

Abstract
Cre recombinase residue Arg259 mediates a canonical bidentate hydrogen-bonded contact with Gua27 of its LoxP DNA substrate. Substituting Cyt8-Gua27 with the three other basepairs, to give LoxAT, LoxTA, and LoxGC, reduced Cre-mediated recombination in vitro, with the preference order of Gua27 >Ade27 ~ Thy27 ≫ Cyt27. While LoxAT and LoxTA exhibited 2.5-fold reduced affinity and 2.5-5-fold slower reaction rates, LoxGC was a barely functional substrate. Its maximum level of turnover was 6-fold reduced over other substrates, and it exhibited 8.5-fold reduced Cre binding and 6.3-fold slower turnover rate. With LoxP, the rate-limiting step for recombination occurs after protein-DNA complex assembly but before completion of the first strand exchange to form the Holliday junction (HJ) intermediate. With the mutant substrates, it occurs after HJ formation. Using an increased DNA-binding E262Q/E266Q “CreQQ” variant, all four substrates react more readily, but with much less difference between them, and maintained the earlier rate-limiting step. The data indicate that Cre discriminates substrates through differences in (i) concentration dependence of active complex assembly, (ii) turnover rate, and (iii) maximum yield of product at saturation, all of which are functions of the Cre-DNA binding interaction. CreQQ suppression of Lox mutant defects implies that coupling between binding and turnover involves a change in Cre subunit DNA affinities during the “conformational switch” that occurs prior to the second strand exchange. These results provide an example of how a DNA-binding enzyme can exert specificity via affinity modulation of conformational transitions that occur along its reaction pathway.
High fidelity substrate recognition is critical for appropriate enzyme function. For site-specific DNA-modifying enzymes, substrate DNA sequences must be differentiated from an excess of similarly structured nonsubstrate sequences to properly control replication, gene expression, base repair, topological state, and recombination. To achieve maximum specificity, these proteins often exhibit multiple levels of DNA discrimination. At a primary level, recognition of double helical structure in conjunction with base- or structure-specific atomic features recruit site-specific enzymes to their target sites (“binding specificity”), and differentiation is exerted via stability differences between cognate and noncognate protein-DNA complexes. However, many enzymes also exert additional specificity in the progression from reactant to product (“kinetic specificity”) (1–6). Thus, specificity-conferring protein-DNA interactions can be categorized as contributing to assembly of active complexes or promoting the required chemical reaction steps (5, 7). For example, interactions of MuA transposase with DNA target recognition elements affect substrate binding and transpososome assembly but not the rate of DNA cleavage and strand transfer (8). In contrast, restriction enzymes and DNA glycosylases exert specificity in the chemical step through transition-state recognition (1–3), so that active-site residues are correctly positioned only if the cognate DNA sequence or three-dimensional structure is present. Because of high transition-state energies, this kinetic specificity provides a powerful differentiation mechanism, which contributes up to 106- to 108-fold to restriction enzyme fidelity (9, 10). Conceivably, any sequence-sensitive energy barrier that occurs after protein-DNA complex assembly could influence substrate discrimination. Therefore, reactions involving multiple steps and conformational changes have many potential points at which DNA sequences can regulate activity.
Bacteriophage P1 Cre protein promotes site-specific recombination at 34 bp1 LoxP sites (Figure 1a) (11–14) via a multistep reaction mechanism that requires protein and DNA conformational changes (Figure 1b) (15). We previously demonstrated that Cre differentiates LoxP from a mutant Lox site via both DNA binding and turnover2 rate (16). In this respect, Cre is unlike MuA transposase, in that its interactions with LoxP enforce both binding and kinetic specificity. In the present study, we studied Cre’s ability to recombine several variant Lox sites that it binds with different affinities and identified three levels at which Cre differentiates substrates. We also bracketed the post-binding step at which kinetic specificity is exerted.
Figure 1.
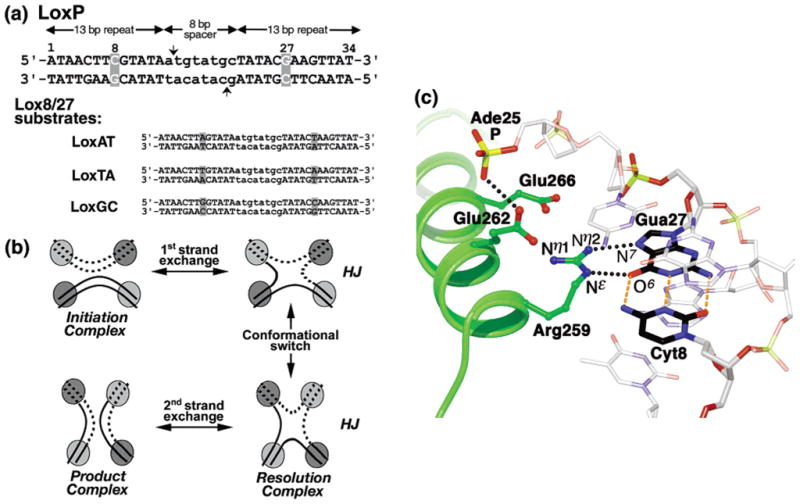
LoxP site, Cre-Lox recombination mechanism and the Arg259-Gua27 protein-DNA interface. (a) LoxP sequence and symmetric 8/27 substitutions. The 34 bp LoxP sequence is shown, with each position numbered 1–34 on the top strand. LoxP is comprised of two 13 bp inverted repeat binding elements (uppercase) surrounding an asymmetric 8 bp spacer (lowercase). Cleavage sites for strand exchange are indicated with vertical arrows. The position 8 and 27 substitution sites are highlighted (gray boxes). The 8/27 base pair substitutions are listed for the substrates LoxAT, LoxTA, and LoxGC. (b) Recombination mechanism. During recombination, four Cre monomers associate with two LoxP sites. Two Cre monomers are in an active “cleaving” conformation (light gray circles), while the other two monomers are in the inactive “noncleaving” conformation (dark gray circles). Strand exchange is accomplished by single strand cleavage to form a covalent phosphotyrosine-DNA intermediate (not shown) followed by strand “swapping” and religation of the homologous strands. The first strand exchange step (“initiation”) forms a Holliday junction (HJ) intermediate complex. During HJ complex isomerization, the Cre subunits undergo a “conformational switch”, in which inactive Cre monomers are now activated for cleavage, and the active monomers are inactivated. In the second strand exchange step (“HJ resolution”), the HJ is resolved to form recombinant products. We hypothesize that the Cre cleaving conformation has higher DNA affinity than the noncleaving conformation. (c) Protein-DNA interactions near the 8/27 substitutions, in a CreWT/LoxP HJ complex (35). Helix J (green ribbons) occupies the LoxP major groove, centered on the Cyt8-Gua27 base-pair (black bonds, interbase hydrogen bonds are indicated by orange dotted lines). Arg259 recognizes Gua27 O6 and N7 atoms via paired hydrogen bonds with the guanidinium Nε and Nη2 atoms, respectively, while Glu262 makes close contact (2.8 Å) with the phosphate of Ade25 (black dotted lines).
The LoxP recombination substrate (Figure 1a) is comprised of two 13 bp inverted repeat Cre binding elements (“13 bp repeat”) separated by an 8 bp asymmetric intervening segment (“8 bp spacer”) that contains the cleavage sites for strand exchange (12, 17). Cre recombination activity is directed solely by its LoxP substrate to insert, excise, or invert DNA segments (12, 18–20). Specific protein-protein and protein-DNA interactions direct assembly of the active recombination complex that contains four Cre monomers and two LoxP sites (15, 21). Substantial DNA bends within the LoxP 8 bp spacer allow formation of the required intersubunit protein-protein contacts, suggesting that active complex stability may be determined by the interplay of favorable intermolecular interactions and unfavorable protein and DNA distortions.
The Cre-Lox conservative recombination mechanism is shared with other members of the tyrosine recombinase family, such as bacteriophage lambda integrase, Escherichia coli XerC/XerD, and Saccharomyces cerevisiae FLP recom-binase (13, 22–25). Recombination occurs in two consecutive single-strand exchanges in which the signatory active-site tyrosine acts as a nucleophile for cleavage and as a leaving group for ligation (Figure 1b) (14). To limit exchange to a single pair of DNA strands, chemically identical Cre subunits are differentiated into two distinct conformations. One pair of monomers is in the “cleaving” conformation that is activated for strand exchange, while the other two monomers have the “noncleaving” inactive conformation (15). The first exchange is preferentially initiated by bottom strand cleavage on the right side of the 8 bp spacer (14, 26–30) (Figure 1a), and the resulting covalent intermediate is converted to the Holliday junction intermediate (HJ1) through ligation to the cleaved homologue strand. To activate second-strand cleavage, the Cre tetramer undergoes “HJ isomerization” in which a “conformational switch” (15) interchanges active and inactive subunit conformations. Second strand exchange is then effected by resolving the HJ into recombinant products via upper strand cleavage and ligation on the left side of the spacer, analogous to a reversal of the first exchange (14, 31). The multistep nature of this reaction and the fact that sequence-specific protein-DNA interactions persist through its entirety (15, 32–35) provide many points for differentiating substrates during and after recombination complex assembly.
