

Abstract
An integral feature of gammaherpesvirus infections is the ability to establish lifelong latency in B cells. During latency, the viral genome is maintained as an extrachomosomal episome, with stable maintenance in dividing cells mediated by the viral proteins Epstein-Barr nuclear antigen 1 (EBNA-1) for Epstein-Barr virus and latency-associated nuclear antigen (LANA) for Kaposi's sarcoma-associated herpesvirus. It is believed that the expression of episome maintenance proteins is turned off in the predominant long-term latency reservoir of resting memory B cells, suggesting that chronic gammaherpesvirus infection is primarily dormant. However, the kinetics of LANA/EBNA-1 expression in individual B-cell subsets throughout a course of infection has not been examined. The infection of mice with murine gammaherpesvirus 68 (MHV68, γHV68) provides a model to determine the specific cellular and molecular events that occur in vivo during lifelong gammaherpesvirus latency. In work described here, we make use of a heterologously expressed enzymatic marker to define the types of B cells that express the LANA homolog (mLANA) during chronic MHV68 infection. Our data demonstrate that mLANA is expressed in a stable fraction of B cells throughout chronic infection, with a prominent peak at 28 days. The expression of mLANA was detected in naïve follicular B cells, germinal-center B cells, and memory B cells throughout infection, with germinal-center and memory B cells accounting for more than 80% of the mLANA-expressing cells during the maintenance phase of latency. These findings suggest that the maintenance phase of latency is an active process that involves the ongoing proliferation or reseeding of latently infected memory B cells.
Gammaherpesviruses such as Epstein-Barr virus (EBV), Kaposi's sarcoma-associated virus (KSHV, HHV-8), and murine gammaherpesvirus 68 (MHV68, γHV68) are associated with lymphoproliferative diseases and a variety of malignancies of both epithelial and lymphoid origin. The strict species specificity exhibited by gammaherpesviruses has limited research on the human viruses primarily to in vitro studies. MHV68 is genetically colinear to the human gammaherpesviruses and exhibits many similar pathogenic features (54, 62). MHV68 is a natural pathogen of rodents (6, 9, 44), making the inoculation of mice with MHV68 a useful small-animal model to study gammaherpesvirus infection in vivo.
A hallmark of gammaherpesvirus infections is the establishment of lifelong latency in B cells. During latency, the viral genome is maintained as an extrachromosomal episome, viral gene expression is highly restricted, and no new progeny virions are generated. The stable maintenance of the episome in dividing cells requires regulated plasmid DNA replication and the efficient partitioning of replicated genomes to daughter cells. These processes are mediated by critical episome maintenance protein Epstein-Barr nuclear antigen 1 (EBNA-1) for EBV and latency-associated nuclear antigen (LANA) for KSHV. EBNA-1 and LANA facilitate the replication of the episomal viral genome in dividing cells by (i) binding to regulatory cis elements in the plasmid origin of replication and recruiting host DNA replication factors, and (ii) tethering episomes to host chromatin during mitosis (5, 13, 27, 43, 67). Consistent with these essential functions of the human gammaherpesvirus proteins, the mutation of the MHV68 LANA homolog (mLANA) severely attenuates the establishment of MHV68 latency in vivo (20, 40). In addition to their essential plasmid maintenance activities, these proteins modulate numerous viral and cellular pathways (reviewed in references 37 and 61). For example, KSHV LANA binds to viral DNA and blocks the expression of Rta, the key transcriptional activator of reactivation from latency (32), induces its own expression (45), and modulates the transcription of multiple cellular genes (47). Both KSHV LANA (21) and MHV68 mLANA (19) dysregulate the activity of the tumor suppressor p53, perhaps in part as a means to promote virus growth and prevent cell death (19). Additionally, recent reports have demonstrated the mLANA-mediated proteosomal degradation of the NF-κB family member p65/RelA (47) and the activation of G1/S cyclin promoters via interaction with cellular bromodomain-containing BET proteins (42).
Thus, the gammaherpesvirus episomal maintenance proteins are multifunctional regulators of virus and host pathways that are required for the efficient establishment of lifelong latency in vivo. Importantly, these are the only viral proteins that are absolutely required for plasmid retention in dividing cells (5, 13, 67) and, as such, are believed to be expressed in the majority of infected cells during the establishment phase of latency. For example, in vivo EBNA-1 is expressed in all known transcriptionally active forms of EBV latency in B cells and in all EBV-associated tumors (56). Similarly, KSHV LANA is expressed in all KSHV-associated malignancies, including primary effusion lymphoma B cells, Kaposi's sarcoma-derived endothelial cells, and B cells from multicentric Castleman's disease (MCD) patients (15, 23, 28, 33, 51). During MHV68 infection, transcripts corresponding to orf73 (encoding mLANA) are detectable by quantitative reverse transcription-PCR (qRT-PCR) in sorted splenic germinal-center (GC) and marginal-zone (MZ) B cells at 14 days postinoculation (38), a time point that corresponds with the peak expansion of latently infected cells (10, 38, 64). Consistent with this finding, by limiting-dilution nested RT-PCR, 5 to 10% of viral genome-positive splenocytes express spliced orf73 transcripts at 16 days postinoculation (3).
Taken together, these reports suggest that the expression of episomal maintenance proteins in dividing cells is a critical aspect of the pathogenesis of chronic gammaherpesvirus infection. The MHV68 system provides a means to systematically dissect mechanisms used by a gammaherpesvirus to establish and maintain long-term latency in vivo. Based on the critical role of the episomal maintenance protein in maintaining stable latency, it stands to reason that kinetic studies of the specific cell types expressing episomal maintenance proteins in vivo will provide fundamental insight into gammaherpesvirus pathogenesis. To identify specific cell types expressing mLANA in vivo during the establishment and maintenance of latency, we have generated a recombinant MHV68 expressing a modified β-lactamase enzymatic marker as a C-terminal fusion to mLANA. The use of this virus in conjunction with a β-lactam-based fluorogenic substrate (69) provides a sensitive means to detect and isolate mLANA-expressing cells from in vivo samples using flow cytometry. In work presented here, we demonstrate that mLANA is expressed in naïve follicular B cells, germinal-center B cells, and memory B cells during early latency, but that expression in naïve follicular B cells wanes over time. The observation that mLANA continues to be expressed in a substantial fraction of memory B cells throughout chronic infection supports the concept that the maintenance of long-term latency is a dynamic process in vivo.
MATERIALS AND METHODS
Mice and cells.
C57BL/6J (B6) mice obtained from Jackson Laboratory (Bar Harbor, ME) were housed and bred in a pathogen-free facility at Louisiana State University Health Sciences Center-Shreveport in accordance with all federal and university guidelines. Six- to 12-week-old mice were used in all experiments. NIH 3T12 murine fibroblasts were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum, 100 U/ml of penicillin, 100 mg/ml streptomycin, and 2 mM l-glutamine.
Generation of MHV68.ORF73βla.
The MHV68.ORF73βla recombinant virus was generated from the parental bacterial artificial chromosome (BAC) (2) using allelic exchange (53). The entire 945-bp orf73 coding region upstream of the stop codon (genome coordinates 103927 to 104871; GenBank accession number U97553) was amplified from wild-type MHV68 BAC DNA (2) by PCR using primers 5′-TGTCTGAGACCCTTGTCCCTGTTGTCC-3′ and 5′-CACCATAATGCCCACATCCCCACCGAC-3′. The resultant amplicon was ligated in-frame into a plasmid that encodes the modified β-lactamase (pcDNA6.2/cGeneBLAzer-GW/D-TOPO; Invitrogen, Carlsbad, CA) to generate pBLAz.ORF73. The adjacent 1,086-bp region including M11 (genome coordinates 102849 to 103934) was amplified from wild-type MHV68 BAC DNA by PCR using primers 5′-GATGGGCATGTAGGCCCTGACC-3′ and 5′-CTCAGACATAAATCACATTCCC-3′ and was ligated into pCR-Blunt (Invitrogen) to generate pCR-Blunt.M11. Because the orf73 and M11 transcripts share a 7-bp overlap at their 3′ termini, the overlap sequence was duplicated during the construction of the orf73 fusion to ensure the integrity of M11. pBLAz.ORF73 then was digested with BglII and NdeI and the 2,561-bp ORF73βla fragment was cloned into the AseI sites of pCR-Blunt.M11, generating pCR-Blunt.M11.ORF73βla. Subsequently, this vector was cut with NotI, yielding a 3,264-bp M11.ORF73βla fragment that was ligated into the NotI site of the pGS284 allelic exchange vector (53).
