

Abstract
Hantaviruses, members of the Bunyaviridae family, are emerging category A pathogens that carry three negative stranded RNA molecules as their genome. Hantavirus nucleocapsid protein (N) is encoded by the smallest S segment genomic RNA (viral RNA). N specifically binds mRNA caps and requires four nucleotides adjacent to the cap for high affinity binding. We show that the N peptide has distinct cap- and RNA-binding sites that independently interact with mRNA cap and viral genomic RNA, respectively. In addition, N can simultaneously bind with both mRNA cap and vRNA. N undergoes distinct conformational changes after binding with either mRNA cap or vRNA or both mRNA cap and vRNA simultaneously. Hantavirus RNA-dependent RNA polymerase (RdRp) uses a capped RNA primer for transcription initiation. The capped RNA primer is generated from host cell mRNA by the cap-snatching mechanism and is supposed to anneal with the 3′ terminus of vRNA template during transcription initiation by single G-C base pairing. We show that the capped RNA primer binds at the cap-binding site and induces a conformational change in N. The conformationally altered N with a capped primer loaded at the cap-binding site specifically binds the conserved 3′ nine nucleotides of vRNA and assists the bound primer to anneal at the 3′ terminus. We suggest that the cap-binding site of N, in conjunction with RdRp, plays a key role during the transcription and replication initiation of vRNA genome.
Keywords: Negative Strand RNA Viruses, Viral Polymerase, Viral Protein, Viral Transcription, Virus Assembly, HPS, Hantavirus Assembly, Hantavirus Cardiopulmonery Syndrome, Hantaviruses, Nucleocapsid Protein
Introduction
Hantaviruses cause two types of serious human illnesses when transmitted to humans from rodent hosts: hemorrhagic fever with renal syndrome and hantavirus cardiopulmonary syndrome (1, 2). The spherical hantavirus particles harbor three negative sense genomic RNA segments (S, L, and M segments) within a lipid bilayer (3). The mRNAs derived from S, L, and M segments encode viral nucleocapsid protein (N), viral RNA-dependent RNA polymerase (RdRp),2 and glycoproteins (G1 and G2), respectively. The characteristic feature of the hantaviral genome is the partially complementary sequence at the 5′ and 3′ termini of each of the three genome segments that undergo base pairing and form panhandle structures (4–6). N is a multifunctional protein playing a vital role in multiple processes of virus replication cycle and has been found to undergo trimerization both in vivo and in vitro (7–19). During encapsidation, the three viral RNA (vRNA) molecules are specifically recognized by N inside the host cell and targeted for packaging. Multiple in vitro studies have shown that N preferentially binds vRNA compared with complementary RNA (cRNA) or nonviral RNA (13, 20–25). It has been proposed that the specific binding of N with either the panhandle or the sequence at the 5′ terminus alone selectively facilitates the encapsidation of vRNA to generate three nucleocapsids that are packaged into infectious virions (25, 26). The RNA-binding domain of Hantaan virus N protein has been mapped to the central conserved region corresponding to amino acids from 175 to 217 (24).
Nucleocapsids composed of vRNA and N protein serve as templates for the transcription and replication of viral genome by the RdRp (27, 28). The RdRp from segmented negative sense RNA viruses requires a capped RNA oligo as a primer to initiate the transcription (29–33). The capped RNA primer is generated from the 5′ terminus of host cell mRNA by the unique “cap-snatching” process that is well characterized in the influenza virus. Although knowledge about the sequence, length, and structure of the 5′ mRNA terminus that donates the primer is rather limited, most common cap donor mRNAs are cleaved 15 nucleotides downstream the cap with a variation of 10–20 nucleotides (30, 31, 34–40). Exceptions have been reported for the members of the Arenaviridae and Nairovirus genus that use relatively shorter primers, varying in length from 1–4 and 5–16 nucleotides, respectively (37, 41, 42). In the case of the influenza virus, the PB2 subunit of heterotrimeric RdRp binds to the 5′ caps of host pre-mRNAs, followed by the endonucleolytic cleavage by the PB1 subunit 10–13 nucleotides downstream from the cap (43, 44). A cap-snatching mechanism similar to the influenza virus has been proposed for all negative sense-segmented RNA viruses, including hantaviruses. However unlike the influenza virus, the hantavirus RdRp is a single peptide of ∼280 kDa that replicates the viral genome in the cytoplasm of infected cells. In addition, the endonuclease activity of hantavirus RdRp is not known, suggesting that the hantavirus cap-snatching mechanism might be different from influenza.
Sequence analysis of many viral mRNA 5′ termini have revealed a nucleotide preference at the 3′ end of the capped primer that have been assumed to reflect the sequence preference for cleavage by the viral endonuclease during cap snatching. For example, in case of the Dugbe virus, endonucleolytic cleavage is assumed to take place after a “C” residue (37), whereas for Bunyamwera virus, a strong preference for cleavage after a “U” residue has been proposed (45). In tomato spotted wilt virus, a preference for an “A” residue has been confirmed (46).
In hantaviruses, the capped primers generated by the cap-snatching mechanism have a 3′ “G” residue that has been proposed to undergo base pairing with one of the C residues at the 3′ terminus of vRNA template during transcription initiation. It has been suggested that the annealed primer is elongated by RdRp during transcription initiation using a “prime and realign” mechanism (14, 41). However, it is not yet clear how a single G-C base pairing between the RNA primer and 3′ terminus of vRNA template stabilizes the primer and favors its annealing. Hantavirus nucleocapsid protein binds the mRNA caps and requires five nucleotides adjacent to the cap for high affinity binding (16). The cap-binding activity of hantavirus N has been suggested to play a role in the cap-snatching process (14). In this manuscript, we report that hantavirus N protein has two distinct binding sites for RNA and mRNA cap. N can simultaneously bind with both the vRNA and mRNA cap. The role of cap- and RNA-binding sites to assist the annealing of a capped RNA primer at the 3′ terminus of vRNA template has been discussed. Because hantaviral RdRp requires N for its function, it is likely that N works in conjunction with RdRp during cap snatching and transcription initiation.
EXPERIMENTAL PROCEDURES
Oligonucleotides and Enzymes
PCR primers were from Integrated DNA Technologies. All restriction enzymes were from New England Biolabs. HotMaster taq polymerase was from Eppendorf. DNase I and T7 transcription reagents were from Promega. RNA purification reagents were from Qiagen and reverse transcription reagents were from Invitrogen. All other chemicals, including bis-ANS and cap analogs (m7GTP, m7GDP, and m7GMP) were obtained from Sigma. Radio isotopes [α-32P]CTP and [α-32P]dATP were from Perkin Elmer.
Expression and Purification of Hantavirus N Protein
A gene encoding N protein was derived from Sin Nombre hantavirus strain 77734 (47, 48) and cloned in pTriEx 1.1 vector to generate the expression vector pTSNV N as described previously (16). N containing a His6 tag at the C terminus was expressed in Escherichia coli and purified as described previously (13, 16). For the expression of an N mutant lacking an RNA binding domain, we PCR-amplified the parent vector that expresses wild type N, using forward primer (5′-CCTGTTATGGGTGTGATTGGCTTTAGTTTCT-3′) and reverse primer (5′-TCTTGGCTTACGTATTCCATTAACT-3′). The PCR product represented the whole expression vector, except for the sequence that encoded amino acids 175–217 of the N gene. The PCR product was self-ligated, generating the circularized plasmid that was transformed into Rossetta DE3 pLacI bacteria (Novagen). Similar to wild type N, the mutant N contained a C-terminal histidine tag that was used for its purification on nickel-nitrilotriacetic acid column.
