

Abstract
Molecular dynamics (MD) simulation is a well-established method for studying protein motion at the atomic scale. However, it is computationally intensive and generates massive amounts of data. One way of addressing the dual challenges of computation efficiency and data analysis is to construct simplified models of long-timescale protein motion from MD simulation data. In this direction, we propose to use Markov models with hidden states, in which the Markovian states represent potentially overlapping probabilistic distributions over protein conformations. We also propose a principled criterion for evaluating the quality of a model by its ability to predict long-timescale protein motions. Our method was tested on 2D synthetic energy landscapes and two extensively studied peptides, alanine dipeptide and the villin headpiece subdomain (HP-35 NleNle). One interesting finding is that although a widely accepted model of alanine dipeptide contains six states, a simpler model with only three states is equally good for predicting long-timescale motions. We also used the constructed Markov models to estimate important kinetic and dynamic quantities for protein folding, in particular, mean first-passage time. The results are consistent with available experimental measurements.
Contact: chiangts@comp.nus.edu.sg
1 INTRODUCTION
Protein motion is the aggregate result of complex interactions among individual atoms of a protein at timescales ranging over several orders of magnitudes. Thermal fluctuations, which occur in picoseconds (10−12 s), are small-amplitude, uncorrelated, harmonic motions of atoms, but they eventually provide the protein enough momentum to overcome energy barriers between metastable states. In contrast, biologically significant conformational motions, which occur in microseconds to milliseconds, are often large-scale, correlated, anharmonic motions between meta-stable states. For example, in a folded protein, they may occur between binding and non-binding states. The wide range of timescales and complex relationships among the motions at different timescales make it difficult to capture the biologically significant, long-timescale dynamics of protein motion in a compact model.
Molecular dynamics (MD) simulation is a well-established method for studying macromolecular motion at the atomic scale (Shea and Brooks III, 2001). However, it requires a detailed energy function and the equations of motion must be integrated with a time step much shorter than the timescale of atomic thermal fluctuations. For many proteins, today's computers can generate roughly a few nanoseconds of simulation trajectories in a day, which is insufficient for capturing events of biological significance. Distributed computing (Pande et al., 2002) and specialized computer architectures (Shaw et al., 2007) speed up MD simulation significantly, but the sheer size of data generated is a major hurdle that prevents biological insights. One way of addressing both the issues of computational efficiency and data analysis is to construct simplified models that capture the essential features of protein motions at long timescales. Markov dynamic models (MDMs) provide a promising direction towards this goal.
An MDM of a system—here, a protein—can be represented as a directed graph. Each node of the graph represents a state s of the system, and each edge represents a transition from state s to s′. An edge (s, s′) is also assigned the probability that the system transitions from s to s′ in one time step. MDMs have several advantages for modeling protein motion. First, they are probabilistic and thus naturally capture the stochasticity of protein motion. Second, MDMs represent states explicitly. This makes them potentially easier to understand and faster to simulate. Finally, there are standard algorithmic tools, e.g. first-step analysis (Taylor and Karlin, 1994), for exploiting MDMs without expensive explicit simulation.
A key question in MDM construction is the choice of states. What are the Markovian states of a protein if we want to model its long-timescale dynamics accurately? One contribution of this work is to have states represent not individual protein conformations (Apaydin et al., 2003; Singhal et al., 2004), not even disjoint regions of the conformation space (Chodera et al., 2007; Ozkan et al., 2002), but overlapping probabilistic distributions over the conformation space. This choice reflects the view that a conformation does not contain enough information to be assigned to a single state. Although this may seem odd at first, it is in fact quite natural in modeling many physical systems. For example, suppose that we want to classify some physical objects into two states, table or chair. For a cubic object one meter in size, if we see a meal on top of it, we may consider it a table; if we see someone seated on it, we consider it a chair. So, a cube in itself cannot be assigned a single state because of insufficient information. Often, acquiring and representing missing information, if at all possible, is more difficult than capturing it in a probabilistic distribution. Hence, our choice of Markovian states that represent probabilistic distributions over the protein conformation space. This choice leads to MDMs with hidden states, formally, hidden Markov models (HMMs). In this article, we present a method to automatically construct an HMM of the long-timescale dynamics of a protein from a dataset of MD simulation trajectories.
Another key question is how to measure the quality of a model. A good model enables us to predict biologically relevant quantities of protein motion accurately and efficiently. However, a particular model may do well for one quantity, but poorly for another. Also, we may not know in advance the quantities to be predicted when constructing a model. Another contribution of this work is to propose a principled criterion for evaluating the quality of a model by its ability to predict long-timescale protein motions, as many interesting kinetic and dynamic properties of proteins ultimately depend on such motions. Specifically, we score an HMM probabilistically by its likelihood for a test dataset of MD trajectories. Using this criterion, we are able to select models that make good predictions on ensemble quantities characterizing the folding of alanine dipeptide and the villin headpiece subdomain (HP-35 NleNle), two extensively studied peptides.
We also present an efficient algorithm for computing mean first-passage time from any conformation of a protein to the folded conformation, using an HMM of protein dynamics.
2 RELATED WORK
2.1 Graphical models of protein motion
Our work proceeds from a series of developments that started with adapting probabilistic roadmap (PRM) planning (Kavraki et al., 1996) from robotics to model molecular motion. PRM is a class of algorithms for controlling the motion of complex robots.
2.1.1 Roadmap models
A PRM for a robot is an undirected graph. Each node q of the graph represents a valid robot configuration sampled randomly from the space of all valid robot configurations, and each edge between two nodes q and q′ represents a valid motion between robot configurations corresponding to q and q′. PRM planning is currently the most successful approach for motion planning of complex robots with many degrees of freedom. The PRM approach was adapted to model and analyze the motion of a flexible ligand binding with a protein (Singh et al., 1999). The modified roadmap is a directed graph, in which each node represents a sampled ligand conformation and each directed edge represents the transition from one ligand conformation to another. Each edge is also assigned a heuristic weight measuring the ‘energetic difficulty’ of the transition. This approach was used to predict active binding sites of a protein (Singh et al., 1999) and the dominant order of secondary structure formation in protein folding (Amato et al., 2003).
