

Abstract
Dyskeratosis congenita (DC) is characterized by multiple features including mucocutaneous abnormalities, bone marrow failure and an increased predisposition to cancer. It exhibits marked clinical and genetic heterogeneity. DKC1 encoding dyskerin, a component of H/ACA small nucleolar ribonucleoprotein (snoRNP) particles is mutated in X-linked recessive DC. Telomerase RNA component (TERC), the RNA component and TERT the enzymatic component of telomerase, are mutated in autosomal dominant DC, suggesting that DC is primarily a disease of defective telomere maintenance. The gene(s) involved in autosomal recessive DC remains elusive. This paper describes studies aimed at defining the genetic basis of AR-DC. Homozygosity mapping in 16 consanguineous families with 25 affected individuals demonstrates that there is no single genetic locus for AR-DC. However, we show that NOP10, a component of H/ACA snoRNP complexes including telomerase is mutated in a large consanguineous family with classical DC. Affected homozygous individuals have significant telomere shortening and reduced TERC levels. While a reduction of TERC levels is not a universal feature of DC, it can be brought about through a reduction of NOP10 transcripts, as demonstrated by siRNA interference studies. A similar reduction in TERC levels is also seen when the mutant NOP10 is expressed in HeLa cells. These findings identify the genetic basis of one subtype of AR-DC being due to the first documented mutations in NOP10. This further strengthens the model that defective telomere maintenance is the primary pathology in DC and substantiates the evidence in humans for the involvement of NOP10 in the telomerase complex and telomere maintenance.
INTRODUCTION
Telomerase is a large ribonucleoprotein complex found in organisms ranging from ciliates to humans that is responsible for the de novo synthesis and the maintenance of telomere ends (1). The complex has two core subunits, telomerase reverse transcriptase (TERT) and the telomerase RNA component (TERC), where TERT copies the short template of TERC into telomeric DNA to extend the 3′ end of the chromosome on the lagging strand (2). In addition to TERC being required for the replication of telomeres, its 3′ end can be folded into a structure reminiscent of box H/ACA small nucleolar RNAs (snoRNA) which have been shown to be involved in the pseudouridylation of ribosomal RNA (3-5). Four common proteins that are associated with H/ACA snoRNAs in humans are dyskerin, GAR1, NHP2 and NOP10 (also known as NOLA4, NOLA1, NOLA2 and NOLA3, respectively) and these four proteins have also been shown to be involved in the telomerase complex (2,6,7). Dyskerin, NHP2 and NOP10 are involved in the stability of the complex and closely interact with each other, whereas the involvement of GAR1 is more remote (8,9).
Dyskeratosis congenita (DC) is a multi-system bone marrow failure syndrome characterized by abnormal skin pigmentation, nail dystrophy, leukoplakia and an increased predisposition to cancer (10). DC is a very heterogeneous disorder both clinically and genetically. The severity, age of onset and range of somatic and haematological abnormalities varies widely as does the age of death. The inheritance of the disease also varies. The most common form is X-linked recessive (MIM #305000), with autosomal dominant (AD-DC, MIM #127550) and autosomal recessive (AR-DC, MIM #224230) forms also recognized. The severity of the mucocutaneous triad and other non-cutaneous features depends, to some extent, on the mode of inheritance; the X-linked recessive form is generally more severe than autosomal DC, with the AD form being less severe, but exceptions to this do exist. Heterogeneity is also observed within affected families and is more pronounced in female cases. Mutations in DKC1 encoding the protein dyskerin have been identified in patients with X-linked DC (11,12) and patients with AD-DC have been shown to have heterozygous mutations in TERC (13). Short telomeres have been observed in patients with mutations in both DKC1 and TERC (14,15). Recently, heterozygous mutations have been described in TERT in patients with both DC and aplastic anaemia and they too have short telomeres (16-21). One TERT mutation has been shown to segregate with the disease in a family with AD-DC (18). The gene(s) involved in AR-DC remains elusive.
Analysis of data obtained from the 270 families registered on the International Dyskeratosis Congenita Registry (DCR) formally at the Hammersmith Hospital, now relocated to The Royal London Hospital (London, UK), shows that mutations in both DKC1 and TERC account for approximately 36% of cases reported (approximately 30% DKC1 and 6% TERC). Two patients in the DCR have been found to have heterozygous missense mutations in TERT (17,19), but in the remaining patients the genetic basis of the disease remains unknown. The aim of this study was therefore to identify the genetic basis of AR-DC in all or some of these families in whom the molecular basis of DC is neither a DKC1, nor a TERC or TERT mutation.
RESULTS
Genotyping and linkage analysis
Homozygosity mapping was performed on 25 DC individuals with a presumed AR mode of inheritance from 16 consanguineous marriages. DNA samples were available for analysis from three affected individuals in two families, two affected individuals in five families and one affected individual in nine families (Fig. 1A). A minimum of 350 autosomal microsatellite markers were typed in all families at an average spacing of 10 cM. A family was scored as being homozygous for a particular marker if all the available affected individuals were homozygous for the same allele at that marker. Therefore, families with multiple affected children had more power than those with only one affected child available for analysis. From this initial screen no single marker was identified as being homozygous in all the families investigated. One marker was homozygous in 14 of 16 families (D14S1051, Fig. 1B), but on further investigation this particular marker was found to be uninformative in the parents of the majority of families studied, so is of limited use in further genetic analysis. Also the reported heterozygosity for this marker is 0.32 which further supports the high level of homozygosity observed. Data from this original genome screen suggests that there are two or more genes involved in the AR form of DC.
Figure 1.

Homozygosity mapping used to identify a locus for autosomal recessive DC. (A) Pedigrees of consanguineous families studied. DNA samples were not available on all individuals shown. Open circles and squares represent normal females and males, respectively. Black circles and squares represent affected females and males, respectively. Affected individuals marked with asterisk were not available for analysis. (B) The five most homozygous markers demonstrate the degree of genetic heterogeneity that exists in AR-DC. Where there is more than one affected individual in a family a consensus for the family is shown. Filled boxes indicate homozygosity for the same alleles within a family. For comparison the homozygosity profile of D15S1007 is also shown.
