

Abstract
Spontaneous DNA damage occurs throughout the genome, requiring that DNA repair enzymes search each nucleotide every cell cycle. This search is postulated to be more efficient if the enzyme can diffuse along the DNA, but our understanding of this process is incomplete. A key distinction between mechanisms of diffusion is whether the protein maintains continuous contact (sliding) or whether it undergoes microscopic dissociation (hopping). We describe a simple chemical assay to detect the ability of a DNA modifying enzyme to hop and have applied it to human alkyladenine DNA glycosylase (AAG), a monomeric enzyme that initiates repair of alkylated and deaminated purine bases. Our results indicate that AAG uses hopping to effectively search both strands of a DNA duplex in a single binding encounter. This raised the possibility that AAG might be capable of circumnavigating blocks such as tightly bound proteins. We tested this hypothesis by binding an EcoRI endonuclease dimer between two sites of DNA damage and measuring the ability of AAG to act at both damaged sites in a single binding encounter. Remarkably, AAG bypasses this roadblock in ∼50% of the binding events. We infer that AAG makes significant excursions from the surface of the DNA, allowing reorientation between strands and the bypass of a bound protein. This has important biological implications for the search for DNA damage because eukaryotic DNA is replete with proteins and only transiently accessible.
The human base excision DNA repair pathway repairs ∼10,000 lesions per cell per day (1). This is a daunting task because these relatively rare lesions must be located from among ∼12,000,000,000 normal nucleotides in the genome. Almost a dozen different human DNA repair glycosylases continuously and independently search the genome for a wide variety of oxidized or alkylated bases. Once a damaged nucleotide has been located, the glycosylase catalyzes the hydrolysis of the N-glycosidic bond to release the damaged base and create an abasic site. This abasic site is further processed to restore the correct DNA sequence using the opposing nucleotide as a template. There is considerable in vitro evidence that glycosylases use thermally-driven linear diffusion to efficiently search for sites of damage, whereby the enzyme diffuses along DNA in a non-directional manner, searching many adjacent sites within a single binding event (2-8). The biological importance of linear diffusion has been confirmed by the findings that mutants of T4 pyrimidine dimer glycosylase and EcoRV endonuclease that are deficient in linear diffusion have decreased activity in vivo (9-11).
The task of locating specific sites within the genome is central to DNA repair and to many other nuclear processes such as DNA replication and transcription (12-14). Two distinct mechanisms are recognized for diffusion along DNA and they are commonly referred to as sliding and hopping (13-22). As illustrated in Scheme 1, sliding involves continuous contact between the protein and the DNA backbone so that transfer occurs between linearly contiguous sites on the same strand. This implies that sliding follows a helical path, and there is experimental evidence of this for several proteins, including DNA glycosylases (3, 15, 23-25). In the alternative mode of translocation, referred to as hopping, a bound protein microscopically dissociates to a point at which it is still very close to the originally bound site and will with high probability re-associate to the same or nearby site on either DNA strand (12, 22). This pathway has been experimentally observed for several proteins (16-22), including E. coli uracil DNA glycosylase (7).
Scheme 1.

Sliding and hopping are distinct mechanisms of linear diffusion
We investigated the mechanism by which alkyladenine DNA glycosylase (AAG), a human DNA glycosylase responsible for the repair of a diverse set of alkylated and deaminated purines, locates damaged nucleotides. The results provide strong evidence for hopping by this single domain protein and demonstrate that AAG simultaneously searches both strands of DNA. Remarkably, a tightly bound protein serves as only a partial block of AAG diffusion. We suggest that hopping plays important roles in the search for DNA damage, allowing greater distances to be covered in a given time, both strands to be sampled, and bound proteins to be circumvented.
Results and Discussion
We previously described a processivity assay that allows the transfer of a glycosylase between two sites on a DNA molecule to be monitored by measuring the correlated cleavage events for a substrate containing two sites of damage (5). Base excision catalyzed by AAG at one of the two sites results in an abasic product and AAG subsequently partitions between action at the second site and dissociation into solution (Figure 1). The fraction of processive events (Fp) is determined by alkaline hydrolysis of abasic sites and initial rates for single and double excision events. These experiments demonstrated that AAG locates sites of damage via linear diffusion and that electrostatic interactions are critical to maintaining contact with DNA. Although it is not absolutely required, the amino terminus of AAG contributes to the processivity of the enzyme (5). These experiments provided insight into the mechanism by which AAG locates sites of damage, but they did not distinguish whether AAG uses hopping, sliding, or a combination of these two modes of diffusion (Scheme 1). The physical and chemical mechanism of linear diffusion is critical to understanding biological processes in the nucleus, such as transcription and DNA repair. In the present work we have varied the distance between the two lesions to investigate the effective searching distance and to gain additional insight into the mechanism of diffusion along DNA.
Figure 1.

Processivity assays to determine the mechanism of linear diffusion by a DNA repair glycosylase. (a) Sequences of oligonucleotides that were employed in this study. All DNA duplexes contained two εA lesions (E) on the same or opposing strands and one or two fluorescein labels (asterisk). The local sequence context for the εA lesions are marked by a solid line if they are identical for a given substrate and the 10 bp palindrome that contains a central EcoRI recognition sequence (GAATTC) is marked by a dashed line. (b) Processivity assays follow events subsequent to an initial base excision event, effectively measuring partitioning between dissociation and correlated excision at the nearby lesion site. Substrates were designed so that AAG randomly binds to and excises either of the two εA lesions to create an abasic site (Ab). AAG release is irreversible under both multiple turnover and pulse-chase conditions, because the excess substrate prevents rebinding to a released intermediate.