Given its simple substrate requirements and high efficiency, Cre-Lox recombination has enjoyed wide use for altering chromosome structure in living cells (36–39). The lack of useful Lox-related sequences in most host genomes has stimulated efforts to alter Cre DNA specificity via protein engineering with some success (40–42). Despite these technological advances, the fundamental mechanisms by which Cre differentiates substrates and nonsubstrates is largely unknown.
Our previous work (16) focused on the effects of a symmetric substitution at positions 8 and 27 ("8/27 basepair") within the 13 bp repeat binding elements to make “LoxAT”3 (Figure 1a). This mutation not only reduced substrate binding but also slowed the turnover rate by wild-type Cre (“CreWT”4). The results suggested that there might be a general relationship between the substrate affinity and the recombination rate, providing a kinetic component to Cre DNA discrimination mechanisms. This connection was supported by the suppression of the LoxAT defects by the CreQQ4 mutant that contains the Glu262 → Gln and Glu266 → Gln substitutions (Figure 1c). CreQQ exhibited enhanced DNA binding and increased reaction rates with both LoxP and LoxAT, but differentially, so that CreQQ was much less able to distinguish between LoxP and LoxAT. The structural basis for this “affinity-kinetic coupling” was not obvious. In the CreWT/LoxAT HJ complex crystal structure (PDB entry 1MA7), reduced Cre binding could be readily rationalized from the local rearrangements in protein-DNA interactions induced by the LoxAT substitutions (16). However, no disruption of the 17–20 Å distant catalytic site was observed.
The Cre interaction with the LoxP 8/27 basepair is mediated by a canonical α-helix/major groove contact involving bidentate hydrogen bonds between Arg259 and Gua275 (Figure 1c). This interaction is critical for substrate recognition, since mutation of Arg259 to Gln, Asn, Asp, and Leu abolished LoxP binding (43). In general, bidentate recognition of Gua N7 and O6 atoms by Arg guanidinium nitrogens occurs frequently in protein-DNA complexes. Our own survey of a culled library of 139 protein/DNA complexes (44) revealed 63 instances of bidentate Gua-Arg interactions (see Experimental Procedures, and Supplemental Table 1, Supporting Information). The Nε-O6/Nη2-N7 interaction (Figure 1c) is observed in 10 of the 63 occurrences. In particular, the yeast Cre homologue FLP recombinase recognizes its LoxP-like FRT target site analogously (45), and this Arg-Gua interaction is also critical to Flp/FRT recognition (46–49). Arg259 is also conserved in a number of other tyrosine recombinases (24). In the lambda integrase/att recognition complex structures, it interacts with an analogous guanine in att sites but only via a single hydrogen bond (50, 51).
The structural specificity and the conservation of this Arg-Gua interaction in related tyrosine recombinases, in conjunction with the structural responses observed in the CreWT/LoxAT complex (16), suggested that Cre should be highly selective for the Cyt8-Gua27 basepair. In this work, we characterized the effects of substituting each of the other basepairs at LoxP position 8/27 to probe both Cre specificity for this site and the generality of our previous conclusions. We addressed the following questions: (1) How does Cre respond to different basepairs at this position in terms of complex assembly efficiency and substrate turnover rate? (2) Is there a persistent connection between turnover rate and DNA-binding affinity? (3) Which reaction step(s) are perturbed to account for the slower turnover rates? Our results indicate that with CreWT each Lox mutation altered complex assembly efficiency and turnover rate to varying degrees. Further, these defects are attributable to weaker Cre binding since they were readily suppressed by the enhanced-affinity CreQQ mutant. The substitutions similarly changed the rate-limiting step for the CreWT reactions, and this change was reversed with CreQQ. Together, the data support the idea that Cre DNA affinity and reaction rate are coupled through affinity-dependent conformational changes that occur during the reaction cycle.
EXPERIMENTAL PROCEDURES
Cre Proteins, Lox Substrates and the Quantitative Recombination Assay
N-terminal His6-tagged CreWT and CreQQ were purified as previously described (16, 52). LoxP and Lox8/27 mutant substrate oligonucleotides were obtained from MWG Biotech, and 32P 5′-end-labeled 220 bp Lox-containing fragments of LoxP and each mutant were prepared as previously described (16, 35). The results for LoxGC were replicated using two different synthetic DNA preparations and multiple 220 bp fragment DNA labeling reactions.
In all single turnover intermolecular recombination reactions (16, 35), synthetic 34 bp Lox was mixed with gel-purified, labeled 220 bp Lox-containing DNA in reaction buffer (300 mM LiOAc, 1 mM Na-EDTA, 20 mM Tris-acetate, pH 8.3) to a total volume of 10–50 μL. Reactions were quenched with loading buffer (final concentrations of 1% SDS, 20 mM DTT, 6% glycerol, 0.5 mg/mL proteinase K, and 0.05% bromophenol blue) and digested for 1 h at 37 °C prior to electrophoresis through SDS/10% polyacrylamide gels. A Fuji Image plate was exposed to the dried gels and scanned using Molecular Dynamics Storm 860 image plate reader. Relative labeled DNA band intensities were quantitated using ImageQuant.
It should be noted that different reaction quench methods can affect HJ accumulation levels. The SDS/proteinase/heat method used here is the most rapid we know of. For others (53–55), reactions quenched with SDS, proteinase, phenol, or heat yielded similar low HJ levels, indicating that the methods used in this work do not introduce unusual bias. In contrast, ethidium bromide quenching trapped more than 50% of the substrate as HJ intermediates (54), suggesting that it altered the on-enzyme equilibria between substrates, intermediates, and products (53–55). In our work, we cannot rule out the idea that SDS/proteinase may dissociate the lower stability mutant Lox complexes more readily, preventing them from reacting further as they might in CreWT/LoxP and CreQQ reactions. However, we have not observed significant post-quench reactivity (data not shown), and we do not see the higher HJ and lower product levels expected if post-quench HJ resolution was blocked. In fact, the Lox 8/27 mutant reactions generally accumulate lower HJ levels while the more tightly binding CreQQ reaction accumulates higher levels (Table 3).
Table 3.
CreWT and CreQQ HJ Formation Kinetic Parameters
| reaction | HJ extenta, f | A | k1 (× 103 s−1) | k2 (× 103 s−1) | t1/2 (s)b |
|---|---|---|---|---|---|
| CreWT-LoxP | 11 ± 1 | 0.63 ± 0.03 | 5.3 ± 0.2 | 49 ± 13 | 53 |
| LoxTA | 12 ± 3 | 0.41 ± 0.17 | 13 ± 3 | 45 ± 6 | 29 |
| LoxAT | 8 ± 1 | 0.47 ± 0.01 | 6.6 ± 0.2 | 32 ± 11 | 41 |
| LoxGCc | 1.2 | 0.74 | 5.8 | 71 | 88 |
| CreQQ-LoxP | 11 ± 1 | 0.50 ± 0.04 | 7.9 ± 1.2 | 89 ± 1 | 21 |
| LoxTA | 24 ± 3 | 0.47 ± 0.05 | 8.5 ± 0.9 | 90 ± 4 | 19 |
| LoxAT | 15 ± 1 | 0.30 ± 0.01 | 3.2 ± 0.1 | 96 ± 1 | 14 |
| LoxGC | 21 ± 2 | 0.34 ± 0.13 | 8.8 ± 1.8 | 68 ± 10 | 17 |
The fit value of "f" describes the maximal amount substrate reacted to HJ complexes only; HJ extent + products extent (Table 4) ≅ extent (Table 2).
Values for t1/2 are calculated based on the averaged curve fits for each reaction (see Figure 5).
Because of low substrate turnover, the parameters reported are from the fit of the averaged data points from four replicate reactions. The data were fit to the sequential a → b → c kinetic model described in Experimental Procedures, for comparison with CreWT/LoxGC product formation.
