

Abstract
The inositol 1,4,5-trisphosphate (InsP3) receptors (InsP3Rs) are a family of Ca2+ release channels localized predominately in the endoplasmic reticulum of all cell types. They function to release Ca2+ into the cytoplasm in response to InsP3 produced by diverse stimuli, generating complex local and global Ca2+ signals that regulate numerous cell physiological processes ranging from gene transcription to secretion to learning and memory. The InsP3R is a calcium-selective cation channel whose gating is regulated not only by InsP3, but by other ligands as well, in particular cytoplasmic Ca2+. Over the last decade, detailed quantitative studies of InsP3R channel function and its regulation by ligands and interacting proteins have provided new insights into a remarkable richness of channel regulation and of the structural aspects that underlie signal transduction and permeation. Here, we focus on these developments and review and synthesize the literature regarding the structure and single-channel properties of the InsP3R.
I. INTRODUCTION
Modulation of cytoplasmic free calcium concentration ([Ca2+]i) is a signaling system involved in the regulation of numerous processes, including transepithelial transport, learning and memory, muscle contraction, membrane trafficking, synaptic transmission, secretion, motility, membrane excitability, gene expression, cell division, and apoptosis. A ubiquitous mechanism of modulating [Ca2+]i involves the activation of phospholipase C (PLC)-β and PLC-γ by a wide variety of stimuli including ligand interaction with G protein- or tyrosine kinase-linked receptors. PLC hydrolyzes the membrane lipid phosphatidylinositol 4,5-bisphosphate, generating inositol 1,4,5-trisphosphate (InsP3) (27, 377). InsP3 diffuses in the cytoplasm and binds to its receptor (InsP3R), which is an intracellular ligand-gated Ca2+ release channel (136, 270) localized primarily in the endoplasmic reticulum (ER) membrane (132, 354, 400). The ER is the major Ca2+ storage organelle in most cells. ER membrane Ca2+-ATP-ases accumulate Ca2+ in the ER lumen to quite high levels. Because the lumen contains high concentrations of Ca2+ binding proteins, the total amount of Ca2+ in the lumen may be >1 mM; the concentration of free Ca2+ has been estimated to be between 100 and 700 μM (8, 21, 69, 330, 355, 373, 491). In contrast, the concentration of Ca2+ in the cytoplasm of unstimulated cells is between 50 and 100 nM, 3–4 orders of magnitude lower than in the ER lumen. This low concentration is maintained by Ca2+ pumps and other Ca2+ transporters located in the ER, as well as plasma, membranes. Upon binding InsP3, the InsP3R is gated open, providing a pathway for Ca2+ to diffuse down this electrochemical gradient from the ER lumen to cytoplasm. Ca2+ in the cytoplasm moves by passive diffusion, at a rate that is reduced by mobile and immobile Ca2+ binding proteins acting as buffers. As a consequence, microdomains with steep Ca2+ concentration gradients can rapidly form and dissipate near the mouth of an InsP3R Ca2+ channel. The Ca2+ concentration adjacent to the open channel may be 100 μM or more, whereas concentrations as close as 1–2 μm from the channel pore may be below 1 μM (342, 343, 390). Therefore, Ca2+ has only a restricted “range of action,” on the order of 5 μm (7). The distribution and concentrations of Ca2+ binding proteins and the release channels, as well as the complex properties of the release channels, enable InsP3R-mediated [Ca2+]i signals to have diverse spatial and temporal properties that can be exploited by cells, making this signaling system remarkably robust. Consequently, despite its expression in probably all cells in the body, this signaling system can provide specific signals that regulate diverse cell physiological processes.
Analyses of InsP3-mediated [Ca2+]i signals in single cells has revealed them to be complex. In the temporal domain, this complexity is manifested as repetitive spikes or oscillations, with frequencies often tuned to levels of stimulation, suggesting that [Ca2+]i signals may be transduced by frequency encoding as well as amplitude. In the spatial domain, [Ca2+]i signals may initiate at specific locations and remain highly localized or propagate as waves (27, 28, 89, 466). Thus InsP3-mediated [Ca2+]i signals are often organized to provide different signals to discrete parts of the cell. High-resolution optical imaging of fluorescent Ca2+ indicator dyes in intact cells have suggested that InsP3-mediated [Ca2+]i signals are organized at three broad levels. Each level can provide different signaling functions and serve as a building block for [Ca2+] signals at the next level (Fig. 1) (26, 49, 358). At the first level, “fundamental” signals result from openings of individual InsP3R Ca2+ channels. Weak activation by low [InsP3] evokes localized elevations of cytoplasmic [Ca2+] that arise stochastically and autonomously at discrete release sites. They have variable size, with the smallest, called “blips” (358), possibly involving Ca2+ flux through one or, more likely, a few InsP3Rs (Fig. 1A). At the next level, “elementary” signals arise from the concerted opening of several channels. Larger events (“puffs”) involve the concerted opening of multiple InsP3R channels organized within a cluster (446). The coordinated opening of several channels is triggered by Ca2+ release from one channel acting as an activating ligand to stimulate gating of nearby channels through a process of Ca2+-induced Ca2+ release (CICR) (see discussion below about activating ligands of the InsP3R channel) (Fig. 1B). Appropriate colocalization with effector proteins enables spatially restricted fundamental and elementary signals to provide specificity of cellular responses (49, 291). At the third level, with higher [InsP3] associated with stronger extra-cellular agonist stimulation, Ca2+ released at one cluster site can trigger Ca2+ release at adjacent sites by CICR, leading to the generation of Ca2+ waves (Fig. 1C) that propagate in a saltatory manner (48, 71, 86) at velocities of a few tens of microns per second by successive cycles of Ca2+ release, diffusion, and CICR (26, 101).
FIG. 1.
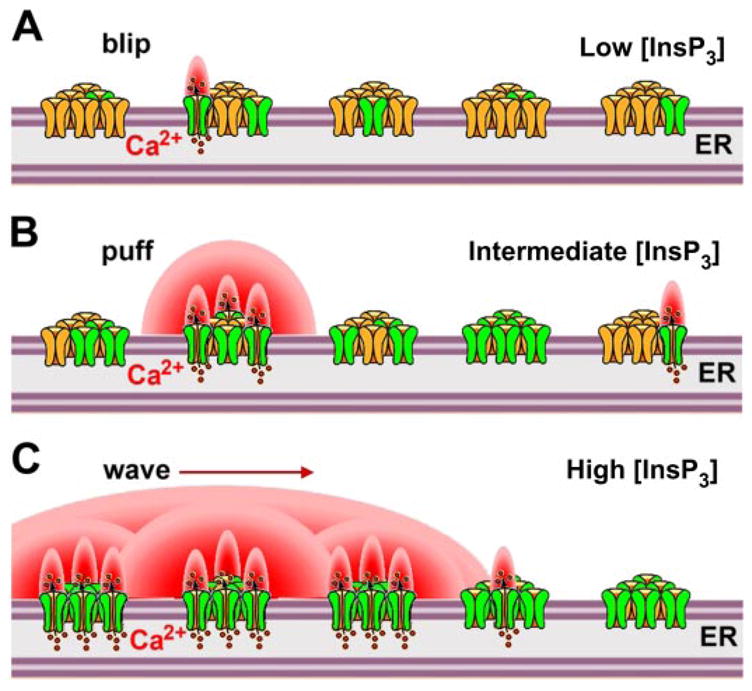
Schematic of the behaviors of inositol trisphosphate receptor (InsP3R) channels in the presence of increasing concentrations of InsP3. InsP3Rs are shown arranged in clusters that form discrete release sites within the continuous endoplasmic reticulum. A: at low [InsP3] during weak agonist stimulation, few receptors (in green) bind InsP3. Others (in yellow) are not InsP3 liganded and therefore not activated. Consequently, highly localized small Ca2+ signals (“blips”) are generated by Ca2+ released through a single or few InsP3R channels raising cytoplasmic Ca2+ concentration (shown in red). B: at higher levels of [InsP3], coordinated openings of several channels (InsP3 liganded) within a cluster is triggered by Ca2+ release from one channel acting as an activating ligand to stimulate gating of nearby channels through a process of Ca2+-induced Ca2+ release (CICR). C: even higher [InsP3] evokes global propagating Ca2+ signals (waves). Ca2+ released at one cluster can trigger Ca2+ release at adjacent clusters by CICR, leading to the generation of Ca2+ waves that propagate by successive cycles of Ca2+ release, diffusion, and CICR. [Figure kindly supplied by I. Parker and N. Callamaras. Adapted from Parker et al. (358).]
The spatial organization of InsP3R channels within clusters and the distribution of clusters, together with the positive regulation of the InsP3R by InsP3 and Ca2+ (CICR), enable local and long-range Ca2+ signals to be constructed from the activities of single InsP3R Ca2+ channels. The cytoplasm has been described as an excitable medium: a collection of Ca2+ release sites coupled by messenger (Ca2+) diffusion and an autocatalytic process (CICR) (248). The InsP3R is the fundamental building block of the excitable medium. Nevertheless, this description does not account for all of the properties of InsP3R-mediated Ca2+ signals in cells. The regenerative action of CICR would normally be expected to lead to all-or-nothing binary cellular Ca2+ responses. Appreciable spacing between release sites may limit the efficacy of CICR, depending on the excitability of the system, but additional mechanisms exist that also play a role in grading Ca2+ release with stimulus intensity, as well as in terminating Ca2+ release, including stochastic attrition (435), Ca2+ feedback inhibition, and inactivation.
InsP3-mediated Ca2+ signals are an example in which finite fluctuations at the microscopic (single channel) level give rise to signals that are observable at the macroscopic (cytoplasmic) level (230). The ability to trigger global signals depends strongly on InsP3R single-channel properties. Detailed knowledge of the microscopic properties of single InsP3R Ca-2+ release channels is therefore necessary for an understanding of the diverse Ca2+ signals elicited by the InsP3 pathway. The focus of this review is on the permeation and gating properties of single InsP3R Ca2+ release channels. As such, we review recent studies that have provided new information regarding structural features of the InsP3R, the mechanisms of ion permeation, and channel gating and its regulation by InsP3, Ca2+, and other cellular factors including interacting proteins.
II. MOLECULAR PROPERTIES OF THE INOSITOL TRISPHOSPHATE RECEPTOR
A. Identification of the InsP3R
The glycoprotein receptor for InsP3 was first purified from rat cerebellum (443). Binding of InsP3 to the purified protein had high affinity (Kd ~ 100 nM) compared with other inositol phosphates and was inhibited by heparin, properties that were similar to those of the InsP3 receptor in crude cerebellar microsomes (520). Electrophoretic analysis revealed that the receptor had an apparent molecular mass of ~260 kDa, whereas gel fractionation indicated a molecular mass of the native protein of ~1 MDa, demonstrating that the receptor was a tetramer (443), a result that was later confirmed by cross-linking (270). Immunostaining of cerebellar Purkinje cells revealed that the receptor was expressed in the ER, nuclear envelope, and portions of the Golgi complex, but not mitochondria or plasma membranes (400). Subsequent studies have indicated that the plasma membrane in some cell types may also contain InsP3R (108, 457). Two groups simultaneously cloned full-length (150) and partial (313) type 1 InsP3R (InsP3R-1) cDNAs from mouse cerebellum. The full-length rat cerebellar InsP3R-1 cDNA was cloned shortly thereafter (311). The full-length mouse cDNA sequence encoded for a protein of 2,749 amino acids with a predicted molecular mass of 313 kDa (150), whereas an additional 2,734-amino acid protein was discovered as a splice variant in the rat (311). Expression of the recombinant proteins enhanced the magnitudes of InsP3 binding and InsP3-induced Ca2+ release from isolated membrane fractions (321). Reconstitution of the purified receptor into lipid vesicles showed that it mediated Ca2+ release in response to InsP3, with half-maximal flux activated by 40–80 nM InsP3 (136, 271). Furthermore, reconstitution of purified InsP3R into planar bilayer membranes resulted in the appearance of Ca2+-permeable ion channels (270, 302). Taken together, the data suggested that the InsP3R was itself an intracellular ligand-gated Ca2+ release channel.
B. InsP3R Diversity
1. Gene expression
Subsequently, it was established that three genes (39, 103, 150, 289, 313, 399, 439) encode for a family of InsP3Rs in mammalian cells, including humans, and other vertebrates. The three full-length amino acid sequences are 60–80% homologous overall, with regions, including the ligand-binding and pore domains (discussed below), having much higher homology (363, 460). In contrast, invertebrates appear to express only a single InsP3R, most closely related to the type 1 isoform (196, 200). In mammals, the InsP3R is ubiquitously expressed, perhaps in all cell types (104, 146, 149, 415, 460). The three channel isoforms have distinct and overlapping patterns of expression, with most cells outside the central nervous system expressing more than one type (68, 104, 105, 340, 345, 418, 451, 460, 493). InsP3R isoform expression levels can be modified during development and differentiation (129, 242, 340, 394, 419, 450, 460) and in response to various normal and pathological stimuli (20, 61, 70, 218, 226, 250, 305, 403, 418, 460, 502, 526). Furthermore, InsP3R protein expression levels can be downregulated by a use-dependent mechanism that involves InsP3- and Ca2+-dependent channel ubiquitination, and subsequent degradation involving the proteasome (9, 10, 35, 515).
2. Alternative splicing
Further diversity of InsP3R expression is created by alternative splicing (99, 138, 311, 340, 348). The type 1 channel has three main splice regions, denoted SI, SII, and SIII. The SI site is located within the core InsP3 binding region, comprising residues 318–332 (Fig. 2B) within a loop connecting β-strands 6 and 7 of the second β-trefoil domain (the structure is discussed in sect. IIIB1). The SII site is located near the middle of the protein sequence in the coupling domain, comprising residues 1693–1731 (Fig. 2B). It was proposed that the SII+ variant (the “long” form) is the neuronal form, while peripheral tissues express primarily the SII− short form (99). The SII sequence is absent in the types 2 and 3 isoforms. SIII corresponds to a 9-amino acid insert after residue 917 (Fig. 2B). Until recently, none of the splice forms had been cloned from tissues. A detailed analysis of the InsP3R-1 transcriptome has revealed a previously unrecognized and remarkable diversity of expression in the brain (386). SII splicing can come in four varieties (339, 386), with the result that the type 1 transcript can vary at six segments within the open reading frame, which can give rise to 48 possible channel subunits. Seventeen variants were detected in cerebellum, with each brain tissue and developmental stage generating 11–13 forms. A biased stochastic model for splicing regulation could quantitatively account for the multiple forms expressed at each developmental stage.
FIG. 2.

Structural determinants of the InsP3R. A: overall domain structure. The InsP3R molecule depicted as a linear amino acid sequence, with the NH2-terminal InsP3 binding region (red), coupling region (yellow), transmembrane region (green), and COOH tail (blue) depicted. B: linear amino acid sequence. Residues are numbered according to the rat type 1 SI+, SII+, SIII− sequence (protein accession no. 121838). Structural features (see section in this review where each element is described) shown are as follows: arm subdomain and β-trefoil in the InsP3-binding suppressor domain (sect. IIIB1); β-trefoil and armadillo repeats in InsP3 binding core domain (sect. IIIB1); armadillo repeats in the coupling domain (sect. IIIB3); alternative splicing regions SI, SII, and SIII (sect. IIB2) for type 1 InsP3R; opt deletion in type 1 InsP3R mutant (sect. IIIB3); ATP-binding sites ATPA, ATPB, and ATPC (sect. VIF4); transmembrane helices TM1–6 and pore-forming P region (sect. IIIB2A) with selectivity filter (sect. VF); linker region (sect. IIIB1); dimerizing region (sect. IIIC1); tetramer forming region (sect. IIIC1). Trypsin proteolysis sites (sect. IIIC3) are indicated by black arrowheads. The caspase 3 cleavage site (sect. IIIC3) is also shown. G25 (sect. IIIB1), S217 (sect. IIIB1), T799 (sect. IIIB3), M837 (sect. IIIB3), C1430 (sect. IIIB3), and G2045 (sect. IIIB3) are highly conserved residues the mutation of which can impact InsP3R channel functions. Mutation of E2100 modifies Ca2+ regulation of InsP3R channel (sect. VIK). R265, L508, and R511 are critically important for InsP3 binding (sect. IIIB1). G2586, T2591, L2592, and F2595 are residues that may be involved in forming the gate of the InsP3R channel (sect. IIIB2B). N2475 and N2503 are glycosylation sites (sect. IIIB). S1589 and S1755 are PKA/PKG phosphorylation sites (sect. VIL, 1 and 2); S2681 is an Akt phosphorylation site (sect. VIL4); Y353 is a Fyn tyrosine kinase phosphorylation site (sect. VIL5); and S421 and T799 are cdc2/CyB phosphorylation sites (sect. VIL6). Sequences involved in interaction of InsP3R channel with the following proteins are also depicted: homer (sect. VIN13); calmodulin (sect. VIN1); CaBP (sect. VIN2); RACK1 (sect. VIN3); IRBIT (sect. VIN4); CIB1 (sect. VIN2); Na+-K+-ATPase (sect. VIN14); COOH terminal of InsP3R in the tetrameric channel (sect. IIIB1); TRPC3 (sect. VIN14); GAPDH (sect. VIN8); AKAP9 [through leucine/isoleucine zipper (LIZ) motif] (sect. VIL1); FKBP12 (sect. VIN7); Erp44 (sect. VIN6); chromogranins (sect. VIN5); cytochrome c (sect. VIN10); HAP1 and Httexp (sect. VIN11); protein 4.1N (sect. VIN13); and PP1 (sect. VIL1).
