

Abstract
Integrase (IN) is the newest validated target against AIDS and retroviral infections. The remarkable activity of raltegravir (Isentress®) led to its rapid approval by the FDA in 2007 as the first IN inhibitor. Several other IN strand transfer inhibitors (STIs) are in development with the primary goal to overcome resistance due to the rapid occurrence of IN mutations in raltegravir-treated patients. Thus, many scientists and drug companies are actively pursuing clinically useful IN inhibitors. The objective of this review is to provide an update on the IN inhibitors reported in the last two years, including second generation strand transfer inhibitors (STI), recently developed hydroxylated aromatics, natural products, peptide, antibody and oligonucleotide inhibitors. Additionally, the targeting of IN cofactors such as LEDGF and Vpr will be discussed as novel strategies for the treatment of AIDS.
Keywords: HIV-1 IN, diketo acids, HIV inhibitors, AIDS, IN inhibitors, peptides, oligonucleotides
Introduction
Approximately 30 years of research have produced 25 FDA approved drugs for the treatment of AIDS, 13 of which target viral replication (reverse transcriptase; RT), 9 target maturation (protease), one targets viral fusion, one viral entry (CCR5 antagonist), and one integration (Table 1). The IN inhibitor raltegravir and the CCR5 antagonist maraviroc were both approved by the FDA in 2007. The success of highly active antiretroviral therapy (HAART) combinations with increasingly potent drugs has improved patient long-term survival and tolerability to drug regimens. The current HAART treatments for new patients consist of three drug combinations targeting just two viral enzymes: RT and protease. They consist of two nucleoside reverse transcriptase inhibitors (NRTIs) combined with either a non-nucleoside reverse transcriptase inhibitor (NNRTI) or a protease inhibitor. However, mutations of the HIV-1 genome confer resistance to those drugs in a large number of patients. Chronic dosing is also commonly associated with side effects that hamper compliance to therapy. Thus, the development of new inhibitors remains essential to reduce side effects and selection of drug-resistant viruses. Simultaneous inhibition of several viral targets remains the best way to circumvent drug resistance because multiple mutations are required for resistance, some of which are bound to be detrimental to viral replication and growth.
Table 1.
Approved anti-AIDS therapies
| FDA Approval | Brand Name | Generic Name | Manufacturer |
|---|---|---|---|
| Nucleosides Reverse Transcriptase Inhibitors (NRTIs) | |||
| 1987 | Retrovir | zidovudine, azidothymidine, AZT, ZDV | GlaxoSmithKline |
| 1991 | Videx | didanosine, dideoxyinosine, ddI | Bristol-Myers Squibb |
| 1992 | Hivid | zalcitabine, dideoxycytidine, ddC | Roche Pharmaceuticals |
| 1994 | Zerit | stavudine, d4T | Bristol-Myers Squibb |
| 1995 | Epivir | lamivudine, 3TC | GlaxoSmithKline |
| 1997 | Combivir | lamivudine + zidovudine | GlaxoSmithKline |
| 1998 | Ziagen | abacavir sulfate, ABC | GlaxoSmithKline |
| 2000 | Trizivir | abacavir+ lamivudine+ zidovudine | GlaxoSmithKline |
| 2000 | Videx EC | enteric coated didanosine, ddI EC | Bristol-Myers Squibb |
| 2001 | Viread | tenofovir disoproxil fumarate, TDF | Gilead Sciences |
| 2003 | Emtriva | emtricitabine, FTC | Gilead Sciences |
| 2004 | Epzicom | abacavir+ lamivudine | GlaxoSmithKline |
| 2004 | Truvada | emtricitabine + tenofovir disoproxil fumarate | Gilead Sciences |
| Non-Nucleosides Reverse Transcriptase Inhibitors (NNRTIs) | |||
| 1996 | Viramune | nevirapine, NVP | Boehringer Ingelheim |
| 1997 | Rescriptor | delavirdine, DLV | Pfizer |
| 1998 | Sustiva | efavirenz, EFV | Bristol-Myers Squibb |
| 2008 | Intelence | etravirine | Tibotec Therapeutics |
| Protease Inhibitors (PIs) | |||
| 1995 | Invirase | saquinavir mesylate, SQV | Roche Pharmaceuticals |
| 1996 | Norvir | ritonavir, RTV | Abbott Laboratories |
| 1996 | Crixivan | indinavir, IDV | Merck |
| 1997 | Viracept | nelfinavir mesylate, NFV | Pfizer |
| 1997 | Fortovase | saquinavir (no longer marketed) | Roche Pharmaceuticals |
| 1999 | Agenerase | amprenavir, APV | GlaxoSmithKline |
| 2000 | Kaletra | lopinavir+ ritonavir, LPV/RTV | Abbott Laboratories |
| 2003 | Reyataz | atazanavir sulfate, ATV | Bristol-Myers Squibb |
| 2003 | Lexiva | fosamprenavir calcium, FOS-APV | GlaxoSmithKline |
| 2005 | Aptivus | tripranavir, TPV | Boehringer Ingelheim |
| 2006 | Prezista | darunavir | Tibotec Therapeutics |
| Fusion Inhibitors | |||
| 2003 | Fuzeon | Enfuvirtide, T-20 | Roche Pharmaceuticals & Trimeris |
| Entry Inhibitors | |||
| 2007 | Selzentry | maraviroc | Pfizer |
| IN Inhibitors | |||
| 2007 | Isentress | raltegravir | Merck |
From http://www.fda.gov
With the approval of raltegravir, the antiretroviral drugs can now target all three viral enzymes: RT, protease and IN (IN). The absence of known human homologue to IN probably accounts for the low toxicity and high selectivity of raltegravir and of the forthcoming IN inhibitors. That statement, however, should be made with caution since diketo acids can inhibit RAG1/2 recombinase, albeit at drug concentrations 20-fold higher than required to inhibit IN 1. RAG recombinases have mechanistic similarities to HIV-1 IN, further suggesting novel IN inhibitors should be tested against RAG1/2 to assess their selectivity and avoid potential toxicity. Another enzyme with structural similarities to IN is RNase H, which is part of RT activity 2, 3. This stresses the value of screening IN inhibitors also against RT-RNase H 4. The present review describes recent advances in the search for HIV-1 IN inhibitors with a focus on the mechanism of inhibition by diketo acids and small molecules, natural products, peptides, antibody and oligonucleotide inhibitors published in the last two years. It is an update of our prior landmark reviews 5, 6 and complements other recent reviews 7–22.
IN Structural Overview
IN is required for three events leading to viral integration [Fig. (1A)]: 1) site-specific endonucleolytic cleavage of the 3’-ends of the viral DNA (3’-processing, 3’-P), 2) assembly of the preintegration complex (PIC) on the ends of the viral DNA, and 3) insertion of the viral DNA into a host chromosomal DNA (strand transfer, ST). The completion of integration requires an additional step with the processing and attachment of the 5’-ends of the viral DNA to the host genome (5’-processing and gap filling) [Fig. (1A)], which is probably catalyzed by cellular enzymes.
Figure 1.

The mechanism of HIV integration. A: The 3’-P and ST reactions result in integration of the viral DNA ends into a host chromosome. B: In vitro assay to analyze IN catalysis and drug inhibition. Dinucleotide cleavage shortens the DNA substrate by two nucleotides (21mer to 19mer) during the 3’-P reaction. The subsequent ST reaction generates bands that migrate slower than the 19mer on a denaturing sequencing gel.
IN is encoded at the 3’-end of the HIV POL gene, which also encodes RT and protease [see scheme in Box 1 p. 240 in ref. 6]. The polyprotein precursor is cleaved by protease during maturation, generating the IN polypeptide, which is packaged within the newly formed HIV virions. HIV-1 IN is a 32,000 Daltons polypeptide of 288 amino acids comprising three functional domains 3, 23. The amino-terminal domain (amino acids 1–50) contains a conserved and essential zinc-binding motif HHCC (histidines 12 and 16, cysteines 40 and 43) that coordinates one zinc atom 24, though the structure of this region does not resemble a zinc finger 25. One known function of the amino-terminal domain region is protein multimerization. The catalytic core domain (amino acids 50–212) contains the catalytic DDE motif, which is conserved among all retroviral INs and consists of the active site residues D64, D116, and E152 in HIV-1 IN (shown in red in Fig. 2). Mutation of any one of these three residues is sufficient to inactivate IN. Crystal structures show that HIV-1 IN binds one magnesium ion between D64 and D116 (pink sphere in Fig. 2A), and that ASV binds an additional Zn2+ or Cd2+ ion between D64 and E157 (the ortholog of E152) 26. Thus, it is likely that the HIV-1 IN active site binds two metal ions (Mg+2 or Mn+2) when complexed with the ends of the viral DNA during the cleavage and joining reactions. Another structural feature of the catalytic core domain is the 10 amino acid flexible loop encompassed between glycine residues G140 and G149. Those two glycines potentially act as hinges for the overall movement of the loop that may serve as a clamp for the binding of the viral DNA ends to the catalytic site of IN. Consistent with this possibility, glutamine 148 (Q148), one of the flexible loop residues has been shown to bind selectively to the penultimate cytosine at the 5’-end of the viral DNA 27. Q148 is also a key residue for IN catalytic activity 28 and resistance to raltegravir and elvitegravir 28. The carboxyl-terminal domain (amino acids 213–288) of HIV-1 IN is important for nonspecific DNA binding of sub-terminal viral DNA and of the host (target) DNA 29–32. Its structure contains an SH2-like motif 3, which can be considered for rational drug design 6. While each of the IN domains forms dimers, IN functions as a tetramer 33–35.
Figure 2.

Panels A, C and D are derived from the crystal structure of the IN core domain complexed with 5CITEP 41. The catalytic amino acids are shown in red, the magnesium ion is colored in magenta and the four coordinating water molecules are yellow. A: 5CITEP interactions within the HIV-1 IN active site. Amino acids with direct interactions with 5CITEP 41 are highlighted in green. The view angle is the same as in panel C. B: Crystal structure of the IN core domain dimer 38. Alpha helices targeted by peptide inhibitors are colored and labeled. Helices 1 (cyan) and 5 (magenta) form the dimerization interface between two IN monomers. Helix 4 (green) is proximal to the active site and includes the catalytic amino acid E152. The three catalytic residues, D64, D116, and E512 are shown in red. C and D: Illustration of the amino acids that are mutated in diketo acid resistant viruses. The side chains of the amino acids conferring resistance to DKA are highlighted in gold. Panel C is a view in the same orientation as in panel A. In panel D, the IN is rotated horizontally 90°.
HIV-1 IN recognizes the specific sequence 5’-GCAGT-3’ at the ends of each viral long terminal repeat (LTR) and binds tightly to those LTR ends [Fig. (1A)]. The association of IN with the host chromosomal (target) DNA is of weaker affinity and specificity 36, which probably explains the integration of viral DNA into many possible genomic regions with relatively little sequence selectivity 37. The integration reaction requires the association of two viral DNA (donor) ends and a host chromosome (acceptor), and an unknown number of IN subunits [at least a tetramer 33, 34]. Unraveling the complexity of the structure of the IN/DNA complex has remained a challenging task.
Several crystal structures comprising different domains of HIV-1 IN have been solved: core domain 38, 39, core and C-terminal domains 40, and core and N-terminal domains 35, but a crystal of the full-length protein with or without DNA remains elusive due to solubility difficulties. Two co-crystal structures with IN inhibitors have been reported: one of HIV-1 IN complexed with 5CITEP 41, and the other of ASV IN complexed with the inhibitor Y-3 42. Additionally, one study reported the proposed binding site of a mononucleotide inhibitor in the IN active site 43 and a more recent study the proposed binding of pyridoxal 5’-phosphate in the C-terminal domain around lysine 244 44. Another drug binding site has been reported for a photoactivatable coumarin derivative with cysteine 130 and tryptophane 132 45. A crystal structure of the full catalytic complex with inhibitor would be of great benefit to IN inhibitor design. For instance, the development of successful HIV-1 protease and RT inhibitors was achieved through structure-based drug design. Adequate structural information defining the interactions of IN with DNA and inhibitors would certainly speed up similar structure-based drug design efforts 46. Solving IN-DNA structures is probably key to the elucidation of the binding site of raltegravir and other strand transfer inhibitors as these drugs are likely to act as interfacial inhibitors 47–49 by binding at the interface of the viral DNA LTR end and the DDE catalytic site of IN and thereby preventing the binding of the acceptor (chromosomal) DNA 6, 19.
Mechanism of Integration
The IN reaction occurs biochemically in two steps following reverse transcription and the binding of IN to the LTR ends with several other proteins within the PIC 22. Assembly of the PIC onto the 5’-GCAGT-3’ LTR sequence of the viral DNA ends forms a complex competent for integration, although in vitro, only IN and DNA are required, Fig. (1A). The first step is an endonucleolytic cleavage, or 3’-P, which results in removal of two nucleotides from the 3’-ends of each viral DNA LTR immediately 3’ from the conserved CA dinucleotide (5’-GCAGT-3’). The phosphodiester bond between the deoxyadenosine and deoxyguanosine is hydrolyzed by water as the nucleophile in the presence of Mg2+ (the presumably physiological metal) or Mn2+ 29. IN can also use glycerol, alcohols, or the 3’ viral terminus as the attacking nucleophile in the presence of Mn2+ 50, 51. The terminal 3’-end of the viral DNA is released beyond the conserved CA dinucleotide [see Fig. (1B)], and generally a 5’-GT-3’ dinucleotide is released (unless the 3’-end has been made longer during reverse transcription 52), resulting in recessed 3’-hydroxyl ends at each terminus of the viral DNA.
Following nuclear translocation of the PIC, integration catalyzes the second transesterification reaction where the 3’-hydroxyl ends of the viral DNA act as nucleophiles to attack the DNA phosphodiester backbone of a host chromosome. HIV-1 IN positions both viral ends to attack both strands of a host chromosome (acceptor DNA) with a five base pair gap on each strand, probably across the major groove of the acceptor DNA [see Fig. 2d in 6]. The result is a staggered insertion that is sealed by host DNA repair enzymes. Raltegravir, elvitegravir (Table 1) and the diketo acid inhibitors reported recently (Table 2) are selective against the ST reaction compared to 3’-P 53, 54. Thus, they are often referred to as STI (strand transfer inhibitors).
Table 2.
The first approved and the most advanced IN inhibitors.
| Name | Structure | IC50 | EC50 | EC95 | CC50 | Ref. |
|---|---|---|---|---|---|---|
| Raltegravir |  |
87 10 |
8.8 | 33 31 |
ND | 20, 28, 58, 61 |
| Elvitegravir |  |
7.2, 28 |
1.1 0.7 0.9 1.8 |
ND | 4000 | 28, 60–62 |
ND means not determined or not disclosed. Values are expressed in nM.