Active Complex Assembly Titrations Assays
To assess the strength of the Cre-Lox interaction, the disappearance of the 220 bp substrate (“substrate turnover”2) was measured as a function of complex concentration, by titrating a fixed amount of nonlimiting labeled 220 bp substrate with different Cre-synthetic Lox concentrations in 16-h endpoint reactions (16). The 220 bp Lox-containing DNA duplex concentration was ~2 nM, while the 4:1 Cre-Lox concentrations were varied for each substrate and are as previously described for LoxP and LoxAT (16). The Cre/Lox complex concentration, defined as 34 bp Lox concentration, differed for each reaction as follows: CreWT/LoxTA: 20, 45, 60, 90, 120, 200, and 400 nM; CreWT/LoxGC: 150, 300, 450, 600, 900, 1200, 1350, 1600, 1800, and 2400 nM; CreQQ/LoxTA and CreQQ/LoxGC: 10, 20, 30, 45, 90, 120, and 200 nM. Complex assembly parameters were determined by fitting the measured amount of substrate turnover (v′) and total complex concentration ([complex]) to the function v′ = (f[complex]α /(KDα + [complex] α). The fit parameters were f, the maximum amount of substrate turnover (“extent”2); KD, the apparent dissociation constant; and α, the apparent Hill coefficient (reported as the average of four independent experiments ± the standard deviation in Table 1).
Table 1.
CreWT and CreQQ Binding Parametersa
| complex | extent, f | KD (nM) | Hill coefficient, α |
|---|---|---|---|
| CreWT-LoxPb | 67 ± 8 | 58 ± 13 | 2.1 ± 0.8 |
| LoxTA | 65 ± 1 | 144 ± 13 | 3.1 ± 0.3 |
| LoxATb | 57 ± 8 | 146 ± 22 | 3.9 ± 0.3 |
| LoxGC | 12 ± 1 | 496 ± 60 | 2.0 ± 0.2 |
| CreQQ-LoxPb | 41 ± 9 | 27 ± 6 | 2.0 ± 0.6 |
| LoxTA | 55 ± 5 | 47 ± 3 | 2.8 ± 0.3 |
| LoxATb | 56 ± 3 | 58 ± 7 | 2.5 ± 0.4 |
| LoxGC | 56 ± 8 | 68 ± 3 | 3.9 ± 0.1 |
The parameters were fit to the modified Hill binding function as described in Experimental Procedures.
Data from ref 16.
Single Turnover Kinetic Assays
In time course reactions (16), the 220 bp Lox-containing DNA was 10 nM, and Cre/Lox concentrations were saturated at 4800 nM Cre and 1200 nM Lox, except for CreWT/LoxGC, which was at 9600 nM CreWT and 2400 nM LoxGC. CreWT/LoxTA, CreWT/LoxGC, CreQQ/LoxTA, and CreQQ/LoxGC reactions were quenched at 30, 60, 120, 240, 480, 960, and 1920 s, with CreWT/LoxGC having an additional time point of 3600 s. Rate parameters were determined by fitting the measured quantities of percent substrate reacted (v′) and reaction time (from reaction start until quenching, t) to the function v′ (t) = f{1 − [Ae−k1t + (1 − A)e−k2t]}. The fit parameters were f, the percent of substrate turnover at t = ∞ (“extent”); A and k1, the amplitude and rate constant for the reaction slow phase; k2, the rate constant for the fast phase (reported as the average of four independent experiments ± the standard deviation in Table 2).
Table 2.
CreWT and CreQQ Kinetic Parametersa: Rates of Substrate Disappearance
| reaction | extent, f | A | k1 (× 103 s−1) | k2 (× 103 s−1) | t1/2 (s)b | RLSc |
|---|---|---|---|---|---|---|
| CreWT-LoxPd | 60 ± 6 | 0.42 ± 0.02 | 4.5 ± 1.3 | 32 ± 3 | 48 | first strand exchange |
| LoxTA | 64 ± 3 | 0.81 ± 0.08 | 4.3 ± 1.0 | 22 ± 3 | 119 | HJ resolution |
| LoxATd | 62 ± 10 | 0.87 ± 0.04 | 2.7 ± 0.6 | 35 ± 6 | 234 | HJ resolution |
| LoxGCe | 7 ± 2 | 0.87 ± 0.06 | 2.1 ± 0.2 | 24 ± 2 | 301 | HJ resolution |
| CreQQ-LoxPd | 42 ± 1 | 0.54 ± 0.05 | 14 ± 0.2 | 120 ± 5 | 19 | first strand exchange |
| LoxTA | 60 ± 2 | 0.27 ± 0.05 | 5.1 ± 0.8 | 54 ± 6 | 19 | first strand exchange |
| LoxATd | 48 ± 2 | 0.43 ± 0.01 | 9.0 ± 0.1 | 93 ± 6 | 17 | first strand exchange |
| LoxGC | 55 ± 2 | 0.28 ± 0.03 | 7.0 ± 3.0 | 57 ± 6 | 18 | first strand exchange |
The parameters were fit to the biphasic kinetic function as described in Experimental Procedures.
Values for t1/2 are calculated based on the averaged curve fits for each reaction (see Figure 4b).
The rate-limiting step (“RLS”) occurs either during the first strand cleavage and exchange to form the HJ intermediate (“first strand exchange”) or during HJ isomerization or the second strand exchange to form recombinant products (“HJ resolution”).
Data from ref 16.
The data were fit to the sequential a → b → c kinetic model described in Experimental Procedures, for comparison with CreWT/LoxGC product formation.
We also fit the time dependence of HJ or product formation in each reaction to the biphasic model, with one exception. Because it was not possible to fit the delayed product appearance in LoxGC reactions to the biphasic model, we fit the data to a three-component sequential kinetic model (a → b → c) by simulation, as previously described (35). The fit parameters are f, the extent of HJ or product formation at t = ∞; A, the fraction of substrate reacting from the a state, k1, rate constant for conversion from a to b, and k2, rate constant conversion of b to c. Under certain conditions of A, k1, and k2, the fit parameters are identical for the two models. For others, adjustments in A, using the biphasic model rate constants, render them superimposable within experimental error. For comparisons to LoxGC, the following A values for the sequential model yield identical progress curves with the biphasic rate constants given in Tables 2–4. For CreWT and each substrate, the A values for total substrate reacted, HJ formation, and product formation, respectively, are LoxP, 0.36, 0.57, and 0.45; LoxTA, 0.65, 0.30, and 0; and LoxAT, 0.80, 0.38, and 0. For LoxGC, a biphasic A value of 0.94 yields the equivalent time course to the sequential time course in which A equals 0.87 for substrate disappearance, while HJ and product formation were only fit to the sequential model.
Table 4.
CreWT and CreQQ Product Formation Kinetic Parameters
| reaction | products extenta, f | A | k1 (× 103 s−1) | k2 (× 103 s−1) | t1/2 (s)b |
|---|---|---|---|---|---|
| CreWT-LoxP | 45 ± 5 | 0.59 ± 0.12 | 6.2 ± 1.9 | 26 ± 6 | 59 |
| LoxTA | 55 ± 2 | 0 | 0 | 3.8 ± 1.0 | 181 |
| LoxAT | 62 ± 11 | 0 | 0 | 3.1 ± 1.0 | 223 |
| LoxGCc | 6.0 | 1 | 6.5 | 2.2 | 482 |
| CreQQ-LoxP | 33 ± 2 | 0.34 ± 0.07 | 5.7 ± 0.6 | 58 ± 5 | 21 |
| LoxTA | 35 ± 3 | 0.24 ± 0.02 | 12 ± 3 | 66 ± 6 | 15 |
| LoxAT | 37 ± 5 | 0.49 ± 0.04 | 7.4 ± 3.5 | 71 ± 2 | 24 |
| LoxGC | 33 ± 2 | 0.24 ± 0.04 | 12 ± 4 | 63 ± 17 | 15 |
The fit value of "f" describes the maximal amount substrate reacted to products (not including HJ intermediates); products extent + HJ extent (Table 3) = extent (Table 2).
Values for t1/2 are calculated based on the averaged curve fits for each reaction (see Figure 5).
There was 60-s lag in product formation in this reaction, and therefore the data were fit to the sequential a → b → c kinetic model described in Experimental Procedures. The parameters reported are from the fit of the averaged data points from four replicate reactions.
Database Analysis of Protein-DNA Interactions
A database of 139 protein-DNA complex structures (44) was analyzed for contacts within 3.2 Å between specific amino acid and base atoms: (a) any Gua-Arg interaction (60 proteins, 98 interactions); (b) all bidentate Arg-Gua interactions (any Nε/Nη2 or Nη1/Nη2 paired with O6 and N7, 43 proteins, 63 interactions); and (c) Arg259-like bidentate interactions (Nε-O6 and Nη2-N7, 9 proteins, 10 interactions). We also identified several acid-phosphate contacts within 3.2 Å (Glu Oε1/Oε2 or Asp Oδ 1/Oδ 2 to phosphate O1P/O2P, 6 interactions in 6 proteins). These results excluded catalytic Glu or Asp residues in 10 metal-dependent endonucleases. Searches were performed using the automated scripts for the PDB file editor EDPDB (56), followed by visual confirmation. The results of this analysis are summarized in Supplemental Tables 1 and 2 (Supporting Information).