2.1.2 From roadmaps to MDMs
To capture the stochasticity of molecular motion, a roadmap model was transformed into an MDM by treating each roadmap node as a state and assigning each edge (q, q′) the transition probability derived from the energetic difference between the conformations corresponding to q and q′ (Apaydin et al., 2003). We call this model a point-based MDM, as each state represents a single conformation. This model was used to compute efficiently the p-fold value, a theoretical measure on the progress of protein folding (Apaydin et al., 2003) and was later improved to predict experimental measures of folding kinetics, such as folding rates and ϕ-values (Chiang et al., 2006). An improved sampling method generates the states of an MDM using MD simulation data (Singhal et al., 2004). It provides better coverage of the biologically relevant part of the protein conformation space.
2.1.3 From point-based to cell-based MDMs
In a point-based MDM, a state represents a conformation. However, a single conformation rarely contains enough information to guarantee the Markovian property, a fundamental model assumption requiring that the future state of a protein depends on its current state only and not on the past history. Consequently a large number of states are needed to construct a good MDM. This drawback led to cell-based MDMs (Chodera et al., 2007), in which each node corresponds to a region (a cell) of the protein's conformation space. A cell roughly matches a basin in the protein's energy landscape and represents a metastable state. The protein interconverts rapidly among different conformations within a basin s before it overcomes the energy barrier and transitions to another basin s′. The assumption is that after many interconversions within s, the protein ‘forgets’ the history of how it entered s and transitions into s′ with probability depending on s only. MD simulation is used to generate the data for building a cell-based MDM (Chodera et al., 2007). To satisfy the Markovian property well, conformations along simulation trajectories are grouped into clusters in such a way that maximizes self-transition probabilities for the states in the MDM. More recent work extended this approach to build MDMs at multiple resolutions through hierarchical clustering (Huang et al., 2010).
A preliminary form of the cell-based MDM was used earlier to analyze a simplified lattice protein model (Ozkan et al., 2002). The data for model construction was obtained by solving the master equation instead of performing MD simulation.
2.2 Other approaches
Various alternative approaches have been explored to model and understand protein motion. See Elber (2005) for a recent survey. Here, we only mention a few that are more closely related to our work.
Normal mode analysis (Levitt et al., 1985) and related approaches, such as elastic network models (Haliloglu et al., 1997), simplify the complex dynamic law that governs protein motion by approximating it near an equilibrium conformation. One advantage is that they capture the geometry and mass distribution of a protein structure compactly in a relatively simple model. However, they are accurate only in the neighborhood of the equilibrium conformation.
Another approach for building simple dynamic models is to find reaction coordinates (Lois et al., 2009). Significant events are described along a carefully chosen one-dimensional reaction coordinate. The choice of this coordinate, however, requires a priori understanding of the protein motion. Furthermore, not all proteins can have their motions described and understood along a single coordinate.
Instead of building simplified dynamic models, one may analyze MD simulation data directly through dimensionality reduction methods (Amadei et al., 1993; Teodoro et al., 2002). Unlike normal mode analysis, this approach provides a global view of protein motion. It may also help to identify a good reaction coordinate. However, this approach does not provide a predictive model that generalizes the simulation data. Nor does it identify interesting states of protein dynamics.
3 MDMs with Hidden States
An MDM Θ of a protein can be represented as a weighted directed graph. A node s of Θ represents a state of the protein, and a directed edge (s, s′) from node s to s′ represents a transition between the corresponding states. Each edge (s, s′) is assigned a weight ass′ representing the probability that the protein in state s transitions to state s′ in a time step of fixed duration h. The probabilities associated with the outgoing edges from any node s must sum up to 1. The duration h is the time resolution of the model.
An MDM describes how the state of the protein changes stochastically over time. Given an initial state s0 of the protein at time 0, an MDM can be used to predict a sequence of future states s1, s2,…, where st is the state of the protein at time t × h for t=1, 2,…. If st=s, then the next state st+1 can be predicted by choosing an outgoing edge (s, s′) from s with probability ass′ and setting st+1=s′. The simple and explicit structure of MDMs allows such predictions to be computed efficiently.
In a point-based MDM, a state represents a single conformation. In a cell-based MDM, a state represents a set of conformations (Section 2.1). The definition of states is crucial. The choice of a single conformation as a state is more precise and informative than the choice of a set of conformations. However, it often causes violation of the Markovian property and consequently reduces the predictive power of the MDM. We now address the delicate question of defining the states.
3.1 Why hidden states?
By defining a state as a subset of the protein conformation space, rather than a single conformation, cell-based MDMs achieve the dual objectives of better satisfying the Markovian assumption and reducing the number of states. This is a major step forward. However, cell-based MDMs still violate the Markovian assumption in a subtle way. Consider a protein at a conformation q near the boundary of a cell. The future state of the protein depends not only on q, but also on the protein's velocity, in other words, on the past history of how the protein reached q. By requiring each conformation to belong to a single state, cell-based MDMs violate the Markovian assumption, especially near the cell boundaries. Similar violations also occur in cells corresponding to shallow energy basins, where the protein's energy landscape is flat.
One way of avoiding such violations is to define more refined states using information on both conformation and conformational velocity. However, this necessarily increases the number of states, thus partially reversing a key advantage of cell-based MDMs. Furthermore, a much larger dataset is needed for model construction in order to capture the detailed transition probabilities among the refined states. In contrast, we propose to assign a conformation to multiple states and use probability to capture the uncertainty of state assignment. This leads to an MDM with hidden states, formally, an HMM. Our HMM for protein dynamics is specified as a tuple Θ=(S, C, Π, A, E):
the set of states S = {si ∣ i=1, 2,…, K};
the conformation space C of a protein;
Π={πi ∣ i=1, 2,…, K}, where πi is the prior probability that the protein is in state si∈S at time t=0;
A={aij ∣ i, j=1, 2,…, K}, where aij=p(sj | si) is the probability of transitioning from state si∈S to sj∈S in a single time step of duration h;
E = {ei ∣ i=1, 2,…, K}, where ei(q)=p(q | si) is the emission probability of observing conformation q∈C when the protein is in state si∈S.
