

Abstract
Transmissible spongiform encephalopathies (TSEs) are lethal, infectious disorders of the mammalian nervous system. A TSE hallmark is the conversion of the cellular protein PrPC to disease-associated PrPSc (named for scrapie, the first known TSE). PrPC is protease-sensitive, monomeric, detergent soluble, and primarily α-helical; PrPSc is protease-resistant, polymerized, detergent insoluble, and rich in β-sheet. The “protein-only” hypothesis posits that PrPSc is the infectious TSE agent that directly converts host-encoded PrPC to fresh PrPSc, harming neurons and creating new agents of infection. To gain insight on the conformational transitions of PrP, we tested the ability of several protein chaperones, which supervise the conformational transitions of proteins in diverse ways, to affect conversion of PrPC to its protease-resistant state. None affected conversion in the absence of pre-existing PrPSc. In its presence, only two, GroEL and Hsp104 (heat shock protein 104), significantly affected conversion. Both promoted it, but the reaction characteristics of conversions with the two chaperones were distinct. In contrast, chemical chaperones inhibited conversion. Our findings provide new mechanistic insights into nature of PrP conversions, and provide a new set of tools for studying the process underlying TSE pathogenesis.
Keywords: transmissible spongiform encephalopathy, aggregation, GroEL, Hsp104
The family of transmissible spongiform encephalopathies (TSEs) include scrapie in sheep, bovine spongiform encephalopathy or “mad cow disease” in cattle, and several rare human neuropathies: Creutzfeld–Jacob disease, fatal familial insomnia, Gertsmann–Straussler–Scheinker syndrome, and kuru (1, 2). A central event in TSE pathogenesis is the accumulation in the nervous system of an abnormally folded version (PrPSc) of a normal cellular protein, PrPC. Griffith (3) first proposed a “protein-only” model to explain the unconventional behavior of the infectious TSE agent. Indeed, the “prion”, a term by which the agent is popularly known today, appears to be almost entirely proteinaceous: consisting primarily of PrPSc (1, 2).
Several lines of evidence show that PrPC is conformationally distinct from PrPSc, although both molecules derive from the same primary sequence and have no detectable posttranslational differences (1, 2, 4–6). The conversion of PrPC to PrPSc appears to involve direct interactions of PrPC with pre-existing PrPSc (1, 2). However, the exact mechanism underlying conversion is not known. Genetic and inhibitor studies have suggested that other cellular factors may influence TSE pathogenesis or serve as regulators of disease (7–12). None have been conclusively identified; however, cellular osmolytes (sometimes called chemical chaperones; ref. 13) and protein chaperones have been frequently speculated to be among them (7, 10–12). The goal of this study was to assess whether or not molecular chaperones, whose known functions are to alter the conformational states of proteins (14–16), regulate the conversion of PrPC to PrPSc.
To test for chaperone involvement, we used a cell-free assay, wherein metabolically labeled [35S]PrPC, purified from cultured cells in an acid-treated state, is converted to a conformational state characteristic of PrPSc (17, 18). In this altered state, PrP is aggregated and a specific portion of the molecule is highly resistant to proteolysis. This simple in vitro conversion reaction faithfully recapitulates several salient TSE features. First, like experimental TSEs, in vitro conversion of PrPC to its protease-resistant form requires pre-existing PrPSc (17–22). Second, strain-specific PrPSc protease digestion properties, specifically those associated with two mink TSE strains—hyper and drowsy—were precisely propagated from PrPSc to radiolabeled PrPC in this assay (19). Third, the known in vivo barriers to transmitting TSEs between different species were reflected well in the efficiencies of in vitro conversion (20, 21). Last, this cell-free assay modeled accurately another in vivo TSE barrier, based on genetic polymorphisms in PrP, which render sheep either highly susceptible, moderately susceptible, or resistant to scrapie (22). Together, these studies provide substantial evidence that in vitro converted, protease-resistant PrP is either authentic PrPSc or has a very similar conformation. However, because neither the putative infectious nature of pure PrPSc protein nor that of the in vitro converted PrP has been demonstrated, we refer to the in vitro converted material operationally as protease-resistant PrP (PrP-res).
Here, we provide the first evidence that molecular chaperones can regulate conformational transitions in PrP. Two protein chaperones, GroEL and Hsp104 (heat shock protein 104), promoted in vitro conversion; in contrast, the chemical chaperones, sucrose, trehalose, and dimethyl sulfoxide (DMSO) inhibited it. Importantly, our results with chaperones demonstrate that in vitro converted PrP-res is a bona fide conformationally altered PrP molecule. Chaperones provide new understanding of the nature of PrP intermediates involved in PrP conversion and provide evidence that the conversion process has two steps. We propose that, if chaperone-like molecules supervise PrPSc formation in TSEs in vivo, such molecules will represent important clinical targets to combat this dreaded disease.
MATERIALS AND METHODS
Chaperone Proteins.