RNA Synthesis by T7 Transcription Reaction
DNA encoding the full-length S segment RNA molecule with a proximally located T7 promoter was generated by PCR using an upstream primer containing a T7 promoter sequence, a downstream primer that overlaps the terminal sequence of the S segment transcription unit, and a pAD-S plasmid as template, as described previously (26). The resulting DNA containing the entire S segment transcription unit was gel-purified and used directly in transcription reactions with T7 RNA polymerase (MBI Fermentase). A short RNA molecule five nucleotides in length with a 5′ terminal cap was synthesized and gel-purified as described previously (16). RNA molecules, 10–20 nucleotides in length with or without 5′ caps, were synthesized by the T7 transcription reaction using short DNA templates containing proximal T7 promoter juxtaposed with a sequence that encodes the RNA of interest. Short DNA templates were generated by PCR amplification of two complementary primers. PCR product was gel-purified and used in T7 transcription reaction as described previously (14). For the synthesis of capped RNA, the m7G cap analog was used in the T7 transcription as described previously (16). Transcripts were radiolabeled by the incorporation of [α-32P]CTP in the transcription reaction. RNA was quantified by determining the percent incorporation of [α-32P]CTP into RNA using trichloroacetic acid precipitation. The total mass of RNA synthesized in T7 transcription reactions was calculated on the basis of the total number of moles of [α-32P]CTP incorporated and the average molecular weight of a nucleotide (320.5 g/mol). The specific activity of the RNA product was determined by trichloroacetic acid precipitation. RNA concentration was verified by A260 measurements.
Fluorescence Spectroscopic Studies
As reported previously (16), binding of cap analogs (m7GTP, m7GDP, and m7GMP) with N was carried out in Shimadzu spectrofluorometer RF-5301PC. Fluorescence spectrum of N from 300–500 nm was recorded in RNA binding buffer (40 mm HEPES, pH 7.4, 80 mm NaCl, 20 mm KCl, and 1 mm dithiothreitol) using an excitation wavelength of 295 nm (excitation slit width, 5 nm; and emission slit width, 10 nm). The steady state value of fluorescence emission at 330 nm indicated the absence of photodegradation of N. All fluorescence binding experiments were carried out at room temperature. To a fixed concentration of N (150 nm), cap analog was added at increasing concentrations, and the fluorescence spectrum of N, at 300–500 nm, was recorded at each input concentration of the cap analog. Because cap analog yields an independent fluorescence signal in the same wavelength range, we recorded the fluorescence signal of free cap analog at each input concentration and subtracted it from the fluorescence spectrum obtained when the cap analog was bound to N. To calculate the dissociation constant (Kd), the fluorescence value at 330 nm was calculated from differential spectra at each input concentration of the ligand (cap analog). The binding profile was generated by plotting the fluorescence value at 330 nm along the Y axis and ligand concentration along the X axis.
The dissociation constant (Kd) for the N-ligand interaction was calculated by a nonlinear curve-fitting analysis of the data points, based on the following equilibrium: L + P ⇔ LP, where L and P represent ligand and N, respectively. Curve fitting was carried out according to Equations 1 and 2 (49, 50). Goodness of fit was ascertained by least square analysis.
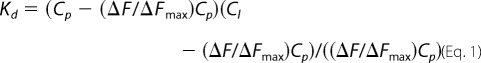 |
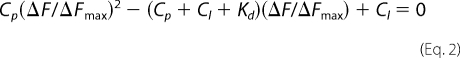 |
where ΔF is the change in fluorescence signal at 330 nm at each addition of the ligand. ΔFmax is the same parameter when N is totally bound to the ligand. Cl is the input concentration of the ligand. Cp is the initial concentration of N. The double reciprocal plot (1/ΔF versus 1/Cl (51)) was used to calculate the value of ΔFmax using Equation 3.
To monitor the conformational changes in N due to the binding of either capped RNA decamer at cap-binding site or S segment vRNA at the RNA-binding site or due to simultaneous binding of both capped decamer and vRNA at their respective binding sites, we monitored the quenching of tryptophan fluorescence of N by a neutral quencher acrylamide before or after the occupation of cap- and RNA-binding sites of N with their respective substrates, according to the Stern-Volmer equation (52) F0/F = 1 + Ksv[Q], where F0 and F are the fluorescence intensities in the absence and presence of the quencher respectively, [Q] is the concentration of the quencher, and Ksv is the Stern-Volmer quenching constant. Small aliquots of acrylamide from a concentrated stock solution were added to 300 μl of N protein solution containing 25 nm N in RNA binding buffer and fluorescence signal of tryptophan at 330 nm was recorded at each input concentration of acrylamide. Similar quenching studies were carried out with N that was preincubated with either 600 nm capped RNA decamer or 125 nm S segment vRNA or both together. If the data points in a plot of F0/F versus [Q] fit to a straight line that indicates a single species of tryptophan residues that are equally accessible to the neutral quencher. However, if the data points in the plot show a biphasic nature that indicates two species of tryptophan residues that are differentially accessible to the quencher. Stern-Volmer quenching constant (Ksv) was calculated from the slope of the straight line fitted to the data points, as reported previously (13, 53).
Fluorescence studies of hydrophobic fluorophore bis-ANS were carried out in Shimadzu spectrofluorometer RF-5301PC. The fluorophore was dissolved in dimethyl sulfoxide, and its concentration was determined from extinction coefficient (Eo32 396 = 2,400 m−1 cm−1). The fluorophore was excited at 399 nm, and emission spectra were recorded at room temperature in RNA binding buffer from 420–600 nm. To a fixed concentration of N (25 nm), small aliquots of bis-ANS were added from a higher concentration stock and fluorescence value at 485 nm was recorded at each input concentration. Similarly, N was first preincubated with either capped RNA decamer (600 nm) or S segment vRNA (125 nm) or both simultaneously at room temperature for 45 min, followed by the addition of small aliquots of bis-ANS from higher concentration stock and fluorescence value at 485 nm was recorded. To determine the change in fluorescence signal of bis-ANS due to binding with N, the fluorescence signal of free bis-ANS in RNA binding buffer without N was subtracted.
RNA Filter Binding
Interaction of N with different radiolabeled RNA molecules including capped or uncapped RNA decamers, SNV S segment RNA, and short RNA oligos (Table 3) was studied by filter binding assay. RNA molecules were synthesized in vitro with T7 transcription reaction and radiolabeled with [α-32P]CTP during synthesis, as described above. All binding reactions were carried out in RNA binding buffer (26) at a constant concentration of RNA (1 pm) with increasing concentrations of N protein. Reaction mixtures were incubated at room temperature for 30–45 min and filtered through nitrocellulose membranes under vacuum. Filters were washed with 10 ml of RNA binding buffer and dried. The amount of RNA retained on the filter at different input concentrations of N was measured using a scintillation counter. Data points were fit to a hyperbolic equation using the program Origin 6 (Microcal). The apparent dissociation constant (Kd) corresponding to the concentration of N protein required to obtain the half-saturation in the binding profile, assuming that the complex formation obeys a simple bimolecular equilibrium. We assumed that the plateau in the binding profile represents complete binding of RNA to allow calculation at half-saturation.
TABLE 3.
Binding of SNV N and N-cap complex with short RNA molecules in RNA binding buffer containing 80 mm NaCl at room temperature
ND, not determined. Due to weak binding Kd values at higher salt concentrations for SNV N with all these sequences were not determined. However, the Kd values at 160 mm and 280 mm NaCl concentrations were 137 ± 4 and 141 ± 3, respectively, for the interaction of the N-cap complex with the sequence 1. Due to weak binding, Kd values at higher salt concentrations for N-cap complex with remaining four sequence were not determined.
| Sequence |
Kd |
|
|---|---|---|
| SNV N | N-cap complex | |
| nm | ||
| 3′ AUCAUCAUCa | 579 ± 7 | 131 ± 2 |
| 3′ UACCUAUCAb | 573 ± 5 | 583 ± 5 |
| 3′ AUCAUCc | ND | ND |
| 3′ CGGCAAUCAUCAUCd | 563 ± 7 | 573 ± 7 |
| 3′ GACUUACAAe | 549 ± 6 | 551 ± 5 |
a The nine-nucleotide-long triplet repeat sequence corresponding to 3′ terminus of vRNA is shown.
b The triplet repeat sequence is randomized.
c Hexanucleotide triplet repeat sequence (same as a) except one of the triplets is deleted.
d A 14-nucleotide-long RNA oligo composed of the 3′ triplet repeat sequence with juxtaposed five nonviral nucleotides.
e a nonviral nine-nucleotide-long RNA oligo.
