

Abstract
Mechanical cues may have important roles in tissue morphogenesis; progression through complex functions like differentiation may be associated with changes in cellular force generation and mechanosensing. To explore this concept, we use elastomer pillar arrays to map forces generated by neural stem cells in vitro, and identify two distinct dynamics of force generation. First, cell generated forces decrease as cells transition from a proliferative mode to differentiation, a process covering several days. This change in force generation correlated with a loss of sensitivity to substrate rigidity over a series of polydimethylsiloxane substrates. Second, neural stem cells exhibit a faster pattern of localized contractions at the cell body and outlying processes; each lasts on the order of minutes, and is not synchronized across the cell. This faster process is reminiscent of migratory behavior observed in vivo, and may be involved in controlling the motion of internal structures such as the cell nucleus. These results together provide new clues into the role of forces during development, and may lead to design principles for materials targeted for use in the central nervous system.
Key terms: cellular traction force, stem cell, microenvironment, differentiation, proliferation
Introduction
Cells have the remarkable ability to sense the rigidity of their environment, which modulates fundamental cell functions including migration, focal adhesion formation, spreading, and stem cell differentiation 10; 35; 36; 21; 33. These responses raise the intriguing possibility that mechanical cues may help coordinate morphogenesis and, conversely, lead to new strategies for improving the design of engineered tissues. In support of such a role in the specific context of the nervous system, Elkin and colleagues 9 report that elastic modulus varies across different regions of the hippocampus, while Saha and colleagues 26 showed that glial vs. neuronal differentiation is sensitive to substrate rigidity. Mechanical forces are key to other aspects of neurogenesis, including complex patterns of migration and nuclear oscillation of radial glial cells 17; 32. It is important to recognize that complex behaviors such as stem cell differentiation involve dramatic changes in cell state. Understanding how cell-material interactions change over the course of such processes is essential for developing a complete model of mechanosensing in tissue morphogenesis.
Here, we seek to identify such a programmatic change in the context of central nervous system neurogenesis. We use a traction force microscopy technique introduced by Tan and colleagues 29 to estimate forces generated by neural stem cells over the course of differentiation. In this approach, arrays of microscale elastomer pillars (μPAs, Figure 1A) serve as the cell culture substrate. Adherent cells deflect these pillars, and the force applied to the pillar can be derived from these deflections (Figure 1B). Image-based measurement of these deflections thus provides a spatially resolved map of the contractile forces. We focus on neural stem cells (NSCs) derived from the subventricular zone (SVZ) of postnatal rats, a site of continued neurogenesis in these animals. Given the importance of laminin in neural tissues 18; 22 and its widespread use as a culture surface for NSCs, we focus predominantly on pillar arrays coated with this extracellular matrix protein. Rigidity sensing as a function of force generation is then tested, supporting a model in which mechanical properties direct neural stem cell function.
Figure 1.
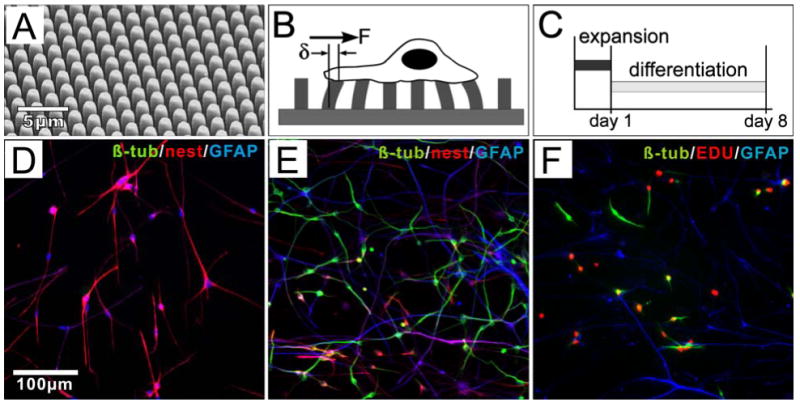
NSCs cultured on μPAs. (A) SEM of a μPA. Pillars measure 1 um in diameter, and are spaced 2 um center-to-center in hexagonal arrays. (B) Schematic of traction force microscopy. (C) Timecourse of standard culture of NSCs. (D-E) Characterization of NSC before/after differentiation, illustrating staining for the phenotypic markers β-tubulin (TuJ1 antibody, green), nestin (red), and GFAP (blue) at 1 DIV (D), and 8 DIV (E). (F) At 4 DIV, a significant population of TuJ1+ (green) cells are also positive for EdU (red). See text for a quantitative discussion of NSC differentiation.
Results
Traction force microscopy of SVZ-derived NSCs
We designed μPAs that are optimized to measure force generation in NSCs, addressing their particular cellular characteristics. First, these cells typically exhibit an elongated, thin, radial-glial-like morphology during expansion culture. Supporting cell attachment and mapping applied forces requires a correspondingly fine spatial resolution. A standard geometry of 1-μm diameter pillars spaced 2 μm center-to-center was chosen to allow close packing while keeping the top surface large enough to accommodate focal adhesion formation. Second, preliminary results showed that the traction forces exerted by NSCs are relatively small, dictating the use of a relatively flexible (long) pillar. A standard pillar height of 7 μm was chosen as these were the shortest pillars for which deflections were reliably detectable by optical microscopy. Assuming a typical bulk elastic modulus of PDMS of 2 MPa, these pillar dimensions provide a lateral “spring constant” of 0.86 nN/μm based on a simple cantilever approximation 29. Unless otherwise specified, the μPAs were prepared for cell culture by coating with laminin, one of the major ECM components in both embryonic and adult neural stem cell microenvironments 18; 22.
