

Abstract
Peptidomics is the identification and study of the in vivo biologically active peptide profile. A combination of high performance liquid chromatography, mass spectrometry, and bioinformatics tools such as database search engines are commonly used to perform the analysis. We report a methodology based on a database system holding the completed translated genome, whereby de novo sequencing and genome-wide database searching are combined. The methodology was applied to the sea urchin neuropeptidome resulting in a 30 percent increase in identification rate.
Keywords: Neuropeptide, peptidomics, sea urchin, de novo sequencing, genome-wide database search, indexed genome
Introduction
Peptidomics is the identification and study of the in vivo biologically active peptide profile at a certain time in a certain tissue or cell type 1-3. A combination of liquid chromatography, mass spectrometry, and bioinformatics tools such as database search engines are commonly used to perform the analysis. Ancillary methodologies have been introduced to optimize database-aided identification. The sequence collections can be improved by limiting them to a set of known peptide precursors which better mimic the peptidome rather than the much larger proteome 4,5. Compiling such subsets is generally achieved by gathering known or orthologous peptide precursors, checking for motif matches 6, and precursor predictions by means of other characteristics such as cleavage site 7 and signal peptide patterns 8.
Nonetheless most analyses still suffer from high failure rates in their attempts to identify mass spectra. Common reasons are the presence of spectra arising from non-peptidic contaminants or concurrent fragmentation of multiple different precursor ions; these problems are impossible to tackle in the analysis step. However, other frequently encountered hurdles can be dealt with, for example: absence of sequences in the database due to genes overlooked by gene prediction algorithms, splice isoforms, and peptides from coding small open reading frames 9,10; furthermore deficiencies in the scoring scheme used to quantify the degree of similarity between the experimental spectrum and those predicted for database peptides, and finally post-translational modifications.
In this work, we present a peptidomics methodology which tries to handle most of the shortcomings mentioned. For this purpose we designed a database system (IggyPep: Indexed Genomes Gracefully Yield Peptide IDs) with advanced indexing and querying strategy, which holds the translated genome in all six reading frames. The system can be queried with full length de novo sequences or partial peptide sequence tags (PSTs). In contrast to other techniques which use de novo derived sequences for database filtration 11-14 or homology-based searches 15-17, our solution directly scans the translated genome (the complete query space for an organism). Two strategies can be followed using this system. On the one hand, a custom database can be created on the fly, based on all open reading frames encompassing reasonably clustered chromosome locations to which automatically derived PSTs map according to IggyPep. In this manner we try to overcome the incompleteness of the query search space typical of more general low-size search engine databases. Furthermore, database hits with an individual peptide score lower than the threshold can be “rescued” thanks to PST overlap and other criteria, thereby circumventing deficiencies in the search engine's scoring scheme. On the other hand, unassigned good quality spectra can be skimmed 11,18,19, followed by manual or automatic de novo sequencing 20,21 and indexed genome querying with IggyPep. In order to do so, we created a web interface (www.iggypep.org) allowing low-throughput genome querying with derived de novo sequences as input.
We chose to apply our methodology to the sea urchin neuropeptidome for validation, since knowledge of neuropeptides from the phylum Echinodermata (of which the sea urchin is a member) is severely limited yet of evolutionary interest as man and sea urchin share a relatively recent common ancestor. The latter lived over 540 million years ago and gave rise to the deuterostomes. All deuterostomes (including sea urchin, man, and all other vertebrates) are more closely related to each other than they are to any other animal not included in the superphylum. As a consequence, among sequenced genomes those of fruit flies and worms appear to be evolutionarily more distant from the sea urchin genome than does the human genome, notwithstanding the seemingly (when looking at morphologies) very low resemblance between man and echinoderms.
Materials and methods
Sample preparation
Adult purple sea urchins (Strongylocentrotus purpuratus) were kindly provided by Charles Hollahan, Santa Barbara Marine Biologicals. The animals were dissected and the radial nerve tissue was isolated. Neuropeptide extraction was performed in acidified acetone or acidified acetonitrile. Organic solvents used during extraction were removed by vacuum drying. The peptides were then resuspended in 9:1 water:acetonitrile solution. In order to remove large proteins, the resuspended peptides were passed through a 10 kDa cutoff filter. The samples were then cleaned using Pepmap spin filter columns, making them ready for MS analysis. For detailed description see supporting information.
Mass spectrometry
The samples were directly analyzed using either an online Capillary LC device connected to a MicroMass ESI-Qtof Mass Spectrometer or, alternatively, fractions were spotted for MALDI-TOF-TOF-MS (Bruker Ultraflex II, Bruker Daltonics, Germany). For detailed description see supporting information.
Peptide characterization
Mass spectra were analyzed using the Mascot search engine (Matrix Science, U.K.) with mass tolerance set to 0.6 Da for peptides and 0.3 Da for fragments in MALDI-MS data. For ESI-Qtof spectrum analysis this was 0.1 Da and 0.1 Da, respectively. All spectra were searched with the following variable post-translational modifications: N-terminal pyroglutamic acid of glutamine and C-terminal amidation. No cleavage enzyme for protein digestion was chosen. A protein search database was downloaded from NCBI (http://www.ncbi.nlm.nih.gov) containing all sea urchin proteins (search criterium txid7668 [Organism:noexp]), resulting in 44039 proteins.
PepNovo v3 (Build 20080724) determined de novo sequences and PSTs as input for IggyPep's batch query mode (see further). The following PepNovo arguments were used: CID_IT_TRYP as probabilistic model, the peptide and fragment mass tolerances described above, no digestion enzyme, no spectrum charge correction, and a PST length of 5 amino acids with a quality cut-off of 0.1. Benchmark experiments indicate that for typical ESI-Qtof runs a threshold of 0.1 removes approximately 40% of the spectra, without a serious loss of identifications: less than 0.5% of the identifiable spectra appear to be lost.