Reverse Transcription Reactions
Reverse transcription reactions using SNV S segment RNA as template and short capped or uncapped RNA oligos (Fig. 6) as primers were carried out according to the manufacturer's protocol (Invitrogen). RNA primers had either three residues or a single G residue at the 3′ terminus complementary to the 3′ terminus of the vRNA template (Fig. 6). 20-μl reactions containing 5 nm SNV S segment RNA, 5 nm RNA primer, and 0.5 mm deoxynucleoside triphosphates with [α-32P]dATP were heated at 65 °C for 5 min and placed on ice to allow the annealing of the RNA primer to the 3′ end of SNV S segment RNA. To determine the effect of N on primer annealing, RNA primers were preincubated with N at room temperature for 20 min followed by further incubation with the vRNA template at room temperature for an additional 20 min without the heating and cooling steps. After incubation, 4 μl of 5× first strand buffer, 2 μl of 0.1 m dithiothreitol and 1 μl of RNase out were added to the reaction mixture, followed by incubation at 37 °C for 2 min. 1 μl of Moloney murine leukemia virus reverse transcriptase was then added and further incubated at 37 °C for 50 min. Reactions were terminated by heating at 70 °C for 5 min, and the samples were analyzed on 12% polyacrylamide gels to characterize the products of reverse transcription.
FIGURE 6.

Sequence of primer-RNA pairs used in reverse transcription reaction (Fig. 7) are shown in panels A–L. Capped RNA primers had either terminal three (A) or a single (B) nucleotide complementary with the 3′ terminus of SNV S segment RNA. C and D are the same as A and B, respectively, except the primers used were uncapped. E and F are the same as A and B, respectively, except the sequence at the 3′ terminus was randomized in italic. G and H are same as A and B, respectively, except the template was a nonviral RNA of the same length as the SNV S segment and contained 3′ terminal nine nucleotides from the 3′ terminus of S segment. I and J are the same as A and B, respectively, except the vRNA template has three nucleotides deleted at the 3′ terminus. K and L are the same as A and B, respectively, except that five extra nucleotides are added to the 3′ terminus of the vRNA template.
RESULTS
Purification of Hantavirus Nucleocapsid Protein
E. coli cells harboring the expression vector pTSNV N were induced with isopropyl 1-thio-β-d-galactopyranoside to express the His6-tagged N protein. The fusion protein was then isolated and purified as mentioned in “Experimental Procedures.” SDS-PAGE analysis (Fig. 1) of purified N protein shows that the protein was largely free of detectable heterologous bacterial proteins and had an electrophoretic mobility consistent with the expected mass of the peptide (52 kDa) as evidenced by comigration relative to that of protein molecular weight markers. Similarly, mutant N lacking RNA binding domain was expressed and purified (Fig. 1, lane 6).
FIGURE 1.
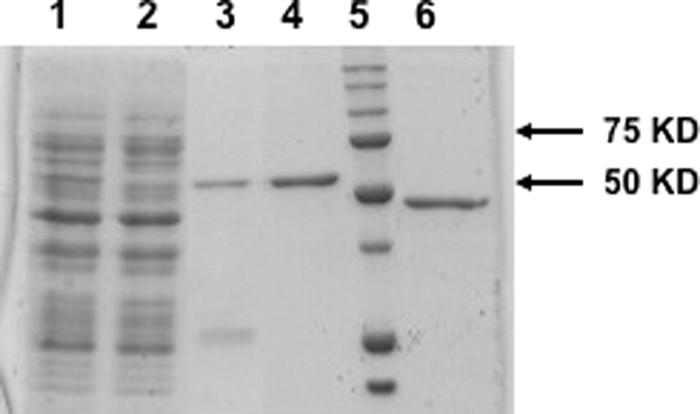
Expression and purification of wild type and mutant N protein. Bacterial lysate containing His-tagged wild type N (lane 1) was loaded onto a nickel-nitrilotriacetic acid column for purification. The flow through the column is shown in lane 2. After washing the column, bound N was eluted by running an imidazole gradient from 40–500 mm. Fractions containing N were pooled (lane 3). Pooled fractions were dialyzed and further purified on a size exclusion column (lane 4). Mutant N was processed similarly, and the final purified fraction of the mutant is shown in lane 6. The size of purified proteins matches correctly with the molecular weight marker (lane 5).
Characterization of Cap-binding Activity of N
We have previously reported that N binds the mRNA 5′ caps with high affinity and facilitates the translation of capped mRNAs (16). Our previous studies showed that N binds the cap analog (m7GTP) with a dissociation constant of ∼50 μm (16). There was a significant increase in the binding affinity when the terminal cap was associated with five adjacent nucleotides (Kd = 0.130 μm) (Table 1). We further carried out the binding studies at different salt concentrations to check the specificity for the interaction between the N and mRNA cap. In addition, we studied the interaction of N with m7GDP and m7GMP to analyze the role of phosphate moieties of the cap, if any, in binding with N. As reported previously, we used fluorescence spectroscopy to analyze the binding affinity between N and the m7G cap. Fig. 2A shows the tryptophan fluorescence spectrum of N protein from 300 nm to 500 nm. As expected from previous studies, we observed a consistent decrease in the fluorescence quantum yield of N at increasing input concentrations of m7GDP. This decrease in fluorescence quantum yield is due the interaction between N and m7GDP. We did not observe a change in the position of the fluorescence peak due to the interaction between N and m7GDP. Fluorescence intensity at 330 nm was recorded at increasing input concentrations of m7GDP to plot a binding profile. Dissociation constant was calculated as described under “Experimental Procedures.” Similarly, the dissociation constant for the interaction of N with m7GMP, m7GTP, and m7GUCUC was calculated at different salt concentrations (Table 1). The binding affinity between N and m7GTP/m7GDP/m7GMP decreased with increasing salt concentrations, indicating nonspecific binding. The phosphate moiety of the cap does not appear to have any role in binding. However, we did not observe a noticeable change in the dissociation constant between N and m7GUCUC at different salt concentrations (Table 1), consistent with specific binding between them.
TABLE 1.
Binding of SNV N with 5′ mRNA cap in RNA binding buffer at room temperature
| Cap analog |
Kd |
||
|---|---|---|---|
| 80 mm NaCl | 160 mm NaCl | 280 mm NaCl | |
| μm | |||
| m7GTP | 50.0 ± 3 | 85.0 ± 2 | 230.0 ± 1 |
| m7GDP | 45.0 ± 2 | 91.0 ± 3 | 250.0 ± 3 |
| m7GMP | 52.0 ± 2 | 89.0 ± 4 | 239.0 ± 3 |
| m7GUCUC | 0.130 ± 3 | 0.135 ± 3 | 0.134 ± 3 |
FIGURE 2.

Fluorescence binding of m7GDP with N protein. A shows the fluorescence spectrum of N protein (150 nm) from 300–500 nm (top spectra) in RNA-binding buffer. Shown is the differential fluorescence spectrum of N, after incubation with 90 nm (middle) and 40 nm (bottom) m7GDP. A fluorescence signal of 40 and 90 nm m7GDP without N was subtracted. Fluorescence intensity at 330 nm was recorded at each input concentration of m7GDP. A plot of fluorescence intensity at 330 nm versus input m7GDP concentration is shown in B. ΔF/ΔFmax was calculated as described under “Experimental Procedures” and plotted against m7G concentration (C). Data points were fitted according to Equations 1 and 2 (under “Experimental Procedures”) for the calculation of Kd. a.u., arbitrary units.