RESULTS
The Effects of LoxP 8/27 Substitutions on Complex Assembly and Turnover Rate
We compared how each Lox 8/27 substitution influences active complex assembly and reaction time course (16). Because recombination complexes dissociate slowly, only single-turnover reactions were utilized. To complete the series of 8/27 substitutions, we generated two new substrates, LoxTA and LoxGC, in which the Cyt8/Gua27 base pair was replaced with Thy-Ade and Gua-Cyt base pairs, respectively (Figure 1a). In the previously established activity assays in vitro (16, 35), recombination of a 32P end-labeled 220 bp Lox-containing DNA duplex with a 34 bp synthetic Lox site yields faster-migrating 141 and 113 bp labeled products, and the slower-migrating HJ intermediate, which are quantitated by SDS-PAGE and autoradiography (Figure 2).
Figure 2.
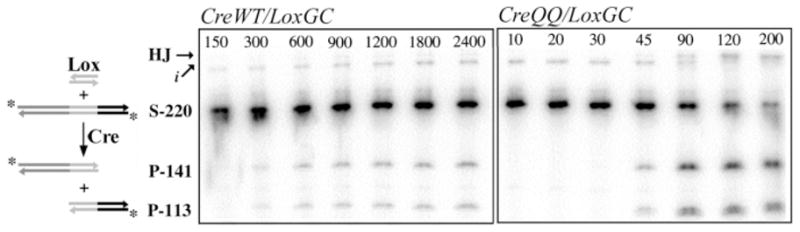
In vitro recombination assay. 34 bp Lox duplex (“Lox”) is recombined with Lox-containing 220 bp duplex DNA, upon addition of CreWT or CreQQ. The Lox-containing DNA is labeled on the 5′ ends with 32P (*). Recombination yields two labeled duplex products, of 141 bp and 113 bp. The reaction mixture is separated by SDS-PAGE, and the unreacted substrate (“S-220”), products (“P-141” and “P-113”), and the slower migrating HJ intermediate (“HJ”) are visualized and quantitated using autoradiography and phosphorimaging. Impurities (“i”, <2% of total counts) were not included in the quantitation. The two gels are representative of the raw data for assembly titration reactions of CreWT/LoxGC (left panel) and CreQQ/LoxGC (right panel), with the complex concentration (in nM) listed above each lane.
To assess effects on active complex assembly, we measured the amount of substrate turnover in 16-h endpoint reactions as a function of complex concentration (representative gels for the LoxGC active complex assembly titration are shown in Figure 2). We define “substrate turnover”2 as the disappearance of labeled substrate, reacted to HJ intermediates or products, and “reaction extent”2 as the maximum percentage of substrate converted. The data were fit using the Hill binding function to yield apparent binding constants and Hill coefficients (described in Experimental Procedures and ref 16). The apparent KD value (Table 1) provides a combined assessment of monomer-binding, dimerization, and tetramerization equilibria that contribute to active Cre4Lox2 complex formation. The physical significance of the Hill coefficients in these reactions is not well understood but may reflect the interplay between DNA binding and subunit interactions (16). The concentration dependence of substrate turnover was plotted for LoxTA and LoxGC (Figure 3a), and titration curves for all substrates are illustrated normalized to their mean reaction extents (Figure 4a).
Figure 3.
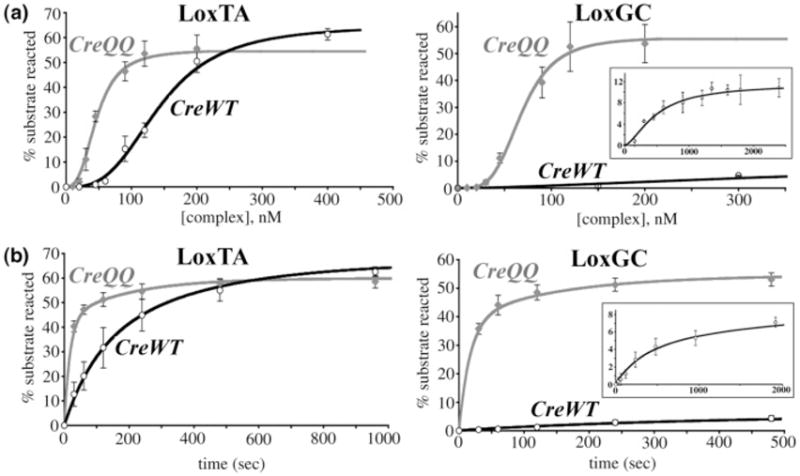
Assembly titration and kinetic assay data for CreWT and CreQQ reactions with LoxTA and LoxGC. (a) Substrate turnover as a function of active Cre4Lox2 complex concentration. Assembly titration data and averaged fit curves are shown for CreWT (black curves) and CreQQ (gray curves) with LoxTA (left plot) and LoxGC (right plot). Error bars denote the standard deviation from four replicate reactions. The inset details the CreWT/LoxGC reaction. LoxP and LoxAT reactions have been reported earlier (16). (b) Substrate turnover as a function of time. Time course data and fit curves are shown for CreWT (black curves) and CreQQ (gray curves) with LoxTA (left plot) and LoxGC (right plot). Error bars denote the standard deviation from four replicate reactions. The inset details the CreWT/LoxGC reaction.
Figure 4.
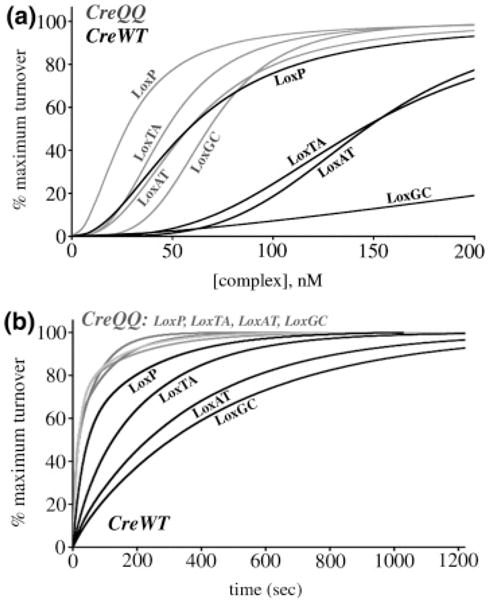
Normalized assembly titration and time course progress curves. The maximum substrate reacted (“extent”) was used as the normalization factor, and curves are drawn using the fit parameters from Tables 1 and 2. Models and fits are described in Experimental Procedures. The substrates are indicated adjacent to each curve. (a) Complex assembly reactions. CreWT (black) and CreQQ (gray) binding curves are determined from an average of four or more reactions. The plot is expanded to 0-200 nM complex concentrations to show the differences between the CreQQ reactions. (b) Time course reactions. CreWT (black) and CreQQ (gray) curves are determined from the average of 3 or more replicates, and the curves from CreQQ reactions with each substrate overlap.
While LoxAT, LoxTA and LoxGC bound poorly in an electrophoretic mobility shift assay (EMSA,1 2–4% of LoxP binding) (57), they formed significant products in our in vitro recombination assay. Compared with LoxP, higher complex concentrations were required to achieve half the maximum amount of substrate turnover (“apparent KD”), and LoxGC was most dramatic in this regard (Figure 4a). Whereas LoxAT and LoxTA increased the apparent KD 2.5-fold compared to LoxP, LoxGC increased the KD 8.5-fold compared to LoxP (Table 1). The apparent Hill coefficients of each of these substrates varied, with LoxAT (16) having the highest value (3.9) and LoxGC having the lowest (2.0), although this value is unreliable due to the low level of LoxGC reaction. Generally, LoxP 8/27 substitutions have varying degrees of influence on the affinity and cooperative assembly required to form an active Cre-Lox synapse. However and unexpectedly, the CreWT/LoxGC reaction extent was reduced to only 12%, compared to LoxP, LoxTA, and LoxAT reaction extents of 57–67% (Table 1).
To assess effects on substrate turnover rates, we measured time-dependent substrate disappearance in single turnover reactions at saturating complex concentrations. Substrate turnover data were plotted for LoxTA and LoxGC (Figure 3b) and normalized single turnover progress curves for all substrates were compared (Figure 4b). As previously described for CreWT/LoxAT reactions (16), either biexponential or nearly-equivalent sequential (a → b → c) kinetic models are required to adequately describe the time-course data (See Experimental Procedures). These four-parameter fits yielded rate constants for “slow” and “fast” phases that differed by factors of 5–13 (Table 2). The correspondences between these fast and slow processes and mechanistic reaction steps are not known but may be related to the formation of two populations of reactive complexes prior to strand exchange, one of which may require a structural rearrangement prior to performing recombination. Multiple kinetic processes can be an indication of a heterogeneous enzyme preparation. However, with some substrates Cre exhibits monophasic reaction kinetics (30, 35), supporting the idea of two populations of complexes rather than two different enzyme activities.