Recombinant IN is catalytically active in biochemical assays, which eases the screening of IN inhibitors 55, 56. In vitro, IN, divalent metal (Mg2+ or Mn2+), and short oligonucleotides derived from the DNA sequence of the U5 or U3 LTR are sufficient for integration. The mechanisms of the in vitro reactions are shown in Fig. (1B). Most of the inhibitors reported were discovered using oligonucleotide-based assays, with either gel or microtiter plate/fluorescence analysis. In this review, IC50 values described as “in vitro” were generated using such biochemical assays, unless a cell culture system is specifically described. In vitro reactions differ from in vivo integration because ST of only one DNA end occurs efficiently in the in vitro reactions using short oligonucleotides. Recent reports describe conditions allowing higher efficiency of full-site (concerted) integration 34, 57. Concerted joining of both viral DNA ends probably occurs more efficiently in vivo due to the presence of long DNA substrates, other PIC components and cellular cofactors.
IN inhibitors in clinical use
Isentres™ (raltegravir, RAL, also known as MK-0518) is the first and still the only IN inhibitor approved by the FDA (in October 2007) for the treatment of patients who are failing HAART (Table 1). Raltegravir is the result of a long-term commitment by Merck and Co. in the development of IN inhibitors 20. Raltegravir is well-tolerated, highly potent, and with a good pharmacokinetics (Table 2) 20, 21, 58. Raltegravir is administered orally at the dose of 400 mg twice daily and leads to viral RNA levels below detection before 48 weeks of treatment 59. It has been difficult to increase raltegravir blood levels by increasing dosing in patients who develop resistance to raltegravir because the raltegravir blood levels remain somewhat limited (see below).
Elvitegravir (EVG = GS-9137 = JTK-303) is the next most advanced IN inhibitor in clinical development (Table 2). It has not yet been approved by the FDA. This quinolone carboxylic acid derivative was originally developed by Japan Tobacco Inc. under the name JTK-303 60. JTK-303 was subsequently licensed to Gilead Sciences under the name GS-9137 for development in all countries except Japan. Elvitegravir, like raltegravir, is a potent antiviral with single digit nanomolar antiviral potency (Table 2) 60–62. Elvitegravir is slightly more potent than raltegravir in vitro and ex vivo 28, 61 but exhibits a potential higher cytotoxicity in non-infected cells 60 (Marchand et al. unpublished observations). One limitation of elvitegravir is that unlike raltegravir, it is metabolized by cytochrome P450 (CYP3A4/5) and secondarily by glucuronidation (UGT1A1/3) to produce metabolites that are less potent than the parent drug 20, 63. Co-administration of ritonavir with elvitegravir can overcome this metabolic inactivation and can increase the plasma levels of raltegravir[NN1] by 10- to 16-fold 63.
Early diketo acid IN inhibitors and mechanisms of action studies
Both raltegravir and elvitegravir are derived from the diketo acid (DKA) (also referred as ketoenol acid 19) family of IN inhibitors. Diketo acids were the first IN inhibitors reported with selectivity for the strand transfer step of the integration reaction (ST-inhibitor, See 6), high specificity for IN, and antiviral activity that could be related to IN inhibition 54. Historically, the two first reported DKA molecules are 5CITEP from Shionogi & Co. Ltd 41 and the L-731,988 series from Merck and Co. 54 (Table 3). 5CITEP has been the only DKA co-crystallized with the catalytic core domain of IN (Fig. 2A)41. 5CITEP is stabilized in the active site by hydrogen bonds with one of the catalytic residues (E152) as well as with other amino acids (T66, N155, Q148) and two lysine residues (K156 and K159) previously reported to be involved in DNA substrate binding 29, 30 and mononucleotide positioning 43 (Fig. 2A). In the 5CITEP-IN co-crystal structure, neither direct contact between the drug and the two other catalytic residues (D64 and D116) nor direct interaction with the magnesium ion could be detected. Only a van der Waals interaction between the indole ring and one of the four water molecules coordinating the magnesium ion seemed plausible (Fig. 2A).
Table 3.
Diketo acid derivatives
| Name | Structure | IC50 | EC50 | EC95 | |
|---|---|---|---|---|---|
| 5CITEP |  |
2000 | NA | NA | 41 |
| L-731,988 |  |
50 | 1000 | ND | 54 |
| L-870,810 |  |
8 | ND | 15 | 73 |
| MK-2048 | 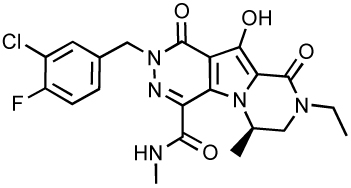 |
ND | ND | 41 | 20 |
| GS-9160 | 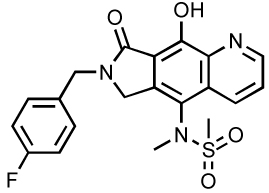 |
28 | 2.1 1.7 8.5 |
4000 | 78, 172, 173 |
| ST1 |  |
15 | ND | ND | 79 |
| RDS1625 |  |
40 60 |
1170 | ND | 67, 80 |
NA means not antiviral, and ND means not determined or not disclosed. Values are expressed in nM.
The DKA derivatives selectively inhibit ST at nanomolar IC50 values and inhibit integration without interfering with viral DNA synthesis in cells 54. Validation of HIV-1 IN as a molecular target was obtained by generation of drug-resistant mutant viruses, which were found to bear single or multiple mutations in their IN coding sequence. The resulting mutations (T66I, L74M, E92Q, G140G/A, Y143R/C, Q148R/H/K, S153Y, M154I, N155H) all cluster around the DDE catalytic triad of amino acids (D64, D116, E152) (Fig. 2C&2D) 54, 64, 65. Consistent with the drug binding within the IN catalytic site, inhibition was found to be metal-dependent 66 and to interfere with binding of the flexible loop to the viral LTR DNA end 67. Selective IN inhibition in HIV infected cells was also suggested by the increased formation of 2-LTR viral circles, which are abortive viral replication intermediates 54. Binding studies revealed that DKA compounds only bind to the catalytic site of HIV-1 IN specifically in the presence of viral LTR DNA 68. Other studies suggested that these compounds also interact with the 5’-end of the viral DNA 53, 69. Mutating one of the HIV IN core catalytic residues is sufficient to abrogate drug binding in the presence of a viral DNA substrate 68. Prior binding of the target DNA substrate to the IN-viral DNA complex prohibits binding and inhibition of integration by DKAs in a concentration-dependent manner, suggesting a competition between diketo acids and DNA in the ST complex 17, 53, 68.
DKA derivatives mediate their antiviral activity through metal chelation 69–71. Although the type and number of metals required for IN activity have still not be determined, it has been postulated that the integration mechanism involves two divalent metals (magnesium) in the catalytic site of the enzyme 69–71. The first magnesium atom, as observed in the different crystal structures, is coordinated by the two catalytic residues D64 and D116 (Fig. 2). The second would be coordinated either by D116 and E152 69–71.
Shionogi & Co. Ltd. has developed IN inhibitors in partnership with GlaxoSmithKline and both went into phase I/II clinical trials with S-1360, a derivative of 5CITEP 72. The compound was active and well tolerated but the trial was terminated for undisclosed reasons 8. Merck & Co. after demonstrating the efficacy of DKA derivatives to suppress SIV viral replication in rhesus macaques 65, developed the napthyridine carboxamides (L-870,810, Table 3), a class of IN inhibitors derived from the DKA family 73. L-870,810 is a potent ST inhibitor with improved bioavailability compared to previously reported DKA. L-870,810 inhibits the IN ST activity in vitro with an IC50 value of 8 nM and is antiviral with an EC95 value of 15 nM in a cell culture assay (Table 3) 73. During clinical trials, L-870,810 was terminated in Phase II due to toxicity in dogs 8.
Resistance to raltegravir and elvitegravir and second generation ST inhibitors
Despite its recent success in the clinic, emergence of resistance leading to treatment failure has already been reported for raltegravir and elvitegravir 20, 28, 61, 62. Consistent with the selective targeting of IN by raltegravir and elvitegravir, resistance pathways have been described in relationship with point mutations of IN residues surrounding the IN catalytic core site. Three main pathways appear to drive resistance. They involve “primary mutations” Q148R/H/K, N155H or Y143R/C 28, 61, 62, 74–77. Each tends to be associated with “secondary mutations” that appear to restore viral fitness in those primary mutants: for instance G140S with Q148H or G140A with Q148R 75, 77(Marchand and Pommier, unpublished). In a recent study, we showed that the resistance profile of elvitegravir closely resembles that of raltegravir, suggesting that elvitegravir is unlikely to overcome resistance to raltegravir 28.
Current research efforts are now devoted to the discovery and the development of novel compounds and model systems to circumvent raltegravir and elvitegravir resistance. Merck has already developed second generation ST-inhibitors such as the tricyclic MK-2048 (Table 3), which exhibits an EC95 value of 41 nM and excellent pharmacokinetics in preclinical species 20. Gilead Sciences is developing its own next generation tricyclic inhibitors. GS-9160 has been designed on the naphtyridine carboxamide scaffold (Table 3) and exhibits a single digit nanomolar antiviral activity with over 3 logs of selectivity index 78. Bristol-Myers Squibb is also committed to develop ST inhibitors with the use of compound ST1 (Table 3) to study integration mechanisms 79. Academic groups are also actively involved in the development of novel IN inhibitors. One example is the quinolinonyl DKA, which shares with elvitegravir the same quinolone structural motif (Table 3) 80 and novel fused DKA compounds (see Table 5) 81–83. The rest of this chapter will be devoted to other compounds, many of which inhibit both 3’-processing and strand transfer.
Table 5.
New catechol derivatives
| Name | Structure | IC50 | EC50 | Ref. | |
|---|---|---|---|---|---|
| 3’-P | ST | ||||
| Dihydroxybe nzoic acid hydrazides |
 |
6.9 | 1.6 | 1.7 | 82 |
| Dihydroxy isoindolones |
 |
13 | 0.16 | 0.48 | 83 |
| Dihydroxy naphthyl derivatives |
 |
1.9 | 0.9 | 46 | 93 |
| Styrylquina- zoline |
 |
21 | ND | ND | 97 |
| Catechol Derivative |
 |
7 | ND | ND | 45 |
| Dinitrorosma rinic acid |
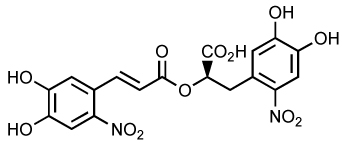 |
52 | 0.07 | 12 | 99 |
| Coumarin Caffeoyl derivative |
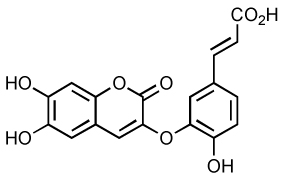 |
1 | ND | 31 | 100 |
ND means not determined or not disclosed. Values are expressed in µM.
L-Chicoric Acid Derivatives
Caffeic acid phenylethyl ester (CAPE) was one of the first reported natural product (from bee hives) inhibitors of IN (Table 4) 84, 85. Furthermore, CAPE was the first compound found to inhibit ST with selectively over 3’-P with IC50 values of 5 µM 84, 85. L-Chicoric acid (L-CA) is a related bidentate catechol that has also been identified as an inhibitor of IN (Table 4). Inhibition has been shown at sub-micromolar concentrations through a noncompetitive and reversible mechanism both in vitro and ex vivo 86, 87. However, L-CA enters cells poorly and its antiviral activity has been proposed to result primarily from inhibition of viral entry by interaction with gp120 88. These two molecules, CAPE and L-CA, have served as parent structures for the identification of new molecules with improved properties. For example, an L-CA derivative (Table 4) has shown improved selectivity against the integration step in cell culture according to quantitative real-time PCR 86, 87. In addition, several L-CA derivatives have shown clinical potential based on their ability to readily enter cells and increased activity against IN. Notably, the clinically relevant IN mutations G140S and F185K confer significant resistance to L-CA and its derivatives 87.
Table 4.
Caffeic acid derivatives and related catechols
| Name | Structure | IC50 | EC50 | Ref. |
|---|---|---|---|---|
| CAPE | 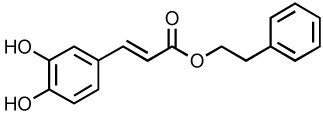 |
5 | ND | 84, 85, 174 |
| L-chicoric Acid (L-CA) |
 |
0.5 | 4.2 | 175 |
| Caffeic acid derivative |
 |
0.73 | > 4.5 | 176 |
| Chicoric acid derivative |
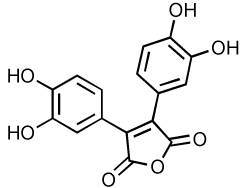 |
3.8 | ND | 90 |
| Caffeoylamino acid derivative |
 |
12 | ND | 91 |
ND means not determined or not disclosed. Values are expressed in µM.
The caffeic acid derivative in which two caffeoyl moieties are bridged by a tartaric acid (Table 4) is active against HIV-1 IN at submicromolar concentrations and against HIV-1 replication at micromolar concentrations 86. Unfortunately, toxicity of this molecule is also in the same concentration range leading to poor selectivity indexes 89. A chicoric acid derivative substituted with maleic anhydride (Table 4) represents a potent inhibitor of IN with an IC50 as low as 3.8 µM 90. However, this compound is unable to inhibit HIV-1 replication under nontoxic concentrations. A series of caffeoylamino acid derivatives have also been generated(Table 4). For example, catechol ring substitutions have shown to reduce toxicity. In addition, substitution of the carboxylic acid with an amide moiety results in a decrease potency in vitro, but has demonstrated improved cell entry 91.
A second set of compounds has been designed based on L-CA and dicaffeoyl derivatives 89, 90, 92. These molecules contain a dicaffeoyl group connected by a furan ring linker containing hydrogen bond accepting or donating groups (Table 4). They exhibit IC50 values in the range of 0.5–12 µM. Unfortunately, these compounds do not inhibit HIV-1 replication in MT-4 cells at non-toxic concentrations.
New Catechol Derivatives
The dihydroxybenzoic acid hydrazides family of IN inhibitors is also derived from a catechol structure (Table 5). Theses compounds are potent IN inhibitors, but do not display selectivity for 3’-P or ST. Inhibition is observed with both Mg2+ and Mn2+ cofactors. The hydrazide containing inhibitors exhibit high inhibitory potency only when Mn+2 is used 82.
Dihydroxyisoindolone derivatives are more potent and selective for ST 82 (Table 5). To improve potency and reduce toxicity, halo-substitutions of the aromatic ring were performed. The 3-chloro-4-fluoro-derivative is the most potent inhibitor with an IC50 for ST of 0.16 µM. It also exerts an 80-fold ST selectivity compared to 3’-P inhibition 81. Antiviral activities have been found at submicromolar concentrations for these inhibitors, but with relatively high cytotoxicity suggesting that they may have relatively unfavorable therapeutic indexes. The dihydroxynaphthyl derivatives have variable selectivity for ST. Specifically, the dihydroxynaphthyl derivative with catechol-DKA hybrids (Table 5) is active against IN at low micromolar concentrations with a two- to three-fold decrease in selectivity for ST compared to DKA 93.