N Has Distinct m7G Cap- and RNA-binding Sites
Because N preferentially binds the mRNA cap and requires four adjacent nucleotides for high affinity binding (Table 1), we asked whether N has a distinct binding domain for m7G cap. We synthesized a RNA decamer (5′GUCUCUCCCA-3′) using T7 RNA polymerase, as described under “Experimental Procedures.” This decamer has a single G residue at the 5′ terminus that can be easily replaced by the m7G cap during synthesis by the incorporation of m7GTP instead of regular GTP in the transcription reaction. This decamer was radiolabeled with [α-32P]CTP during synthesis and purified on denaturing acrylamide gel as described previously (14, 16). We used filter binding assays to study the interaction of N with both capped and uncapped decamers (Fig. 3A). Binding reactions were carried out at three different salt concentrations to evaluate the binding specificity. As expected, N bound tightly with capped decamer compared with uncapped decamer at low salt concentrations (Table 2). The binding affinity of N for capped decamer (Kd = 119 nm) is ∼4-fold higher than uncapped decamer (Kd = 510 nm) at 80 mm NaCl concentration. With the increasing salt concentration, the binding affinity of N for uncapped decamer consistently decreased and was fairly low beyond 160 mm NaCl concentrations, suggesting nonspecific binding of N with uncapped decamer. In comparison, the binding affinity of N for capped decamer remained unaltered with increasing salt concentrations suggesting specific binding. Unlike uncapped decamer, N interacted specifically with viral S segment RNA, as is evident from unaltered high affinity binding at high salt concentrations (Table 2). Due to nonspecific binding, further studies with uncapped decamer were avoided.
FIGURE 3.

Filter binding of different radiolabeled RNA molecules with N. A, results of a filter binding assay for the interaction of radiolabeled capped (filled squares) and uncapped (open circles) RNA decamer with N. Radiolabeled RNA decamers were incubated with increasing concentrations of N at room temperature and filtered through a nitrocellulose filter. The percentage of hot RNA retained on the filter is plotted versus increasing input concentration of N to generate the binding profile for the calculation of dissociation constants (shown in Table 2). Similarly, the binding profiles for the interaction of wild type N (open squares) and mutant N (filled squares) with SNV S segment RNA and capped RNA decamer, respectively, were generated (B). The binding profile for N-capped decamer interaction is replotted for comparison. C, in a competition assay, a fixed concentration of N (530 nm) was incubated with 0.01 nm radiolabeled capped decamer at increasing input concentrations of either cold capped decamer (filled squares) or cold viral S segment RNA (open squares) and filtered through a nitrocellulose filter. The percentage of hot capped decamer retained on the filter is plotted versus competitor RNA concentration. D, a filter binding assay was carried out in which a fixed concentration of radiolabeled SNV S segment was incubated with increasing concentrations of N-cap complex. The N-cap complex was generated by incubating N with saturating concentrations of cold capped RNA decamer. (see “Experimental Procedures” for details). Reaction mixtures were filtered through a nitrocellulose filter, and the percentage of hot S segment RNA retained on the filter is plotted versus input concentrations N-cap complex to generate the binding profile for the calculation of Kd. a.u., arbitrary units.
TABLE 2.
Binding of SNV N with different RNA molecules in RNA binding buffer at room temperature
| RNA decamer |
Kd |
||
|---|---|---|---|
| 80 mm NaCl | 160 mm NaCl | 280 mm NaCl | |
| nm | |||
| Capped decamer | 119.0 ± 3 | 120.0 ± 2 | 122.0 ± 1 |
| Uncapped decamer | 510.0 ± 2 | ND | ND |
| SNV S segment RNA | 27.0 ± 3 | 25.0 ± 2 | 28.0 ± 1 |
| SNV S segment RNAa | 29.0 ± 3 | 27.0 ± 2 | 32.0 ± 1 |
| Capped decamerb | 125 ± 2 | 129 ± 3 | 131 ± 3 |
a Shown is the binding of the SNV S segment RNA with the N-cap complex. The complex was generated by incubated N with cold capped RNA decamer (see text for details).
b Shown is the binding of mutant N that lacks RNA binding domain with capped RNA decamer.
We used a competitive binding assay to check whether capped decamer and viral S segment RNA bind to the same or different sites on N protein. 530 nm N was incubated with 0.01 nm radiolabeled capped decamer at different input concentrations of either cold capped decamer or viral S segment RNA. As expected, cold capped decamer efficiently competed with the hot capped decamer for binding with N. Surprisingly, even an 1,000-fold excess of S segment RNA failed to compete with hot capped decamer for binding with N (Fig. 3C). We observed a 99% binding of hot capped decamer with N, although cold S segment RNA was 1,000-fold higher in concentration in the binding reaction (Fig. 3C). This suggests that binding sites for capped decamer and viral S segment RNA are different.
To further confirm that N has different binding sites for capped decamer and viral S segment RNA, we asked whether a complex of N with capped decamer is still able to interact with viral S segment RNA. This interaction is possible only if N has different binding sites for capped decamer and S segment viral RNA. We incubated N with cold capped decamer at a molar ratio of 1:100 of N:cold capped decamer to allow the complex formation. This complex was tested for the binding with hot S segment viral RNA. Using filter binding assay, a fixed concentration of hot S segment RNA was incubated with the increasing input concentrations of the complex. Reaction mixtures were filtered through nitrocellulose filter and the retention of hot S segment RNA on the filter was monitored. Fig. 3 depicts that the N-capped decamer complex is able to interact with radiolabeled S segment RNA with the same affinity as free N (compare S segment binding from Fig. 3, B and D). This is further evident from the similar dissociation constants (Table 2). This observation verifies the presence of different binding sites on N for m7G capped decamer and viral RNA.
Hantaviruses have a highly conserved RNA binding domain located at 175–217 amino acids (24). Deletion of this RNA-binding domain abrogates the ability of N to interact with viral S segment RNA (24, 54). We generated a deletion mutant of N that lacks the RNA binding domain and was purified as described in “Experimental Procedures” (Fig. 1). We hypothesized that if N has different binding sites for cap and viral RNA, then mutant N should be able to interact with the capped decamer. Filter binding assay was used to check the interaction of mutant N with capped RNA decamer. Binding reactions were carried out at three different salt concentrations to analyze the binding specificity. As expected, this mutant N was able to interact specifically with the capped decamer with similar affinity as wild type N (Fig. 3, A and B). This is evident with unaltered binding affinity at different salt concentrations (Table 2). These observations clearly demonstrate that N has distinct binding sites for cap and viral RNA.
Induction of Conformational Change in N Due to m7G Cap and RNA Binding
To further characterize the distinctive nature of cap- and RNA-binding sites on N, we compared fluorescence quenching of N-associated tryptophan residues in the absence and presence of either capped decamer or SNV S segment RNA or both. Fig. 4A depicts the effect of SNV S segment RNA on the fluorescence quenching of purified N with increasing concentrations of acrylamide, an uncharged quencher that tends to quench the fluorescence signal of accessible tryptophan residues. This analysis yielded a Stern-Volmer quenching constant (Ksv) of 4.25 for free N. However, when N is bound to the SNV S segment RNA, the tryptophan fluorescence was rapidly quenched at each input concentration of acrylamide, resulting in a Ksv of 6.25. This is consistent with S segment viral RNA-induced conformational change in N, such that tryptophan residues are exposed on the surface of the protein and easily accessible to the quencher. Similarly, the Ksv value for the binding of N with capped RNA decamer was calculated (Fig. 4B). We observed a decreased Ksv value of 3.36, consistent with a cap-induced conformational change in N such that tryptophan residues are sequestered away from the protein surface following the binding with capped RNA decamer. This difference in Ksv values suggests that binding of the substrate to the cap- and RNA-binding sites differentially alter the conformation of N protein, consistent with the distinctive nature of the two binding sites. We next determined the Ksv for N after its simultaneous binding with both capped decamer and viral S segment RNA (Fig. 4C). We observed a biphasic quenching pattern of tryptophan fluorescence. A fraction of tryptophan residues was freely accessible and thus rapidly quenched by the quencher, yielding a Ksv value of 21.87. Another fraction was least accessible and thus slowly quenched by the quencher, yielding a Ksv value of 2.53. This is consistent with a different conformational state of N after its simultaneous binding with both capped RNA decamer and viral S segment RNA. Taken together, these data suggest that N protein is in different conformational states when its cap- and RNA-binding sites are occupied with their substrates either individually or both simultaneously.
FIGURE 4.