The mouse type 2 isoform was recently shown to have a splice variant (SIm2) comprising residues 176–208, within the first β-trefoil of the InsP3-binding region in the so-called suppressor domain (discussed in sect. IIIB1) (199). The deletion of this sequence eliminates two of the β-strands of the domain, which would be expected to severely disrupt its structure. The SIm2− mRNA comprised 7–20% of the total type 2 transcripts in various mouse tissues, with the submandibular salivary gland expressing it at 41% (199). Another mouse type 2 splice variant termed TIPR was detected in skeletal and heart muscle that codes for a truncated protein of only the NH2-terminal 181 residues (151). It shares the splice acceptor site with the SIm2 variant.
Although invertebrates appear to express only a single InsP3R isoform, the Caenorhabditis elegans channel exists as six alternatively spliced forms (22, 159) and the Drosophila channel exists as two (427).
3. Heteroligomerization
A final level of channel diversity is generated by heteroligomeric interactions among different isoforms. The InsP3Rs are ~2,700–2,800 amino acid intracellular membrane proteins that exist as homo- or heterotetramers (209, 210, 212, 270, 311, 328, 363, 443, 536). By analogy with other cation channels and some structural information, the ion-conducting pore is believed to be created at the central axis of the tetrameric structure (Fig. 3). Evidence for the existence of heterotetramers of two isoforms has come primarily from the ability of isoform-specific antibodies to coprecipitate other isoforms (363). Cross-linking studies (352) and the ability of mutant channels to exert dominant negative effects (40, 433) also support the existence of heterotetrameric channels. The results to date indicate that two different InsP3R forms can exist in the same tetramer. Whether all three isoforms and/or multiple splice variants ever exist in a single tetrameric complex is however unknown. If they do, then the diversity of channels could be quite impressive. For example, adult cerebellum, a source of InsP3R for many biochemical and functional studies, expressed 13 splice forms of the InsP3R-1. Twenty percent of the transcripts were SI+, whereas 98% contained three of four possible SII varieties, and SIII was absent in 73% of transcripts. For tetrameric channels with no bias against heteromultimerization among different forms, the presence of 12 transcript variants is predicted to give rise to nearly 5,900 channel isoforms (386)!
FIG. 3.

The InsP3R Ca2+ release channel. Cartoon depicting three of four InsP3R molecules (in different colors) in a single tetrameric channel structure. Part of the luminal loop connecting transmembrane helices 5 and 6 of each monomer dips into the fourfold symmetrical axis, creating the permeation pathway for Ca2+ efflux from the lumen of the endoplasmic reticulum.
4. Functional implication of InsP3R diversity
A) GENERAL CONSIDERATIONS
This diversity of channel expression is impressive, but the functional implications of this diversity, both at the single-channel as well as cellular levels, are still only poorly explored. This diversity suggests that cells require distinct InsP3Rs to regulate specific functions. Cerebellar Purkinje neurons express the type 1 isoform predominately (although many different splice variants of it), whereas insulin-secreting β-cells express primarily the type 3 channel (460), and cardiac myocytes express predominately the type 2 isoform (256, 368). Genetic knockout of the mouse type 1 receptor causes neurological defects and early death (297), consistent with the dominant expression of the type 1 isoform in the cerebellum. Similarly, genetic deletion of the mouse type 2 receptor abolishes the positive ionotropic and arrhythmogenic effect of endothelin in cardiac atrial myocytes (253) and endothelin-induced HDAC5 nuclear translocation in ventricular myocytes (521). It is therefore perhaps surprising that genetic diseases as a direct consequence of mutations of the human InsP3R have not yet been discovered, whereas several (malignant hyperthermia, central core disease, arrhythmogenic right ventricular cardiomyopathy, catecholaminergic polymorphic ventricular tachycardia) have been identified as consequences of mutations in ryanodine receptors (RyR), the other major family of intracellular Ca2+ release channels (reviewed in Ref. 376). It is possible that disease-causing InsP3R mutations will be discovered and that some InsP3R mutations are invariably embryonically lethal. On the other hand, the lack of identified InsP3R mutations in humans may suggest that expression of multiple InsP3R isoforms in most cell types provides functional redundancy that is necessary because of the critical importance of InsP3-mediated Ca2+ signaling for many cell physiological processes. Indeed, evidence suggests that there is functional redundancy among the InsP3R isoforms in cells that express more than one. For example, knock-out of both types 2 and 3 isoforms together were required to create a pancreatic acinar cell secretion phenotype (152), genetic deletion of the type 1 isoform was without effect on T-cell Ca2+ signaling or development (188), and genetic deletion of all three InsP3R isoforms was necessary to generate an apoptosis phenotype in chicken DT40 B cells (440).
B) ISOFORM DIFFERENCES
Despite these considerations, the molecular diversity of InsP3R expression nevertheless suggests that it is likely that InsP3R isoform-specific permeation, gating, or localization and their regulation by ligands or interacting proteins confers specificity required for specific cell physiological processes. Many different biochemical, Ca2+ release, and channel properties have been proposed to exist among the different isoforms. For example, the three channel isoforms may be differentially sensitive to activation-induced, ubiquitin-mediated down-regulation (516). It has been proposed that different channel isoforms have distinct InsP3 binding properties. However, the order of InsP3 affinity is variable among studies [type 2 > type 1 > type 3 (122, 318, 345, 439); type 1 > type 2 > type 3 (517); type 3 > type 2 > type 1 (344)], and the differences in affinity among the isoforms in some of the studies are modest. Thus the physiological relevance of different InsP3 affinities among isoforms has not been clearly established. It has been proposed that cADP-ribose and the oxidizing agent thimerosal regulate InsP3 binding or Ca2+ release differentially among the three channel isoforms (67, 488, 489) and that InsP3 binding to different isoforms is differentially regulated by [Ca2+]i or calmodulin (77, 487, 530). Nevertheless, the literature regarding all these studies is conflicting (reviewed in Ref. 461). In part, the divergent results likely reflect the different preparations used, for example, fragments of the recombinant InsP3R as bacterially expressed fusion proteins versus full-length channels in microsomes, as well as the different assays used, for example, InsP3 binding to microsomes versus Ca2+ flux from permeabilized cells. Differences among isoforms in Ca2+ release rates may not necessarily reflect intrinsic differences in the properties of the channels, since the state of phosphorylation or association with interacting proteins are usually unknown and are likely different in different cell preparations. It has also been suggested that divergent results could arise from “dominance” of a single subunit within a heterotetramer (461). In addition, the variability of published results likely stems from the presence of different conformational states of the channel present in the different studies. As a highly allosteric protein that is regulated by several heterotropic ligands (including InsP3, Ca2+, ATP, H+, and interacting proteins) as well as by redox and phosphorylation status, apparent binding affinities of each ligand will be strongly influenced by the conformational state of the channel, which is in turn dependent on the state of binding of all the other ligands. Even under identical experimental conditions, apparent differences in otherwise identical ligand binding properties between isoforms may be caused by such allosteric effects. Consequently, various reported ligand-binding properties may have been strongly influenced by the channel conformational state, which could be different among studies. It is therefore quite difficult to interpret much of the literature that has attempted to compare channel isoforms when only limited sets of experimental conditions are employed, as in most published studies.
C) SPLICE VARIANTS
A consistent observation relates to the effects of the SII splice site on the ability of the type 1 channel to be phosphorylated by protein kinase A (PKA) (99, 497, 498). Deletion of the SII region creates a novel ATP binding site (137) (ATPC; Fig. 2B). ATP binding to that site modulates the ability of the channel to be phosphorylated by PKA (496). Channel phosphorylation in turn allosterically modifies the channel sensitivity to InsP3 (497, 498, 541) (discussed in sect. VIL). The SII+ channel as well as the types 2 and 3 channels lack this ATP binding site and are therefore expected to respond differently in response to elevated cAMP, although both the types 2 and 3 channels can be phosphorylated by PKA at different sites (432, 436, 518).
D) PROTEIN INTERACTIONS
Many protein interactions with InsP3R have been described (discussed in sect. VIN and reviewed in Refs. 267, 365). Most interactions have been examined for only one isoform. Some proteins, including CaBP/caldendrin (527), Bcl-2-related proteins (83, 349, 508), and Na+-K+-ATPase α-subunit (540) have been shown to interact with all three InsP3R isoforms, whereas others, including AKAP9 (480) and protein 4.1N (301, 544), appear to interact specifically with the type 1 isoform. Because the stoichiometry of these interactions is unknown, it is unknown whether isoform-specific interactions can also form with heterotetrameric channels containing fewer than four copies of the type 1 channel molecule. It seems likely that many more isoform-specific protein interactions will be discovered and that these interactions may in turn be regulated. Thus a diversity of regulated isoform-specific protein interactions may confer yet further InsP3R channel diversity through mechanisms involving InsP3R localization and gating.
E) BIOPHYSICAL PROPERTIES
Finally, the different channel isoforms and their splice variants may have different biophysical properties related to gating and permeation. As discussed in detail in section V, the permeation properties of different InsP3R channel isoforms are quite similar, likely reflecting the conserved amino acid sequences in the pore region of the different isoforms. The gating properties of different isoforms of homotetrameric InsP3R channels have either been inferred from Ca2+ release studies or examined directly by single-channel electrophysiology. By analysis of Ca2+ release and agonist-induced Ca2+ signals in DT40 chicken B cells with either one or two InsP3R isoforms genetically deleted, it was concluded that the type 2 isoform is required for long-lasting regular [Ca2+]i oscillations, that the type 1 receptor mediated less regular [Ca2+]i oscillations, and that the type 3 channel generated only monophasic [Ca2+]i responses (318). Knockdown of the type 1 channel by RNA interference in HeLa and COS-7 cells abolished agonist-induced [Ca2+]i oscillations, whereas knockdown of the type 3 resulted in long-lasting [Ca2+]i oscillations (173). It was concluded that the two receptors have opposite roles in generating Ca2+ signals (173). Three points should be noted regarding these studies. First, even if these types of measurements provide insights into roles of different isoforms in generating distinct Ca2+ signals, they provide little mechanistic insight into the molecular features that distinguish the different channels. Gating of the InsP3R channel involves channel activation, inhibition, inactivation, stochastic attrition, and sequestration, and all these processes are complicated functions of ligand (Ca2+, InsP3, ATP) sensitivities and concentrations, interactions with proteins, phosphorylation state, etc. (discussed in sect. VI). Which gating properties in channels formed by different isoforms can account for different Ca2+ release behaviors have not been elucidated in these studies. Second, it is also important to note that, because the curves that describe the transient kinetics of Ca2+ release observed in cells and rapid perfusion experiments are reminiscent of the biphasic curves that describe the cytoplasmic Ca2+ concentration dependencies of steady-state channel activity, there has been a tendency in such cell studies to equate the two and to account for kinetic features of Ca2+ signals in terms of observed effects of Ca2+ concentration on steady-state single-channel gating activity. For example, the purported lack of high-Ca2+ inhibition of type 2 or type 3 InsP3R steady-state channel gating (163, 382) has been invoked to account for either the presence or absence of [Ca2+]i oscillations in such studies (173, 318). However, as pointed out (430), the bell-shaped or otherwise biphasic shape of the steady-state open probability (Po) versus [Ca2+]i curve has “very little, if anything” to do with the fact that the InsP3R exhibits complex rapid kinetic behaviors. Third, complex Ca2+ signals in cells are determined not only by “intrinsic” permeation and gating features of each isoform, but by many other factors as well, including the absolute channel density and the spatial distribution of the channels, and the influence of these factors within the context of complex cellular machinery, including pumps and buffers, that participate in regulating cytoplasmic Ca2+ concentration. Thus interpretation regarding the roles of, and differences among, different channel isoforms in such Ca2+ release/Ca2+ signaling studies is complicated and requires considerable caution.
More detailed mechanistic insights into the intrinsic differences among different channel isoforms can be obtained from single-channel recordings. Unfortunately, the channel properties observed in planar bilayer reconstitution studies have been quite variable among studies, even for the same isoforms from the same laboratories, and different from those observed by nuclear patch-clamp electrophysiology. This experimental variability limits the degree to which generalizations can be made regarding distinctions among different channel isoforms or splice variants. In patch-clamp studies of isolated Xenopus oocyte nuclei, it was concluded that the permeation and gating properties of the expressed recombinant rat type 3 channel were similar to those of the endogenous type 1 channel (284), but that the Ca2+ activation properties of the two channels uniquely distinguished them (283). In reconstitution studies of Sf9 cell-produced recombinant rat types 1, 2, and 3 channels, numerous differences were noted (482). First, the maximum channel open probability (Pmax) in 1 mM ATP was considerably smaller for type 3 channels (<5%) compared with the other two isoforms (30%). However, the Po for the type 1 channel recorded in this study was considerably higher than that observed in other reconstitution studies (generally <5%). Furthermore, another study found that the Pmax of the type 2 channel exceeded that of the type 1 channel (382). Of note, the maximum Po values for all three channels in all reconstitution studies were considerably lower than that measured in the patch-clamp studies (80%) (42, 196, 278, 282, 283). Second, the InsP3 sensitivities differed over fourfold, with the order as follows: type 2 > 1 > 3. In addition, the isoforms had different apparent sensitivities to ATP free acid. It is important to note that the determinations of channel Po in these studies were made at a single Ca2+ concentration (200 nM). However, as discussed in detail in section VIF, the interplay between the effects on channel Po of the cytoplasmic concentrations of Ca2+ and InsP3 or ATP as heterotropic ligands of the InsP3R, an allosteric protein, can be manifested as apparent differences in ligand sensitivity depending on the concentration of the other heterotropic ligands (282). Therefore, to determine the effective ligand sensitivity of a channel isoform, it is necessary to examine the effect of InsP3 or ATP concentrations on the Ca2+ concentration dependence of channel Po over a wide range of cytoplasmic Ca2+ concentrations.
The Ca2+ concentration dependencies of the channel Po of the different reconstituted InsP3R isoforms were narrow bell-shaped, centered around 200 nM for all three channels (481). In agreement, patch-clamp electrophysiology of the endogenous Xenopus (282) and Sf9 (196) and recombinant rat type 1 (42) and type 3 (283) channels indicated that their steady-state activities are indeed both activated and inhibited by Ca2+, but the high [Ca2+]i inhibition was exerted at much higher concentrations in the patch-clamp studies (half-maximal inhibition ~20–40 μM in patch-clamp experiments versus ~0.5–1 μM in bilayers). However, very different cytoplasmic Ca2+ concentration dependencies of channel Po have been observed in other single-channel reconstitution studies. In contrast to the observations in Reference 481, it was reported that the activities of the type 3 (163) and type 2 (380, 382) channels are monophasic functions of cytoplasmic Ca2+ concentration, with little or no evidence of high-[Ca2+]-mediated inhibition. Furthermore, the “width” of the biphasic Po versus [Ca2+] curve was distinct for the same reconstituted type 1 SII+ isoform obtained from different sources by the same lab (229, 478), with the reasons for the variability not clear to the authors (478). The reasons for the widely divergent and inconsistent permeation and gating properties and their regulation observed in InsP3R channel reconstitution studies are unclear, but are important to resolve to bring clarity to the field.
In summary, a remarkable diversity of InsP3R isoforms exists, but insights into the functional implications of this diversity are still rudimentary. Although single-channel electrophysiology promises to provide the most detailed insights into the distinct properties of different isoforms, the divergent results obtained within different studies of reconstituted channels and from nuclear patch-clamp studies indicate a need to define more optimal systems for expression and recording of different single InsP3R variants. Furthermore, it is expected that appreciation of the molecular diversity of InsP3R will likely also be enhanced by use of other approaches that address channel localization and interaction with molecular partners in protein complexes.