We then concentrated on the largest family with multiple affected individuals (family DCR 148), in order to identify a locus in this family. Samples were available on eight individuals (three affected, three unaffected and their parents). We used autozygosity mapping to try and identify a candidate gene locus. Autozygosity is defined as homozygosity in which the two alleles are identical by descent, i.e. they are copies of an ancestral gene; therefore people with rare recessive disorders in consanguineous families are likely to be autozygous for markers linked to the disease locus. All homozygous markers in all three affected individuals were typed in the whole family. Of the 18 homozygous markers, 15 were uninformative i.e. both parents were homozygous for the same allele. For two of the remaining markers one unaffected sibling was also homozygous rendering those particular loci unlikely. In the final marker, D15S1007, none of the unaffected siblings were homozygous and both parents were fully informative (Fig. 2A). A logarithm of odds (LOD) score was obtained from all the available data for chromosome 15q14 and this gave a maximum LOD score of 2.7 at 22 cM (Fig. 2B); indicating that the likelihood of this combination of alleles occurring by chance is 1 in 500. These data suggested the causative gene for DC in this family is on chromosome 15.
Figure 2.

Localization of NOP10 as a candidate gene in a family with AR-DC. (A) Pedigree from family DCR 148 showing partial chromosome 15 genotype data. Open circles and squares represent normal females and males, respectively. Black circles and squares represent affected females and males, respectively. The black and white bars highlight the parental disease carrying haplotypes (black, maternal; white, paternal). (B) LOD score graph for chromosome 15 identifying a potential candidate locus. This also shows the approximate location of NOP10 relative to the microsatellite markers.
A search for candidate genes in the region between D15S1007 (20.5 cM) and D15S118 (27.9 cM) revealed the gene encoding NOP10 is located in this candidate interval. NOP10 is an ideal candidate as it is known to be associated with the telomerase complex and it is associated with H/ACA snoRNPs in humans. NOP10 is composed of two exons which encode 64 amino acids and both were sequenced in all family members. A homozygous C to T change (c.100 C> T) in exon 2, resulting in an arginine (R) to tryptophan (W) substitution (p.R34W) was found in all three affected individuals (Fig. 3A). All the remaining members of the family were heterozygous for the mutation which therefore segregates fully with the disease phenotype. This mutation would be predicted to have a significant functional consequence as it occurs in a highly conserved region of the protein (Fig. 3B) and the amino acid change is from one that is basic (i.e. R) to one with an aromatic ring (i.e. W), which is predicted to change the structure of the protein.
Figure 3.
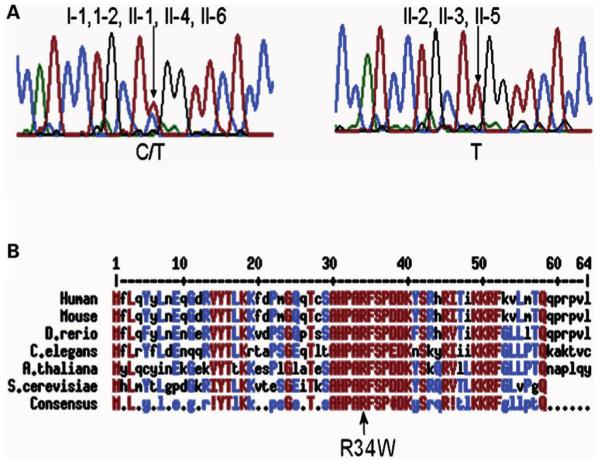
Identification of a homozygous mutation in NOP10. (A) Examples of sequence traces showing the C to T change at nucleotide 100 which causes an amino acid change at codon 34 from arginine to tryptophan. Both parents and the three unaffected children (I-1, I-2, II-1, II-4, II-6) are heterozygous for this mutation (C/T, left panel trace shown from II-1), whereas all three patients (II-2, II-3 and II-5) are homozygous for the mutation (T/T, right panel trace shown from II-5). (B) NOP10 is highly conserved. Human NOP10 was aligned with Nop10 of the mouse, Danio rerio, Caenorhabditis elegans, Arabidopsis thaliana and Saccharomyces cerevisiae. The p.R34W mutation is in a highly conserved region of the molecule. Highly conserved residues are shown in red and moderately conserved residues in blue. Exclamation mark equals I or V; hash symbol equals D, E, N or Q; bullets equal no significant homology between codons. The alignment was obtained with the MultAlin program (43).
Mutation screening of NOP10
Homozygosity at marker D15S1007 was not seen in any of the other families in the initial homozygosity screen suggesting that this mutation is a rare cause of AR-DC (Fig. 1B). To determine if this p.R34W or any other mutation is present in any other family, we sequenced samples from the index case of 171 uncharacterized families (the majority being sporadic cases) on the DCR. A total of nine different sequence variations were seen in this group of patients, six of which (c.34G> C, IVS1 + 21G> A, IVS1-15C> G, c.* 30A> G, c.*31C> T, c.*45G> T, where IVS1 is intron 1 and asterisk indicates after the stop codon) have been described previously as polymorphisms (16). c.34G> C causes an aspartic acid to histidine substitution at amino acid 12 and was observed as a heterozygous change in one individual but has also been reported by Yamaguchi et al. (16) in a screen of 282 healthy subjects. Three sequence variations are novel (IVS1 + 192 C> A, c.*136T> C, c.*149G> A). These changes are also thought to be non-pathogenic due to their frequency (frequency of the minor allele in each case, 0.306, 0.121 and 0.064, respectively) and due to their location, in the middle of an intron or in the 3′-UTR. It was interesting to note that the genetic background for the p.R34W mutation had a unique haplotype compared with all the other individuals typed (data not shown). The p.R34W substitution was not detected in a screen of 56 ethnically matched healthy individuals indicating that this change is not present at a polymorphic frequency in this population.