The styroquinolines (SQL) (Table 5) are IN inhibitors with specificity for 3’-P 94. SQL block viral replication at nontoxic concentrations 95 and inhibit 3'-P activity in vitro by competing with binding of the viral DNA substrate 96. Recently more potent SQL derivatives have been produced replacing the C-8 methoxy by a hydroxy group (Table 5) 97. Inhibition of 3’-P by styrylquinazoline was 6-fold increased compared to the initial SQL. The cellular target of SQLs was confirmed by the appearance of mutations in the IN gene when viruses were grown in the presence of increasing concentrations of SQLs 98. Finally, these mutations led to SQL-resistant viruses when introduced into the wild-type sequence.
Another catechol derivative, the dihydroxyindole dimeric with piperazine (Table 5) inhibits IN in vitro with an IC50 value at 7 µM and exhibits less toxicity than other catechols with a CC50 value above 20 µM 45, 92.
Dinitrorosmarinic acid (Table 5) is a very potent IN inhibitor in vitro with a high selectivity for ST 99. The IC50 for 3’-P and ST are 52 µM and 70 nM, respectively. The mechanism most likely involves the chelation of the divalent cation. This molecule also inhibits viral replication in MT-4 cells with EC50 at 12 µM but its therapeutic index is only 3.8.
The coumarin caffeoyl derivative (Table 5) exhibits potent inhibitory activity at micromolar concentration in vitro 100. In cells, this molecule is less effective with an EC50 value of 31 µM. Interestingly, some coumarin derivatives can interact directly to IN in vitro in the catalytic core region between residues 128AACWWAGIK136 45. This peptide segment is located close to the dimer interface of IN. Thus, coumarin-containing molecules represent a new type of inhibitors that inhibit IN in vitro by shifting its oligomerization state.
Salicylhydrazide Derivatives
Salicylhydrazides (Table 6) were previously reported as an IN inhibitor. However, inhibition occurs in vitro only in the presence of manganese 101, which, unlike magnesium is believed not to be the relevant divalent catalytic metal. Due to the high toxicity of the molecule (CC50 = 0.1 µM), the antiviral properties could not be determined. Two new derivatives (Table 6) inhibit IN in the presence of either manganese or magnesium 102. These compounds displayed low micromolar activity against IN in vitro (range 3–13 µM) and are several hundred-fold less cytotoxic then the parent molecules (CC50 around 20–30 µM). Experiments performed with the integrase mutant C65S and molecular modeling studies suggested that mercaptosalicylhydrazide (MSH) forms a disulfide bond with C65 in the IN active site 102. This interaction has been proposed to facilitate positioning of MSH to allow divalent metal chelation.
Table 6.
Salicylhydrazide derivatives
| Name | Structure | IC50 | Ref. | |
|---|---|---|---|---|
| 3’-P (Mg2+) |
ST (Mg2+) |
|||
| Salicylhydrazide | 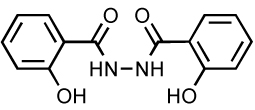 |
2 | 0.7 >1000 |
102 |
| Salicylhydrazide |  |
11 | 5 | 177 |
| Mercapto- salicylhydrazide |
 |
ND | 10.5 | 102 |
ND means not determined or not disclosed. Values are expressed in µM.
Dual inhibitors of IN and RT
Some IN inhibitors such as V-165 (Table 7) have been described to have some activity against HIV-1 reverse transcriptase (RT) 98, 103, 104. This RT inhibition can be due in some cases to a specific inhibition of the RNase H domain of RT that shares structural similarities with IN 105. Even if it is commonly admitted that a good inhibitor must be highly specific and selective for a single target, it may be valuable to develop IN-RT dual inhibitors as an antiretroviral therapy for the treatment of HIV and AIDS.
Table 7.
Dual inhibitors of HIV-1 RT and IN.
| Name | Structure | IC50 | EC | Ref. | |
|---|---|---|---|---|---|
| 3’-P | ST | ||||
| V-165 |  |
0.9 | 16.1 | 8.9 | 103, 178 |
| β- thujaplicinol |
 |
117 | 22 | ND | 106–108 |
| MHL 2e | 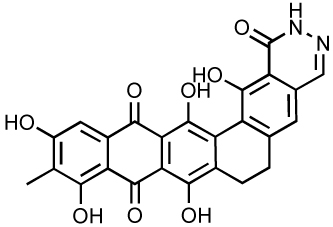 |
ND | 4.06 | ND | 4 |
| Compound 2j |
 |
ND | 1.24 | 22 | 109 |
| Portmanteau 8 |
 |
ND | 2.4 | 0.0033 | 110–112 |
ND means not determined or not disclosed. Values are expressed in µM.
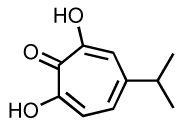
Since 2004, several other dual inhibitors have been reported. Beta-thujaplicinol, a natural tropolone derivative, inhibit IN and RNase H probably by metal chelation 106–108. More recently, madurahydroxylactone and hydroxyisoquinoline dione derivatives (Table 7) have also been reported as dual inhibitors of IN and RNase H 4, 109. An interesting approach in the development of such dual inhibitors consists of rationally designing a chimeric drug based on the fusion of an RT-specific and an IN specific scaffold. The resulting “portmanteau compounds” (Table 7) exhibits nanomolar antiviral activity 110–112.
Peptide Inhibitors from screening
The first IN peptide inhibitor reported was the hexapeptide HCKFWW 113. This peptide (Table 8), discovered by systematic screening of peptide libraries inhibits HIV IN 3’-P and ST with an IC50 value of 2 µM. Dimerization of this peptide via a thioether or a dithiomethylene linker lowers the IC50 values of both 3’-P and ST as much as 10–20-fold 114, 115.
Table 8.
Peptide inhibitors from screening.
| Peptide Name | Sequence | IC50 | Ref. |
|---|---|---|---|
| HCKFWW-NH2b | 2, 3’-P/ST |
113 | |
| EBR28 | YQLLIRMIYKNI | 5, 3’-P | 116 |
| Integramide A: R=H B: R=CH3 |

|
A=17 B= 10 |
117 |
| Indolicidin | ILPWKWPWWPWRR | ND | 119, 121, 179 |
| pep1 | FHNHGKQGGGS | 70, ST | 122 |
| PNA4 B=thymin-1-yl R1=CH3(CH2)16- |
 |
22, 3’-P/ST |
180 |
PNA = Peptide Nucleic Acid. ND means not determined or not disclosed. Values are expressed in µM.
Using the yeast 2-hybrid system, a 33 amino acids long peptide was isolated with an IC50 of 9 µM for 3’-P and for ST 116. The amino-terminal portion of the peptide, EBR28 (Table 8), shows increase activity with an IC50 of 5 µM against 3’-P. This peptide forms an amphipathic alpha helix in solution and interacts with the catalytic domain to block DNA binding. EBR28 also inhibits HIV-1 infectivity of CD4+ HeLa cells with an IC50 of 40 µM and apparently low toxicity.
Integramides A and B (Table 8) are non-ribosomal peptides isolated from Dendrodochium sp. with IC50 values of 17 and 10 µM, respectively 117. Antiviral activity could not be tested because these compound exhibited toxicity at IC50 levels. Recently, a total chemical synthesis was performed and the synthetic product was comparable to the natural product 118.
Indolicidin (Table 8) is an antimicrobial peptide isolated from the large granules of bovine neutrophils with virucidal properties against HIV-1 119. The complete effect was obtained at 174 µM and the 50% inhibitory dose was between 39 and 58µM. In vitro activities are in the same range of concentration with IC50 value of around 60 µM for both 3’-P and ST 120 and result from interference in the DNA-IN complex formation by direct binding of indolicidin to DNA 121. Peptide toxicity is important with a CC50 value around 20 µM; but by connecting monomers with a lysine linker, in vitro activities can be increased. The most potent derivative is a tetramer with an IC50 value of 0.6 µM for both 3’-P and ST 120.
Using phage display, several small peptides were isolated including Pep1 (Table 8). This peptide is able to bind specifically to the C-terminus of IN and inhibits only the ST reaction with an IC50 value of 70 µM 122. The postulated mechanism was an inhibition of the pre-cleaved substrate binding.
Peptide nucleic acid (PNA) are DNA analog in which sugar-phosphate skeleton are replaced by peptidic group as [N-(2-amino-ethyl)-glycine]. The resulting molecules (Table 8) are not sensitive to nucleases but can still interact tightly with DNA in a Watson-Crick manner. Recently, a N-terminal lipid functionalized PNA, PNA4 (Table 8) was reported to be antiviral with inhibition of both RT and IN 123. In vitro, this molecule inhibits both 3’P and ST with an IC50 of 22 µM.
Peptide Inhibitors derived from IN
The continued development of peptide inhibitors of HIV-1 IN has provided biochemical data helping to further define the structure and dimerization properties of the enzyme. Several peptide inhibitors of HIV-1 IN have been derived from polypeptide helices found within the IN structure. These helices are highlighted in Fig. (3B), which shows a crystal structure of the core domain dimer of IN 38. The inhibitory data for the polypeptide inhibitors are summarized in Table 9.
Table 9.
Peptide inhibitors derived from IN.
| Peptide Name |
Sequence | IC50 | Ref. |
|---|---|---|---|
| INH1 | ATGQETAYFLLKLAGKA-CONH2 | 250, 3’-P 150, ST |
124 |
| INH5 | DQAEHLKTAVQMAVFIHNYKA-CONH2 | 0.085, 3’-P 0.060, ST |
124 |
| K159 | SQGVVESMNKELKKIIGQVRDQAEHLKTAY | 600, ST | 125 |
Values are expressed in µM.
Alpha helices 1 and 5 form the dimerization interface between the core domains from different IN subunits. Synthetic peptides INH1 and INH5 derived from α1 helix (amino acids 93–107) and α5 helix/loop (amino acids 167–187), respectively, inhibit HIV-1 IN in the nanomolar range 124. INH5 is the most active of these peptides with IC50 values of 0.085 µM against 3’-P and 0.060 µM against ST in vitro. Fluorescence spectroscopy and size exclusion chromatography showed that INH5 binds the core domain in the full-length protein and induces the dissociation of IN oligomers.
Helix 4 of IN [amino acids 151–172, shown in green in Fig. (2B)] is an amphipathic alpha helix containing several amino acids shown to interact with the HIV-1 LTR 29–31 and nucleotides 43 including G152, K156, and K159. A peptide inhibitor (K159) derived from the α4 helix of IN was reported to inhibit HIV-1 IN in vitro with a relatively low potency (IC50 of 600 µM and complete inhibition at 1.2 mM) 125. Circular dichroism and chemical cross-linking showed auto-association of K159. It has been suggested that the inhibitory activity of the peptide results from a coiled-coil interaction stabilized through a hydrophobic interface with the peptide and IN helix 4, blocking the catalytic residue E152 126. Mutagenesis of K159 leads to a new peptide, EAA26, with increased helical properties and enhanced potency against IN compared to K159 peptide 127.
Subsequently, a monoclonal antibody against K159 (anti-K159) was developed and characterized 128. The functional epitope was identified as amino acids 163–175, in a helical form important for DNA binding. Filter binding studies showed that interaction between anti-K159 and IN decreases DNA binding. The reported IC50 for inhibition of DNA binding is approximately 0.6 µM for anti-K159 and the IC50 values for ST and 3’-P are respectively 25 nM and 16 nM.
Peptide Inhibitors derived from viral or cellular cofactors
Throughout the replication process in cells, viral DNA is part of a large nucleoprotein complex, which moves from the reverse transcription complex to the pre-integration complex (PIC) by gain and lost of viral and cellular factors. PICs have been difficult to fully resolve due to their potentially dynamic composition and to the fact that cofactors can vary depending on the purification protocol.
Using the yeast 2-hybrid system, IN was found to interact with Ini1 (IN interactor 1), also known as SNF5, and part of the SWI/SNF complex 129,130. This interaction could be implicated in the targeting of the integration and so as a possible therapeutic target. Fragment S6 (Table 10) of Ini1 (aa 183–294) is the minimal interacting sequence with IN. S6 when expressed in cells exhibits a trans-dominant effect and inhibits viral production and replication. S6 does not exhibit apparent toxicity and should not be immunogenic due to its host origin 131. It appears that Ini1 is important for early steps of replication 132, 133 and S6 inhibition is dependent on its oligomerization state 134.
Table 10.
Peptide inhibitors derived from other proteins.
| Peptide Name |
Sequence | IC50 | Ref. |
|---|---|---|---|
| S6 | Ini1 183–294 | 60, ST | 181 |
| IBD | LEDGF 355–377 | ND | 135 |
| 4286 (RT palm) |
KILEPFRKQNPDIVIYQYMD | 4.8, 3’-P 4.5, ST |
140 |
| 4321 (RNaseH) |
ELVNQIIEQLIKKEKVYLAW | 6.9, 3’-P 5, ST |
140 |
| Rev13–23 | LKTVRLIKFLY | 25 | 182 |
| Rev53–67 | RSISGWILSTYLGRP | 25 | 182 |
| Vpr 61–75 | IRILQQLLFIHFRIG | 1.3, 3’-P 1, ST |
145 |
Ini1 = IN interactor 1 ; IBD = IN Binding Domain ; LEDGF = Lens Epithelium-Derived Growth Factor. ND means not determined or not disclosed. Values are expressed in µM.
Searching into cells with an IN-flag allowed the isolation of a large complex composed by a tetramer of IN bound to a 76 kDa nuclear protein previously known as Lens-Epithelium Derived Growth Factor (LEDGF/p75) 33. The IN binding domain (IBD) corresponds to amino acids 347–429 of LEDGF (Table 10) and is composed of 4 or 5 alpha helix 135. A deletion of LEDGF in cells by siRNA or knock down induces a three to five fold decrease of viral replication due to a lack of integration 136. On the other hand, when IBD is over-expressed in cells, HIV-1 infection is strongly inhibited after DNA translocation into the nucleus but before integration 137. The LEDGF-IN interaction is attractive as therapeutic target because disruption of the LEDGF-IN interaction induces a failure in viral replication. But LEDGF is a cellular factor, so side effects can be important when modifying its expression or activity. A number of small molecules have been reported for their capacity to specifically interfere with the LEDGF-IN interaction. CHIBA-3003 (2-hydroxy-4-(-4-hydroxy-1H-indol-3-yl)-4-oxobut-2-enoic acid) inhibits the LEDGF-IN complex formation in vitro with an IC50 of 35 µM 138.
Another way to disrupt an important interaction with IN without risk of side effect is to target a viral co-factor. To date, several proteins have been described in the PICs including MA, Vpr, or RT 139. By screening a complete library of RT-, Rev- and Vpr-derived peptides, several peptides were isolated with IN inhibitory activity (Table 10). Peptides 4286 and 4321 are derived from reverse transcriptase palm (166–185) and RNase H domain (514–528), respectively 140. Both 3’-P and ST are inhibited with IC50 between 4.5 and 6.9 µM. Rev13–23 and Rev 53–67 inhibit both 3’-P and ST in vitro with an IC50 of 25 µM and inhibit IN ex vivo at low micromolar concentrations 141. In fact, Rev-derived peptides act as shiftide in vitro resulting in the tetramerization and de facto inhibition of 3’-P 142. This mechanism of inhibition can also be reproduced with unrelated peptides like IN1 (WQCLTLTHRGFVLLTITVLR), which was isolated by yeast 2-hybrid and found to inhibit IN with IC50 at 38 (3’-P) and 12 µM (ST) 143. C-terminal domain of Vpr is also able to bind and stimulate ST in vitro 144. Derived peptides can bind and/or inhibit both RT (polymerase and RNase H activities) and IN (3’-P and ST) 145. The most potent peptide, Vpr 61–75, exhibits IC50 at single digit micromolar concentration for all these activities.