MS-Filter 11 and spectrumQuality 18 were used to assess spectrum quality and detect unassigned high quality spectra to be marked for further detailed analysis.
Indexed genome database (IggyPep) overview
The latest assembly of the sea urchin genome was used to build the system (version 2.1; http://www.hgsc.bcm.tmc.edu/projects/seaurchin/). The genomic sequence was translated in all six reading frames and cut up into three amino acid (AA) sized pieces by means of custom PERL scripts (available on request). This allows us to query genomic data with amino acid tags of length three and onwards, ensuring that less accurate mass spectra resulting in shorter reliable de novo tags can be queried. On the other hand, very accurate mass spectrometers such as FT-ICR generally yield longer correct sequence tags (4 or more AA), as opposed to Qtof, but long tags can be queried by combining two or more short tags, whether or not with a partial overlap. Genomic sequences were not pre-processed to warrant that no query results are missed, since we focus on possible novelties like splice-isoforms or peptides from coding small open reading frames (sORFs) 9,10.
Chopping up the genomic sequence in such a way and storing it in a table blows up the table size to approximately six billion records per genome, which normally leads to very poor query performance. A state-of-the-art relational database system (Oracle 10gR2) is used to house these cut-up genomic data. Current database technologies allow us to dramatically increase query performance on tables with such immense numbers of tuples (database rows). Table partitioning and compression, B-tree indexing, and query hints – techniques very often used in data warehouse environments – helped to optimize processing speed in our setup (see Figure 1).
Figure 1.
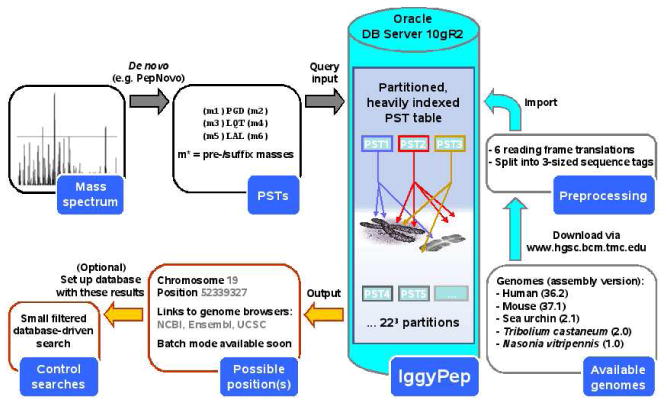
Simplified schematic overview of the IggyPep system: building its components (right), how it interacts with peptide sequence tags (PSTs) as input (top left), and the information contained in the output (bottom left). When one or more plausible loci are found to match with the PST input, these can be brought together on the fly in a small, FASTA-formatted, custom database for crosschecking with database-driven methods.
Our system uses a complete indexation strategy for every amino acid of the six reading frame translations. This contrasts with e.g. the protein BLAT server 22 which covers the non-overlapping 4-mers while excluding repetitive regions. In practice the protein BLAT server will find sequences of 18 or more amino acids long, whereas IggyPep will pinpoint sequences as short as three amino acids. Furthermore, any combination of PSTs can be queried provided that the user includes information on 1) the position relative to another PST and/or 2) the search window size. A sequence search is actually translated into a database SQL query. This complete indexing strategy, allowing overlapping tags, delivers higher sensitivity at the cost of speed which is nevertheless desirable for our application. The system has therefore been configured to attain an optimal balance between sensitivity and searching speed, so that on average a query returns its results within 0.5-2 second(s).
Although other string indexing methods exist to search for (non-)exact matching 23, we used a database system for several reasons. Firstly, despite the disk-based suffix tree technique having been scaled up for indexing complete genomes 24,25, it is not yet adapted for the translated genome: expanding the alphabet from four letters (nucleotides, untranslated) to roughly twenty (AA, translated codons) still caused practical issues. Secondly, within the database system it is comparatively easier to perform breakdown queries, e.g. when up to five PSTs together with their relative position or a search window need to be mapped.
Batch query versus detail query mode
In this study, IggyPep was applied both in batch query and detail query mode. The idea of the former is to batch process all mass spectra, reducing manual interaction. The method can be subdivided into the following steps: 1) run a de novo algorithm (in our case pepNovo v3 build 20080724) on the mass spectra, resulting in a set of reliable sequence tags for each spectrum; 2) pinpoint all possible genome locations matching these sequence tags; 3) determine the open reading frame amino acid sequences comprising these tags, e.g. by applying the EMBOSS getorf program 26; 4) compile a FASTA-formatted sequence file for database engine search. The main goal of this approach is to construct a custom sequence database, based on de novo sequences derived from the spectra that can be used as a “complete” search database in database-driven methods. Our objective is to work out a publicly available solution integrating several types of de novo sequencing tools with IggyPep. For the moment our focus is on integration with pepNovo 11,27,28. Although IggyPep's batch mode is not yet publicly available, custom databases can already be compiled on demand with PepNovo results as input.
For the detail query mode a web interface has been built to run searches with manually or in silico derived de novo sequences. Two query options are available. The first accepts full de novo sequences. Herewith two wildcards are allowed: “X” can be used for an unknown amino acid, “?” stands for a gap of unknown length. The second option (breakdown input) accepts multiple PSTs (up to five tags of three AA) to build the query. In addition this requires either the position relative to another tag or a search window size. In the latter case, an extra option handling frame shifts (e.g. due to introns) is also available.
The query results are shown in a tabular fashion, with hyperlinks to the existing genome browsers: GBrowse, Ensembl, UCSC, and NCBI 29-32. Furthermore it is possible to download the results as a comma-separated values file (CSV) or as a FASTA file holding all open reading frames (obtained by the EMBOSS getorf program) overarching the resulting chromosome positions and query sequence (see Supplemental Data 6).