III. STRUCTURE OF THE INOSITOL TRISPHOSPHATE RECEPTOR
A. Overview
Each InsP3R molecule contains ~2,700 amino acids with a molecular mass of ~310 kDa. Structurally, each InsP3R molecule contains a cytoplasmic NH2 terminus comprising ~85% of the protein mass, a hydrophobic region predicted to contain six membrane-spanning helices that contribute to the ion-conducting pore of the InsP3R channel, and a relatively short cytoplasmic COOH terminus (Fig. 2A). Functionally, the NH2-terminal region can be divided into a proximal InsP3 binding domain and a more distal “regulatory”/“coupling” domain (Fig. 2A). InsP3 binding to the InsP3R is stoichiometric and localized by mutagenesis and an X-ray crystal structure to a region within residues 226–578 (Fig. 2B). Because of the similarity among channel isoforms, to facilitate discussion of various structural aspects of the InsP3R in this review, we refer throughout to specific amino acids in the sequence involved in ligand binding, protein interactions, etc., using numbering based on the rat type 1 SI+, SII+, SIII− InsP3R sequence. The InsP3R channel is a tetramer of four InsP3R molecules (Fig. 3). Approximately 2,000 amino acids separate the InsP3-binding domain from the pore. This intervening region between the InsP3 binding domain and the pore contains consensus sequences for phosphorylation and binding by nucleotides and various proteins. It may function to integrate, through allosteric coupling, other signaling pathways or metabolic states with the gating of the InsP3R.
B. Structural Properties of the InsP3R Molecule
1. InsP3 binding region
The localization of the InsP3 binding region to the NH2 terminus of the InsP3R was first proposed by Mignery and co-workers based on the discovery that deletion of the first 410 residues of the protein completely eliminated InsP3 binding (311) and that soluble monomeric proteins with COOH-terminal boundaries between residues 519 and 788, that lacked the transmembrane regions, efficiently bound InsP3 (312). Similar experiments subsequently established the NH2 terminus as the site of InsP3 binding in all three isoforms (289, 322, 439). Binding of InsP3 to the receptor is stoichiometric (271, 443) with an apparent Kd usually in the range of 10–80 nM. Binding of InsP3 to recombinant InsP3R proteins containing only the NH2-terminal 586 residues had similar affinity, pH sensitivity, and inositol phosphate selectivity as the native channel (345). Of note, deletions from either the NH2 or COOH terminus of this construct eliminated binding, indicating that this region contained the complete InsP3 binding domain (345). Further deletion mutagenesis confirmed that even small NH2-terminal deletions abolished InsP3 binding to the ligand-binding region. However, binding was restored when more substantial deletions were made, with a mutant construct with the first 225 residues deleted having 10- to 100-fold higher affinity than the full-length construct (538). Thus the region encompassing residues 226–576 was sufficient for InsP3 binding, forming an InsP3 binding “core,” whereas the region containing residues 1–225 was referred to as the “suppressor” domain (538) (Fig. 2B). Within the core domain, site-directed mutagenesis identified 10 conserved arginine and lysine residues distributed throughout the domain as playing important roles in InsP3 binding, with residues Arg-265, Lys-508, and Arg-511 critically important (538).
Crystal structures of both the core (52) and suppressor (54) domains of the mouse type 1 InsP3R have been solved (Fig. 4A). In the 2.2-Å resolution structure of the core domain in complex with InsP3, two distinct domains are present at right angles to each other in an elongated L-shaped structure. The region from 225–436 constitutes a β-sheet-rich β-trefoil domain, whereas the region from 436–600 is α-helical, comprised of two partial and one complete armadillo repeats. InsP3 is present in the structure at the interface of the domains, in a deep cleft with important binding determinants contributed by both. The cleft is lined with basic residues that anchor InsP3 to the protein. The phosphates in the 1 (P1) and 5 (P5) positions of InsP3 interact primarily with residues from the α-helical domain, whereas the phosphate at the 4 position (P4) interacts with the β-trefoil domain (Fig. 4A). The most extensive interactions involve P4 and P5 through hydrogen bonding primarily with several basic residues, although nonbasic residues as well as water are also involved. P1 interacts with only two basic residues.
FIG. 4.
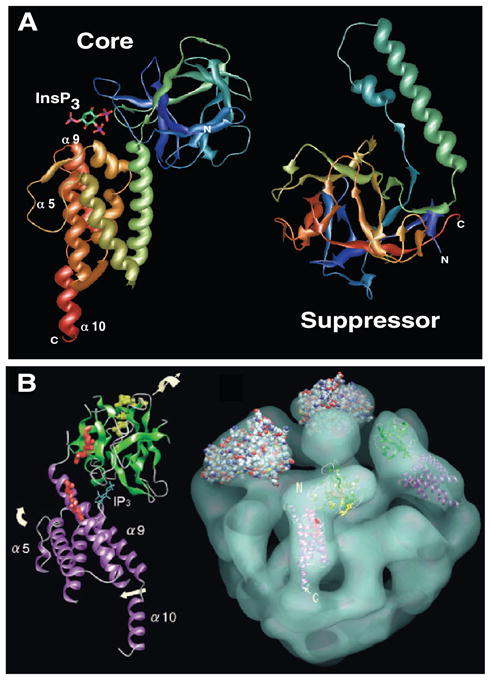
Structures of the InsP3R. A: crystal structures of the core InsP3 binding domain (left) and suppressor domain (right). InsP3 present in the core domain structure coordinated in a cleft created by an NH2-terminal β-sheet-rich β-trefoil domain and an α-helical armadillo-repeat domain. Suppressor domain is comprised entirely of a β-trefoil domain (head) with a helical insert (arm). Structures solved in Refs. 52, 54. B: cryoelectron microscopic single-particle reconstruction of InsP3R (right, tilted with respect to the plane of the page with cytoplasmic aspect facing upwards toward viewer with InsP3 binding core domain density fitted into an L-shaped density). For a better fit, various parts of the InsP3 binding core domain were rotated as indicated by the arrows with respect to the crystal structure shown in A. N and C refer to NH2 and COOH termini of each domain in A and B. [From Sato et al. (407), with permission from Elsevier.]
Adenophostin A (AdA), a fungal glyconucleotide metabolite (448), and its analogs (23, 290, 420) are potent agonists of the InsP3R. Although their molecular structures are significantly different from those of InsP3 and its analogs (198), they activate the channel by interactions with the InsP3 binding site (157). Molecular docking of AdA into the core domain crystal structure was consistent with experimental structure-activity relationships and provided some possible clues to the mechanisms involved in the high affinity of AdA for the InsP3R (397).
NMR studies of the core domain revealed well-resolved peaks when the core domain protein was complexed with InsP3, whereas many broadened peaks in the spectrum appeared in the absence of InsP3 (53). It was suggested that a dynamic equilibrium might exist in the ligand-free domain as a result of motions around the hinge region that connects the two subdomains. InsP3 binding to this region stabilizes the conformational relationship of the two domains with each other, consistent with earlier studies that indicated that InsP3 binding to an NH2-terminal 1,800-residue fragment of the receptor caused a conformational change (312). However, these studies of the isolated core domain may not reflect the behavior of this region within the context of the whole channel, where interactions with other parts of the protein, for example, the suppressor domain, or other structural features may constrain the mobility of this region. Nevertheless, InsP3 binding undoubtedly stabilizes the observed structure of the two domains. By analogy with the mechanism of glutamate binding to its bidentate binding pocket in the glutamate receptor (269), it has been speculated that InsP3 might bind primarily to either the β-trefoil or armadillo-repeat domain first, and then recruit and stabilize the other domain in the structure observed in the crystal (459).
The suppressor domain, encompassing residues 2–223 of the mouse type 1 InsP3R, was resolved at 1.8 Å (54) (Fig. 4A). The structure is comprised of a typical β-trefoil domain, referred to as the “head” subdomain, that contains an unusual helix-loop-helix insert that protrudes away from the structure, referred to as the “arm” subdomain, with the overall appearance reminiscent of a hammerlike structure. Thus the complete ligand-binding region (1–586) contains a proximal pair of β-trefoil domains and a distal armadillo repeat region. Whereas the sequence similarity between the two β-trefoil domains is low, their structures superimpose well, excluding the helix-loop-helix insert in the first domain, and a long loop in the second domain that contains the SI splice site (54). Before the structure of the 2–223 suppressor domain fragment was solved, it had been noted (375) that this region has repeats that are recapitulated in what is now recognized as the (second) 225–436 β-trefoil domain, so the discovery of the suppressor domain as a β-trefoil domain was somewhat anticipated. It had been similarly noted that the NH2 terminus of the RyR also contains the same repeats (375). Molecular modeling is consistent with the presence of tandem β-trefoil domains similarly present in the RyR (54). The ug3 mutation (109, 216) in the single Drosophila InsP3R gene (3, 490), a missense mutation that changes a serine to phenylalanine at position 217 (Fig. 2B) near the COOH terminus of the suppressor domain, enhances the sensitivity of activation, but not the binding affinity, of the reconstituted Drosophila InsP3R to InsP3 (434), suggesting that the suppressor region may allosterically couple InsP3 binding to gating activation. Interestingly, mutations within these domains in the RyR cause central core disease and malignant hyperthermia (54). Some of the residues are predicted to contribute to the β-trefoil fold, so their mutation might be expected to have structural implications for the entire domain. Others, however, were predicted to be located in surface-exposed loops, suggesting that they are importantly involved in channel function. It is quite interesting that such striking structural homology between the two families of Ca2+ release channels, if confirmed, should be present in this region of the channels, since the InsP3 binding function in the InsP3R is a main feature that distinguishes InsP3R from RyR.
Although deletion of the suppressor domain enhances InsP3 affinity of the core domain, Ca2+ release activity of the channel could not be elicited by InsP3, suggesting that the suppressor domain is required for normal channel activation (486). It has been proposed that the suppressor domain may therefore couple InsP3 binding in the core domain to other regions of the channel that impinge on the gating mechanisms (486). A critical next step is to resolve details regarding the structures of the three domains together, both in the presence and absence of InsP3 and Ca2+. The mechanisms by which the suppressor domain modulates the affinity of the channel for InsP3 are not elucidated by these structures. A logical hypothesis is that the suppressor domain interacts directly with the core binding domain. Deletion of the unusual helix-loop-helix arm subdomain was without major effect on the ability of a recombinant NH2-terminal 604 residue ligand-binding domain to bind to InsP3, suggesting that it was not critical for the suppressor function of the suppressor domain (54). It was noted that one surface of the suppressor domain contains several conserved residues (54). Mutagenesis of particular residues located within the surface enhanced the InsP3 affinity of the recombinant binding domain (54), consistent with the notion that this region of the head might participate in a protein interaction with another region that modulates InsP3 binding affinity of the core domain. Genetic studies have shown that the single C. elegans InsP3R gene, itr-1 (22, 88), is important to the ultradian rhythm underlying defecation (97). One of two InsP3R alleles identified that disrupt the defecation cycle, n2559, characterized as a loss-of-function mutation because the defecation cycle was eliminated, was mapped to residue 103 as a missense alteration of Gly to Glu (G103E), corresponding to Gly-25 (Fig. 2B). This residue is located immediately adjacent to the residues identified (54) whose mutations enhance InsP3 binding. This result suggests that whereas this region might participate in regulating the InsP3 binding properties of the core domain, the suppressor domain is also required for the channel to function, consistent with the loss of channel activation by InsP3 binding when the entire domain is deleted (486).
Inspection of the two crystal structures could not identify a potential binding interface within the core domain that might constitute the interaction region with the suppressor domain (54). However, other regions of the InsP3R molecule have also been proposed to interact with the ligand-binding region. First, a direct association between the NH2-terminal 340 residues and the COOH terminus has been observed (40, 214). The 340-residue NH2-terminal construct contains the suppressor domain and part of the second β-trefoil domain. Because truncation of the construct in the middle of the β-trefoil domain likely severely disrupts its structure, the suppressor domain probably mediates the interaction with the COOH terminus. Thus the conserved patch observed in the crystal structure of the suppressor domain (54) could possibly be involved in interactions with the COOH terminus. Recently, the NH2-terminal interacting region in the COOH terminus of the channel was localized to the cytoplasmic linker that connects transmembrane helices 4 and 5 (S4-S5 linker) (411). The possible implications of this interaction for activation gating of the channel are discussed in section IIIB2B.
2. The transmembrane region
A) THE PORE
A six transmembrane topology of the InsP3R was established by immunocytochemical techniques and N-linked glycosylation analyses of full-length and truncated proteins (153, 309). These studies, together with analogy modeling of InsP3R and RyR with well-characterized cation channels, suggested that COOH-terminal transmembrane helices are involved in ion permeation, with helices 5 and 6 and intervening sequences in InsP3R critical for creating the basic pore structure (309, 459, 514) (Fig. 3). Deletion of the first four transmembrane helices from InsP3R, leaving transmembrane helices 5 and 6, resulted in a channel with normal conductance and selectivity properties (383), consistent with this model. Site-directed mutagenesis of two residues between TM5 and 6, and believed to be located in the putative selectivity filter (43), also suggested that such a model provides a rational basis for considering the roles of particular residues that contribute to conductance and selectivity properties of the InsP3R permeation pathway. Furthermore, homology of RyR and InsP3R sequences in the putative pore region suggests that insights from studies of the RyR can provide insights into important molecular determinants in InsP3R.
The bacterial K+ channel KcsA has been used as a template to successfully model the pore region of the InsP3R and RyR (414, 507). Based on homology, predicted secondary structure, surface area, hydrophobicity, and electrostatic potential, the assembled tetrameric TM5/6 region of RyR2 adopted an equivalent structure to that of KcsA (507). The validity of the model was demonstrated by its ability to quantitatively predict in molecular dynamics simulations empirical permeation results for RyR2. Recent electron microscopic structures of the RyR resolved at 13.6 Å (405) and ~10 Å (261) provide details regarding the membrane domain, including the pore. In these studies, some helices were resolved that could be well fitted with the pore helices from crystal structures of bacterial K+ channels. These studies reinforce the hypotheses based on homology modeling that the pore of RyR and, by extrapolation because of their sequence, secondary structural and functional similarities, the InsP3R as well, are constructed in a manner believed to be similar in many types of cation channels (266). Additional details regarding the functional and structural properties of the InsP3R pore are discussed in section V that focuses on ion permeation properties of the channel.
B) THE GATE
InsP3 binding to the NH2 terminus of the channel induces conformational changes that are transduced to the activation gate that then enables ion flow through the channel. The molecular identity of the gate is unknown, and the mechanisms that couple ligand binding to opening and closing of the gate are unknown as well. Structural and functional studies in other cation channels indicate that activation gating can reside at two locations. First, the inner helices associated with the pore cross each other near the cytoplasmic surface of the membrane, at a so-called bundle crossing (115). The bundle crossing appears to either provide too narrow a passage for ion translocation or it is lined with hydrophobic residues at the narrow point that act effectively as a barrier to ion flow (115, 243, 416). The structures of bacterial MthK and KvAP K+ channels (205, 206), and functional accessibility and structural studies in other channels (106, 369, 370), indicate that activation gating is associated with bending and rotation of the inner helix, with consequent widening of the pore access region, creating the inner vestibule. Helix bending is conferred by highly conserved glycine residues located above the helix bundle crossing (205, 206, 266), or by inner-helix proline residues in some cases (106, 244). Alternatively, the activation gate appears to be located at the selectivity filter in some ion channels, including inward-rectifier and small-conductance K+ channels and CNG channels (62, 142, 143, 257, 260, 550). Furthermore, the selectivity filter may undergo conformational changes during gating (36, 143). It has been speculated that distinct channel kinetic states in Kir channels reflect gating at the two different gates (36).
Analyses and modeling of single-channel gating kinetics of patch-clamped InsP3R indicate that besides the ligand (InsP3 and Ca2+)-regulated gating mechanism, the channel has a ligand-independent gating mechanism responsible for maximum channel Po being less than unity in saturating InsP3 and optimal cytoplasmic Ca2+ concentrations (285). By analogy, therefore, it is possible that the two activation gating kinetics observed in the InsP3R are localized to the inner helix (TM6) and selectivity filter as well.