The state space S is discrete, while the conformation space C is continuous. Intuitively each state si∈S loosely matches an energy basin of the protein, and the corresponding emission probability ei(q)=p(q|si) connects states with conformations by modeling the distribution of protein conformations within the basin.
In an HMM, we cannot assign a unique state for a given conformation q. Instead, we calculate p(si | q), the probability that q belongs to a state si. The uncertainty in state assignment arises because at a conformation q, the protein may have different velocities, as well as other differences that we choose not to model or do not know about. We model the uncertainty due to this lack of information with the emission probability distributions.
In contrast, a cell-based MDM partitions C into disjoint regions C1, C2,…, and each state si represents a region Ci. So we can assign a conformation q to a unique state. If we define ei as a step function such that ei(q) is a strictly positive constant for q∈Ci and 0 otherwise, then the states are no longer hidden, and our model degenerates into a cell-based MDM. Our distribution-based models are therefore more general than cell-based MDMs.
Hidden states was used to model protein structure before (Hirsch and Habeck, 2008), but the goal there was to capture compactly the variations in an ensemble of protein structures obtained from NMR experiments, rather than the dynamics.
3.2 What is a good model?
Another difficulty with cell-based MDMs is the lack of a principled criterion for evaluating model quality. Cell-based MDMs are constructed to maximize the self-transition probabilities for the states in the model (Chodera et al., 2007). This criterion, however, results in the paradoxical conclusion that a trivial one-state model is perfect, as all transitions are self-transitions. Since simple models are generally preferred, how do we decide that a simple model, such as the one-state model, is (not) as good as a more complex one?
Our goal is to build a model Θ of the long-timescale dynamics of a protein from a given dataset D of MD simulation trajectories. The model Θ is then used to predict the protein's kinetic and dynamic properties, such as mean first-passage times (MFPTs; Leach, 2001), p-fold values (Du et al., 1998), transition state ensembles (Leach, 2001), etc. A model Θ1 has stronger predictive power than a model Θ2, if Θ1 predicts the kinetic and dynamic properties more accurately than Θ2. Clearly, it is impossible to check the predictive power of a model Θ on all such properties, as we may not even know all of them in advance. However, since many kinetic and dynamic properties are determined by protein motion trajectories, we can check instead the ability of Θ to predict these trajectories. In our HMM framework, we do this by calculating the likelihood p(D | Θ), which is the probability that a dataset D of MD simulation trajectories occur under the model Θ. The likelihood p(D|Θ) measures the quality of Θ.
Specifically, let D={Di ∣ i=1, 2,…} be a dataset of trajectories. Each trajectory Di is a sequence of protein conformations (q0, q1,…, qT), where qt is the protein conformation at time t × h. The likelihood of Θ for Di is
| (1) |
where st is the state of the protein at time t × h and p(s0), p(st|st−1) and p(qt|st) are given by the model parameters Π, A and E of Θ, respectively (Bishop, 2007). The summation ∑Q is performed over all possible state assignments Q=(s0, s1,…, sT)∈STa to the conformations (q0, q1,…, qT) in Di. The likelihood of Θ for the entire dataset D is simply p(D|Θ)=∏i p(Di|Θ).
In contrast to the cell-based MDM, the likelihood p(D|Θ) provides a quantitative measure of model quality and enables us to compare models with different number of states. This is possible, because our model uses emission probabilities ei(q)=p(q | si) to connect states with conformations, while a cell-based MDM does not. The likelihood criterion shows that a single-state MDM is in fact not good. Although the transition probabilities p(st | st−1)=1 for all t, the emission probabilities p(qt|st) are small, because the model relies on a single state to capture variability over the entire conformation space. Hence, the overall likelihood p(D|Θ) is small.
3.3 Benefits and limitations
One goal of model construction is to predict a protein's kinetic and dynamic properties. Since our model is constructed from MD simulation data (Section 5), a basic question is ‘How can the model provide better predictions than the simulation trajectories themselves?’ The answer is that the model generalizes the data under the Markovian property and thus contains a lot more trajectories than the dataset used in the model construction. Consider, for example, a dataset contains two trajectories with state sequences (s0, s1, s2) and (s0′, s1, s2′). Using the Markovian property, the model assumes that two additional state sequences (s0, s1, s2′) and (s0′, s1, s2) are also valid. By combining the trajectories, the model generates exponentially more trajectories than the dataset contains. If the assumption of the Markovian property is valid, then the model is a more accurate approximation of the underlying protein dynamics and can predict kinetic and dynamic properties better.
A related question is ‘With MD simulation data at the nanosecond scale, how can the model predict events at the microsecond or millisecond scale?’ Again using the Markovian property, the model concatenates short simulation trajectories into much longer ones (Chodera et al., 2006, 2007) and uses them to predict long-timescale kinetic and dynamic properties. This approach can succeed even for large proteins, if the transitions between metastable states are relatively fast (Henzler-Wildman and Kern, 2007).
At the same time, our model cannot have state transitions not implied by the simulation trajectories in the original dataset and thus does not address the question of conformation space sampling, which is difficult, but has seen rapid progress in recent years (e.g. Raveh et al., 2009; Singhal et al., 2004). Advances in sampling methods will provide better simulation data and improve the quality of the resulting models.
4 MODEL EXPLOITATION
We now illustrate the use of our model in the context of protein folding. However, our approach is general and can be used to study the dynamics of a folded protein as well.
First, our MDM is a graphical model. We can gain various insights of the underlying folding process by inspecting the structure and the edge weights of the graph. We give an example in Section 6.