Evaluating the Sliding-Only Model. There is evidence that many proteins use a sliding mode to diffuse along DNA (3, 15, 16, 24). Sliding relies on simple diffusion and does not have a directional bias. Therefore, a bound protein has equal probability of moving to either adjacent position and the mean position of the sliding protein will not vary from its starting point (14, 19, 20, 26). However, the distribution around the mean will broaden with time, with a translocation of n base pairs from the mean requiring n2 single base pair steps (designated as N). The probability of a protein reaching a position n base pairs away (Pn) is given by equation 1, in which P1 is the probability that the enzyme will move one step along the DNA without dissociating, ks is the rate constant for sliding by one base pair and koff is the rate constant for dissociation into solution (13, 19, 20). If protein translocation is monitored by enzymatic activity at the target site, the efficiency of action (E) must also be considered (eq 2), because some binding encounters might be unproductive (7).
| (1) |
| (2) |
The processivity of AAG is decreased by increasing the ionic strength or by truncation of the poorly conserved amino terminus (5). The theoretical model derived for a sliding-only model (eq 2) applies at any ionic strength, and changes in Fp must be attributed to the ionic strength dependence of one or more of these terms. Experimental and theoretical work suggests that ks is insensitive to changes in ionic strength (21, 27). Therefore, the observed ionic strength dependence of Fp could be due to changes in E or koff. It is known that the efficiency of action of εA is relatively insensitive to ionic strength (28), whereas the rate of dissociation of AAG from DNA is strongly dependent upon the ionic strength (5, 29). These considerations suggest that the rate of dissociation controls the processivity of AAG across a wide range of salt concentrations. Truncation of the amino terminus (Δ80) appears to decrease the processivity by increasing the rate of dissociation (5).
To test whether AAG moves predominantly by sliding, we compared the relative processivity on substrates that have a different number of base pairs (bp) between the two sites (Figure 1). The microscopic rate constants ks and koff and the value of E are identical for the two substrates. Therefore, the partition function (ks/(ks+koff)) is a constant for a given condition and can be used to predict how the Fp value would change with sliding distance according to the sliding-only model. Multiple turnover processivity assays for substrates with lesions separated by 25 bp (47εA2F2) and 50 bp (72εA2F2) were performed for both full-length and Δ80 AAG and the results are presented in Figure 2. The data for the 25 bp separation are in excellent agreement with previous data obtained at 10-fold higher substrate concentration (5). We used the purely sliding model to predict the theoretical effect of increasing the searching distance from 25 to 50 bp (Figure 2, dashed lines; see Supporting Information). It is notable that for both enzymes and all ionic strength conditions there is no significant difference between the two substrates, in contrast to the large difference that is predicted from a sliding-only model (22). This suggests that AAG employs a mode of diffusion other than/or in addition to sliding, such as hopping. Below we describe assays designed to directly detect hopping.
Figure 2.
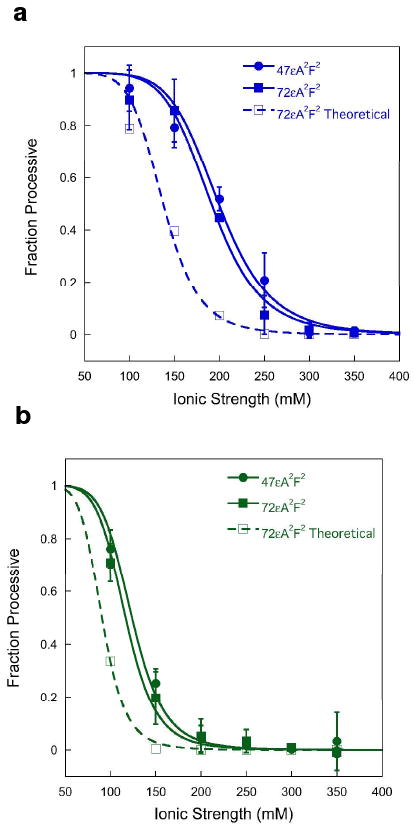
Ionic strength dependence for the processivity of AAG is inconsistent with a purely sliding model. Multiple turnover processivity assays were performed with either full-length (a) or Δ80 AAG (b), using substrates that contained two εA lesions separated by 25 bp (47εA2F2, ●) or 50 bp (72εA2F2, ■). The fraction processive was calculated as described in the Methods and the ionic strength dependence was fit by a cooperative model with 7 inhibitory sodium ions (5). Each data point reflects the mean value from at least two independent experiments and the error bars indicate one standard deviation (n ≥ 4). The theoretical processivity for the 72mer (□, dashed lines) was calculated from the data for the 47mer, using the sliding model described in the text (See Supporting Information).