By further analogy with results from experimental studies in other cation channels, some sequence features also suggest that TM6 might function as the ligand activation gate. First, examination of TM6 of InsP3R and RyR reveals a highly conserved glycine (Gly-2586) located approximately halfway down the helix (Fig. 2B). It is the only glycine (in InsP3R), and there is no proline in the TM6 helix, so this might be a gating hinge. Second, located five residues down from Gly-2586 is a threonine (Thr-2591) in InsP3R and alanine in RyR. It has been noted that alanine is often five residues down from the gating hinge, and it was speculated that its small side chain is less likely to interfere with ion conduction (205). The threonine in the InsP3R does not fit the model, but it is interesting to note that a mutation in the RyR that changes the alanine to threonine, to conform to the InsP3R sequence, causes central core disease (263). It is tempting to speculate that this residue indeed plays a role in regulating ion access to the selectivity filter, with the larger conductance in RyR compared with InsP3R due in part to the presence of alanine instead of threonine, and that restricted access associated with the mutation to threonine in RyR is the basis for central core disease.
But where is the gate? In the crystal structure of the closed KirBac1.1 channel, the side chains of phenylalanine (Phe-146) located four to five residues before the end of the inner helix blocked the conduction pathway (243). It was noted that residues with large hydrophobic aromatic or aliphatic side chains are favored in that position. It was therefore concluded that this residue constituted the activation gate. In the acetylcholine receptor, leucine side chains rotate into the center of the pore in the closed state (324). It is possible that InsP3R utilizes similarly localized hydrophobic residues to block the pore in the channel closed state and that conserved Phe-2592 or Leu-2595 constitute the ligand-dependent activation gate in InsP3R. Accordingly, activation gating by ligand binding might be caused by conformational changes that are transduced, ultimately, into a mechanical force on TM6 that pulls the helices laterally, separating these hydrophobic “plugs,” thereby opening the inner vestibule to ion conduction. However, experimental support for these speculations is currently lacking.
As discussed above, the suppressor domain-interacting region in the COOH terminus of the InsP3R was localized to the S4-S5 linker (411). In the crystal structure of the two-transmembrane helix KirBac1.1 K+ channel, the COOH and NH2 termini were coupled by interactions mediated in part by a short so-called slide helix located immediately before the outer helix (243). Similarly, in the structure of the six transmembrane Kv1.2 K+ channel, a short helix is present immediately preceding helix 5, the equivalent outer helix (259). In each case, the helices are amphipathic with their hydrophobic faces associated with the inner surface of the plasma membrane. For both, it was proposed that gating activation involves a lateral movement of the slide helix, resulting in displacement of the outer helix, enabling the inner helix to move out of the conduction pathway, allowing ion flow. The S4-S5 linker sequence is highly conserved among InsP3R as well as RyR. Secondary structural analysis suggests that it contains a conserved short helical region that is amphipathic in both channel types, as it is in KirBac1.1 and Kv1.2. This conserved primary, secondary, and tertiary structure that mirrors the structure and location of the slide helix in the Kir and Kv1.2 channels suggest that it is functionally important. Thus the gating of the InsP3R channel might possibly involve lateral movement of the S4-S5 linker, possibly through interactions with the NH2-terminal suppressor domain. Again, experimental support is lacking, and much more work will be required to understand the molecular details of gating in the InsP3R channel.
3. The coupling region
Between the InsP3 binding domain and the membrane region (586–2276) is a stretch of ~1,700 residues (Fig. 2A). Sequence analysis suggests that the region spanning residues 460–1500 is predominately α-helical with the region between residues 760 and 1740 possibly containing several armadillo repeats (52). It was suggested that this may provide a long arm with a length of ~200 Å and a diameter of 35 Å that may correspond to the rodlike arm observed in low-resolution electron microscopic structures of the InsP3R (discussed below) (52). Not surprisingly, gross deletions of residues within this region disrupt channel function, although deletion of residues 1692–1731, the SII splice region, left the channel functional (486). A naturally occurring deletion of residues 1732–1839 in the type 1 channel, immediately after the SII region, in the opt mouse (437) also leaves the channel functional (437, 478; unpublished results). InsP3-mediated Ca2+ release is still present in cerebellar Purkinje neurons from opt mice (437), and single-channel analyses of a reconstituted recombinant opt InsP3R-1 confirmed that the channel is functional, although it had an apparent diminished ATP sensitivity compared with wild-type channels (478). The opt deleted region of the InsP3R contains a putative ATP binding site (so-called ATPA site) that may account for the reduced ATP responsiveness (discussed in sect. VIF4). Interestingly, the phenotype of the opt mouse is similar to that of the type 1 InsP3R knock-out mouse. Both mice lack normal locomotor behaviors, display seizures at ~2 wk of life, and then die by 3–4 wk of age (297, 437). The mutant protein is expressed at lower levels than wild-type protein. Thus it is possible that either reduced protein expression and/or altered ATP sensitivity accounts for the severe phenotype observed in the opt mouse.
Clues to important functional regions of the coupling domain have been revealed by mutations in this region that have been identified in C. elegans and Drosophila. An InsP3R allele that disrupted the defecation cycle in C. elegans, sa73, is a reduction-of-function mutation that lengthens the defecation cycle time. It is also associated with reduced brood size and reduced gonadal sheath contractility (529). The mutation has been mapped to residue 1571 as a missense alteration of Cys to Tyr (C1571T), in the coupling domain near the middle of the linear amino acid sequence. This residue, equivalent to Cys-1430 (Fig. 2B), is conserved from human to C. elegans and in all isoforms. However, the role of this residue and the effects of the mutation on either the localization or single-channel properties of the InsP3R are unknown. Five other InsP3R mutant alleles were identified by suppression of sterility in let-23 mutants. These are, presumably, gain-of-function mutants. sy328 and sy327, corresponding to S900F and L945R, are equivalent to Thr-799 and Met-837 (Fig. 2B), respectively. The Ser-900 residue is conserved as either Thr or Ser across species; the Leu-945 residue is conserved as a hydrophobic residue across species. The effects of these mutations on the InsP3R are unknown. A putative loss of function InsP3R allele, wc703, was created by chemical mutagenesis of Drosophila (216). This allele corresponded to G2117E, equivalent to Gly-2045 (Fig. 2B), which is highly conserved across species and isoforms as well as in the RyR. Electrophysiology of reconstituted recombinant channels indicated that the bell-shaped Ca2+ dependence of channel activity was narrower compared with the wild-type channel (434).
4. The COOH-terminal tail
The COOH-terminal tail of the InsP3R extends from the end of TM6 to the COOH terminus, encompassing ~150 residues (Fig. 2). Secondary structural analysis suggests the presence of an extended α-helix from TM6 is followed by three additional helical regions. Up to the last helical region, there is sequence and predicted secondary-structure homology with the COOH terminus of the RyR. The final α-helical region, while conserved within the InsP3R family, is absent in RyR. Sequence conservation among the different InsP3R isoforms becomes more divergent towards the COOH terminus. An antibody directed against an epitope comprising the COOH-terminal 11 residues of the type 1 channel blocked InsP3-mediated Ca2+ release (337). On the other hand, deletion of these residues did not inhibit Ca2+ release (486). As discussed in section VIN, the COOH-terminal tail has been shown to interact with several proteins. These interactions have functional effects, most prominently to enhance the apparent sensitivity of the channel to InsP3. This suggests an allosteric influence of the COOH terminus on the mechanism that couples InsP3 binding to opening of the channel gate. Steric interference with this role of the COOH terminus may account for the inhibitory effects of the antibody despite binding to a sequence that is dispensable for normal channel function.
5. Regulatory Ca2+ binding sites
Ca2+ is a critical modulator of InsP3R channel function. The steady-state gating activity of the InsP3-liganded channel is regulated by Ca2+ with a biphasic Ca2+ concentration dependence (34, 42, 196, 282, 283). The InsP3R is, most fundamentally, a Ca2+-activated ion channel. As discussed in detail in section VIC1, the primary functional effect of InsP3 is to relieve Ca2+ inhibition of the channel, enabling Ca2+ activation sites to gate it (282). In essence, Ca2+ is the true channel ligand. Experimental results and insights that have emerged from patch-clamp studies of the InsP3R, together with molecular modeling, indicate that Ca2+ regulation of the channel is very complex, involving several distinct Ca2+ binding sites (discussed in sect. VI, B and C).
Where are these Ca2+ binding sites in the InsP3R structure and sequence? Here, there is very little information available. Eight glutathione S-transferase (GST)-fused denatured peptide fragments of the InsP3R located throughout the linear sequence were found to bind Ca2+ in gel overlays (421, 422), and although the biochemical detection of several sites is consistent with the presence of multiple Ca2+ binding sites inferred from kinetic studies of single-channel gating (above), the physiological implications of such data are unclear. Mutagenesis of a conserved glutamic acid residue in InsP3R (Glu-2100) affected [Ca2+]i signals (319) and shifted the apparent Ca2+ dependence of reconstituted channel Po by ~5-fold, from 0.2 to 1 μM (479). A peptide spanning residues Glu-1932-Arg-2270 bound Ca2+ with an apparent affinity of 160 nM as measured by tryptophan fluorescence, which was decreased to ~1 μM when Glu-2100 was mutated (479). Although it has been concluded that this residue and region are important for Ca2+ regulation (319, 479), there are significant caveats. First, it was not determined whether the observed effects on channel function were due to modification of one of the Ca2+ binding sites discussed above, or whether they were secondary effects caused by long-range allosteric mechanisms. Second, metal binding sites in proteins generally comprise several interacting residues that help to coordinate and stabilize the ion in a binding pocket. Ca2+ binding sites usually consist of six or seven coordinating oxygen atoms provided by side-chain carboxyls, main chain carbonyls, and water (528). However, additional residues that might interact with Glu-2100 to coordinate and bind Ca2+ have not been identified. With these caveats in mind, this region of the channel may play a role in Ca2+ sensing, but further experimentation is necessary to determine whether it is a Ca2+ binding site, and which of the functional sites it represents (see sect. VIK).
Another region of the receptor that has been considered to be involved in Ca2+ regulation of channel gating is the ligand-binding domain. Two surface acidic clusters were observed in the crystal structure of the InsP3-bound 225–604 fragment (52). Site 1 was contained completely within the β-trefoil domain, whereas site 2 was located across the two domains. Both sites had been shown previously to bind Ca2+ in gel overlay experiments (422). The residues that contribute to the acidic patches are highly conserved among isoforms. Site 1 consists of residues Glu-246, Glu-425, Asp-426, and Glu-428. Site 2 is composed of residues Glu-283, Glu-285, Asp-444, and Asp-448. It was noted that site 2, which spans the β-trefoil and armadillo-repeat domains, overlaps with a surface patch of particularly high homology among isoforms. It was speculated that this site might be involved in protein-protein interactions and that InsP3 binding might relieve a conformational constraint involving this interface that then enables Ca2+ to bind there and activate the channel (52). Nevertheless, mutagenesis studies failed to provide evidence for a role for residues in either site in Ca2+ activation, since none of the mutations affected the ability of the channel to function at low cytoplasmic Ca2+ concentrations in Ca2+ release assays (211). However, it should be noted that only single point mutations were examined in that study. The relatively low resolution of Ca2+ release assays for measuring detailed channel properties may require multiple residues in a Ca2+ binding motif to be mutated to observe functional consequences. Single-channel studies of these mutant channels may reveal more subtle effects on the Ca2+ dependence of gating.
C. Structural Properties of the Tetrameric InsP3R Channel
1. The tetrameric structure
InsP3R channels are tetramers of InsP3R molecules (Fig. 3). Electron microscopy of purified InsP3R revealed them to be 20- to 25-nm pinwheel- (80) or square-shaped (271) particles. The pinwheel structure was more commonly observed when the purified particles were incubated in the presence of 1 mM Ca2+, whereas the square form was common when the particles were incubated in 0 Ca2+ (169, 170). Native InsP3R in cerebellar neurons were square-shaped with ~12-nm sides (228). More recent single-particle three-dimensional electron microscopic analyses suggest a square structure at the widest region with dimensions ranging from 17 to 25 nm (96, 170, 204, 407, 412) (discussed in sect. IIIC4). The observed fourfold symmetries, together with biochemical techniques discussed earlier, indicate that the InsP3R channel is a tetramer.
The structural requirements for tetramerization reside primarily in the COOH region of the protein. A receptor lacking the transmembrane domain but containing the cytoplasmic COOH terminus was monomeric (312). Expression of a truncated InsP3R that contained only the transmembrane domain region and the cytoplasmic COOH terminus formed tetramers (409). In vitro translation studies indicated that the region between TM5 and the COOH terminus is required for homoligomerization (209). In support, formation of a functional ion channel was observed from a construct that lacked the first four transmembrane helices (383). A detailed examination revealed that channels having a contiguous pair of transmembrane helices could form tetramers, but those that contained transmembrane helices 5 and 6 formed tetramers most efficiently (153). The presence of the cytoplasmic COOH terminus enhanced tetramerization (153, 154). Truncations from the COOH terminus revealed that residues 2629–2654 (Fig. 2B) were important for this effect (154). Interestingly, this sequence mediated dimerization of InsP3R subunits, suggesting that together with the membrane-spanning region, particularly TM5 and TM6, the tetrameric channel may be formed as a dimer of dimers (154). In support, a construct that lacked the transmembrane helices and COOH terminus could not be cross-linked, whereas a construct that similarly lacked the transmembrane domain but contained the COOH terminus could be cross-linked as dimers (322). The tetramer may be further stabilized by more distal sequences, since the region encompassing residues 2694–2721, the last conserved region before the divergent more distal COOH residues, forms tetramers in vitro (411). Of note however, whereas the COOH-terminal regions of the InsP3R and RyR have strong homology and predicted secondary structure, this last predicted helical region is absent in RyR. Taken together, these results suggest that important oligomerization determinants reside primarily in the pore-forming domain with contributions from more distal cytoplasmic sequences.
2. ER localization determinants
The sequences that specify ER localization of the tetrameric channel also reside in the membrane-spanning domain. A full-length protein truncated immediately before the last transmembrane helix targeted to the ER (409, 449). Any pair of contiguous TM helices by themselves were sufficient to target and retain the expressed proteins in ER membranes (357). The InsP3R appears to possess redundant signals that ensure a primarily ER localization.
3. Structural insights from limited proteolysis
The spatial arrangements of regions of the InsP3R in the quaternary structure of the tetrameric channel have been explored by proteolysis. Limited trypsin digestion of mouse cerebellar membrane fractions revealed five major trypsin-resistant fragments that accounted for the entire sequence (537). Fragment 1 extended from the NH2 terminus to residue 346, near the SI splice site; fragment 2 extended from 347 to 922, near the SII splice site; fragment 3 extended from 923 to 1582; fragment 4 extended from 1583 to 1932; and fragment 5 extended from 1933 to the COOH terminus (Fig. 2B). In addition, it was noted that the most distal portion of the COOH terminus was also susceptible to trypsin cleavage. It was concluded that each monomer in the channel had four exposed or disordered regions that were susceptible to trypsin cleavage, with five well-folded structural elements. In retrospect, this conclusion is not entirely accurate, since the first trypsin site at residue 346 occurs in the middle of a well-defined β-trefoil domain, such that the first and second fragments each contain a portion of a domain in addition to any full-domain structures. Fragment 5 contained the membrane-spanning domain as well as the COOH terminus. Interestingly, after limited trypsin proteolysis, fragments 1–4 remained associated with fragment 5 by noncovalent direct or indirect interactions (537). The trypsinized channel retained the ability to respond to InsP3 by InsP3-mediated Ca2+ release from microsomes (537). The function of these interactions is presumably not to mediate oligomerization, which is mediated by the transmembrane domain (above). Interactions both within and between monomers in the tetramer is expected, since it is likely that modulation of these interactions accounts for the allosteric effects on channel gating observed in response to mediators (Ca2+, ATP) and protein interactions (see sect. VI).
InsP3 did not affect the proteolytic pattern observed in Reference 214, nor did it perturb the association of the trypsin fragments in Reference 537. However, these results argue neither for nor against the possibility that InsP3 binding causes conformational changes in the protein. In contrast, lysyl endopeptidase proteolytic fragments of purified cerebellar InsP3R were different in the presence versus the absence of Ca2+(170). The apparent Ca2+ sensitivity of the generation of a 38-kDa fragment was between 10 and 100 nM. Electron microscopic analysis of negatively stained InsP3R revealed a Ca2+ dependence of the prevalence of a windmill form with similar Ca2+ sensitivity (170), suggesting that high-affinity Ca2+ binding to the InsP3-unliganded channel can induce conformational alterations that modify protease sensitivity.