NOP10 mutation causes short telomeres
In order to determine if p.R34W has any effect in the telomerase complex in terms of telomere maintenance, telomere length was measured in all family members except II-2 (sample limitation). Telomere length was measured as a terminal restriction fragment (TRF) which also includes approximately 7.6 kb of sub-telomeric DNA. The length obtained was age-adjusted by comparing it with the line-of-best-fit through the measurements of 112 healthy individuals (delta Tel). The telomere lengths of the two affected individuals were exceptionally short when compared with healthy individuals (delta Tel P < 0.001; Mann–Whitney U test). Other family members who were heterozygous for the mutation also had significantly shortened telomeres when compared with the 112 controls (delta Tel P < 0.01), but as can be seen from Figure 4 they are not as short as in individuals with disease. These delta Tel measurements are shown relative to previously reported and now updated data from other patients with DC (13,17,19,22). As previously demonstrated, patients with DKC1 mutations have significantly shortened telomeres when compared with healthy individuals (n = 67; P < 0.00001). This is also true for those patients with TERC mutations (n = 35; P < 0.0001). Although shorter after age adjustment, in a small group of patients with TERT mutations (n = 7) we do not see significantly shortened telomeres when compared with the healthy individuals.
Figure 4.

Delta Tel measurements in dyskeratosis congenita (DC) patients. Age-adjusted telomere length measurements from whole blood are shown for different subtypes of DC emphasizing how short telomeres are in the homozygous p.R34W individuals relative to both the heterozygous individuals, healthy controls and other DC types. P-values are expressed relative to the healthy individual group; NS, P-value not significant.
NOP10 mutation results in reduced TERC levels
An earlier study by Wong et al. (23) demonstrated that the presence of a mutation in dyskerin causes a reduction in the levels of detectable TERC in one patient. Like dyskerin, NOP10 is known to associate directly with TERC and this suggested that telomere maintenance may also be affected through reduction in TERC levels in patients with homozygous p.R34W NOP10 mutation. To examine this further we measured TERC levels in cDNA samples from whole blood from individuals with the NOP10 p.R34W mutation. TERC levels were also measured in 22 cDNA samples from healthy individuals and normalized against the house-keeping ABL gene. From the data shown in Figure 5, the TERC/ABL ratio is significantly lower in patients with homozygous NOP10 mutations as well as the heterozygous non-affected family members when each group is compared with the healthy group (P = 0.03 and P = 0.01, respectively; Mann–Whitney U test). The presence of the p.R34W change decreases the level of TERC available, with the homozygous mutation having a more pronounced effect than the heterozygous change implying that this is a result of the p.R34W mutation in this family.
Figure 5.

Effect of p.R34W on TERC levels. TERC was quantitated and expressed relative to ABL in all individuals with NOP10 p.R34W. TERC/ABL ratios were also determined for DC patients with other mutations from whole blood. P-values are expressed relative to the healthy individual group. Unknown, the genetic basis of the disease remains unidentified. NS, P-value not significant.
We also measured TERC levels in cDNA samples of whole blood from other DC patients with mutations in DKC1 (n = 27), TERC (n = 9), TERT (n = 4) as well as those for whom the genetic basis is still unknown (n = 28). TERC/ABL ratio is highly significantly reduced in patients with DKC1 mutations (P < 0.0001) supporting the earlier observation by Wong et al. (23) as well as in patients with TERC mutations (P = 0.005). Although some patients with heterozygous TERT mutations had a slightly reduced TERC/ABL ratio, this was not significant, nor is the TERC/ABL ratio generally reduced in those DC patients whose genetic basis of disease is unknown, suggesting that a reduced TERC level is not a uniform feature of DC.
To demonstrate that the reduction in TERC levels observed in patient material were related to NOP10, we investigated whether changes in NOP10 could directly impact on TERC expression levels. NOP10 was knocked down in HeLa cells using two different siRNAs against NOP10. HeLa cells were allowed to grow for up to 72 h after transfection with the siRNA. Cells were harvested at 24, 48 and 72 h and cDNA prepared. Expression of both NOP10 and TERC were measured in samples at each time point and all were expressed relative to the untransfected 24 h time point. Both siRNAs were highly effective, reducing NOP10 mRNA levels by over 90% at all three time points (Fig. 6A). This translated into a reduced level of TERC at 48 and 72 h (Fig. 6B). Although the levels of both TERC and NOP10 vary in the mock-transfected and the negative siRNA transfection controls, the levels of reduction are greater in the cells treated with either of the siRNA constructs.
Figure 6.

Effects of siRNAs against NOP10 in HeLa cells in a transient system. (A) Levels of NOP10 expression are reduced by siRNA I and II at all time points studied when compared with mock-transfected and negative siRNA transfected cells (24 h, solid black bar; 48 h, grey bar; 72 h, solid white bar). (B) TERC levels are reduced from 48 h in the siRNA-treated cells when compared with controls.
To investigate whether the p.R34W mutation has a similar effect, we stably transfected HeLa cells with plasmids containing either wild-type NOP10, or NOP10 with the c.100C>T mutation (p.R34W) or with no insert. After two weeks of selection to enrich for plasmid-derived NOP10 expression, endogenous NOP10 was knocked down using siRNA I. This siRNA specifically knocks down the endogenous transcript by targeting the 3′-UTR which is absent from the plasmid derived NOP10. Cells were harvested at 48 h after transfection and were analysed as before. As demonstrated in the transient system, the levels of NOP10 were reduced in the empty vector cell line when treated with the siRNA against endogenous NOP10 (Fig. 7A). A reduction was also seen in cell lines with both the wild-type and the mutant NOP10 after siRNA treatment but this was not as pronounced indicating that NOP10 is being expressed from the plasmids. TERC expression levels were also reduced in the empty vector cell line after knockdown by the siRNA. When wild-type NOP10 is expressed from the plasmid, TERC levels are partially rescued, but this effect is much less pronounced in the cell line expressing the mutant NOP10 (Fig. 7B). These data indicate that a reduction in the expression of NOP10 leads to a reduction in TERC accumulation and this reduction can be partially rescued when wild-type NOP10 is expressed in the cells, but not with p.R34W NOP10. This in turn supports the idea that p.R34W in NOP10 directly impacts on the levels of TERC, as seen in the patient samples.