Yeast Hsp40 (Ydj1), Hsp70 (ssa1/ssa2), and Hsp104 [wild type (WT) and mutant] were purified as described (23–25) and were generous gifts of J. R. Glover and Y. Kimura (University of Chicago, IL). Bacterial GroES and GroEL (WT and mutant) were kindly provided by A. L. Horwich (Yale University, New Haven, CT). Hsp26 was a generous gift of T. Suzuki and E. Vierling (University of Arizona, Tucson), and yeast Hsp90 was kindly provided by J. Buchner (Universität Regensburg, Regensburg, Germany).
Chaperone Folding Assays.
Hsp104 promoted the refolding of kinetically trapped, denatured luciferase, but only when Hsp40, Hsp70, and ATP were also present (J. R. Glover and S.L., unpublished data). The function of other chaperones were assessed by using previously published procedures. GroEL and GroES activities were measured by the refolding of denatured rhodanese (26); Hsp90 suppressed the aggregation of β-galactosidase (27); and Hsp26 activity was measured by the suppression of aggregation of malate dehydrogenase (28).
PrP Purification.
PrPSc was purified from hamsters infected with 263K strain of scrapie as described (17). Hamster [35S]PrPC and [35S]PrPGPI- proteins were purified from cultured cells by a procedure described by Caughey et al. (18), except that radiolabeled proteins were eluted with 0.1 M acetic acid at 22°C for 30 min and stored at 4°C before use. To obtain nonglycosylated [35S]PrPC, cultured cells were pre-incubated and 35S-labeled in the presence of 2 μg/ml tunicamycin (Boehringer Mannheim), an inhibitor of glycosylation (18).
Cell-Free PrP Conversion.
Unless otherwise stated, all reactions were performed by using the same modification of a published procedure (18). [35S]PrPC (20,000 cpm, ≈3 ng) denatured in 0.1 M acetic acid was diluted into 1× conversion buffer [CB; 50 mM sodium citrate-HCl (pH 6.0) supplemented with 1% N-lauryl sarkosine]. PrPSc (100 ng) was incubated with [35S]PrPC (20 μl volume) at 37°C for 24 hr. When indicated, PrPSc was pretreated for 1 hr with either 2 M Gdn⋅HCl at 37°C or 4 M urea at 22°C; in conversion reactions, Gdn⋅HCl and urea were present at 0.2 M and 0.4 M, respectively. In chaperone-mediated conversions, chaperones (1 μM, unless otherwise stated) were added to CB prior to the addition of [35S]PrPC and PrPSc. Reactions with chaperones contained 10 mM MgCl2, 1.5 mM NaCl, and 140 mM KCl, and unless otherwise stated, 5 mM ATP. All reactions with ATP included an ATP regenerating system containing 20 mM phosphocreatine and 10 μg/ml creatine phosphokinase. These supplements did not affect PrP conversion (data not shown). For each reaction, one-tenth to one-fifth of the sample was left untreated for determination of percent conversion of [35S]PrPC to PrP-res (18). The remainder was digested with proteinase K (PK; 80 μg/ml) for 1 hr at 37°C, and both PK-untreated and PK-treated samples were prepared for SDS/PAGE (18). [35S]PrP products were visualized in dried gels by phosphorimaging and quantified with imagequant software (Molecular Dynamics).
RESULTS
Chaperones Alone Do Not Convert PrPC to PrP-res.
We first examined the ability of major cellular chaperones GroES (Hsp10), Hsp26, Hsp40, GroEL (Hsp60), Hsp70, Hsp90, and Hsp104, to promote [35S]PrPC conversion in the absence of PrPSc. These chaperones were chosen because they employ different mechanisms to affect the conformation and physical state of other proteins (14–16). In separate experiments, these same chaperone preparations functioned appropriately in a variety of protein folding assays (data not shown). Yet, over a broad range of concentrations, alone and in various combinations, with (Fig. 1A) or without ATP (data not shown), none of these chaperones promoted the conversion of PrPC to PrP-res in the absence of PrPSc. This observation strongly underscores the importance of pre-existing PrPSc in the conversion of PrPC.
Figure 1.