SVZ-derived NSCs on these μPAs exhibited a pattern of differentiation similar to that on flat cell culture surfaces. In the standard experimental procedure used throughout this study (Figure 1C), proliferative cells were seeded onto the arrays and maintained in FGF-supplemented expansion media for a short time (1 day, unless otherwise stated), allowing cells to become established on the culture surface. Cells were then switched to differentiation media which is FGF-free but supplemented with forskolin and retinoic acid, and promotes neuronal differentiation. In expansion media, NSCs express high levels of the intermediate filament protein nestin, a marker of neural stem cells (Figure 1D), along with moderate levels of glial fibrillary acidic protein (GFAP) 7. Following the switch to differentiation media, cells gradually reduce nestin expression, with concurrent emergence of lineage-specific protein markers. The neuronal specific marker βIII-tubulin (β-tub, detected using the TuJ1 clone) is evident in cells after 3 days in differentiation media (4 days in vitro). The percentage of cells undergoing neuronal differentiation was typically measured at 8 days in vitro (Figure 1E), at which expression of nestin was minimal. At that time point, the fraction of TuJ1+ cells on μPAs of 7-μm height was similar to that on flat, planar substrates of laminin-coated PDMS, which served as reference controls for substrate topology (53.7±15.5% and 56.5±7.4%, mean ±s.d, on μPA and flat PDMS substrates, respectively; n > 500 cells per surface over three replicate experiments). Thus, the architecture of these μPAs did not influence cell differentiation, validating their use for measuring the contractile force of these NSCs. On all surfaces, the remaining, non-TuJ1+ cells consisted of mature astrocytes and a small percentage (< 2%) of oligodendrocytes as indicated by high levels of GFAP and O4, respectively. To confirm the origin of neuronal cells, differentiating NSC cultures were incubated with the EdU nucleoside overnight (to minimize any effect on cell function) prior to fixation and staining for lineage markers (on 4 DIV, Figure 1F). Of cells staining positive for TuJ1, 56.4±5.4% (mean±s.d. from three independent experiments) were also positive for EdU, indicating that a large population of neuronal cells derive from a proliferating pool of NSCs. The remaining TuJ1+ cells were NSC-derived and either not undergoing DNA replication during the brief EdU loading period or had committed by 3 DIV or were committed before transfer to differentiation media. Lastly, TuJ1+ cells were present as both individual cells and as clusters, migrating over each other in a pulsate mode resembling “chain migration” of neuroblasts (see Movie S1 in the Supplementary Material), which has been observed both in vitro and in vivo 34.
Force generation is modulated over multiple temporal scales
Live cell microscopy was used to examine the interaction of NSCs with underlying μPAs, revealing complex and dynamic patterns of force application by the cells. Following seeding onto an array, NSCs generated forces that were organized into local areas of contraction – one under the cell body and one or occasionally two near a process tip as illustrated in Figure 2A; time-series movies of additional representative cells are included in the Supplementary Material, S2. The specific frame illustrated in Figure 2A illustrates both sites of contraction, but as will be discussed later in this section, force generation at each site changed rapidly over the course of minutes, and was not coordinated between locations. We first focus on an additional pattern of force generation that changed over the course of days.
Figure 2.
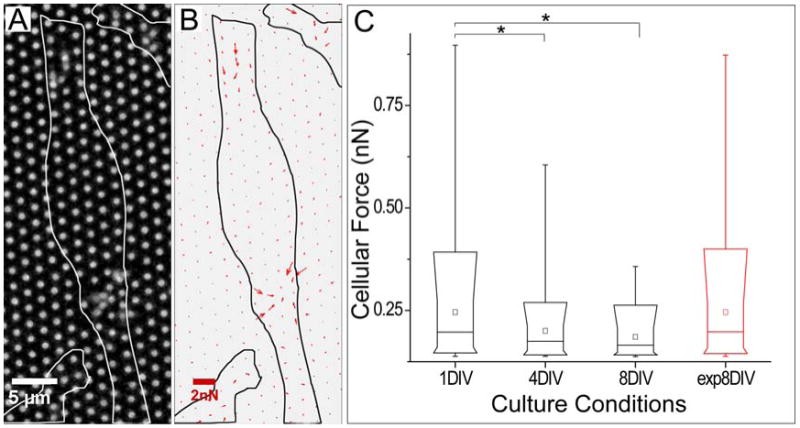
Force generation by NSCs decreases with differentiation. (A) Deflection of pillars by NSCs 6 hours after seeding. Outlines of cells are indicated by the white profiles. (B) Force vector field measured from the image in panel A, illustrating the presence of two separate sites of contraction. (C) Comparison of traction force magnitudes at different stages of differentiation (0, 3, 7 days of differentiation, corresponding to 1, 4, and 8 DIV). The final column (exp8DIV, red) indicates cells maintained in expansion media for 8 days. The parts of the box plots indicate 10, 25, 50, 75, and 90 percentiles, while the whiskers indicate 1 and 99%. The square box indicates mean of the data. * α <0.05, compared using Krusal-Wallis analysis (n = 7, 20, 34, 33 cells for conditions of 1DIV, 4DIV, 8DIV, exp8DIV respectively, representing over 300 pillars per timepoint and covering two independent timecourse experiments).
Specifically, the forces applied by NSCs at each site decreases as cell differentiation progresses in vitro. The maximal force generated by NSCs during expansion was estimated for individual pillars by collecting time series stacks of cells on μPAs, taken at 30 second intervals over 10-15 minutes. Maximal deflections were extracted from these series and used to estimate an applied force using a linear cantilever model 29. Data from different conditions (Figure 2C) were compared using Kruskal-Wallis methods (α = 0.05, n = 7, 20, 34, 33 cells for conditions of 1DIV, 4DIV, 8DIV, exp8DIV respectively, representing over 300 pillars per timepoint and covering two independent timecourse experiments). During the first day in culture (in expansion media), cells applied maximal forces in the sub-nanoNewton range. However, by 7 days in differentiation media (8 DIV) only minor deflections could be identified. Applied forces at either 1, 4, or 8 DIV are statistically different from the other two time points. Differentiation of SVZ-derived NSCs in vitro thus correlates with a decrease in the magnitude of contractile forces exerted on substrates. As a control, forces exerted by cells cultured on the pillar arrays for 8 days in expansion media was found to be comparable to that of cells in expansion culture for 1 day (Figure 2C), supporting the concept that differentiation, and not simply time in culture, is associated with decreasing force generation.