Allelic exchange was performed using pGS284.M11.ORF73βla+ S17λpir and MHV68.BAC+ RecA+ GS500 Escherichia coli, as previously described (53). Following positive and negative selection, diagnostic restriction digests were performed on multiple clones to determine the integrity of the viral genome (Fig. 1 B), and Southern blots using a β-lactamase probe were performed to determine the correct insertion of the β-lactamase reporter (data not shown). In addition, the region of interest was directly sequenced, and the correct sequence was confirmed (data not shown). BAC DNA from one verified MHV68.ORF73βla clone was transfected into NIH 3T12 murine fibroblasts to generate high-titer viral stocks, as has been described previously (1). The resulting MHV68.ORF73βla.BAC+ virus was serially passaged in NIH 3T12 cells stably expressing Cre recombinase, resulting in the removal of the loxP-flanked BAC sequence and the generation of the MHV68.ORF73βla.BAC− virus, termed MHV68.ORF73βla.
FIG. 1.

Generation of the MHV68.ORF73βla marker virus. (A) Schematic of MHV68.ORF73βla construction. The marker virus was constructed using allelic exchange with a bacterial artificial chromosome (BAC) containing the complete wild-type MHV68 genome. orf73 was amplified by PCR with the omission of the stop codon and was fused in-frame upstream of the β-lactamase gene. (B) DNA digestion analysis. DNA was digested with the restriction enzyme AseI, KpnI, or NcoI and subjected to agarose gel electrophoresis with ethidium bromide visualization to confirm the insertion of the marker molecule and assess the integrity of the viral genome. Shifts in DNA fragment size due to the insertion of the β-lactamase marker are indicated.
Viruses and infections.
Wild-type MHV68 clone WUMS originally was obtained from the American Type Culture Collection (ATCC VR1465). BAC-derived MHV68 (MHV68.BAC) has been described previously (1). BAC-deleted stocks of MHV68 and MHV68.ORF73βla were generated from multiple passages on Cre-expressing NIH 3T12 cells, generating MHV68.BAC− and MHV68.ORF73βla.BAC− viruses. For intraperitoneal (i.p.) infections, mice were infected with 104 PFU of virus in 500 μl serum-free DMEM. For intranasal (i.n.) infections, mice were anesthetized with isofluorane and inoculated with 104 PFU of virus in 30 μl of serum-free DMEM. At indicated time points, spleens from three to five mice per experimental group were harvested and pooled, unless otherwise stated.
Plaque assays.
Plaque assays were performed as previously described (59, 60). Briefly, harvested spleens were placed in sterile 2-ml screw-cap tubes containing 1 ml of DMEM and 100 μl of 1-mm-diameter zirconia-silica beads (BioSpec Products, Inc., Bartlesville, OK) and stored at −80°C. Samples were thawed on ice and tissues homogenized using a Mini BeadBeater (Biospec). Samples were serially diluted 10-fold prior to the infection of NIH 3T12 monolayers. Infected monolayers were overlaid with methylcellulose (Sigma), and plaques were visualized by neutral red staining at day 7. The limit of detection was 50 PFU.
Limiting-dilution nested PCR analyses.
Single-copy-sensitive nested PCR was used on serial dilutions of harvested cells to determine the frequency of cells harboring viral genome, as previously described (35, 58). In brief, cell samples were washed, resuspended in an isotonic solution, counted, and serially diluted 3-fold in a background of uninfected RAW 264.7 murine macrophages such that a total of 104 cells were present in each reaction mixture. Cells were plated in a 96-well PCR plate at 12 wells per dilution. Positive control reaction mixtures contained 10, 1, or 0.1 copies of an MHV68 ORF72 plasmid in a background of RAW 264.7 cells, and negative control reaction mixtures of RAW 264.7 cells only were included on all plates. Cells were lysed using overnight digestion with proteinase K at 56°C, followed by enzyme deactivation at 95°C. Nested PCR was performed using primers specific for MHV68 ORF72, and 195-bp amplicons were visualized on a 3% agarose gel with ethidium bromide staining.
Flow cytometry.
Following spleen harvest, single-cell suspensions were prepared and red blood cells were lysed in red blood cell lysis buffer (144 mM NH4Cl and 17 mM Tris, pH 7.2) for 7 min at 37°C. For cell surface marker staining, harvested splenocytes were washed twice in cold complete DMEM following red blood cell lysis. Cells were adjusted to 2 × 107 cells/ml in blocking buffer (phosphate-buffered saline, pH 7.2, 5% bovine serum albumin, and 10% normal rat serum). All samples were blocked with purified anti-mouse CD16/CD32 (Fc block; clone 2.4G2; BD Biosciences, San Jose, CA) followed by antibody staining. Cells then were stained for 30 min on ice in the dark with rat anti-mouse allophycocyanin (APC)/Cy7-conjugated CD19 (clone 1D3; BD Biosciences), rat anti-mouse APC-conjugated CD38 (clone 90; eBioscience, San Diego, CA), and rat anti-mouse biotin-conjugated IgD (clone 11-26c; eBioscience) in blocking buffer. Alexa fluor 700-conjugated streptavidin (Invitrogen, Carlsbad, CA) was used as a secondary reagent to detect bound anti-IgD antibody. For the flow-cytometric analysis of ORF73βla expression, cells were washed twice and adjusted to 2 × 106 cells/ml in PBS. Freshly prepared 6× coumarin cephalosporin fluorescein acetoxymethyl ester (CCF2/AM) β-lactamase substrate (Invitrogen) was added for 1 h at room temperature. Cells then were washed twice in PBS and resuspended in PBS containing 10% fetal calf serum. Unstained and isotype-staining controls were included in each experiment. Flow-cytometric analysis was performed on an LSR II flow cytometer, and fluorescent-activated cell sorting (FACS) was performed on a FACS Aria flow cytometer (BD Biosciences). Data were analyzed using FACSDiva software (BD Biosciences).
Statistical analyses.
All data were analyzed using GraphPad Prism software (GraphPad Software, San Diego, CA). The frequencies of viral genome-positive cells were determined from the nonlinear regression analysis of sigmoidal dose-response best-fit curve data. Based on Poisson distributions, the frequency at which at least one event in a given population is present occurs at the point where the regression analysis line intersects 63.2%.
RESULTS
Generation of the MHV68.ORF73βla marker virus.
To generate a recombinant MHV68 that would facilitate the detection of cells expressing mLANA in vivo, we fused a gene encoding a modified β-lactamase molecule to the C terminus of the orf73 coding sequence (Fig. 1A). Although such a fusion could interfere with the functions of mLANA during viral infection, previous work has demonstrated that the fusion of a heterologous protein epitope to the C terminus had no significant effect on latency establishment (7, 52). Although the MHV68 mLANA protein has not been carefully mapped, the orf73 coding region is conserved among the gamma-2-herpesviruses KSHV, herpesvirus saimiri (HVS), and MHV68 (25), and the products of these genes share C-terminal homology with EBNA-1 (20, 25). The episomal tethering function of KSHV LANA is mediated by portions of both the N and C termini, which bind nucleosomes and viral terminal repeats, respectively (24, 36, 43). The central repeat regions of LANA and EBNA-1 are believed to be responsible for cis-acting cytotoxic T-cell evasion by inhibiting translation and proteosomal degradation (7, 34, 48, 68), and fusion to the mLANA C terminus appears to prevent the proteosomal degradation of heterologous fusion proteins (7, 52).
The bacterial enzyme β-lactamase was chosen as a reporter molecule due to its ability to cleave the β-lactam ring of a passively diffusing, membrane-permeant fluorogenic substrate (69), allowing a versatile and sensitive means to detect mLANA expression in living cells. The advantage of this system over other potential markers is that the catalytic activity of β-lactamase allows (i) the detection of cells in which as few as 10 copies of the marker are present, and (ii) detection at frequencies as low as 1 β-lactamase-positive cell in a background of 106 negative cells (29). Thus, the use of this marker as an in-frame fusion should allow the detection of mLANA-expressing cells even when mLANA is expressed at a low level in rarely infected cells. To our knowledge, this is the first use of this β-lactamase marker/fluorogeneic substrate reporter system in the context of an intact virus.