Direct Evidence for Hopping
If the search for DNA damage is restricted to sliding, then only one strand of DNA could be searched during an individual DNA binding encounter. However, if the protein were capable of microscopic dissociation and re-association (i.e., hopping), then the protein could switch between searching one or the other strand. Therefore, we tested to what extent AAG acts processively on substrates that contained lesions on opposing strands. A key feature of this experimental design is that the two strands are connected via a hairpin so that action at both sites is intramolecular and strand switching can be monitored. To evaluate the possible effects of the DNA ends, we varied the position of the fluorescein labels (internal or external) and compared T5 (polyT) and GNRA hairpin structures (Figure 1A). GNRA hairpins are known to form compact three dimensional structures in DNA or RNA (30). In contrast, a polyT loop is expected to be more flexible.
Under conditions of low ionic strength, full-length AAG was highly processive on the GNRAEF substrate that had lesions on opposing strands and the processivity was indistinguishable from that observed for the substrates with lesions on the same strand (see Supporting Information). This demonstrates that AAG is able to hop between strands at least once so that both lesions are excised prior to macroscopic dissociation and rebinding to another DNA molecule. However, a decrease in the processivity would be difficult to detect under these conditions because the residence time of AAG on the DNA is very long. The maximum sensitivity would be observed when 50% of the binding events are processive (i.e., Fp = 0.5). These conditions are obtained at an ionic strength of 200 mM for full-length and 115 mM for the truncated form of AAG (Figure 2). Therefore, multiple turnover processivity assays were carried out under these conditions for each of the hairpin substrates with both forms of AAG and the results are summarized in Figure 3.
Figure 3.
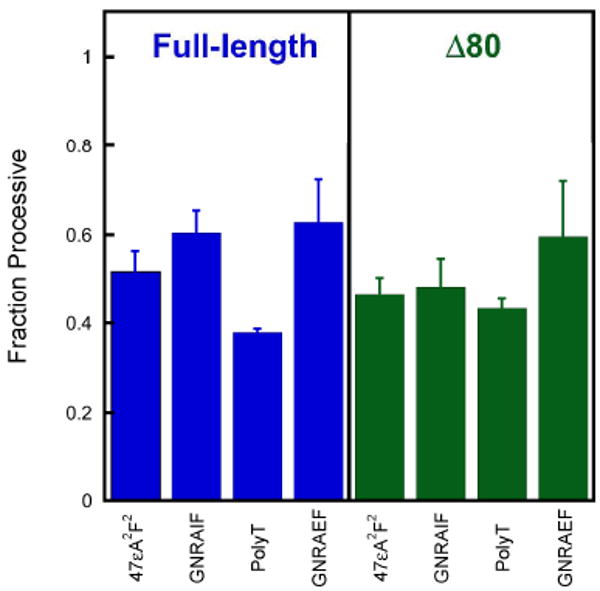
AAG searches both strands of DNA. To test whether hopping contributes to the searching mechanism of AAG, we measured the processivity for substrates in which lesions are on the opposing strands and compared this to a substrate in which the lesions are on the same strand (47εA2F2). See Figure 1 for the DNA sequences. Multiple-turnover processivity assays were performed at an ionic strength of 200 mM for full-length AAG (blue) or 115 mM for Δ80 AAG (green). Each column represents the average of at least two independent experiments with error bars indicating one standard deviation from the mean (n ≥ 4).
In all cases, the processivity of AAG on a substrate with lesions on opposing strands was similar to the processivity on a substrate with lesions on the same strand. Similar values were obtained with different hairpins, GNRA versus polyT, and with different positions of the fluorescein label, suggesting that strand switching occurs at internal sites rather than at the hairpin or blunt ends. These results are indicative of frequent hopping events that allow AAG to simultaneously search both strands of an exposed DNA substrate. Furthermore, it is clear that the amino terminus of AAG is not required for strand switching (Figure 3).
Although our results seem to be at odds with recent reports that several glycosylases and other single domain enzymes rotate as they diffuse along DNA (15, 24), it should be noted that rotational diffusion (due to sliding along one strand) does not exclude hopping. Indeed, a recent report showed that E. coli uracil DNA glycosylase is capable of frequent hops (7). These differences between experiments conducted at the ensemble and single molecule level can be explained by a majority of steps being classified as sliding, with less frequent hopping steps. As glycosylases can only sample one nucleotide of a base pair, but lesions can occur in either strand, strand switching via hopping events is expected to greatly increase the efficiency of the search for DNA damage.
AAG Can Bypass a Protein Roadblock
A criticism of in vitro studies of linear diffusion of proteins on naked DNA is that DNA is expected to be only transiently accessible in the cell. DNA binding proteins bind densely to chromosomal DNA and those with high affinity can have lifetimes of hours. Previous studies have revealed that tightly bound proteins pose a barrier to sliding by endonuclease EcoRI (25), to translocation by mismatch repair proteins (31), and to transcription by RNA polymerase (32). In contrast, we hypothesized that microscopic dissociation followed by re-association might allow a protein such as AAG to hop past a tightly bound protein.