Focusing on force generation at each contraction site, we found a faster dynamic on the timescale of minutes, illustrated in Figure 3A and the time series movies in Supporting Material S2. Within each site, traction forces were centripetal and pulsatile, with each contraction lasting 4.1 ± 1.7 min (mean ± s.d., n = 245 contraction events, Figure 3B,C), a timescale that is fast compared to traction dynamics in the leading edge of typical fibroblasts (typically 15-20 minutes) 1. Moreover, contraction of pillars under the cell body (nearer the trailing edge of a cell) was independent to that of the more distal position along the cell process (Figure 3B). As a quantitative measurement of this lack of synchronization, we calculated a Pearson's correlation coefficient (see Methods), comparing groups of pillars underlying a specific cell into the two identified regions. We measured a correlation coefficient between these sites of -0.13 ± 0.24 (mean ± s.d., n=7), indicating no correlation between contractions at these positions in the cell; this coefficient ranges from 1 to -1, associated with strong correlation and anticorrelation, respectively.
Figure 3.
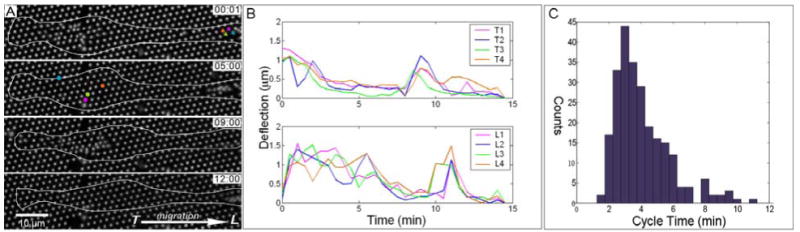
Dynamic contraction by NSCs in expansion media. (A) Time sequence illustrating periodic, sequential application of force by cells at two localized sites, indicated by circles at the trailing edge and arrows at the leading edge. (B) Magnitudes of deflections for the color-coded pillars as a function of time. (C) Histogram of the duration of individual, pulsatile contractions measuring 4.1 ± 1.7 min (mean ± s.d., n = 245 contraction events).
The role of this dynamic force generation during expansion in coordinating cell function is currently unclear, but may correlate with movement of centrosome and nucleus in neural progenitors 31. Specifically, radial glial cells exhibit pulsatile nuclear migration during neural tissue development 32, a behavior that has also been observed in vitro 5. Such nuclear oscillations require cytoskeletal mediated force generation and contributes to the control of cell-cycle progression and division 16. The periodic contractions we observed under the cell bodies suggest that cells may use cell-matrix interactions to propel intracellular reorganization of these cells. Notably, we observed that external force generation and nuclear translocation were sequential and not concurrent, with periods of strong force generation often preceding large changes of nuclear location (illustrated by the two strong contraction events in Supplementary Material S3 at 9 and 12 minutes of this time series).
Changes in molecular architecture parallel force generation
Adherent cells generate tension via contraction of the actin cytoskeleton and transmit this force via focal adhesions to the underlying substrate 2. Towards an insight into the mechanisms by which force generation changes over time, we examined the distribution of molecules involved in cytoskeletal and substrate-anchorage assemblies. Figure 4A illustrates the distribution of actin along the cell surface facing the pillar array at 1, 4, and 8 DIV. At 1 DIV, actin was observed throughout the cellular processes and in small, punctuate clusters resting on and across the pillar tops (higher-detail images illustrating the pillar location are shown in Supporting Material S4). The presence of actin decreased by 4 DIV and was minimal at 8 DIV, restricted to occasional small clusters localized at areas of cell-cell contact. Similar concentrations of active myosin were reported in the migrating neuronal precursors from SVZ explants 28. The distribution of focal adhesion proteins followed a similar change in distribution. Specifically, we followed the distribution of the β1 integrin subunit, which is expressed at high levels in neural stem cells and plays a role in their maintenance 4; 18; 19, as well as paxillin. Cells in expansion media (1 DIV) exhibited clusters of β1-integrin that colocalized with paxillin, indicative of focal adhesions, as illustrated in the top row of Figure 4B. By the end of differentiation culture, β1-integrin expression in the bipolar neuroblast-like precursor cells was minimal, and paxillin clusters were absent, bottom row of Figure 4B. These data indicate that NSCs decrease their cell-ECM contacts over the course of differentiation, potentially switching to a cell-cell communication and adhesion mode. We also recognize that the specific nature of the cell-ECM interaction may be altered over the course of differentiation. We chose laminin for use in coating the μPAs due to its widespread use as a NSC cell culture surface. However, degradation and replacement of this ECM, as a result of metalloprotease activity, for example, may change the local ECM composition. As such, future experiments using more firmly bound proteins or specifically tethered protein fragments may shed new light onto the issue of cell-ECM interaction on neural stem cell function.
Figure 4.
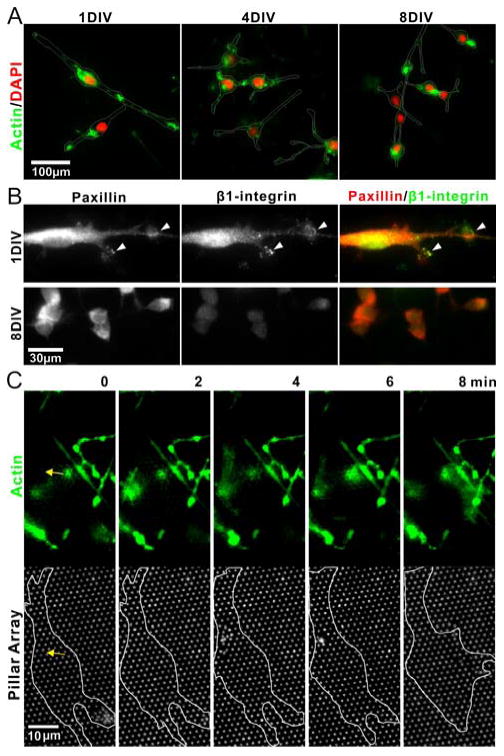
Changes in expression of molecular structures parallel that of force generation. (A) Timecourse of actin (green) distribution on the basal surface of cells. Cells were counterstained for nuclei using DAPI (red). The outlines of cells, derived from differential interference contrast images, are added to help delineate the borders of the cell. (B) Localization of the anchorage-associated proteins paxillin (red) and β1-integrin subunit (green). (C) Fast dynamics of actin assembly, preceding the application of force to the pillar arrays. The outline indicates the boundary of a representative cell which is the focus of this time series, but additional actin-containing processes from other cells are visible in the top row. The yellow arrow indicates the same position in the two rows, corresponding to a site of contraction, as detailed in the text.