The MHV68.ORF73βla recombinant virus was generated from the parental MHV68 BAC using allelic exchange, as described in Materials and Methods. Following selection, diagnostic restriction digests were performed on multiple clones to determine the integrity of the viral genome and correct the insertion of the β-lactamase reporter (Fig. 1B). In addition, the region of interest was directly sequenced, and the correct sequence was confirmed (data not shown). BAC DNA from one verified MHV68.ORF73βla clone was used to generate high-titer viral stocks on NIH 3T12 murine fibroblasts, and the resulting MHV68.ORF73βla.BAC+ virus was serially passaged in NIH 3T12 cells stably expressing Cre recombinase, resulting in the removal of the loxP-flanked BAC sequence and the generation of the MHV68.ORF73βla.ΔBAC virus, hereafter termed MHV68.ORF73βla.
MHV68.ORF73βla undergoes normal lytic and latent infection.
Because mLANA plays multiple roles in MHV68 infection, we questioned whether the fusion of a marker to the mLANA C terminus would compromise lytic replication or latency. For example, previous studies have demonstrated that the mutation of MHV68 mLANA results in delayed lytic replication and a severe defect in latency establishment (1, 40). To determine whether β-lactamase fusion to mLANA interferes with lytic replication, we first performed single-step (Fig. 2 A) and multistep (Fig. 2B) growth curve analyses in vitro, as previously described (59, 60). No significant difference between the MHV68 and MHV68.ORF73βla viruses was detected at any time point in single-step growth curves, indicating that the insertion of β-lactamase does not interfere with the ability of the recombinant MHV68 to undergo lytic replication. In multistep growth curves, although the peak viral titers were nearly identical for MHV68 and MHV68.ORF73βla, MHV68.ORF73βla titers were decreased at 24 and 48 h postinoculation. This finding is consistent with the reported lytic replication defect of a mutant MHV68 lacking the expression of mLANA (19, 40), suggesting that the C-terminal fusion of β-lactamase alters lytic replication functions of mLANA. The discrepancy between single-step (no defect) and multistep (delayed replication) growth of MHV68.ORF73βla suggests that the C-terminal fusion of β-lactamase to mLANA causes a modest delay in virus spread. To determine whether acute lytic replication is altered in vivo, C57Bl/6J mice were infected with 104 PFU MHV68 or MHV68.ORF73βla, spleens were harvested at 7 days postinoculation, and viral titers were determined by plaque assay. There was no significant difference in the ability of the MHV68.ORF73βla virus to replicate in vivo compared to that of MHV68 (Fig. 2C), indicating that any defect of the recombinant virus in lytic replication is minor.
FIG. 2.

Lytic MHV68.ORF73βla virus replication in vitro and in vivo. (A) Single-step growth curve. NIH 3T12 murine fibroblasts were infected in vitro with MHV68 or MHV68.ORF73βla at an MOI of 5. Cells and supernatants were harvested at the indicated time points, and viral titers were determined by plaque assay (n = 2). (B) Multistep growth curve. NIH 3T12 murine fibroblasts were infected in vitro with MHV68 or MHV68.ORF73βla at an MOI of 0.05. Cells and supernatants were harvested at the indicated time points, and viral titers were determined by plaque assay (n = 2). (C) In vivo acute infection. C57BL/6J mice were infected with 104 PFU of MHV68 or the MHV68.ORF73βla virus. Spleens were harvested at 7 days postinoculation, and viral titers were determined by plaque assay (n = 8).
Given the essential role of mLANA in the establishment and maintenance of latency, we also determined whether the C-terminal fusion of β-lactamase to mLANA interfered with the critical episomal viral genome maintenance function of the protein. Mice were infected i.n. with wild-type MHV68 or MHV68.ORF73βla, and spleens were harvested at 16, 28, 42, and 90 days postinoculation. The frequency of splenocytes harboring viral genome was determined using limiting-dilution nested PCR analysis using primers specific for MHV68 orf72, as previously described (58). Poisson distribution analysis was used to determine the frequency of cells that harbor viral genome at each time point. As expected, the frequency was highest at 16 days (Fig. 3). Importantly, similar frequencies of splenocytes were positive for viral genome during wild-type MHV68 infection (1 in 140) and MHV68.ORF73βla infection (1 in 130). Frequencies also were similar after i.p. inoculation (1 in 300 genome-positive splenocytes for MHV68 and 1 in 310 genome-positive splenocytes for MHV68.ORF73βla; data not shown). The frequencies of genome-positive cells decreased significantly by 28 days, as has been observed previously (64), but did not differ significantly between the two viruses at any of the time points: 28 days (1 in 1,300 for MHV68 versus 1 in 1,100 for MHV68.ORF73βla), 42 days (1 in 2,400 for MHV68 versus 1 in 2,800 for MHV68.ORF73βla), or 90 days (1 in 4,400 for MHV68 versus 1 in 5,600 for MHV68.ORF73βla). These data indicate that MHV68.ORF73βla establishes and maintains latent infection at levels comparable to those of wild-type MHV68. Taken together, these results demonstrate that the C-terminal fusion of β-lactamase to mLANA does not significantly alter the normal progression of MHV68 acute replication or latency.
FIG. 3.
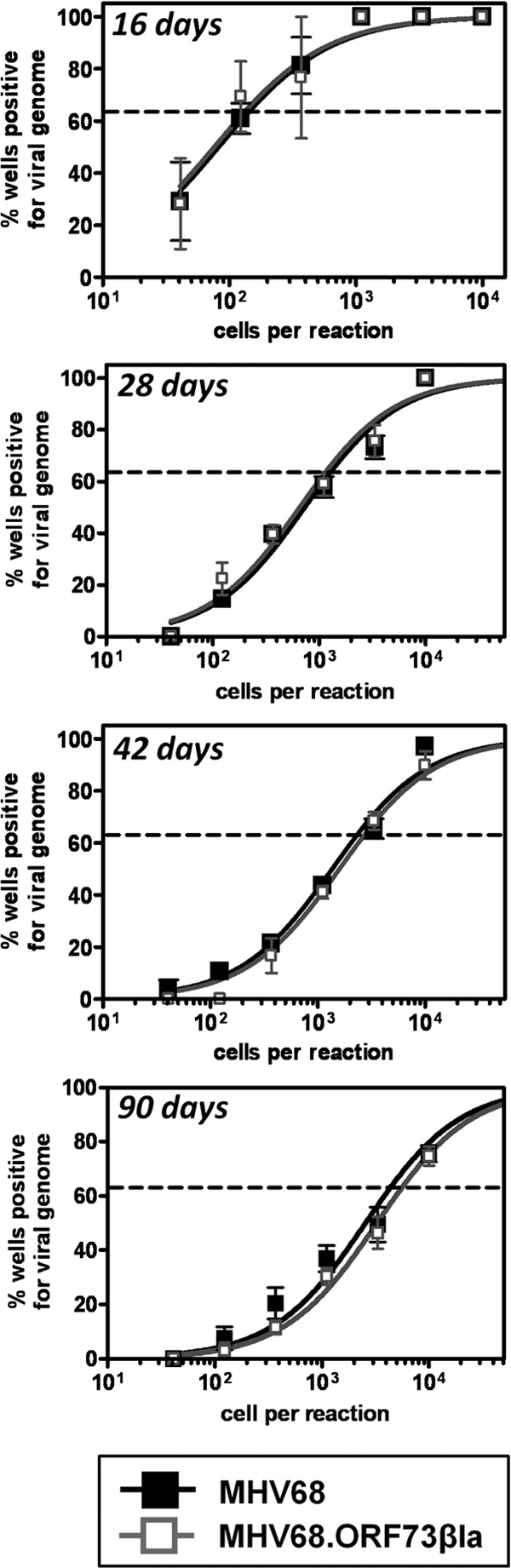
Limiting-dilution nested PCR analysis of splenocytes from MHV68.ORF73βla-infected mice during chronic infection. Single-copy sensitive limiting-dilution nested PCR analysis was performed on splenocytes harvested at 16, 28, 42, and 90 days postinoculation. Harvested cells were serially diluted 3-fold in a background of uninfected RAW264.7 murine macrophages, plated at 12 reactions per cell dilution, and then subjected to lysis followed by nested PCR specific for viral genome. The frequency of cells harboring viral DNA was determined at 63.2% (indicated by the horizontal line) using Poisson distribution (n = 3).
mLANA is expressed in a stable fraction of latently infected B cells during chronic infection.