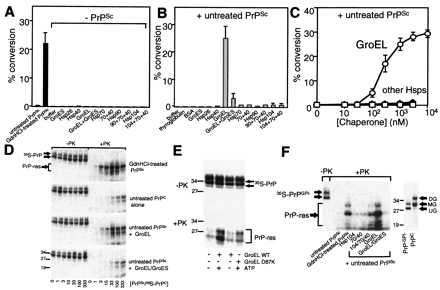
Effects of chaperones on cell-free conversion of [35S]PrPC to its protease-resistant form. (A) Conversions (as percent of total [35S]PrPC) obtained after 24 hr either with PrPSc or without PrPSc (100 ng), but with the indicated chaperones (each at 5 μM, with 5 mM ATP), by using the standard assay described. In indicated reaction (second from left), PrPSc was partially denatured with guanidinium hydrochloride (Gdn⋅HCl). Identical results were obtained, over a broad range of chaperone concentrations, with or without ATP (data not shown). (B) Conversions performed as in A, with the addition of untreated PrPSc. Mean values are from three to six experiments, with standard errors. Buffers for storing various chaperones differed slightly in salt and glycerol content, but none affected conversion (data not shown). (C) Concentration-dependent effects of chaperones in promoting conversion with untreated PrPSc. Other heat shock proteins were tested as in A. (D) SDS/PAGE phosphorimage of [35S]PrPC products from representative conversion reactions obtained with 3 ng [35S]PrPC and increasing amounts of PrPSc (3–1,000 ng). One-tenth of each reaction was left untreated (−PK); the remainder was digested with proteinase K (+PK). GroEL and GroES were at 1 μM. When indicated, PrPSc was partially denatured with Gdn⋅HCl. PrPSc fold represents the ratio of PrPSc/[35S]PrPC in the reaction. (E) ATP dependence of GroEL-mediated conversions. SDS/PAGE phosphorimages of representative conversion reactions obtained with untreated PrPSc and GroEL (WT and mutant D87K), with or without ATP. Both proteinase K-treated (+PK; Lower) and untreated samples (−PK, one-fifth sample; Upper) are shown. (F) [35S]PrPGPI- conversions with or without chaperones. Reactions contained either untreated PrPSc or Gdn⋅HCl-treated PrPSc, and a variant PrP missing the GPI anchor, [35S]PrPGPI-. [35S]PrPGPI- and [35S]PrPC preparations are compared (Right): UG, unglycosylated; MG, monoglycosylated; and DG, diglycosylated PrP species as indicated.
GroEL Promotes Conversion in Reactions Nucleated with Untreated PrPSc.
Next, we asked whether chaperones influenced [35S]PrPC conversion in the presence of PrPSc. To date, efficient in vitro conversion of PrPC to PrP-res has usually required partial chemical denaturation of PrPSc (Fig. 1A, left bars; refs. 17 and 18). Untreated and completely denatured PrPSc (6 M Gdn⋅HCl pretreatment) have little (Fig. 1D) and no converting ability, respectively (17, 18). We first asked whether chaperones influenced conversion with PrP-res that was not subjected to partial denaturation. Several chaperones produced reproducible, but very small increases in conversion (Fig. 1 B and D). One, however, facilitated conversion at a high level (Fig. 1 A and B). With GroEL, typically 25–30%, and occasionally 50–100%, of converted [35S]PrPC.
Notably GroEL not only reduced by 10-fold the quantity of PrPSc required for detectable conversion, but also increased by more than 10-fold the maximal levels of conversion attained, compared with reactions nucleated with the same preparation of untreated PrPSc, but not with GroEL (Fig. 1D). These effects of GroEL were dose-dependent (Fig. 1C).
GroEL Effects Require ATP, But Not GroES.
GroEL-promoted protein folding usually, but not always, requires the cochaperone GroES and ATP (14, 15). PrP conversion was not observed in the absence of ATP (Fig. 1E). Moreover, two point mutants of GroEL, which block release of substrate (D87K and 337/349; ref. 29), strongly reduced conversion (Fig. 1E, and data not shown). Surprisingly, however, the stimulating effects of GroEL on [35S]PrPC conversion were consistently eliminated by GroES (Fig. 1D). This inhibition was caused by an effect of GroES on GroEL, rather than on PrP, because GroES did not inhibit the denaturant-promoted conversion of [35S]PrPC that occurs in the absence of GroEL (data not shown).
Posttranslational PrP Modifications Modestly Affect Chaperone-Promoted Conversions.
We used a PrP mutant that lacks the glycosylphosphatidylinositol (GPI) anchor (PrPGPI-; refs. 17 and 18) and accumulates in mono- and unglycosylated form (Fig. 1F Right) to determine whether these natural modifications affect chaperone-mediated conversion. Again, of the various chaperones tested, GroEL was the only one that efficiently stimulated conversion in the presence of untreated PrPSc (Fig. 1F, and data not shown). And once again, GroEL-promoted effects were ablated in the absence of ATP (data not shown) and inhibited by GroES (Fig. 1F). With this form of PrP, however, conversion was more efficient (typically 30–40%). Moreover, conversion was also achieved with a combination of Hsp104, Hsp70, and Hsp40, albeit less consistently and less strongly than with GroEL (Fig. 1F). Results similar to those obtained with PrPGPI- were also obtained with unglycosylated [35S]PrPC purified from cells cultured with tunicamycin (data not shown). Therefore, the ability of the chaperones to mediate the conversion of [35S]PrPC to PrP-res was modestly facilitated by the absence of N-linked sugars or the GPI anchor.
Conversion Kinetics Reveal a Two-Step Process.