Live cell microscopy, using GFP-tagged actin, revealed detail into the dynamics of force generation by NSCs in expansion phase, as shown in the representative cell in Figure 4C. The yellow arrow indicates the site of a contraction that will occur over the series of these images, while the end of a second, previous event can be seen in the lower right part of this figure. A cloud of actin accumulates near the site of contraction prior to the event. Contraction of pillars coincides with a slight spread of actin, which then disperses following the event. The molecular processes driving this process remain unclear.
Finally, we examined the role of myosin type II in generation of the observed cell traction forces. Application of the myosin II inhibitor blebbistatin to NSCs in expansion media caused rapid (< 30 minutes) loss of local contraction and micropillar deflection, along with rounding up of the cell body (Figure 5A-C, Supplementary Material S5). Application of blebbistatin also severely impaired nuclear translocation. Before administration of blebbistatin, 74.1% cells on flat PDMS substrates moved their soma more than 50 μm over 30 minutes (Figure 5D, n=58 cells). Thirty minutes after application of blebbistatin (and observing cells over the next 30 minutes), this portion of cells dropped to only 18.3% (Figure 5E, n=71 cells). Together, these data suggest that myosin II activity is a critical component of the multiple forms of traction force generation and cellular reorganization observed in NSCs during expansion.
Figure 5.
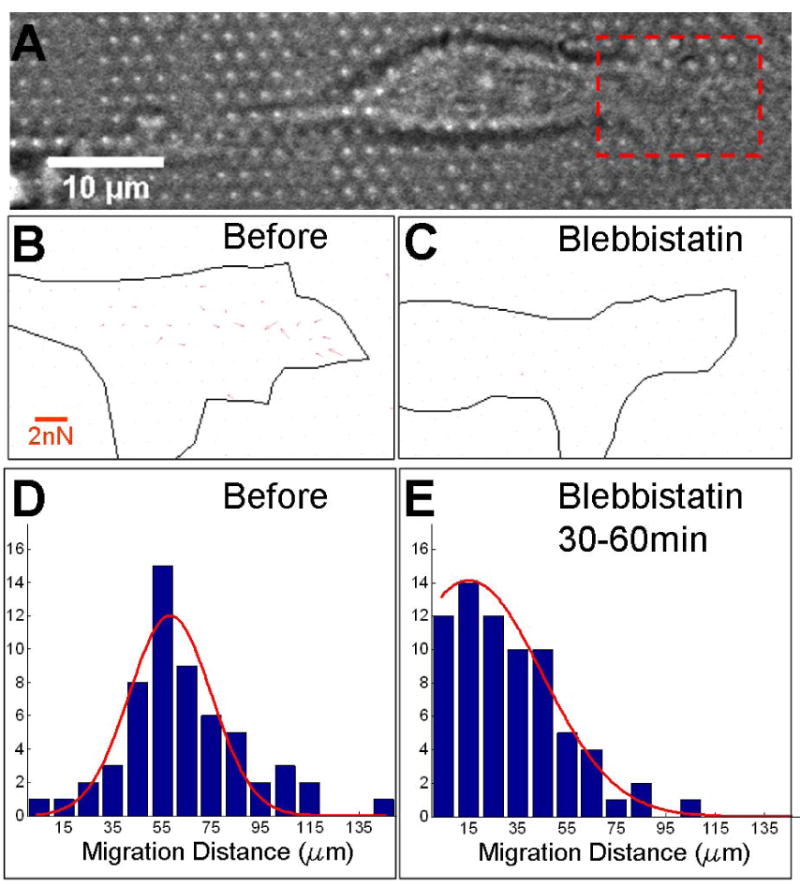
Inhibition of contractile function. (A-C) Map of cell traction forces before (B) and 30 minutes after (C) application of 50 μM blebbistatin, focusing on the region outlined in panel A. (D-E) Distance of nucleus migration over a 30 minute interval either before (D) or 30 minutes after (E) application of blebbstatin.
Modulation of cell function correlates with force generation as a function of time
An underlying assumption in many models of rigidity sensing is that cells must exert a force on their environment to test this mechanical property. Given the changes in maximal force generation that were observed over the course of differentiation, we set out test whether specific cellular functions associated with separate stages of this process are differentially sensitive to rigidity. For these studies, cells were cultured on planar PDMS substrates with Young's modulus of 25kPa, 500Kpa, 2MPa, created by changing the ratio of base : curing agent in the elastomer formulation; while these substrates are of relatively high rigidity compared to that associated with tissues, these surfaces present the same chemistry as the μPAs, and the ability of cells to sense changes in rigidity in these higher ranges is increasingly recognized 30.
We first examined cell proliferation, associated with growth of cells in expansion media and correlating to periods of high contraction force generation. Cell proliferation was quantified by comparing the growth rate of cells on each surface over a period of 4 days, cultured in expansion media for the duration of the experiment. Cells on substrates of 2MPa modulus exhibited a higher proliferation rate than those on softer substrates (n=4, >800 cells examined for each condition, Figure 6A), demonstrating an effect of substrate rigidity during the expansion process.
Figure 6.
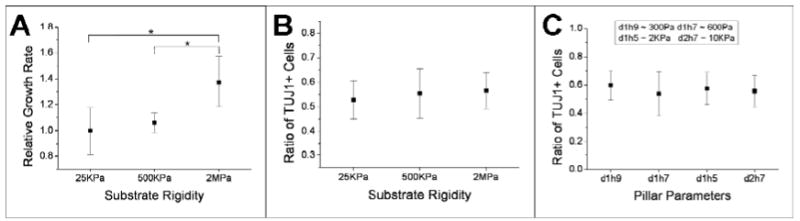
Rigidity sensing correlates with force generation. (A) Proliferation of SVZ NSCs on planar substrates of varying rigidity. Cells were maintained in expansion media for the entire 4-day experiment. (B) Differentiation of NSCs on planar substrates, as measured by the fraction of TuJ1+ cells. (C) Differentiation of NSC on pillar arrays of different dimension, d = center-to-center distance, h = pillar height; i.e., d1h9 indicates pillars of 1 μm diameter and 9 μm height. All data are mean ± s.d., n=4, > 800 cells examined each condition, and were analyzed using ANOVA methods. * p < 0.05.