The detail query mode web interface can be used as a cross-validation tool for peptides identified in a database-driven search. More interestingly, new peptides corresponding to unpredicted genes or unanticipated alternative splice forms can be identified using full length de novo sequences or multiple partial PSTs.
Results and discussion
In the present report, we describe a methodology to detect and identify endogenous peptides by means of a hybrid de novo genome-wide database search (batch query mode), followed by further analysis of the remaining unassigned high quality spectra (detailed query mode). The methodology was coined IggyPep, and was validated by two mass spectrometry data sets from a sea urchin nervous system. All identified peptides are listed in Table 1. For comparison, a Mascot search against the complete sea urchin NCBI protein database was conducted with both the Maldi TOF-TOF and ESI-Qtof mass spectra, revealing 34 unique neuropeptides cleaved from 11 different precursor proteins. IggyPep was able to confirm respectively 30 and 8 of these neuropeptides and precursor proteins, but in addition 15 new neuropeptides from 6 different prohormones were identified (Figure 2), some of which were predicted for the first time.
Table 1.
Neuropeptides identified from the sea urchin radial nerve tissue by combined database search and hybrid approach.
| Protein/EST Id | Description | Sequence | Ptm | Mascot ProtDBa | Rescueb | Custom DB based on Maldi/Qtof MSc | Detail IggyPepd | Masco t score | Mascot score threshold | delta mass | charge |
|---|---|---|---|---|---|---|---|---|---|---|---|
| XP_001176287 | hypothetical protein | R.GFETPASSRIN.S | √ | -/√ | 43 | 35 | 0.0151 | 2 | |||
| XP_001176287 | hypothetical protein | R.GFETPASSRINS.R | √ | √/√ | 46 | 37 | 0.0314 | 2 | |||
| XP_001176287 | hypothetical protein | R.GFRVLPQLNDDD.N | √ | √/- | 40 | 39 | 0.2654 | 2 | |||
| XP_001176287 | hypothetical protein | R.GFRVLPQLNDDDN.- | √ | √/√ | 73 | 39 | 0.0251 | 2 | |||
| XP_001176287 | hypothetical protein | A.EDGMELTHDEQPLNAMEI.R | √ | √ | √/- | 26 | 37 | 0.3249 | 2 | ||
| XP_001176287 | hypothetical protein | R.SPQDEQIDLRYLLQNFLDNRDT.R | √ | √/- | 48 | 27 | 0.4845 | 2 | |||
| XP_785647 | similar to pedal peptide precursor protein | K.GFNSGAMEPLGAGFF.K | √ | √/√ | 30 | 26 | 0.036 | 2 | |||
| XP_785647 | similar to pedal peptide precursor protein | K.GFHNGAMEPLKSGFL.K | √ | √/√ | 51 | 31 | 0.0990 | 2 | |||
| XP_785647 | similar to pedal peptide precursor protein | K.RFLTGALEPLSSGFI.K | √ | √/√ | 56 | 36 | 0.0269 | 2 | |||
| XP_785647 | similar to pedal peptide precursor protein | K.GFNTGAMEPLGSGFI.K | √ | √ | -/√ | 11 | 24 | 0.0157 | 2 | ||
| XP_785647 | similar to pedal peptide precursor protein | R.FLTGALEPLSSGFI.K | √ | -/√ | 42 | 27 | 0.0811 | 2 | |||
| XP_785647 | similar to pedal peptide precursor protein | K.DFNTGAMEPLGSGFI.K | √ | √ | -/√ | 11 | 22 | 0.0239 | 2 | ||
| XP_785647 | similar to pedal peptide precursor protein | K.GFHAGAMEPLSSGFIDG.K | Am | √ | √/√ | 85 | 34 | 0.3258 | 2 | ||
| XP_785647 | similar to pedal peptide precursor protein | R.GFYNGAMEPLSAGFHQG.K | Am | √ | √/- | 69 | 32 | 0.3522 | 2 | ||
| XP_785647 | similar to pedal peptide precursor protein | K.GFNSGAMEPLGSGFI.K | √ | √/√ | 36 | 33 | 0.0871 | 2 | |||
| XP_785647 | similar to pedal peptide precursor protein | R.GFYNGAMEPLSAGFHQG.K | Am | √ | √/√ | 69 | 31 | 0.3426 | 2 | ||
| XP_001175944 | hypothetical protein | S.LQFETTQDRVPA.K | √ | √/√ | 87 | 44 | 0.2271 | 2 | |||
| XP_001175944 | hypothetical protein | C.SLQFETTQDRVPA.K | √ | √/√ | 80 | 46 | 0.2941 | 2 | |||
| XP_799858 | hypothetical protein | K.APVYSGAKPIM.- | √ | √/√ | 62 | 52 | 0.2208 | 2 | |||
| XP_799858 | hypothetical protein | R.SINSYLPGDMVRHVS.K | √ | √/- | 39 | 26 | 0.0071 | 2 | |||
| XP_799858 | hypothetical protein | R.SLKNRQLFTQTRNKY.S | √ | √ | √/- | 18 | 31 | 0.5412 | 4 | ||