When [35S]PrPC converts to PrP-res, it becomes associated with PrPSc, which is a pelletable aggregate (refs. 18 and 30, and data not shown). To gain insight into the chaperone-mediated conversion process, we analyzed the kinetics of conversion, monitoring both protease resistance and insolubility. GroEL promoted the acquisition of both of these signature features of PrPSc in [35S]PrP (Fig. 2 A and B). In reactions driven with untreated PrPSc and GroEL, protease resistance was acquired at a pace similar to that observed in reactions nucleated with partially denatured PrPSc in the absence of GroEL (Fig. 2A). Moreover, in both sets of reactions, protease-resistant radioactivity was found only in pelletable material (Fig. 2C). Surprisingly, however, when the rate at which [35S]PrP became insoluble was examined, the chaperone-driven reaction showed very different kinetics than those driven by partially denatured PrPSc. No pelletable radioactivity was detected at two hours in reactions driven by partially denatured PrPSc (Fig. 2 A and B). In striking contrast, in chaperone-driven reactions, the conversion of PrP to a pelletable form was virtually complete in 2 hr. This conversion occurred long before [35S]PrP converted to its characteristic protease-resistant form (Fig. 2 A and B). This pelleting of [35S]PrPC was almost certainly caused by an association with pre-existing PrPSc, because in parallel reactions with GroEL, but without PrPSc, most [35S]PrPC remained soluble (Fig. 2B).
Figure 2.
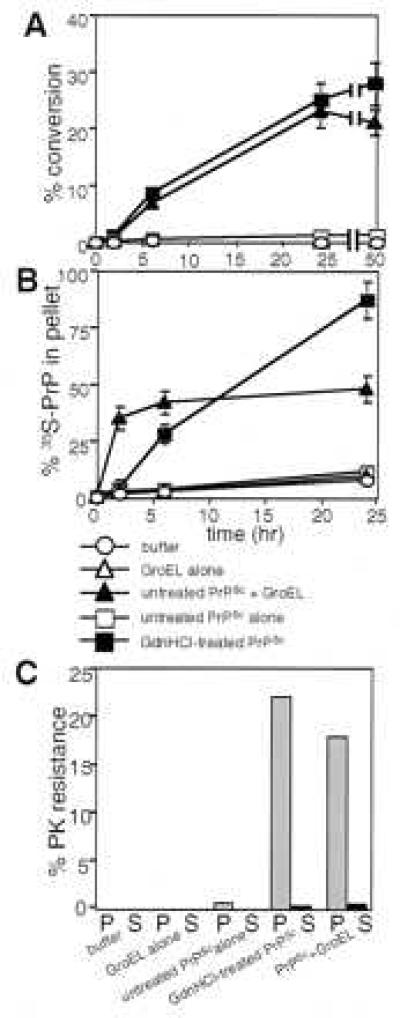
Time course of conversion with or without chaperone. (A) Appearance of PrP-res at 2, 6, 24, and 48 hr, in reactions treated with proteinase K and analyzed by quantitative phosphorimaging of SDS/PAGE. Mean values are from three independent measurements, with standard errors. (B) Pelletable [35S]PrP determined by quantitative phosphorimaging of SDS/PAGE. At the indicated times, [35S]PrP reaction products were centrifuged at 15,000 × g for 30 min at 22°C. After separating the supernatant fraction (S), the pelletable fraction (P) was resuspended in conversion buffer, and both fractions were prepared for SDS/PAGE. Mean values are from three independent experiments, with standard errors. (C) Protease-resistant [35S]PrP in pellet (P) and supernatant (S) fractions quantified from SDS/PAGE phosphorimages of 24-hr reactions. Averages are of two independent experiments.
In Reactions Nucleated with Partially Denatured PrPSc, Hsp104 also Promotes Conversion.
Although we did not detect a substantial activity for other chaperones in promoting conversion with untreated PrPSc, another chaperone was effective in reactions seeded with partially denatured PrPSc. For these reactions, a milder denaturant, urea, was used because some chaperones are sensitive to inhibition by Gdn⋅HCl (ref. 31; J. R. Glover and S.L., unpublished observations with Hsp104). Moreover, the lower basal rate of conversion obtained with urea (Fig. 3A, buffer) allowed us to test the ability of other chaperones to either inhibit or stimulate conversion. None inhibited (Fig. 3A). Several stimulated, but only to a small degree (Fig. 3A). Strikingly, under these conditions, in addition to GroEL, Hsp104 strongly stimulated conversion (Fig. 3A). With Hsp104, typically 20–30%, occasionally more than 50% of total [35S]PrPC converted. The stimulatory effects of Hsp104 required partial denaturation of PrPSc, with pretreatments in 3–4 M urea being optimal (Fig. 3B).
Figure 3.