We next examined differentiation of NSCs on substrates of different rigidity, staining these cells for neuronal markers after 7-days in differentiation media. Cells were in expansion media only 1 day after seeding (and then switched to differentiation media), minimizing the effect of rigidity-dependent proliferation in these experiments. As shown in Figure 6B, the fraction of TuJ1+ cells was similar across the samples, indicating that this aspect of neurogenesis by SVZ NSCs is not sensitive to substrate rigidity at the range examined.
We note that our results of cell differentiation are different from Saha and colleagues 26. However, that report is based on polyacrylamide gels, which are of generally much lower elastic modulus (100 - 10KPa) than the PDMS surfaces used here and closer to that associated with neural tissues. To better align these studies, we also examined NSC differentiation on pillar arrays of different heights (Figure 6C) in order to bridge these systems, correlating the spring constant of the pillars with an equivalent substrate rigidity 8; 14; a constant pillar diameter of 1 μm was maintained across a key set of these surfaces to ensure that cells see the same amount and local organization of ECM protein. We again observed that the fraction of cells expressing a neuronal phenotype (TuJ1+) was not statistically different across the arrays of 1-μm pillars of various heights. A final sample based on 2-μm diameter pillars, included to reach higher equivalent rigidities, also had similar neuronal differentiation, further supporting the idea that substrate rigidity does not direct cell differentiation. However, it should be appreciated that the pillar arrays present a very different form of rigidity than a slab of varying rigidity. Specifically, the individual pillars are locally stiff on the scale of the pillar tops, appearing softer only over a larger physical range associated with the dimensions of multiple pillars. Reconciling the use of elastomer pillar arrays in this context, as well as understanding the complex response of the native extracellular environment, remain active areas of research.
Discussion
In this report, we used the high spatial and temporal resolution of the μPA platform to explore the dynamics of force generation by NSCs onto an underlying substrate. Most prominently, we demonstrate that forces exerted by NSCs onto laminin-coated pillars drops dramatically over the course of days, paralleling an increase of expression of phenotypic markers of neural stem cell differentiation. This drop in force generation may represent a change from cell-ECM contact to cell-cell contact, following changes in cell association observed with these cells both in vitro and in vivo. We also demonstrate that at short timescales of minutes, NSCs apply localized, puslatile patterns of forces to an underlying substrate. This shares some similarity with dynamics observed in fibroblasts, whose migration is closely coupled with protrusion and retraction of processes 6; 12; 15; 24, as forces were localized to the tips of processes elaborated by these cells. However, NSCs also exhibit a localized contraction underlying cells, not normally associated with force generation by cells in culture. These forces have strong correlations with the pulsatile motion of neural precursors observed in vitro, providing potentially a new system for studying this specialized motion.
The temporal dynamics of these pulsatile contraction sites share other similarities with non-neuronal cell systems. In particular, the Sheetz group has identified and analyzed cyclic, oscillatory waves of lamellipodia extension and contraction in early spreading of a range of cells, including fibroblasts, wing-disk cells, and T cells, posing a strategy by which these cells measure the mechanical properties of their environment 3; 11; 15. While sharing the transient property of the contraction sites observed in the present study, the waves associated with cell spreading are organized around the periphery of the cell, rather than under the cell processes and body. Moreover, the localized contraction sites underlying NSCs showed no correlation in contraction; in contrast, the membrane waves studied by the Sheetz group demonstrate different degrees of spatial synchronization around the cell periphery. The roles of regulatory signaling proteins that are proposed to regulate the membrane waves, such as Rac and cofilin, may provide important insights for understanding force generation in NSCs.
The role of matrix rigidity in local direction of neural development and stem cell function remains a provocative yet incompletely understood question. Our results indicate that proliferation of NSCs is sensitive to substrate rigidity to a larger extent than differentiation, suggesting that local stiffness of the niche may play a role in coordinating the cell-by-cell regulation of proliferation vs. differentiation required for proper tissue organization. However, we recognize that other groups 26 have observed changes in neural stem cell differentiation as a function of rigidity on softer surfaces than the planar PDMS used in this report. Together, these data support a model in which the magnitude of applied force may correlate with the range of rigidities over which cells can differentially respond. It is notable that recent reports by Ladoux and colleagues 14; 25 provides evidence that across substrates of different stiffness, cells will apply forces to a substrate that will induce a specific degree of deformation, up to a characteristic limit; this limit, and the deformation, are cell specific. Our results, building on this model, suggest that these characteristics may change as a function of differentiation or cell state, allowing cells to respond to specific ranges of matrix stiffness. Further investigations are needed to test the intriguing aspects of rigidity in directing NSC growth, leading to improved understanding of neural development and design of materials that interact with the nervous system.
Materials and Methods
Preparation of poly(dimethylsiloxane) μPAs
Established microfabrication techniques were used to create primary masters from which the elastomer μPAs were ultimately produced 8; 13; 29. Briefly, a deep plasma etching process was used to create arrays of pillars in a silicon wafer. The resultant master was silanized with tridecafluoro-trichlorosilane in vapor phase to facilitate release of elastomer. PDMS elastomer is then poured on the silicon master and cured for 12 hours before being peered off to form a secondary PDMS master, which was further silanized with perfluorooctyl-trichlorosilane before use. Arrays were produced by placing a small drop uncured PDMS on the secondary PDMS master, and pressed against a coverslip. This construct was then incubated at 85 °C for 12 hours before releasing, yielding a final μPA attached to a coverslip. This approach yields a thin PDMS layer underlying the pillars, which assists in conducting microscopy with the short-working distance objectives typically used for high-resolution microscopy.
Before seeding of cells, the μPA was coated with natural mouse laminin (Invitrogen) overnight at a concentration of 10 μg/ml. A small fraction of laminin was labeled with Cy5 dye (GE Healthcare) and mixed 1:5 with unlabeled protein, in order to fluorescently visualize the pillars while minimizing the effect of labeling on cell response.
Preparation of variable rigidity surfaces
Planar PDMS substrates of different rigidities (25kPa, 500Kpa, 2MPa) were prepared by varying the ratio of curing agent to base (1:50, 1:25 and 1:10, respectively). These substrates were then coated with laminin at 10 μg/ml overnight before use; the per-area concentration of protein absorbed to these substrates is independent of formulation 30.