Antibody Inhibitors
A library of monoclonal antibodies against HIV-1 IN was developed 146. Characterization of these antibodies has provided information on their mechanism of inhibition of IN through interaction with protein domains, as well as insight into subunit arrangement of the enzyme. Four monoclonal antibodies (Table 11) have been characterized: mAb17 binds to the N-terminal domain, mAb4 binds to the catalytic domain, and mAb32 and mAb33 bind to the C-terminal domain of IN. Three of these antibodies inhibit IN in vitro (Table 11) 147. MAb17 and Fab17 inhibit 3’-P with IC50 values of 0.4 and 4.5 µM, respectively 148. These antibodies bind the N-terminal domain and inhibits the enzyme possibly by interfering with protein-protein interactions or conformation, without disturbing dimerization or DNA binding.
Table 11.
Antibody inhibitors of HIV-1 IN.
| Antibody | Target Domain | IC50 3’- P | Ref. |
|---|---|---|---|
| mAb4 | Catalytic core | ND | 146 |
| mab17 | N-terminal | 0.4 | 148 |
| mAb32 | C-terminal | 2 | 149 |
| mAb33 | C-terminal | 0.18 | 149, 150 |
| IgG | ND | ND | 151 |
ND means not determined or not disclosed. Values are expressed in µM.
Mab32 has a weak inhibition against IN 3’-P (IC50 of 2 µM) and Fab32 lacks inhibitory activity. The most potent antibody, mAb33 and the corresponding isolated Fab33 fragment, bind to IN C-terminal domain to DNA (220–270) inhibiting DNA binding 149,150. The IC50 values for 3’-P are 0.18 and 1.8 µM for mAb33 and Fab33, respectively. An IN conformational change that occurs upon metal binding is prevented by the addition of mAb33, and if the conformational change already occurred, antibody binding is blocked 147. Therefore, mAb33 may act by locking the enzyme in the catalytically inactive conformation prior to metal binding.
Recently, it was shown that IgG isolated from HIV-infected patients can act as abzyme and hydrolyse specifically IN 151. The mechanism involved is similar to autoimmune proteolytic abzyme but the potential role of these immunoglobulins during infection is not known. Nevertheless, hydrolyzing antibodies can be useful for diagnosis purposes.
The potential for antibody-based therapeutics may be realized using a gene therapy approach. Intracellular expression of a recombinant antibody gene has been reported to inhibit effectively many HIV-1 proteins including reverse transcriptase, REV, IN, and CXCR4 152–156. Transfection of human peripheral blood monocytic cells (PBMCs) with retroviral vectors expressing the single chain variable (SFv) region of mAb33 (IN#33) dramatically reduces HIV-1 replication 154. Further studies showed reduced HIV-1 infection and reduced virion infectivity in T cells stably transfected with a Vpr-IN-SFv fusion construct 153. Greater than 95% of IN#33 transduced CD34+ progenitor T cells from rhesus macaque and human fetal bone marrow cells expressed the IN antibody, and high expression continued after transplantation into SCID mice 157. These or similar stably transduced genes may be useful in treating diseases residing in CD34+ cells. Recently, a reduced number of HIV-1 infected thymocytes in human thymic grafts was demonstrated in a mouse model, suggesting SV40-derived vectors may provide efficient in vivo therapeutic delivery of genes encoding IN antibodies 158.
Update on Oligonucleotide Inhibitors
Zintevir™ (AR177), a guanosine quartet (G4)-forming oligonucleotide 159–161 was the first anti-IN drug tested in clinical trials by Aronex Pharmaceuticals. Mono- and di-nucleotides IN inhibitors were the focus of prior reviews 5, 9, 18 and are not discussed further here. An oligonucleotide containing 6-oxocytosine has been reported to inhibit IN in the sub-micromolar range 162. The best oligonucleotide inhibitor described contains the sequence 5’-AGAGATTTTC*C*, where C* indicates 6-oxocytosine substitution for cytosine (Table 12). This oligonucleotide inhibits IN with IC50 values of 0.3 µM for both 3’-P and ST. The sequence is identical to the sequence beginning seven bases from the 5’ end of the non-cleaved strand of the U5 LTR. Substitution of two cytosines with 6-oxocytosine residues results in a potent IN inhibitor even though the unmodified oligonucleotide has similar affinity for IN but fails to inhibit IN activity 163. Inhibition was dependent on sequence, presence of 6-oxocytosine, and positioning the 6-oxocytosine at the terminus of the oligonucleotide 162, 163. The 6-oxycytosine-containing oligonucleotide inhibits binding of IN to substrate DNA, when added either before or after binding of the substrate DNA 163.
Table 12.
Oligonucleotide inhibitors.
| Name | Sequence | IC50 | EC50 | Ref. |
|---|---|---|---|---|
| 6-oxycytosine (*) | 5’-AGAGATTTTC*C*-3’ | 500, ST | ND | 163 |
| GTGT-Acr (* acridine) |
5’-GGTTTTTGTGT*-3’ | 2500, 3’-P 2000, ST |
ND | 164 |
| 6-oxycytosine (*) | 5’-AGAGATTTTC*C*-3’ | 500, ST | ND | 163 |
|
T30177 (*phosphorothioate) |
5’-G*TGGTGGGTGGGTGGG*T-3’ | 79, 3’-P 49, ST |
75 | 159 |
| T30923 | 5’-GGGTGGGTGGGTGGGT-3’ | 75, 3’P 80, ST |
65 | 183 |
| 93del | 5’-GGGGTGGGAGGAGGGT-3’ | 42, 3’-P 166, ST |
20 | 167 |
| 60del | 5’-GGGGGGGCCAGGCCATGG-3’ | ≈100, 3’-P ≈100, ST |
ND | 169 |
ND means not determined or not disclosed. Values are expressed in nM.
Based on this same principle, small single stranded DNA harboring a 3’-acridine group are able to inhibit IN activities in vitro in the low micromolar concentrations 164. This GTGT-Acr dissociates the IN-DNA complex and the acridine modification seems to be the dissociating factor while the DNA sequence only acts a targeting factor.
Oligonucleotides as potential therapeutics have several limitations. Nucleic acids in a cellular context can be subject to degradation and chemical modifications unless they are modified as phosphorothioates or with other special structural modifications to confer resistance to nucleases. Guanosine quadruplexes (G-quartets) are especially stable and efficient inhibitors of HIV-1 IN in vitro 159. The G-quartet T30177 (Table 12) inhibits IN 3’-P and ST with IC50 values of 0.079 and 0.049 µM, respectively 159. HIV-1 replication is also inhibited with an IC50 of 0.075 µM 161 and, as mentioned at the beginning of this section, a derivative of T30177 was tested as the first IN inhibitor in clinical trials in 1996 as Zintevir™ by Aronex Pharmaceuticals. However, T30177 was also shown to inhibit gp120 165 and resistant viruses obtained during T30177 treatment exhibited mutations in the gp120 gene 166. Increasing the length and sequence of the G-quartet did not decrease inhibition efficiency and modeling suggested the G-quartet structure interacted with IN in a “face-to-face” orientation occurring between the G-quartet loops and the IN active site 160.
Oligonucleotides selected against and inhibitors of RNase H are also able to inhibit IN 167. 93del (Table 12) forms specific dimeric G-quartets 168 and inhibits recombinant HIV-1 IN with IC50 values in the nanomolar range. Consistent with the formation of G4s, IN inhibition is significantly improved in the presence of potassium ion. This aptamer blocks HIV-1 replication in human P4 cells with EC50 values of 20 nM. However this oligonucleotide does not inhibit cell fusion, suggesting that IN may be the primary target of this second generation G4 inhibitors. Selecting against the isolated RNase H domain, other G-rich oligonucleotides, including ODN 60del (Table 12), have been selected. They are not inhibitors of RNase H in the RT context but inhibit IN in vitro 169. The IC50 of 60del against the 3’-P and ST is quite higher than those of 93del and other G-quartet but still remain around 100 nM for both reactions. According to the high specificity of aptamers and the stability of G-quadraduplexes, it is reasonable to think that oligonucleotides can be a good alternative to treat HIV.
Intracellular penetration of the G-quartets such as T30177 or 93del has been considered a potential limitation. Nevertheless early studies with 32P-radiolabeled T30177 demonstrated this not to be case 161. Delivery of T40214 was achieved using a liposomal delivery system 170. T40214 enters the nuclei of 3T3, CEMSS and MT4 cells and decreases HIV-1 replication with an EC50 of 0.2 µM, suggesting that IN can be targeted intracellularly by G-quartets. Inhibition of IN in cells has not yet been demonstrated. Recenty, T30923 and 93del were used to study the cellular uptake 171. It appears that both 93del and T30923 are able to enter different cell lines including lymphocyte cells at nanomolar concentrations. This entrance is enhanced using transfection agent and can be directed into a cell compartment depending on the cationic lipid used. In presence of HIV-1 particles, these G-rich forming quartet oligonucleotides exhibit also a very high entrance, comparable to a transfection experiment, which highlights a probable favoring role of the virus in the oligonucleotide uptake. Further experiments have to be done to discriminate the exact role of the virus; the mechanism is apparently gp120-independent as VSV-G pseudo-typed viruses induce the same pattern of oligonucleotide entry. Further studies are therefore warranted to elucidate whether other targets besides IN exist in HIV-infected cells.
Conclusions
The approval and remarkable activity and tolerance of raltegravir, and the clinical activity of elvitegravir have demonstrated that targeting IN can lead to effective therapies against AIDS and HIV infections. Judging from the other HIV targets, RT and protease, it is expected that many more drugs targeting IN are on their way to the clinic. The availability of the raltegravir-resistant IN mutants and viruses should enable the selection and development of drugs that will overcome such resistance. Thus, IN inhibitors are likely to have a major impact on AIDS and HIV infections. It is also possible that IN could be used as a target for other retroviral infections.
Acknowledgements
This work was supported by the Center for Cancer Research, an Intramural Division of the National Cancer Institute, National Institutes of Health. We wish to thank Dr. Thomas Dexheimer for his careful reading and suggestion for this review. We also wish to thank all the other members of the Laboratory of Molecular Pharmacology for stimulating discussions.
