674(79)90212-5. [DOI] [PubMed] [Google Scholar]
- 10.Wiener F, Babonits M, Bregula U, Klein G, Leonard A, Wax JS, Potter M. High resolution banding analysis of the involvement of strain BALB/c- and AKR-derived chromosomes No. 15 in plasmacytoma-specific translocations. J. Exp. Med. 1984;159:276–291. doi: 10.1084/jem.159.1.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shen-Ong GL, Keath EJ, Piccoli SP, Cole MD. Novel myc oncogene RNA from abortive immunoglobulin-gene recombination in mouse plasmacytomas. Cell. 1982;31:443–452. doi: 10.1016/0092-8674(82)90137-4. [DOI] [PubMed] [Google Scholar]
- 12.Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RD, Brinster RL. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318:533–538. doi: 10.1038/318533a0. [DOI] [PubMed] [Google Scholar]
- 13.Suda Y, Aizawa S, Hirai S, Inoue T, Furuta Y, Suzuki M, Hirohashi S, Ikawa Y. Driven by the same Ig enhancer and SV40 T promoter ras induced lung adenomatous tumors, myc induced pre-B cell lymphomas and SV40 large T gene a variety of tumors in transgenic mice. EMBO J. 1987;6:4055–4065. doi: 10.1002/j.1460-2075.1987.tb02751.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmidt EV, Pattengale PK, Weir L, Leder P. Transgenic mice bearing the human c-myc gene activated by an immunoglobulin enhancer: a pre-B-cell lymphoma model. Proc. Natl. Acad. Sci. USA. 1988;85:6047–6051. doi: 10.1073/pnas.85.16.6047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yukawa K, Kikutani H, Inomoto T, Uehira M, Bin SH, Akagi K, Yamamura K, Kishimoto T. Strain dependency of B and T lymphoma development in immunoglobulin heavy chain enhancer (Eμ)-myc transgenic mice. J. Exp. Med. 1989;170:711–726. doi: 10.1084/jem.170.3.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Butzler C, Zou X, Popov AV, Bruggemann M. Rapid induction of B-cell lymphomas in mice carrying a human IgH/c-mycYAC. Oncogene. 1997;14:1383–1388. doi: 10.1038/sj.onc.1200968. [DOI] [PubMed] [Google Scholar]
- 17.Palomo C, Zou X, Nicholson IC, Butzler C, Bruggemann M. B-cell tumorigenesis in mice carrying a yeast artificial chromosome-based immunoglobulin heavy/c-myc translocus is independent of the heavy chain intron enhancer (Eμ) Cancer Res. 1999;59:5625–5628. [PubMed] [Google Scholar]
- 18.Madisen L, Groudine M. Identification of a locus control region in the immunoglobulin heavy-chain locus that deregulates c-myc expression in plasmacytoma and Burkitt’s lymphoma cells. Genes Dev. 1994;8:2212–2226. doi: 10.1101/gad.8.18.2212. [DOI] [PubMed] [Google Scholar]
- 19.Janz S. Myc translocations in B cell and plasma cell neoplasms. DNA Repair. 2006;5:1213–1224. doi: 10.1016/j.dnarep.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 20.Cheung WC, Kim JS, Linden M, Peng L, Van Ness B, Polakiewicz RD, Janz S. Novel targeted deregulation of c-Myc cooperates with Bcl-xL to cause plasma cell neoplasms in mice. J. Clin. Invest. 2004;113:1763–1773. doi: 10.1172/JCI20369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grandori C, Cowley SM, James LP, Eisenman RN. The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu. Rev. Cell Dev. Biol. 2000;16:653–699. doi: 10.1146/annurev.cellbio.16.1.653. [DOI] [PubMed] [Google Scholar]
- 22.Hipfner DR, Cohen SM. Connecting proliferation and apoptosis in development and disease. Nat. Rev. Mol. Cell Biol. 2004;5:805–815. doi: 10.1038/nrm1491. [DOI] [PubMed] [Google Scholar]