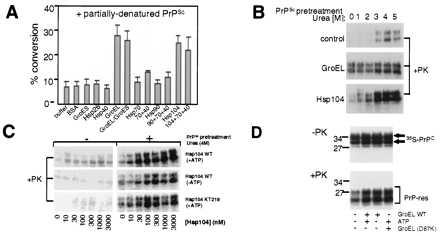
Combined effects of chaperones and partially denatured PrPSc on conversion. (A) Conversions obtained with partially denatured PrPSc (4 M urea pretreatment) with buffer alone, or with the indicated chaperones and control proteins (each at 5 μM). Mean values are from three to six independent measurements, with standard errors. (B) SDS/PAGE phosphorimage of representative conversion reactions obtained with untreated PrPSc (0) or PrPSc partially denatured in the presence of increasing urea concentrations (1–5 M), with or without chaperone (Hsp104 or GroEL, 3 μM). Only proteinase K-treated (+PK) samples are shown. (C) SDS/PAGE phosphorimage of representative conversion reactions obtained with Hsp104 (WT or mutant KT218), with or without ATP, and untreated or partially denatured PrPSc (4 M urea pretreatment). Only proteinase K-treated samples (+PK) are shown. (D) SDS/PAGE phosphorimage of representative conversion reactions obtained with partially denatured PrPSc (4 M urea pretreatment), with or without ATP, and with or without GroEL (WT or mutant D87K). Both proteinase K-treated (+PK; Lower) and untreated samples (−PK, one-fifth sample; Upper) are shown.
Folded State of PrPSc Governs Properties of Chaperone-Promoted Conversion.
Although some Hsp104 functions require ATP (16, 32), in these reactions nucleotide was somewhat stimulatory but was not required (Fig. 3C). Furthermore, two ATPase-deficient Hsp104 mutants (KT218 and KT620; ref. 33) promoted [35S]PrPC conversion nearly as well as wild-type Hsp104 (Fig. 3C, and data not shown).
Remarkably, the use of partially denatured PrPSc changed the character of conversions promoted by GroEL as well. These conversions lost ATP-dependence (Fig. 3D). Moreover, they became refractory to GroES inhibition (Fig. 3A). Thus, chaperone-mediated conversions are mechanistically distinct in reactions nucleated with partially denatured PrPSc, and those nucleated by untreated PrPSc.
Chemical Chaperones Inhibit Conversion.
We also tested the effects of several small organic molecules (or chemical chaperones) known to affect protein folding: sucrose, glycerol, trehalose, DMSO, and the cyclodextrin compounds (8, 13, 34). None of the compounds we tested affected [35S]PrPC conversions in reactions without PrPSc, nor in reactions seeded with untreated PrPSc (data not shown). In reactions seeded with partially denatured PrPSc, DMSO had a complex dose-dependent effect, intermediate levels (1–3%) stimulated conversion 2- to 3-fold and higher levels (up to 30%) virtually eliminated conversion (Fig. 4). Glycerol (Fig. 4) and cyclodextrin compounds (α-, β-, γ-forms; data not shown) had no effect. Sucrose and trehalose inhibited conversion. This inhibition was observed only at high concentrations, but is physiologically relevant because these osmolytes are known to accumulate to such levels in vivo under stressful conditions (34).
Figure 4.
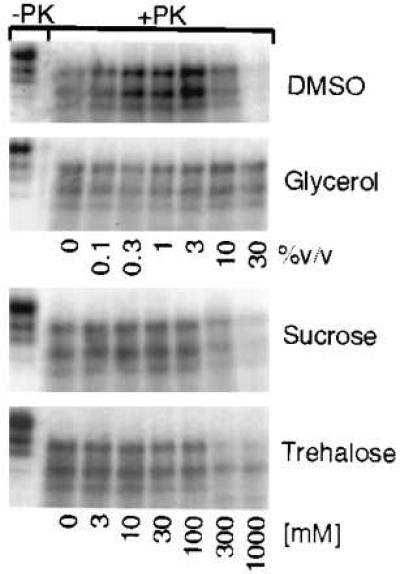
Conversion of [35S]PrPC in the presence of chemical chaperones. SDS/PAGE phosphorimages of representative conversion reactions obtained with partially denatured PrPSc (4 M urea pretreatment) in the presence of increasing concentrations of DMSO, glycerol, sucrose, or trehalose.
DISCUSSION
Recently, protein chaperones and small organic molecules have figured prominently among cellular factors speculated to influence conversion of PrPC to PrPSc (7, 8, 11, 12). In scrapie-infected cells, some of the same organic molecules we tested have been shown to reduce the rate of PrPSc formation (8). We provide the first evidence that protein chaperones and small organic molecules can directly affect conformational transitions of PrP. Our findings, along with the accompanying study (35), also provide the first direct demonstration that chaperone Hsp104 can alter the conformation state of another protein.
In studying the conversion of PrPC to PrP-res, we employed previously characterized chaperones from bacteria and the eukaryotic cytosol because protein chaperones have not yet been identified in compartments where PrPC converts to PrPSc. Indeed, the site where conversion occurs is still unclear. WT PrPC is thought to convert extracellularly, within endosomes, or in caveolae (36–39). Mutant PrP, proposed to model inherited TSEs, can acquire certain PrPSc-like properties spontaneously in the ER/Golgi complex (40). Of the chaperones we tested, only GroEL and Hsp104 affected conversion. Our results indicate that such chaperone interactions in vivo, if they occur, are likely to be highly specific. Clearly, the elucidation of PrP chaperone interactions in vivo are of great import as they provide potential targets for therapeutic intervention.