Abbreviations
- PIC
pre-integration complex
- LTR
long terminal repeat
- L-CA
L-chicoric acid
- 3’-P
3’-processing
- ST
strand transfer
References
- 1.Melek M, Jones JM, O'Dea MH, Pais G, Burke TR, Jr, Pommier Y, Neamati N, Gellert M. Effect of HIV integrase inhibitors on the RAG1/2 recombinase. Proc Natl Acad Sci U S A. 2002;99:134–137. doi: 10.1073/pnas.012610699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rice P, Craigie R, Davies DR. Retroviral integrases and their cousins. Current Opinion in Structural Biology. 1996;6:76–83. doi: 10.1016/s0959-440x(96)80098-4. [DOI] [PubMed] [Google Scholar]
- 3.Chiu TK, Davies DR. Structure and function of HIV-1 integrase. Curr Top Med Chem. 2004;4:965–977. doi: 10.2174/1568026043388547. [DOI] [PubMed] [Google Scholar]
- 4.Marchand C, Beutler JA, Wamiru A, Budihas S, Mollmann U, Heinisch L, Mellors JW, Le Grice SF, Pommier Y. Madurahydroxylactone derivatives as dual inhibitors of human immunodeficiency virus type 1 integrase and RNase H. Antimicrob Agents Chemother. 2008;52:361–364. doi: 10.1128/AAC.00883-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson AA, Marchand C, Pommier Y. HIV-1 integrase inhibitors: a decade of research and two drugs in clinical trial. Curr Top Med Chem. 2004;4:1059–1077. doi: 10.2174/1568026043388394. [DOI] [PubMed] [Google Scholar]
- 6.Pommier Y, Johnson AA, Marchand C. Integrase inhibitors to treat HIV/AIDS. Nat Rev Drug Discov. 2005;4:236–248. doi: 10.1038/nrd1660. [DOI] [PubMed] [Google Scholar]
- 7.Marchand C, Johnson AA, Semenova EA, Pommier Y. Mechanism and inhibition of HIV integration. Drug Discov Today: Disease Mechanisms. 2006;3:253–260. doi: 10.1016/j.ddmec.2006.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Semenova EA, Johnson AA, Marchand C, Pommier Y. Integration of human immunodeficiency virus as a target for antiretroviral therapy. Curr Opin HIV AIDS. 2006;1:380–387. doi: 10.1097/01.COH.0000239850.14991.f9. [DOI] [PubMed] [Google Scholar]
- 9.Nair V. HIV integrase as a target for antiviral chemotherapy. Reviews in Medical Virology. 2002;12:179–193. doi: 10.1002/rmv.350. [DOI] [PubMed] [Google Scholar]
- 10.Tarrago-Litvak L, Andreola ML, Fournier M, Nevinsky GA, Parissi V, Soultrait VR, Litvak S. Inhibitors of HIV-1 Reverse Transcriptase and Integrase: Classical and Emerging Therapeutical Approaches. Curr Pharm Des. 2002;8:595–614. doi: 10.2174/1381612024607162. [DOI] [PubMed] [Google Scholar]
- 11.d'Angelo J, Mouscadet JF, Desmaele D, Zouhiri F, Leh H. HIV-1 integrase: the next target for AIDS therapy? Pathol Biol (Paris) 2001;49:237–246. doi: 10.1016/s0369-8114(01)00135-3. [DOI] [PubMed] [Google Scholar]
- 12.Pommier Y, Marchand C, Neamati N. Retroviral integrase inhibitors year 2000: update and perspectives. Antiviral Res. 2000;47:139–148. doi: 10.1016/s0166-3542(00)00112-1. [DOI] [PubMed] [Google Scholar]
- 13.Pais GCG, Burke TR., Jr Novel aryl diketo-containing inhibitors of HIV-1 integrase. Drugs of the Future. 2002;27:1101–1111. [Google Scholar]
- 14.Dayam R, Deng J, Neamati N. HIV-1 integrase inhibitors: 2003–2004 update. Med Res Rev. 2006;26:271–309. doi: 10.1002/med.20054. [DOI] [PubMed] [Google Scholar]
- 15.Deng J, Dayam R, Al-Mawsawi LQ, Neamati N. Design of second generation HIV-1 integrase inhibitors. Curr Pharm Des. 2007;13:129–141. doi: 10.2174/138161207779313687. [DOI] [PubMed] [Google Scholar]
- 16.Busschots K, De Rijck J, Christ F, Debyser Z. In search of small molecules blocking interactions between HIV proteins and intracellular cofactors. Mol Biosyst. 2009;5:21–31. doi: 10.1039/b810306b. [DOI] [PubMed] [Google Scholar]
- 17.Grobler JA, Stillmock KA, Hazuda DJ. Scintillation proximity assays for mechanistic and pharmacological analyses of HIV-1 integration. Methods. 2009;47:249–253. doi: 10.1016/j.ymeth.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 18.Nair V, Chi G. HIV integrase inhibitors as therapeutic agents in AIDS. Rev Med Virol. 2007;17:277–295. doi: 10.1002/rmv.539. [DOI] [PubMed] [Google Scholar]
- 19.Savarino A. A historical sketch of the discovery and development of HIV-1 integrase inhibitors. Expert Opin Investig Drugs. 2006;15:1507–1522. doi: 10.1517/13543784.15.12.1507. [DOI] [PubMed] [Google Scholar]
- 20.Pace P, Rowley M. Integrase inhibitors for the treatment of HIV infection. Curr Opin Drug Discov Devel. 2008;11:471–449. [PubMed] [Google Scholar]
- 21.Al-Mawsawi LQ, Al-Safi RI, Neamati N. Clinical progress of HIV-1 integrase inhibitors. Expert Opin Emerging Drugs. 2008;13:213–225. doi: 10.1517/14728214.13.2.213. [DOI] [PubMed] [Google Scholar]
- 22.Vandegraaff N, Engelman A. Molecular mechanisms of HIV integration and therapeutic intervention. Expert Rev Mol Med. 2007;9:1–19. doi: 10.1017/S1462399407000257. [DOI] [PubMed] [Google Scholar]
- 23.Jaskolski M, Alexandratos JN, Bujacz G, Wlodawer A. Piecing together the structure of retroviral integrase, an important target in AIDS therapy. Febs Journal. 2009;276:2926–2946. doi: 10.1111/j.1742-4658.2009.07009.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng R, Jenkins TM, Craigie R. Zinc folds the N-terminal domain of HIV-1 integrase, promotes multimerization, and enhances catalytic activity. Proc. Natl. Acad. Sci. USA. 1996;93:13659–13664. doi: 10.1073/pnas.93.24.13659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lodi PJ, Ernst JA, Kuszewski J, Hickman AB, Engelman A, Craigie R, Clore GM, Gronenborn AM. Solution structure of the DNA binding domain of HIV-1 integrase. Biochemistry. 1995;34:9826–9833. doi: 10.1021/bi00031a002. [DOI] [PubMed] [Google Scholar]
- 26.Bujacz G, Alexandratos J, Wlodawer A, Merkel G, Andrake M, Katz RA, Skalka AM. Binding of different divalent cations to the active site of avian sarcoma virus integrase and their effects on enzymatic activity. J. Biol. Chem. 1997;272:18161–18168. doi: 10.1074/jbc.272.29.18161. [DOI] [PubMed] [Google Scholar]
- 27.Johnson AA, Santos W, Pais GC, Marchand C, Amin R, Burke TR, Jr, Verdine G, Pommier Y. Integration Requires a Specific Interaction of the Donor DNA Terminal 5'-Cytosine with Glutamine 148 of the HIV-1 Integrase Flexible Loop. J Biol Chem. 2006;281:461–467. doi: 10.1074/jbc.M511348200. [DOI] [PubMed] [Google Scholar]
- 28.Marinello J, Marchand C, Mott BT, Bain A, Thomas CJ, Pommier Y. Comparison of raltegravir and elvitegravir on HIV-1 integrase catalytic reactions and on a series of drug-resistant integrase mutants. Biochemistry. 2008;47:9345–9354. doi: 10.1021/bi800791q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Esposito D, Craigie R. Sequence selectivity of viral end DNA binding by HIV-1 integrase reveals critical regions for protein-DNA interactions. EMBO J. 1998;17:5832–5843. doi: 10.1093/emboj/17.19.5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jenkins TM, Esposito D, Engelman A, Craigie R. Critical contacts between HIV-1 integrase and viral DNA identified by structure-based analysis and photo-crosslinking. EMBO J. 1997;16:6849–6859. doi: 10.1093/emboj/16.22.6849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heuer TS, Brown PO. Mapping features of HIV-1 integrase near selected sites on viral and target DNA molecules in an active enzyme-DNA complex by photo-cross-linking. Biochemistry. 1997;36:10655–10665. doi: 10.1021/bi970782h. [DOI] [PubMed] [Google Scholar]
- 32.Heuer TS, Brown PO. Photo-Cross-Linking Studies Suggest a Model for the Architecture of an Active Human Immunodeficiency Virus Type 1 Integrase-DNA Complex. Biochemistry. 1998;37:6667–6678. doi: 10.1021/bi972949c. [DOI] [PubMed] [Google Scholar]
- 33.Cherepanov P, Maertens G, Proost P, Devreese B, Van Beeumen J, Engelborghs Y, De Clercq E, Debyser Z. HIV-1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells. J Biol Chem. 2002;278:372–381. doi: 10.1074/jbc.M209278200. [DOI] [PubMed] [Google Scholar]
- 34.Li M, Mizuuchi M, Burke TR, Jr, Craigie R. Retroviral DNA integration: reaction pathway and critical intermediates. EMBO J. 2006;25:1295–1304. doi: 10.1038/sj.emboj.7601005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang JY, Ling H, Yang W, Craigie R. Structure of a two-domain fragment of HIV-1 integrase: implications for domain organization in the intact protein. Embo J. 2001;20:7333–7343. doi: 10.1093/emboj/20.24.7333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ellison V, Brown PO. A stable complex between integrase and viral DNA ends mediates human immunodeficiency virus integration in vitro. Proc. Natl. Acad. Sci. U.S.A. 1994;91:7316–7320. doi: 10.1073/pnas.91.15.7316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maxfield LF, Fraize CD, Coffin JM. Relationship between retroviral DNA-integration-site selection and host cell transcription. Proc Natl Acad Sci U S A. 2005;102:1436–1441. doi: 10.1073/pnas.0409204102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dyda F, Hickman AB, Jenkins TM, Engelman A, Craigie R, Davies DR. Crystal structure of the catalytic domain of HIV-1 integrase: similarity of other polynucleotide transferases. Science. 1994;266:1981–1986. doi: 10.1126/science.7801124. [DOI] [PubMed] [Google Scholar]
- 39.Goldgur Y, Dyda F, Hickman AB, Jenkins TM, Craigie R, Davies DR. Three new structures of the core domain of HIV-1 integrase: An active site that binds magnesium. Proc Natl Acad Sci U S A. 1998;95:9150–4. doi: 10.1073/pnas.95.16.9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen JC, Krucinski J, Miercke LJ, Finer-Moore JS, Tang AH, Leavitt AD, Stroud RM. Crystal structure of the HIV-1 integrase catalytic core and C-terminal domains: a model for viral DNA binding. Proc Natl Acad Sci U S A. 2000;97:8233–8238. doi: 10.1073/pnas.150220297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goldgur Y, Craigie R, Cohen GH, Fujiwara T, Yoshinaga T, Fujishita T, Sugimoto H, Endo T, Murai H, Davies DR. Structure of the HIV-1 integrase catalytic domain complexed with an inhibitor: A platform for antiviral drug design. Proc Natl Acad Sci U S A. 1999;96:13040–13043. doi: 10.1073/pnas.96.23.13040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lubkowski J, Yang F, Alexandratos J, Wlodawer A, Zhao H, Burke TR, Neamati N, Pommier Y, Merkel G, Skalka AM. Structure of the catalytic domain of avian sarcoma virus integrase with a bound HIV-1 integrase-targeted inhibitor. Proc. Natl. Acad. Sci. U.S.A. 1998;95:4831–4836. doi: 10.1073/pnas.95.9.4831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Drake R, Neamati N, Hong H, Pilon AA, Sunthankar P, Hume SD, Milne GWA, Pommier Y. Identification of a nucleotide binding site in HIV-1 integrase. Proc. Natl. Acad. Sci. USA. 1998;95:4170–4175. doi: 10.1073/pnas.95.8.4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Williams KL, Zhang Y, Shkriabai N, Karki RG, Nicklaus MC, Kotrikadze N, Hess S, Le Grice SF, Craigie R, Pathak VK, Kvaratskhelia M. Mass spectrometric analysis of the HIV-1 integrase-pyridoxal 5'-phosphate complex reveals a new binding site for a nucleotide inhibitor. J Biol Chem. 2005;280:7949–7955. doi: 10.1074/jbc.M413579200. [DOI] [PubMed] [Google Scholar]
- 45.Al-Mawsawi LQ, Fikkert V, Dayam R, Witvrouw M, Burke TR, Jr, Borchers CH, Neamati N. Discovery of a small-molecule HIV-1 integrase inhibitor-binding site. Proc Natl Acad Sci U S A. 2006;103:10080–10085. doi: 10.1073/pnas.0511254103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Neamati N. Structure-based HIV-1 integrase inhibitor design: a future perspective. Expert Opin Investig Drugs. 2001;10:281–296. doi: 10.1517/13543784.10.2.281. [DOI] [PubMed] [Google Scholar]
- 47.Pommier Y, Cherfils J. Interfacial protein inhibition: a nature's paradigm for drug discovery. Trends Pharmacol. Sci. 2005;28:136–145. doi: 10.1016/j.tips.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 48.Pommier Y, Marchand C. Interfacial inhibitors of protein-nucleic acid interactions. Curr. Med. Chem. Anti-Canc. Agents. 2005;5:421–429. doi: 10.2174/1568011054222337. [DOI] [PubMed] [Google Scholar]
- 49.Marchand C, Antony S, Kohn KW, Cushman M, Ioanoviciu A, Staker BL, Burgin AB, Stewart L, Pommier Y. A novel norindenoisoquinoline structure reveals a common interfacial inhibitor paradigm for ternary trapping of topoisomerase I-DNA covalent complexes. Mol Cancer Ther. 2006;5:287–295. doi: 10.1158/1535-7163.MCT-05-0456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Engelman A, Mizuuchi K, Craigie R. HIV-1 DNA integration: mechanism of viral DNA cleavage and DNA strand transfer. Cell. 1991;67:1211–1221. doi: 10.1016/0092-8674(91)90297-c. [DOI] [PubMed] [Google Scholar]
- 51.Vink C, Oude Groeneger AM, Plasterk RH. Identification of the catalytic and DNA-binding region of the human immunodeficiency virus type I integrase protein. Nucleic Acids Res. 1993;21:1419–1425. doi: 10.1093/nar/21.6.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yoder KE, Bushman FD. Repair of gaps in retroviral DNA integration intermediates. J Virol. 2000;74:11191–11200. doi: 10.1128/jvi.74.23.11191-11200.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marchand C, Zhang X, Pais GC, Cowansage K, Neamati N, Burke TR, Jr, Pommier Y. Structural determinants for HIV-1 integrase inhibition by beta-diketo acids. J Biol Chem. 2002;277:12596–12603. doi: 10.1074/jbc.M110758200. [DOI] [PubMed] [Google Scholar]