- 23.Nilsson JA, Cleveland JL. Myc pathways provoking cell suicide and cancer. Oncogene. 2003;22:9007–9021. doi: 10.1038/sj.onc.1207261. [DOI] [PubMed] [Google Scholar]
- 24.O’Connor BP, Raman VS, Erickson LD, Cook WJ, Weaver LK, Ahonen C, Lin LL, Mantchev GT, Bram RJ, Noelle RJ. BCMA is essential for the survival of long-lived bone marrow plasma cells. J. Exp. Med. 2004;199:91–98. doi: 10.1084/jem.20031330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Claudio JO, Masih-Khan E, Tang H, Goncalves J, Voralia M, Li ZH, Nadeem V, Cukerman E, Francisco-Pabalan O, Liew CC, et al. A molecular compendium of genes expressed in multiple myeloma. Blood. 2002;100:2175–2186. doi: 10.1182/blood-2002-01-0008. [DOI] [PubMed] [Google Scholar]
- 26.Novak AJ, Darce JR, Arendt BK, Harder B, Henderson K, Kindsvogel W, Gross JA, Greipp PR, Jelinek DF. Expression of BCMA, TACI, and BAFF-R in multiple myeloma: a mechanism for growth and survival. Blood. 2004;103:689–694. doi: 10.1182/blood-2003-06-2043. [DOI] [PubMed] [Google Scholar]
- 27.O’Connor BP, Cascalho M, Noelle RJ. Short-lived and long-lived bone marrow plasma cells are derived from a novel precursor population. J. Exp. Med. 2002;195:737–745. doi: 10.1084/jem.20011626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’Connor BP, Vogel LA, Zhang W, Loo W, Shnider D, Lind EF, Ratliff M, Noelle RJ, Erickson LD. Imprinting the fate of antigen-reactive B cells through the affinity of the B cell receptor. J. Immunol. 2006;177:7723–7732. doi: 10.4049/jimmunol.177.11.7723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Billadeau D, Ahmann G, Greipp P, Van Ness B. The bone marrow of multiple myeloma patients contains B cell populations at different stages of differentiation that are clonally related to the malignant plasma cell. J. Exp. Med. 1993;178:1023–1031. doi: 10.1084/jem.178.3.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yaccoby S, Epstein J. The proliferative potential of myeloma plasma cells manifest in the SCID-hu host. Blood. 1999;94:3576–3582. [PubMed] [Google Scholar]
- 31.Pilarski LM, Belch AR. Clonotypic myeloma cells able to xeno-graft myeloma to nonobese diabetic severe combined immunodeficient mice co-purify with CD34+ hematopoietic progenitors. Clin. Cancer Res. 2002;8:3198–3204. [PubMed] [Google Scholar]
- 32.Eischen CM, Woo D, Roussel MF, Cleveland JL. Apoptosis triggered by Myc-induced suppression of Bcl-xL or Bcl-2 is bypassed during lymphomagenesis. Mol. Cell. Biol. 2001;21:5063–5070. doi: 10.1128/MCB.21.15.5063-5070.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jacobsen KA, Prasad VS, Sidman CL, Osmond DG. Apoptosis and macrophage-mediated deletion of precursor B cells in the bone marrow of Eμ-myc transgenic mice. Blood. 1994;84:2784–2794. [PubMed] [Google Scholar]
- 34.Maclean KH, Keller UB, Rodriguez-Galindo C, Nilsson JA, Cleveland JL. c-Myc augments γ irradiation-induced apoptosis by suppressing Bcl-xL. Mol. Cell. Biol. 2003;23:7256–7270. doi: 10.1128/MCB.23.20.7256-7270.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.George TC, Basiji DA, Hall BE, Lynch DH, Ortyn WE, Perry DJ, Seo MJ, Zimmerman CA, Morrissey PJ. Distinguishing modes of cell death using the ImageStream multispectral imaging flow cytometer. Cytometry. 2004;59:237–245. doi: 10.1002/cyto.a.20048. [DOI] [PubMed] [Google Scholar]