To assess whether mLANA/β-lactamase expression could be detected ex vivo from latently infected splenocytes, we initially harvested spleens 16 days postinoculation with MHV68 or MHV68.ORF73βla and loaded cells with the permeable, fluorogenic β-lactamase substrate coumarin cephalosporin fluorescein acetoxymethyl ester (CCF2/AM; Invitrogen). CCF2/AM is composed of a 7-hydroxycoumarin (coumarin) molecule linked to a fluorescein molecule by a β-lactam ring; in the presence of β-lactamase, the β-lactam ring joining the fluorescence resonance energy transfer (FRET) donor coumarin and the FRET acceptor fluorescein is hydrolyzed, disrupting FRET and resulting in fluorescence from the coumarin moiety under violet laser excitation. This detectable shift in fluorescence emission makes it possible to distinguish β-lactamase-expressing cells from non-β-lactamase-expressing cells by fluorescence microscopy or flow cytometry. After CCF2/AM loading, samples were analyzed flow cytometrically to quantify the frequency of cells that displayed β-lactamase activity (Fig. 4 A). At 16 days after i.n. inoculation, 0.10% of splenocytes from MHV68.ORF73βla-infected mice displayed β-lactamase activity, corresponding to a frequency of approximately 1 in 1,120. Similarly, following i.p. inoculation with MHV68.ORF73βla, 0.12% of splenocytes were positive for β-lactamase activity, corresponding to an approximate frequency of 1 in 1,510. In contrast to these results, less that 0.0002% of splenocytes from wild-type MHV68-infected mice were detected in the coumarin gate, demonstrating that the spontaneous cleavage of CCF2/AM is negligible. These results demonstrate that the detection of coumarin emission directly correlates with the expression of the mLANA-β-lactamase fusion.
FIG. 4.

Detection of splenocytes expressing mLANA. (A) Detection of mLANA expression using flow cytometry. Spleens were harvested at 16 days after i.n. (top panel) or i.p. (bottom panel) inoculation. Single-cell suspensions were loaded with the CCF2/AM β-lactamase substrate and subjected to flow-cytometric analysis. mLANA/βla-positive cells (blue) are indicated by the boxed gate. (B) Limiting-dilution nested PCR for viral genome. Sorted mLANA+ and mLANA− cell populations were subjected to PCR analyses as described for Fig. 3. The frequency of cells harboring viral DNA was determined at 63.2% (indicated by the horizontal line) using Poisson distribution (n = 6). (C) Limiting-dilution analyses for ex vivo reactivation and preformed infectious virus. For reactivation assays, sorted mLANA+ and mLANA− cell populations were subjected to serial dilution and then plated on macrophage monolayers to assess cytopathic effect following spontaneous reactivation from latency. The frequency of reactivating cells was determined at 63.2% (indicated by the horizontal line) using Poisson distribution (n = 3). (D) Kinetics of mLANA expression in splenocytes. Splenocytes were harvested from MHV68.ORF73βla-infected mice at the indicated time points. Single-cell suspensions were loaded with the CCF2/AM substrate and subjected to flow-cytometric analysis (n = 3 to 5). The x axis is the reciprocal frequency of mLANA-positive cells. The y axis is the number of days postinoculation. (E) Ratio of mLANA-positive to viral genome-positive splenocytes over time. The ratio at each time point was calculated based on the frequency of mLANA-positive cells determined for Fig. 4D and the frequency of genome-positive cells determined for Fig. 3.
To determine whether cells expressing mLANA were latently infected, we used limiting-dilution assays of sorted cells to quantify the frequencies of mLANA+ cells that harbored viral genome and spontaneously reactivated ex vivo, as previously described (57, 64). Sixteen days postinfection, splenocytes were harvested, loaded with CCF2/AM, and flow sorted for mLANA+ and mLANA− cells. Subsequently, sorted populations were subjected to limiting-dilution PCR analysis to detect the presence of viral genome (Fig. 4B). The frequency of mLANA+ cells that harbored viral genome was nearly 1 in 1, demonstrating that mLANA expression correlates with an infected cell population. The frequency of genome-positive cells in the mLANA− population was substantially lower (1 in 24,000). The presence of viral genome-positive cells in this population likely indicates the presence of MHV68-infected cells that either do not express mLANA or express mLANA below the level of detection. Bulk populations of latently infected cells typically exhibit the spontaneous reactivation of MHV68 ex vivo but are free of preformed or infectious virus particles (57, 64). To verify that mLANA+ cells were latently infected, we examined the sorted populations of mLANA+ or mLANA− cells for reactivation and preformed virus (Fig. 4C). No preformed infectious virus was detected in mLANA+ cells, while approximately 1 in 290 mLANA+ cells spontaneously reactivated ex vivo. A similarly low proportion of mLANA− cells reactivated ex vivo. Taken together, these data indicate that mLANA+ cells are latently infected in vivo.
To determine the frequency of splenocytes that express mLANA throughout chronic infection, C57BL/6J mice were infected i.n. with MHV68.ORF73βla, and the frequency of splenocytes displaying β-lactamase activity was assessed from 7 to 90 days postinoculation (Fig. 4D). The frequency of mLANA-positive cells was highest at 16 days (1 in 1,120), a time point that is coincident with the peak of MHV68 latent cell expansion (Fig. 3 and references 10, 38, and 64). Consistent with the drop in genome-positive cells over time that we observed, the frequency of cells expressing mLANA decreased to approximately 1 in 18,600 by 42 days and 1 in 30,800 by 90 days. Overall, these data indicate that a significant proportion of genome-positive cells express mLANA even during long-term infection, including 18.1% as late as 90 days postinoculation (Fig. 4E). Interestingly, the highest percentage of mLANA-expressing cells (41.6%) occurred at 28 days, indicating that this time point represents a key stage during latency establishment.
mLANA is expressed in germinal-center and memory B cells during chronic infection.
MHV68 establishes latent infection in vivo in multiple B-cell subsets, including naïve follicular, germinal-center, and memory B cells (16, 18, 38, 50, 66). To determine in which of these cell populations mLANA is expressed during latency establishment and maintenance, we used flow cytometry to examine the surface phenotype of β-lactamase-expressing cells. Splenocytes were harvested at 16 days postinoculation, and cell samples were stained with antibodies to CD19, IgD, and CD38. While CD19 is a pan-B-cell marker, naïve B cells are the only B subset that express surface IgD (aside from a minor antigen-experienced memory population). In mice, CD38 is expressed at high levels on naïve follicular B cells and memory B cells but is downregulated on germinal-center B cells (46). Thus, we defined naïve B cells as CD19+ IgD+ CD38+, germinal-center B cells as CD19+ IgD− CD38low, and memory B cells as CD19+ IgD− CD38hi. Following antibody staining, cells were loaded with CCF2/AM dye, and the distribution of naïve follicular B cells, germinal-center B cells, and memory B cells expressing mLANA was assessed using flow cytometry (Fig. 5). At 16 days, all three B-cell subsets expressed mLANA, a finding that is consistent with previous observations that each of these cell types harbors genome during latency establishment (18, 38, 66).
FIG. 5.

Detection of mLANA expression in mature B-cell subsets. Spleens from MHV68.ORF73βla-infected mice were harvested 16 days after intranasal (A) or intraperitoneal (B) inoculation. Single-cell suspensions were stained with antibodies to cell surface markers and then loaded with CCF2/AM β-lactamase substrate and subjected to flow-cytometric analysis. Naïve follicular B cells (CD19+ IgD+ CD38+), germinal-center B cells (CD19+ IgD− CD38low), and memory B cells (CD19+ IgD− CD38high) are depicted in the bottom panels, with mLANA/βla-positive cells (blue) indicated by the boxed gate. Data are representative of five individual experiments.
To gain further insight into the dynamics of mLANA expression in these populations over time, we performed similar experiments throughout a 90-day course of infection (Fig. 6 and Table 1 ). Seven days postinoculation, a time point at which acute replication is apparent (Fig. 1C), the frequencies of mLANA-positive cells in all three subsets were less than 1 in 30,000. Following the clearance of acute replication in the spleen, the number of mLANA-expressing splenocytes increased significantly. At 16 days, a time point that coincides with early latency establishment and correlates with the highest frequency of genome-positive cells during latency (10, 18, 63), the frequencies of germinal-center (1 in 330) and memory (1 in 210) B cells expressing mLANA were significantly higher than that of naïve B cells (1 in 1,190). Although mLANA expression in all three populations diminished by 42 days, expression in the germinal-center and memory B-cell subsets maintained 10-fold higher frequencies than naïve B cells at this time point (1 in 2,500 for germinal-center and 1 in 5,000 for memory versus <1 in 55,000 for naïve cells), and these levels did not decrease through 90 days (1 in 4,300 for germinal-center and 1 in 3,800 for memory cells). In contrast, mLANA expression in naïve B cells waned over time, with less than 1 in 200,000 cells displaying β-lactamase activity 90 days postinoculation. These results therefore demonstrate that mLANA is expressed in all three subsets of mature B cells during early MHV68 latency, and that the expression of mLANA is maintained in a substantial fraction of germinal-center and memory B cells during chronic MHV68 infection.