Stern-Volmer plots of N under different conditions. Stern-Volmer plots were generated by quenching the tryptophan fluorescence of N with acrylamide. A fixed concentration of N in RNA-binding buffer was excited at 295 nm, and the emission was recorded at 330 nm. The fluorescence studies were carried out at room temperature. The fluorescence signal from free binding buffer in absence of N was subtracted whenever required. The fluorescence intensity at 330 nm was determined at different input concentrations of acrylamide. Plots of F0/F as a function of acrylamide concentration (Stern-Volmer plot) for free N protein is shown by filled square in all three panels. Open squares show the Stern-Volmer plot of N bound to viral S segment RNA (A), capped decamer (B), and both the capped decamer and S segment vRNA simultaneously (C).
We next employed bis-ANS as the probe to monitor the difference in conformational changes in N that take place due to the simultaneous binding of the two substrates at their respective binding sites or when the sites are individually occupied with their substrates. Results are presented in Fig. 5. The increase in fluorescence signal of bis-ANS upon association with N is due to the binding of the probe at the hydrophobic pockets of the protein. In comparison with free N, the fluorescence signal of bis-ANS increases upon association with N, that has been preincubated with viral S segment RNA (Fig. 5). This is consistent with a vRNA-induced conformational change in N that increases the number of binding sites for the hydrophobic probe bis-ANS. However, in comparison with free N, bis-ANS fluorescence decreases upon association with N that has been preincubated with capped decamer, suggesting a decrease in the number of hydrophobic binding sites for bis-ANS due to the cap-induced conformation change in N. These observations suggest that binding of the substrate to either cap- or RNA-binding sites induce conformational changes in N protein that are different in nature, again signifying the distinctive nature of the two sites. When both cap- and RNA-binding sites are saturated with their respective substrates simultaneously, N protein is structurally altered into a different conformational state, as evident from the significant increase in the fluorescence signal of bound bis-ANS (Fig. 5).
FIGURE 5.
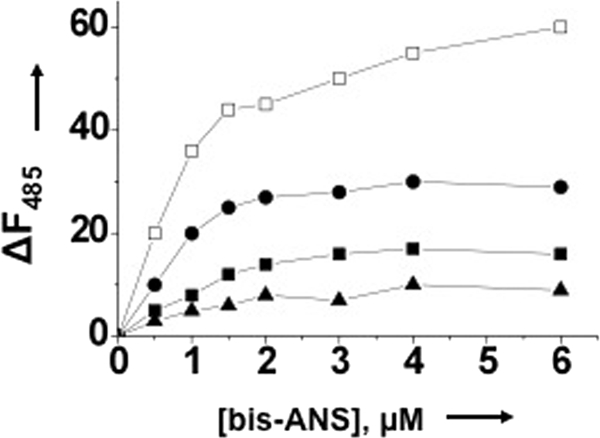
Fluorescence titration of N with hydrophobic fluorophore (bis-ANS) in RNA-binding buffer at room temperature under different conditions. The fluorophore was excited at 399 nm, and emission was recorded at 485 nm. Shown is the titration curve of bis-ANS binding with either free N (filled squares) or N that was preincubated with either S segment RNA (filled circles) or capped RNA decamer (filled triangles) or simultaneously with both capped decamer and S segment vRNA (open squares). For details, see under “Experimental Procedures.”
Cap-binding Site of N Is Involved in the Transcription and Replication Initiation of vRNA
In hantaviruses, the capped RNA primer has a 3′ G residue, that has been proposed to form a base pair with a single C residue at the 3′ terminus of the vRNA template during transcription initiation. After annealing, the primer is elongated by RdRp to initiate transcription. It is interesting to understand how single G-C base pairing between the primer and the vRNA template facilitates annealing of the primer.
Because N has distinct cap- and RNA-binding sites and also interacts simultaneously with both the cap and vRNA, we asked whether cap binding activity of N is involved in annealing the primer at the 3′ terminus of vRNA template. Using T7 RNA polymerase, we synthesized an 18-nucleotide-long capped RNA oligo that either had three nucleotides or a single G residue at the 3′ terminus complementary with the 3′ terminus of SNV S segment vRNA (Fig. 6, A and B). SNV S segment vRNA was synthesized by T7 transcription reaction (see “Experimental Procedures”). The two capped RNA oligos were used as primers in a reverse transcription reaction using SNV S segment RNA as template. Reverse transcripts generated from two different primers in the presence or absence of N, were compared. In two separate reactions, the primer and vRNA template were mixed and heated at 90 °C for 3 min, followed by cooling to allow the annealing of the primer with the template. In a control reaction, the primers were incubated with the template at room temperature without heating and cooling steps. MMLV reverse transcriptase was added to the reaction mixture to generate the reverse transcript. We used MMLV reverse transcriptase because hantaviral RdRp is difficult to express in bacteria due to its huge size. As expected, we did not see any reverse transcription product, likely due the failure of the primer to anneal with the template (Fig. 7, lanes 1–4). Further reactions were carried out at room temperature without heating and cooling steps. However, when primers were preincubated with N prior to further incubation with the vRNA template at room temperature, we observed that a significant amount of reverse transcription product was generated by MMLV reverse transcriptase from both primers (Fig. 7, lanes 5 and 6). This observation clearly indicated that N helps in the generation of reverse transcription product likely by facilitating the annealing of the primer at the 3′ terminus of vRNA template. We next synthesized the same primers without the 5′ terminal cap (Fig. 6, C and D) and preincubated them with N, followed by further incubation with the template at room temperature. As expected, MMLV reverse transcriptase did not generate the reverse transcription product as the uncapped primers fail to bind at the cap-binding site of N, implying that the cap-binding site is required for N-mediated primer annealing. To check whether the RNA-binding site of N is involved in annealing the primer with the vRNA template, we used an N mutant lacking the RNA-binding domain along with capped primers in reverse transcription experiments. We observed that MMLV reverse transcriptase did not generate the reverse transcription product in the presence of mutant N, indicating that the RNA-binding site is also required in annealing the primer with the template (Fig. 7, lanes 9 and 10).
FIGURE 7.

Reverse transcription reactions. Reverse transcription reactions using primer-template pairs (A–L) from Fig. 6 were carried out as described in detail under “Experimental Procedures.” Reverse transcription products were radiolabeled during synthesis and run on 12% SDS-PAGE. The primer-template pairs used in reverse transcription reactions are shown at the top of the gel. In lanes 1 and 2, the primer and template were heated at 90 °C followed by cooling at room temperature to allow the annealing of the primer with the template. In lanes 3–18, the primer and template were incubated at room temperature without heating and cooling steps. All lanes from 5 to 18 (except lanes 9 and 10) contained wild type N. Lanes 9 and 10 contained mutant N that lacked the RNA-binding domain.
Hantaviruses have a highly conserved sequence at the 5′ and 3′ terminus of their genome. We next wanted to know whether the 3′ conserved sequence, comprised of a nine-nucleotide-long triplet repeat (3′AUCAUCAUC) has any role in N-mediated primer annealing. Without altering the primer sequence, we randomized the sequence of the terminal nine nucleotides of the vRNA template (Fig. 6, E and F) and used them in reverse transcription reaction along with N. Although both primers may anneal with the randomized template via G-C base pairing, MMLV reverse transcriptase failed to generate the product, indicating that the sequence at the 3′ terminus is required for the proper annealing of the primer by N. To further demonstrate the requirement of terminal conserved nine nucleotides for N-mediated primer annealing, we synthesized a nonviral RNA that contained terminal conserved nine nucleotides from the 3′ terminus of S segment vRNA (Fig. 6, G and H). This nonviral RNA, similar in length as SNV S segment RNA, was used as a template in the reverse transcription reaction along with N and capped primers with a different sequence (Fig. 6, G and H). Reverse transcriptase generated the product, suggesting that primers were properly annealed at the 3′ terminus of nonviral RNA that contained a hantaviral nine-nucleotide triplet repeat at the 3′ terminus. A controlled reaction lacking N protein failed to generate the reverse transcription product (data not shown). This clearly suggests that conserved nucleotides located at the 3′ terminus of hantaviral genomic RNA play a role in N-mediated primer annealing.