Overall, the substituted substrates had reduced turnover rates compared to LoxP (Figure 4b). LoxGC, with the lowest apparent affinity, had the most substantially reduced turnover rate (Table 2). Using t1/2 estimates, the time required to turn over half the maximum amounts of substrate calculated from the kinetic model, LoxP reacts the fastest, LoxTA is 2.5-fold slower, LoxAT is 4.9-fold slower, and LoxGC is 6.3-fold slower.
As was previously observed with LoxAT (16), the apparent rate constants for disappearances of each substrate varied only maximally by a factor of 2, while the primary difference between substrates was the partitioning between the fast and slow processes. Generally, the mutant substrates exhibited a large fraction of complexes that reacted via the slow phase, 0.81–0.87, while LoxP partitioned more equally between the slow and fast phases. As with LoxAT (16), Cre discriminates between LoxP, LoxTA, and LoxGC at the levels of both active complex assembly and turnover rate. The low level of LoxGC reaction extent represents an additional and previously un-described level of substrate differentiation. This behavior may be related to the fact that LoxGC is the lowest affinity substrate.
CreQQ Suppresses Lox8/27 Substitution Effects on Binding and Turnover
In all CreWT-DNA complex structures determined to date, Glu262 makes an unfavorable contact with the phosphate of Ade25 of Lox substrates (Figure 1c). When Glu262 is substituted to Gln, we hypothesized that the unfavorable contact is replaced by a favorable amide-phosphate interaction that greatly enhances DNA-binding (16). Indeed, CreQQ exhibited increased affinity and reaction rates in vitro for both LoxP and LoxAT but reduced specificity between these substrates (16). The Glu266 → Gln mutation does not significantly affect DNA binding but improves Cre/Lox complex solubility (data not shown).
As with LoxAT, CreQQ also displayed increased binding with LoxTA and LoxGC, having 3–7-fold higher affinity than CreWT (Table 1). CreQQ had similar substrate preferences as CreWT, in that LoxP had the lowest apparent KD and LoxGC had the highest apparent KD (Figure 4a), but it exhibited much lower specificity. CreQQ had only a 2.5-fold higher KD with LoxGC compared to LoxP, whereas CreWT had an 8.5-fold difference. Significantly, CreQQ also rescued the reduction in LoxGC reaction extent. While CreWT had up to 8.6-fold reduced reaction extent of LoxGC compared to LoxP in 30 min reactions (Table 2; 5.6-fold reduced extent in 16-h reactions, see Table 1), CreQQ maintained a similar reaction extent with each substrate (Tables 1 and 2). The Hill coefficients followed a different trend than with CreWT. With CreQQ, LoxGC had the largest Hill coefficient (3.9), and LoxP had the smallest (2.0).
CreQQ also accelerated recombination rates with all substrates compared to CreWT (Figure 4b). The rate constants for both reaction phases increased up to 3.8-fold, with favored partitioning toward the fast phase with each substrate (Table 2). As a result, CreQQ did not distinguish kinetically between the substrates, and the fit-derived t1/2 values were within experimental error (Table 2).
Binding Affinity Influences the Rate-Limiting Reaction Step
To determine the effects of Lox 8/27 mutations on each of the two strand exchanges, we tested whether the substitutions differentially altered the HJ and product formation rates. We followed HJ and product formation in the same reactions and compared rate constants (Tables 3 and 4) and progress curves (Figure 5). When HJ and product appearance exhibit similar time courses, the first strand exchange is rate-limiting. Indeed, for CreWT/LoxP (Figure 5a), HJ and product formation curves essentially overlap. Protein-DNA association is not the slowest step in this case because the rate constants do not change when Cre/Lox concentrations are halved (data not shown). Thus, the rate-limiting step occurs after active complex formation but before the completion of the first strand exchange.
Figure 5.

Normalized HJ and product formation in time course reactions. The representative curves are normalized to 100%, using the maximum substrate turned over to HJ or products in each reaction as the normalization factor. The fit parameters are listed in Tables 3 and 4. (a) CreWT reactions. The time dependence of HJ formation is shown as gray curves/gray diamonds, and product formation as black curves/open circles. (b) Comparison of the time dependence of HJ formation (left panel) or product formation (right panel) in CreWT reactions. (c) CreQQ reactions. HJ formation is shown as gray curves/gray diamonds, and product formation as black curves/open circles.
However, in CreWT reactions with the Lox8/27 substrates, HJ formation is clearly faster than product formation (Figure 5a), indicating that weaker binding influences the rate-limiting step in these reactions. In fact, HJ formation is more rapid for LoxAT and LoxTA, compared to LoxP, while LoxGC is only 1.7-fold slower (Figure 5b and Table 3). Rate-limiting product formation in LoxAT and LoxTA reactions was 3.8-fold slower than LoxP (Figure 5b) and could essentially be described by a single rate constant (Table 4). In the extreme case of CreWT/LoxGC, product formation was delayed 60 s while HJ intermediates were observed at the earliest sample time of 30 s. The difference in the apparent number of processes involved in the first and second strand exchange (Tables 3 and 4) implies that, generally, biphasic substrate disappearance kinetics arise from assembly of two initial complexes, one that reacts immediately, and another that undergoes a rearrangement to the rapidly acting form prior to the first strand exchange.
Since HJ formation is faster than product formation for the Lox8/27 substrates, HJ isomerization and/or the second strand exchange are rate-limiting. In contrast to the CreWT reactions with 8/27 substrates, analogous CreQQ reactions exhibit the early rate-limiting step (Tables 3 and 4): HJ and product formation progress curves overlap as they do with CreWT/LoxP (Figure 5c). Thus, the delay in product formation in the mutant substrates is likely to be a result of less tight DNA binding by CreWT. The results also suggest that the Cre-DNA affinity switch during HJ isomerization is the driving force for the second strand exchange.
DISCUSSION
Cre Exhibits Multiple Levels of Substrate Discrimination
Our data suggest that Cre’s recombination specificity is manifested at three levels, each of which are related to its affinity for the 13 bp repeat. At the level of active complex assembly, increases in half-saturating concentrations and sigmoidicity effectively discriminated against reduced-binding LoxAT (16) and LoxTA substrates at low concentrations but this discrimination vanished at high concentrations. However, the most severely perturbed LoxGC substrate produced little product, even at saturation, suggesting that DNA-binding free energy is also linked to assembly of recombination-competent complexes. At the kinetic level and consistent with our previous observations, Cre-mutant Lox complexes exhibited reduced turnover rates that varied with DNA affinity, albeit nonlinearly. Together, the two concentration-independent influences, that is reductions in turnover rate and reaction extent, contribute 35–60-fold to discrimination between LoxP and LoxGC. The connection between these three seemingly distinct specificity mechanisms and Cre substrate affinity is further supported by their suppression by tighter-binding CreQQ protein.
Mechanistically, increased sigmoidicity of the assembly titration curves might result from thermodynamic linkage of favorable protein-DNA and intersubunit contacts with unfavorable energetic barriers to assembling active recombination complexes. The likely origins of these adverse influences are specific bending of the Lox 8 bp spacer DNA (58) and protein conformational strain. In particular, the low LoxGC reaction extent may reflect the effects of this “assembly strain”. With insufficient DNA binding affinity, Cre-DNA complexes may have a greater proclivity to be trapped in alternate misassembled forms. This idea is supported by the recovery of high product levels in CreQQ/LoxGC reactions. The structural nature of these mis-assembled complexes is not known, but they may lack the complete complement of intermolecular interactions, or possess higher conformational barriers to strand exchange. However, the lack of a correlation between apparent DNA affinity and Hill number suggests a more detailed explanation, specific to the substitutions, is required. For example, the origin of the low sigmoidicity for LoxGC concentration-dependence is not known. This property may reflect the inaccuracies of measurements for low turnover reactions but also may be a result of turnover by a subpopulation of complexes assembled from higher-order Cre oligomers, which tend to form from free Cre protein under the reaction conditions (16, 52).
Generality of Affinity-Kinetic Coupling
The results reported here coincide with our previous observations, suggesting that Cre generally recombines weaker-binding substrates more slowly. Other reduced-binding Lox substrates with differently located substitutions also exhibit this behavior, but in these cases direct active site perturbation cannot be ruled out (data not shown). Cre apparently expresses its kinetic specificity differently from restriction enzymes and DNA glycosylases since the distant 8/27 substitutions are not likely to perturb the active site. While the 3–7-fold kinetic discrimination factors with 8/27-substituted substrates are small compared to restriction enzyme fidelity levels, they can be potentially significant in vivo where they can be amplified by non-steady-state conditions. In E. coli, Cre-mediated marker excision is markedly slower with LoxAT and LoxGC compared to LoxP (S. Martin and E. Baldwin, unpublished data).