Neural Stem Cells
All culture reagents were from Invitrogen unless otherwise stated. SVZ NSCs were generated and maintained in adherent culture following established methods with modifications 20; 23;. Brain tissue was isolated from juvenile rats (post-natal day 11, Taconic), from which a ∼400 um coronal slice of the forebrain was obtained and placed in cold PBS supplemented with 1.26 mM Ca2+ and 4.5 g/L glucose (GPBS). A stripe of SVZ tissue was cut along the lateral ventricle wall and kept in cold GPBS. Tissue was minced and treated with papain (Sigma) for 30 min before being transferred to DMEM media supplemented with 10% FBS. Tissue was mechanically dissociated by repeated trituration. The cell solution was passed through a 40-μm pore nylon mesh to remove large tissue debris, and seeded onto petri-dishes which were sequentially coated with polyornithine (Sigma) and laminin. These primary cells were maintained in DMEM with 10% FBS for 24 hours. The media was then replaced with serum free expansion media consisting of DMEM/F12 (1:1), B27 supplements without Vitamin A, 2 μg/ml heparin, 20 ng/ml bFGF (Upstate) and antibiotics. Half of the media was replaced every other day. To promote neuronal differentiation, expansion media was replaced with DMEM/F12 containing B27 supplements, 1μM retinoic acid, 2μM forskolin and antibiotics. When cultured on μPA, NSCs were seeded in expansion media at a density of 5000-8000/cm2, and kept overnight before switching to differentiation media. Half of the media was replaced every 3 days. As specified for specific experiments, blebbistatin was included in the cell culture media at a concentration of 50 μM.
Immunocytochemistry
Cell morphology and differentiation were determined using standard immunocytochemical techniques. At specific time points (1, 4, 8 days) in culture, cells were fixed for 15 min in 4% paraformaldehyde in PBS, permeabilized in 0.25% Triton X-100 for 10 min, and then blocked with 4% BSA in PBS for either 1 hour at 37°C or overnight at 4°C. Samples were incubated with primary antibodies in 4% BSA in PBS for 1 hour at room temperature as indicated in the text and figures: anti-βIII-tubulin (1:2000, Covance), anti-nestin (1:500, R&D Systems), anti-GFAP (1:750, Millipore). Samples were then rinsed with PBS, incubated with secondary antibodies labeled with either Alexa 488, Alexa 568, or Alexa 647 (Invitrogen), and diluted 1:500 in 4% BSA solution for 30 minutes, rinsed with PBS and then stained for nucleus with DAPI (Invitrogen). Imaging and recording were done by using a Hamamatsu C9100-02 CCD camera attached to an Olympus IX71 microscope equipped with fluorescence imaging capabilities, and MetaMorph image analysis software (Universal Imaging).
Proliferating cells were identified using the Click-iT EdU (5-ethynyl-2′-deoxyuridine) kit (Invitrogen). One day before fixation, cells were treated with EdU containing media at 0.5 μM overnight. After fixation permeablization as described above, newly synthesized DNA was detected using Alexa Fluor 488 azide, part of the Click-iT EdU kit. The sample was then counterstained for lineage markers as described above. As an alternative approach that provides a more direct measure of functional proliferation, we compared the per-area density of cells in culture, on the same culture, over a period of 4 days.
Proliferation assay
To quantify the growth rates of NSC in expansion media, the number of cells per area on each surface was counted at 24 hour intervals. The data was then fit with an exponential growth model to obtain the growth rate r, where 1/r is the population doubling time. These data were normalized against average growth rate of the softest planar substrate we examined (PDMS of 25KPa Young's modulus).
Live Cell Microscopy
Live cells were observed by fluorescent and bright-field microscopy using a 100× oil objective (NA 1.45) on an inverted Olympus IX71 microscope. The temperature, CO2 and humidity level were maintained at 37 °C, 5% and 70% respectively using a live-cell imaging chamber (Pathology Devices, MD). Unless otherwise noted, images were collected at 30 second intervals. Pillars were imaged through the μPA (as opposed to through the cells) to minimize artifacts associated with refraction of light by cellular structures. Cells forming extensive cell-cell contacts were excluded from these analyses. However, cells formed extensive contacts between processes in normal culture; the formation of such process-process contact did not exclude cells from analysis.
The force applied by cells to individual pillars was estimated by measuring the deflection of each pillar tip. Displacement of each pillar from its original position was extracted from a time series of fluorescent images using the MetaMorph software suite. The trajectory of each pillar tip over time was generated from these displacements using an ImageJ plug-in 27 that outputs the coordinates of every pillar in each frame. These deflections were converted to estimated forces using a linear, infinitesimal deflection cantilever equation 29; F = (3*E*I/L3)*δ, where E = bulk Young's modulus of the material, I = second moment of inertia of the cantilever cross section, and L = pillar length. Pillars deflecting less than 2 pixels (a threshold established from observing the random fluctuations of pillars away from the cells) were not included in analysis. Comparisons of forces applied by cells under different conditions were compared using Kruskal-Wallis methods, α = 0.05.
To identify the maximum forces reported in Fig. 2, stacks of images taken at 30 second intervals and covering a total of 10-15 minutes were collected for cells on the pillar arrays. The maximum deflection of each pillar was determined from these stacks. The typical duration of a contraction event (Fig. 3) was also determined from image stacks covering 10-15 minutes. Plots of pillar deflection as a function of time were derived from these stacks, and a contraction was defined as the time over which the deflection was over 20% of the maximal value for that event. Coordination of contractions at different sites underlying a single cell was analyzed by calculating a Pearson's correlation coefficient, r, over these stacks. Traces of pillar deflections under a cell were divided into two groups, x and y, each representing a different contraction site and containing n timepoints. For each cell, a correlation coefficient was calculated as r = [Σ(xi−x̄)(yi−ȳ)]/[(n−1)σxσy];i = 1..n. Values of 1 and -1 indicate strong correlation and anticorrelation, respectively, while values of 0 indicate no correlation.