To further confirm the role of these conserved nucleotides in primer annealing, we next deleted one triplet repeat from the 3′ terminus of vRNA template (Fig. 6, I and J) and used it in reverse transcription reaction with same primers along with N. Again, we did not see any reverse transcription product. This suggests that all nine nucleotide are required for N-mediated primer annealing and transcription initiation.
To determine whether terminal placement of the conserved triplet repeat is necessary for N-mediated primer annealing, SNV S segment RNA was synthesized with extra nonviral five nucleotides upstream of the conserved triplet repeat sequence (Fig. 6, K and L). This modified vRNA was used as template in reverse transcription reaction along with N and capped primers (Fig. 6, K and L). MMLV reverse transcriptase did not generate the product, suggesting that terminal placement of conserved triplet repeat is necessary for N-mediated primer annealing.
Taken together, these data suggest that N facilitates the annealing of a capped RNA primer, which has a single terminal G residue complementary to one of the C residues of the conserved triplet repeat, at the 3′ terminus of vRNA template. For successful annealing, both cap- and RNA-binding domains of N are required. In addition, the conserved 3′ terminal sequence is necessary for N-mediated primer annealing.
N-Cap Complex Preferentially Binds the Conserved Triplet Repeat Sequence at the 3′ Terminus of vRNA
Reverse transcription experiments suggest that simultaneous binding of N with the capped primer and conserved triplet repeat sequence might help to anneal the primer at the 3′ terminus of vRNA template.
Using T7 RNA polymerase, we synthesized and radiolabeled the 3′ nine-nucleotide triplet repeat sequence of vRNA and then carried out the filter binding experiments to check its interaction with N. We found that N weakly interacted with the triplet repeat sequence at low salt concentrations (Fig. 8 and Table 3a). Higher salt concentrations abrogated the interaction between N and the triplet repeat sequence, suggesting the nonspecific interaction between the two. Because N has distinct cap- and RNA-binding sites and simultaneously interacts with both the cap and vRNA, we asked whether a complex of N with capped RNA primer can bind the conserved triplet repeat sequence. Both capped primers from Fig. 6 (A and B) were used to generate the N-cap complex. N and cold capped primer were mixed at a molar ratio of 1:100 of N:primer and were incubated at room temperature for 1 h. The N-cap complex was titrated against a hot triplet repeat sequence to generate the binding profile (Fig. 8). Interestingly, the binding affinity of N-cap complex with the 3′ triplet repeat sequence significantly increased and did not change at high salt concentrations (Fig. 8, Table 3). Similar to the wild type triplet repeat sequence, N interacted nonspecifically with a randomized triplet repeat sequence or hexanucleotide triplet repeat sequence obtained by deleting one of the triplets or a 14-nucleotide-long RNA oligo composed of the 5′ triplet repeat sequence with five juxtaposed nonviral nucleotides or a nine-nucleotide-long nonviral RNA (Table 3). However, unlike the wild type triplet repeat sequence, the N-cap complex failed to interact with these control sequences (Table 3). Similar results were obtained with both the primers used to generate the N-cap complex (data not shown). Taken together, these data suggest that a complex of N with capped RNA primer specifically interacts with the conserved nine-nucleotide-triplet repeat sequence at the 3′ terminus of hantaviral RNA. These binding data are in agreement with the reverse transcription experiments (compare Fig. 7 and Table 3). By abrogating the interaction between the 3′ triplet repeat sequence and N-cap complex by either randomizing the 3′ terminal triplet repeat sequence or deleting one of the triplet repeats or by adding extra nucleotides at the 3′ terminus results in the abrogation of N-mediated primer annealing and failure to generate the reverse transcript.
FIGURE 8.

Filter binding assay showing the interaction of N with 3′ terminal nine nucleotides of SNV S segment RNA. Binding reactions were carried out as described in detail under “Experimental Procedures.” A shows a representative binding profile of N with a 3′ terminal nine-nucleotide-long triplet repeat sequence (5′-CUACUACUA-3′) of SNV S segment vRNA. The binding reaction was carried out in RNA-binding buffer containing 80 mm NaCl. In B, N protein was preincubated with saturating concentrations of cold capped RNA primer (shown in Fig. 6A) to generate the N-cap complex. The increasing concentrations of the complex were incubated with radiolabeled triplet repeat sequence and filtered through nitrocellulose filter. Radioactive signal retained on the filter at different input concentrations was used to generate the binding profile for the calculation of dissociation constants, shown in Table 3.
DISCUSSION
It is well understood that caps at the 5′ termini of mRNA are required for their efficient translation and also protect the mRNA from degradation by exonucleases. Cap-binding proteins in eukaryotes have been well characterized, for example, cap-binding protein CBP20 facilitates the mRNA transport from the nucleus to the cytoplasm of a cell. Another eukaryotic cap-binding protein eukaryotic translation initiation factor 4E (eIF4E) is required to program the translation initiation of mRNA. Viruses have evolved unique strategies to cap their mRNAs, for example, retroviruses use host cell capping machinery to cap their mRNAs, and many other viruses, such as poxyviruses, coronaviruses, flaviviruses, and reoviruses, have evolved their own capping mechanisms. Negative-stranded RNA viruses replicating either in the cell nucleus (orthomyoxviruses) or in the cell cytoplasm (bunyaviruses) have not evolved capping enzymes as such. They snatch caps from the host cell mRNAs by a novel cap-snatching mechanism. We have recently found that the hantavirus N protein binds mRNA caps and requires five nucleotides adjacent to the 5′ cap for high affinity binding. We have suggested that the cap-binding activity of N is involved in hantavirus cap snatching (16).
Here, we carried out multiple experiments to demonstrate the role of triphosphate moiety of m7GTP in binding with N. Similar to the well characterized vaccinia virus protein 39 (VP39) (55–57) we found that N binds weakly with m7GTP with a dissociation constant in μm range. In comparison, eIF4E binds m7GTP with a dissociation constant of 260–280 nm (58), and CBP has the strongest affinity for m7GTP with a dissociation constant of 10 nm (59). Unlike eIF4E, N and VP39 bind m7GDP or m7GMP with a similar weak affinity, suggesting that the triphosphate moiety of the terminal cap does not play a major role during binding. However, the role of triphosphate moiety in the binding of cap with eIF4E is well understood (55–57). Previous studies have also reported that nucleotides adjacent to the 5′ cap affect the cap binding with eIF4E, CBP, and VP39 in a different manner. 20 nucleotides downstream the 5′ cap enhance its binding affinity with VP39 by 100-fold (60), whereas the binding affinity of eIF4E and CBP was increased by only ∼6-fold (61). Similar to VP39, we noticed that four nucleotides adjacent to the 5′ cap significantly enhanced its ability to interact with N (16). These observations suggest that the cap-binding pocket of N and VP39 might have similar dimensions, and these two proteins might have evolved a common cap-binding mechanism.
Structural studies of evolutionary unrelated cap binding proteins including eIF4E, CBP20, VP39, and the PB2 subunit of influenza virus RdRp, have revealed that specific recognition of the m7G cap occurs by sandwiching the guanine base between two aromatic amino acid residues (44, 61–63). Because these evolutionary unrelated proteins have conserved aromatic cap binding pockets, it has been suggested that a convergent evolution of the cap-binding pocket has occurred. These cap-binding proteins do not share a sequence homology; however, the aromatic cap binding pocket is structured with aromatic amino acids that accommodate the m7G aromatic ring stacked between two aromatic residues. The aromatic residues of the cap-binding pocket are located at different position in the linear amino acid sequence. For example, Trp102/Trp56 in eIF4E, Tyr20/Tyr43 in CBP20, Tyr22/Phe180 in VP39 and Phe404/Phe323 in the PB2 subunit of influenza RdRp constitute the pairs of aromatic amino acid residues that form a sandwich with the guanine base of the cap stacked between them. In addition, the delocalized positive charge of m7G purine ring enables the formation of salt bridge with an acidic residue located in the acidic cavity of the cap binding pocket. The acidic cavity contains at least one critical acidic residue. For instance, Glu103 of eIF4E, Asp116 of CBP20, and Glu233 of VP39 are involved in the salt bridge interaction with the m7G cap (44, 61–63). We aligned the sequence of N with eIF4E, CBP20, VP30, and the PB2 subunit and, as expected, did not observe any sequence homology. However, N has two aromatic amino acid residues, Trp119 and Trp105, that are separated by 46 amino acids similar to eIF4E. In addition, similar to eIF4E, N contains an adjacent acidic residue (Glu166). It is likely that Trp119 and Trp105 are involved in a specific interaction with the m7G cap by sandwiching the guanine base, and Glu166 forms the salt bridge to stabilize the interaction.