- 54.Hazuda DJ, Felock P, Witmer M, Wolfe A, Stillmock K, Grobler JA, Espesath A, Gabryelski L, Schlelf W, Blau C, Miller MD. Inhibitors of strand transfer that prevent integration and inhibit HIV-1 replication in cells. Science. 2000;287:646–650. doi: 10.1126/science.287.5453.646. [DOI] [PubMed] [Google Scholar]
- 55.Marchand C, Neamati N, Pommier Y. In vitro human immunodeficiency virus type 1 integrase assays. Methods Enzymol. 2001;340:624–633. doi: 10.1016/s0076-6879(01)40446-0. [DOI] [PubMed] [Google Scholar]
- 56.Debyser Z, Cherepanov P, Pluymers W, De Clercq E. Assays for the evaluation of HIV-1 integrase inhibitors. Methods Mol Biol. 2001;160:139–155. doi: 10.1385/1-59259-233-3:139. [DOI] [PubMed] [Google Scholar]
- 57.Sinha S, Pursley MH, Grandgenett DP. Efficient concerted integration by recombinant human immunodeficiency virus type 1 integrase without cellular or viral cofactors. J Virol. 2002;76:3105–3113. doi: 10.1128/JVI.76.7.3105-3113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Iwamoto M, Wenning LA, Petry AS, Laethem M, De Smet M, Kost JT, Merschman SA, Strohmaier KM, Ramael S, Lasseter KC, Stone JA, Gottesdiener KM, Wagner JA. Safety, tolerability, and pharmacokinetics of raltegravir after single and multiple doses in healthy subjects. Clin Pharmacol Ther. 2008;83:293–299. doi: 10.1038/sj.clpt.6100281. [DOI] [PubMed] [Google Scholar]
- 59.Markowitz M, Nguyen BY, Gotuzzo E, Mendo F, Ratanasuwan W, Kovacs C, Prada G, Morales-Ramirez JO, Crumpacker CS, Isaacs RD, Gilde LR, Wan H, Miller MD, Wenning LA, Teppler H. Rapid and durable antiretroviral effect of the HIV-1 Integrase inhibitor raltegravir as part of combination therapy in treatment-naive patients with HIV-1 infection: results of a 48-week controlled study. J Acquir Immune Defic Syndr. 2007;46:125–133. doi: 10.1097/QAI.0b013e318157131c. [DOI] [PubMed] [Google Scholar]
- 60.Sato M, Motomura T, Aramaki H, Matsuda T, Yamashita M, Ito Y, Kawakami H, Matsuzaki Y, Watanabe W, Yamataka K, Ikeda S, Kodama E, Matsuoka M, Shinkai H. Novel HIV-1 integrase inhibitors derived from quinolone antibiotics. J Med Chem. 2006;49:1506–1508. doi: 10.1021/jm0600139. [DOI] [PubMed] [Google Scholar]
- 61.Kobayashi M, Nakahara K, Seki T, Miki S, Kawauchi S, Suyama A, Wakasa-Morimoto C, Kodama M, Endoh T, Oosugi E, Matsushita Y, Murai H, Fujishita T, Yoshinaga T, Garvey E, Foster S, Underwood M, Johns B, Sato A, Fujiwara T. Selection of diverse and clinically relevant integrase inhibitor-resistant human immunodeficiency virus type 1 mutants. Antiviral Res. 2008;80:213–222. doi: 10.1016/j.antiviral.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 62.Shimura K, Kodama E, Sakagami Y, Matsuzaki Y, Watanabe W, Yamataka K, Watanabe Y, Ohata Y, Doi S, Sato M, Kano M, Ikeda S, Matsuoka M. Broad antiretroviral activity and resistance profile of the novel human immunodeficiency virus integrase inhibitor elvitegravir (JTK-303/GS-9137) J Virol. 2008;82:764–774. doi: 10.1128/JVI.01534-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ramanathan S, Shen G, Hinkle J, Enejosa J, Kearney BP. Pharmacokinetics of coadmistered ritonavir-boosted elvitegravir and zidovirine, didanosine, stavudine or abacavir. J. Acquir. Immune Defic. Syndr. 2007;46:160–166. doi: 10.1097/QAI.0b013e318151fd9a. [DOI] [PubMed] [Google Scholar]
- 64.Witvrouw M, Fikkert V, Van Maele B, Pannecouque C, Neamati N, Burke TR, Pais G, De Clercq E, Debyser Z. Antiviral Resistance to Diketo Acids is Associated with Mutations T66I, L74M and S230R in the HIV Integrase Gene presented at the 15th International Conference on Antiviral Research; Antiviral Res; Prague, Czech Republic. 2002. p. A18. abstract 45. [Google Scholar]
- 65.Hazuda DJ, Young SD, Guare JP, Anthony NJ, Gomez RP, Wai JS, Vacca JP, Handt L, Motzel SL, Klein HJ, Dornadula G, Danovich RM, Witmer MV, Wilson KA, Tussey L, Schleif WA, Gabryelski LS, Jin L, Miller MD, Casimiro DR, Emini EA, Shiver JW. Integrase inhibitors and cellular immunity suppress retroviral replication in rhesus macaques. Science. 2004;305:528–532. doi: 10.1126/science.1098632. [DOI] [PubMed] [Google Scholar]
- 66.Marchand C, Johnson AA, Karki RG, Pais GC, Zhang X, Cowansage K, Patel TA, Nicklaus MC, Burke TR, Jr, Pommier Y. Metal-Dependent Inhibition of HIV-1 Integrase by {beta}-Diketo Acids and Resistance of the Soluble Double-Mutant (F185K/C280S) Mol Pharmacol. 2003;64:600–609. doi: 10.1124/mol.64.3.600. [DOI] [PubMed] [Google Scholar]
- 67.Johnson AA, Marchand C, Patil SS, Costi R, Di Santo R, Burke TR, Jr, Pommier Y. Probing HIV-1 integrase inhibitor binding sites with position-specific integrase-DNA cross-linking assays. Mol Pharmacol. 2007;71:893–901. doi: 10.1124/mol.106.030817. [DOI] [PubMed] [Google Scholar]
- 68.Espeseth AS, Felock P, Wolfe A, Witmer M, Grobler J, Anthony N, Egbertson M, Melamed JY, Young S, Hamill T, Cole JL, Hazuda DJ. HIV-1 integrase inhibitors that compete with the target DNA substrate define a unique strand transfer conformation for integrase. Proc Natl Acad Sci U S A. 2000;97:11244–11249. doi: 10.1073/pnas.200139397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pais GCG, Zhang X, Marchand C, Neamati N, Cowansage K, Svarovskaia ES, Pathak VK, Tang Y, Nicklaus MC, Pommier Y, Burke TR., Jr Structure Activity of 3-aryl-1,3-diketo-Containing Compounds as HIV-1 Integrase Inhibitors. J Med Chem. 2002 doi: 10.1021/jm020037p. In Press. [DOI] [PubMed] [Google Scholar]
- 70.Grobler JA, Stillmock K, Hu B, Witmer M, Felock P, Espeseth AS, Wolfe A, Egbertson M, Bourgeois M, Melamed J, Wai JS, Young S, Vacca J, Hazuda DJ. Diketo acid inhibitor mechanism and HIV-1 integrase: implications for metal binding in the active site of phosphotransferase enzymes. Proc Natl Acad Sci U S A. 2002;99:6661–6666. doi: 10.1073/pnas.092056199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Marchand C, Johnson AA, Karki RG, Pais GCG, Zhang X, Cowansage K, Patel TA, Nicklaus MC, Burke TR, Jr, Pommier Y. Metal-dependent inhibition of HIV-1 integrase by beta-diketo acids and resistance of the soluble double-mutant (F185K/C280S) Mol Pharmacol. 2003;64:600–609. doi: 10.1124/mol.64.3.600. [DOI] [PubMed] [Google Scholar]
- 72.Yoshinaga TSA, Fujishita T, Fujiwara T. In Vitro Activity of a New HIV-1 Integrase Inhibitor in Clinical Development. 9th Conference on Retroviruses and Opportunistic Infections; Seattle, USA. 2002. [Google Scholar]
- 73.Hazuda DJ, Anthony NJ, Gomez RP, Jolly SM, Wai JS, Zhuang L, Fisher TE, Embrey M, Guare JP, Jr, Egbertson MS, Vacca JP, Huff JR, Felock PJ, Witmer MV, Stillmock KA, Danovich R, Grobler J, Miller MD, Espeseth AS, Jin L, Chen IW, Lin JH, Kassahun K, Ellis JD, Wong BK, Xu W, Pearson PG, Schleif WA, Cortese R, Emini E, Summa V, Holloway MK, Young SD. A naphthyridine carboxamide provides evidence for discordant resistance between mechanistically identical inhibitors of HIV-1 integrase. Proc Natl Acad Sci U S A. 2004;101:11233–11238. doi: 10.1073/pnas.0402357101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Malet I, Delelis O, Valantin MA, Montes B, Soulie C, Wirden M, Tchertanov L, Peytavin G, Reynes J, Mouscadet JF, Katlama C, Calvez V, Marcelin AG. Mutations associated with failure of raltegravir treatment affect integrase sensitivity to the inhibitor in vitro. Antimicrob Agents Chemother. 2008;52:1351–1358. doi: 10.1128/AAC.01228-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Delelis O, Malet I, Na L, Tchertanov L, Calvez V, Marcelin AG, Subra F, Deprez E, Mouscadet JF. The G140S mutation in HIV integrases from raltegravir-resistant patients rescues catalytic defect due to the resistance Q148H mutation. Nucleic Acids Res. 2009;37:1193–1201. doi: 10.1093/nar/gkn1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sichtig N, Sierra S, Kaiser R, Daumer M, Reuter S, Schulter E, Altmann A, Fatkenheuer G, Dittmer U, Pfister H, Esser S. Evolution of raltegravir resistance during therapy. J Antimicrob Chemother. 2009;64:25–32. doi: 10.1093/jac/dkp153. [DOI] [PubMed] [Google Scholar]
- 77.Malet I, Delelis O, Soulie C, Wirden M, Tchertanov L, Mottaz P, Peytavin G, Katlama C, Mouscadet JF, Calvez V, Marcelin AG. Quasispecies variant dynamics during emergence of resistance to raltegravir in HIV-1-infected patients. J Antimicrob Chemother. 2009;63:795–804. doi: 10.1093/jac/dkp014. [DOI] [PubMed] [Google Scholar]
- 78.Jones GS, Yu F, Zeynalzadegan A, Hesselgesser J, Chen X, Chen J, Jin H, Kim CU, Wright M, Geleziunas R, Tsiang M. Preclinical evaluation of GS-9160, a novel inhibitor of human immunodeficiency virus type 1 integrase. Antimicrob Agents Chemother. 2009;53:1194–1203. doi: 10.1128/AAC.00984-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dicker IB, Samanta HK, Li Z, Hong Y, Tian Y, Banville J, Remillard RR, Walker MA, Langley DR, Krystal M. Changes to the HIV long terminal repeat and to HIV integrase differentially impact HIV integrase assembly, activity, and the binding of strand transfer inhibitors. J Biol Chem. 2007;282:31186–31196. doi: 10.1074/jbc.M704935200. [DOI] [PubMed] [Google Scholar]
- 80.Di Santo R, Costi R, Roux A, Miele G, Crucitti GC, Iacovo A, Rosi F, Lavecchia A, Marinelli L, Di Giovanni C, Novellino E, Palmisano L, Andreotti M, Amici R, Galluzzo CM, Nencioni L, Palamara AT, Pommier Y, Marchand C. Novel quinolinonyl diketo acid derivatives as HIV-1 integrase inhibitors: design, synthesis, and biological activities. J Med Chem. 2008;51:4744–4750. doi: 10.1021/jm8001422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhao XZ, Maddali K, Marchand C, Pommier Y, Burke TR., Jr Diketoacid-genre HIV-1 integrase inhibitors containing enantiomeric arylamide functionality. Bioorg Med Chem. 2009 doi: 10.1016/j.bmc.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhao XZ, Semenova EA, Vu BC, Maddali K, Marchand C, Hughes SH, Pommier Y, Burke TR., Jr 2,3-Dihydro-6,7-dihydroxy-1H-isoindol-1-one-Based HIV-1 Integrase Inhibitors. J Med Chem. 2008;51:251–259. doi: 10.1021/jm070715d. [DOI] [PubMed] [Google Scholar]
- 83.Zhao XZ, Maddali K, Vu BC, Marchand C, Hughes SH, Pommier Y, Burke TR., Jr Examination of halogen substituent effects on HIV-1 integrase inhibitors derived from 2,3-dihydro-6,7-dihydroxy-1H-isoindol-1-ones and 4,5-dihydroxy-1H-isoindole-1,3(2H)-diones. Bioorg Med Chem Lett. 2009;19:2714–2717. doi: 10.1016/j.bmcl.2009.03.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fesen MR, Kohn KW, Leteurtre F, Pommier Y. Inhibitors of human immunodeficiency virus integrase. Proc. Natl. Acad. Sci. U.S.A. 1993;90:2399–2403. doi: 10.1073/pnas.90.6.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fesen MR, Pommier Y, Leteurtre F, Hiroguchi S, Yung J, Kohn KW. Inhibition of HIV-1 integrase by flavones, caffeic acid phenethyl ester (CAPE) and related compounds. Biochem. Pharmacol. 1994;48:595–608. doi: 10.1016/0006-2952(94)90291-7. [DOI] [PubMed] [Google Scholar]
- 86.Reinke RA, King PJ, Victoria JG, McDougall BR, Ma G, Mao Y, Reinecke MG, Robinson WE., Jr Dicaffeoyltartaric Acid Analogues Inhibit Human Immunodeficiency Virus Type 1 (HIV-1) Integrase and HIV-1 Replication at Nontoxic Concentrations. J Med Chem. 2002;45:3669–3683. doi: 10.1021/jm010359d. [DOI] [PubMed] [Google Scholar]
- 87.Reinke RA, Lee DJ, McDougall BR, King PJ, Victoria J, Mao Y, Lei X, Reinecke MG, Robinson WE., Jr L-chicoric acid inhibits human immunodeficiency virus type 1 integration in vivo and is a noncompetitive but reversible inhibitor of HIV-1 integrase in vitro. Virology. 2004;326:203–219. doi: 10.1016/j.virol.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 88.Pluymers W, Pannecouque C, Fikkert V, Neamati N, Marchand C, Burke TR, Jr, Pommier Y, Schols D, De Clercq E, Debyser Z, Witvrouw M. Viral entry as the primary target for the anti-HIV activity of chicoric acid and its tetra-acetyl esters. Mol. Pharmacol. 2000;38:641–648. doi: 10.1124/mol.58.3.641. [DOI] [PubMed] [Google Scholar]
- 89.Queffelec C, Bailly F, Mbemba G, Mouscadet JF, Debyser Z, Witvrouw M, Cotelle P. The total synthesis of fukiic acid, an HIV-1 integrase inhibitor. Eur J Med Chem. 2008;43:2268–2271. doi: 10.1016/j.ejmech.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 90.Lee JY, Yoon KJ, Lee YS. Catechol-substituted L-chicoric acid analogues as HIV integrase inhibitors. Bioorg Med Chem Lett. 2003;13:4331–4334. doi: 10.1016/j.bmcl.2003.09.046. [DOI] [PubMed] [Google Scholar]
- 91.Lee SU, Shin CG, Lee CK, Lee YS. Caffeoylglycolic and caffeoylamino acid derivatives, halfmers of L-chicoric acid, as new HIV-1 integrase inhibitors. Eur J Med Chem. 2007;42:1309–1315. doi: 10.1016/j.ejmech.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 92.Sechi M, Angotzi G, Dallocchio R, Dessi A, Carta F, Sannia L, Mariani A, Fiori S, Sanchez T, Movsessian L, Plasencia C, Neamati N. Design and synthesis of novel dihydroxyindole-2-carboxylic acids as HIV-1 integrase inhibitors. Antivir Chem Chemother. 2004;15:67–81. doi: 10.1177/095632020401500203. [DOI] [PubMed] [Google Scholar]
- 93.Maurin C, Bailly F, Mbemba G, Mouscadet JF, Cotelle P. Design, synthesis, and anti-integrase activity of catechol-DKA hybrids. Bioorg Med Chem. 2006;14:2978–2984. doi: 10.1016/j.bmc.2005.12.039. [DOI] [PubMed] [Google Scholar]