Caspase 3 is a protease that becomes activated during programmed cell death. The type 1 InsP3R contains a consensus caspase cleavage site in the middle of the coupling domain (Fig. 2B) that is a physiological target for caspase 3-mediated cleavage (189). Caspase 3-mediated cleavage was associated with loss of InsP3-mediated Ca2+ release from isolated microsomes (189). It was subsequently demonstrated that expression of an InsP3R lacking the region NH2-terminal to the caspase 3 cleavage site was associated with depletion of ER Ca2+ stores (341). Although expression of the same construct did not deplete ER Ca2+ stores in another study (14), it was concluded in both studies that the caspase 3-cleaved channel in vivo may become “leaky” and contribute to elevated [Ca2+]i during apoptosis. However, in light of the ability of the channel to remain “intact” after limited trypsin proteolysis, it is unclear if the caspase-proteolyzed full-length channel has InsP3-independent or otherwise altered functions. Despite speculations to the contrary (447), at this time there is no evidence that the InsP3R becomes an unregulated Ca2+ leak channel under any physiological circumstance.
4. Three-dimensional structures
Five three-dimensional structures of purified InsP3R have been resolved by electron microscopic single-particle analyses, an approach that has been used to resolve structures of RyR Ca2+ release channels at up to 10- Å resolution. Four of the structures had purported resolutions of 24–34 Å (96, 170, 204, 412), whereas the most recent study reported a resolution of 15–20 Å (407) (Fig. 4B). The five structures share some basic similarities but differ considerably in the details and resolution. All five structures reveal two large domains, interpreted to be the membrane-spanning region and the large cytoplasmic domain, although the dimensions differ in the different structures. In all studies, the channels were completely unliganded, since InsP3 was absent and Ca2+ was removed at all stages of purification. In one study, however, the channel was exposed as well to a solution that contained Ca2+ (170).
Before the structures are discussed, it should be noted that we lack information regarding to which channel conformations, as measured by functional techniques such as single-channel electrophysiology, any of these structures correspond. Channels purified and resolved in the complete absence of Ca2+ and InsP3 may adopt structures that do not correspond to those of the channel in physiologically relevant conditions. For example, electrophysiological studies of Xenopus type 1 and rat type 3 InsP3R channels revealed that the presence of unphysiologically low Ca2+ concentrations (<10 nM) in the absence of InsP3 caused the channels to have a finite probability of opening (spontaneous openings), whereas channels incubated in more physiological Ca2+ concentrations (25–50 nM) did not exhibit spontaneous openings (285). Channels exposed for a few minutes to low Ca2+ concentrations (<10 nM) in the absence of InsP3 lose high-[Ca2+] inhibition of steady-state gating (286). Channels exposed to Ca2+ in the absence of InsP3 may adopt structures that correspond to activated or inhibited or inactivated or sequestered conformations (196). The diversity of the structures revealed in the published single-particle electron microscopic analyses may be related to not only technical issues having to do with channel purification and handling, image processing, and resolution, as discussed in Reference 407, but also to the presence of distinct, and possibly unphysiological, channel conformations. The diversity of the published InsP3R three-dimensional structures limits the insights and conclusions that can be comfortably drawn from them. Without higher resolutions, more consistent results, and observations of changes in structure related to understood modifications, for example, binding of a protein to a known sequence in the channel, as done for the RyR (495), the diverse structures can be open to a corresponding diversity of interpretation.
With these caveats in mind, we will briefly review the results of these early efforts. As noted above, one study revealed that the presence of Ca2+ led to a predominance of a pinwheel structure, whereas a square form was common when the particles were incubated in the absence of Ca2+ (169, 170). Two other structures also revealed prominent pinwheel-like aspects, despite the absence of Ca2+ in the protein preparation. In one study of purified InsP3R from mouse cerebellum with purported 24-Å resolution, the cytoplasmic domain resembled a bulb with four small arms that protruded laterally by ~50 Å (204). A 30-Å structure of the InsP3R purified from bovine cerebellum exhibited a fourfold symmetrical pinwheel of radial arms projecting from a central square mass (412). Although all three structures reveal pinwheel-like features, there are pronounced differences in the details of the size and structures and arrangements within the pinwheel regions among them. The best fit in a computerized docking of the crystal structure of the core InsP3-binding domain into the structure of Reference 412 placed it in the pinwheel structure. This assignment is consistent with the observation that heparin-gold binds near the tips of the windmill structure of purified InsP3R observed in the presence of Ca2+ (169). However, another structure assigned the InsP3-binding region to a more central location (96). This 30-Å resolution structure of purified rat cerebellar InsP3R had an overall more square aspect, with a suggested flower-like appearance, with a central hourglass-shaped mass with square ends, referred to as the stigma, surrounded by four lobes, referred to as the petals, at each corner (96). The lobes (petals) are somewhat reminiscent of the pinwheel arms in the structure of Reference 412. Nevertheless, the InsP3 binding region was localized to the central stigma region, based on previous studies in which binding by dimers of InsP3 molecules suggested that the InsP3 binding sites are separated by no more than 20 Å (389). Assigning the InsP3 binding region to the petals would be inconsistent with these data. Nevertheless, similar electron microscopic structural studies of RyR also provide, albeit even less direct, support for the idea that the InsP3 binding domain is likely to be more peripherally located. As discussed earlier, the sequence homology of the InsP3R and RyR in their NH2-terminal regions, and molecular modeling, have revealed that the NH2-terminal regions of the two channels have strong structural homology (53). The NH2-terminal region of the RyR has been localized to the so-called clamp domains, which are located at the corners of the square channel structure (495). In addition, the best fit of the core InsP3 binding domain into the 14-Å resolved RyR structure assigned it to a peripheral region associated with the clamp domain (413).
The most recent InsP3R structure, resolved at 15–20 Å, reveals considerably more detail than the previous structures and is dissimilar from them (407) (Fig. 4B). The overall shape is that of a hot air balloon, with the cytoplasmic domain forming the balloon, and the membrane domain forming the hanging basket. An outer shell of densities with many cavities forms the balloon shape and surrounds an inner continuous square-shaped tubular density. The crystal structure of the core InsP3 binding-domain was fitted into each of four L-shaped densities at the top of the balloon located most distal to the plane of the membrane, with the hinge that forms the InsP3 binding cleft located at the elbow of the L (Fig. 4B). Fitting required reorientation of the β-trefoil domain with respect to the armadillo-repeat domain, as well as some reorientations of α-helices in the latter. As a result, acidic residues that had been assigned previously to two putative Ca2+ binding regions (Ca2+-I and Ca2+-II; see sect. IIIB1) that were not proximal to each other in the crystal structure, became more closely associated in the reoriented hypothetical structure. It was speculated that Ca2+ binding to this region, that we refer to here as Ca2+-III, may stabilize a conformation of the channel that has low InsP3 affinity, and that InsP3 binding involves a conformational change in the β-trefoil domain that involves its rotation relative to the armadillo-repeat domain (407). Such an InsP3R-induced reorientation would disrupt Ca2+-III, inhibiting Ca2+ binding there. Interestingly, this hypothesis is consistent with the proposed mechanism of InsP3 activation gating based on single-channel studies. As discussed in section VIC1, nuclear patch-clamp studies have indicated that InsP3 activates channel gating by apparent relief of Ca2+ inhibition. Molecular modeling indicated that this was caused by InsP3-mediated transformation of a high-affinity inhibitory Ca2+ binding site into a lower-affinity Ca2+ activation site (285). The structural model proposed by Sato et al. (407) may therefore suggest that the high-affinity inhibitory Ca2+ binding site is Ca2+-III, whereas the lower affinity activating site would be either Ca2+-I or Ca2+-II that become fully formed in the InsP3-liganded state. This hypothesis will need to be tested by examining the properties of appropriately mutated channels.
IV. ELECTROPHYSIOLOGICAL STUDIES OF INOSITOL TRISPHOSPHATE RECEPTOR CHANNELS
The amount of Ca2+ that flows through an open InsP3R channel depends on the rate of Ca2+ flow through the channel when it is open (the Ca2+ current) and how much time the channel spends in the open, conducting state. The magnitude of the current through the open channel is a function of the electrochemical driving force for Ca2+, the properties of the channel pore, and the presence of ions and other molecules that interfere with Ca2+ flux through the pore, either by competition for binding sites in the pore (permeant ion block) or by interactions with the channel in other parts of the permeation pathway (e.g., blockers). The amount of time that the channel spends in the conducting state, the Po, is the target of most physiological regulation by agonists and antagonists of channel activity. Analysis of the steady-state gating of the channel can determine the details of the patterns of opening, i.e., stationary, modal, bursting, as well as the dwell times of particular channel conformations, thereby providing insights into how channel modulators affect Po, for example, by stabilizing or destabilizing either open or closed channel states. Thus electrophysiological studies of single InsP3R offer unique detailed molecular insights into the behavior of the channel.
Electrophysiological studies of the detailed permeation and gating properties of single InsP3R ion channels have been studied with two distinct approaches, described below. It is important to note that, whereas the goal in such studies is to understand at a molecular level the properties of the InsP3R channel, in both types of studies the channel is studied outside of its normal cellular context in solutions that are simple compared with the cytoplasm. Normal cytoplasm contains 100–140 mM K+, 10 mM Na+, 5–70 mM Cl−, 1 mM free Mg2+, 5–10 mM MgATP, 50–100 nM free Ca2+, and a crowded mixture of proteins and other cellular constituents, including proteins that physically interact with the channel. In all single-channel studies, it is necessary to ask whether observed behaviors reflect those that occur in the cell.
Because intracellular membranes in situ are not accessible to patch pipettes (although see Ref. 208), single intracellular membrane localized ion channels have been studied traditionally following their isolation (enrichment or purification) and reconstitution into artificial lipid planar bilayer membranes. The first electrophysiological recordings of single InsP3R channels were made using this approach (34, 124, 270, 505), and some laboratories continue to use this technique to study both endogenous as well as recombinant InsP3R channels (379, 470, 482). All RyR single-channel studies use this approach (139). As in all in vitro reconstitution systems, there is a concern that the isolation, purification, and reconstitution protocols may disrupt normal functional properties of the channels. Furthermore, the in vitro environment used for channel current recordings, including the bilayer lipid composition and composition of the bathing solutions, may alter the normal channel permeation or gating properties.
The second approach employs the patch-clamp technique and enables InsP3R channels to be recorded in their native ER membrane environment (Fig. 5). Since the ER is continuous with the outer membrane of the nuclear envelope (113), ER-localized ion channels such as the InsP3R are also expected to be present in the nuclear envelope outer membrane (Fig. 5). Single InsP3R channels can be recorded in their native membrane environment by patch clamping isolated nuclei (Fig. 5). This approach has successfully recorded single InsP3R channels using nuclei isolated from a variety of cells, including Xenopus oocytes (277), insect Sf9 cells (196, 508) (Fig. 5), and CHO, COS-7, DT40, HeLa, and primary rat parotid acinar cells (unpublished results), suggesting that nuclear patch-clamp electrophysiology is a general approach that can be applied to many cell types. It is surprising that formation of seals with giga-ohm resistances, a prerequisite for studying ion channel activity, can be achieved with very high frequency despite the high density of nuclear pores in the nuclear envelope (158). It must be assumed, without understanding the mechanisms involved, that the nuclear pores are all closed and impermeable in these nuclear membrane patch-clamp studies. Studies over the past several years (42, 43, 276, 278–286, 527, 542) have shown that InsP3R channels recorded in their native ER membranes behave far more robustly (higher Po, higher consistency among studies) and very differently (responses to ligands and kinetic features) from those observed in reconstitution studies. Nevertheless, there are important technical limitations associated with this approach, including the inability to readily exchange the solution bathing the cytoplasmic face of the membrane patch, the presence of unknown luminal factors unless the patch is excised, and the fact that the protein is in a membrane of unknown composition with possible interactions with unknown factors, including other proteins. Furthermore, the consistency of results obtained using this technique may reflect the fact that the studies have largely come from one laboratory (ours).
FIG. 5.
Nuclear patch-clamp electrophysiology. A: schematic of cell nucleus, illustrating that the outer membrane of the double-membrane nuclear envelope is continuous with the endoplasmic reticulum (ER), with the lumen between the two membranes continuous with the ER lumen. Patch-clamping isolated Xenopus oocyte nucleus (B) and insect Sf9 cell nucleus (C) visualized on the stage of a patch-clamp microscope, with patch pipettes forming giga-ohm seals on the outer nuclear membrane. Horizontal shadow over the Xenopus nucleus is the edge of a stabilizing piece of coverslip. Intact Sf9 cell is also present in C. [B modified from Mak et al. (287).]
Different types of recording solutions have been used in various studies of single InsP3R channels. In many bilayer experiments, 50 mM divalent cation (Ca2+ or Ba2+) is present on the luminal side of the channel, with the solutions on both sides containing Tris-HEPES as the only other major component. The absence of more physiologically relevant ions is necessary to prevent conduction through contaminating ion channels that complicate the recordings. The measured currents are therefore divalent cation currents. Because the function of the InsP3R is to conduct Ca2+, these recordings might possibly be considered physiological. However, this conclusion must be tempered by two considerations. First, the InsP3R is also highly permeable to monovalent cations and Mg2+. Because K+ is present at over 100 mM, and free Mg2+ is present at ~1 mM, their fluxes through the open InsP3R in vivo will be considerable. Ca2+ currents measured in their absence are therefore not physiological Ca2+ currents. Second, since the activities of InsP3R channel are sophisticatedly regulated by cytoplasmic Ca2+ concentration, one of the major advantages of single-channel studies of InsP3R channel activities (compared with Ca2+ concentration or flux measurements) is that the Ca2+ concentration on one (in on-nuclear patch-clamp experiments) or both (in excised nuclear patch-clamp and lipid bilayer experiments) sides of the InsP3R channel studied can be rigorously controlled in the experiments. However, to utilize this advantage, the concentrations of free Ca2+ in experimental solutions must be properly ascertained. Use of Ca2+ as the current carrier may cause the local Ca2+ concentration near the mouth of an open channel to change during the experimental recording, particularly when used at concentrations (50 mM) that are at least 50 times greater than those that exist physiologically.
Furthermore, in a significant fraction of published single-channel studies that used Ca2+ as the current carrier, or that altered Ca2+ concentrations on the cytoplasmic side of the bilayer during recordings, the free Ca2+ concentration in the experimental solutions was simply calculated from the total amount of Ca2+ and Ca2+ chelator present in the solutions, rather than directly measured. For this to be accurate, Ca2+ chelator(s) with the appropriate Ca2+ affinity must be used in suitable quantities to properly buffer the free Ca2+ concentrations in the solutions to the desired level. A high-affinity Ca2+ chelator like EGTA with Kd for Ca2+ of ~100 nM (477) cannot provide adequate buffering for solutions with free Ca2+ concentration >3 μM (163, 219, 438) because over 99.9% of the EGTA is bound to Ca2+ at that level and there is little buffering capacity left. Furthermore, because the Ca2+ affinities of chelators are affected by physical characteristics of the solution (ionic strength, temperature, pH), with some chelators having stronger dependence on such characteristics than others (29), addition of components, e.g., ATP, to the experimental solutions can substantially change the Ca2+ affinity of the chelator (219). Such effects must be carefully taken into consideration for the calculations to give good estimates of the actual free Ca2+ concentrations in the experimental solutions (29). Finally, even if the effects of all measurable physical characteristics (temperature, pH, ionic strength) are properly factored into the calculations, it is not easy to ascertain the purity of the chelator and its efficacy to chelate Ca2+. Thus it is more reliable to determine the free Ca2+ concentrations in experimental solutions directly using either Ca2+-selective electrodes (42, 279, 280, 283, 286, 368, 380–383) (method described in Ref. 29) or fluorimetry (196) (method described in Ref. 220).
In some planar bilayer experiments, cesium methanesulfonate solutions have been used on both sides of the bilayer, with Ca2+ on either side buffered to ~250 nM, measured with electrodes (368, 379–383). Here, the rationale is that methanesulfonate is impermeant through anion channels and that Cs+ blocks contaminating cation K+ channels but is nevertheless permeant through the InsP3R. Similarly, nuclear patch-clamp studies have employed potassium chloride solutions (with Ca2+ concentration measured by either electrodes or fluorimetry) because contaminating K+ or Cl− conductances are observed only infrequently in these studies. Use of the monovalent cations (K+, Cs+) as current carriers in the absence of normal luminal 500 μM Ca2+ on the luminal side is not physiological, but it ensures that Ca2+ concentration near the mouth of the channel does not change when the channel opens. Thus Ca2+ regulation of channel gating can be studied under rigorously controlled conditions.