Imaging of cytoskeleton dynamics
NSCs were transduced with actin-GFP using the Cellular Light baculovirus system (Invitrogen). Briefly, cells were cultured to 80% confluence, then culture media was replaced by the Cellular Light reagent diluted in PBS and incubated with gentle rotation for 1 hour at room temperature. Cells were further cultured in media containing enhancer for 2 hours before being changed back to normal expansion media. The transduced cells were collected by trypsin treatment, seed onto laminin coated μPA substrates and imaged the next day.
Supplementary Material
Supplementary Material S1. Cells differentiated from SVZ-NSCs were present as clusters migrating over each other in a pulsate mode, resembling “chain migration” of neuroblasts in the SVZ stem cell niche.
Supplementary Material S2. Transient, multisite force generation by NSCs in culture. Each time series was collected at 30 second intervals (showed at 1 minute interval), covering a 10-minute observation period.
Supplementary Material S3. Pulsatile nuclear migration by radial-glia-like NSCs in vitro. The position of the nucleus in each frame was determined by fitting a oval to the cell body; the centroid of this structure is indicated by the green dot in each frame. The bottom frame compares average force generation under the nucleus and nucleus position as a function of time.
Supplementary Material S4. Detail of molecular architecture shown in Figure 4A, 1DIV. μPAs were coated with fluorescently-labeled laminin (blue). The μPA array is the standard dimensions of 1 μm diameter, 2 μm center-to-center spacing, 7 μm height; the field of view of this image measures 133 μm across. Actin cytoskeleton and cell nuclei were stained with phalloiding (green) and DAPI (red) respectively. Image stacks were collected in standard epifluorescence mode then deconvoluted and projected onto the indicated visualization planes using standard image processing software.
Supplementary Material S5. Application of blebbstatin to cells in expansion media induces rapid changes in morphology. Corresponding effects on nuclear migration are quantified in Figure 5 D&E.
Acknowledgments
This work was funded by the National Institutes of Health through the NIH Roadmap for Medical Research (PN2 EY016586).
References
- 1.Beningo KA, Dembo M, Kaverina I, Small JV, Wang YL. Nascent focal adhesions are responsible for the generation of strong propulsive forces in migrating fibroblasts. J Cell Biol. 2001;153:881–888. doi: 10.1083/jcb.153.4.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burridge K, Chrzanowska-Wodnicka M. Focal adhesions, contractility, and signaling. Annu Rev Cell Dev Biol. 1996;12:463–518. doi: 10.1146/annurev.cellbio.12.1.463. [DOI] [PubMed] [Google Scholar]
- 3.Cai Y, Biais N, Giannone G, Tanase M, Jiang G, Hofman JM, Wiggins CH, Silberzan P, Buguin A, Ladoux B, Sheetz MP. Nonmuscle myosin IIA-dependent force inhibits cell spreading and drives F-actin flow. Biophys J. 2006;91:3907–3920. doi: 10.1529/biophysj.106.084806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Campos LS, Leone DP, Relvas JB, Brakebusch C, Fassler R, Suter U, ffrench-Constant C. Beta1 integrins activate a MAPK signalling pathway in neural stem cells that contributes to their maintenance. Development. 2004;131:3433–3444. doi: 10.1242/dev.01199. [DOI] [PubMed] [Google Scholar]
- 5.Conti L, Pollard SM, Gorba T, Reitano E, Toselli M, Biella G, Sun Y, Sanzone S, Ying QL, Cattaneo E, Smith A. Niche-independent symmetrical self-renewal of a mammalian tissue stem cell. PLoS Biol. 2005;3:e283. doi: 10.1371/journal.pbio.0030283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dembo M, Wang YL. Stresses at the cell-to-substrate interface during locomotion of fibroblasts. Biophys J. 1999;76:2307–2316. doi: 10.1016/S0006-3495(99)77386-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doetsch F, Caille I, Lim DA, Garcia-Verdugo JM, Alvarez-Buylla A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell. 1999;97:703–716. doi: 10.1016/s0092-8674(00)80783-7. [DOI] [PubMed] [Google Scholar]
- 8.du Roure O, Saez A, Buguin A, Austin RH, Chavrier P, Silberzan P, Ladoux B. Force mapping in epithelial cell migration. Proceedings of the National Academy of Science, USA. 2005;102:2390–2395. doi: 10.1073/pnas.0408482102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elkin BS, Azeloglu EU, Costa KD, Morrison B., 3rd Mechanical heterogeneity of the rat hippocampus measured by atomic force microscope indentation. J Neurotrauma. 2007;24:812–822. doi: 10.1089/neu.2006.0169. [DOI] [PubMed] [Google Scholar]
- 10.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 11.Galbraith CG, Sheetz MP. A micromachined device provides a new bend on fibroblast traction forces. Proc Natl Acad Sci U S A. 1997;94:9114–9118. doi: 10.1073/pnas.94.17.9114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Galbraith CG, Yamada KM, Sheetz MP. The relationship between force and focal complex development. J Cell Biol. 2002;159:695–705. doi: 10.1083/jcb.200204153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghassemi S, Biais N, Maniura K, Wind SK, Sheetz MP, Hone J. Fabrication of elastomer Pillar Arrays with Modulated Stiffness for Cellular Force Measurements. Journal of Vacuum Science & Technology B. 2008;26:2549–2553. doi: 10.1116/1.3013424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghibaudo M, Saez A, Trichet L, Xayaphoummine A, Browaeys J, Silberzan P, Buguin A, Ladoux B. Traction forces and rigidity sensing regulate cell functions. Soft Matter. 2008;4:1836–1843. [Google Scholar]
- 15.Giannone G, Dubin-Thaler BJ, Dobereiner HG, Kieffer N, Bresnick AR, Sheetz MP. Periodic lamellipodial contractions correlate with rearward actin waves. Cell. 2004;116:431–443. doi: 10.1016/s0092-8674(04)00058-3. [DOI] [PubMed] [Google Scholar]
- 16.Gotz M, Huttner WB. The cell biology of neurogenesis. Nat Rev Mol Cell Biol. 2005;6:777–788. doi: 10.1038/nrm1739. [DOI] [PubMed] [Google Scholar]