- 94.Mekouar K, Mouscadet JF, Desmaele D, Subra F, Leh H, Savoure D, Auclair C, d'Angelo J. Styrylquinoline derivatives: a new class of potent HIV-1 integrase inhibitors that block HIV-1 replication in CEM cells. J Med Chem. 1998;41:2846–2857. doi: 10.1021/jm980043e. [DOI] [PubMed] [Google Scholar]
- 95.Zouhiri F, Mouscadet JF, Mekouar K, Desmaele D, Savoure D, Leh H, Subra F, Le Bret M, Auclair C, d'Angelo J. Structure-Activity Relationships and Binding Mode of Styrylquinolines as Potent Inhibitors of HIV-1 Integrase and Replication of HIV-1 in Cell Culture. J Med Chem. 2000;43:1533–1540. doi: 10.1021/jm990467o. [DOI] [PubMed] [Google Scholar]
- 96.Mousnier A, Leh H, Mouscadet JF, Dargemont C. Nuclear import of HIV-1 integrase is inhibited in vitro by styrylquinoline derivatives. Mol Pharmacol. 2004;66:783–788. doi: 10.1124/mol.104.001735. [DOI] [PubMed] [Google Scholar]
- 97.Lee JY, Park JH, Lee SJ, Park H, Lee YS. Styrylquinazoline derivatives as HIV-1 integrase inhibitors. Arch Pharm (Weinheim) 2002;335:277–282. doi: 10.1002/1521-4184(200208)335:6<277::AID-ARDP277>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 98.Bonnenfant S, Thomas CM, Vita C, Subra F, Deprez E, Zouhiri F, Desmaele D, D'Angelo J, Mouscadet JF, Leh H. Styrylquinolines, integrase inhibitors acting prior to integration: a new mechanism of action for anti-integrase agents. J Virol. 2004;78:5728–5736. doi: 10.1128/JVI.78.11.5728-5736.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dubois M, Bailly F, Mbemba G, Mouscadet JF, Debyser Z, Witvrouw M, Cotelle P. Reaction of rosmarinic acid with nitrite ions in acidic conditions: discovery of nitro- and dinitrorosmarinic acids as new anti-HIV-1 agents. J Med Chem. 2008;51:2575–2579. doi: 10.1021/jm7011134. [DOI] [PubMed] [Google Scholar]
- 100.Bailly F, Queffelec C, Mbemba G, Mouscadet JF, Cotelle P. Synthesis and HIV-1 integrase inhibitory activities of caffeic acid dimers derived from Salvia officinalis. Bioorg Med Chem Lett. 2005;15:5053–5056. doi: 10.1016/j.bmcl.2005.07.091. [DOI] [PubMed] [Google Scholar]
- 101.Neamati N, Hong H, Owen JM, Sunder S, Winslow HE, Christensen JL, Zhao H, Burke TR, Milne GWA, Pommier Y. Salicylhydrazine-containing inhibitors of HIV-1 integrase: implication for a selective chelation in the integrase active site. J. Med. Chem. 1998;41:3202–3209. doi: 10.1021/jm9801760. [DOI] [PubMed] [Google Scholar]
- 102.Neamati N, Lin Z, Karki RG, Orr A, Cowansage K, Strumberg D, Pais GC, Voigt JH, Nicklaus MC, Winslow HE, Zhao H, Turpin JA, Yi J, Skalka AM, Burke TR, Jr, Pommier Y. Metal-dependent inhibition of HIV-1 integrase. J Med Chem. 2002;45:5661–5670. doi: 10.1021/jm0201417. [DOI] [PubMed] [Google Scholar]
- 103.Pannecouque C, Pluymers W, Van Maele B, Tetz V, Cherepanov P, De Clercq E, Witvrouw M, Debyser Z. New class of HIV integrase inhibitors that block viral replication in cell culture. Curr Biol. 2002;12:1169–1177. doi: 10.1016/s0960-9822(02)00952-1. [DOI] [PubMed] [Google Scholar]
- 104.Shaw-Reid CA, Munshi V, Graham P, Wolfe A, Witmer M, Danzeisen R, Olsen DB, Carroll SS, Embrey M, Wai JS, Miller MD, Cole JL, Hazuda DJ. Inhibition of HIV-1 ribonuclease H by a novel diketo acid, 4-[5-(benzoylamino)thien-2-yl]-2,4-dioxobutanoic acid. J Biol Chem. 2003;278:2777–2780. doi: 10.1074/jbc.C200621200. [DOI] [PubMed] [Google Scholar]
- 105.Rice PA, Baker TA. Comparative architecture of transposase and integrase complexes. Nat Struct Biol. 2001;8:302–307. doi: 10.1038/86166. [DOI] [PubMed] [Google Scholar]
- 106.Budihas SR, Gorshkova I, Gaidamakov S, Wamiru A, Bona MK, Parniak MA, Crouch RJ, McMahon JB, Beutler JA, Le Grice SF. Selective inhibition of HIV-1 reverse transcriptase-associated ribonuclease H activity by hydroxylated tropolones. Nucleic Acids Res. 2005;33:1249–1256. doi: 10.1093/nar/gki268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Didierjean J, Isel C, Querre F, Mouscadet JF, Aubertin AM, Valnot JY, Piettre SR, Marquet R. Inhibition of human immunodeficiency virus type 1 reverse transcriptase, RNase H, and integrase activities by hydroxytropolones. Antimicrob Agents Chemother. 2005;49:4884–4894. doi: 10.1128/AAC.49.12.4884-4894.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Semenova EA, Johnson AA, Marchand C, Davis DA, Yarchoan R, Pommier Y. Preferential inhibition of the magnesium-dependent strand transfer reaction of HIV-1 integrase by alpha-hydroxytropolones. Mol Pharmacol. 2006;69:1454–1460. doi: 10.1124/mol.105.020321. [DOI] [PubMed] [Google Scholar]
- 109.Billamboz M, Bailly F, Barreca ML, De Luca L, Mouscadet JF, Calmels C, Andreola ML, Witvrouw M, Christ F, Debyser Z, Cotelle P. Design, synthesis, and biological evaluation of a series of 2-hydroxyisoquinoline-1,3(2H,4H)-diones as dual inhibitors of human immunodeficiency virus type 1 integrase and the reverse transcriptase RNase H domain. J Med Chem. 2008;51:7717–7730. doi: 10.1021/jm8007085. [DOI] [PubMed] [Google Scholar]
- 110.Wang Z, Bennett EM, Wilson DJ, Salomon C, Vince R. Rationally designed dual inhibitors of HIV reverse transcriptase and integrase. J Med Chem. 2007;50:3416–3419. doi: 10.1021/jm070512p. [DOI] [PubMed] [Google Scholar]
- 111.Wang Z, Vince R. Design and synthesis of dual inhibitors of HIV reverse transcriptase and integrase: introducing a diketoacid functionality into delavirdine. Bioorg Med Chem. 2008;16:3587–3595. doi: 10.1016/j.bmc.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 112.Wang Z, Vince R. Synthesis of pyrimidine and quinolone conjugates as a scaffold for dual inhibitors of HIV reverse transcriptase and integrase. Bioorg Med Chem Lett. 2008;18:1293–1296. doi: 10.1016/j.bmcl.2008.01.025. [DOI] [PubMed] [Google Scholar]
- 113.Puras Lutzke RA, Eppens NA, Weber PA, Houghten RA, Plasterk RH. Identification of a hexapeptide inhibitor of the human immunodeficiency virus integrase protein by using a combinatorial chemical library. Proc Natl Acad Sci U S A. 1995;92:11456–11460. doi: 10.1073/pnas.92.25.11456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Roller PP, Long Y-Q, Lung F-DT, Pommier Y, Neamati N. Tryptophan-rich integrase inhibitory peptides and peptidomimetics. In: Martinez JaF, J-A, editors. Peptides 2000. Paris: EDK; 2001. pp. 725–726. [Google Scholar]
- 115.Krajewski K, Long YQ, Marchand C, Pommier Y, Roller PP. Design and synthesis of dimeric HIV-1 integrase inhibitory peptides. Bioorg Med Chem Lett. 2003;13:3203–3205. doi: 10.1016/s0960-894x(03)00679-6. [DOI] [PubMed] [Google Scholar]
- 116.de Soultrait VRCA, Parissi V, Morellet N, Ventura M, Lenoir C, Litvak S, Fournier M, Roques BP. A Novel Short Peptide is a Specific Inhibitor of the Human Immunodeficiency Virus Type 1 Integrase. J Mol Biol. 2002;318:45–58. doi: 10.1016/S0022-2836(02)00033-5. [DOI] [PubMed] [Google Scholar]
- 117.Singh SB, Herath K, Guan Z, Zink DL, Dombrowski AW, Polishook JD, Silverman KC, Lingham RB, Felock PJ, Hazuda DJ. Integramides A and B, two novel non-ribosomal linear peptides containing nine C(alpha)-methyl amino acids produced by fungal fermentations that are inhibitors of HIV-1 integrase. Org Lett. 2002;4:1431–1434. doi: 10.1021/ol025540a. [DOI] [PubMed] [Google Scholar]
- 118.De Zotti M, Formaggio F, Kaptein B, Broxterman QB, Felock PJ, Hazuda DJ, Singh SB, Bruckner H, Toniolo C. Complete absolute configuration of integramide A, a natural, 16-mer peptide inhibitor of HIV-1 integrase, elucidated by total synthesis. Chembiochem. 2009;10:87–90. doi: 10.1002/cbic.200800443. [DOI] [PubMed] [Google Scholar]
- 119.Robinson WE, Jr, McDougall B, Tran D, Selsted ME. Anti-HIV-1 activity of indolicidin, an antimicrobial peptide from neutrophils. J Leukoc Biol. 1998;63:94–100. doi: 10.1002/jlb.63.1.94. [DOI] [PubMed] [Google Scholar]
- 120.Krajewski K, Marchand C, Long YQ, Pommier Y, Roller PP. Synthesis and HIV-1 integrase inhibitory activity of dimeric and tetrameric analogs of indolicidin. Bioorg Med Chem Lett. 2004;14:5595–5598. doi: 10.1016/j.bmcl.2004.08.061. [DOI] [PubMed] [Google Scholar]
- 121.Marchand C, Krajewski K, Lee HF, Antony S, Johnson AA, Amin R, Roller P, Kvaratskhelia M, Pommier Y. Covalent binding of the natural antimicrobial peptide indolicidin to DNA abasic sites. Nucleic Acids Res. 2006;34:5157–5165. doi: 10.1093/nar/gkl667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Desjobert C, de Soultrait VR, Faure A, Parissi V, Litvak S, Tarrago-Litvak L, Fournier M. Identification by phage display selection of a short peptide able to inhibit only the strand transfer reaction catalyzed by human immunodeficiency virus type 1 integrase. Biochemistry. 2004;43:13097–13105. doi: 10.1021/bi049385e. [DOI] [PubMed] [Google Scholar]
- 123.Thomas JA, Gorelick RJ. Nucleocapsid protein function in early infection processes. Virus Res. 2008;134:39–63. doi: 10.1016/j.virusres.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Maroun RG, Gayet S, Benleulmi MS, Porumb H, Zargarian L, Merad H, Leh H, Mouscadet JF, Troalen F, Fermandjian S. Peptide inhibitors of HIV-1 integrase dissociate the enzyme oligomers. Biochemistry. 2001;40:13840–13848. doi: 10.1021/bi011328n. [DOI] [PubMed] [Google Scholar]
- 125.Sourgen F, Maroun RG, Frere V, Bouziane M, Auclair C, Troalen F, Fermandjian S. A synthetic peptide from the human immunodeficiency virus type-1 integrase exhibits coiled-coil properties and interferes with the in vitro integration activity of the enzyme. Correlated biochemical and spectroscopic results. Eur. J. Biochem. 1996;240:765–773. doi: 10.1111/j.1432-1033.1996.0765h.x. [DOI] [PubMed] [Google Scholar]
- 126.Fermandjian S, Maroun RS, Amekraz B, Jankowski CK. Self-association of an amphipathic helix peptide inhibitor of HIV-1 integrase assessed by electro spray ionization mass spectrometry in trifluoroethanol/water mixtures. Rapid Commun Mass Spectrom. 2001;15:320–324. doi: 10.1002/rcm.231. [DOI] [PubMed] [Google Scholar]
- 127.Krebs D, Maroun RG, Sourgen F, Troalen F, Davoust D, Fermandjian S. Helical and coiled-coil-forming properties of peptides derived from and inhibiting human immunodeficiency virus type 1 integrase assessed by 1H- NMR--use of NH temperature coefficients to probe coiled-coil structures. Eur J Biochem. 1998;253:236–244. doi: 10.1046/j.1432-1327.1998.2530236.x. [DOI] [PubMed] [Google Scholar]
- 128.Maroun RG, Krebs D, Roshani M, Porumb H, Auclair C, Troalen F, Fermandjian S. Conformational aspects of HIV-1 integrase inhibition by a peptide derived from the enzyme central domain and by antibodies raised against this peptide. Eur J Biochem. 1999;260:145–155. doi: 10.1046/j.1432-1327.1999.00130.x. [DOI] [PubMed] [Google Scholar]
- 129.Kalpana GV, Goff SP. Protein-protein interactions of HIV-1 IN: INI-1 (IN Interactor-1), a novel human gene with sequence similarity to yeast transcription factor SNF5. J. Cell. Biochem. 1994;18B:27. [Google Scholar]
- 130.Kalpana GV, Marmon S, Wang W, Crabtree GR, Goff SP. Binding and stimulation of HIV-1 integrase by a human homolog of yeast transcription factor SNF5. Science. 1994;266:2002–2006. doi: 10.1126/science.7801128. [DOI] [PubMed] [Google Scholar]
- 131.Yung E, Sorin M, Pal A, Craig E, Morozov A, Delattre O, Kappes J, Ott D, Kalpana GV. Inhibition of HIV-1 virion production by a transdominant mutant of integrase interactor 1. Nat Med. 2001;7:920–926. doi: 10.1038/90959. [DOI] [PubMed] [Google Scholar]
- 132.Sorin M, Yung E, Wu X, Kalpana GV. HIV-1 replication in cell lines harboring INI1/hSNF5 mutations. Retrovirology. 2006;3:56. doi: 10.1186/1742-4690-3-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Maroun M, Delelis O, Coadou G, Bader T, Segeral E, Mbemba G, Petit C, Sonigo P, Rain JC, Mouscadet JF, Benarous R, Emiliani S. Inhibition of early steps of HIV-1 replication by SNF5/Ini1. J Biol Chem. 2006;281:22736–22743. doi: 10.1074/jbc.M604849200. [DOI] [PubMed] [Google Scholar]
- 134.Das S, Cano J, Kalpana GV. Multimerization and DNA binding properties of INI1/hSNF5 and its functional significance. J Biol Chem. 2009 doi: 10.1074/jbc.M808141200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Cherepanov P, Devroe E, Silver PA, Engelman A. Identification of an evolutionarily conserved domain in human lens epithelium-derived growth factor/transcriptional co-activator p75 (LEDGF/p75) that binds HIV-1 integrase. J Biol Chem. 2004;279:48883–48892. doi: 10.1074/jbc.M406307200. [DOI] [PubMed] [Google Scholar]
- 136.Vandekerckhove L, Christ F, Van Maele B, De Rijck J, Gijsbers R, Van den Haute C, Witvrouw M, Debyser Z. Transient and stable knockdown of the integrase cofactor LEDGF/p75 reveals its role in the replication cycle of human immunodeficiency virus. J Virol. 2006;80:1886–1896. doi: 10.1128/JVI.80.4.1886-1896.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.De Rijck J, Vandekerckhove L, Gijsbers R, Hombrouck A, Hendrix J, Vercammen J, Engelborghs Y, Christ F, Debyser Z. Overexpression of the lens epithelium-derived growth factor/p75 integrase binding domain inhibits human immunodeficiency virus replication. J Virol. 2006;80:11498–11509. doi: 10.1128/JVI.00801-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.De Luca L, Barreca ML, Ferro S, Christ F, Iraci N, Gitto R, Monforte AM, Debyser Z, Chimirri A. Pharmacophore-Based Discovery of Small-Molecule Inhibitors of Protein-Protein Interactions between HIV-1 Integrase and Cellular Cofactor LEDGF/p75. ChemMedChem. 2009 doi: 10.1002/cmdc.200900070. [DOI] [PubMed] [Google Scholar]