| XP_001175555 | hypothetical protein | L.YEEPIRQEGGD.K | √ | -/- | √ | 35 | 29 | 0.0221 | 2 | ||
| XP_001199000 | similartoarginine/serine-richsplicingfactor4 | T.SIKADGEVTEDVDK.R | √ | √ | -/√ | 21 | 34 | 0.0383 | 2 | ||
| XP_001199000 | similartoarginine/serine-richsplicingfactor4 | R.ANMFRSRLRGNG.K | Am | √ | √ | -/√ | 9 | 24 | 0.0445 | 2 | |
| XP_001199000 | similartoarginine/serine-richsplicingfactor4 | R.ANYFRGRGRKPG.K | Am | √ | √ | -/√ | 11 | 28 | 0.035 | 2 | |
| XP_001199000 | similartoarginine/serine-richsplicingfactor4 | R.ANMFRSRLRGKG.K | Am | √ | √ | √/√ | 18 | 25 | 0.0454 | 2 | |
| XP_001199000 | similartoarginine/serine-richsplicingfactor4 | R.DDPDAAEALVPGGDLSEE.K | √ | √ | √/√ | 19 | 33 | 0.0311 | 2 | ||
| XP_001199000 | similartoarginine/serine-richsplicingfactor4 | R.DDPDAALVDEFMDEE.K | √ | √/√ | 42 | 29 | 0.0316 | 2 | |||
| ABU55286 | GnRH-like tetrapeptide | R.QFVGGELIPSPEL.R | pQ | √ | -/√ | 29 | 27 | 0.0256 | 2 | ||
| AAC26833 | thymosin beta | N.TLPTKETIEQEKTA.- | √ | -/- | √ | 43 | 25 | 0.0113 | 2 | ||
| XP_001175484 | hypothetical protein | A.APAPYFDEDAMDLMDPVFNFKDDSAV.K | √ | -/- | 25 | 22 | 0.523 | 2 | |||
| XP_001186882 | similar to LFRFa precursor | R.PHGGSAFVFG.R | Am | √ | √ | -/- | 25 | 42 | 0.1743 | 2 | |
| XP_001186882 | similar to LFRFa precursor | R.DWAPREQDFANAAEESGPY.K | √ | √ | -/- | 23 | 27 | 0.3151 | 2 | ||
| XP 001175691 | hypothetical protein | R.GAAENALDEQEIYEIIESLEHAM.S | √ | -/- | 35 | 34 | 0.4755 | 3 | |||
| New_Precursor_GSTPEDIA | R.GSTPEDIAELVSRN.R | √/√ | 92 | 49 | 0.2919 | 2 | |||||
| New_Precursor_GSTPEDIA | Q.QKQDLAAILDQLHNTYQM.A | pQ | √/√ | 70 | 57 | 0.4383 | 2 | ||||
| CD294941 | New_Precursor_FGGANEPM | K.NFGGSMEPMQSGFY.K | √ | √/- | 18 | 32 | 0.2296 | 2 | |||
| CD294941 | New_Precursor_FGGANEPM | K.NFGSGLNMEPMQSGFY.K | -/√ | 35 | 34 | 0.017 | 2 | ||||
| CD294941 | New_Precursor_FGGANEPM | R.FGGANEPMRSGFF.K | √/√ | 86 | 50 | 0.298 | 2 | ||||
| CD294941 | New_Precursor_FGGANEPM | R.FGSGLDSMQSGFY.K | √ | -/√ | 21 | 25 | 0.0326 | 2 | |||
| CD294941 | New_Precursor_FGGANEPM | R.FGSGSLEPMSSGFY.K | √ | -/√ | 24 | 35 | 0.2291 | 2 | |||
| CD294941 | New_Precursor_FGGANEPM | R.FGSMNMEPLVSGFY.K | √/√ | 45 | 33 | 0.2724 | 2 | ||||
| CD294941 | New_Precursor_FGGANEPM | T.LPIEDKDGLDIEDQE.E | -/√ | 54 | 32 | 0.0481 | 2 | ||||
| CD294941 | New_Precursor_FGGANEPM | T.LPIEDKDGLDIEDQEE.A | √/√ | 53 | 40 | 0.2881 | 2 | ||||
| CD294941 | New_Precursor_FGGANEPM | T.LPIEDKDGLDIEDQEEAE.K | -/√ | 54 | 27 | 0.0303 | 2 | ||||
| New_Precursor_GYPRNSVV | R.GYPRNSVVADPVL.R | √/√ | 57 | 40 | 0.2945 | 2 | |||||
| New_Precursor_DAGPAWYG | R.DAGPHAWYGTGMFG.K | Am | √/- | 40 | 30 | 0.2646 | 2 | ||||
| New_Precursor_LPANLARE | S.LPANLARE.R | √ | 0.0076 | 2 | |||||||
| New_Precursor_LYDALKNA | R.LYDALKNAQV.K | √ | 0.017 | 2 |
Neuropeptides identified with a Mascot database search against the NCBI Sea Urchin protein database containing 44039 sequences.
Neuropeptides having a Mascot score below the threshold are “rescued” based on multiple criteria: 1) one or more de novo derived tags coincide with the Mascot sequence or the peptide is exclusively found with IggyPep, 2) the precursor protein received a good Mascot score, 3) manual inspection of Δmass, fragmentation pattern, and error distribution between experimental and predicted fragment ions. Only peptides that meet the neuropeptide precursor characteristics (basic cleavage patterns, leading signal peptide) are considered for “rescuing”.
Neuropeptides identified with a Mascot database search against a custom database, compiled using all open reading frames sharing overlap with the chromosome locations where de novo derived tags map (IggyPep batch mode).Data shown for both Maldi TOF-TOF and ESI-Qtof spectra.
Neuropeptides identified from high quality unassigned spectra using the IggyPep web interface (www.iggypep.org) after manual or automatic de novo sequencing (IggyPep detailed mode).
Figure 2.