Several previous studies have employed EcoRI as a block, because it binds tightly as a homodimer and makes intimate contacts with both strands of the DNA (25, 31, 32). Therefore, we tested whether EcoRI blocks the diffusion of AAG (Figure 4). The 72 bp oligonucleotide that was employed is shorter than the persistence length of DNA, rendering intersegmental transfer unlikely. EcoRI has been reported to bend DNA by ∼50° (33), but this is insufficient to juxtapose the two lesions. AAG does not require Mg2+, and this allowed us to use wildtype EcoRI since it binds with high affinity to its recognition site in the absence of Mg2+ (34). The use of wildtype EcoRI has an advantage over the use of an inactive mutant, because its presence at its recognition site can be directly quantified by the rapid DNA cleavage that occurs upon the addition of Mg2+.
Figure 4.

Testing the effect of a protein roadblock on linear diffusion by AAG. a) The 72mer substrate is depicted with an EcoRI dimer (green) bound to the central recognition site, and with an AAG monomer (blue) bound to the abasic product from the first excision reaction. If AAG is able to bypass the tightly bound protein (dashed arrow), then processive excision of the second εA will be observed. b) Structure of the EcoRI•DNA complex (41) is from the pdb (1ERI). The surfaces of the two EcoRI monomers are shown in blue and green and the DNA is depicted as a cartoon with the backbone in orange and the central EcoRI recognition sequence in yellow. c) The structure of the complex of the catalytic domain of AAG bound to εA-DNA (42) is from the pdb (1F4R). Images were rendered with Pymol (http://www.pymol.org).
Control experiments confirmed that the 72εA2F2 substrate, which contains a single EcoRI recognition site, could be bound completely at a 1:1 stoichiometry of EcoRI dimer to DNA under the glycosylase assay conditions (see Supporting Information). Multiple turnover processivity experiments were performed in the presence of EcoRI with full-length AAG at both 100 and 200 mM ionic strength (Figure 5). The addition of one equivalent of EcoRI dimer decreased the processivity of AAG by ∼50% under both conditions. Addition of another equivalent of EcoRI did not have any further effect on the processivity of AAG, confirming that the DNA was saturated with EcoRI. Thus, a bound dimer of EcoRI appears to be only a partial block of the processive action of AAG.
Figure 5.
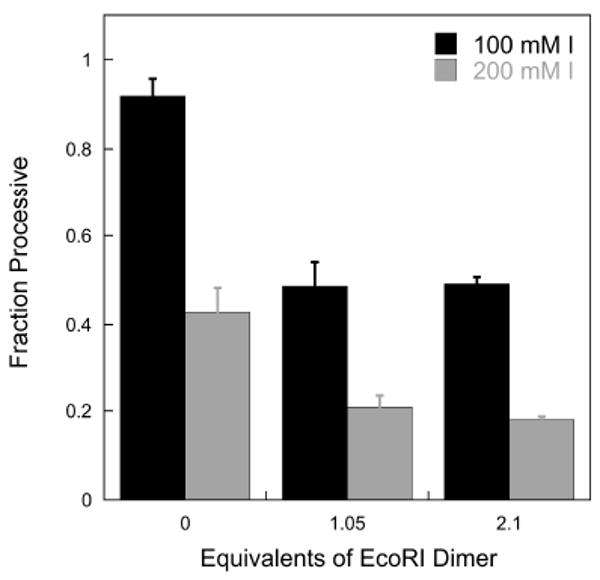
Effect of bound EcoRI on the processivity of AAG. Multiple turnover processivity assays were performed with 200 nM 72εA2F2, 2 nM full-length AAG, and 0, 210, and 420 nM EcoRI dimer at both 100 and 200 mM ionic strength and the calculated processivity values are shown. The presence of the tightly bound EcoRI dimer reduces the processivity of AAG by ∼50% at ionic strengths of 100 mM (black bars) and 200 mM (gray bars).
However, the long assay time required for multiple turnover assays poses a problem for the roadblock assay because EcoRI might diffuse away from its recognition site or dissociate into solution. This limitation was overcome with pulse-chase experiments that limit the processivity assay to a single encounter of AAG with DNA. In this approach AAG is incubated with labeled DNA substrate, then immediately chased with an excess of unlabeled substrate. AAG can dissociate directly, or excise either one or two εA lesions before dissociating. Due to the presence of chase DNA, once AAG has dissociated it has a very low probability of rebinding a labeled substrate. We used an unlabeled DNA chase that is otherwise identical to the labeled substrate, and therefore it captures any AAG or EcoRI molecules that dissociate during the experiment.
To confirm the validity of this approach, we first performed the pulse chase experiment in the absence of EcoRI (ionic strength = 100 mM; Figure 6). A single turnover of substrate equal to the amount of AAG was completed within 25 minutes (Figure 6b), in agreement with the single-turnover rate constant of 0.20 min-1 (5). Quantification of the intermediates (single εA excised) and products (two εA excised) allowed the processivity to be calculated (eq 5). The resulting processivity from the pulse chase method is identical within error to the value determined for the multiple turnover processivity assay for both full-length (0.89 ± 0.01 versus 0.90 ± 0.1) and Δ80 AAG (0.74 ± 0.04 versus 0.71 ± 0.07). Thus, the pulse-chase assay measures correlated events occurring within a single binding encounter.
Figure 6.