V. PERMEATION PROPERTIES OF THE INOSITOL TRISPHOSPHATE RECEPTOR
A. Overview
The InsP3R is itself a ligand-gated ion channel, as demonstrated first by measurements of tracer Ca2+ fluxes following reconstitution of purified cerebellar type 1 receptors into liposomes (136), and subsequently by electrical recordings following reconstitution into planar bi-layer membranes (270, 302). The amount and kinetics of Ca2+ flux through an InsP3R depends on both its permeation properties and its gating kinetics. The detailed permeation properties of the InsP3R have been directly examined by reconstitution of channels into planar bilayer membranes and by nuclear envelope patch-clamp electrophysiology. The methodology involved in these techniques was described in the previous section.
B. Monovalent Cation Conductance Properties
Monovalent cation conductance properties have been examined for endogenous as well as recombinant InsP3Rs. In patch-clamp studies of InsP3R channels recorded in the outer membrane of the nuclear envelope of isolated Xenopus oocytes, with the channel exposed to symmetric 140 mM K+ solutions in the absence of Mg2+, the current-voltage (I–V) relationship is linear over a range of voltages (−40 to +60 mV) with a slope conductance of ~320–360 pS for both endogenous Xenopus type 1 (276) and expressed recombinant rat type 3 (284) channels. A similar K+ conductance (360–390 pS) was recorded for expressed recombinant rat InsP3R-1 present in COS-7 cell nuclei (42) and for an endogenous rat cerebellar Purkinje cell InsP3R recorded in the inner nuclear membrane (294).
Rat cardiac and recombinant InsP3R-2 reconstituted into planar bilayer membranes and recorded in symmetric 220 mM Cs+ had a slope conductance of 275 pS (368, 380, 382). The smaller conductance recorded for the type 2 channel may reflect a true difference in its conduction properties compared with the Xenopus and rat types 1 and 3 channels, or be due to the different membrane environment in planar bilayers and nuclear envelopes. More likely, it is caused by the use of Cs+ rather than K+ as the permeant ion. Although the relative conductances of Cs+ and K+ in the InsP3R have not been determined, Cs+ currents through RyR channels are only 60% as large as those carried by K+ (255). Given the strong structural and functional similarities of the pore properties of InsP3R and RyR (discussed earlier and in additional detail below), it is possible that Cs+ currents are smaller in InsP3R channels as well, and that the type 2 channel K+ conductance is therefore similar to that of the other isoforms. Indeed, parallel measurements of the bovine cerebellar type 1 InsP3R in the same Cs+ bathing solutions yielded a single-channel conductance of 280 pS, the same as the type 2 channel (382).
Endogenous insect Sf9 InsP3R recorded in isolated cell nuclei in symmetric 140 mM K+ had a slope conductance of ~480 pS (196). This larger conductance may be due to the membrane environment of the Sf9 cell, or to molecular differences between the insect and mammalian InsP3Rs. Although the amino acid sequence of the Sf9-InsP3R is not known, all known invertebrate InsP3R pore selectivity filter sequences contain a GGIGD motif (similar to the RyR), whereas the vertebrate InsP3R sequence is GGVGD. By site-directed mutagenesis of rat InsP3R-1, it was demonstrated that this sequence is involved in ion conductance and selectivity (43). A mutant mammalian InsP3R channel with the pore Val replaced with Ile to resemble the invertebrate InsP3R had a higher conductance (490 ± 13 pS) (43), close to that (~480 pS) of the Sf9-InsP3R. Thus other invertebrate InsP3R channels may also have higher single-channel conductance than their mammalian counterparts. On the other hand, recordings of reconstituted Drosophila InsP3R revealed that single-channel Ba2+ conductance was similar to that of the rat type 1 channel recorded similarly (434). Further studies of other invertebrate InsP3R channels will be required to determine if a systematic difference exists in the conductance properties between vertebrate and invertebrate channels.
C. Ion Permeability of the InsP3R
The relative ion permeabilities of the InsP3R in nuclear patch-clamp studies have been estimated by reversal potential measurements in symmetrical 140 mM K+ baths before and after addition of divalent cation (usually 10 mM) to the luminal face of the channel in excised patches. These studies, summarized in Table 1, indicate that the permeability properties of different InsP3R isoforms from different species are well conserved and that the InsP3R channel is a divalent-selective cation channel, with a selectivity for Ca2+ and Mg2+ over K+ of ~8 and 5, respectively, with relatively little selectivity among different divalent cations. The permeability sequence recorded in the InsP3R channel is similar to that of the RyR channel determined under comparable ionic conditions (Table 1). The permeability of a channel, as determined by reversal potential measurements, reflects the relative ease of an ion to enter the permeation pathway. Thus divalent cations enter the pore more readily than monovalent cations, suggesting that a divalent cation-binding site is associated with the permeation pathway. Because the energy of dehydration of Mg2+ is much higher than that of Ca2+ and Ba2+ (185), the relatively poor discrimination of this site between Ca2+ and Mg2+ suggests that ion dehydration does not play a major role in divalent cation permeation.
TABLE 1.
Permeability and conductance properties of the InsP3R
| InsP3R Channel | Permeability (P) Sequence |
Reference Nos. | ||||
|---|---|---|---|---|---|---|
| PCa: | PBa: | PMg: | PK: | PCl | ||
| Xenopus InsP3R-1* | 7.6: | 4.3: | 3.2: | 1: | 0.23 | 276, 277 |
| Sf9 InsP3R* | 10: | 6.8: | 1: | 0.2 | 196 | |
| Rat InsP3R-1† | 5.2: | 5.7: | 1 | 294 | ||
| Rat InsP3R-1† | 6.3: | 1 | 33 | |||
| Recombinant rat InsP3R-1‡ | 4.3: | 1: | 0.07 | 42 | ||
| Recombinant rat InsP3R-3‡ | 5.6: | 1: | 0.2 | 284 | ||
| Sheep RyR2 | 6.5: | 5.8: | 5.9: | 1 | 473 | |
| InsP3R Channel | Conductance (G) Sequence, pS |
Reference Nos. | ||||
|---|---|---|---|---|---|---|
| GBa: | GSr: | GCa: | GMg: | GMn | ||
| Rat InsP3R-1† | 85: | 77: | 53: | 42: | 17 | 33, 438 |
Endogenous channel in nuclear envelope.
Reconstituted from cerebellar microsomes; presumably the type 1 isoform.
Expressed recombinant channel recorded in nuclear envelope.
D. Divalent Cation Conductance
In contrast to permeability, the conductance of a channel reflects the ease of permeation through the channel. Divalent cation conductance was first examined for InsP3R from canine or mouse cerebellar microsomes reconstituted into planar bilayer membranes. In these studies, the trans side of the channel, equivalent to the ER luminal aspect, was exposed to 50–55 mM divalent cation and 250 mM HEPES-Tris, with the cis (cytoplasmic) side exposed to 250 mM HEPES-Tris with 200 nM free Ca2+. With 55 mM Ca2+ on the luminal side, the unitary Ca2+ conductance of the canine InsP3R ranged in different studies between 10 and 125 pS (33, 34, 124, 505). Ca2+ conductance of mouse cerebellar InsP3R recorded under similar conditions was 100 pS (310) or 26 pS (270), similar to the 32 pS recorded for InsP3R purified from bovine aortic smooth muscle (302).
The reasons for the wide range of values reported for reconstituted channels are unclear. Many subconductance states were observed in these studies, with the fully conducting state observed relatively rarely (505). Subconductances have also been observed for endogenous and recombinant InsP3R recorded by patch-clamp electrophysiology of nuclear envelopes (42, 196, 277, 278, 284), but they are very rare, accounting for <1% of opening transitions. It is possible, therefore, that the main state conductance was underestimated in some of the studies of reconstituted channels. The Ba2+ conductance of reconstituted channels recorded under similar conditions by the same laboratory (Bezprozvanny) for InsP3R from canine cerebellum (33) and recombinant rat type 1 InsP3R expressed in either HEK (229) or Sf9 (478, 482) cells were all within a narrower range of 80–95 pS. The Ca2+ conductance determined by this group, ~50 pS, would therefore appear to be the most reliable estimate available.
The divalent cation conductance selectivity sequence of the InsP3R (Table 1) corresponds to the mobility of these ions in free solution, suggesting that divalent cations may not be fully dehydrated in the pore. Accordingly, cross-sectional area of the selectivity filter of the channel pore is at least 10 Å 2 (229). The apparent large diameter of the pore is consistent with the extremely high single-channel divalent and monovalent cation conductances observed. In highly selective cation channels, high ion throughput is the result of ion-ion electrostatic repulsive interactions by multi-ion occupancy of the pore (115). The “anomalous mole fraction effect” was not observed with mixtures of Mg2+ and Ba2+ in reconstituted canine cerebellar InsP3R (229), suggesting, although not proving, that the open channel pore is occupied by only a single ion. Thus the high rates of permeation are likely mediated by mechanisms that are distinct from those used in highly selective monovalent and divalent cation channels (115, 182).
An increase in Mg2+ concentration decreased the K+ conductance of the endogenous Xenopus InsP3R-1 in nuclear patch-clamp studies (276). The K+ conductance around 0 mV was reduced from 320–360 pS in 0 Mg2+ to 115 pS in the presence of 1.5 mM Mg2+. Simultaneously, the I–V relation of the channel, which was linear in the absence of Mg2+, became symmetrically rectified in the presence of free Mg2+ on either or both sides of the channel, with the slope conductance increasing with high applied voltages. The effect of Mg2+ could not be accounted for by electrostatic screening by Mg2+ of surface charges around the channel pore or competitive block of the channel pore by impermeant Mg2+ present on one side of the channel (276). The effects of Mg2+ are reminiscent of the effects of divalent cations on the monovalent cation conductance of the RyR (471, 472). The RyR has a high Mg2+ permeability (473), similar to the InsP3R. There, it has been proposed that divalent cations experience low energy barriers to entry into the pore of the RyR, which enables them to move into the conduction pathway with relatively high permeabilities compared with monovalent cations (471, 472). However, since the divalent ions bind tightly in a potential well inside the channel pore, they therefore permeate through the channel pore more slowly than monovalent ions. Because the InsP3R and RyR are probably single-ion occupancy channels (229, 473), they are effectively nonconducting when occupied by a divalent cation. Thus the slow passage of divalent cations through the channel reduces the monovalent cation conductance as well. High applied voltages alleviate the block by increasing the rate at which divalent ions move through the channel, thus generating nonlinearity in the I–V relation. The conversion of the linear monovalent cation I–V relation in the absence of divalent cations to the rectified I–V relations in their presence (both Mg2+ and Ba2+) observed in both the InsP3R and RyR (276, 471, 472) strongly suggests that this model for ionic conduction is applicable to the InsP3R. These results indicate that the InsP3R has a pore which possesses lower energy barriers for divalent (Ca2+ and Mg2+) relative to monovalent (K+) cation entry, and therefore higher divalent cation permeabilities, and relatively stronger divalent cation binding sites, which cause divalent ion blocking of the channel (276). Mg2+ inhibited K+ conductance of the InsP3R with an apparent dissociation constant of 560 μM (276), indicating that the divalent binding site has high affinity that enables it to bind divalent cations at physiologically important concentrations.
The symmetrical nonlinear I–V relation of the IP3R in the presence of symmetric divalent ions (276) suggests that the energy profile experienced by divalent ions in the channel pore is symmetrical about a central axis (472). Rectification of the I–V relation occurs at both positive and negative applied voltages with Mg2+ present on only one side of the channel (276). Thus the polarity of the applied voltage does not affect significantly the movement of Mg2+ from either side of the channel into those binding sites that cause channel block and consequent rectification. This implies that a divalent ion binding site is located a short electrical distance from the mouth of the pore on each side of the channel. This feature is again reminiscent of the RyR conduction pathway, which has been modeled with divalent ion binding sites 10 and 90% of the way across the potential drop through the channel besides a central binding site (472).
E. Physiological Ca2+ Current Through the InsP3R
Because of the high concentrations of K+ and free Mg2+ in the cytoplasm, and the relatively high permeability of the InsP3R to Mg2+ and K+, an InsP3R channel in situ must be blocked to Ca2+ flow for a significant portion of its open time due to the occupation of the channel by Mg2+ and K+ that bind in the permeation pathway. This suggests that the magnitude of the Ca2+ current passing through single open InsP3R channels under physiological ionic conditions will be substantially lower than that measured in the absence of Mg2+ and K+. Unfortunately, measurements of Ca2+ currents through the InsP3R in the presence of physiologically relevant ionic conditions have not yet been performed. An accurate determination of the physiological Ca2+ current through an open InsP3R is important for interpreting local Ca2+ signaling events in cells, including blips and puffs, and estimating the number of open release channels that contribute to them. Measurements of Ba2+ currents in the presence of symmetrical 110 mM K+ and 2.5 mM luminal Ba2+ revealed a current amplitude of 3.4 pA (229). With the assumption of a similar Ca2+ affinity and concentration, the predicted Ca2+ current was estimated to be 0.5 pA (229). However, because the studies were performed in the absence of Mg2+, this value is certainly an overestimate of the Ca2+ current through an InsP3R in vivo. Nevertheless, it is possible to estimate the magnitude of the Ca2+ current through an open InsP3R channel under physiological ionic conditions by considering data from RyR channels. The magnitude of the Ca2+ current through RyR has been measured in the presence of symmetrical 150 mM K+ and 1 mM Mg2+ with 1 mM luminal free Ca2+, conditions close to the physiological situation. Under these conditions, the Ca2+ current was <0.5 pA (231). The relative magnitudes of the Ca2+ currents through InsP3R and RyR in the absence of K+ and Mg2+ can be estimated from References 229 and 231 to be ~2.85 (4.4 pA for RyR; 1.4 pA for InsP3R). With the use of this factor, the Ca2+ current through the InsP3R under physiological ionic conditions is predicted to be nearly threefold less than through RyR, or ~0.1–0.2 pA (30.5/2.85).
F. Molecular Models of the InsP3R Pore
The InsP3R and RyR are distinct among cation channels in having extremely large single-channel monovalent ion conductances: 320–500 pS for InsP3R (in 140 mM K+, 0 Mg2+) (42, 196, 277, 284, 368); up to 900 pS for RyR (in symmetrical 250 mM K+ with 0 Mg2+) (514). Both channels function physiologically as Ca2+ channels, yet they lack the exquisite Ca2+ selectivity of plasma membrane voltage-gated Ca2+ channels, discriminating relatively poorly among cations, with both channels exhibiting a selectivity ratio PCa:PK of ~6–8 (42, 277, 514). The InsP3R and RyR share significant homology in the COOH-terminal pore region, and the basic ion permeation and selectivity properties of InsP3R and RyR appear to be similar, although the mechanisms of permeation have been more extensively studied for RyR (reviewed in Ref. 514). A description of the RyR pore has been developed to account for the enormous conductance of the channel and pharmacological and electrophysiological analyses of a wide range of permeant and impermeant molecules (514). In this model, the RyR pore has a central binding site located 50% of the distance of the electrical field and accommodates only one ion in the permeation pathway. Thus high throughput of ions through the RyR/InsP3R pore does not rely on electrostatic repulsion among several ions simultaneously present there, as it does in many other cation and anion channels (115, 121, 266, 548). Consequently, it is likely that the pore is short with a large capture radius (514), the area through which a diffusing ion can enter the channel. Divalent cation permeation does not appear to require disruption of the inner hydration shell of the ions (514). Thus the channel does not need to replace shed waters with dipole groups in the pore wall, which is again distinct from the mechanism observed in K+ channels (115). The sequences for monovalent and divalent cation conductances suggest that permeant ions bind to a high field strength central site where the energetic difference between ion-site interaction and the energy of dehydration favors binding of ions with smaller crystal radii (472). The affinity of the central site is higher for divalent than for monovalent cations. The selectivity filter is believed to be localized close to the luminal end of the bilayer membrane (514). Its width has been estimated at ~7 Å (475), considerably wider than that observed for the KcsA K+ channel (~3 Å) (115). This large size likely contributes to the prodigious rates of ion translocation in RyR/InsP3R. It has been estimated, based on the voltage dependence of TEA penetration into the RyR pore, that the narrowest part of the selectivity filter is only 1 Å long (474). In addition, two additional binding sites are located on either side of the central site, ~10% and 90% of the distance through the electric field. A large capture radius might be achieved by maximizing the diameter of vestibules continuous with the bulk solutions that lead to the pore on either surface of the channel. In addition, the capture radius could be enhanced by negative charges near the entrances to the pore.