FIG. 6.

mLANA expression in mature B-cell subsets over time. Spleens from MHV68.ORF73βla-infected mice were harvested at multiple time points, stained with antibodies to cell surface markers, and then loaded with CCF2/AM β-lactamase substrate. Cells were subjected to flow-cytometric analysis, and the reciprocal frequencies of mLANA+ cells within the naïve follicular, germinal-center, and memory B-cell compartments were calculated. Data points represent the means from three to five individual experiments (detailed in Table 1).
TABLE 1.
Results of detection of mLANA+ cells
| Cell type and inoculation route (n) | % of population in spleena | Total no. of cells per spleenb | % mLANA+ in populationc | Approx frequency of mLANA+ in populationd | Approx no. of mLANA+ cells in spleene |
|---|---|---|---|---|---|
| 7 dpi | |||||
| i.n. (3) | |||||
| Total | 100 | 52.1 × 106 | 0.006 | 18,300 | 3,100 |
| CD19+ | 55.6 | 29.2 × 106 | 0.004 | 33,700 | 1,000 |
| Naïve | 30.9 | 16.3 × 106 | 0.003 | 39,000 | 500 |
| GC | 9.0 | 4.7 × 106 | 0.004 | 35,200 | 200 |
| Memory | 13.2 | 6.9 × 106 | 0.006 | 28,400 | 300 |
| 16 dpi | |||||
| i.n. (5) | |||||
| Total | 100 | 102.5 × 106 | 0.10 | 1110 | 110,500 |
| CD19+ | 51.3 | 52.8 × 106 | 0.18 | 680 | 98,300 |
| Naïve | 34.7 | 35.6 × 106 | 0.04 | 2640 | 14,600 |
| GC | 5.4 | 5.7 × 106 | 0.25 | 530 | 13,200 |
| Memory | 9.7 | 9.9 × 106 | 0.63 | 210 | 65,300 |
| i.p. (3) | |||||
| Total | 100 | 106.9 × 106 | 0.11 | 1500 | 118,600 |
| CD19+ | 44.1 | 47.2 × 106 | 0.26 | 770 | 111,800 |
| Naïve | 24.2 | 25.8 × 106 | 0.20 | 950 | 48,200 |
| GC | 4.8 | 5.2 × 106 | 0.63 | 360 | 20,600 |
| Memory | 13.9 | 14.9 × 106 | 0.14 | 1830 | 21,400 |
| 28 dpi | |||||
| i.n. (3) | |||||
| Total | 100 | 82.3 × 106 | 0.04 | 3180 | 28,600 |
| CD19+ | 47.4 | 39 × 106 | 0.07 | 1750 | 25,100 |
| Naïve | 36.4 | 30 × 106 | 0.01 | 9710 | 3400 |
| GC | 4.2 | 3.5 × 106 | 0.39 | 310 | 13,300 |
| Memory | 6.8 | 5.6 × 106 | 0.13 | 990 | 7100 |
| 42 dpi | |||||
| i.n. (4) | |||||
| Total | 100 | 88.1 × 106 | 0.004 | 25,700 | 3600 |
| CD19+ | 49.5 | 43.6 × 106 | 0.006 | 20,600 | 2400 |
| Naïve | 37.55 | 33.2 × 106 | 0.001 | 116,500 | 400 |
| GC | 3.5 | 3.0 × 106 | 0.040 | 2800 | 1200 |
| Memory | 7.5 | 6.6 × 106 | 0.014 | 9700 | 800 |
| 90 dpi | |||||
| i.n. (3) | |||||
| Total | 100 | 56.2 × 106 | 0.003 | 40,200 | 1500 |
| CD19+ | 45.7 | 25.6 × 106 | 0.004 | 26,500 | 1000 |
| Naïve | 34.9 | 19.6 × 106 | 0.001 | 116,600 | 200 |
| GC | 3.1 | 1.7 × 105 | 0.038 | 4200 | 500 |
| Memory | 7.0 | 3.9 × 106 | 0.008 | 14100 | 300 |
Percentage of total splenocytes that are CD19+, naïve follicular (CD19+ IgD+ CD38+), germinal-center (CD19+ IgD− CD38low), and memory (CD19+ IgD− CD38high) B cells.
Estimated total number of CD19+, naïve follicular, germinal-center, and memory B cells per spleen based on the total number of harvested splenocytes and the percentage of analyzed cells in each population.
Percentage of mLANA+ cells in each population based on the number of detected mLANA+ cells in a population divided by the total number of cells detected in that population multiplied by 100.
Approximate frequency of mLANA+ cells in each population based on the total number of cells detected in a population divided by the number of mLANA+ cells in that population.
Approximate frequency of mLANA+ cells in each population based on the total number of cells in a population multiplied by the percent of mLANA+ cells in that population.
DISCUSSION
In work presented here, we describe the use of a novel enzymatic marker to define the types of B cells that express the critical latency protein mLANA during chronic MHV68 infection. The use of this recombinant marker virus presents a unique opportunity to address fundamental questions about the virus-host relationship, in particular about the role played by mLANA in this relationship, during latent infection in vivo. A primary function of MHV68 mLANA and its homologs KSHV LANA and EBV EBNA-1 is the maintenance of the viral episome during mitosis. It has been shown that EBNA-1 expression is induced during cell cycle progression (14), and that EBNA-1 is expressed in latently infected memory B cells upon division (26). Although B-cell subsets that express EBNA-1 have been defined from human samples during chronic infection (4), the kinetics of LANA/EBNA-1 expression during different stages of latency (i.e., establishment versus maintenance) and the subsets of latently infected B cells that express this protein as a function of time have not been determined previously. This information could provide critical insight into the dynamic nature of gammaherpesvirus latency.
Our data demonstrate that mLANA is expressed in a significant proportion of infected B cells throughout latent infection. Interestingly, the highest frequency of infected cells expressing mLANA (42%) occurred at 28 days (Fig. 4C), a time at which germinal-center and effector T-cell responses peak despite the significant loss of cells harboring viral genome (30). In contrast, only 14.6% of genome-positive cells expressed mLANA at 16 days, a time point that is coincident with the peak of MHV68 latent cell expansion (10, 38, 64), and therefore might have been predicted to be the most active with regard to mLANA expression. Thus, these data point to 28 days as a previously unrecognized critical stage for the establishment of stable long-term latency. Although the absolute number of cells expressing mLANA diminished significantly during the first 42 days of infection (Fig. 7 A), this decrease paralleled the loss of genome-positive cells that occurs during this time frame. As a result, the ratio of mLANA-positive to genome-positive cells was 15% or higher at every time point tested (Fig. 4C). These percentages are higher than the 5 to 10% previously reported following limiting-dilution nested RT-PCR analyses (3), perhaps due to the detection of multiple mLANA transcript variants by the β-lactamase fusion marker. There are multiple mLANA transcript variants that originate from promoters just upstream of the orf73 coding region (p3), in a unique region between orf75a and the terminal repeats (p2), and in the terminal repeats themselves (p1) (3, 11). Thus, mLANA transcript variants include (i) the orf73 coding region (E3) alone, (ii) E3 plus an exon downstream of p2 (E2), or (iii) E3, E2, and an additional exon downstream of p1 (E1). The previous experiments to detect mLANA by RT-PCR used primers designed to detect transcripts spliced from exon 2 and thus would not have detected alternatively spliced mLANA transcripts or mLANA transcripts originating at a promoter directly upstream of the orf73 coding region. In contrast, we fused the β-lactamase gene to the C terminus of the orf73-coding exon to incorporate all potential mLANA transcript variants, potentially explaining the higher frequency of mLANA-expressing latently infected cells detected in our studies. Although these numbers are higher than those previously recorded, it is likely that the β-lactamase fusion marker system is not sufficient to detect all mLANA-expressing cells. Previous pharmacological studies have demonstrated that as few as 10 β-lactamase copies per cell are detectable by flow cytometry using the CCF2/AM system (29); however, in all likelihood the expression of the mLANA/β-lactamase fusion marker is not sufficiently sensitive to detect every cell that expresses mLANA in the context of viral infection.