Pulse-chase processivity assays indicate that AAG can bypass a bound EcoRI dimer. a) The experimental design is depicted. Fluorescein-labeled substrate (72εA2F2) was incubated with or without EcoRI for 1 hour (t1), after which AAG was added. AAG was incubated for 40 seconds (t2), before mixing with excess unlabeled substrate (72εA2). Incubations continued for 50 minutes (t3) and aliquots were removed and analyzed by the gel-based glycosylase assay. The ratio of AAG to labeled substrate to unlabeled chase was 1:10:260. b) A representative time course for full-length AAG was performed in duplicate at an ionic strength of 100 mM. Substrate depletion is fit to a single exponential (kobs = kchem). The amplitude of ∼10% disappearance of substrate confirms that AAG was bound and excised at least one εA lesion prior to dissociation. No further glycosylase activity was observed up to 50 minutes, confirming that adequate chase was used. The build-up of products (red) and intermediates (black) were fit to single-exponentials solely to show trends. c) The fraction processive was calculated from the burst amplitudes in panel b and from additional experiments for both full-length (blue) and Δ80 AAG (green). Each column represents the average of two independent experiments with error bars representing one standard deviation from the mean (n ≥ 4). The column labeled “Control” is from reactions in which EcoRI was first bound to the unlabeled substrate instead of the labeled substrate. The final reaction conditions are identical to the +EcoRI reactions, but the endonuclease and glycosylase are bound on different DNA molecules. Therefore, the decrease in processivity is due to a direct block of AAG diffusion as opposed to an artifact of some other component of the EcoRI sample.
We next evaluated the effectiveness of the EcoRI block with the pulse-chase assay. A greater amount of intermediates were released by AAG when EcoRI was bound, indicating a modest decrease in processivity (Figure 6b). This was quantified and the results for both full-length and Δ80 AAG are shown in Figure 6c. EcoRI decreased the processivity of both full-length and Δ80 AAG by approximately 50%. This is identical within error to the results from multiple turnover assays (Figure 5). We confirmed that the decrease in processivity requires EcoRI to be pre-bound to the labeled substrate, because control reactions in which EcoRI was first bound to the unlabeled chase did not show any decrease in processivity (Figure 6c). Additional controls ruled out the trivial possibility that partial blockage was due to incomplete saturation by EcoRI or dissociation of EcoRI on the time scale of the experiment. To address both points, we added Mg2+ after the AAG burst was over and measured cleavage by EcoRI. The results demonstrated that less than 2% of the EcoRI had dissociated (Supplementary Figure S9).
These experiments establish that EcoRI remains bound to the same DNA molecule for the entire assay. However, because EcoRI locates and leaves its recognition sequence predominantly by sliding, these data don't address transient excursions of EcoRI away from its specific recognition site (31, 35-38). Such excursions have been observed on the same time scale (t1/2∼40 minutes) as our processivity assay (31). However, the short substrate that we employed is constrained by the ends of the DNA so that EcoRI cannot diffuse past the εA lesion to allow access by AAG. Although EcoRI spends most of its time bound to the specific recognition site, and bypass by AAG most likely occurs at this site, we cannot rule out the possibility that AAG and EcoRI transiently pass each other at an adjacent nonspecific site. Several pathways can be envisioned for the bypass of EcoRI by AAG (Figure 4). (i) Hopping by AAG may occur over a sufficient distance to allow AAG to dissociate and re-associate on the other side of the bound EcoRI. (ii) AAG may be able to diffuse over the surface of EcoRI, possibly facilitated by electrostatic interactions between the positively charged AAG and the negatively charged solution-exposed face of EcoRI. (iii) AAG may be able to navigate past EcoRI by multiple strand switching events, perhaps facilitated by breathing of EcoRI. Additional experiments will be required to distinguish these possibilities. Regardless of the exact pathway(s), it is notable that a tightly bound protein hinders, but does not completely block linear diffusion by AAG.
Implications
Recent ensemble and single-molecule studies have focused on elucidating the contributions of each mode of translocation to the searching mechanism of DNA glycosylases to gain insight into how these enzymes rapidly and efficiently locate rare sites of DNA damage. Theoretical analysis suggests that the rate of target site location is optimized by a combination of sliding and hopping (13, 39). However, a mechanism comprised almost exclusively of rotation-coupled sliding over hundreds of base pairs of nonspecific DNA has been suggested for human OGG1, E. coli MutM, and B. stearothermophilus MutY (3, 15). By maintaining constant contact with the DNA backbone, redundant sliding over relatively short stretches of nonspecific DNA allows each nucleotide of the bound strand to be encountered multiple times and increases the probability that a lesion is recognized. However, reliance on sliding would slow down searches over longer distances and would allow only a single strand to be searched. Hopping can optimize the rate of target site location by allowing a sliding enzyme to escape from redundantly scanned stretches of DNA and access new sites (3, 13, 19). Our results indicate that diffusion by AAG involves a significant contribution from hopping and are in good agreement with the conclusion that diffusion by E. coli uracil DNA glycosylase is dominated by hopping over long distances with local sliding contributing to damage recognition (7). The ability to hop to the opposing strand allows the rapid and essentially simultaneous search of both strands of a given segment of DNA. We suggest that this ability also enables AAG to bypass a tightly bound protein, which has important implications for the biological search for DNA damage.