Of more immediate application, chaperones provide new tools for probing the basic nature of PrP conversion. Here, they have yielded several novel insights. First, chaperones provide a strong demonstration of the importance of PrPSc in creating a template for PrPC conversion. The chaperones we tested interact with different folding intermediates, bind them in different ways, and promote conformational changes by distinct mechanisms (14–16). Yet none could promote the conversion of acid-treated PrPC to PrP-res in the absence of PrPSc.
Second, the specific effects of different chaperones provide information about the nature of PrP intermediates on the pathway of PrPSc formation. On the one hand, the ability of osmolytes to inhibit PrP conversion correlates with their known ability to stabilize proteins in the folded state (13, 34). On the other, the unique ability of GroEL and Hsp104, among protein chaperones, to promote refolding of PrPC to PrP-res correlates well with their ability to speed the refolding of kinetically trapped intermediates (refs. 14–16; J. R. Glover and S.L., unpublished observations with Hsp104).
With untreated PrPSc, only GroEL stimulated [35S]PrP conversions, but with partially denatured PrPSc, GroEL, GroEL/GroES, and Hsp104 were stimulated. Presumably, partial denaturation allows PrPSc to accept [35S]PrP in a broader variety of conformational intermediates. Indeed, GroES is known to promote the release of substrates from GroEL in a state that is more committed to folding (14, 15). Thus, the inhibitory effects of GroES on GroEL-mediated conversion nucleated with untreated PrPSc indicates that such committed [35S]PrP molecules are less likely to interact productively with this form of PrPSc.
Third, kinetic analysis with GroEL suggests that conversion is a two-step process with GroEL specifically increasing the rate at which PrPC assumes a pelletable conformation. Because conversion of [35S]PrPC to a pelletable state requires PrPSc, this process most likely involves recruitment of a conformational [35S]PrP intermediate, generated by the chaperone, into a PrPSc polymer. Once PrP has converted to a pelletable state, conversion to PrP-res follows at a slower pace. Based on these observations, we propose a model (Fig. 5).
Figure 5.
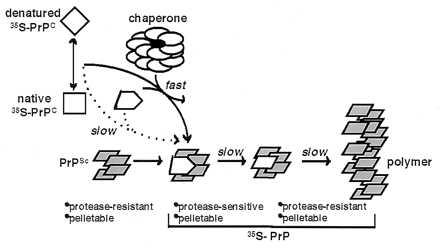
Model for chaperone-supervised PrP conversion. Conversion of [35S]PrPC to PrP-res in vitro requires pre-existing PrPSc (refs. 17–22, and this study). Without chaperone, conversion is slow and inefficient likely because [35S]PrP intermediates that productively associate with PrPSc are sparsely populated. The chaperone likely recognizes and binds near-native and nonnative intermediates derived from acid-treated [35S]PrPC, alters their conformation, and releases them in states that associate productively with PrPSc. Thereby, the chaperone facilitates the first step in conversion: specific binding of [35S]PrP to PrPSc. In this stage, [35S]PrP is pelletable, but remains protease-sensitive. A second slower step then follows, wherein PrPSc-bound [35S]PrP undergoes a second conformational transition to form PrP-res, the converted state with protease digestion properties strikingly similar to PrPSc.
Fourth, our findings provide a “proof of principle” that the acquisition of protease resistance in PrP-res results from an authentic conformational change in PrP. Despite the high degree of specificity (17, 19–22, 30), the in vitro conversion assay has been subject to the criticism that the protease-resistance of [35S]PrP results from nonspecific aggregation or association with PrPSc. That chaperones, which alter the conformation states of other proteins, promote the conversion of PrPC to a pelletable form, and a second step must ensue to generate the specific protease-resistant form of PrP, further establishes that PrP-res is a conformationally altered molecule and not simply a nonspecific aggregate.
Fifth, the ability of chaperones to enhance at least one step in the conversion process may provide an avenue for generating sufficient quantities of PrPSc in vitro to test the “protein-only” hypothesis.
Finally, our observations provide a unifying biochemical connection between mammalian TSEs (the so-called prion diseases) and [PSI+], a genetic element in yeast (sometimes called a “yeast prion;” ref. 41). The proposed “mammalian prion” determinant PrPSc, and the “yeast prion” determinant Sup35, are functionally unrelated and share no sequence identity. Also, [PSI+] produces a heritable change in metabolism rather than a lethal infection. However, both mammalian and yeast prions apparently share a common mode of transmission based on self-propagating changes in protein conformation (42–44). Among yeast chaperones, the striking specificity of Hsp104 for PrP conversions, and its known in vivo specificity in regulating [PSI+] (41–44) suggest that conformations of PrP and Sup35 share an underlying biochemical similarity that allows for recognition by particular chaperones and prion-like conformational transitions. In added support of this notion, the accompanying study (35) provides evidence for specific interactions of Hsp104 with PrP and Sup35 proteins with circular dichroism and ATP hydrolysis measurements.