What is the mechanism of this affinity-kinetic coupling for CreWT? The rate-limiting step changes from occurring before the first strand exchange with LoxP, to occurring after the first strand exchange with the lower affinity 8/27-substituted substrates. Interestingly, HJ formation rates are comparable to that for LoxP, but product formation was significantly slower and delayed with LoxGC (Figure 5b). This affinity-dependent inhibition of HJ resolution could be manifested in any of several reaction steps after HJ formation. We previously suggested that poorer DNA binding allows the noncleaving Cre subunit to allosterically down-regulate the cleaving subunit active site via the 198–208 loop (16). Chemical steps during the second strand exchange could also be directly perturbed by rearrangements or order changes in the active site. In either case, structural differences between initiation and HJ-resolution complexes (Figure 1b) would need to be invoked to rationalize a differential effect on the second strand exchange.
A simpler alternative “conformational coupling model” is that HJ isomerization itself is inhibited; that is, poorer DNA binding slows the switch between noncleaving and cleaving Cre subunit conformations. HJ isomerization may be retarded because of an overall change in Cre affinity for DNA that occurs upon conformational switching. This affinity change is suggested by ordering and positioning differences of Cre protomers in complex crystal structures (16), with the more well-ordered and tightly associated cleaving subunits binding DNA more strongly than the poorly ordered and loosely associated noncleaving subunits. In this view, the driving force for HJ isomerization is preferential association of the cleaving subunit with the Lox arm that is cleaved during HJ resolution (Figure 1b) (29, 35). Reduced DNA affinity renders this interconversion less favorable, so that it becomes rate-limiting. With particularly poor-binding substrates, a delay in product appearance might be expected, as is observed with LoxGC. When protein-DNA affinity is restored using CreQQ, events prior to HJ formation again become rate-limiting. Apparently, DNA release by the cleaving subunit during isomerization is not limiting, or CreQQ would inhibit HJ resolution.
The conformational coupling model described above is an example of how altering protein-DNA interactions can influence multistep reactions. Conformational differences lead to differential stability changes in reaction intermediates and transition states in response to mutations, affecting reaction progress and outcome. For example, differential stabilization of the initiation complex and first-strand exchange intermediates over the second exchange intermediates and product complexes could reduce the maximum amount of substrate turnover as observed with LoxGC. Although LoxGC turns over much less substrate the HJ to product ratio is similar to that for LoxP (Tables 3 and 4), suggesting that free energy differences in initiation complex formation rather than differential stabilization of reaction intermediates is responsible for the low yields. To a first approximation the conformational coupling model proposed above provides a ready explanation for the gross reactivity differences based on DNA affinity, but the complete explanation may be more complex. Clearly, the differences in HJ accumulation levels and reaction parameters between similarly binding LoxAT and LoxTA substrates (Tables 1–4) require a more detailed description and may reflect a number of energy shifts along the reaction coordinate.
Arg259 Makes a Preferential Interaction with Gua27 of LoxP
The bidentate hydrogen-bond donors of Arg259 guanidinium Nε and Nη1 are ideally configured for specific interaction with Gua27 O6 and N7, although this particular recognition mode is somewhat less common than “end-on” recognition by Arg Nη1 and Nη2 atoms (see Database Analysis of Protein-DNA Interactions in Experimental Procedures). Data presented here and elsewhere (16, 41, 43, 57, 59) indicate that the Arg259-Gua27 interaction is a significant contributor to Cre-LoxP recognition, and substituting the Cyt8-Gua27 basepair with the three other basepairs significantly weakens Cre binding. The preference order observed here, Gua >Ade ~ Thy ≫ Cyt, and, in particular, the severe disruption by the Cyt substitution, suggests unique structural responses to the substitutions. The particularly deleterious LoxGC effect on recombination was not indicated by the similar Cre binding reductions for all 8/27 substitutions assessed by EMSA (57). This discrepancy underscores the need to examine the complete reaction to evaluate mutational consequences, since EMSA results are sensitive to complex kinetic stability and other factors.
The structural reasons for the extreme behavior of LoxGC await the CreWT/LoxGC complex structure solution. In the CreWT/LoxAT complex, Arg259 does not interact with Thy27 O4 because of steric hindrance with 5-methyl group (16), and the similar affinity of LoxTA may reflect a similar loss of the Arg-Gua interaction. The LoxGC effect likely involves more than the simple inability to provide hydrogen-bond acceptors for Arg259. Other mechanisms, such as an unfavorable cation-pi interaction (60), may contribute to particular discrimination against Cyt29. Interestingly, Flp recombinase has a different recombination preference order for analogous FRT substitutions in vitro, Gua >Thy >Cyt >Ade (49), suggesting that context contributions beyond the Arg-Gua interaction are important.
Role of Glu262 in Substrate Discrimination
The unfavorable Glu262-phosphate interaction has a key role in sequence discrimination by Cre. Conversion to a favorable contact in CreQQ suppressed the binding and kinetic defects of all the 8/27-substituted Lox sites; that is, they behaved more similarly and, in particular, recombination rates were indistinguishable.
The Glu262 → Gln mutation is also associated with increased Cre reactivity toward Lox sites containing a variety of substitutions (40, 42, discussed in ref 16). At moderately high concentrations (>150 nM LoxP/600 nM CreQQ), CreQQ will even recombine the triply substituted LoxM7 (41), which shows no activity with CreWT at complex concentrations up to 1200 nM LoxP/4800 nM CreWT (S. Martin and E. Baldwin, unpublished data).
The manner in which CreQQ accelerates reactions is not simply related to its global affinity, since it binds LoxGC about as tightly as CreWT binds LoxP, but it recombined LoxGC more than twice as rapidly. Local immobilization of the DNA backbone imposed by the Gln262 amide-phosphate interaction alone cannot explain this effect. Although CreQQ reacted with a LoxP half-site suicide substrate ("l*/r" in ref 35) at most 30–40% faster compared to CreWT (data not shown), this increase is significantly below the 2–4-fold increases in rate constants and 2.5-17- fold reductions in t1/2 values for CreQQ reactions with full-site substrates.
The absence of base contacts in the favorable Gln262-backbone interaction would seem to suggest that CreQQ affinity increases should be DNA sequence-independent. Indeed, CreQQ more or less preserves the CreWT specificity order in the 8/27 mutant series, arguing for a nonspecific enhancement mechanism. However, protein and DNA substitutions at this interface have previously been associated with significant shifts in the DNA backbone (16, 59), and Gln262 might be expected to interact differently in such contexts. This “indirect readout” may account for the discrepancy between the 3-fold enhancement for LoxTA binding and the 7-fold enhancement with LoxGC. Structural comparisons of CreQQ and CreWT complexes with Lox8/27 mutants will address the underlying mechanism for this differential effect.
Interplay between Substrate Affinity and Reaction Specificity
A key implication of these results is that Cre’s overall substrate affinity is finely tuned for specificity. Surprisingly, the apparent role of Glu262 is to attenuate Cre binding, acting as a kind of “gate-keeper” that sets the affinity threshold for viable substrates. Because of the high degree of binding cooperativity mediated through protein oligomerization, promiscuous binding by Cre subunits might be expected to more drastically reduce specificity than would be the case for a monomeric protein. Several other protein-DNA complexes depict similar carboxylate-phosphate interactions (see Experimental Procedures and Supplemental Table 2, Supporting Information), providing evidence that this discrimination strategy could be generally utilized. The repulsive interaction may also facilitate protein movement along DNA by lowering the activation barrier for dissociation, suggested by the occurrence of such carboxylate interactions between T7 polymerase or HIV-reverse transcriptase with their double-stranded templates.
Another implication is that Cre mutants with altered specificity for the 13 bp repeat should have no intrinsic limit to high activity, and recombination defects could be suppressed by simple affinity increases. In this regard, CreQQ or the Gln262 substitution may prove generally useful for enhancing recombination of poor substrates in vivo, such as the partially inactivated Lox sites used to promote unidirectional recombination (61). However, it does not appear likely that affinity increases will ameliorate defects from 8 bp spacer substitutions, which can severely alter the course and outcome of the reaction (30). Similarly, 13 bp repeat DNA substitutions near the active site (Lox nucleotides 10–13 and 20–23), and the corresponding compensating Cre substitutions that restore binding might also be disruptive for catalysis.