Methods
Proteins
Full-length and truncated recombinant human AAG were purified and the concentration of active AAG was determined by burst analysis as previously described (5). EcoRI was expressed and purified as previously described (40) and the concentration of active dimer was determined by burst analysis (See Supporting Information). T4 DNA Ligase was from New England Biolabs.
Oligonucleotides
DNA substrates were synthesized by Integrated DNA Technologies or the Keck Center at Yale University and purified by denaturing PAGE as previously described (5). For the GNRAIF and PolyT hairpin substrates, the DNA was synthesized in fragments and each DNA fragment was purified. DNA fragments for each respective substrate were then annealed in annealing buffer (10 mM NaMES, pH 6.5, 50 mM NaCl) and ligated for 24 hrs at 16 °C using T4 DNA Ligase in the recommended NEB Ligase Buffer. Ligation reactions were quenched after 24 hrs with 20 mM EDTA and complete ligation products were purified. The concentration of single-stranded DNA was determined from the absorbance at 260 nm using the calculated extinction coefficients, and the concentration of duplex (hairpin) DNA was determined from the extinction coefficient at 495 nm for fluorescein (E495 = 7.5×105 M-1cm-1).
Glycosylase Activity Assay
Reactions were carried out at 37 °C in 50 mM NaMES, pH 6.1, 1 mM EDTA, 1 mM DTT, 10% glycerol, 0.1 mg mL-1 BSA and the ionic strength was adjusted with NaCl. Reactions were initiated by adding enzyme (2 – 7.5 nM final concentration) to obtain a reaction volume of 50 – 100 µL that contained 75 - 200 nM fluorescein-labeled DNA. Aliquots were withdrawn at various times and quenched with 2 volumes of 0.3 M NaOH or 0.5 volumes of 0.6 M NaOH to yield a final concentration of 0.2 M. Samples were heated to 70 °C for 15 minutes, formamide was added to 65%, and the DNA fragments were resolved on 14% (w/v) polyacrylamide gels containing 8 M urea as previously described (5). Gels were scanned with a Typhoon Trio+ fluorescence imager (GE Healthcare) to detect fluorescein (excitation at 488 nM and emission with 520BP40 filter). The resulting fluorescent signal was quantified using ImageQuant TL and corrected for background signal. The intensity of each DNA band was converted into a fraction by dividing its intensity by the sum of the intensities for all of the DNA species present.
Multiple Turnover Kinetics
Steady state kinetics for the dual lesion substrates were measured with 100-fold excess of substrate (200 nM) over enzyme (2 nM) as previously described (5). The initial rates were calculated from the first 10-15% of the reaction and were linear in all cases. Values for both kcat and Fp were calculated and the ionic strength dependence of both were fit with a cooperative model (5). The value of Fp was calculated according to equation 3, in which Vi and Vp are the initial rates for the formation of intermediates (retaining an εA lesion) and products (both εA excised), respectively. The processivity equation takes into account the fact that a single excision event gives rise to one product and one intermediate. Approximately 2% of εA nucleotides are damaged during synthesis, deprotection and gel purification, and therefore the maximal processivity value that is expected is 0.92 (5). The minimal value that could be observed is ∼0.05 for a completely distributive mechanism, that is attributed to rebinding of AAG. Therefore the effective range of experimental Fp values is 0.87 (0.92-0.05). To facilitate the comparison with the theoretical Fp values based upon the sliding model, we have corrected all of the observed processivity values (eq 4).
| (3) |
| (4) |
Pulse-Chase Processivity Assay
To test whether AAG can bypass a protein roadblock, we incubated labeled substrate (72εA2F2) with either 0 or 1.5-fold excess EcoRI endonuclease as indicated in Figure 6. The final concentration of 72εA2F2 was 75nM, AAG was 7.5 nM, EcoRI dimer was 110 nM, and 72εA2 chase DNA was 2 µM. As a control to establish that occupancy of the recognition sequence by EcoRI was responsible for the decrease in processivity, a reaction was carried out in which 72εA2F2 was incubated in the absence of EcoRI. After dilution and the addition of AAG to the labeled substrate, unlabeled substrate (72εA2) that had been pre-incubated with EcoRI was added. Glycosylase activity was measured as described above.
The amplitude of the burst phase for each DNA fragment was determined by the Y-intercept of a linear fit (slope = 0) to the data after the burst phase was complete (25-50 min). The burst amplitude of the substrate matched the expected burst size, and the burst rate constant was the same within error as the previously determined single turnover rate constant for excision of εA (5). The value of Fp was calculated from the concentration of products and intermediates that were formed during the burst phase, by substituting the initial rates in equation 3 with the corresponding concentrations of products (p) and intermediates (i) to give equation 5. The range of Fp values in the pulse-chase assay with 10-fold excess of DNA are identical to the range in the multiple turnover assay with 100-fold excess of DNA. The upper limit is set to 0.92 due to the damaged εA sites, whereas the lower limit of 0.05 is due to the 1% probability that two AAG molecules will bind to the same DNA molecule.
| (5) |
Supplementary Material
Acknowledgments
We thank P. Modrich for providing the expression plasmid for E. coli EcoRI endonuclease and members of the O'Brien lab for helpful discussions. This work was supported by a grant from the National Institutes of Health (CA122254).