FIG. 7.

Analysis of the absolute numbers of splenocytes that express mLANA and the distribution of mLANA-expressing B cells over time. (A) The absolute numbers of cells that expressed mLANA versus cells that harbored viral genome. For each time point, the absolute number of splenocytes that expressed mLANA (black columns) was calculated from the percentage of mLANA-positive cells per spleen and the total number of splenocytes, as detailed in Table 1. Also shown are the absolute number of viral-genome-positive cells, as determined for Fig. 3, and the total number of splenocytes, as detailed in Table 1. (B) The distribution of CD19+ B cells that expressed mLANA. The percentage of CD19+ mLANA+ cells that displayed markers of naïve, germinal-center, or memory B cells was calculated based on the absolute number of mLANA+ cells in each population, as detailed in Table 1. Cells that were not accounted for in these calculations were termed “other.”
Our data also indicate that the distribution of B-cell subsets expressing mLANA is relatively stable during long-term infection. Taking into account the absolute number of each B-cell subset expressing mLANA at each time point (Table 1 and Fig. 7B), it is apparent that naïve B cells represent only a minority of mLANA-expressing cells in all phases of latency. In contrast, memory B cells surprisingly represent the vast majority of mLANA-expressing cells at 16 days, while germinal-center B cells represent the majority of mLANA-expressing cells during chronic infection. Together, germinal-center and memory B cells accounted for more than 80% of mLANA-expressing cells throughout the maintenance phase of latency. These observations are somewhat surprising considering the perception of gammaherpesvirus latency as a dormant state. Although it is well established that B cells are the primary reservoir, it remains unclear whether life-long latency represents a truly quiescent form of infection in which the virus silently resides in resting memory B cells, or whether long-term latency is a dynamic process involving the active turnover of latently infected cells. The prevailing paradigm for EBV is that following the primary infection of naïve B cells, the virus provides surrogate signals to drive these cells through germinal-center reactions and into the resting, long-lived memory B-cell compartment. Consistent with such a model, MHV68 genome is detected in a high frequency of naïve, germinal-center, and memory B cells during the early stages of latency, but it transitions to the major reservoirs of class-switched B cells during long-term latency (18, 38, 66). While these observations seem to favor a model in which latency is maintained in a dormant state, our findings instead imply that long-term MHV68 latency is maintained through an active process involving the constant generation of new latently infected cells through the division of infected germinal-center and/or memory B cells. In support of this argument, proliferation is critical for long-term latency in B cells (41), and MHV68 genome is detectable in both resting and proliferating germinal-center and memory B cells at 90 days postinfection (18, 41).
The finding that a significant proportion of mLANA-expressing latently infected cells are memory B cells is particularly surprising, because these cells are considered to be primarily resting and therefore presumably would not require the presence of mLANA for the maintenance of the viral genome. There are multiple explanations for our observations. First, it is possible that the memory B cells detected in this assay are indeed undergoing cell division, either as a consequence of virus-driven or antigen-specific activation, or were detected prior to their entering into a resting state after the differentiation of de novo-infected naïve or germinal-center B cells. Consistent with this idea, EBV EBNA-1 is expressed in circulating memory B cells only when they are undergoing proliferation (26). Interestingly, memory B cells undergo clonal expansion in response to stimulation by cognate antigen and prior to differentiation to plasma cells (8), and the infection of plasma B cells by MHV68 has been described very recently (12, 49), supporting the possibility that mLANA-expressing cells are transitioning to the plasma B-cell compartment. Second, it is formally possible that memory B-cell proliferation occurs in response to nonspecific inflammatory stimuli, which is a long-standing proposal; however, we feel that this explanation is unlikely in light of recent work indicating that memory B-cell proliferation and differentiation is a tightly regulated process that is induced by cognate antigen but not bystander inflammatory signals (8). Third, it is possible that mLANA performs a critical function separate from episome maintenance that is required in a subset of nondividing memory B cells. Finally, it is conceivable that some memory B cells were detected in this assay due to the slow decay of mLANA following germinal-center B-cell differentiation; however, this is not likely to be the sole explanation, as the half-life of EBNA-1 and LANA are in the range of 20 to 48 h (14, 22, 31, 34, 55).
It is important to note that these experiments did not examine mLANA expression in other cell populations or at other anatomical locations. For the purpose of this study, we chose to focus on the major mature B-cell subsets in the spleen that have been reported previously to be latently infected (18, 38, 66). However, other B-cell subsets have been demonstrated to harbor viral genome during chronic infection, including plasma cells (12, 49), marginal-zone B cells, and newly formed B cells (38). In addition, dendritic cells (16) and macrophages (65) are reservoirs for MHV68 latency. Notably, up to 5.2% (16 and 28 days) of CD19+ cells that expressed mLANA were not accounted for in our conservative gating for naïve, germinal-center, and memory B-cell populations (Fig. 7B), suggesting that other B-cell populations express mLANA at those time points. In addition, B cells, macrophages, and dendritic cells are latently infected at other sites, including the blood, lymph nodes, lung, and peritoneal cavity (16, 17, 39, 54, 65). Future experiments will be critical to determine whether mLANA is expressed in other major infected cell populations.
This work demonstrates the utility of a heterologously expressed enzymatic marker to identify gammaherpesvirus-infected cells that express a critical latency protein in vivo. Naïve, germinal-center, and memory B cells expressed mLANA during chronic infection, supporting previous reports that these cell types are reservoirs for long-term latency (18, 38, 66). Notably, a stable proportion of memory B cells expressed mLANA during chronic infection, suggesting that the maintenance of latency is an active process, perhaps involving the proliferation of latently infected memory B cells or the de novo infection and generation of new latently infected memory B cells. Future work should uncover other cell types that express mLANA during chronic infection and determine whether mLANA expression in individual cell types correlates with proliferative capacity.
Acknowledgments
This work was supported by NIH grant CA139984 and NIH COBRE Center for Molecular and Tumor Virology grant P20-RR018724. M.N. was supported by an American Heart Association Predoctoral Fellowship (0815151E).
We thank Robert Chervenak, Deborah Chervenak, and Shannon Mumphrey for expert assistance with flow cytometry experiments. We thank Doug White for providing the CRE-NIH 3T12 cell line and Yali Jia for assistance with viral DNA digests.
Footnotes
Published ahead of print on 19 May 2010.