Figure 7.

Stable expression of mutant and wild-type NOP10. (A) Endogenous NOP10 expression is reduced with siRNA treatment but modest plasmid-derived NOP10 is detected in cell lines containing either wild-type or mutant plasmid. NOP10 expression in stable cell lines is shown relative to time = 0 in the cell line with the empty vector (empty vector, dotted bar; wild-type plasmid, vertical lines; mutant plasmid, horizontal lines). (B) TERC levels are partially rescued in the cell line expressing wild-type NOP10 but not in the cell line expressing the mutant NOP10. TERC expression is shown relative to time = 0 for each cell line.
DISCUSSION
In this study we have shown that AR-DC is a genetically heterogeneous disease and have identified a mutation in NOP10 in one specific family. From the homozygosity mapping undertaken in 16 consanguineous AR-DC families it was not possible to identify a single disease locus. Although several highly homozygous markers were identified, further work is required to determine whether any of these represent any potential disease loci. The data from D14S1051 also highlights the potential pitfalls of just considering the degree of homozygosity without considering the level of expected heterozygosity for that marker. Highly homozygous markers may be due to the lack of heterozygosity rather than true allele combination i.e. homozygous by state rather than by descent. From these observations it is apparent that there are at least two causal genes for this genetic subtype of DC demonstrating that AR-DC is genetically heterogeneous.
To investigate this further we concentrated on one large family with three affected individuals. Autozygosity mapping identified a locus on chromosome 15 which gave an LOD score of 2.7. This is highly suggestive of a disease locus being in this area and NOP10 was identified as a candidate gene. Mutational analysis identified an arginine (R) to tryptophan (W) substitution at amino acid 34 that fully segregates with disease. The mutation itself causes a major change in amino acid structure, by the addition of an aromatic ring and as it occurs in a very highly conserved region of the molecule (Fig. 3), it would be predicted to have major functional consequences.
We have shown that telomere lengths are significantly shorter, in individuals with this mutation even when it is present in the heterozygous form. This observation is of particular note as the patients had normal blood counts and therefore the telomere shortening was not as a consequence of bone marrow failure as is sometimes observed. The absence of bone marrow failure in two of the three affected individuals is not unusual as there are many families on our registry demonstrating considerable phenotypic heterogeneity within affected members of the same family. Previous studies (19) demonstrate that, in general, DC patients have shorter telomeres than healthy controls, although there is considerable variation in the range of telomeres in different patients as shown in Figure 4. The two patients homozygous for the p.R34W mutation have comparable lengths to those observed in patients with DKC1 mutations and severe disease. Furthermore, patients with the shortest telomeres tend to have severest phenotype. In autosomal dominant DC, disease anticipation is observed and it is associated with telomere shortening through the generations. Again this shortness of telomeres is consistent with the idea that a defective gene in the telomerase complex results in impaired telomerase function.
It is clear that AR-DC is genetically heterogeneous and that homozygous mutations in NOP10 are disease-causing in this large family. This is analogous to the situation in Fanconi anaemia where only one family has been described with a mutation in the complementation group M (24). We have also observed that the individuals with heterozygous p.R34W mutation have telomere lengths that are statistically lower than normal and these individuals are asymptomatic. This has been observed in other haematological recessive diseases such as beta thalassaemia major, where in some heterozygous individuals minor abnormalities in blood counts are observed, but there are no overt clinical features of the disease (25).
Quantitative TaqMan® analysis of cDNA samples from the affected family shows that the presence of the p.R34W substitution, again also in the heterozygous form, is associated with low levels of TERC RNA. Reduced TERC levels are observed in DC patients with DKC1 and TERC mutations; however this is not the case in all DC patients. Mutations in TERT or in the genetically uncharacterized patients do not consistently show the same reduction in TERC implying that reduced TERC is not a universal feature of DC. However, from the mutations identified so far in DC patients, those who have mutations in a component of the H/ACA snoRNP associated with TERC i.e. dyskerin, TERC and now NOP10 generally appear to have reduced telomere lengths and reduced TERC levels. Furthermore, the finding of lower TERC levels even in individuals with the heterozygous p.R34W mutation demonstrates a direct biological link in vivo between the number of mutant NOP10 alleles, TERC levels and telomere length.
In addition, siRNA studies in HeLa cells show that when NOP10 expression is knocked down, there is a subsequent reduction in the amount of TERC. By enriching for cells in which either wild-type or mutant NOP10 are expressed from plasmids which escape this knockdown, we have demonstrated that the presence of the p.R34W mutation may be responsible for the reduction seen, as cells expressing wild-type NOP10 were able to partially rescue the levels of TERC expression but this was not the case in cells expressing the p.R34W the mutation. How this happens is unknown but it is likely that the reduced levels of NOP10 when translated into protein result in reduced stability of TERC. This may result from a direct interaction, as it has been suggested that Nop10p can interact directly with RNA in yeast as well as acting via the snoRNP complex (26), or the effect could result from a stoichiometric relationship in the complex as a whole. Once snoRNP proteins are assembled on the RNA they form a stable complex and do not exchange their components suggesting that the formation of additional snoRNPs requires de novo synthesis (9). Therefore, if there is less NOP10 available, this could impact on the formation of the snoRNP complex and all its functions.
NOP10 is one of the core proteins associated with the H/ACA snoRNPs and together with dyskerin, NHP2 and GAR1 are essential for several functions including eukaryotic ribosome biogenesis, pre-mRNA splicing and telomere maintenance (27). NOP10 is evolutionarily conserved and is found in all classes of organisms except bacteria (28). Although the precise role of NOP10 in the snoRNP complex is unclear, information regarding its structure has been recently elucidated from structural analysis of the interactions between aCbf5 (archaeal dyskerin homologue) and aNop10 (archaeal NOP10 homologue) from Methanococcus jannaschii. The N-terminal region appears to be structured with a zinc-binding motif and two β-sheets with an α-helix domain present at the C-terminus. The domains are connected by a 10 amino acid random coil and both domains contact the aCbf5 catalytic domain (26,29,30). This direct interaction between Cbf5 and Nop10 appears to be universal among all archaeal and eukaryotic H/ACA snoRNPs (26). Immunoprecipitation studies have shown that NOP10 alone is co-precipitated with NAP57 (rat homologue of dyskerin). Precipitation of NHP2 requires the presence of both NAP57 and NOP10, suggesting that these three molecules form a trimer and it has been suggested that they are in the ratio of two NOP10 and possibly two NAP57 per one NHP2 in each core trimer (9). GAR1 is more loosely associated with this trimer and it has been suggested that GAR1 only associates with the complex once it has been transported from the site of transcription to the nucleus or nucleolus, whereas the other components of the complex associate at the site of transcription (31). GAR1 is not required for binding to RNA, also the absence of GAR1 has no effect on the accumulation of mature Terc in yeast (32).