This latter strategy was shown to be part of the mechanism by which maxi-K (BK, slo) channels achieve high conductance (~250 pS). BK channels have one or two conserved acidic residues near the end of the pore inner helix that is absent in lower conductance K+ channels. Neutralization of these residues greatly reduced single-channel conductance, whereas introduction of the acidic residues into KcsA enhanced channel conductance into a range characteristic of BK channels (58, 346). The magnitude of the conductance was correlated with the amount of negative charge in the inner vestibule. Recent mutagenesis studies suggest that this mechanism may also play a role in contributing to the large K+ conductance of the RyR (504, 524). However, it remains to be determined whether similar mechanisms apply to the InsP3R.
Cyclic nucleotide-gated (CNG) channels are similar to InsP3R/RyR channels in having divalent permeability with considerable monovalent permeability, with little selectivity among monovalent alkali cations (see references in Ref. 125). Comparison of the pore regions of CNG channels and K+ channels reveals that two residues in the K+ channel selectivity filter are absent in CNG channels (GYGD in K+ channels, G - - D in CNG channels) (181). Substitution of the CNG channel selectivity sequence into Shaker K+ channels caused the K+ channel to lose K+ selectivity and acquire high-affinity permeant divalent cation block, both features of the CNG channel (181), indicating that the chimeric K+ channel mimicked the CNG channel pore. The acidic Asp acid residue was critical for conveying the divalent cation sensitivity (125, 181). These results suggest that the -YG- motif in K+ channel selectivity filters is critical for conferring K+ selectivity, whereas the adjacent acidic residue is important for divalent cation binding in the absence of such a motif. It is interesting to note that highly divalent-cation selective voltage-gated Ca2+ channels contain a Gly-Glu pair (G - - E) in three of the four channel domains. Of note, whereas mutations of these residues in CNG channels have demonstrated their key roles in selectivity, they had little effect on the rate of ion translocation (channel conductance). Thus the molecular determinants of selectivity and conductance are likely distinct in divalent cation permeant channels. Experimental support for this conclusion has been obtained in InsP3R channels. The predicted selectivity filter sequence in InsP3R and RyR is GGVGD2550 and GGIGD, respectively. The sequences are similar to that of CNG channels in that the YG motif is absent, yet they differ in that they contain two residues between Gly and Asp, as in the K+ channels. On the basis of these considerations, the lack of a YG motif in the InsP3R/RyR sequences is consistent with the channels’ lack of K+ selectivity, and the presence of the Asp residue is consistent with the idea that it provides a divalent ion binding site that conveys divalent permeability. A conservative mutation of Asp-2550 to Glu was without effect on K+ conductance (391 ± 4 versus 364 ± 5 pS, respectively), whereas it altered cation selectivity, with PCa:PK = 1.25, significantly reduced compared with the normal 6–8. In contrast, channels with mutation of Val-2548 to Ile had increased K+ conductance (490 pS) but retained normal Ca2+ selectivity (43). Thus the InsP3R pore has distinct sites that control monovalent permeation and divalent selectivity.
Most of the current knowledge regarding the atomic determinants of permeation in the InsP3R are inferences based on homology modeling of the InsP3R pore region with K+ channel structures and analogies based on insights regarding the function of the homologous region of the RyR. Only a few studies have used mutagenesis and functional recording of single recombinant InsP3R channels to probe the molecular determinants of permeation and selectivity. As indicated above, transmembrane deletion analysis localized the permeation pathway to a region containing the two COOH helices (383), and site-directed mutagenesis of residues in the putative selectivity filter altered channel monovalent conductance and divalent selectivity (43). The lack of more substantial data in this area begs for additional experimental effort. A number of mutations in the pore region of the RyR have either been described (central core disease mutations) or engineered (114, 116, 117, 155, 217, 304, 500, 501, 504, 522, 524, 546). The pore region of the cardiac RyR2 channel has been modeled onto the KcsA pore structure (507). These studies will be helpful in guiding future efforts to define the molecular basis of ion permeation in InsP3R channels.
VI. REGULATION OF INOSITOL TRISPHOSPHATE RECEPTOR CHANNEL GATING
A. Overview
As a crucial element in the generation and modulation of intracellular Ca2+ signals, the activity of the InsP3R as a Ca2+ release channel in the ER Ca2+ store is regulated by a wide range of ligands. The most important ligands regulating InsP3R channel activity are InsP3 and Ca2+, its physiological permeant ion, although it is important to note that InsP3, and other ligands such as ATP, regulate channel activity mainly by modifying the sensitivity of the channels to Ca2+ regulation. Generally, Ca2+ regulates steady-state InsP3R channel gating with a biphasic concentration dependence: Ca2+ at low concentrations activates the channel and increases its Po, whereas at higher concentrations, Ca2+ inhibits the channel. In the presence of saturating concentrations of InsP3, Ca2+ activation has been observed consistently in all InsP3R channels under various experimental conditions with similar apparent Ca2+ affinities, and mostly as a positively cooperative process. On the other hand, the presence and characteristics of high-Ca2+ inhibition have been highly variable among studies. In some studies, InsP3R channels in saturating concentrations of InsP3 exhibited a low sensitivity to inhibition by high cytoplasmic Ca2+ concentrations, with broad, plateau-shaped Ca2+ dependence curves and robust maximum Po close to 1. In contrast, other studies revealed InsP3R channels with a significantly higher sensitivity to Ca2+ inhibition that therefore displayed narrow, bell-shaped Ca2+ dependence curves with maximum Po significantly lower than 1. Finally, under certain experimental conditions and in some studies of some InsP3R isoforms, Ca2+ inhibition was either severely reduced or even totally absent, with the InsP3R channels remaining active even at very high Ca2+ concentrations.
The most systematic studies of InsP3R activities have revealed that InsP3, as well as AdA and its analogs, regulates the InsP3R channel by allosterically modulating the sensitivity of the channels to Ca2+ inhibition, with little effect on Ca2+ activation properties of the channels. Nevertheless, InsP3R channels observed to lack Ca2+ inhibition are still InsP3 dependent, with the maximum Po of the channels being increased by increases in InsP3 concentration. Molecular modeling indicates that the InsP3 sensitivity of channels that lack Ca2+ inhibition is due to the presence in the InsP3R of two distinct Ca2+ inhibition sites, with only one of them InsP3 sensitive. Channels that lack Ca2+ inhibition specifically lack a functional InsP3-insensitive inhibitory Ca2+ binding site.
Whereas InsP3 and Ca2+ are essential for InsP3R channel activation, other physiological ligands, such as ATP, are not. Increases in concentrations of free ATP (not coordinated with divalent ions) potentiate InsP3 channel activity allosterically by enhancing the sensitivity of the channel to Ca2+ activation. Regulation of InsP3R channel activity by its natural ligands (Ca2+, InsP3, and ATP) is due primarily to effects of ligand concentrations on the duration the channel stays closed between two successive channel openings. The mean duration of a channel opening remains remarkably constant over most [InsP3], [Ca2+]i, and [ATP] investigated. Thus channel activity is largely regulated by modulation of the channel opening rate.
In addition to steady-state channel gating kinetics, other aspects of InsP3R channel activity are also regulated by Ca2+ and InsP3. In the constant presence of InsP3, InsP3R channel activity inevitably terminates. This reversible, InsP3-induced inactivation of the channel progresses faster in high Ca2+ concentrations and in subsaturating concentrations of InsP3. In addition, channel recruitment, mediated by a process of ligand-sensitive channel sequestration, is a distinct process in which suboptimal Ca2+ concentrations (too low or too high) and subsaturating InsP3 concentrations activate fewer InsP3R channels than optimal concentrations of either ligand.
Thus ligand regulation of the InsP3R channel is complex, with Ca2+ as the major determinant of the channel properties. Ca2+ modulates channel activity by binding to several apparently distinct binding sites that regulate Ca2+ activation, InsP3-dependent Ca2+ inhibition, InsP3-independent Ca2+ inhibition, channel inactivation, and channel recruitment. In addition, a separate Ca2+ binding site appears to regulate the properties of the InsP3-independent Ca2+ inhibition site. Ca2+ regulation of the channel is impinged on by other channel regulators, including InsP3 and ATP, as allosteric modulators.
B. Cytoplasmic Ca2+ Regulation of InsP3R Channels
1. Biphasic regulation of InsP3R channel gating by Ca2+
The most widely studied aspect of InsP3R channel activity regulation is that by Ca2+. The level of InsP3R channel activity is quantified by its Po. As discussed earlier, direct single-channel measurements of the gating properties have been made using two approaches: planar bilayer reconstitutions and nuclear patch-clamp electrophysiology. Unfortunately, the results from these distinct approaches have been divergent in some important respects. As we discuss the regulation of InsP3R channel gating, we will attempt to point out these discrepancies and offer some insights into them.
Patch-clamp experiments on outer membranes of isolated nuclei of different InsP3R isoforms from different species (Xenopus type 1, rat type 1, rat type 3, and InsP3R from insect Spodoptera) (42, 196, 282, 283) have revealed that in the presence of saturating concentrations of InsP3, InsP3R channels have very low activity in resting cytoplasmic Ca2+ concentrations (~50 nM), but that as Ca2+ is raised through the submicromolar range (<1 μM), InsP3R channel activity increases to a maximum level with Po ~ 0.8 (Fig. 6). Thus Ca2+ in submicromolar levels activates InsP3R channels. The channel Po remains at the maximal level over a wide range of Ca2+ concentrations before further increases begin to inhibit the channel, at Ca2+ concentrations >10 μM (Fig. 6). Thus the Ca2+ dependence of channel activity for these InsP3R channels is biphasic, with remarkably similar broad, plateau-shaped Po versus [Ca2+]i curves (Fig. 7).
FIG. 6.

Typical single-channel current traces of X-InsP3R-1 in various cytoplasmic Ca2+ concentrations and saturating 10 μM InsP3. Current traces were recorded during nuclear patch-clamp experiments at cytoplasmic Ca2+ concentrations as tabulated, in 0.5 mM free ATP. All current traces in this and other graphs were recorded at 20 mV. Arrows indicate closed-channel current level in all current traces. Channel open probability (Po) was evaluated for the single-channel patch-clamp experiments yielding the current traces shown in A, B, C, and D of 0.008, 0.50, 0.89, and 0.002, respectively. [Modified from Mak et al. (282).]
FIG. 7.

[Ca2+]i and [InsP3] regulation of InsP3R channel activity. A: [Ca2+]i dependence of mean Po of endogenous X-InsP3R-1 channels (solid symbols) in various [InsP3] as tabulated. [Modified from Mak et al. (282).] Each data point in this and subsequent Po versus [Ca2+ ]i plots is the average of channel Po from at least 4 experiments using the same ligand concentrations. The curves are least-squares fit of the data points using the biphasic Ca2+ regulation Hill equation (Eq. 1) with parameters as tabulated. The large open circles represent Po for recombinant rat InsP3R-1 channels in various [Ca2+]i in saturating 10 μM InsP3. [Modified from Boehning et al. (42).] Inset: plot of Kinh derived from the biphasic Hill equation fit of Po data versus [InsP3] used. The curve is the least-squares fit of the Kinh values using the activation Hill equation with parameters as tabulated. B: [Ca2+]i dependence of mean Po of recombinant r-InsP3R-3 channels in various [InsP3] as tabulated. [Modified from Mak et al. (283).] Data points and fitted curves are obtained as described for A. C: [Ca2+]i dependence of mean Po of endogenous InsP3R channels from Sf9 cells in various [InsP3] as tabulated. [Modified from Ionescu et al. (196).] Data points, fitted curves, and inset graph are obtained as described for A. D: [Ca2+]i dependence of mean Po of X-InsP3R-1 InsP3R channels that have been exposed to bath solution with very low [Ca2+]i (< 5 nM) for a few minutes before the patch-clamp experiments, in various [InsP3] as tabulated. The curves are least-squares fits to the data using activation Hill equation Po = PHill{1 + (Kact/[Ca2+]i)Hact}−1 with parameters as tabulated. [Modified from Mak et al. (286).]
Although the general shapes of the channel Po versus [Ca2+]i curves in these patch-clamp studies are similar, the Ca2+ dependencies have minor differences among the different InsP3R channels. These differences may have physiological relevance. Ca2+ activation of type 1 InsP3R channels is positively cooperative, enabling channel Po to increase sharply and reach the maximum value within a narrow range of [Ca2+]i (Fig. 7A). Such a behavior is ideal for CICR. In contrast, Po of the type 3 InsP3R channel increases over a broader range of Ca2+ concentrations and with a higher Ca2+ affinity (Fig. 7B). Such a behavior is ideal as a trigger that responds to low InsP3 concentrations. Ca2+ inhibition of the vertebrate InsP3R channels in these studies is highly cooperative, so channel Po decreases rapidly over a narrow range of Ca2+ concentrations (Fig. 7, A and B), whereas Ca2+ inhibition of the insect Sf9 cell channel exhibits no cooperativity, and channel Po decreases gently over a broader range of Ca2+ concentrations (Fig. 7C). The physiological relevance of these differences is not clear.
In contrast, other single-channel studies have revealed radically different Ca2+ concentration dependencies. In some (294, 381, 382, 438, 478, 479, 481), InsP3R channels in saturating InsP3 exhibited substantially higher sensitivity to Ca2+ inhibition than observed in the patch-clamp studies, so that their biphasic Po versus [Ca2+]i curves are narrow and bell-shaped (Fig. 8). In other studies, high Ca2+ inhibition of channel activity was significantly reduced or completely absent (163, 286, 380, 382) so that the channel remained active in physiologically attainable supramicromolar Ca2+ concentrations (>500 μM) (Figs. 7D and 8).
FIG. 8.

[Ca2+]i dependence of vertebrate InsP3R channel activity in saturating [InsP3] observed in various single-channel studies. Biphasic Hill equation curves shown are generated either using parameters provided by studies cited below, or by fitting data provided in those studies with the Hill equation. Entries denoted by red letters are data from endogenously expressed InsP3R channels; other entries denoted by black letters are from recombinant homotetrameric InsP3R channels. Entries marked with asterisks are obtained by nuclear patch-clamp experiments; others are from InsP3R channels reconstituted into planar lipid bilayers. All data were observed in the presence of 0.5–1 mM Na2 ATP on the cytoplasmic side of the channel unless stated otherwise. A: canine cerebellar (438). B: bovine cerebellar in 0 ATP (382). C: rat type 1 SI+ SII+ in COS cells in 0 ATP (381). D: rat cerebellar (478). E: rat type 1 SI− SII+ in Sf9 cells (478). F: rat type 1 SI− SII+ in Sf9 cells (479). G: Xenopus oocyte (282). H: rat cerebellar (294). I: ferret cardiac ventricular myocyte in 0 ATP (382). J: rat type 2 in COS cells in 0 ATP (380). K: rat type 2 in Sf9 cells (481). L: rat pancreatic RIN-m5F cells (163). M: rat type 3 in Sf9 cells in 5 mM Na2ATP (481). N: rat type 3 in Xenopus oocytes (283).
2. Po exhibited by maximally activated InsP3R channels
In patch-clamp studies of channels of different InsP3R isoforms (type 1 and 3) from different species (rat, Xenopus, and insect Spodoptera) (42, 196, 282, 283), the values of Pmax, the maximum channel Po observed in optimally stimulating ligand conditions, are consistently high (~0.8, see Fig. 7, A–C). In contrast, a wide range of different Pmax for InsP3R channel have been reported in other studies (Fig. 8). Part of this diversity is due to the variable sensitivities to Ca2+ inhibition of the InsP3R channels in these studies. Channels that exhibit higher sensitivity to Ca2+ inhibition and display narrow, bell-shaped Ca2+ dependence curves may start to be inhibited by Ca2+ even before they are fully activated by Ca2+, and will therefore have lower Pmax (Fig. 8). However, this cannot account for all the diversity in Pmax observed because channels that lacked high Ca2+ inhibition were still observed with low Pmax (163), and channels in different studies with similar sensitivities to Ca2+ inhibition nevertheless exhibited very different Pmax (438, 478).
It is therefore likely that Pmax of InsP3R channels is affected not only by the sensitivity of the channel to high Ca2+ inhibition, but also to factors extrinsic to the channel, such as lipid composition of the membrane (75), presence of phosphatidylinositol 4,5-bisphosphate in the InsP3-binding site (262), integrity of the InsP3R after isolation and reconstitution, and loss of interacting proteins and factors as the channel is isolated from its cytoplasmic environment. Although these factors are mentioned as possible reasons for divergent observed results, they may have relevance for physiological modulation of InsP3R channel activity.
Given the high variability of Pmax observed in different experimental systems, we would encourage experimentalists to clearly provide the absolute Pmax in each study so that results from different experiments can be more readily compared.