Next, our MDM is generative and can be used for simulation. To generate a simulation trajectory of length T, we first sample a state sequence (s0, s1,…, sT) from the model. We sample the initial state s0 according to a prior distribution adapted to the environmental condition of the biological events under study. We then sample each subsequent state st conditioned on the previous state st−1 according to the transition probabilities A. Next, we generate the trajectory (q0, q1,…, qT) by sampling each qt conditioned on st with probability p(qt | st) according to the emission probabilities E.
Furthermore, an important advantage of MDMs is that they can be analyzed systematically without explicitly generating simulation trajectories. Specifically, our model allows for efficient computation of ensemble properties of protein folding. Ensemble properties, such as MFPT, characterize the average behavior of a folding process over myriad pathways at the microscopic level. In principle, we can compute ensemble properties by simulating many individual pathways and then averaging over them, but explicit simulation is computationally expensive. In the following, we describe a more efficient algorithm that computes MFPT using our model. The p-fold value and other ensemble properties can be computed similarly.
The MFPT of a conformation q is the expected time for a protein to reach a folded conformation, starting from q. A straightforward way of estimating the MFPT of q is to simulate many folding trajectories, each starting from q and terminating upon reaching a folded conformation. The estimated MFPT is then the average duration of these trajectories. This approach typically requires a huge number of simulation trajectories to get a reliable estimate for a single conformation q. Instead, we apply first-step analysis (Taylor and Karlin, 1994) from Markov chain theory to our model. It implicitly simulates infinitely many trajectories (Section 3.3), resulting in much faster and reliable computation of MFPTs.
Our computation proceeds in two stages. First, we compute the MFPTs for all the states in S. Let CF ⊂ C be the subset of folded conformations of a protein. Let γi be the first-passage time (FPT) of a folding trajectory that starts in state si. Consider what happens in the very first time step of the folding trajectory:
If the initial conformation q0 ∈ CF, then obviously γi=0. This event happens with probability ei(CF)=∫CF ei(q)dq.
If q0 ∉ CF, then γi depends on the MFPT of the state that the trajectory reaches after a one-step transition. This event happens with probability 1 − ei(CF).
The MFPT for si is 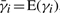 , where the expectation is taken over all trajectories that start in si and end in CF. By conditioning on the events in the first time step, we obtain the following equation for
, where the expectation is taken over all trajectories that start in si and end in CF. By conditioning on the events in the first time step, we obtain the following equation for 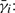 :
:
| (2) |
The transition probabilities p(sj|si) are model parameters. The only unknowns in (2) are the MFPTs 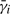 for i = 1, 2,…, K. Since there is one such equation for each
for i = 1, 2,…, K. Since there is one such equation for each 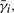 , we get a linear system of K equations with K unknowns, which can be solved efficiently using standard numerical methods. The algebraic process of solving the linear system implicitly enumerates all possible state sequences of the folding trajectories in an efficient way.
, we get a linear system of K equations with K unknowns, which can be solved efficiently using standard numerical methods. The algebraic process of solving the linear system implicitly enumerates all possible state sequences of the folding trajectories in an efficient way.
Next, we compute the MFPT for a given conformation q0. Let γ be the FPT of a folding trajectory that starts at q0. Conditioning on the initial state s0 at t = 0, we see that the MFPT of q0 is given by
| (3) |
We calculate p(s0 | q0) using the Bayes rule:
| (4) |
where p(s0) and p(q0| s0) can be obtained from the prior probabilities Π and the emission probabilities E of the model, respectively. Calculating E(γ|q0, s0) is more subtle. Suppose that the initial state s0 is some particular state si∈S. It is tempting to think that 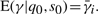 . This is incorrect, because
. This is incorrect, because 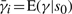 and the additional information provided by q0 may alter the expected value of γ. To calculate E(γ|q0, s0), we condition once more on the state s1 at time t=1:
and the additional information provided by q0 may alter the expected value of γ. To calculate E(γ|q0, s0), we condition once more on the state s1 at time t=1:
| (5) |
| (6) |
where the last line follows from the conditional independence properties of HMMs (Bishop, 2007). Now the values for E(γ|s1) can be obtained from the MFPTs 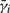 , and the values for p(s1|s0), from the transition probabilities A of the model. Substituting (4) and (6) into (3) gives us the desired result.
, and the values for p(s1|s0), from the transition probabilities A of the model. Substituting (4) and (6) into (3) gives us the desired result.
In practice, when we compare with experimental measures, we are interested in the MFPT for a region C′ of C rather than a single conformation q0∈C. To calculate E(γ|C′), we need to modify (3), (4), and (6) slightly by integrating q0 over C′.
5 MODEL CONSTRUCTION
Under the likelihood criterion, we want to construct a model Θ that maximizes p(D|Θ) for a given dataset D of MD simulation trajectories. Expectation maximization (EM) is a standard algorithm for such optimization problems. However, EM is computationally intensive. It may also get stuck in a local maximum and fail to find the model with maximum likelihood. To alleviate these difficulties, we proceed in three steps. First, we preprocess the input trajectories to remove the ‘noise’, i.e. motions at timescales much shorter than that of interest. Next, we use K-medoids clustering to build an initial model Θ0. Since clustering is much faster than EM, we run the clustering algorithm multiple times and choose the best result as Θ0. This reduces the chance of ending up with a bad local maximum. Finally, we initialize EM with Θ0 and search for the model with maximum p(D|Θ). Since both K-medoids clustering and EM are well known algorithms (see, e.g. Bishop, 2007), we only describe the relevant details of these steps below.
Data preparation: the time resolution h of the model should be compatible with the timescale of biological events under study. If h is too large, the resulting model may miss the events under study. If h is too small, the model will try to capture fine details at uninteresting short timescales and become unnecessarily complex with reduced predictive power. In our tests, a relatively wide range of h values led to models with similar predictive power. We typically set h to be 1/100 to 1/10 the timescale of interest. We then apply standard signal processing techniques (Oppenheim and Schafer, 2009) to smooth and downsample each trajectory in D so that the time duration between any two successive conformations along a trajectory is exactly h.