Further structural analysis of the interactions between aCbf5 and aNop10 show that amino acids 33–36 of aNop10 stabilize the active site of aCbf5 by buttressing residues in a sequence element conserved throughout pseudouridine synthases that is important for protein stability (33) and that archaeal R34 in aNop10 interacts directly with aCbf5 (29). It has also been demonstrated that R34 in aNop10 is one of the highly conserved residues that binds directly to the P2 region of RNA component of the H/ACA RNP as well as directly interacting with aCbf5; furthermore, the long side chain of the R34 bridges two phosphate groups across the RNA major groove (34). It is likely that the addition of an aromatic ring to the protein structure at this point (observed in the family described in this study) would interrupt the interactions between dyskerin/NOP10 and the guide RNA and thereby result in reduced biological activity of the whole complex.
In summary, linkage analysis in AR-DC families has highlighted that this form of the disease is genetically heterogeneous and there is more than one gene responsible for this subcategory of DC. Secondly, we have demonstrated that TERC levels are reduced in some subtypes of DC, but this is not a universal feature of the disease. Finally, we have shown that in one large family with AR-DC the disease is due to homozygous mutations in NOP10. Affected individuals have very short telomeres and reduced TERC levels. We have also shown that a knockdown of NOP10 mRNA results in a reduction in TERC levels and the p.R34W mutation identified in this work is responsible for this reduction. These findings identify the first gene responsible for AR-DC suggest that NOP10 has a critical role in telomere maintenance in humans and also strengthen the model that DC is primarily a disease of defective telomere maintenance.
MATERIALS AND METHODS
Participants and consent
The study was approved by the Local Research Ethics Committee. Informed consent was obtained from all participants.
Clinical presentation of patients in DCR 148
This family from Saudi Arabia presented with three affected individuals, six unaffected siblings (samples available on three) and their parents who were first cousins. All the unaffected siblings as well as the parents showed no overt features of the disease. The ages at diagnosis of the three affected individuals were II-2 16 years, II-3 20 years and II-5 15 years. All three affected individuals had reticular skin pigmentation over the neck and upper extremities as well as thickening of the skin over the palms and soles. Dystrophic changes were seen on the nails and all three had abnormal dentition, although II-3 and II-5 were less severe than II-2. Hypocellular bone marrow with peripheral pancytopenia was only observed in II-2. Leukoplakia was absent in all three individuals.
Genotyping and logarithm of odds score calculation
Genomic DNA was extracted from peripheral blood using Puregene DNA isolation kits (Gentra Systems). A genome-wide search for linkage was performed using 350 LMS-MD 10 ABI PRISM microsatellite markers (Applied Biosystems, Foster City, CA, USA). DNA was analysed from 25 affected individuals from 16 consanguineous-marriage families. PCR products were analysed using a 3730 DNA analyser and scored using GeneMapper software program (Applied Biosystems). LOD score analysis was performed using GeneHunter (35) assuming 100% penetrance, AR inheritance and a rare prevalence (0.005) of the disease allele. PCR conditions are available upon request.
Sequence analysis of nop10
Primers were designed to specifically amplify both exons of NOP10 as listed: Exon 1 forward 5′-ATT GAC GAA CAC GTG ACG C-3′ Exon 1 reverse 5′-GCG AAC TCC TGA GCT AAG-3′ Exon 2 forward 5′-GAC CAT TGC ATC TTC TAT TTT C-3′ Exon 2 reverse 5′-CCT TTA TGG GTG ATG CCA C-3′. The resulting products were visualized on a 1.8% agarose gel and the resultant band was excised and purified using QIAquick gel extraction kit (QIAgen GmbH, Germany). The purified products were subjected to direct sequence analysis by BIGDYE™ chain termination cycle sequencing and fragment analysis on the 3700 DNA analyser (Applied Biosystems Inc.). PCR conditions are available upon request.
Screening for p.R34W mutation in ethnically matched individuals
In order to determine whether p.R34W is a mutation or a rare polymorphism, DNA samples were obtained from 56 individuals from either Saudi Arabia or the United Arab Emirates to check for the presence of the rare T allele in an ethnically matched population. Samples were genotyped using a tetra-primer sequence-specific assay, a method that uses two primer pairs to specifically amplify the normal and mutant sequences plus a positive control in a single reaction (36) using sequence-specific primer PCR methodology (SSP–PCR) (37). The control band was provided by the primers for exon 2 forward and exon 2 reverse. To amplify the wild-type allele (C) specifically, exon 2 reverse was combined with a forward wild-type specific primer—5′-CTC AGC CCA TCC TGC TC-3′. The mutant allele (T) was specifically amplified using exon 2 forward with a reverse mutant-specific primer—5′-TTG TCA TCT GGG GAG AAC CA-3′, in both cases the specific base of interest is underlined. PCR conditions are available upon request.
Telomere length measurement
Genomic DNA, extracted from whole blood, was digested with BamHI and analysed by Southern blot analysis using 0.75% agarose gels and the sub-telomeric probe pTelBam8 (38,39). Telomere lengths were measured as the size of the fragment of peak signal intensity, which includes 7.6 kb of sub-telomeric DNA. Sizes were determined with reference to the same standards run on each gel using the Gel Blot-Pro software from UVP (Upland). Data on control samples was collected using the same image analysis system as the patient group. Because telomere lengths shorten with age, an age-adjusted value (delta Tel) is obtained by drawing a line-of-best-fit through all control sample measurements plotted against age. A predicted telomere length value for any age is then obtained from this line and this is subtracted from the measurement obtained by hybridization for any subject to give a delta Tel (the difference between the actual value and the predicted value) for each individual.