- 36.Suzuki A, Kaisho T, Ohishi M, Tsukio-Yamaguchi M, Tsubata T, Koni PA, Sasaki T, Mak TW, Nakano T. Critical roles of Pten in B cell homeostasis and immunoglobulin class switch recombination. J. Exp. Med. 2003;197:657–667. doi: 10.1084/jem.20021101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Erickson LD, Durell BG, Vogel LA, O’Connor BP, Cascalho M, Yasui T, Kikutani H, Noelle RJ. Short-circuiting long-lived humoral immunity by the heightened engagement of CD40. J. Clin. Invest. 2002;109:613–620. doi: 10.1172/JCI14110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Slifka MK, Antia R, Whitmire JK, Ahmed R. Humoral immunity due to long-lived plasma cells. Immunity. 1998;8:363–372. doi: 10.1016/s1074-7613(00)80541-5. [DOI] [PubMed] [Google Scholar]
- 39.Manz RA, Thiel A, Radbruch A. A lifetime of plasma cells in the bone marrow. Nature. 1997;388:133–134. doi: 10.1038/40540. [DOI] [PubMed] [Google Scholar]
- 40.Allman D, Lindsley RC, DeMuth W, Rudd K, Shinton SA, Hardy RR. Resolution of three nonproliferative immature splenic B cell subsets reveals multiple selection points during peripheral B cell maturation. J. Immunol. 2001;167:6834–6840. doi: 10.4049/jimmunol.167.12.6834. [DOI] [PubMed] [Google Scholar]
- 41.Loder F, Mutschler B, Ray RJ, Paige CJ, Sideras P, Torres R, Lamers MC, Carsetti R. B cell development in the spleen takes place in discrete steps and is determined by the quality of B cell receptor-derived signals. J. Exp. Med. 1999;190:75–89. doi: 10.1084/jem.190.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martin F, Oliver AM, Kearney JF. Marginal zone and B1 B cells unite in the early response against T-independent blood-borne particulate antigens. Immunity. 2001;14:617–629. doi: 10.1016/s1074-7613(01)00129-7. [DOI] [PubMed] [Google Scholar]
- 43.Lalor PA, Nossal GJ, Sanderson RD, McHeyzer-Williams MG. Functional and molecular characterization of single, (4-hydroxy-3-nitrophenyl)acetyl (NP)-specific, IgG1+ B cells from antibody-secreting and memory B cell pathways in the C57BL/6 immune response to NP. Eur. J. Immunol. 1992;22:3001–3011. doi: 10.1002/eji.1830221136. [DOI] [PubMed] [Google Scholar]
- 44.Maizels N, Bothwell A. The T-cell-independent immune response to the hapten NP uses a large repertoire of heavy chain genes. Cell. 1985;43:715–720. doi: 10.1016/0092-8674(85)90244-2. [DOI] [PubMed] [Google Scholar]
- 45.Li SC, Rothman PB, Zhang J, Chan C, Hirsh D, Alt FW. Expression of Iμ-Cγ hybrid germline transcripts subsequent to immunoglobulin heavy chain class switching. Int. Immunol. 1994;6:491–497. doi: 10.1093/intimm/6.4.491. [DOI] [PubMed] [Google Scholar]
- 46.Esser C, Radbruch A. Immunoglobulin class switching: molecular and cellular analysis. Annu. Rev. Immunol. 1990;8:717–735. doi: 10.1146/annurev.iy.08.040190.003441. [DOI] [PubMed] [Google Scholar]
- 47.Shapiro-Shelef M, Lin KI, McHeyzer-Williams LJ, Liao J, McHeyzer-Williams MG, Calame K. Blimp-1 is required for the formation of immunoglobulin secreting plasma cells and pre-plasma memory B cells. Immunity. 2003;19:607–620. doi: 10.1016/s1074-7613(03)00267-x. [DOI] [PubMed] [Google Scholar]
- 48.Calame KL, Lin KI, Tunyaplin C. Regulatory mechanisms that determine the development and function of plasma cells. Annu. Rev. Immunol. 2003;21:205–230. doi: 10.1146/annurev.immunol.21.120601.141138. [DOI] [PubMed] [Google Scholar]