REFERENCES
- 1.Adler, H., M. Messerle, and U. H. Koszinowski. 2001. Virus reconstituted from infectious bacterial artificial chromosome (BAC)-cloned murine gammaherpesvirus 68 acquires wild-type properties in vivo only after excision of BAC vector sequences. J. Virol. 75:5692-5696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adler, H., M. Messerle, M. Wagner, and U. H. Koszinowski. 2000. Cloning and mutagenesis of the murine gammaherpesvirus 68 genome as an infectious bacterial artificial chromosome. J. Virol. 74:6964-6974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allen, R. D., S. Dickerson, and S. H. Speck. 2006. Identification of spliced gammaherpesvirus 68 LANA and v-cyclin transcripts and analysis of their expression in vivo during latent infection. J. Virol. 80:2055-2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Babcock, G. J., D. Hochberg, and D. A. Thorley-Lawson. 2000. The expression pattern of Epstein-Barr virus latent genes in vivo is dependent upon the differentiation stage of the infected B cell. Immunity 13:497-506. [DOI] [PubMed] [Google Scholar]
- 5.Ballestas, M. E., P. A. Chatis, and K. M. Kaye. 1999. Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science 284:641-644. [DOI] [PubMed] [Google Scholar]
- 6.Becker, S. D., M. Bennett, J. P. Stewart, and J. L. Hurst. 2007. Serological survey of virus infection among wild house mice (Mus domesticus) in the UK. Lab. Anim. 41:229-238. [DOI] [PubMed] [Google Scholar]
- 7.Bennett, N. J., J. S. May, and P. G. Stevenson. 2005. Gamma-herpesvirus latency requires T cell evasion during episome maintenance. PLoS Biol. 3:e120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Benson, M. J., R. Elgueta, W. Schpero, M. Molloy, W. Zhang, E. Usherwood, and R. J. Noelle. 2009. Distinction of the memory B cell response to cognate antigen versus bystander inflammatory signals. J. Exp. Med. 206:2013-2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blasdell, K., C. McCracken, A. Morris, A. A. Nash, M. Begon, M. Bennett, and J. P. Stewart. 2003. The wood mouse is a natural host for Murid herpesvirus 4. J. Gen. Virol. 84:111-113. [DOI] [PubMed] [Google Scholar]
- 10.Cardin, R. D., J. W. Brooks, S. R. Sarawar, and P. C. Doherty. 1996. Progressive loss of CD8+ T cell-mediated control of a gamma-herpesvirus in the absence of CD4+ T cells. J. Exp. Med. 184:863-871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coleman, H. M., S. Efstathiou, and P. G. Stevenson. 2005. Transcription of the murine gammaherpesvirus 68 ORF73 from promoters in the viral terminal repeats. J. Gen. Virol. 86:561-574. [DOI] [PubMed] [Google Scholar]
- 12.Collins, C. M., J. M. Boss, and S. H. Speck. 2009. Identification of infected B-cell populations by using a recombinant murine gammaherpesvirus 68 expressing a fluorescent protein. J. Virol. 83:6484-6493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cotter, M. A., and E. S. Robertson. 1999. The latency-associated nuclear antigen tethers the kaposi's sarcoma-associated herpesvirus genome to host chromosomes in body cavity-based lymphoma cells. Virology 264:254-264. [DOI] [PubMed] [Google Scholar]
- 14.Davenport, M. G., and J. S. Pagano. 1999. Expression of EBNA-1 mRNA is regulated by cell cycle during Epstein-Barr virus type I latency. J. Virol. 73:3154-3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dupin, N., C. Fisher, P. Kellam, S. Ariad, M. Tulliez, N. Franck, E. van Marck, D. Salmon, I. Gorin, J. Escande, R. A. Weiss, K. Alitalo, and C. Boshoff. 1999. Distribution of human herpesvirus-8 latently infected cells in Kaposi's sarcoma, multicentric Castleman's disease, and primary effusion lymphoma. Proc. Natl. Acad. Sci. USA 96:4546-4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flaño, E., S. M. Husain, J. T. Sample, D. L. Woodland, and M. A. Blackman. 2000. Latent murine gammaherpesvirus infection is established in activated B cells, dendritic cells, and macrophages. J. Immunol. 165:1074-1081. [DOI] [PubMed] [Google Scholar]
- 17.Flaño, E., I. Kim, J. Moore, D. L. Woodland, and M. A. Blackman. 2003. Differential gamma-herpesvirus distribution in distinct anatomical locations and cell subsets during persistent infection in mice. J. Immunol. 170:3828-3834. [DOI] [PubMed] [Google Scholar]
- 18.Flaño, E., I. Kim, D. L. Woodland, and M. A. Blackman. 2002. Gammaherpesvirus latency is preferentially maintained in splenic germinal center and memory B cells. J. Exp. Med. 196:1363-1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Forrest, J. C., C. R. Paden, R. D. Allen, J. Collins, and S. H. Speck. 2007. ORF73-null murine gammaherpesvirus 68 reveals roles for mLANA and p53 in virus replication. J. Virol. 81:11957-11971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fowler, P., S. Marques, J. P. Simas, and S. Efstathiou. 2003. ORF73 of murine herpesvirus-68 is critical for the establishment and maintenance of latency. J. Gen. Virol. 84:3405-3416. [DOI] [PubMed] [Google Scholar]
- 21.Friborg, J., W. Kong, M. O. Hottiger, and G. J. Nabel. 1999. p53 inhibition by the LANA protein of KSHV protects against cell death. Nature 402:889-894. [DOI] [PubMed] [Google Scholar]
- 22.Gao, J., J. M. Coulson, A. Whitehouse, and N. Blake. 2009. Reduction in RNA levels rather than retardation of translation is responsible for the inhibition of major histocompatibility complex class I antigen presentation by the glutamic acid-rich repeat of herpesvirus saimiri open reading frame 73. J. Virol. 83:273-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao, S. J., L. Kingsley, M. Li, W. Zheng, C. Parravicini, J. Ziegler, R. Newton, C. R. Rinaldo, A. Saah, J. Phair, R. Detels, Y. Chang, and P. S. Moore. 1996. KSHV antibodies among Americans, Italians and Ugandans with and without Kaposi's sarcoma. Nat. Med. 2:925-928. [DOI] [PubMed] [Google Scholar]
- 24.Garber, A. C., J. Hu, and R. Renne. 2002. Latency-associated nuclear antigen (LANA) cooperatively binds to two sites within the terminal repeat, and both sites contribute to the ability of LANA to suppress transcription and to facilitate DNA replication. J. Biol. Chem. 277:27401-27411. [DOI] [PubMed] [Google Scholar]
- 25.Grundhoff, A., and D. Ganem. 2003. The Latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus permits replication of terminal repeat-containing plasmids. J. Virol. 77:2779-2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hochberg, D., J. M. Middeldorp, M. Catalina, J. L. Sullivan, K. Luzuriaga, and D. A. Thorley-Lawson. 2004. Demonstration of the Burkitt's lymphoma Epstein-Barr virus phenotype in dividing latently infected memory cells in vivo. Proc. Natl. Acad. Sci. USA 101:239-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kanda, T., M. Otter, and G. M. Wahl. 2001. Coupling of mitotic chromosome tethering and replication competence in Epstein-Barr virus-based plasmids. Mol. Cell. Biol. 21:3576-3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kedes, D. H., E. Operskalski, M. Busch, R. Kohn, J. Flood, and D. Ganem. 1996. The seroepidemiology of human herpesvirus 8 (Kaposi's sarcoma-associated herpesvirus): distribution of infection in KS risk groups and evidence for sexual transmission. Nat. Med. 2:918-924. [DOI] [PubMed] [Google Scholar]
- 29.Knapp, T., E. Hare, L. Feng, G. Zlokarnik, and P. Negulescu. 2003. Detection of beta-lactamase reporter gene expression by flow cytometry. Cytometry 51A:68-78. [DOI] [PubMed] [Google Scholar]
- 30.Krug, L. T., C. M. Collins, L. M. Gargano, and S. H. Speck. 2009. NF-κB p50 plays distinct roles in the establishment and control of murine gammaherpesvirus 68 latency. J. Virol. 83:4732-4748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kwun, H. J., S. R. Da Silva, I. M. Shah, N. Blake, P. S. Moore, and Y. Chang. 2007. Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen 1 mimics Epstein-Barr virus EBNA1 immune evasion through central repeat domain effects on protein processing. J. Virol. 81:8225-8235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lan, K., D. A. Kuppers, S. C. Verma, and E. S. Robertson. 2004. Kaposi's sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen inhibits lytic replication by targeting Rta: a potential mechanism for virus-mediated control of latency. J. Virol. 78:6585-6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lennette, E. T., D. J. Blackbourn, and J. A. Levy. 1996. Antibodies to human herpesvirus type 8 in the general population and in Kaposi's sarcoma patients. Lancet 348:858-861. [DOI] [PubMed] [Google Scholar]
- 34.Levitskaya, J., M. Coram, V. Levitsky, S. Imreh, P. M. Steigerwald-Mullen, G. Klein, M. G. Kurilla, and M. G. Masucci. 1995. Inhibition of antigen processing by the internal repeat region of the Epstein-Barr virus nuclear antigen-1. Nature 375:685-688. [DOI] [PubMed] [Google Scholar]