RNA extraction and cDNA synthesis
RNA was prepared from whole blood using QIAamp RNA blood mini kit (QIAgen GmbH) or from HeLa cells using the RNeasy mini kit (QIAgen GmbH). After DNaseI treatment to remove any possible genomic DNA contamination, the RNA was reverse-transcribed by M-MLV (Invitrogen, UK) into cDNA with random hexamer primers (Qiagen) using manufacturer’s protocol.
Transient siRNA transfections
All siRNA experiments were performed on HeLa cells cultured at 37°C and 5% CO2 in DMEM (Cambrex, UK) supplemented with l-glutamine and 10% fetal calf serum (FCS). Cells were seeded into six well plates at a density of 2 × 105 per well 24 h before treatment. Transfections were carried out using Lipofectamine 2000 (Invitrogen) and pEGFP-C1 (Clontech, USA) plasmid DNA as a carrier according to the manufacturer’s protocol. Cells were transfected with two commercially available siRNAs to NOP10 (siRNA I sense GGGAACACAUUUGAUUUUtt, antisense AAAAGUCAAAUGUGUUCCCtt; siRNA II sense CCCAUAAAGGGAACACAUUtt, antisense AAUGUGUUCC CUUUAUGGGtg; Ambion, UK) at 50 nm, a commercially available negative control siRNA (Negative control no. 1 siRNA 5 nm; Ambion, UK) at 30 nm and a mock transfection with pEGFP-C1 at 400 ng. Cells were harvested at 24, 48 and 72 h time points and RNA extracted as described.
NOP10 expression constructs
NOP10 coding sequence was cloned from human cDNA by PCR and ligated into pCR Blunt II-TOPO (Invitrogen). Plasmid clones were sequenced to ensure the NOP10 sequence acquired was identical to that in the published literature. NOP10 was then subcloned into the multiple cloning site of pEF1/V5-His (Invitrogen) as an EcoRI restriction fragment. The R34W mutation was introduced into the NOP10 coding sequence by site-directed mutagenesis using the QuikChange Site-Directed Mutagenesis Kit (Stratagene).
Stable cell line construction and siRNA knockdown
HeLa cells were transfected with pEF1/NOP10 mutant and wild-type plasmids using Lipofectamine 2000 (Invitrogen) and maintained in OptiMEM (Invitrogen). pEF1/V5-His was used as an ‘empty-vector’ control. Twenty-four hours post-transfection, cells were split into 1:4 and re-seeded into DMEM (Cambrex) supplemented with 10% FCS and penicillin/streptomycin. Forty-eight hours later, G418 sulphate was added to the medium to a final concentration of 8 μg/ml. Cells were maintained in G418-supplemented medium for two weeks without passage until clear antibiotic-resistant foci developed. The resultant stably transfected clones were pooled for use in siRNA transfection studies. Cells from each line (empty vector, wild-type and mutant) were seeded into six well plates at a density of 2 × 105 per well 24 h before treatment. One well for each cell type was treated with lipofectamine alone (mock), transfected with negative control siRNA or siRNA I or left untreated. Cells were harvested at 48 h and RNA was extracted as for the transient siRNA transfections. RNA was also collected before cells were transfected giving a time=0 point. It is worth noting that in this case the siRNA used is specific to the endogenous NOP10 as it targets the 3′-UTR region of the gene, which is not present in the transcript derived from the plasmid NOP10.
Quantitative real-time PCR
Expression of total NOP10, TERC and ABL were determined by quantitative RT-PCR using the ABI PRISM 3700 sequence detection system (Applied Biosystems). NOP10 primers and probes were obtained as a commercially available gene expression assay (Applied Biosystems) and do not distinguish between endogenous and plasmid-derived NOP10. TERC primers and probes were manufactured according to Yajima et al. (40) and ABL primers and probe according to Chen et al. (41). Twenty-five microlitre reactions were performed using TaqMan® Universal PCR master mix, 2 μl cDNA with 300 nm of TERC primers and 200 nm TERC probe or 100 nm ABL primers and 300 nm ABL probe. NOP10 primers and probe were used as described in the manufacturer’s protocol. ABL expression was used as the endogenous cDNA quality control for all samples. TERC and NOP10 expression were normalized to ABL for all analyses (42). For patient material standard curves were generated and expression was calculated as a TERC/ABL ratio. For the siRNA experiments the ΔCt method was used (ABI user bulletin no. 2).
Statistical analysis
All differences between healthy controls and the various mutation groups for Delta Tel and TERC/ABL ratio were compared using the Mann–Whitney U test.
ACKNOWLEDGEMENTS
We wish to thank all clinicians and patients for providing samples, N. Killeen for technical assistance, R. Szydlo for statistical advice and the Wellcome Trust for funding. The ABL primers and probes were a kind gift from Dr J. Kaeda of the Minimal Residual Disease laboratory, Hammersmith Hospital London, UK. The initial linkage analysis was performed at the Linkage Hotel, MRC UK HGMP Resource Centre.
Footnotes
Conflict of Interest statement. None declared.