RNA filter binding studies have shown that N has distinctive cap- and RNA-binding domains. The RNA-binding domain of another hantavirus (Hantaan virus) has been mapped to the central conserved region of the N peptide (amino acids 175–217) (54). N binds the viral genomic RNA with high affinity (Kd ∼ 25 nm). In comparison with viral genomic RNA, N weakly interacts with a capped RNA decamer (Kd ∼ 120 nm). As anticipated, an N mutant lacking the RNA binding-domain failed to interact with vRNA with specificity and high affinity. However, it was able to interact with capped RNA decamer with similar affinity as wild type N. In addition, the complex generated between N and vRNA, when the RNA-binding site of N was saturated with vRNA, was still able to interact with the capped decamer with similar affinity as free N. The simultaneous binding of N with both the capped decamer and vRNA suggests that unlike other cap-binding proteins, N has distinct cap- and RNA-binding sites.
N undergoes a conformational change after an interaction with either vRNA or capped decamer or due to simultaneous binding with both capped decamer and vRNA. However, the conformational changes induced in N due to the substrate binding at either cap- or RNA-binding site are different in nature. In addition, N is in a different conformational state when both the cap- and RNA-binding sites are occupied with their respective substrates simultaneously, as evident from a significantly high Ksv value and biphasic nature of the Stern-Volmer plot. The differential nature of conformational changes in N peptide is consistent with the idea that N harbors distinct binding sites for vRNA and the terminal mRNA cap. The distinctive nature of two binding sites is further supported by the bis-ANS binding data. The conformational change induced in N due to the binding of vRNA at the RNA-binding site increases the number of accessible hydrophobic binding sites for the hydrophobic bis-ANS fluorescent probe. In comparison, the binding of capped decamer at the cap-binding site induces a different conformation change in N peptide that reduces the number of hydrophobic binding sites for bis-ANS. Again, bis-ANS binding shows that N is in a different conformational state when both cap- and RNA-binding sites are simultaneously occupied with their respective substrates.
It is well understood that the RNA-binding site of N is required for packaging the vRNA during virus assembly. However, the role of cap-binding site is not yet clear. We have previously reported that in the cell cytoplasm, N localizes in cellular P-bodies where it prevents host mRNA degradation from 5′ terminus due to the binding with mRNA 5′ caps. The rescued 5′ capped oligos are stored in P-bodies by N, which are used later on as primers by viral RdRp during transcription initiation (14). Unlike the influenza virus, the negative-stranded RNA viruses replicating in the cell cytoplasm have to compete with cellular decapping machinery to protect the mRNA caps from degradation to generate capped RNA primers during cap snatching. We have shown that the cap-binding domain of N serves that purpose in hantaviruses. We suggest that other RNA viruses, which replicate in cell cytoplasm and use the cap-snatching mechanism, have evolved cap-binding proteins to encounter cellular decapping machinery for the protection of host mRNA caps from degradation.
A prime and realign mechanism has been proposed for the transcription and replication initiation of vRNA by RdRp. This mechanism proposes the base pairing between the terminal G residue of the capped primer and one of the three C residues of a nine-nucleotide triplet repeat sequence (AUCAUCAUC) at the 3′ terminus of the vRNA template. After single G-C base pairing, the primer is slightly elongated by the RdRp. The elongated primer dissociates from the template and is realigned at the absolute 3′ terminus, followed by elongation by the RdRp to generate the cRNA that has complementary nine-nucleotide triplet repeat sequence at the 5′ terminus. It is interesting to understand how the initial G-C base pairing stabilizes the RNA primer at the 3′ terminus of vRNA template. We have shown that the inbuilt cap- and RNA-binding sites enable N to interact with both the capped primer and vRNA template simultaneously. N undergoes a conformational change after binding to the capped primer. The conformationally altered N, loaded with capped primer at the cap-binding site, specifically recognizes highly conserved triplet repeat sequence at the 3′ terminus of the vRNA template. In the absence of capped primer, N fails to recognize the conserved 3′ sequence with specificity and high affinity. As depicted in the proposed model (Fig. 9), we suggest that simultaneous binding of N with the capped primer and 3′ triplet repeat sequence stabilizes the primer and facilitates the annealing.
FIGURE 9.

Model showing the role of N in primer annealing during transcription initiation. Hantaviral RNA, enveloped with N in viral capsids, is used as a template by viral RdRp during transcription and replication. Trimeric N binds the panhandle that is formed by the base pairing of complementary nucleotides at the 5′ and 3′ termini of vRNA. After binding, N unwinds the panhandle and remains attached with the 5′ terminus and leaves 3′ terminus accessible for RdRp. N with a capped RNA primer loaded at its cap binding site specifically recognizes the 3′ terminus of vRNA and assists the annealing of the bound primer.
Hantaviral genomic RNAs have a partially complementary 3′ and 5′ nucleotide sequence that undergoes base pairing to form a panhandle structure. We have previously shown that trimeric N recognizes the panhandle with specificity. N is also an RNA chaperone that unwinds double-stranded RNA helices. Our previous studies demonstrated that after specific recognition, N unwinds the panhandle and remains attached with the 5′ terminus leaving the 3′ terminus single stranded. This renders the 3′ terminus accessible for both annealing of the primer and transcription initiation by the RdRp (15). In the current model (Fig. 9), we suggest that after unwinding the panhandle, a different N molecule with a bound capped primer specifically recognizes the single-stranded 3′ terminus of vRNA and facilitates the annealing of the primer. The annealed primer is elongated by RdRp to initiate the transcription. Our studies suggest that distinct cap- and RNA-binding sites of N have unique roles in the virus replication cycle.
This work was supported, in whole or in part, by National Institutes of Health Research Grant RAI083672Z. This work was also supported by COBRE Grant P20 RR016443 from the Department of Microbiology, Molecular Genetics and Immunology, Kansas University Medical Center.
- RdRp
- RNA-dependent RNA polymerase
- vRNA
- viral RNA
- ANS
- 1-anilino-8-naphthalenesulfonate
- MMLV
- Moloney murine leukemia virus
- eIF4E
- eukaryotic translation initiation factor 4E
- VP39
- vaccinia virus protein 39
- bis-ANS
- bis-1-anilino-8-naphthalenesulfonate
- SNV
- Sin Nombre virus.