- 35.Li, H., K. Ikuta, J. W. Sixbey, and S. A. Tibbetts. 2008. A replication-defective gammaherpesvirus efficiently establishes long-term latency in macrophages but not in B cells in vivo. J. Virol. 82:8500-8508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lim, C., H. Sohn, D. Lee, Y. Gwack, and J. Choe. 2002. Functional dissection of latency-associated nuclear antigen 1 of Kaposi's sarcoma-associated herpesvirus involved in latent DNA replication and transcription of terminal repeats of the viral genome. J. Virol. 76:10320-10331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lindner, S. E., and B. Sugden. 2007. The plasmid replicon of Epstein-Barr virus: mechanistic insights into efficient, licensed, extrachromosomal replication in human cells. Plasmid 58:1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marques, S., S. Efstathiou, K. G. Smith, M. Haury, and J. P. Simas. 2003. Selective gene expression of latent murine gammaherpesvirus 68 in B lymphocytes. J. Virol. 77:7308-7318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Milho, R., C. M. Smith, S. Marques, M. Alenquer, J. S. May, L. Gillet, M. Gaspar, S. Efstathiou, J. P. Simas, and P. G. Stevenson. 2009. In vivo imaging of murid herpesvirus-4 infection. J. Gen. Virol. 90:21-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moorman, N. J., D. O. Willer, and S. H. Speck. 2003. The gammaherpesvirus 68 latency-associated nuclear antigen homolog is critical for the establishment of splenic latency. J. Virol. 77:10295-10303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moser, J. M., J. W. Upton, R. D. Allen, C. B. Wilson, and S. H. Speck. 2005. Role of B-cell proliferation in the establishment of gammaherpesvirus latency. J. Virol. 79:9480-9491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ottinger, M., D. Pliquet, T. Christalla, R. Frank, J. P. Stewart, and T. F. Schulz. 2009. The interaction of the gammaherpesvirus 68 orf73 protein with cellular BET proteins affects the activation of cell cycle promoters. J. Virol. 83:4423-4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Piolot, T., M. Tramier, M. Coppey, J. C. Nicolas, and V. Marechal. 2001. Close but distinct regions of human herpesvirus 8 latency-associated nuclear antigen 1 are responsible for nuclear targeting and binding to human mitotic chromosomes. J. Virol. 75:3948-3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rajcáni, J., D. Blaskovic, J. Svobodová, F. Ciampor, D. Hucková, and D. Staneková. 1985. Pathogenesis of acute and persistent murine herpesvirus infection in mice. Acta Virol. 29:51-60. [PubMed] [Google Scholar]
- 45.Renne, R., C. Barry, D. Dittmer, N. Compitello, P. O. Brown, and D. Ganem. 2001. Modulation of cellular and viral gene expression by the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus. J. Virol. 75:458-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ridderstad, A., and D. M. Tarlinton. 1998. Kinetics of establishing the memory B cell population as revealed by CD38 expression. J. Immunol. 160:4688-4695. [PubMed] [Google Scholar]
- 47.Rodrigues, L., J. Filipe, M. P. Seldon, L. Fonseca, J. Anrather, M. P. Soares, and J. P. Simas. 2009. Termination of NF-κB activity through a gammaherpesvirus protein that assembles an EC5S ubiquitin-ligase. EMBO J. 28:1283-1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sharipo, A., M. Imreh, A. Leonchiks, S. Imreh, and M. G. Masucci. 1998. A minimal glycine-alanine repeat prevents the interaction of ubiquitinated I kappaB alpha with the proteasome: a new mechanism for selective inhibition of proteolysis. Nat. Med. 4:939-944. [DOI] [PubMed] [Google Scholar]
- 49.Siegel, A. M., U. S. Rangaswamy, R. J. Napier, and S. H. Speck. 2010. Blimp-1 dependent plasma cell differentiation is required for efficient maintenance of murine gammaherpesvirus latency and antiviral antibody responses. J. Virol. 84:674-685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Simas, J. P., S. Marques, A. Bridgeman, S. Efstathiou, and H. Adler. 2004. The M2 gene product of murine gammaherpesvirus 68 is required for efficient colonization of splenic follicles but is not necessary for expansion of latently infected germinal centre B cells. J. Gen. Virol. 85:2789-2797. [DOI] [PubMed] [Google Scholar]
- 51.Simpson, G. R., T. F. Schulz, D. Whitby, P. M. Cook, C. Boshoff, L. Rainbow, M. R. Howard, S. Gao, R. A. Bohenzky, P. Simmonds, C. Lee, A. de Ruiter, A. Hatzakis, R. S. Tedder, I. V. Weller, R. A. Weiss, and P. S. Moore. 1996. Prevalence of Kaposi's sarcoma associated herpesvirus infection measured by antibodies to recombinant capsid protein and latent immunofluorescence antigen. Lancet 348:1133-1138. [DOI] [PubMed] [Google Scholar]
- 52.Smith, C. M., G. Rosa, J. May, N. Bennett, A. Mount, G. Belz, and P. Stevenson. 2006. CD4+ T cells specific for a model latency-associated antigen fail to control a gammaherpesvirus in vivo. Eur. J. Immunol. 36:3186-3197. [DOI] [PubMed] [Google Scholar]
- 53.Smith, G. A., and L. W. Enquist. 1999. Construction and transposon mutagenesis in Escherichia coli of a full-length infectious clone of pseudorabies virus, an alphaherpesvirus. J. Virol. 73:6405-6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sunil-Chandra, N. P., S. Efstathiou, and A. A. Nash. 1992. Murine gammaherpesvirus 68 establishes a latent infection in mouse B lymphocytes in vivo. J. Gen. Virol. 73(Pt 12):3275-3279. [DOI] [PubMed] [Google Scholar]
- 55.Tellam, J., G. Connolly, K. J. Green, J. J. Miles, D. J. Moss, S. R. Burrows, and R. Khanna. 2004. Endogenous presentation of CD8+ T cell epitopes from Epstein-Barr virus-encoded nuclear antigen 1. J. Exp. Med. 199:1421-1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thorley-Lawson, D. A., and A. Gross. 2004. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N. Engl. J. Med. 350:1328-1337. [DOI] [PubMed] [Google Scholar]
- 57.Tibbetts, S. A., L. F. van Dyk, S. H. Speck, and H. W. Virgin. 2002. Immune control of the number and reactivation phenotype of cells latently infected with a gammaherpesvirus. J. Virol. 76:7125-7132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tibbetts, S. A., J. S. McClellan, S. Gangappa, S. H. Speck, and H. W. Virgin. 2003. Effective vaccination against long-term gammaherpesvirus latency. J. Virol. 77:2522-2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van Berkel, V., B. Levine, S. B. Kapadia, J. E. Goldman, S. H. Speck, and H. W. V. Iv. 2002. Critical role for a high-affinity chemokine-binding protein in γ-herpesvirus-induced lethal meningitis. J. Clin. Investig. 109:905-914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.van Dyk, L. F., H. W. Virgin, and S. H. Speck. 2000. The murine gammaherpesvirus 68 v-cyclin is a critical regulator of reactivation from latency. J. Virol. 74:7451-7461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Verma, S. C., K. Lan, and E. Robertson. 2007. Structure and function of latency-associated nuclear antigen. Curr. Top. Microbiol. Immunol. 312:101-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Virgin, H. W., P. Latreille, P. Wamsley, K. Hallsworth, K. E. Weck, A. J. Dal Canto, and S. H. Speck. 1997. Complete sequence and genomic analysis of murine gammaherpesvirus 68. J. Virol. 71:5894-5904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Weck, K. E., M. L. Barkon, L. I. Yoo, S. H. Speck, and H. W. Virgin IV. 1996. Mature B cells are required for acute splenic infection, but not for establishment of latency, by murine gammaherpesvirus 68. J. Virol. 70:6775-6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weck, K. E., S. S. Kim, H. W. Virgin, and S. H. Speck. 1999. B cells regulate murine gammaherpesvirus 68 latency. J. Virol. 73:4651-4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Weck, K. E., S. S. Kim, H. W. Virgin, and S. H. Speck. 1999. Macrophages are the major reservoir of latent murine gammaherpesvirus 68 in peritoneal cells. J. Virol. 73:3273-3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Willer, D. O., and S. H. Speck. 2003. Long-term latent murine gammaherpesvirus 68 infection is preferentially found within the surface immunoglobulin D-negative subset of splenic B cells in vivo. J. Virol. 77:8310-8321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yates, J. L., N. Warren, and B. Sugden. 1985. Stable replication of plasmids derived from Epstein-Barr virus in various mammalian cells. Nature 313:812-815. [DOI] [PubMed] [Google Scholar]
- 68.Zaldumbide, A., M. Ossevoort, E. J. H. J. Wiertz, and R. C. Hoeben. 2007. In cis inhibition of antigen processing by the latency-associated nuclear antigen I of Kaposi sarcoma herpes virus. Mol. Immunol. 44:1352-1360. [DOI] [PubMed] [Google Scholar]
- 69.Zlokarnik, G., P. A. Negulescu, T. E. Knapp, L. Mere, N. Burres, L. Feng, M. Whitney, K. Roemer, and R. Y. Tsien. 1998. Quantitation of transcription and clonal selection of single living cells with beta-lactamase as reporter. Science 279:84-88. [DOI] [PubMed] [Google Scholar]