Acknowledgments
We thank R. Kascsak for anti-PrP antibodies, R. Roos for use of cell culture facilities, and M. Singer, M. M. Patino, and T. Serio for helpful comments on the manuscript. S.K.D.B. is a Howard Hughes Medical Institute Fellow of the Life Sciences Research Foundation and has also been supported by a University of Chicago Cancer Biology postdoctoral fellowship. This work was supported by the National Institutes of Health and the Howard Hughes Medical Institute.
ABBREVIATIONS
- TSEs
transmissible spongiform encephalopathies
- PrPC
cellular prion protein
- PrPSc
protease-resistant prion protein purified from scrapie-infected hamsters
- PrP-res
in vitro-converted protease-resistant prion protein
- Hsp104
heat shock protein 104
- DMSO
dimethyl sulfoxide
- WT
wild type
- GPI
glycosylphosphatidylinositol
References
- 1.Caughey B, Chesebro B. Trends Cell Biol. 1997;7:56–62. doi: 10.1016/S0962-8924(96)10054-4. [DOI] [PubMed] [Google Scholar]
- 2.Prusiner S B. Trends Biochem Sci. 1996;21:482–487. doi: 10.1016/s0968-0004(96)10063-3. [DOI] [PubMed] [Google Scholar]
- 3.Griffith J S. Nature (London) 1967;215:1043–1044. doi: 10.1038/2151043a0. [DOI] [PubMed] [Google Scholar]
- 4.Caughey B, Dong A, Bhat K S, Ernst D, Hayes S F, Caughey W S. Biochemistry. 1991;30:7672–7680. doi: 10.1021/bi00245a003. [DOI] [PubMed] [Google Scholar]
- 5.Pan K-M, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Mehlhorn I, Huang Z, Fletterick R J, Cohen F E, Prusiner S B. Proc Natl Acad Sci USA. 1993;90:10962–10966. doi: 10.1073/pnas.90.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Riek R, Hornemann S, Wider G, Billeter M, Glockshuber R, Wuthrich K. Nature (London) 1996;382:180–184. doi: 10.1038/382180a0. [DOI] [PubMed] [Google Scholar]
- 7.Kenward N, Landon M, Laszlo L, Mayor R J. Cell Stress Chaperones. 1996;1:18–22. doi: 10.1379/1466-1268(1996)001<0018:hspmca>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Talzelt J, Prusiner S B, Welch W J. EMBO J. 1996;15:6363–6373. [PMC free article] [PubMed] [Google Scholar]
- 9.Carlson G A, Goodman P A, Lovett M, Taylor B, Marshall S T, Peterson-Torchia M, Westaway D, Prusiner S B. Mol Cell Biol. 1988;8:5528–5540. doi: 10.1128/mcb.8.12.5528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caughey B, Brown K, Raymond G J, Katzenstein G E, Tresher W. J Virol. 1994;68:2135–2141. doi: 10.1128/jvi.68.4.2135-2141.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Telling G C, Scott M, Mastrianni J, Gabizon R, Torchia M, Cohen F E, DeArmond S J, Prusiner S B. Cell. 1995;83:79–90. doi: 10.1016/0092-8674(95)90236-8. [DOI] [PubMed] [Google Scholar]
- 12.Edenhofer F, Reiger R, Famulok M, Weiss S, Winnacker E L. J Virol. 1996;70:4724–4728. doi: 10.1128/jvi.70.7.4724-4728.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Welch W J, Brown C R. Cell Stress Chaperones. 1996;1:109–115. doi: 10.1379/1466-1268(1996)001<0109:iomacc>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hartl F U. Nature (London) 1996;381:571–580. doi: 10.1038/381571a0. [DOI] [PubMed] [Google Scholar]
- 15.Buchner J. FASEB J. 1996;10:10–19. [PubMed] [Google Scholar]
- 16.Parsell D A, Lindquist S L. Annu Rev Genet. 1993;27:437–496. doi: 10.1146/annurev.ge.27.120193.002253. [DOI] [PubMed] [Google Scholar]
- 17.Kocisko D A, Come J H, Priola S A, Chesebro B, Raymond G J, Lansbury P T, Jr, Caughey B. Nature (London) 1994;370:471–474. doi: 10.1038/370471a0. [DOI] [PubMed] [Google Scholar]
- 18.Caughey B, Kocisko D A, Raymond G J, Lansbury P T. Chem Biol. 1995;2:807–817. doi: 10.1016/1074-5521(95)90087-x. [DOI] [PubMed] [Google Scholar]
- 19.Bessen R A, Kocisko D A, Raymond G J, Nandan S, Lansbury P T, Jr, Caughey B. Nature (London) 1995;375:698–700. doi: 10.1038/375698a0. [DOI] [PubMed] [Google Scholar]