- 49.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 50.McHeyzer-Williams LJ, Cool M, McHeyzer-Williams MG. Antigen-specific B cell memory: expression and replenishment of a novel B220− memory B cell compartment. J. Exp. Med. 2000;191:1149–1166. doi: 10.1084/jem.191.7.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Erickson LD, Vogel LA, Cascalho M, Wong J, Wabl M, Durell BG, Noelle RJ. B cell immunopoiesis: visualizing the impact of CD40 engagement on the course of T cell-independent immune responses in an Ig transgenic system. Eur. J. Immunol. 2000;30:3121–3131. doi: 10.1002/1521-4141(200011)30:11<3121::AID-IMMU3121>3.0.CO;2-M. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Laszlo G, Hathcock KS, Dickler HB, Hodes RJ. Characterization of a novel cell-surface molecule expressed on subpopulations of activated T and B cells. J. Immunol. 1993;150:5252–5262. [PubMed] [Google Scholar]
- 53.Han S, Dillon SR, Zheng B, Shimoda M, Schlissel MS, Kelsoe G. V(D)J recombinase activity in a subset of germinal center B lymphocytes. Science. 1997;278:301–305. doi: 10.1126/science.278.5336.301. [DOI] [PubMed] [Google Scholar]
- 54.McHeyzer-Williams LJ, McHeyzer-Williams MG. Antigen-specific memory B cell development. Annu. Rev. Immunol. 2005;23:487–513. doi: 10.1146/annurev.immunol.23.021704.115732. [DOI] [PubMed] [Google Scholar]
- 55.Guzman ML, Li X, Corbett CA, Rossi RM, Bushnell T, Liesveld JL, Hebert J, Young F, Jordan CT. Rapid and selective death of leukemia stem and progenitor cells induced by the compound 4-benzyl,2-methyl, 1,2,4-thiadiazolidine-3,5-dione (TDZD-8) Blood. 2007;110:4436–4444. doi: 10.1182/blood-2007-05-088815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Slifka MK, Matloubian M, Ahmed R. Bone marrow is a major site of long-term antibody production after acute viral infection. J. Virol. 1995;69:1895–1902. doi: 10.1128/jvi.69.3.1895-1902.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Seshasayee D, Valdez P, Yan M, Dixit VM, Tumas D, Grewal IS. Loss of TACI causes fatal lymphoproliferation and autoimmunity, establishing TACI as an inhibitory BLyS receptor. Immunity. 2003;18:279–288. doi: 10.1016/s1074-7613(03)00025-6. [DOI] [PubMed] [Google Scholar]
- 58.Yan M, Wang H, Chan B, Roose-Girma M, Erickson S, Baker T, Tumas D, Grewal IS, Dixit VM. Activation and accumulation of B cells in TACI-deficient mice. Nat. Immunol. 2001;2:638–643. doi: 10.1038/89790. [DOI] [PubMed] [Google Scholar]
- 59.Sakurai D, Kanno Y, Hase H, Kojima H, Okumura K, Kobata T. TACI attenuates antibody production costimulated by BAFF-R and CD40. Eur. J. Immunol. 2007;37:110–118. doi: 10.1002/eji.200636623. [DOI] [PubMed] [Google Scholar]
- 60.Spets H, Stromberg T, Georgii-Hemming P, Siljason J, Nilsson K, Jernberg-Wiklund H. Expression of the bcl-2 family of pro- and anti-apoptotic genes in multiple myeloma and normal plasma cells: regulation during interleukin-6 (IL-6)-induced growth and survival. Eur. J. Haematol. 2002;69:76–89. doi: 10.1034/j.1600-0609.2002.01549.x. [DOI] [PubMed] [Google Scholar]
- 61.Zhang B, Potyagaylo V, Fenton RG. IL-6-independent expression of Mcl-1 in human multiple myeloma. Oncogene. 2003;22:1848–1859. doi: 10.1038/sj.onc.1206358. [DOI] [PubMed] [Google Scholar]
- 62.Puthier D, Derenne S, Barille S, Moreau P, Harousseau JL, Bataille R, Amiot M. Mcl-1 and Bcl-xL are co-regulated by IL-6 in human myeloma cells. Br. J. Haematol. 1999;107:392–395. doi: 10.1046/j.1365-2141.1999.01705.x. [DOI] [PubMed] [Google Scholar]
- 63.Ursini-Siegel J, Zhang W, Altmeyer A, Hatada EN, Do RK, Yagita H, Chen-Kiang S. TRAIL/Apo-2 ligand induces primary plasma cell apoptosis. J. Immunol. 2002;169:5505–5513. doi: 10.4049/jimmunol.169.10.5505. [DOI] [PubMed] [Google Scholar]