Footnotes
Supporting Information Available: This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 2.Bennett SE, Sanderson RJ, Mosbaugh DW. Processivity of Escherichia coli and rat liver mitochondrial uracil-DNA glycosylase is affected by NaCl concentration. Biochemistry. 1995;34:6109–6119. doi: 10.1021/bi00018a014. [DOI] [PubMed] [Google Scholar]
- 3.Blainey PC, van Oijen AM, Banerjee A, Verdine GL, Xie XS. A base-excision DNA-repair protein finds intrahelical lesion bases by fast sliding in contact with DNA. Proc Natl Acad Sci U S A. 2006;103:5752–5757. doi: 10.1073/pnas.0509723103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Francis AW, David SS. Escherichia coli MutY and Fpg utilize a processive mechanism for target location. Biochemistry. 2003;42:801–810. doi: 10.1021/bi026375+. [DOI] [PubMed] [Google Scholar]
- 5.Hedglin M, O'Brien PJ. Human alkyladenine DNA glycosylase employs a processive search for DNA damage. Biochemistry. 2008;47:11434–11445. doi: 10.1021/bi801046y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Higley M, Lloyd RS. Processivity of uracil DNA glycosylase. Mutat Res. 1993;294:109–116. doi: 10.1016/0921-8777(93)90019-d. [DOI] [PubMed] [Google Scholar]
- 7.Porecha RH, Stivers JT. Uracil DNA glycosylase uses DNA hopping and short-range sliding to trap extrahelical uracils. Proc Natl Acad Sci U S A. 2008;105:10791–10796. doi: 10.1073/pnas.0801612105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sidorenko VS, Zharkov DO. Correlated cleavage of damaged DNA by bacterial and human 8-oxoguanine-DNA glycosylases. Biochemistry. 2008;47:8970–8976. doi: 10.1021/bi800569e. [DOI] [PubMed] [Google Scholar]
- 9.Dowd DR, Lloyd RS. Biological consequences of a reduction in the non-target DNA scanning capacity of a DNA repair enzyme. J Mol Biol. 1989;208:701–707. doi: 10.1016/0022-2836(89)90160-5. [DOI] [PubMed] [Google Scholar]
- 10.Dowd DR, Lloyd RS. Biological significance of facilitated diffusion in protein-DNA interactions. Applications to T4 endonuclease V-initiated DNA repair. J Biol Chem. 1990;265:3424–3431. [PubMed] [Google Scholar]
- 11.Jeltsch A, Wenz C, Stahl F, Pingoud A. Linear diffusion of the restriction endonuclease EcoRV on DNA is essential for the in vivo function of the enzyme. EMBO J. 1996;15:5104–5111. [PMC free article] [PubMed] [Google Scholar]
- 12.Berg OG, Winter RB, von Hippel PH. Diffusion-driven mechanisms of protein translocation on nucleic acids. 1. Models and theory. Biochemistry. 1981;20:6929–6948. doi: 10.1021/bi00527a028. [DOI] [PubMed] [Google Scholar]
- 13.Halford SE, Marko JF. How do site-specific DNA-binding proteins find their targets? Nucleic Acids Res. 2004;32:3040–3052. doi: 10.1093/nar/gkh624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gorman J, Greene EC. Visualizing one-dimensional diffusion of proteins along DNA. Nat Struct Mol Biol. 2008;15:768–774. doi: 10.1038/nsmb.1441. [DOI] [PubMed] [Google Scholar]
- 15.Blainey PC, Luo G, Kou SC, Mangel WF, Verdine GL, Bagchi B, Xie XS. Nonspecifically bound proteins spin while diffusing along DNA. Nat Struct Mol Biol. 2009;16:1224–1229. doi: 10.1038/nsmb.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bonnet I, Biebricher A, Porte PL, Loverdo C, Benichou O, Voituriez R, Escude C, Wende W, Pingoud A, Desbiolles P. Sliding and jumping of single EcoRV restriction enzymes on non-cognate DNA. Nucleic Acids Res. 2008;36:4118–4127. doi: 10.1093/nar/gkn376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gowers DM, Halford SE. Protein motion from non-specific to specific DNA by three-dimensional routes aided by supercoiling. EMBO J. 2003;22:1410–1418. doi: 10.1093/emboj/cdg125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gowers DM, Wilson GG, Halford SE. Measurement of the contributions of 1D and 3D pathways to the translocation of a protein along DNA. Proc Natl Acad Sci U S A. 2005;102:15883–15888. doi: 10.1073/pnas.0505378102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Halford SE. An end to 40 years of mistakes in DNA-protein association kinetics? Biochem Soc Trans. 2009;37:343–348. doi: 10.1042/BST0370343. [DOI] [PubMed] [Google Scholar]
- 20.Halford SE, Szczelkun MD. How to get from A to B: strategies for analysing protein motion on DNA. Eur Biophys J. 2002;31:257–267. doi: 10.1007/s00249-002-0224-4. [DOI] [PubMed] [Google Scholar]