REFERENCES
- 1.Cech TR. Beginning to understand the end of the chromosome. Cell. 2004;116:273–279. doi: 10.1016/s0092-8674(04)00038-8. [DOI] [PubMed] [Google Scholar]
- 2.Collins K, Mitchell JR. Telomerase in the human organism. Oncogene. 2002;21:564–579. doi: 10.1038/sj.onc.1205083. [DOI] [PubMed] [Google Scholar]
- 3.Mitchell JR, Cheng J, Collins K. A box H/ACA small nucleolar RNA-like domain at the human telomerase RNA 3′ end. Mol. Cell. Biol. 1999;19:567–576. doi: 10.1128/mcb.19.1.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kiss T. Small nucleolar RNAs: an abundant group of noncoding RNAs with diverse cellular functions. Cell. 2002;109:145–148. doi: 10.1016/s0092-8674(02)00718-3. [DOI] [PubMed] [Google Scholar]
- 5.Filipowicz W, Pogacic V. Biogenesis of small nucleolar ribonucleoproteins. Curr. Opin. Cell. Biol. 2002;14:319–327. doi: 10.1016/s0955-0674(02)00334-4. [DOI] [PubMed] [Google Scholar]
- 6.Henras A, Henry Y, Bousquet-Antonelli C, Noaillac-Depeyre J, Gelugne JP, Caizergues-Ferrer M. Nhp2p and Nop10p are essential for the function of H/ACA snoRNPs. EMBO J. 1998;17:7078–7090. doi: 10.1093/emboj/17.23.7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pogacic V, Dragon F, Filipowicz W. Human H/ACA small nucleolar RNPs and telomerase share evolutionarily conserved proteins NHP2 and NOP10. Mol. Cell. Biol. 2000;20:9028–9040. doi: 10.1128/mcb.20.23.9028-9040.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Henras AK, Capeyrou R, Henry Y, Caizergues-Ferrer M. Cbf5p, the putative pseudouridine synthase of H/ACA-type snoRNPs, can form a complex with Gar1p and Nop10p in absence of Nhp2p and box H/ACA snoRNAs. RNA. 2004;10:1704–1712. doi: 10.1261/rna.7770604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang C, Meier UT. Architecture and assembly of mammalian H/ACA small nucleolar and telomerase ribonucleoproteins. EMBO J. 2004;23:1857–1867. doi: 10.1038/sj.emboj.7600181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dokal I. Dyskeratosis congenita in all its forms. Br. J. Haematol. 2000;110:768–779. doi: 10.1046/j.1365-2141.2000.02109.x. [DOI] [PubMed] [Google Scholar]
- 11.Heiss NS, Knight SW, Vulliamy TJ, Klauck SM, Wiemann S, Mason PJ, Poustka A, Dokal I. X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nat. Genet. 1998;19:32–38. doi: 10.1038/ng0598-32. [DOI] [PubMed] [Google Scholar]
- 12.Mitchell JR, Wood E, Collins K. A telomerase component is defective in the human disease dyskeratosis congenita. Nature. 1999;402:551–555. doi: 10.1038/990141. [DOI] [PubMed] [Google Scholar]
- 13.Vulliamy T, Marrone A, Goldman F, Dearlove A, Bessler M, Mason PJ, Dokal I. The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature. 2001;413:432–435. doi: 10.1038/35096585. [DOI] [PubMed] [Google Scholar]
- 14.Vulliamy TJ, Knight SW, Mason PJ, Dokal I. Very short telomeres in the peripheral blood of patients with X-linked and autosomal dyskeratosis congenita. Blood Cells Mol. Dis. 2001;27:353–357. doi: 10.1006/bcmd.2001.0389. [DOI] [PubMed] [Google Scholar]
- 15.Vulliamy T, Marrone A, Dokal I, Mason PJ. Association between aplastic anaemia and mutations in telomerase RNA. Lancet. 2002;359:2168–2170. doi: 10.1016/S0140-6736(02)09087-6. [DOI] [PubMed] [Google Scholar]
- 16.Yamaguchi H, Calado RT, Ly H, Kajigaya S, Baerlocher GM, Chanock SJ, Lansdorp PM, Young NS. Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N. Engl. J. Med. 2005;352:1413–1424. doi: 10.1056/NEJMoa042980. [DOI] [PubMed] [Google Scholar]
- 17.Vulliamy TJ, Walne A, Baskaradas A, Mason PJ, Marrone A, Dokal I. Mutations in the reverse transcriptase component of telomerase (TERT) in patients with bone marrow failure. Blood Cells Mol. Dis. 2005;34:257–263. doi: 10.1016/j.bcmd.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 18.Armanios M, Chen JL, Chang YP, Brodsky RA, Hawkins A, Griffin CA, Eshleman JR, Cohen AR, Chakravarti A, Hamosh A, et al. Haploinsufficiency of telomerase reverse transcriptase leads to anticipation in autosomal dominant dyskeratosis congenita. Proc. Natl Acad. Sci. USA. 2005;102:15960–15964. doi: 10.1073/pnas.0508124102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vulliamy TJ, Marrone A, Knight SW, Walne A, Mason PJ, Dokal I. Mutations in dyskeratosis congenita: their impact on telomere length and the diversity of clinical presentation. Blood. 2006;107:2680–2685. doi: 10.1182/blood-2005-07-2622. [DOI] [PubMed] [Google Scholar]
- 20.Liang J, Yagasaki H, Kamachi Y, Hama A, Matsumoto K, Kato K, Kudo K, Kojima S. Mutations in telomerase catalytic protein in Japanese children with aplastic anemia. Haematologica. 2006;91:656–658. [PubMed] [Google Scholar]
- 21.Xin ZT, Beauchamp AD, Calado RT, Bradford JW, Regal JA, Shenoy A, Liang Y, Lansdorp PM, Young NS, Ly H. Functional characterization of natural telomerase mutations found in patients with hematological disorders. Blood. 2007;109:524–532. doi: 10.1182/blood-2006-07-035089. [DOI] [PubMed] [Google Scholar]
- 22.Vulliamy T, Marrone A, Szydlo R, Walne A, Mason PJ, Dokal I. Disease anticipation is associated with progressive telomere shortening in families with dyskeratosis congenita due to mutations in TERC. Nat. Genet. 2004;36:447–449. doi: 10.1038/ng1346. [DOI] [PubMed] [Google Scholar]
- 23.Wong JM, Kyasa MJ, Hutchins L, Collins K. Telomerase RNA deficiency in peripheral blood mononuclear cells in X-linked dyskeratosis congenita. Hum. Genet. 2004;115:448–455. doi: 10.1007/s00439-004-1178-7. [DOI] [PubMed] [Google Scholar]