- 64.Underhill GH, George D, Bremer EG, Kansas GS. Gene expression profiling reveals a highly specialized genetic program of plasma cells. Blood. 2003;101:4013–4021. doi: 10.1182/blood-2002-08-2673. [DOI] [PubMed] [Google Scholar]
- 65.Gauthier ER, Piche L, Lemieux G, Lemieux R. Role of bcl-xL in the control of apoptosis in murine myeloma cells. Cancer Res. 1996;56:1451–1456. [PubMed] [Google Scholar]
- 66.Ramiro AR, Jankovic M, Eisenreich T, Difilippantonio S, Chen-Kiang S, Muramatsu M, Honjo T, Nussenzweig A, Nussenzweig MC. AID is required for c-myc/IgH chromosome translocations in vivo. Cell. 2004;118:431–438. doi: 10.1016/j.cell.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 67.Dorsett Y, Robbiani DF, Jankovic M, Reina-San-Martin B, Eisenreich TR, Nussenzweig MC. A role for AID in chromosome translocations between c-myc and the IgH variable region. J. Exp. Med. 2007;204:2225–2232. doi: 10.1084/jem.20070884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jankovic M, Nussenzweig A, Nussenzweig MC. Antigen receptor diversification and chromosome translocations. Nat. Immunol. 2007;8:801–808. doi: 10.1038/ni1498. [DOI] [PubMed] [Google Scholar]
- 69.Pasqualucci L, Bhagat G, Jankovic M, Compagno M, Smith P, Muramatsu M, Honjo T, Morse HC, 3rd, Nussenzweig MC, Dalla-Favera R. AID is required for germinal center-derived lymphomagenesis. Nat. Genet. 2008;40:108–112. doi: 10.1038/ng.2007.35. [DOI] [PubMed] [Google Scholar]
- 70.Strasser A, Harris AW, Bath ML, Cory S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature. 1990;348:331–333. doi: 10.1038/348331a0. [DOI] [PubMed] [Google Scholar]
- 71.Bouchard C, Thieke K, Maier A, Saffrich R, Hanley-Hyde J, Ansorge W, Reed S, Sicinski P, Bartek J, Eilers M. Direct induction of cyclin D2 by Myc contributes to cell cycle progression and sequestration of p27. EMBO J. 1999;18:5321–5333. doi: 10.1093/emboj/18.19.5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hermeking H, Rago C, Schuhmacher M, Li Q, Barrett JF, Obaya AJ, O’Connell BC, Mateyak MK, Tam W, Kohlhuber F, et al. Identification of CDK4 as a target of c-Myc. Proc. Natl. Acad. Sci. USA. 2000;97:2229–2234. doi: 10.1073/pnas.050586197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Roussel MF, Cleveland JL, Shurtleff SA, Sherr CJ. Myc rescue of a mutant CSF-1 receptor impaired in mitogenic signalling. Nature. 1991;353:361–363. doi: 10.1038/353361a0. [DOI] [PubMed] [Google Scholar]
- 74.Seoane J, Le HV, Massague J. Myc suppression of the p21Cip1 Cdk inhibitor influences the outcome of the p53 response to DNA damage. Nature. 2002;419:729–734. doi: 10.1038/nature01119. [DOI] [PubMed] [Google Scholar]
- 75.Staller P, Peukert K, Kiermaier A, Seoane J, Lukas J, Karsunky H, Moroy T, Bartek J, Massague J, Hanel F, Eilers M. Repression of p15INK4b expression by Myc through association with Miz-1. Nat. Cell Biol. 2001;3:392–399. doi: 10.1038/35070076. [DOI] [PubMed] [Google Scholar]
- 76.Wu S, Cetinkaya C, Munoz-Alonso MJ, von der Lehr N, Bahram F, Beuger V, Eilers M, Leon J, Larsson LG. Myc represses differentiation-induced p21CIP1 expression via Miz-1-dependent interaction with the p21 core promoter. Oncogene. 2003;22:351–360. doi: 10.1038/sj.onc.1206145. [DOI] [PubMed] [Google Scholar]
- 77.Yang W, Shen J, Wu M, Arsura M, FitzGerald M, Suldan Z, Kim DW, Hofmann CS, Pianetti S, Romieu-Mourez R, et al. Repression of transcription of the p27Kip1 cyclin-dependent kinase inhibitor gene by c-Myc. Oncogene. 2001;20:1688–1702. doi: 10.1038/sj.onc.1204245. [DOI] [PubMed] [Google Scholar]