Emission probability distributions: the emission probability ei models the distribution of protein conformations in state si. We approximate ei with a Gaussian distribution:
| (7) |
where d(q, μi) denotes a suitable distance measure between the conformations q and μi. Other approximating distributions are possible. There are two main considerations in choosing the distribution: it should match the distribution of conformations in si and be simple enough to be learned effectively with a limited amount of data.
Initialization: the states in our model roughly correspond to energy basins. Within a basin, a protein interconverts rapidly, which allows interstate protein motions to satisfy the Markovian property. Rapid interconversion results in a high-density cluster of protein conformations inside the basin. So, to get an initial estimate of the states, we treat the input dataset D as a set of conformations and use the K-medoids algorithm to partition the conformations in D into K clusters, where K is a prespecified parameter. K-medoids forms compact clusters by minimizing the sum of intracluster distances between conformations (Bishop, 2007) under the same distance d as that in (7). The center of a cluster B is a conformation q∈B that minimizes the sum of distances from q to other conformations in B.
Each cluster becomes a state of our initial model Θ0. Using the cluster labels of the conformations in D, we can easily compute the prior probabilities Π and transition probabilities A for Θ0 by simply counting. To get the emission probability ei(q)=N(q|μi, σi2), we set μi to the center of the cluster corresponding to state si and σi2 to the variance of conformations in this cluster.
Optimization: we use Θ0 to initialize the EM algorithm and search for a K-state HMM Θ that maximizes the likelihood p(D| Θ). EM iterates over two steps, expectation and maximization, and improves the current model until no further improvement is possible. Inspection of (1) shows that our main difficulty is the summation of all possible state assignments to the conformations (q0, q1,…, qT) along a trajectory Di. Performing this summation by brute force takes time O(KT), which is exponential in the length T of the trajectory. EM overcomes this difficulty through dynamic programming. See Bishop (2007) for details.
The number of states: the number of states K controls the model complexity. It must be specified in both K-medoids clustering and EM. A complex model with many states in principle fits the data better, thus achieving higher likelihood. However, it may suffer from overfitting when there is insufficient data. A complex model is also more difficult to analyze and understand. Typically, a simple model is preferred when it does not sacrifice much predictive power. To choose a suitable K value, we pick a small random subset D′ of D as a test dataset. We start with a small K value and gradually increases it until the likelihood p(D′|Θ) levels off. It is important to note that we can perform such a search over model complexity because our likelihood criterion enables us to compare models with different number of states.
6 RESULTS
6.1 Synthetic energy landscapes
Synthetic energy landscapes are useful for testing our algorithms in controlled settings where the ground truth is known. In particular, we want to examine whether our likelihood criterion and model construction algorithm can identify simple models with strong predictive power.
We created a series of five energy landscapes in two dimensions (Fig. 1). Landscapes A and B each contains one energy basin, but B's basin is slightly more elongated. Landscapes C, D and E each contains two basins with varying amount of separation. For each landscape, we used Langevin dynamics to generate 1000 trajectories of 200 time steps each. We set aside half of the trajectories as the training dataset for model construction and the other half as the test dataset D′ for checking the quality of the model constructed.
Fig. 1.

Five synthetic energy landscapes and the corresponding models.
For each landscape, we built models with increasing number of states. In all the models, the resolution h is 10 simulation time steps. The distance measure d used in defining the emission probabilities is the Euclidean distance in the plane.
Figure 2 plots the scores of all the models. The score is the average log-likelihood of a model for a single transition step along a trajectory. It is computed by dividing the log-likelihood of a model given D′ by the total number of conformations in D′. Figure 2 shows that for landscape A, which contains only 1 basin, the 1-state model is slightly better than the 2-state model. As we move from landscape A to E, the predictive power of the 1-state model degrades. The 2-state model performs fairly well on all five energy landscapes. Figure 1 shows the differences between the 1- and 2-state models by simulating them and plotting the resulting conformations. Figure 2 also shows that increasing the number of states beyond two has negligible benefit. Although these results are not surprising, they highlight the importance of a principled criterion for evaluating the model quality.
Fig. 2.
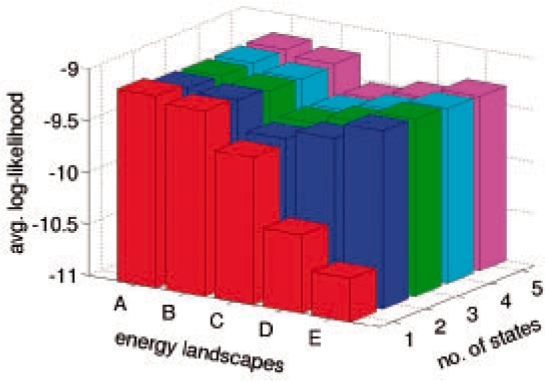
Average log-likelihood scores of the models for synthetic energy landscapes.
6.2 Alanine dipeptide
Alanine dipeptide (Ace-Ala-Nme) is a small molecule widely used for studying biomolecular motion, as it is simple and exhibits an extensive range of torsional angles. We use the same dataset as that from a previous study (Chodera et al., 2007). It consists of 1000 MD simulation trajectories, each roughly 20 ps in duration. Again, we divide them equally into training and test datasets.
We built models with up to seven states. They are named A1 to A7. As alanine dipeptide is very small, its motion is fast. So the time resolution h of the models is set to 1.0 ps. A conformation of alanine dipeptide is specified by three backbone torsional angles (ϕ, ψ, ω), and the distance between two conformations is defined as the root sum squared angular differences between the corresponding torsional angles.