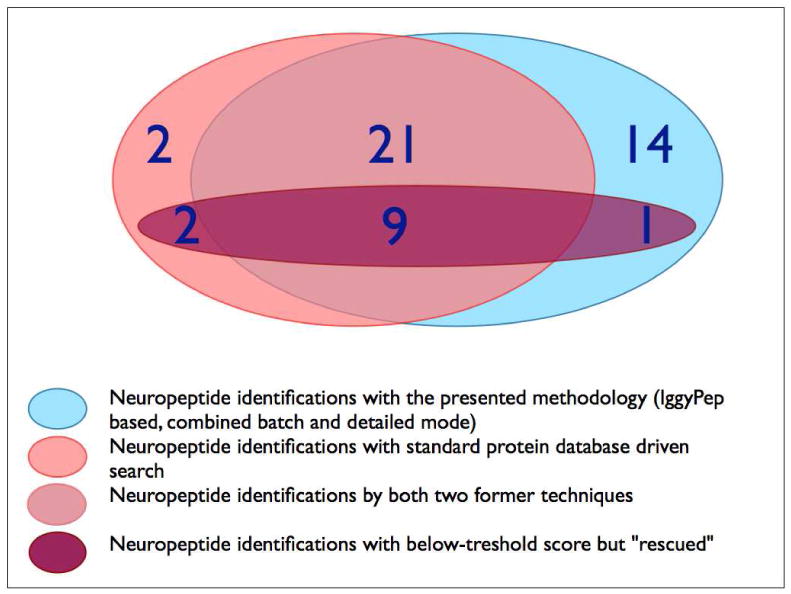
Venn diagram depicting the number of neuropeptides identified by the different methods.
The Mascot searches against the protein databases had false discovery rates below 5% (as compared with the built-in decoy database search). The peptide threshold score suggested by the search engine served as a first verification step to evaluate the search result, which showed 35 identifications. Several other peptides had a below-threshold score but were nevertheless retained (14 cases, see Figure 2). Such a “rescue” operation is useful and even advisable in the field of peptidomics, where peptides usually are not produced by strict enzyme rules, causing the Mascot-suggested threshold to be typically but undesirably much higher than in proteomics 33. Only below-threshold peptides with typical bio-active hallmarks were picked for this operation: 1) they must show basic cleavage site patterns 34-36, 2) their precursors must have an N-terminal signal peptide 8. The “rescue” operation itself becomes successful if several extra criteria are met: 1) a de novo derived tag coincides with the Mascot sequence, 2) the precursor protein comes with an above threshold score, 3) the Δmass between experimental and theoretical parent ion is within limits, 4) the error distributions of the Δmass between experimental and theoretical fragment ions are manually inspected, as is done for 5) the fragmentation patterns. A table listing these characteristics for all “rescued” peptides is provided as Supplemental Data 5. One could still argue the validity of certain below-threshold peptide identifications, but it was decided to retain the aforementioned 14 peptides, on the grounds that they all show typical neuropeptide hallmarks and meet several other quality criteria. It is our belief that neuropeptide prediction programs 34-36 would definitely pick up these “rescued” peptides and that they show enough identification evidence.
The threshold score for identification (p<0.05) in the Mascot searches against the compiled ORF databases (from both types of MS data) was higher than that against the sea urchin protein database due to the size difference of the databases (respectively around 1,183,000 and 44,000 sequences), which led to a higher false negative score 4. Again, some of the below-threshold peptides with typical bio-active hallmarks were investigated and “rescued” if quality criteria as outlined above were met.
Almost 90% of the peptides identified with the standard protein database-driven search were found back with IggyPep, four identifications were missed. This can be attributed to characteristics of the de novo algorithm. In this case PepNovo (version 3) uses a probabilistic model learned from a training data set. Several parameters influence this model and its outcome as reflected by the derived de novo tags. To begin with, although it is possible to specify “no cleavage enzyme” as an input parameter while running PepNovo, the applied model does assume tryptically cleaved peptides are in effect. Secondly, the training data was produced by an LTQ (Linear Trap Quadrupole), a different instrument than the ones used in this study. Training a model with bio-active peptide fragmentation data (which would be ideal) is unfortunately very hard since bio-active peptide MS/MS datasets are very small as compared to proteomics data sets. Another way to improve the hit rate with IggyPep could be to incorporate peptide sequence tags obtained from an extra de novo algorithm such as for example Peaks 20. Comparing the outcome of four different de novo peptide sequencing algorithms showed remarkable differences among the result sets 28. Combining multiple de novo algorithms would give a more comprehensive set of peptide sequence tags for querying a genome with IggyPep.
High quality spectra that remained unidentified were scrutinized in more detail. Figure 4 shows the spectra which resulted in extra peptide identifications upon manual de novo sequencing combined with indexed genome searching by IggyPep. From the first spectrum the peptide sequence tag LxYDALx was distilled from the y-ions series. Additionally, the tag AQV was obtained from the high mass b-ion peaks, as confirmed by the corresponding y-ions. Querying the sea urchin indexed genome with this tag pair yielded exactly one chromosome location. By contrast, the tag LxPANLxA derived from the second spectrum yielded 200 plausible loci; these were brought together in a custom database and crosschecked with a Mascot search. The latter reliably identified the peptide LPANLARE. In spectrum three, the tag YEEPLxR was obtained from the high mass y-ion peaks. When fed to the IggyPep system this tag proved to be embedded in seven different loci, narrowing down the flanking amino acid possibilities to Lys, Thr, Gln, Arg, and His. The de novo readout from the b-ion peaks showed a mass peak enabling us to further lengthen the derived tag with a glutamine residue. A nearly complete de novo sequence, TLxPTKETLxEQEK, could be obtained from the last spectrum. No corresponding loci were discovered when querying this tag, implying either an error in the de novo sequence or the occurrence of a splice site. Since our system uses raw translated genomic sequence, it is incapable of mapping cross splice site tags. Further refinement of the query by consecutively adding one amino acid and using the window query option resulted in successful pinpointing of the peptide to the thymosin beta gene (see table 1).
The gene prediction program, Fgenesh++ (www.softberry.com), was applied to the genomic sequences corresponding to the newly identified peptides. The softberry gene finding software has a model trained for the sea urchin. The predicted genes were checked for prohormone features (basic cleavage patterns, signal peptide) and expression evidence (tblastn against NCBI EST database). Annotated precursor sequences can be found in Supplemental Data 1.