- 139.Zeinalipour-Loizidou E, Nicolaou C, Nicolaides A, Kostrikis LG. HIV-1 integrase: from biology to chemotherapeutics. Curr HIV Res. 2007;5:365–388. doi: 10.2174/157016207781023965. [DOI] [PubMed] [Google Scholar]
- 140.Oz Gleenberg I, Avidan O, Goldgur Y, Herschhorn A, Hizi A. Peptides derived from the reverse transcriptase of human immunodeficiency virus type 1 as novel inhibitors of the viral integrase. J Biol Chem. 2005;280:21987–21996. doi: 10.1074/jbc.M414679200. [DOI] [PubMed] [Google Scholar]
- 141.Rosenbluh J, Hayouka Z, Loya S, Levin A, Armon-Omer A, Britan E, Hizi A, Kotler M, Friedler A, Loyter A. Interaction between HIV-1 Rev and Integrase proteins: A basis for the development of anti-HIV peptides. J Biol Chem. 2007 doi: 10.1074/jbc.M609864200. [DOI] [PubMed] [Google Scholar]
- 142.Hayouka Z, Rosenbluh J, Levin A, Loya S, Lebendiker M, Veprintsev D, Kotler M, Hizi A, Loyter A, Friedler A. Inhibiting HIV-1 integrase by shifting its oligomerization equilibrium. Proc Natl Acad Sci U S A. 2007 doi: 10.1073/pnas.0700781104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Armon-Omer A, Levin A, Hayouka Z, Butz K, Hoppe-Seyler F, Loya S, Hizi A, Friedler A, Loyter A. Correlation between shiftide activity and HIV-1 integrase inhibition by a peptide selected from a combinatorial library. J Mol Biol. 2008;376:971–982. doi: 10.1016/j.jmb.2007.11.095. [DOI] [PubMed] [Google Scholar]
- 144.Bischerour J, Tauc P, Leh H, Rocquigny Hd H, Roques B, Mouscadet JF. The (52–96) C-terminal domain of Vpr stimulates HIV-1 IN-mediated homologous strand transfer of mini-viral DNA. Nucleic Acids Res. 2003;31:2694–2702. doi: 10.1093/nar/gkg364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gleenberg IO, Herschhorn A, Hizi A. Inhibition of the activities of reverse transcriptase and integrase of human immunodeficiency virus type-1 by peptides derived from the homologous viral protein R (Vpr) J Mol Biol. 2007;369:1230–1243. doi: 10.1016/j.jmb.2007.03.073. [DOI] [PubMed] [Google Scholar]
- 146.Bizub-Bender D, Kulkosky J, Skalka AM. Monoclonal antibodies against HIV type 1 integrase: clues to molecular structure. AIDS. Res. Hum. Retrov. 1994;10:1105–1115. doi: 10.1089/aid.1994.10.1105. [DOI] [PubMed] [Google Scholar]
- 147.Asante-Appiah E, Skalka AM. A metal-induced conformational change and activation of HIV-1 integrase. J. Biol. Chem. 1997;272:16196–16205. doi: 10.1074/jbc.272.26.16196. [DOI] [PubMed] [Google Scholar]
- 148.Yi J, Arthur JW, Dunbrack RL, Jr, Skalka AM. An inhibitory monoclonal antibody binds at the turn of the helix-turn- helix motif in the N-terminal domain of HIV-1 integrase. J Biol Chem. 2000;275:38739–38748. doi: 10.1074/jbc.M005499200. [DOI] [PubMed] [Google Scholar]
- 149.Yi J, Cheng H, Andrake MD, Dunbrack RL, Jr, Roder H, Skalka AM. Mapping the epitope of an inhibitory monoclonal antibody to the C- terminal DNA-binding domain of HIV-1 integrase. J Biol Chem. 2002;277:12164–12174. doi: 10.1074/jbc.M105072200. [DOI] [PubMed] [Google Scholar]
- 150.Ramcharan J, Colleluori DM, Merkel G, Andrake MD, Skalka AM. Mode of inhibition of HIV-1 Integrase by a C-terminal domain-specific monoclonal antibody. Retrovirology. 2006;3:34. doi: 10.1186/1742-4690-3-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Baranova SV, Buneva VN, Kharitonova MA, Sizyakina LP, Calmels C, Andreola ML, Parissi V, Nevinsky GA. HIV-1 integrase-hydrolyzing antibodies from sera of HIV-infected patients. Biochimie. 2009 doi: 10.1016/j.biochi.2009.06.018. [DOI] [PubMed] [Google Scholar]
- 152.BouHamdan M, Duan LX, Pomerantz RJ, Strayer DS. Inhibition of HIV-1 by an anti-integrase single-chain variable fragment (SFv): delivery by SV40 provides durable protection against HIV-1 and does not require selection. Gene Ther. 1999;6:660–666. doi: 10.1038/sj.gt.3300864. [DOI] [PubMed] [Google Scholar]
- 153.BouHamdan M, Kulkosky J, Duan LX, Pomerantz RJ. Inhibition of HIV-1 replication and infectivity by expression of a fusion protein, VPR-anti-integrase single-chain variable fragment (SFv): intravirion molecular therapies. J Hum Virol. 2000;3:6–15. [PubMed] [Google Scholar]
- 154.Levy-Mintz P, Duan L, Zhang H, Hu B, Dornadula G, Zhu M, Kulkosky J, Bizub-Bender D, Skalka AM, Pomerantz RJ. Intracellular expression of single-chain variable fragments to inhibit early stages of the viral life cycle by targeting human immunodeficiency virus type 1 integrase. J. Virol. 1996;70:8821–8832. doi: 10.1128/jvi.70.12.8821-8832.1996. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 155.Ho WZ, Lai JP, Bouhamdan M, Duan L, Pomerantz RJ, Starr SE. Inhibition of HIV type 1 replication in chronically infected monocytes and lymphocytes by retrovirus-mediated gene transfer of anti-Rev single- chain variable fragments. AIDS Res Hum Retroviruses. 1998;14:1573–1580. doi: 10.1089/aid.1998.14.1573. [DOI] [PubMed] [Google Scholar]
- 156.BouHamdan M, Strayer DS, Wei D, Mukhtar M, Duan LX, Hoxie J, Pomerantz RJ. Inhibition of HIV-1 infection by down-regulation of the CXCR4 co- receptor using an intracellular single chain variable fragment against CXCR4. Gene Ther. 2001;8:408–418. doi: 10.1038/sj.gt.3301411. [DOI] [PubMed] [Google Scholar]
- 157.Strayer DSP, R J, Yu M, Rosenzweig M, BouHamdan M, Yurasov S, Johnson RP, Goldstein H. Efficient gene transfer to hematopoietic progenitor cells using SV40-derived vectors. Gene Therapy. 2000;7:886–895. doi: 10.1038/sj.gt.3301159. [DOI] [PubMed] [Google Scholar]
- 158.Goldstein H, Pettoello-Mantovani M, Anderson CM, Cordelier P, Pomerantz RJ, Strayer DS. Gene therapy using a simian virus 40-derived vector inhibits the development of in vivo human immunodeficiency virus type 1 infection of severe combined immunodeficiency mice implanted with human fetal thymic and liver tissue. J Infect Dis. 2002;185:1425–1430. doi: 10.1086/340210. [DOI] [PubMed] [Google Scholar]
- 159.Mazumder A, Neamati N, Ojwang JO, Sunder S, Rando RF, Pommier Y. Inhibition of human immunodeficiency virus type 1 integrase by guanosine quartet structures. Biochemistry. 1996;43:13762–13771. doi: 10.1021/bi960541u. [DOI] [PubMed] [Google Scholar]
- 160.Jing N, Marchand C, Liu J, Mitra R, Hogan ME, Pommier Y. Mechanism of inhibition of HIV-1 integrase by G-tetrad-forming oligonucleotides in Vitro. J Biol Chem. 2000;275:21460–21467. doi: 10.1074/jbc.M001436200. [DOI] [PubMed] [Google Scholar]
- 161.Rando RF, Ojwang J, Elbaggari A, Reyes GR, Tinder R, McGrath MS, Hogan ME. Suppression of human immunodeficiency virus type 1 activity in vitro by oligonucleotides which form intramolecular tetrads. J. Biol. Chem. 1995;270:1754–1760. doi: 10.1074/jbc.270.4.1754. [DOI] [PubMed] [Google Scholar]
- 162.Brodin P, Pinskaya M, Parsch U, Bischerour J, Leh H, Romanova E, Engels JW, Gottikh M, Mouscadet JF. 6-oxocytidine containing oligonucleotides inhibit the HIV-1 integrase in vitro. Nucleosides Nucleotides Nucleic Acids. 2001;20:481–486. doi: 10.1081/NCN-100002322. [DOI] [PubMed] [Google Scholar]
- 163.Brodin P, Pinskaya M, Buckle M, Parsch U, Romanova E, Engels J, Gottikh M, Mouscadet JF. Disruption of HIV-1 integrase-DNA complexes by short 6-oxocytosine-containing oligonucleotides. Biochemistry. 2002;41:1529–1538. doi: 10.1021/bi015732y. [DOI] [PubMed] [Google Scholar]
- 164.Pinskaya M, Romanova E, Volkov E, Deprez E, Leh H, Brochon JC, Mouscadet JF, Gottikh M. HIV-1 integrase complexes with DNA dissociate in the presence of short oligonucleotides conjugated to acridine. Biochemistry. 2004;43:8735–8743. doi: 10.1021/bi049706m. [DOI] [PubMed] [Google Scholar]
- 165.Cherepanov P, Este JA, Rando RF, Ojwang JO, Reekmans G, Steinfeld R, David G, De Clercq E, Debyser Z. Mode of interaction of G-quartets with the integrase of human immunodeficiency virus type 1. Mol Pharmacol. 1997;52:771–780. doi: 10.1124/mol.52.5.771. [DOI] [PubMed] [Google Scholar]
- 166.Este JA, Cabrera C, Schols D, Cherepanov P, Gutierrez A, Witvrouw M, Pannecouque C, Debyser Z, Rando RF, Clotet B, Desmyter J, De Clercq E. Human immunodeficiency virus glycoprotein gp120 as the primary target for the antiviral action of AR177 (Zintevir) Mol Pharmacol. 1998;53:340–345. doi: 10.1124/mol.53.2.340. [DOI] [PubMed] [Google Scholar]
- 167.de Soultrait VR, Lozach PY, Altmeyer R, Tarrago-Litvak L, Litvak S, Andreola ML. DNA aptamers derived from HIV-1 RNase H inhibitors are strong anti-integrase agents. J Mol Biol. 2002;324:195–203. doi: 10.1016/s0022-2836(02)01064-1. [DOI] [PubMed] [Google Scholar]
- 168.Phan AT, Kuryavyi V, Ma JB, Faure A, Andreola ML, Patel DJ. An interlocked dimeric parallel-stranded DNA quadruplex: a potent inhibitor of HIV-1 integrase. Proc Natl Acad Sci U S A. 2005;102:634–639. doi: 10.1073/pnas.0406278102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Metifiot M, Leon O, Tarrago-Litvak L, Litvak S, Andreola ML. Targeting HIV-1 integrase with aptamers selected against the purified RNase H domain of HIV-1 RT. Biochimie. 2005;87:911–919. doi: 10.1016/j.biochi.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 170.Jing N, Xiong W, Guan Y, Pallansch L, Wang S. Potassium-dependent folding: a key to intracellular delivery of G- quartet oligonucleotides as HIV inhibitors. Biochemistry. 2002;41:5397–5403. doi: 10.1021/bi0120401. [DOI] [PubMed] [Google Scholar]
- 171.Metifiot M, Faure A, Guyonnet-Duperat V, Bellecave P, Litvak S, Ventura M, Andreola ML. Cellular uptake of ODNs in HIV-1 human-infected cells: a role for viral particles in DNA delivery? Oligonucleotides. 2007;17:151–165. doi: 10.1089/oli.2006.0061. [DOI] [PubMed] [Google Scholar]
- 172.Jin H, Metobo S, Jabri S, Mish M, Lansdown R, Chen X, Tsiang M, Wright M, Kim CU. Tricyclic HIV integrase inhibitors V. SAR studies on the benzyl moiety. Bioorg Med Chem Lett. 2009;19:2263–2265. doi: 10.1016/j.bmcl.2009.02.092. [DOI] [PubMed] [Google Scholar]
- 173.Daelemans D, Lu R, De Clercq E, Engelman A. Characterization of a replication-competent, integrase-defective human immunodeficiency virus (HIV)/simian virus 40 chimera as a powerful tool for the discovery and validation of HIV integrase inhibitors. J Virol. 2007;81:4381–4385. doi: 10.1128/JVI.02637-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Sechi M, Derudas M, Dallocchio R, Dessi A, Bacchi A, Sannia L, Carta F, Palomba M, Ragab O, Chan C, Shoemaker R, Sei S, Dayam R, Neamati N. Design and synthesis of novel indole beta-diketo acid derivatives as HIV-1 integrase inhibitors. J Med Chem. 2004;47:5298–5310. doi: 10.1021/jm049944f. [DOI] [PubMed] [Google Scholar]
- 175.Charvat TT, Lee DJ, Robinson WE, Chamberlin AR. Design, synthesis, and biological evaluation of chicoric acid analogs as inhibitors of HIV-1 integrase. Bioorg Med Chem. 2006;14:4552–4567. doi: 10.1016/j.bmc.2006.02.030. [DOI] [PubMed] [Google Scholar]
- 176.Queffelec C, Bailly F, Mbemba G, Mouscadet JF, Hayes S, Debyser Z, Witvrouw M, Cotelle P. Synthesis and antiviral properties of some polyphenols related to Salvia genus. Bioorg Med Chem Lett. 2008;18:4736–4740. doi: 10.1016/j.bmcl.2008.06.063. [DOI] [PubMed] [Google Scholar]
- 177.Al-Mawsawi LQ, Dayam R, Taheri L, Witvrouw M, Debyser Z, Neamati N. Discovery of novel non-cytotoxic salicylhydrazide containing HIV-1 integrase inhibitors. Bioorg Med Chem Lett. 2007;17:6472–6475. doi: 10.1016/j.bmcl.2007.09.102. [DOI] [PubMed] [Google Scholar]
- 178.Hombrouck A, Hantson A, van Remoortel B, Michiels M, Vercammen J, Rhodes D, Tetz V, Engelborghs Y, Christ F, Debyser Z, Witvrouw M. Selection of human immunodeficiency virus type 1 resistance against the pyranodipyrimidine V-165 points to a multimodal mechanism of action. J Antimicrob Chemother. 2007;59:1084–1095. doi: 10.1093/jac/dkm101. [DOI] [PubMed] [Google Scholar]
- 179.Krajewski K, Marchand C, Long YQ, Pommier Y, Roller PP. Synthesis and HIV-1 integrase inhibitory activity of dimeric and tetrameric analogs of indolicidin. Bioorg Med Chem Lett. 2004;14:5595–5598. doi: 10.1016/j.bmcl.2004.08.061. [DOI] [PubMed] [Google Scholar]
- 180.Thomas MG, Maga G, Mouscadet JF, Howarth NM. Investigation of novel lipid-functionalized PNA monomers as potential HIV-1 non-nucleoside reverse transcriptase and/or integrase inhibitors. Nucleosides Nucleotides Nucleic Acids. 2007;26:1063–1066. doi: 10.1080/15257770701513414. [DOI] [PubMed] [Google Scholar]
- 181.Yung E, Sorin M, Pal A, Craig E, Morozov A, Delattre O, Kappes J, Ott D, Kalpana GV. Inhibition of HIV-1 virion production by a transdominant mutant of integrase interactor 1. Nat Med. 2001;7:920–926. doi: 10.1038/90959. [DOI] [PubMed] [Google Scholar]
- 182.Rosenbluh J, Hayouka Z, Loya S, Levin A, Armon-Omer A, Britan E, Hizi A, Kotler M, Friedler A, Loyter A. Interaction between HIV-1 Rev and integrase proteins: a basis for the development of anti-HIV peptides. J Biol Chem. 2007;282:15743–15753. doi: 10.1074/jbc.M609864200. [DOI] [PubMed] [Google Scholar]
- 183.Jing N, Marchand C, Guan Y, Liu J, Pallansch L, Lackman-Smith C, De Clercq E, Pommier Y. Structure-activity of inhibition of HIV-1 integrase and virus replication by Gquartet oligonucleotides. DNA Cell Biol. 2001;20:499–508. doi: 10.1089/104454901316976136. [DOI] [PubMed] [Google Scholar]