- 21.Komazin-Meredith G, Mirchev R, Golan DE, van Oijen AM, Coen DM. Hopping of a processivity factor on DNA revealed by single-molecule assays of diffusion. Proc Natl Acad Sci U S A. 2008;105:10721–10726. doi: 10.1073/pnas.0802676105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stanford NP, Szczelkun MD, Marko JF, Halford SE. One- and three-dimensional pathways for proteins to reach specific DNA sites. EMBO J. 2000;19:6546–6557. doi: 10.1093/emboj/19.23.6546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sakata-Sogawa K, Shimamoto N. RNA polymerase can track a DNA groove during promoter search. Proc Natl Acad Sci U S A. 2004;101:14731–14735. doi: 10.1073/pnas.0406441101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin Y, Zhao T, Jian X, Farooqui Z, Qu X, He C, Dinner AR, Scherer NF. Using the bias from flow to elucidate single DNA repair protein sliding and interactions with DNA. Biophys J. 2009;96:1911–1917. doi: 10.1016/j.bpj.2008.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jeltsch A, Alves J, Wolfes H, Maass G, Pingoud A. Pausing of the restriction endonuclease EcoRI during linear diffusion on DNA. Biochemistry. 1994;33:10215–10219. doi: 10.1021/bi00200a001. [DOI] [PubMed] [Google Scholar]
- 26.Zharkov DO, Grollman AP. The DNA trackwalkers: principles of lesion search and recognition by DNA glycosylases. Mutat Res. 2005;577:24–54. doi: 10.1016/j.mrfmmm.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 27.Kochaniak AB, Habuchi S, Loparo JJ, Chang DJ, Cimprich KA, Walter JC, van Oijen AM. Proliferating cell nuclear antigen uses two distinct modes to move along DNA. J Biol Chem. 2009;284:17700–17710. doi: 10.1074/jbc.M109.008706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wolfe AE, O'Brien PJ. Kinetic mechanism for the flipping and excision of 1,N(6)-ethenoadenine by human alkyladenine DNA glycosylase. Biochemistry. 2009;48:11357–11369. doi: 10.1021/bi9015082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baldwin MR, O'Brien PJ. Human AP Endonuclease I Stimulates Multiple-Turnover Base Excision by Alkyladenine DNA Glycosylase. Biochemistry. 2009;48:6022–6033. doi: 10.1021/bi900517y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Varani G. Exceptionally stable nucleic acid hairpins. Annu Rev Biophys Biomol Struct. 1995;24:379–404. doi: 10.1146/annurev.bb.24.060195.002115. [DOI] [PubMed] [Google Scholar]
- 31.Pluciennik A, Modrich P. Protein roadblocks and helix discontinuities are barriers to the initiation of mismatch repair. Proc Natl Acad Sci U S A. 2007;104:12709–12713. doi: 10.1073/pnas.0705129104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pavco PA, Steege DA. Elongation by Escherichia coli RNA polymerase is blocked in vitro by a site-specific DNA binding protein. J Biol Chem. 1990;265:9960–9969. [PubMed] [Google Scholar]
- 33.Thompson JF, Landy A. Empirical estimation of protein-induced DNA bending angles: applications to lambda site-specific recombination complexes. Nucleic Acids Res. 1988;16:9687–9705. doi: 10.1093/nar/16.20.9687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Terry BJ, Jack WE, Rubin RA, Modrich P. Thermodynamic parameters governing interaction of EcoRI endonuclease with specific and nonspecific DNA sequences. J Biol Chem. 1983;258:9820–9825. [PubMed] [Google Scholar]
- 35.Jack WE, Terry BJ, Modrich P. Involvement of outside DNA sequences in the major kinetic path by which EcoRI endonuclease locates and leaves its recognition sequence. Proc Natl Acad Sci U S A. 1982;79:4010–4014. doi: 10.1073/pnas.79.13.4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pingoud A, Jeltsch A. Structure and function of type II restriction endonucleases. Nucleic Acids Res. 2001;29:3705–3727. doi: 10.1093/nar/29.18.3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rau DC, Sidorova NY. Diffusion of the Restriction Nuclease EcoRI along DNA. J Mol Biol. 2009 doi: 10.1016/j.jmb.2009.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wright DJ, Jack WE, Modrich P. The kinetic mechanism of EcoRI endonuclease. J Biol Chem. 1999;274:31896–31902. doi: 10.1074/jbc.274.45.31896. [DOI] [PubMed] [Google Scholar]
- 39.Givaty O, Levy Y. Protein sliding along DNA: dynamics and structural characterization. J Mol Biol. 2009;385:1087–1097. doi: 10.1016/j.jmb.2008.11.016. [DOI] [PubMed] [Google Scholar]
- 40.Cheng SC, Kim R, King K, Kim SH, Modrich P. Isolation of gram quantities of EcoRI restriction and modification enzymes from an overproducing strain. J Biol Chem. 1984;259:11571–11575. [PubMed] [Google Scholar]
- 41.Kim YC, Grable JC, Love R, Greene PJ, Rosenberg JM. Refinement of Eco RI endonuclease crystal structure: a revised protein chain tracing. Science. 1990;249:1307–1309. doi: 10.1126/science.2399465. [DOI] [PubMed] [Google Scholar]
- 42.Lau AY, Wyatt MD, Glassner BJ, Samson LD, Ellenberger T. Molecular basis for discriminating between normal and damaged bases by the human alkyladenine glycosylase, AAG. Proc Natl Acad Sci U S A. 2000;97:13573–13578. doi: 10.1073/pnas.97.25.13573. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.