The conformation space of alanine dipeptide has been manually decomposed into six disjoint regions, each corresponding to a meta-stable state. This well-accepted decomposition has led to several dynamic models of alanine dipeptide (Chodera et al., 2006, 2007). For comparison, we built a 6-state model based on the same manual decomposition. During the model construction, instead of applying K-medoids, we group conformations into a cluster if they belong to the same disjoint region of the manual decomposition. Other steps of the construction algorithm remain the same. The resulting model is named M6.
Figure 3 plots the average log-likelihood scores of all the models constructed. Models A3–A7 all achieve scores comparable with that of M6. The interesting finding is that although the score jumps substantially as we move from A1 to A3, the score of A3 is almost as good as those of A6 and M6. This indicates that for predicting the motion of alanine dipeptide, the simpler 3-state model A3 is almost as good as the 6-state model M6, which is obtained from the well-accepted manual decomposition of the alanine dipeptide conformation space!
Fig. 3.
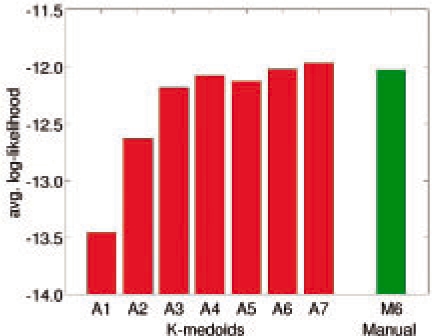
Average log-likelihood scores of alanine dipeptide models.
To see the differences between A3 and M6, we simulated the two models and plotted the resulting conformations (Fig. 4). Both models capture accurately the frequently visited regions of the conformation space, shown in red and blue in Figure 4. These densely sampled regions correspond to energy basins that dominate the long-timescale dynamics, and accurate modeling of these regions is crucial. For A3, the conformations shown in green capture a large, but less frequented region of the conformation space. Although M6 models the same region as two closely spaced clusters of conformations, the overall density and the location of the conformations are similar in both models. M6 also models the rarely visited region between 0 <ϕ< 90. Due to the transient nature of the protein in these conformations, the additional model complexity contributes little to the observable long-term dynamical phenomena. Therefore, the average log-likelihood score levels off when the number of states in the model surpasses 3.
Fig. 4.

Conformations generated from the 3-state model A3 (a) and the 6-state model M6 (b).
To further validate our models, we used both A3 and M6 to compute MFPTs between the αR and β/C5 regions of the conformation space. We designate conformations with (ϕ = −70 ± 15, ψ = −40 ± 15, ω=180 ± 15) to be within the αR region, and conformations with (ϕ=-140 ± 15, ψ=160 ± 15, ω=180 ± 15) to be within the β/C5 region. Although the results for A3 and M6 differ somewhat in details, they are consistent (Table 1). Both indicate that the transition from αR to β/C5 is roughly an order of magnitude faster than the reverse transition. This matches well with the results reported by Chekmarev et al. (2004).
Table 1.
Estimated MFPTs between αR and β/C5 regions of the alanine dipeptide conformation space
| MFPT (ps) |
||
|---|---|---|
| A3 | M6 | |
| αR → β/C5 | 26.5 | 28.5 |
| β/C5 → αR | 187.0 | 124.0 |
To assess the efficiency of our algorithm for MFPT computation (Section 4), we also computed the MFPTs by explicitly generating simulation trajectories from our constructed models. It took our algorithm 1 s to compute one MFPT, as the alanine dipeptide models are all very simple. In comparison, it took 120 s to generate a sufficiently large number of simulation trajectories from the same HMM in order to bring the standard deviation of the MFPT estimate down to 1% of its value.
6.3 Villin
The data for the fast-folding variant of the villin headpiece (HP-35 NleNle) was generated by the Folding@home project. It consists of 410 MD trajectories, initiated from nine unfolded conformations denoted by Ik, k=0,…, 8. Each trajectory is 1 μs in duration on the average.
The training dataset contains a huge number of conformations. For computational efficiency, we cluster the conformations to form microstates in the conformation space. We sample 8000 conformations uniformly along the trajectories in the dataset as the microstate centers. The remaining conformations in the dataset are then clustered to the nearest microstate centers according to the root mean square deviation (RMSD) of all heavy atoms in the peptide. Earlier work (Plaku and Kavraki, 2007) then indicates that we may assume that the peptide transits directly between microstates that are close according to the RMSD between microstate centers and define a graph that approximates the dynamics of the peptide accordingly. Each node of this graph is a microstate and is connected to a small number of other nodes close by in RMSD. An edge of the graph is assigned a weight equal to the RMSD between the end nodes. The distance between two microstates is defined as the length of the shortest path between them in the graph. For large proteins, this graph-based distance metric better captures the dynamics than the RMSD metric.
We applied our model construction algorithm over the set of microstates and built models with increasing number of states, all at h=5 ns. The average log-likelihood score (Fig. 5) improves significantly when the number of states grows from 1 to 20. It improves more gradually between 20 and 200 states. Beyond 200 states, the score remains approximately constant.
Fig. 5.
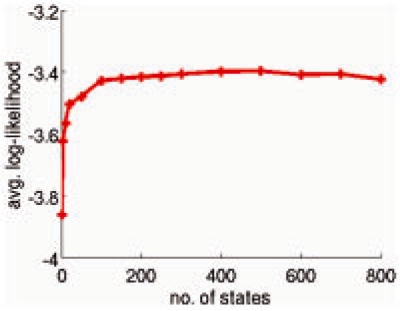
Average log-likelihood scores for the villin headpiece models.
To examine the dynamics of this peptide visually, consider the 20-state model. Figure 6a shows that state 7, 12, 13, 15 and 18 are the most frequently visited states and thus significantly influence the long-term dynamics. By calculating the probabilities p(s | q), we infer that the initial conformations most likely belong to state 12 and the native conformation most likely belongs to state 15. States 12, 7 and 18 form a cycle and transit among themselves with high probability. Although the conformations in state 12 may possess a significant degree of helical structure, helix 1 is often oriented in the wrong direction. From state 12, the peptide transits to states 7 and 18 by attaining additional helical structure (helix 3). In state 18, the peptide loses significant portions of helix 1 and 2. However, it can regain them relatively easily by transiting directly to states 7 and 12. It is interesting to observe the transition from states 12 to 13, which corrects the orientation of helix 1. From state 13, the peptide proceeds to state 15, the folded state, with very high probability. The model also shows that it is much more difficult for the peptide to get out of state 15 than to get in. Consequently, state 15 is also the most frequently visited state and dominates the long-term dynamics, as expected.