- 17.Higginbotham HR, Gleeson JG. The centrosome in neuronal development. Trends Neurosci. 2007;30:276–283. doi: 10.1016/j.tins.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 18.Lathia JD, Patton B, Eckley DM, Magnus T, Mughal MR, Sasaki T, Caldwell MA, Rao MS, Mattson MP, ffrench-Constant C. Patterns of laminins and integrins in the embryonic ventricular zone of the CNS. J Comp Neurol. 2007;505:630–643. doi: 10.1002/cne.21520. [DOI] [PubMed] [Google Scholar]
- 19.Leone DP, Relvas JB, Campos LS, Hemmi S, Brakebusch C, Fassler R, Ffrench-Constant C, Suter U. Regulation of neural progenitor proliferation and survival by beta1 integrins. J Cell Sci. 2005;118:2589–2599. doi: 10.1242/jcs.02396. [DOI] [PubMed] [Google Scholar]
- 20.Lim DA, Alvarez-Buylla A. Interaction between astrocytes and adult subventricular zone precursors stimulates neurogenesis. Proc Natl Acad Sci U S A. 1999;96:7526–7531. doi: 10.1073/pnas.96.13.7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin YW, Cheng CM, LeDuc PR, Chen CC. Understanding Sensory Nerve Mechanotransduction through Localized Elastomeric Matrix Control. PLoS ONE. 2009;4:e4293. doi: 10.1371/journal.pone.0004293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mercier F, Kitasako JT, Hatton GI. Anatomy of the brain neurogenic zones revisited: fractones and the fibroblast/macrophage network. J Comp Neurol. 2002;451:170–188. doi: 10.1002/cne.10342. [DOI] [PubMed] [Google Scholar]
- 23.Palmer TD, Markakis EA, Willhoite AR, Safar F, Gage FH. Fibroblast growth factor-2 activates a latent neurogenic program in neural stem cells from diverse regions of the adult CNS. J Neurosci. 1999;19:8487–8497. doi: 10.1523/JNEUROSCI.19-19-08487.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. Cell migration: integrating signals from front to back. Science. 2003;302:1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 25.Saez A, Buguin A, Silberzan P, Ladoux B. Is the mechanical activity of epithelial cells controlled by deformations or forces? Biophys J. 2005;89:L52–54. doi: 10.1529/biophysj.105.071217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saha K, Keung AJ, Irwin EF, Li Y, Little L, Schaffer DV, Healy KE. Substrate modulus directs neural stem cell behavior. Biophys J. 2008;95:4426–4438. doi: 10.1529/biophysj.108.132217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sbalzarini IF, Koumoutsakos P. Feature point tracking and trajectory analysis for video imaging in cell biology. J Struct Biol. 2005;151:182–195. doi: 10.1016/j.jsb.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 28.Schaar BT, McConnell SK. Cytoskeletal coordination during neuronal migration. Proc Natl Acad Sci U S A. 2005;102:13652–13657. doi: 10.1073/pnas.0506008102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tan JL, Tien J, Pirone DM, Gray DS, Bhadriraju K, Chen CS. Cells lying on a bed of microneedles: an approach to isolate mechanical force. Proc Natl Acad Sci U S A. 2003;100:1484–1489. doi: 10.1073/pnas.0235407100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsai J, Kam L. Rigidity-dependent cross talk between integrin and cadherin signaling. Biophys J. 2009;96:L39–41. doi: 10.1016/j.bpj.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsai JW, Bremner KH, Vallee RB. Dual subcellular roles for LIS1 and dynein in radial neuronal migration in live brain tissue. Nat Neurosci. 2007;10:970–979. doi: 10.1038/nn1934. [DOI] [PubMed] [Google Scholar]
- 32.Tsai JW, Chen Y, Kriegstein AR, Vallee RB. LIS1 RNA interference blocks neural stem cell division, morphogenesis, and motility at multiple stages. J Cell Biol. 2005;170:935–945. doi: 10.1083/jcb.200505166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ulrich TA, de Juan Pardo EM, Kumar S. The mechanical rigidity of the extracellular matrix regulates the structure, motility, and proliferation of glioma cells. Cancer Res. 2009;69:4167–4174. doi: 10.1158/0008-5472.CAN-08-4859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wichterle H, Garcia-Verdugo JM, Alvarez-Buylla A. Direct evidence for homotypic, glia-independent neuronal migration. Neuron. 1997;18:779–791. doi: 10.1016/s0896-6273(00)80317-7. [DOI] [PubMed] [Google Scholar]
- 35.Wozniak MA, Desai R, Solski PA, Der CJ, Keely PJ. ROCK-generated contractility regulates breast epithelial cell differentiation in response to the physical properties of a three-dimensional collagen matrix. J Cell Biol. 2003;163:583–595. doi: 10.1083/jcb.200305010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zajac AL, Discher DE. Cell differentiation through tissue elasticity-coupled, myosin-driven remodeling. Curr Opin Cell Biol. 2008;20:609–615. doi: 10.1016/j.ceb.2008.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material S1. Cells differentiated from SVZ-NSCs were present as clusters migrating over each other in a pulsate mode, resembling “chain migration” of neuroblasts in the SVZ stem cell niche.
Supplementary Material S2. Transient, multisite force generation by NSCs in culture. Each time series was collected at 30 second intervals (showed at 1 minute interval), covering a 10-minute observation period.
Supplementary Material S3. Pulsatile nuclear migration by radial-glia-like NSCs in vitro. The position of the nucleus in each frame was determined by fitting a oval to the cell body; the centroid of this structure is indicated by the green dot in each frame. The bottom frame compares average force generation under the nucleus and nucleus position as a function of time.
Supplementary Material S4. Detail of molecular architecture shown in Figure 4A, 1DIV. μPAs were coated with fluorescently-labeled laminin (blue). The μPA array is the standard dimensions of 1 μm diameter, 2 μm center-to-center spacing, 7 μm height; the field of view of this image measures 133 μm across. Actin cytoskeleton and cell nuclei were stained with phalloiding (green) and DAPI (red) respectively. Image stacks were collected in standard epifluorescence mode then deconvoluted and projected onto the indicated visualization planes using standard image processing software.
Supplementary Material S5. Application of blebbstatin to cells in expansion media induces rapid changes in morphology. Corresponding effects on nuclear migration are quantified in Figure 5 D&E.