- 20.Kocisko D A, Priola S A, Raymond G J, Chesebro B, Lansbury P T, Jr, Laughey B. Proc Natl Acad Sci USA. 1995;92:3923–3927. doi: 10.1073/pnas.92.9.3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raymond G J, Hope J, Kocisko D A, Priola S A, Raymond L D, Bossers A, Ironside J, Will R G, Chen S G, Peterson R B, Gambetti P, Rubenstein R, Smits M A, Lansbury P T, Jr, Caughey B. Nature (London) 1997;388:285–288. doi: 10.1038/40876. [DOI] [PubMed] [Google Scholar]
- 22.Bossers S A, Belt P B G M, Raymond G J, Caughey B, de Vries R, Smits M A. Proc Natl Acad Sci USA. 1997;94:4931–4936. doi: 10.1073/pnas.94.10.4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cyr D M, Lu X, Douglas M G. J Biol Chem. 1992;267:20927–20931. [PubMed] [Google Scholar]
- 24.Zeigelhoffer T, Lopez-Buesa P, Craig E A. J Biol Chem. 1995;270:10412–10419. doi: 10.1074/jbc.270.18.10412. [DOI] [PubMed] [Google Scholar]
- 25.Parsell D A, Kowal A S, Lindquist S. J Biol Chem. 1993;269:4480–4487. [PubMed] [Google Scholar]
- 26.Mendosa J A, Rogers E, Lorimer G H, Horowitz P H. J Biol Chem. 1991;266:13044–13049. [PubMed] [Google Scholar]
- 27.Freeman B C, Morimoto R I. EMBO J. 1996;15:2969–2979. [PMC free article] [PubMed] [Google Scholar]
- 28.Lee G J, Pokala N, Vierling E. J Biol Chem. 1995;270:10432–10438. doi: 10.1074/jbc.270.18.10432. [DOI] [PubMed] [Google Scholar]
- 29.Bruston S G, Weissman J S, Farr G W, Fenton W A, Horwich A L. Nature (London) 1996;383:96–99. doi: 10.1038/383096a0. [DOI] [PubMed] [Google Scholar]
- 30.Bessen R A, Raymond G J, Caughey B. J Biol Chem. 1997;272:15227–15233. doi: 10.1074/jbc.272.24.15227. [DOI] [PubMed] [Google Scholar]
- 31.Todd M J, Lorimer G H. J Biol Chem. 1995;270:5388–5394. doi: 10.1074/jbc.270.10.5388. [DOI] [PubMed] [Google Scholar]
- 32.Schirmer E C, Glover J R, Singer M, Lindquist S. Trends Biochem Sci. 1996;21:289–296. [PubMed] [Google Scholar]
- 33.Parsell D A, Kowal A S, Singer M A, Lindquist S. Nature (London) 1991;353:270–272. [Google Scholar]
- 34.Yancey P H, Clark M E, Hand S C, Bowlus R D, Somero G N. Science. 1982;217:1214–1222. doi: 10.1126/science.7112124. [DOI] [PubMed] [Google Scholar]
- 35.Schirmer E C, Lindquist S L. Proc Natl Acad Sci USA. 1997;94:13932–13937. doi: 10.1073/pnas.94.25.13932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Caughey B, Raymond G J. J Biol Chem. 1991;266:18217–18233. [PubMed] [Google Scholar]
- 37.Borchelt D M, Taraboulos A, Prusiner S B. J Biol Chem. 1992;267:16188–16199. [PubMed] [Google Scholar]
- 38.Vey M, Pilkuhn S, Wille H, Nixon R, DeArmond S J, Smart E, Anderson R G W, Taraboulos A, Prusiner S B. Proc Natl Acad Sci USA. 1996;93:14945–14949. doi: 10.1073/pnas.93.25.14945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Caughey B, Raymond G J, Ernst D, Race R. J Virol. 1991;65:6597–6603. doi: 10.1128/jvi.65.12.6597-6603.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Daude N, Lehmann S, Harris D A. J Biol Chem. 1997;271:1633–1637. doi: 10.1074/jbc.271.3.1633. [DOI] [PubMed] [Google Scholar]
- 41.Wickner R B. Science. 1994;264:566–569. doi: 10.1126/science.7909170. [DOI] [PubMed] [Google Scholar]
- 42.Glover J R, Kowal A S, Schirmer E C, Patino M M, Liu J J, Lindquist S. Cell. 1997;89:811–819. doi: 10.1016/s0092-8674(00)80264-0. [DOI] [PubMed] [Google Scholar]
- 43.Chernoff Y O, Lindquist S L, Ono B-I, Inge-Vechetomov S G, Liebman S W. Science. 1995;268:880–884. doi: 10.1126/science.7754373. [DOI] [PubMed] [Google Scholar]
- 44.Patino M M, Liu J J, Glover J R, Lindquist S L. Science. 1996;273:622–626. doi: 10.1126/science.273.5275.622. [DOI] [PubMed] [Google Scholar]