Fig. 6.

(a) Main state transitions of the 20-state villin headpiece model. The size of each node is proportional to the probability of the corresponding state in the stationary distribution. The width of each edge is proportional to the transition probability. States with probability <0.01 in the stationary distribution, self-transitions and transitions with probability <0.002 are not shown to avoid cluttering the diagram. The initial conformations most likely belong to state 12, and the native conformation most likely belongs to state 15. (b) Example conformations from states 7, 12, 13, 15 and 18. The residues forming helix 1 are drawn in red. (c) The most likely state transition sequences from states 12 to 15.
Our model suggests that attaining both the structure and the correct orientation for helix 1 is likely a significant folding barrier. This is consistent with earlier work suggesting that the ease of attaining helix 1 is one of the factors allowing certain initial conformations to fold faster than others (Ensign et al., 2007).
We also computed the MFPTs for the initial conformations I0 to I8 (Table 2). The results lie in the same microsecond range as the experimental measurements of 4.3 μs from laser temperature jump (Kubelka et al., 2003) and 10 μs from NMR line-shape analysis (Wang et al., 2003). In addition, the MFPTs for I4 and I7 are slightly smaller, which is consistent with the computational analysis of Ensign et al. in (2007).
Table 2.
Estimated MFPTs for nine initial conformations of the villin headpiece (HP-35 NleNle)
| MFPT (μs) | ||||||||
|---|---|---|---|---|---|---|---|---|
| I0 | I1 | I2 | I3 | I4 | I5 | I6 | I7 | I8 |
| 5.89 | 5.87 | 5.86 | 5.88 | 5.84 | 5.84 | 5.85 | 5.84 | 5.86 |
For comparison, we also tried to compute the MFPTs by explicitly generating trajectories from the constructed models. However, after 30 min of computation, the estimated MFPTs are still two orders of magnitude below the microsecond range. In comparison, the results in Table 2 were obtained in <1 min of computation.
7 DISCUSSION
The past decade has witnessed an increasing interest in graphical models of protein dynamics at long timescales. Most recently, the focus has been on cell-based MDMs built from precomputed MD simulation data. Existing methods, however, suffer from two main shortcomings. First, defining states by partitioning the protein conformation space into disjoint cells causes violation of the Markovian property. Second, there is no systematic criterion for evaluating the model quality. Our work addresses these two shortcomings by defining states as probability distributions of conformations. This reflects the view that a single conformation does not contain enough information to be assigned a unique state. The resulting HMM-based modeling framework evaluates the model quality by the likelihood of a model given a test dataset of simulation trajectories. In contrast with the cell-based MDMs, our approach enables us to compare models with different number of states and choose the best one according to the likelihood criterion. The results on synthetic energy landscapes and alanine dipeptide illustrate this benefit.
In general, MDMs have several advantages over direct data analysis of MD simulation trajectories (Amadei et al., 1993; Teodoro et al., 2002), using techniques such as singular value decomposition (SVD). MDMs generalize the simulation data used in constructing them. They not only identify the important states, but also assemble them together to provide a global view of the underlying stochastic protein dynamics. Section 4 shows various ways of exploiting MDMs. Such tasks are difficult or impossible with direct data analysis. At the same time, these two approaches are complementary. When simulation data is limited, it may be more effective and simpler to perform data analysis directly. Furthermore, we may use SVD to perform dimensionality reduction on the MD simulation data in a preprocessing step before running our model construction algorithm.
One important remaining issue is to scale up our approach to handle large proteins. MD simulation is computationally expensive, but advances in computer technology are making it more affordable than before, and large simulation data repositories will become readily available over time. Increasingly, the future challenge will be to gain biological insights from this data by building simple and yet powerful models. As we scale up to larger proteins, the dynamics of protein motion also becomes more complex. For large proteins, it is likely that motions at different timescales contribute to different biological functions. A hierarchy of MDMs constructed at different timescales may capture such multi-timescale dynamics.
Finally, it will be interesting to apply our approach to model the dynamics of a folded protein. The conformational flexibility of a folded protein is critical to some of its functions (Henzler-Wildman and Kern, 2007), such as allosteric interactions. Here, our approach is likely to scale up well to larger proteins, as transitions between the folded states are often fast and hence more easily captured by short MD simulations.
ACKNOWLEDGEMENTS
We thank Vijay Pande and Nina Singhal, who provided us MD simulation data for alanine dipeptide and villin.
Funding: AcRF grant R-252-000-350-112 from the Ministry of Education, Singapore (to D.H., in parts). National Science Foundation grant DMS-0443939 and two grants from the Academic Excellence Alliance program between King Abdullah University of Science & Technology (KAUST) and Stanford University (to J-C.L., in parts)
Conflict of Interest: none declared.