- 78.Guan KL, Jenkins CW, Li Y, Nichols MA, Wu X, O’Keefe CL, Matera AG, Xiong Y. Growth suppression by p18, a p16INK4/MTS1- and p14INK4B/MTS2-related CDK6 inhibitor, correlates with wild-type pRb function. Genes Dev. 1994;8:2939–2952. doi: 10.1101/gad.8.24.2939. [DOI] [PubMed] [Google Scholar]
- 79.Hirai H, Roussel MF, Kato JY, Ashmun RA, Sherr CJ. Novel INK4 proteins, p19 and p18, are specific inhibitors of the cyclin D-dependent kinases CDK4 and CDK6. Mol. Cell. Biol. 1995;15:2672–2681. doi: 10.1128/mcb.15.5.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Morse L, Chen D, Franklin D, Xiong Y, Chen-Kiang S. Induction of cell cycle arrest and B cell terminal differentiation by CDK inhibitor p18INK4c and IL-6. Immunity. 1997;6:47–56. doi: 10.1016/s1074-7613(00)80241-1. [DOI] [PubMed] [Google Scholar]
- 81.Tourigny MR, Ursini-Siegel J, Lee H, Toellner KM, Cunningham AF, Franklin DS, Ely S, Chen M, Qin XF, Xiong Y, et al. CDK inhibitor p18INK4c is required for the generation of functional plasma cells. Immunity. 2002;17:179–189. doi: 10.1016/s1074-7613(02)00364-3. [DOI] [PubMed] [Google Scholar]
- 82.Kulkarni MS, Daggett JL, Bender TP, Kuehl WM, Bergsagel PL, Williams ME. Frequent inactivation of the cyclin-dependent kinase inhibitor p18 by homozygous deletion in multiple myeloma cell lines: ectopic p18 expression inhibits growth and induces apoptosis. Leukemia. 2002;16:127–134. doi: 10.1038/sj.leu.2402328. [DOI] [PubMed] [Google Scholar]
- 83.Tasaka T, Berenson J, Vescio R, Hirama T, Miller CW, Nagai M, Takahara J, Koeffler HP. Analysis of the p16INK4A, p15INK4B and p18INK4C genes in multiple myeloma. Br. J. Haematol. 1997;96:98–102. doi: 10.1046/j.1365-2141.1997.8552482.x. [DOI] [PubMed] [Google Scholar]
- 84.Packham G, White EL, Eischen CM, Yang H, Parganas E, Ihle JN, Grillot DA, Zambetti GP, Nunez G, Cleveland JL. Selective regulation of Bcl-xL by a Jak kinase-dependent pathway is bypassed in murine hematopoietic malignancies. Genes Dev. 1998;12:2475–2487. doi: 10.1101/gad.12.16.2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Benson MJ, Dillon SR, Castigli E, Geha RS, Xu S, Lam KP, Noelle RJ. Cutting edge: the dependence of plasma cells and independence of memory B cells on BAFF and APRIL. J. Immunol. 2008;180:3655–3659. doi: 10.4049/jimmunol.180.6.3655. [DOI] [PubMed] [Google Scholar]
- 86.Moreaux J, Cremer FW, Reme T, Raab M, Mahtouk K, Kaukel P, Pantesco V, De Vos J, Jourdan E, Jauch A, et al. The level of TACI gene expression in myeloma cells is associated with a signature of microenvironment dependence versus a plasmablastic signature. Blood. 2005;106:1021–1030. doi: 10.1182/blood-2004-11-4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Moreaux J, Hose D, Jourdan M, Reme T, Hundemer M, Moos M, Robert N, Moine P, De Vos J, Goldschmidt H, Klein B. TACI expression is associated with a mature bone marrow plasma cell signature and c-MAF overexpression in human myeloma cell lines. Haematologica. 2007;92:803–811. doi: 10.3324/haematol.10574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yaccoby S, Pennisi A, Li X, Dillon SR, Zhan F, Barlogie B, Shaughnessy JD., Jr. Atacicept (TACI-Ig) inhibits growth of TACI(high) primary myeloma cells in SCID-hu mice and in coculture with osteoclasts. Leukemia. 2008;22:406–413. doi: 10.1038/sj.leu.2405048. [DOI] [PMC free article] [PubMed] [Google Scholar]