REFERENCES
- 1.Schmaljohn C. M. (1996) Molecular Biology of Hantaviruses, pp. 1–20, Plenum Press, New York [Google Scholar]
- 2.Schmaljohn C. S., Hooper J. W. (2001) in Fields Virology (Howley K. A. ed.) pp. 1581–1602, Lippencott, Williams, and Wilkins, Philadelphia: [Google Scholar]
- 3.Schmaljohn C. S., Jonsson C. B. (2001) in Hantaviruses (Nichol S. A. ed) pp. 15–32, Springer-Verlag, Berlin [Google Scholar]
- 4.Obijeski J. F., Bishop D. H., Murphy F. A., Palmer E. L. (1976) J. Virol. 19, 985–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pettersson R. F., von Bonsdorff C. H. (1975) J. Virol. 15, 386–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raju R., Kolakofsky D. (1989) J. Virol. 63, 122–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alfadhli A., Love Z., Arvidson B., Seeds J., Willey J., Barklis E. (2001) J. Virol. 75, 2019–2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alfadhli A., Steel E., Finlay L., Bächinger H. P., Barklis E. (2002) J. Biol. Chem. 277, 27103–27108 [DOI] [PubMed] [Google Scholar]
- 9.Blakqori G., Kochs G., Haller O., Weber F. (2003) J. Gen. Virol. 84, 1207–1214 [DOI] [PubMed] [Google Scholar]
- 10.Bridgen A., Elliott R. M. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 15400–15404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ikegami T., Peters C. J., Makino S. (2005) J. Virol. 79, 5606–5615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kohl A., Hart T. J., Noonan C., Royall E., Roberts L. O., Elliott R. M. (2004) J. Virol. 78, 5679–5685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mir M. A., Brown B., Hjelle B., Duran W. A., Panganiban A. T. (2006) J. Virol. 80, 11283–11292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mir M. A., Duran W. A., Hjelle B. L., Ye C., Panganiban A. T. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 19294–19299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mir M. A., Panganiban A. T. (2006) Rna 12, 272–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mir M. A., Panganiban A. T. (2008) EMBO J. 27, 3129–3139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pinschewer D. D., Perez M., de la Torre J. C. (2003) J. Virol. 77, 3882–3887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taylor S. L., Frias-Staheli N., García-Sastre A., Schmaljohn C. S. (2009) J. Virol. 83, 1271–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taylor S. L., Krempel R. L., Schmaljohn C. S. (2009) Ann. N.Y. Acad. Sci. 1171, E86–93 [DOI] [PubMed] [Google Scholar]
- 20.Gött P., Stohwasser R., Schnitzler P., Darai G., Bautz E. K. (1993) Virology 194, 332–337 [DOI] [PubMed] [Google Scholar]
- 21.Jonsson C. B., Gallegos J., Ferro P., Severson W., Xu X., Schmaljohn C. S., Fero P. (2001) Protein Expr. Purif. 23, 134–141 [DOI] [PubMed] [Google Scholar]
- 22.Osborne J. C., Elliott R. M. (2000) J. Virol. 74, 9946–9952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Severson W., Partin L., Schmaljohn C. S., Jonsson C. B. (1999) J. Biol. Chem. 274, 33732–33739 [DOI] [PubMed] [Google Scholar]
- 24.Severson W., Xu X., Kuhn M., Senutovitch N., Thokala M., Ferron F., Longhi S., Canard B., Jonsson C. B. (2005) J. Virol. 79, 10032–10039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Severson W. E., Xu X., Jonsson C. B. (2001) J. Virol. 75, 2646–2652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mir M. A., Panganiban A. T. (2004) J. Virol. 78, 8281–8288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raju R., Kolakofsky D. (1986) Virus Res. 5, 1–9 [DOI] [PubMed] [Google Scholar]
- 28.Raju R., Kolakofsky D. (1987) J. Virol. 61, 667–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Braam J., Ulmanen I., Krug R. M. (1983) Cell 34, 609–618 [DOI] [PubMed] [Google Scholar]
- 30.Caton A. J., Robertson J. S. (1980) Nucleic Acids Res. 8, 2591–2603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dhar R., Chanock R. M., Lai C. J. (1980) Cell 21, 495–500 [DOI] [PubMed] [Google Scholar]
- 32.Plotch S. J., Bouloy M., Ulmanen I., Krug R. M. (1981) Cell 23, 847–858 [DOI] [PubMed] [Google Scholar]
- 33.Ulmanen I., Broni B. A., Krug R. M. (1981) Proc. Natl. Acad. Sci. U.S.A. 78, 7355–7359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bishop D. H., Gay M. E., Matsuoko Y. (1983) Nucleic Acids Res. 11, 6409–6418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bouloy M., Pardigon N., Vialat P., Gerbaud S., Girard M. (1990) Virology 175, 50–58 [DOI] [PubMed] [Google Scholar]
- 36.Eshita Y., Ericson B., Romanowski V., Bishop D. H. (1985) J. Virol. 55, 681–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jin H., Elliott R. M. (1993) J. Gen. Virol. 74, 2293–2297 [DOI] [PubMed] [Google Scholar]
- 38.Patterson J. L., Holloway B., Kolakofsky D. (1984) J. Virol. 52, 215–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Patterson J. L., Kolakofsky D. (1984) J. Virol. 49, 680–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Simons J. F., Pettersson R. F. (1991) J. Virol. 65, 4741–4748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Garcin D., Lezzi M., Dobbs M., Elliott R. M., Schmaljohn C., Kang C. Y., Kolakofsky D. (1995) J. Virol. 69, 5754–5762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Raju R., Raju L., Hacker D., Garcin D., Compans R., Kolakofsky D. (1990) Virology 174, 53–59 [DOI] [PubMed] [Google Scholar]
- 43.Sugiyama K., Obayashi E., Kawaguchi A., Suzuki Y., Tame J. R., Nagata K., Park S. Y. (2009) EMBO J. 28, 1803–1811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guilligay D., Tarendeau F., Resa-Infante P., Coloma R., Crepin T., Sehr P., Lewis J., Ruigrok R. W., Ortin J., Hart D. J., Cusack S. (2008) Nat. Struct. Mol. Biol. 15, 500–506 [DOI] [PubMed] [Google Scholar]
- 45.Jin H., Elliott R. M. (1993) J. Virol. 67, 1396–1404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Duijsings D., Kormelink R., Goldbach R. (2001) EMBO J. 20, 2545–2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Botten J., Mirowsky K., Kusewitt D., Ye C., Gottlieb K., Prescott J., Hjelle B. (2003) J. Virol. 77, 1540–1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Botten J., Mirowsky K., Kusewitt D., Bharadwaj M., Yee J., Ricci R., Feddersen R. M., Hjelle B. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 10578–10583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Padmanabhan U., Dasgupta S., Biswas B. B., Dasgupta D. (2001) J. Biol. Chem. 276, 43635–43644 [DOI] [PubMed] [Google Scholar]
- 50.Mir M. A., Dasgupta D. (2001) Biochemistry 40, 11578–11585 [DOI] [PubMed] [Google Scholar]
- 51.Wang J. L., Edelman G. M. (1971) J. Biol. Chem. 246, 1185–1191 [PubMed] [Google Scholar]
- 52.Lakowicz J. R. (1999) in Principles of Fluorescence Spectroscopy, pp. 237–265, Plenum Press, New York [Google Scholar]
- 53.Mir M. A., Panganiban A. T. (2005) J. Virol. 79, 1824–1835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu X., Severson W., Villegas N., Schmaljohn C. S., Jonsson C. B. (2002) J. Virol. 76, 3301–3308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hu G., Gershon P. D., Hodel A. E., Quiocho F. A. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 7149–7154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hooker L., Sully R., Handa B., Ono N., Koyano H., Klumpp K. (2003) Biochemistry 42, 6234–6240 [DOI] [PubMed] [Google Scholar]
- 57.Cai A., Jankowska-Anyszka M., Centers A., Chlebicka L., Stepinski J., Stolarski R., Darzynkiewicz E., Rhoads R. E. (1999) Biochemistry 38, 8538–8547 [DOI] [PubMed] [Google Scholar]
- 58.Wilson K. F., Fortes P., Singh U. S., Ohno M., Mattaj I. W., Cerione R. A. (1999) J. Biol. Chem. 274, 4166–4173 [DOI] [PubMed] [Google Scholar]
- 59.Niedzwiecka A., Marcotrigiano J., Stepinski J., Jankowska-Anyszka M., Wyslouch-Cieszynska A., Dadlez M., Gingras A. C., Mak P., Darzynkiewicz E., Sonenberg N., Burley S. K., Stolarski R. (2002) J. Mol. Biol. 319, 615–635 [DOI] [PubMed] [Google Scholar]
- 60.Lockless S. W., Cheng H. T., Hodel A. E., Quiocho F. A., Gershon P. D. (1998) Biochemistry 37, 8564–8574 [DOI] [PubMed] [Google Scholar]
- 61.Mazza C., Ohno M., Segref A., Mattaj I. W., Cusack S. (2001) Mol. Cell 8, 383–396 [DOI] [PubMed] [Google Scholar]
- 62.Marcotrigiano J., Gingras A. C., Sonenberg N., Burley S. K. (1997) Cell 89, 951–961 [DOI] [PubMed] [Google Scholar]
- 63.Hodel A. E., Gershon P. D., Shi X., Wang S. M., Quiocho F. A. (1997) Nat. Struct. Biol. 4, 350–354 [DOI] [PubMed] [Google Scholar]