REFERENCES
- Amadei A, et al. Essential dynamics of proteins. Prot. Struct. Funct. Genet. 1993;17:412–425. doi: 10.1002/prot.340170408. [DOI] [PubMed] [Google Scholar]
- Amato NM, et al. Using motion planning to map protein folding landscapes and analyze folding kinetics of known native structures. J. Comput. Biol. 2003;10:239–255. doi: 10.1089/10665270360688002. [DOI] [PubMed] [Google Scholar]
- Apaydin MS, et al. Stochastic roadmap simulation: an efficient representation and algorithm for analyzing molecular motion. J. Comput. Biol. 2003;10:257–281. doi: 10.1089/10665270360688011. [DOI] [PubMed] [Google Scholar]
- Bishop G. Pattern Recognition and Machine Learning. New York: Springer; 2007. [Google Scholar]
- Chekmarev DS, et al. Long-time conformational transitions of alanine dipeptide in aqueous solution: continuous and discrete-state kinetic models. J. Phys. Chem. B. 2004;108:19487–19495. [Google Scholar]
- Chiang TH, et al. Proceedings of the ACM International Conference on Research in Computational Molecular Biology (RECOMB) Venice, Italy: 2006. Predicting experimental quantities in protein folding kinetics using stochastic roadmap simulation. [Google Scholar]
- Chodera J, et al. Long-time protein folding dynamics from short-time molecular dynamics simulations. Multiscale Model. Simul. 2006;5:1214–1226. [Google Scholar]
- Chodera J, et al. Automatic discovery of metastable states for the construction of markov models of macromolecular conformational dynamics. J. Chem. Phys. 2007;126:155101. doi: 10.1063/1.2714538. [DOI] [PubMed] [Google Scholar]
- Du R, et al. On the transition coordinate for protein folding. J. Chem. Phys. 1998;108:334–350. [Google Scholar]
- Elber R. Long-timescale simulation methods. Curr. Opin. Struct. Bio. 2005;15:151–156. doi: 10.1016/j.sbi.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Ensign DL, et al. Heterogeneity even at the speed limit of folding: large-scale molecular dynamics study of a fast-folding variant of the villin headpiece. J. Mol. Biol. 2007;374:806–816. doi: 10.1016/j.jmb.2007.09.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haliloglu T, et al. Gaussian dynamics of folded proteins. Phys. Rev. Lett. 1997;79:3090–3093. [Google Scholar]
- Henzler-Wildman K, Kern D. Dynamic personalities of proteins. Nature. 2007;450:964–972. doi: 10.1038/nature06522. [DOI] [PubMed] [Google Scholar]
- Hirsch M, Habeck M. Mixture models for protein structure ensembles. Bioinformatics. 2008;24:2184–2192. doi: 10.1093/bioinformatics/btn396. [DOI] [PubMed] [Google Scholar]
- Huang X, et al. Proceedings of the Pacific Symposium on Biocomputing. Hawaii, USA: 2010. Constructing multi-resolution markov state models (MSMs) to elucidate rna hairpin folding mechanisms. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavraki LE, et al. Probabilistic roadmaps for path planning in high-dimensional configuration spaces. IEEE Trans. Robotics Automat. 1996;12:66–580. [Google Scholar]
- Kubelka J, et al. Experimental tests of villin subdomain folding simulations. J. Mol. Biol. 2003;329:625–630. doi: 10.1016/s0022-2836(03)00519-9. [DOI] [PubMed] [Google Scholar]
- Leach AR. Molecular Modeling: Principles and Applications. London: Prentice Hall; 2001. [Google Scholar]
- Levitt M, et al. Protein normal-mode dynamics: Trypsin inhibitor, crambin, ribonuclease and lysozyme. J. Mol. Biol. 1985;181:423–447. doi: 10.1016/0022-2836(85)90230-x. [DOI] [PubMed] [Google Scholar]
- Lois G, et al. The free energy reaction path theory of reliable protein folding. Biophys. J. 2009;96:589a–590a. doi: 10.1529/biophysj.108.133132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheim AV, Schafer RW. Discrete-Time Signal Processing. 3rd. London: Prentice Hall; 2009. [Google Scholar]
- Ozkan SB, et al. Fast-folding protein kinetics, hidden intermediates, and the sequential stabilization model. Prot. Sci. 2002;11:1958–1970. doi: 10.1110/ps.0207102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pande VS, et al. Atomistic protein folding simulations on the hundreds of microsecond timescale using worldwide distributed computing. Biopolymers. 2002;68:91–109. doi: 10.1002/bip.10219. [DOI] [PubMed] [Google Scholar]
- Plaku E, Kavraki LE. SIAM International Conference on Data Mining. Minnesota, USA: 2007. Nonlinear dimensionality reduction using approximate nearest neighbors; pp. 180–191. [Google Scholar]
- Raveh B, et al. Rapid sampling of molecular motions with prior information constraints. PLoS Comput. Biol. 2009;5:e1000295. doi: 10.1371/journal.pcbi.1000295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw DE, et al. Proceedigs of the International Symposium on Computer Architecture. California, USA: 2007. Anton, a special-purpose machine for molecular dynamics simulation. [Google Scholar]
- Shea J.-E, Brooks C.L., III From folding theories to folding proteins: A review and assessment of simulation studies of protein folding and unfolding. Annu. Rev. Phy. Chem. 2001;52:499–535. doi: 10.1146/annurev.physchem.52.1.499. [DOI] [PubMed] [Google Scholar]
- Singh AP, et al. Proceedings of the International Conference on Intelligent Systems for Molecular Biology (ISMB) Heidelberg, Germany: 1999. A motion planning approach to flexible ligand binding; pp. 252–261. [PubMed] [Google Scholar]
- Singhal N, et al. Using path sampling to build better Markovian state models: Predicting the folding rate and mechanism of a tryptophan zipper beta hairpin. J. Chem. Phys. 2004;121:415–425. doi: 10.1063/1.1738647. [DOI] [PubMed] [Google Scholar]
- Taylor H, Karlin S. An Introduction to Stochastic Modeling. New York: Academic Press; 1994. [Google Scholar]
- Teodoro M, et al. Proceedings of the ACM International Conference on Computational Molecular Biology (RECOMB) Washington, DC, USA: ACM; 2002. A dimensionality reduction approach to modeling protein flexibility; pp. 299–308. [Google Scholar]
- Wang M, et al. Dynamic NMR line-shape analysis demonstrates that the villin headpiece subdomain folds on the microsecond time scale. J. Am. Chem. Soc. 2003;125:6032–6033. doi: 10.1021/ja028752b. [DOI] [PubMed] [Google Scholar]