The compilation of the custom databases for the 530 Maldi-TOF-TOF and 895 ESI-Qtof spectra took approximately 4 hours, which makes this methodology suitable for looking into smaller mass spectrometry data sets. Pinpointing good quality unassigned mass spectra beyond traditional database approaches, or performing detailed small-scale searches in peptidomics studies, are but two examples of problems for which the IggyPep methodology is very well suited, i.e. whenever the focus lies on the completeness of the search rather than its performance. For this study PepNovo tags with length 5 were used to build the custom databases within the batch query mode. Other peptide sequence lengths were tested, but length 5 proved to be optimal at warranting high identification rates and acceptable database size. Note that shorter tags with length 4 map to many more chromosome locations, producing a database of unmanageable size. The 30 best ranked tags (as determined by PepNovo rank score) were kept and further completed with their reverse complements. PepNovo uses the spectrum graph method to derive PSTs, making it likely that both a correct tag and its mirror are predicted. The mirror tag is the tag obtained when the roles of the b and y-ions are reversed 27. The resulting tags (12994 for Maldi TOF-TOF and 9521 for ESI-Qtof MS/MS) were automatically submitted to the IggyPep batch module, followed by open reading frame sequence retrieval by means of the getorf EMBOSS program.
This paper reports significant increase in peptide identification rate as compared to routinely executed analysis; furthermore it also partly validates the results of a recent neuropeptidomics analysis of the sea urchin 37. Herein both database searches and elaborate manual de novo sequencing on experimental mass spectrometry data as well as sequence similarity searches and motif searches based on orthologous sequences has been performed.
Conclusions
The results from this study clearly prove that for MS identification in peptidomics studies, common database search strategies are readily outperformed by our new strategy which unveiled 15 extra neuropeptides from six extra precursors. The specific gain of the approach lies in compiling a complete genome-wide search space rather than working with a database of limited size. As a consequence, new unannotated genes (for instance short peptides or splice isoforms) can be discovered. Detailed queries via the web interface using unassigned high quality spectra derived tags further increase the identification rate.
In the light of the imminent emergence and spread of alternative de novo partial sequencing techniques for peptides and proteins, we believe that the development of the underlying software tool IggyPep is timely and worthwhile to be continued. At present no batch module is publicly available to process larger-scale analyses. Our objective is to work out a solution integrating several types of de novo sequencing tools with IggyPep. Compiling custom databases based on PepNovo output is already available on request. The IggyPep tool can be accessed at www.iggypep.org and can be used for detailed queries consisting of short amino acid stretches or combinations thereof which may either entail new identifications or validate existing ones. Indexed genomes other than that of sea urchin are also being made available: mouse, man, roundworm, honey bee, parasitoid wasp, and red flour beetle.
In the future a solution will be worked out to avoid the time-consuming final database search step. As for now, this step is still indispensable in the identification process, all the more since a very wide database search space is being compiled upon de novo tag derivation. Further integration of spectrum analysis and the indexed genome search should lead to less spurious chromosome locations, ultimately consolidating into one or only a few possible peptide results. More accurate mass spectra from Fourier Transform Ion Cyclotron Resonance (FT-ICR) and Orbitrap analysis will undoubtedly facilitate taking this hurdle.
Supplementary Material
1. Annotated neuropeptide precursor sequences.
2. Peaklist generation and database searching details.
3. Detailed information on sample preparation and combined LC-MS/MS.
4. MS/MS spectra annotated with masses observed and peptide assignment.
5. Table holding all “rescued” below-threshold scoring peptides. All characteristics relevant to the rescuing operation are listed.
6. Screenshot of the IggyPep web interface.
Figure 3.
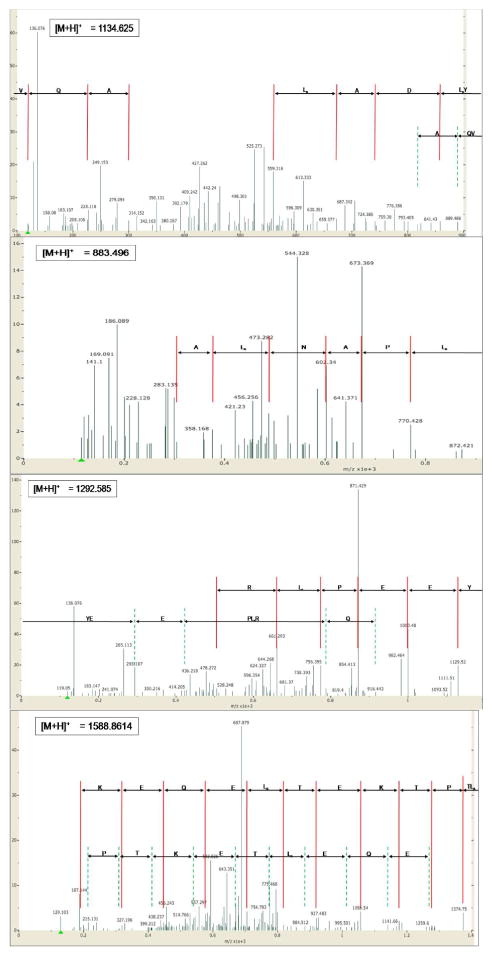
MS spectra identified after manual de novo sequencing in combination with detailed indexed genome querying using the web interface (www.iggypep.org). The peptides obtained are respectively LYDALKNAQV, LPANLARE, YEEPIRQEGGD, and TLPTKETIEQEKTA.
Acknowledgments
This work was supported by an SBO grant (IWT-50164) of the Institute for the Promotion of Innovation by Science and Technology in Flanders (IWT) and by the US National Institutes of Health through P30DA018310. The authors would like to thank Ari Frank for assisting with the PepNovo software tool.
Footnotes
This information is available free of charge via the Internet at http://pubs.acs.org.
Contributor Information
Gerben Menschaert, Email: Gerben.Menschaert@ugent.be.
Tom T.M. Vandekerckhove, Email: Tom.Vandekerckhove@ugent.be.
Geert Baggerman, Email: Geert.Baggerman@bio.kuleuven.be.