Supplementary Material
Acknowledgments
Protein purifications were carried out in the W. M. Keck Protein Expression Facility at U. C. Davis.
Footnotes
This work was funded by the National Institutes of Health, General Medical Sciences, Research Grant RO1-GM63109-01A1 and Training Grant T32-GM07377-25.
bp, number of basepairs; HJ, Holliday junction (intermediate); EMSA, electrophoretic mobility shift assay.
Substrate turnover, the combined conversion of substrate to reaction intermediates and products; reaction extent, the maximum percentage of substrate reacted to form HJs, products, or both in the in vitro recombination reactions, derived from binding or kinetic models.
A8/T27 (LoxAT), T8/A27 (LoxTA), and G8/C27 (LoxGC), Lox sites containing substitutions of Cyt-Gua and Gua-Cyt basepairs at positions 8 and 27 in Lox, with Ade-Thy and Thy-Ade basepairs, Thy-Ade, and Ade-Thy basepairs, and Gua-Cyt and Cyt-Gua basepairs, respectively.
CreWT, wild-type Cre recombinase with a His6 N-terminal tag; CreQQ, CreWT in which glutamate residues at positions 262 and 266 have been substituted with glutamine.
Amino acids and deoxynucleotides are referred to by their three-letter code.
SUPPORTING INFORMATION AVAILABLE
A list of the Gua-Arg interactions similar to that observed between Gua27-Arg259 of LoxP-Cre, the 6 acid-phosphate contacts identified from a database of 139 protein-DNA complexes, and the PDB entries for the 139 complexes. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lesser DR, Kurpiewski MR, Jen-Jacobson L. The energetic basis of specificity in the Eco RI endonuclease-DNA interaction. Science. 1990;250:776–786. doi: 10.1126/science.2237428. [DOI] [PubMed] [Google Scholar]
- 2.Jen-Jacobson L. Protein-DNA recognition complexes: conservation of structure and binding energy in the transition state. Biopolymers. 1997;44:153–180. doi: 10.1002/(SICI)1097-0282(1997)44:2<153::AID-BIP4>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 3.Pingoud A, Jeltsch A. Structure and function of type II restriction endonucleases. Nucleic Acids Res. 2001;29:3705–3727. doi: 10.1093/nar/29.18.3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hollis T, Lau A, Ellenberger T. Crystallizing thoughts about DNA base excision repair. Prog Nucleic Acid Res Mol Biol. 2001;68:305–314. doi: 10.1016/s0079-6603(01)68109-1. [DOI] [PubMed] [Google Scholar]
- 5.Kraut DA, Carroll KS, Herschlag D. Challenges in enzyme mechanism and energetics. Annu Rev Biochem. 2003;72:517–571. doi: 10.1146/annurev.biochem.72.121801.161617. [DOI] [PubMed] [Google Scholar]
- 6.Kurpiewski MR, Engler LE, Wozniak LA, Kobylanska A, Koziolkiewicz M, Stec WJ, Jen-Jacobson L. Mechanisms of coupling between DNA recognition specificity and catalysis in EcoRI endonuclease. Structure. 2004;12:1775–1788. doi: 10.1016/j.str.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 7.Narlikar GJ, Herschlag D. Direct demonstration of the catalytic role of binding interactions in an enzymatic reaction. Biochemistry. 1998;37:9902–9911. doi: 10.1021/bi980495t. [DOI] [PubMed] [Google Scholar]
- 8.Goldhaber-Gordon I, Early MH, Baker TA. MuA transposase separates DNA sequence recognition from catalysis. Biochemistry. 2003;42:14633–14642. doi: 10.1021/bi035360o. [DOI] [PubMed] [Google Scholar]
- 9.Taylor JD, Halford SE. Discrimination between DNA sequences by the EcoRV restriction endonuclease. Biochemistry. 1989;28:6198–6207. doi: 10.1021/bi00441a011. [DOI] [PubMed] [Google Scholar]
- 10.Yang CC, Baxter BK, Topal MD. DNA cleavage by NaeI: protein purification, rate-limiting step, and accuracy. Biochemistry. 1994;33:14918–14925. doi: 10.1021/bi00253a031. [DOI] [PubMed] [Google Scholar]
- 11.Sternberg N, Hamilton D, Austin S, Yarmolinsky M, Hoess R. Site-specific recombination and its role in the life cycle of bacteriophage P1. Cold Spring Harb Symp Quant Biol. 1981;45(Pt 1):297–309. doi: 10.1101/sqb.1981.045.01.042. [DOI] [PubMed] [Google Scholar]
- 12.Hoess RH, Ziese M, Sternberg N. P1 site-specific recombination: nucleotide sequence of the recombining sites. Proc Natl Acad Sci USA. 1982;79:3398–3402. doi: 10.1073/pnas.79.11.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoess RH, Abremski K. Mechanism of strand cleavage and exchange in the Cre-lox site-specific recombination system. J Mol Biol. 1985;181:351–362. doi: 10.1016/0022-2836(85)90224-4. [DOI] [PubMed] [Google Scholar]
- 14.Hoess R, Wierzbicki A, Abremski K. Isolation and characterization of intermediates in site-specific recombination. Proc Natl Acad Sci USA. 1987;84:6840–6844. doi: 10.1073/pnas.84.19.6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo F, Gopaul DN, van Duyne GD. Structure of Cre recombinase complexed with DNA in a site-specific recombination synapse. Nature. 1997;389:40–46. doi: 10.1038/37925. [DOI] [PubMed] [Google Scholar]
- 16.Martin SS, Chu VC, Baldwin E. Modulation of the active complex assembly and turnover rate by protein-DNA interactions in Cre-LoxP recombination. Biochemistry. 2003;42:6814–6826. doi: 10.1021/bi0272306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoess RH, Abremski K. Interaction of the bacteriophage P1 recombinase Cre with the recombining site loxP. Proc Natl Acad Sci USA. 1984;81:1026–1029. doi: 10.1073/pnas.81.4.1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abremski K, Hoess R. Bacteriophage P1 site-specific recombination. Purification and properties of the Cre recombinase protein. J Biol Chem. 1984;259:1509–1514. [PubMed] [Google Scholar]
- 19.Abremski K, Hoess R, Sternberg N. Studies on the properties of P1 site-specific recombination: evidence for topologically unlinked products following recombination. Cell. 1983;32:1301–1311. doi: 10.1016/0092-8674(83)90311-2. [DOI] [PubMed] [Google Scholar]
- 20.Sternberg N, Hamilton D. Bacteriophage P1 site-specific recombination. I Recombination between loxP sites. J Mol Biol. 1981;150:467–486. doi: 10.1016/0022-2836(81)90375-2. [DOI] [PubMed] [Google Scholar]
- 21.Mack A, Sauer B, Abremski K, Hoess R. Stoichiometry of the Cre recombinase bound to the lox recombining site. Nucleic Acids Res. 1992;20:4451–4455. doi: 10.1093/nar/20.17.4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Esposito D, Scocca JJ. The integrase family of tyrosine recombinases: evolution of a conserved active site domain. Nucleic Acids Res. 1997;25:3605–3614. doi: 10.1093/nar/25.18.3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grainge I, Jayaram M. The integrase family of recombinases: organization and function of the active site. Mol Microbiol. 1999;33:449–456. doi: 10.1046/j.1365-2958.1999.01493.x. [DOI] [PubMed] [Google Scholar]
- 24.Nunes-Duby SE, Kwon HJ, Tirumalai RS, Ellenberger T, Landy A. Similarities and differences among 105 members of the Int family of site-specific recombinases. Nucleic Acids Res. 1998;26:391–406. doi: 10.1093/nar/26.2.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sadowski PD. The Flp recombinase of the 2-microns plasmid of Saccharomyces cerevisiae. Prog Nucleic Acid Res Mol Biol. 1995;51:53–91. [PubMed] [Google Scholar]
- 26.Hoess R, Wierzbicki A, Abremski K. Synapsis in the Cre-lox Site-specific Recombination System. In: Sarma R, Sarma M, editors. Structure and Methods: Human Genome Initiative and DNA Recombination. Adenine Press; Schenectady, NY: 1990. pp. 203–213. [Google Scholar]
- 27.Shaikh A, Sadowski P. Trans complementation of variant Cre proteins for defects in cleavage and synapsis. J Biol Chem. 2000;275:30186–30195. doi: 10.1074/jbc.M005256200. [DOI] [PubMed] [Google Scholar]
- 28.Lee L, Sadowski PD. Sequence of the loxP site determines the order of strand exchange by the Cre recombinase. J Mol Biol. 2003;326:397–412. doi: 10.1016/s0022-2836(02)01429-8. [DOI] [PubMed] [Google Scholar]