- 24.Meetei AR, Medhurst AL, Ling C, Xue Y, Singh TR, Bier P, Steltenpool J, Stone S, Dokal I, Mathew CG, et al. A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nat. Genet. 2005;37:958–963. doi: 10.1038/ng1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weatherall DJ. Single gene disorders or complex traits: lessons from the thalassaemias and other monogenic diseases. BMJ. 2000;321:1117–1120. doi: 10.1136/bmj.321.7269.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khanna M, Wu H, Johansson C, Caizergues-Ferrer M, Feigon J. Structural study of the H/ACA snoRNP components Nop10p and the 3′ hairpin of U65 snoRNA. RNA. 2006;12:40–52. doi: 10.1261/rna.2221606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meier UT. The many facets of H/ACA ribonucleoproteins. Chromosoma. 2005;114:1–14. doi: 10.1007/s00412-005-0333-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Watanabe Y, Gray MW. Evolutionary appearance of genes encoding proteins associated with box H/ACA snoRNAs: cbf5p in Euglena gracilis, an early diverging eukaryote, and candidate Gar1p and Nop10p homologs in archaebacteria. Nucleic Acids Res. 2000;28:2342–2352. doi: 10.1093/nar/28.12.2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rashid R, Liang B, Baker DL, Youssef OA, He Y, Phipps K, Terns RM, Terns MP, Li H. Crystal structure of a Cbf5–Nop10–Gar1 complex and implications in RNA-guided pseudouridylation and dyskeratosis congenita. Mol. Cell. 2006;21:249–260. doi: 10.1016/j.molcel.2005.11.017. [DOI] [PubMed] [Google Scholar]
- 30.Manival X, Charron C, Fourmann JB, Godard F, Charpentier B, Branlant C. Crystal structure determination and site-directed mutagenesis of the Pyrococcus abyssi aCBF5–aNOP10 complex reveal crucial roles of the C-terminal domains of both proteins in H/ACA sRNP activity. Nucleic Acids Res. 2006;34:826–839. doi: 10.1093/nar/gkj482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Darzacq X, Kittur N, Roy S, Shav-Tal Y, Singer RH, Meier UT. Stepwise RNP assembly at the site of H/ACA RNA transcription in human cells. J. Cell Biol. 2006;173:207–218. doi: 10.1083/jcb.200601105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dez C, Henras A, Faucon B, Lafontaine D, Caizergues-Ferrer M, Henry Y. Stable expression in yeast of the mature form of human telomerase RNA depends on its association with the box H/ACA small nucleolar RNP proteins Cbf5p, Nhp2p and Nop10p. Nucleic Acids Res. 2001;29:598–603. doi: 10.1093/nar/29.3.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hamma T, Reichow SL, Varani G, Ferre-D’Amare AR. The Cbf5–Nop10 complex is a molecular bracket that organizes box H/ACA RNPs. Nat. Struct. Mol. Biol. 2005;12:1101–1107. doi: 10.1038/nsmb1036. [DOI] [PubMed] [Google Scholar]
- 34.Li L, Ye K. Crystal structure of an H/ACA box ribonucleoprotein particle. Nature. 2006;443:302–307. doi: 10.1038/nature05151. [DOI] [PubMed] [Google Scholar]
- 35.Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES. Parametric and nonparametric linkage analysis: a unified multipoint approach. Am. J. Hum. Genet. 1996;58:1347–1363. [PMC free article] [PubMed] [Google Scholar]
- 36.Jones AV, Kreil S, Zoi K, Waghorn K, Curtis C, Zhang L, Score J, Seear R, Chase AJ, Grand FH, et al. Widespread occurrence of the JAK2 V617F mutation in chronic myeloproliferative disorders. Blood. 2005;106:2162–2168. doi: 10.1182/blood-2005-03-1320. [DOI] [PubMed] [Google Scholar]
- 37.Bunce M, O’Neill CM, Barnardo MC, Krausa P, Browning MJ, Morris PJ, Welsh KI. Phototyping: comprehensive DNA typing for HLA-A, B, C, DRB1, DRB3, DRB4, DRB5 & DQB1 by PCR with 144 primer mixes utilizing sequence-specific primers (PCR-SSP) Tissue Antigens. 1995;46:355–367. doi: 10.1111/j.1399-0039.1995.tb03127.x. [DOI] [PubMed] [Google Scholar]
- 38.Notaro R, Cimmino A, Tabarini D, Rotoli B, Luzzatto L. In vivo telomere dynamics of human hematopoietic stem cells. Proc. Natl Acad. Sci. USA. 1997;94:13782–13785. doi: 10.1073/pnas.94.25.13782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brown WR, MacKinnon PJ, Villasante A, Spurr N, Buckle VJ, Dobson MJ. Structure and polymorphism of human telomere-associated DNA. Cell. 1990;63:119–132. doi: 10.1016/0092-8674(90)90293-n. [DOI] [PubMed] [Google Scholar]
- 40.Yajima T, Yagihashi A, Kameshima H, Kobayashi D, Furuya D, Hirata K, Watanabe N. Quantitative reverse transcription-PCR assay of the RNA component of human telomerase using the TaqMan fluorogenic detection system. Clin. Chem. 1998;44:2441–2445. [PubMed] [Google Scholar]
- 41.Chen ZX, Kaeda J, Saunders S, Goldman JM. Expression patterns of WT-1 and Bcr-Abl measured by TaqMan quantitative real-time RT-PCR during follow-up of leukemia patients with the Ph chromosome. Chin. Med. J. (Engl.) 2004;117:968–971. [PubMed] [Google Scholar]
- 42.Gabert J, Beillard E, van d.V.V., Bi W, Grimwade D, Pallisgaard N, Barbany G, Cazzaniga G, Cayuela JM, Cave H, et al. Standardization and quality control studies of ‘real-time’ quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia—a Europe Against Cancer program. Leukemia. 2003;17:2318–2357. doi: 10.1038/sj.leu.2403135. [DOI] [PubMed] [Google Scholar]
- 43.Corpet F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1988;16:10881–10890. doi: 10.1093/nar/16.22.10881. [DOI] [PMC free article] [PubMed] [Google Scholar]