Bart Landuyt, Email: Bart.Landuyt@bio.kuleuven.be.
Jonathan V. Sweedler, Email: JSweedle@uiuc.edu.
Liliane Schoofs, Email: Liliane.Schoofs@bio.kuleuven.be.
Walter Luyten, Email: Walter.Luyten@bio.kuleuven.be.
Wim Van Criekinge, Email: Wim.Vancriekinge@ugent.be.
References
- 1.Baggerman G, Verleyen P, Clynen E, Huybrechts J, De LA, Schoofs L. Peptidomics. J Chromatogr B Analyt Technol Biomed Life Sci. 2004;803(1):3–16. doi: 10.1016/j.jchromb.2003.07.019. [DOI] [PubMed] [Google Scholar]
- 2.Boonen K, Baggerman G, D'Hertog W, Husson SJ, Overbergh L, Mathieu C, Schoofs L. Neuropeptides of the islets of Langerhans: a peptidomics study. Gen Comp Endocrinol. 2007;152(23):231–241. doi: 10.1016/j.ygcen.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 3.Schoofs L, Baggerman G. Peptidomics in Drosophila melanogaster. Brief Funct Genomic Proteomic. 2003;2(2):114–120. doi: 10.1093/bfgp/2.2.114. [DOI] [PubMed] [Google Scholar]
- 4.Falth M, Skold K, Svensson M, Nilsson A, Fenyo D, Andren PE. Neuropeptidomics strategies for specific and sensitive identification of endogenous peptides. Mol Cell Proteomics. 2007;6(7):1188–1197. doi: 10.1074/mcp.M700016-MCP200. [DOI] [PubMed] [Google Scholar]
- 5.Richmond TA, Baggerman G, Vandekerckhove TTM, Menschaert G, Husson SJ, Verleyen P, Schoofs L, Van Criekinge W. Bioinformatic Strategies for More Complete and Accurate Identification of Neuropeptides. ASMS Conference 2008; 1-6-2008. [Google Scholar]
- 6.Liu F, Baggerman G, Schoofs L, Wets G. Uncovering conserved patterns in bioactive peptides in Metazoa. Peptides. 2006;27(12):3137–3153. doi: 10.1016/j.peptides.2006.08.021. [DOI] [PubMed] [Google Scholar]
- 7.Southey BR, Amare A, Zimmerman TA, Rodriguez-Zas SL, Sweedler JV. NeuroPred: a tool to predict cleavage sites in neuropeptide precursors and provide the masses of the resulting peptides. Nucleic Acids Res. 2006;34(Web Server):W267–W272. doi: 10.1093/nar/gkl161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bendtsen JD, Nielsen H, von HG, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol. 2004;340(4):783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- 9.Galindo MI, Pueyo JI, Fouix S, Bishop SA, Couso JP. Peptides encoded by short ORFs control development and define a new eukaryotic gene family. PLoS Biol. 2007;5(5):e106. doi: 10.1371/journal.pbio.0050106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Savard J, Marques-Souza H, Aranda M, Tautz D. A segmentation gene in tribolium produces a polycistronic mRNA that codes for multiple conserved peptides. Cell. 2006;126(3):559–569. doi: 10.1016/j.cell.2006.05.053. [DOI] [PubMed] [Google Scholar]
- 11.Frank AM, Savitski MM, Nielsen ML, Zubarev RA, Pevzner PA. De novo peptide sequencing and identification with precision mass spectrometry. J Proteome Res. 2007;6(1):114–123. doi: 10.1021/pr060271u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mann M, Wilm M. Error-tolerant identification of peptides in sequence databases by peptide sequence tags. Anal Chem. 1994;66(24):4390–4399. doi: 10.1021/ac00096a002. [DOI] [PubMed] [Google Scholar]
- 13.Tanner S, Shu H, Frank A, Wang LC, Zandi E, Mumby M, Pevzner PA, Bafna V. InsPecT: identification of posttranslationally modified peptides from tandem mass spectra. Anal Chem. 2005;77(14):4626–4639. doi: 10.1021/ac050102d. [DOI] [PubMed] [Google Scholar]
- 14.Tabb DL, Saraf A, Yates JR., III GutenTag: high-throughput sequence tagging via an empirically derived fragmentation model. Anal Chem. 2003;75(23):6415–6421. doi: 10.1021/ac0347462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Han Y, Ma B, Zhang K. SPIDER: software for protein identification from sequence tags with de novo sequencing error. J Bioinform Comput Biol. 2005;3(3):697–716. doi: 10.1142/s0219720005001247. [DOI] [PubMed] [Google Scholar]
- 16.Searle BC, Dasari S, Turner M, Reddy AP, Choi D, Wilmarth PA, McCormack AL, David LL, Nagalla SR. High-throughput identification of proteins and unanticipated sequence modifications using a mass-based alignment algorithm for MS/MS de novo sequencing results. Anal Chem. 2004;76(8):2220–2230. doi: 10.1021/ac035258x. [DOI] [PubMed] [Google Scholar]
- 17.Halligan BD, Ruotti V, Twigger SN, Greene AS. DeNovoID: a web-based tool for identifying peptides from sequence and mass tags deduced from de novo peptide sequencing by mass spectroscopy. Nucleic Acids Res. 2005;33(Web Server):W376–W381. doi: 10.1093/nar/gki461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flikka K, Martens L, Vandekerckhove J, Gevaert K, Eidhammer I. Improving the reliability and throughput of mass spectrometry-based proteomics by spectrum quality filtering. Proteomics. 2006;6(7):2086–2094. doi: 10.1002/pmic.200500309. [DOI] [PubMed] [Google Scholar]