- 29.Lee L, Sadowski PD. Strand Selection by the Tyrosine Recombinases. In: Moldave K, editor. Prog Nucleic Acid Res Mol Biol. Academic Press; New York: 2005. pp. 1–42. [DOI] [PubMed] [Google Scholar]
- 30.Gelato KA, Martin SS, Baldwin EP. Reversed DNA Strand Cleavage Specificity in Initiation of Cre-LoxP Recombination Induced by the His289Ala Active-site Substitution. J Mol Biol. 2005;354:233–245. doi: 10.1016/j.jmb.2005.08.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Duyne GD. A structural view of cre-loxp site-specific recombination. Annu Rev Biophys Biomol Struct. 2001;30:87–104. doi: 10.1146/annurev.biophys.30.1.87. [DOI] [PubMed] [Google Scholar]
- 32.Ennifar E, Meyer JE, Buchholz F, Stewart AF, Suck D. Crystal structure of a wild-type Cre recombinase-loxP synapse reveals a novel spacer conformation suggesting an alternative mechanism for DNA cleavage activation. Nucleic Acids Res. 2003;31:5449–5460. doi: 10.1093/nar/gkg732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gopaul DN, Guo F, Van Duyne GD. Structure of the Holliday junction intermediate in Cre-loxP site-specific recombination. EMBO J. 1998;17:4175–4187. doi: 10.1093/emboj/17.14.4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo F, Gopaul DN, Van Duyne GD. Asymmetric DNA bending in the Cre-loxP site-specific recombination synapse. Proc Natl Acad Sci USA. 1999;96:7143–7148. doi: 10.1073/pnas.96.13.7143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martin SS, Pulido E, Chu VC, Lechner TS, Baldwin EP. The order of strand exchanges in Cre-LoxP recombination and its basis suggested by the crystal structure of a Cre-LoxP Holliday junction complex. J Mol Biol. 2002;319:107–127. doi: 10.1016/S0022-2836(02)00246-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kolb AF. Genome engineering using site-specific recombinases. Cloning Stem Cells. 2002;4:65–80. doi: 10.1089/153623002753632066. [DOI] [PubMed] [Google Scholar]
- 37.Tronche F, Casanova E, Turiault M, Sahly I, Kellendonk C. When reverse genetics meets physiology: the use of site-specific recombinases in mice. FEBS Lett. 2002;529:116–121. doi: 10.1016/s0014-5793(02)03266-0. [DOI] [PubMed] [Google Scholar]
- 38.Branda CS, Dymecki SM. Talking about a revolution: The impact of site-specific recombinases on genetic analyses in mice. Dev Cell. 2004;6:7–28. doi: 10.1016/s1534-5807(03)00399-x. [DOI] [PubMed] [Google Scholar]
- 39.Gilbertson L. Cre-lox recombination: Creative tools for plant biotechnology. Trends Biotechnol. 2003;21:550–555. doi: 10.1016/j.tibtech.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 40.Buchholz F, Stewart AF. Alteration of Cre recombinase site specificity by substrate-linked protein evolution. Nat Biotechnol. 2001;19:1047–1052. doi: 10.1038/nbt1101-1047. [DOI] [PubMed] [Google Scholar]
- 41.Santoro SW, Schultz PG. Directed evolution of the site specificity of Cre recombinase. Proc Natl Acad Sci USA. 2002;99:4185–4190. doi: 10.1073/pnas.022039799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rufer AW, Sauer B. Noncontact positions impose site selectivity on Cre recombinase. Nucleic Acids Res. 2002;30:2764–2771. doi: 10.1093/nar/gkf399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim ST, Kim GW, Lee YS, Park JS. Characterization of Cre-loxP interaction in the major groove: hint for structural distortion of mutant Cre and possible strategy for HIV-1 therapy. J Cell Biochem. 2001;80:321–327. [PubMed] [Google Scholar]
- 44.Lejeune D, Delsaux N, Charloteaux B, Thomas A, Brasseur R. Protein-nucleic acid recognition: statistical analysis of atomic interactions and influence of DNA structure. Proteins. 2005;61:258–271. doi: 10.1002/prot.20607. [DOI] [PubMed] [Google Scholar]
- 45.Chen Y, Narendra U, Iype LE, Cox MM, Rice PA. Crystal structure of a Flp recombinase-Holliday junction complex: assembly of an active oligomer by helix swapping. Mol Cell. 2000;6:885–897. [PubMed] [Google Scholar]
- 46.Andrews BJ, McLeod M, Broach J, Sadowski PD. Interaction of the FLP recombinase of the Saccharomyces cerevisiae 2 micron plasmid with mutated target sequences. Mol Cell Biol. 1986;6:2482–2489. doi: 10.1128/mcb.6.7.2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McLeod M, Craft S, Broach JR. Identification of the crossover site during FLP-mediated recombination in the Saccharomyces cerevisiae plasmid 2 microns circle. Mol Cell Biol. 1986;6:3357–3367. doi: 10.1128/mcb.6.10.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Prasad PV, Horensky D, Young LJ, Jayaram M. Substrate recognition by the 2 micron circle site-specific recombinase: effect of mutations within the symmetry elements of the minimal substrate. Mol Cell Biol. 1986;6:4329–4334. doi: 10.1128/mcb.6.12.4329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Senecoff JF, Rossmeissl PJ, Cox MM. DNA recognition by the FLP recombinase of the yeast 2 mu plasmid. A mutational analysis of the FLP binding site. J Mol Biol. 1988;201:405–421. doi: 10.1016/0022-2836(88)90147-7. [DOI] [PubMed] [Google Scholar]
- 50.Biswas T, Aihara H, Radman-Livaja M, Filman D, Landy A, Ellenberger T. A structural basis for allosteric control of DNA recombination by lambda integrase. Nature. 2005;435:1059–1066. doi: 10.1038/nature03657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kwon HJ, Tirumalai R, Landy A, Ellenberger T. Flexibility in DNA recombination: structure of the lambda integrase catalytic core. Science. 1997;276:126–131. doi: 10.1126/science.276.5309.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Woods KC, Martin SS, Chu VC, Baldwin EP. Quasi-equivalence in site-specific recombinase structure and function: crystal structure and activity of trimeric Cre recombinase bound to a three-way Lox DNA junction. J Mol Biol. 2001;313:49–69. doi: 10.1006/jmbi.2001.5012. [DOI] [PubMed] [Google Scholar]
- 53.Kilbride E, Boocock MR, Stark WM. Topological selectivity of a hybrid site-specific recombination system with elements from Tn3 res/resolvase and bacteriophage P1 loxP/Cre. J Mol Biol. 1999;289:1219–1230. doi: 10.1006/jmbi.1999.2864. [DOI] [PubMed] [Google Scholar]
- 54.Kilbride EA, Burke ME, Boocock MR, Stark WM. Determinants of product topology in a hybrid Cre-Tn3 resolvase site-specific recombination system. J Mol Biol. 2006;355:185–195. doi: 10.1016/j.jmb.2005.10.046. [DOI] [PubMed] [Google Scholar]
- 55.Barre FX, Aroyo M, Colloms SD, Helfrich A, Cornet F, Sherratt DJ. FtsK functions in the processing of a Holliday junction intermediate during bacterial chromosome segregation. Genes Dev. 2000;14:2976–2988. doi: 10.1101/gad.188700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang XJ, Matthews BW. EDPDB: a multifunctional tool for protein structure analysis. J Appl Crystallogr. 1995;28:624–630. [Google Scholar]
- 57.Hartung M, Kisters-Woike B. Cre mutants with altered DNA binding properties. J Biol Chem. 1998;273:22884–22891. doi: 10.1074/jbc.273.36.22884. [DOI] [PubMed] [Google Scholar]
- 58.Lee L, Chu LC, Sadowski PD. Cre induces an asymmetric DNA bend in its target loxP site. J Biol Chem. 2003;278:23118–23129. doi: 10.1074/jbc.M302272200. [DOI] [PubMed] [Google Scholar]
- 59.Baldwin EP, Martin SS, Abel J, Gelato KA, Kim H, Schultz PG, Santoro SW. A specificity switch in selected cre recombinase variants is mediated by macromolecular plasticity and water. Chem Biol. 2003;10:1085–1094. doi: 10.1016/j.chembiol.2003.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wintjens R, Lievin J, Rooman M, Buisine E. Contribution of cation-pi interactions to the stability of protein-DNA complexes. J Mol Biol. 2000;302:395–410. doi: 10.1006/jmbi.2000.4040. [DOI] [PubMed] [Google Scholar]
- 61.Albert H, Dale EC, Lee E, Ow DW. Site-specific integration of DNA into wild-type and mutant lox sites placed in the plant genome. Plant J. 1995;7:649–659. doi: 10.1046/j.1365-313x.1995.7040649.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.