- 19.Nesvizhskii AI, Roos FF, Grossmann J, Vogelzang M, Eddes JS, Gruissem W, Baginsky S, Aebersold R. Dynamic spectrum quality assessment and iterative computational analysis of shotgun proteomic data: toward more efficient identification of post-translational modifications, sequence polymorphisms, and novel peptides. Mol Cell Proteomics. 2006;5(4):652–670. doi: 10.1074/mcp.M500319-MCP200. [DOI] [PubMed] [Google Scholar]
- 20.Ma B, Zhang K, Hendrie C, Liang C, Li M, Doherty-Kirby A, Lajoie G. PEAKS: powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Commun Mass Spectrom. 2003;17(20):2337–2342. doi: 10.1002/rcm.1196. [DOI] [PubMed] [Google Scholar]
- 21.Johnson RS, Taylor JA. Searching sequence databases via de novo peptide sequencing by tandem mass spectrometry. Mol Biotechnol. 2002;22(3):301–315. doi: 10.1385/MB:22:3:301. [DOI] [PubMed] [Google Scholar]
- 22.Kent WJ. BLAT--the BLAST-like alignment tool. Genome Res. 2002;12(4):656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gusfield D. Suffix trees (and relatives) come of age in bioinformatics. 2002 [Google Scholar]
- 24.Phoophakdee B, Zaki MJ. TRELLIS+: an effective approach for indexing genome-scale sequences using suffix trees. Pac Symp Biocomput. 2008:90–101. [PubMed] [Google Scholar]
- 25.Tian Y, Tata S, Hankins R, Patel J. Practical methods fro constructing suffix trees. VLDB Journal. 2005;14(3):281–299. [Google Scholar]
- 26.Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16(6):276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 27.Frank A, Tanner S, Bafna V, Pevzner P. Peptide sequence tags for fast database search in mass-spectrometry. J Proteome Res. 2005;4(4):1287–1295. doi: 10.1021/pr050011x. [DOI] [PubMed] [Google Scholar]
- 28.Frank A, Pevzner P. PepNovo: de novo peptide sequencing via probabilistic network modeling. Anal Chem. 2005;77(4):964–973. doi: 10.1021/ac048788h. [DOI] [PubMed] [Google Scholar]
- 29.Stein LD, Mungall C, Shu S, Caudy M, Mangone M, Day A, Nickerson E, Stajich JE, Harris TW, Arva A, Lewis S. The generic genome browser: a building block for a model organism system database. Genome Res. 2002;12(10):1599–1610. doi: 10.1101/gr.403602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Flicek P, Aken BL, Beal K, Ballester B, Caccamo M, Chen Y, Clarke L, Coates G, Cunningham F, Cutts T, Down T, Dyer SC, Eyre T, Fitzgerald S, Fernandez-Banet J, Graf S, Haider S, Hammond M, Holland R, Howe KL, Howe K, Johnson N, Jenkinson A, Kahari A, Keefe D, Kokocinski F, Kulesha E, Lawson D, Longden I, Megy K, Meidl P, Overduin B, Parker A, Pritchard B, Prlic A, Rice S, Rios D, Schuster M, Sealy I, Slater G, Smedley D, Spudich G, Trevanion S, Vilella AJ, Vogel J, White S, Wood M, Birney E, Cox T, Curwen V, Durbin R, Fernandez-Suarez XM, Herrero J, Hubbard TJ, Kasprzyk A, Proctor G, Smith J, Ureta-Vidal A, Searle S. Ensembl 2008. Nucleic Acids Res. 2008;36(Database):D707–D714. doi: 10.1093/nar/gkm988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karolchik D, Kuhn RM, Baertsch R, Barber GP, Clawson H, Diekhans M, Giardine B, Harte RA, Hinrichs AS, Hsu F, Kober KM, Miller W, Pedersen JS, Pohl A, Raney BJ, Rhead B, Rosenbloom KR, Smith KE, Stanke M, Thakkapallayil A, Trumbower H, Wang T, Zweig AS, Haussler D, Kent WJ. The UCSC Genome Browser Database: 2008 update. Nucleic Acids Res. 2008;36(Database issue):D773–D779. doi: 10.1093/nar/gkm966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wolfsberg TG. Using the NCBI Map Viewer to browse genomic sequence data. Curr Protoc Bioinformatics. 2007;Chapter 1 doi: 10.1002/0471250953.bi0105s16. Unit. [DOI] [PubMed] [Google Scholar]
- 33.Savitski MM, Nielsen ML, Kjeldsen F, Zubarev RA. Proteomics-grade de novo sequencing approach. J Proteome Res. 2005;4(6):2348–2354. doi: 10.1021/pr050288x. [DOI] [PubMed] [Google Scholar]
- 34.Fricker LD. Neuropeptide-processing enzymes: applications for drug discovery. AAPS J. 2005;7(2):E449–E455. doi: 10.1208/aapsj070244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seidah NG, Prat A. Precursor convertases in the secretory pathway, cytosol and extracellular milieu. Essays Biochem. 2002;38:79–94. doi: 10.1042/bse0380079. [DOI] [PubMed] [Google Scholar]
- 36.Zhou A, Webb G, Zhu X, Steiner DF. Proteolytic processing in the secretory pathway. J Biol Chem. 1999;274(30):20745–20748. doi: 10.1074/jbc.274.30.20745. [DOI] [PubMed] [Google Scholar]
- 37.Amare A, Monroe EB, Annangudi SP, Sweedler JV, Richmond T, Schoofs L, Baggerman G. Combination of proteomics and bioinformatics for the identification of (neuro)peptides in the sea urchin Strongylocentrotus purpuratus. Genome Research Submitted [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1. Annotated neuropeptide precursor sequences.
2. Peaklist generation and database searching details.
3. Detailed information on sample preparation and combined LC-MS/MS.
4. MS/MS spectra annotated with masses observed and peptide assignment.
5. Table holding all “rescued” below-threshold scoring peptides. All characteristics relevant to the rescuing operation are listed.
6. Screenshot of the IggyPep web interface.