3. Use of biphasic Hill equation to describe Ca2+ regulation of InsP3R channel activity
A convenient way to quantify Ca2+ regulation of InsP3R channel activity is to fit the Po versus [Ca2+]i data with an empirical, model-independent Hill equation. The biphasic equation
| (1) |
has been used to describe the Po of channels that are both activated and inhibited by Ca2+. Ca2+ dependence of the channel can then be characterized in terms of the five Hill equation parameters: PHill, Kact, Hact, Kinh, Hinh, for easy comparisons among various studies. (These parameters are tabulated in the Po versus [Ca2+]i curves shown in this review; Figs. 7, 10, A–C, and 12, A and B.) For InsP3R channels that exhibit significantly different sensitivities to Ca2+ activation and Ca2+ inhibition, and therefore have a broad, plateau-shaped Po versus [Ca2+]i curve in saturating [InsP3] (196, 282, 283), the experimental data can be fitted by the Hill equation with a unique set of parameters, provided that enough data points have been acquired. Each of the parameters describes one aspect of the Ca2+ dependence of the channel. PHill is the maximum Po that the channel would be activated to if there was no Ca2+ inhibition. Because of the low sensitivity of the channel to Ca2+ inhibition in saturating InsP3, the channel is fully activated before Ca2+ starts to inhibit the channel. Thus the experimentally observed Pmax under optimal ligand conditions is equal to PHill. The parameters Kact and Kinh are then EC50 and IC50 for Ca2+, respectively, i.e., the [Ca2+]i at which Po = 0.5 PHill. They are inversely related to the functional sensitivity of the channel to Ca2+ activation and inhibition, respectively. Hact and Hinh describe the level of cooperativity of Ca2+ activation and inhibition, respectively.
FIG. 10.

Regulation of InsP3R channel activity by ATP. A: [Ca2+]i dependence of mean Po of endogenous X-InsP3R-1 channels in the presence of various [ATP]free as tabulated. [Modified from Mak et al. (281).] Solid symbols represent data obtained in the absence of Mg2+. Open circles represent data obtained in 3 mM Mg2+ and 0 ATP. Open squares represent data obtained in 3 mM Mg2+ and 0.5 mM total [ATP], with [ATP]free = 12 μM calculated by MaxChelator. The curves are fits using either the biphasic Hill equation (Eq. 1) to the data in [ATP]free = 0 (blue) or 0.5 mM (black); or the activating Hill equation (see legend for Fig. 7) to the data in other [ATP]free (10 nM < [Ca2+]i < 1 μM). Biphasic Hill equation parameters fitting data in 0 [ATP]free (blue curve) are tabulated. Inset: plot of the Hill equation parameter Kact versus [ATP]free used. The curve is a fit to the Kact values using the modified Michaelis-Menten equation with parameters as tabulated. B: [Ca2+]i dependence of mean Po of recombinant r-InsP3R-3 channels in the presence of various [ATP]free as tabulated. [Modified from Mak et al. (280).] Solid symbols, open circles, and open squares represent data in a similar convention as described for A. Solid curves are either biphasic Hill equation fit or activation Hill equation fit to the data in various [ATP]free as described for A. The thick dashed blue curve is the biphasic Hill equation fit to channel Po for X-InsP3R-1 in the absence of ATP plotted for comparison. Parameters are tabulated for biphasic Hill equation fit to r-InsP3R-3 channel Po in 0 ATP (in blue), as well as those for activating Hill equation fit to r-InsP3R-3 channel Po in [ATP]free = 0.3 mM (in yellow) and 0.5 mM (in black) ([Ca2+]i < 1 μM). C: [Ca2+]i dependence of mean Po of endogenous X-InsP3R-1 channels in 0 ATP in the presence of various [InsP3] as tabulated. The curves are fits to the data using biphasic Hill equation (Eq. 1) with parameters as tabulated. [Modified from Mak et al. (279).] D: channel Po versus [ATP]free curves calculated for X-InsP3R-1 channels in 10 μM InsP3 and various [Ca2+]i as labeled, using the biphasic Hill equation (Eq. 1) and the modified Michaelis-Menten equation in A.
FIG. 12.

InsP3R channel activities activated by adenophostin A (AdA). [Ca2+]i dependence of mean Po of endogenous X-InsP3R-1 channels in the presence of various ligand (AdA or InsP3) concentrations as tabulated in 0.5 mM [ATP]free (A) or 0 [ATP]free (B). The curves are biphasic Hill equation (Eq. 2) fit to the data using parameters as tabulated. Typical single-channel current traces of X-InsP3R-1 in outer nuclear membrane recorded at optimal [Ca2+]i and saturating 100 nM AdA in 0.5 mM [ATP]free with channel Po of 0.72 (C) and 0 [ATP]free with channel Po of 0.39 (D). [Modified from Mak et al. (279).]
However, InsP3R channels that have similar sensitivities to Ca2+ activation and Ca2+ inhibition display narrow and bell-shaped biphasic Po versus [Ca2+]i curves. Po of these channels does not clearly flatten out at some maximum value because the channel is not yet fully activated by Ca2+ when Ca2+ inhibition starts to reduce Po. In such cases, the set of Hill equation parameters that provide a good Hill equation fit to the data is not unique, i.e., different sets of Hill equation parameters can provide indistinguishable fits to the experimental data (Fig. 9A). Because very different values of Kact and Kinh can be used to describe one set of data equally well, no physical significance can be assigned to one particular set of Hill equation parameters. Conversely, even if a set of Hill equation parameters can fit very well a collection of channel Po data, Kact may not reflect the functional sensitivity of the channel for Ca2+ activation. Likewise, the value of Kinh may not be an appropriate indication of the functional sensitivity of the channel for Ca2+ inhibition. Conclusions drawn from the values of Hill equation parameters that describe bell-shaped Ca2+ dependencies (479) can be highly questionable. Therefore, it can be very misleading to compare narrow bell-shaped Ca2+ dependencies of different InsP3R channels by simply comparing Hill equation parameters. Accordingly, a graph showing the biphasic Hill equation fits to data from various studies (Fig. 8) is used here for comparison of Ca2+ regulation of InsP3R channels.
FIG. 9.

Biphasic Hill equation fits to InsP3R channel Po versus [Ca2+]i data. Open squares are experimental data, and smooth curves are Hill equation fit to the data using parameters as tabulated. A: channel Po data for recombinant rat type 1 E2100D mutant InsP3R expressed in Sf9 cells were fitted (black curve) using modified biphasic Hill equation (Eq. 2) with parameters given in Ref. 479, tabulated in black. An alternative Hill equation fit (thick yellow curve) using the same equation with a different set of parameters (tabulated in yellow) is effectively indistinguishable from the fit provided in Ref. 479. B: channel Po data for recombinant wild-type Drosophila InsP3R channel expressed in Sf9 cells were fitted (black curve) using modified biphasic Hill equation (Eq. 2) with parameters given in Ref. 434, tabulated in black. The more general Hill equation (Eq. 1) gives a better fit to the data (red curve) using parameters tabulated in red.
Because InsP3 regulates InsP3R channels by modifying their sensitivity to Ca2+ inhibition (see later discussion), even channels with broad, plateau-shaped Po versus [Ca2+]i curves will exhibit narrow bell-shaped Po versus [Ca2+]i curves at subsaturating concentrations of InsP3 (see Fig. 1B with 10 or 20 nM InsP3), with Pmax significantly less than the PHill that is observed in saturating [InsP3]. However, it has been demonstrated that Po at subsaturating InsP3 concentrations was still well fitted by the same biphasic Hill equation (Eq. 1) assuming that only Kinh was affected by [InsP3] (282, 283). Thus a set of Hill equation parameters can be uniquely defined with correct physical meanings for such narrow, bell-shaped Po versus [Ca2+]i curves observed in subsaturating [InsP3] by making the reasonable assumption that PHill remains the same for all [InsP3]. However, PHill must be accurately determined.
A variation of the biphasic Hill equation
| (2) |
has been used to fit channel Po versus [Ca2+]i data in many channel reconstitution studies (34, 229, 434, 438, 478, 479, 481). The only difference between Eqs. 1 and 2 is that the Hill coefficients Hinh and Hact in Eq. 1 are assumed to be the same (Hact = Hinh = H) in Eq. 2. Although Eq. 2 can provide good fit to a number of data sets (34, 438, 478), there is no a priori reason to assume that Ca2+ activation and inhibition of the InsP3R channel have the same degree of cooperativity. Thus when Eq. 2 cannot provide a good fit to a data set (Fig. 3B), the more general Eq. 1 should be used. Furthermore, characterizing the Ca2+ dependence of an InsP3R channel with parameters from this form of the Hill equation suffers from the same problem as using Eq. 1 if the channel has a bell-shaped Ca2+ dependence curve, as observed in many reconstituted InsP3R channel studies.
It should also be pointed out that Hill equations are empirical equations not based on any specific model for ligand regulation of channel gating. With the equations, the gating behaviors of the InsP3R channel in a wide range of cytoplasmic Ca2+ concentrations can be characterized adequately and conveniently with a small number of parameters, provided that the Ca2+ dependence curve has a plateau shape with a well-defined PHill value. However, the empirical fit does not provide any insight into the molecular mechanism(s) responsible for the ligand regulation of the InsP3R channel. For instance, the parameters Hact and Hinh do not have a direct relation with the number of activating or inhibitory Ca2+ binding sites in the channel.
4. Ca2+ activation of InsP3R channels
Despite the highly variable shapes of the channel Po versus [Ca2+]i curves observed in these different studies (Fig. 8), Ca2+ activation of channel activity has been consistently observed with comparable functional sensitivity to Ca2+. This suggests that the characteristics of Ca2+ activation are likely determined by intrinsic features of the InsP3R molecule that are conserved throughout evolution and among the various isoforms and splice variants. This conservation probably exists because the activation of InsP3R channels by a rise of Ca2+ from resting levels (~50 nM) to hundreds of nanomolar plays a vital role in intracellular Ca2+ signaling. As discussed earlier, in the presence of activating levels of InsP3, elevations of [Ca2+]i due to Ca2+ released by one InsP3R channel can in turn activate InsP3R channels nearby to release more Ca2+ in a positive feedback. CICR mediated by InsP3R channels acts as a critical communication mechanism between channels to coordinate their activities, generating large-scale Ca2+ signals (puffs and waves) from elementary Ca2+ release events emanating from individual InsP3R channels (26).
Ca2+ activation of InsP3R channels is mostly positively cooperative (Fig. 8), enabling the channels to be activated sharply by Ca2+ within a narrow range. This property likely contributes to the fine-tuning of the channel by cytoplasmic Ca2+ concentration. However, nonco-operative Ca2+ activation has been observed in some InsP3R channels (163, 283, 382), suggesting that cooper ativity of InsP3R channel activation by Ca2+ may not be an essential feature for Ca2+ signaling. Rather, differences in the degree of cooperativity among channels may provide diversity in cytoplasmic Ca2+ signals generated by different isoforms and under different conditions. For example, ATP regulation of the Ca2+ activation properties of the type 3 channel was associated with changes in the degree of cooperativity (283), as discussed in detail later.
InsP3R isoforms with higher sensitivity to Ca2+ activation (especially type 3 InsP3R, see Fig. 8) have higher Po at resting cytoplasmic Ca2+ concentrations when InsP3 is present, than do channels formed by other isoforms. Thus, when an extracellular stimulus elicits production of InsP3 in a resting cell, such channels will have a higher probability of opening to release Ca2+ from the ER. They can therefore act as triggers to initiate InsP3-induced Ca2+ release (IICR) (283). Ca2+ released by these triggers may then raise local cytoplasmic Ca2+ concentration sufficiently to cause nearby InsP3R channels to release Ca2+ through CICR, thus propagating the Ca2+ signals.
5. Ca2+ inhibition of InsP3R channels
Whereas Ca2+ activation of the InsP3R channel is consistently observed in all single-channel experiments with comparable functional sensitivity to Ca2+ and largely similar levels of cooperativity, the sensitivity of InsP3R channels to Ca2+ inhibition is highly variable even in saturating InsP3 so that a wide range of different shapes of channel Po versus [Ca2+]i curves has been observed (Fig. 8).
What accounts for such diversity of observations and why is it important? Ca2+ inhibition can possibly serve as a negative-feedback mechanism to either terminate or prevent Ca2+ release as the local cytoplasmic Ca2+ concentration is raised by InsP3R-mediated Ca2+ release during Ca2+ signaling, even in the continuous presence of InsP3. This process may play a significant role in the generation of Ca2+ spikes and oscillations, as well as the generation of highly localized Ca2+ signals, by preventing runaway Ca2+ release due to the positive feedback of CICR. Thus it is critical to ascertain the presence or absence of Ca2+ inhibition of an InsP3R channel. Furthermore, the range of Ca2+ concentrations over which inhibition is exerted is also important, because it defines the spatial domain over which the channels may experience such regulation. For example, if the apparent affinity of the inhibitory Ca2+ binding site is 20 μM, only channels quite close to an open Ca2+ channel will experience feedback inhibition. On the other hand, if the apparent affinity of the inhibitory Ca2+ binding site is in the submicromolar range, then channels throughout the cytoplasm, even those that are quite far from individual release sites, will experience inhibitory Ca2+ concentrations.
The diversity of properties of Ca2+ inhibition of InsP3R channels observed in different studies may reflect a range of physiologically relevant modifications of Ca2+ inhibition by environmental factors that provide additional regulation of InsP3R-mediated Ca2+ signals. Alternatively, such diversity may reflect a lack of control of important experimental variables during the preparation or recording of the channels. There is no clear correlation between the sensitivity of an InsP3R channel to Ca2+ inhibition and the molecular structure of the InsP3R. Nuclear patch-clamp studies revealed very similar Ca2+ inhibition characteristics (low Ca2+ sensitivity and high level of cooperativity) for homotetrameric channels formed by type 1 or type 3 InsP3R isoforms (282, 283). On the other hand, whereas similar Ca2+ inhibition characteristics for different splice variants of the same isoform of InsP3R (381, 478) or different isoforms (481) have been observed in some reconstitution studies, the same isoforms displayed very different Ca2+ inhibition characteristics in other studies (see Fig. 2 and cf. Refs. 282, 481). Ca2+ inhibition was absent for both type 2 (380) and type 3 (163) InsP3R channels in some planar bilayer reconstitution studies, but not in others (481). Furthermore, the Ca2+ inhibition properties are largely, but not completely, distinct between the reconstitution and patch-clamp studies. For example, similar high sensitivity to Ca2+ inhibition was observed for InsP3R channels formed by the same isoform studied using nuclear patching (294) or bilayer reconstitutions (478).
It is highly likely that various forms of regulation, both physiological and nonphysiological, impinge on the Ca2+ inhibition properties of the channel. The sensitivity of recombinant rat InsP3R-3 to Ca2+ inhibition was radically altered by cytoplasmic ATP (481). Also, Ca2+ inhibition of channel activity is regulated by InsP3, as discussed below. Thus different InsP3 sensitivities of channels in different studies may contribute to different Ca2+ inhibition properties. Different InsP3 sensitivities could in turn be generated by extrinsic factors such as lipid composition of the membrane (75), covalent modifications of the channel (see sect. VIL), presence or absence of interacting proteins (see sect. VIN), or to intrinsic differences in the channel properties between species and/or isoforms. Even a simple nonphysiological maneuver can radically alter the channel’s Ca2+ inhibition properties. Exposure of InsP3R channels to a very low concentration of Ca2+ (<10 nM) for a few minutes before it is exposed to InsP3 completely and reversibly relieves Ca2+ inhibition of types 1 and 3 InsP3R channels (286). Thus the Ca2+ inhibition properties of the channel are regulated by a distinct Ca2+ binding site(s), which could be a locus for either regulation or disruption of Ca2+ inhibition. It is possible that this site was disrupted during protocols involved in channel reconstitution in those studies of the types 2 (380) and 3 (163) channel isoforms that failed to observe high-[Ca2+] inhibition.
One possible mechanism of regulating Ca2+ inhibition of InsP3R channels that has been repeatedly invoked is through interaction of the channel with the protein calmodulin. However, single-channel studies of InsP3R were very much consistent with the absence of any regulatory role for calmodulin in Ca2+ inhibition of InsP3R. A full discussion of the calmodulin interaction with InsP3R is provided in section VIN1. To date, no factor extrinsic to the InsP3R channel has been positively identified to confer Ca2+ inhibition of the channel.
C. InsP3 Activation of InsP3R Channels
1. InsP3 regulates InsP3R channel activity through modulation of its sensitivity to Ca2+ inhibition
In our previous discussions of Ca2+ regulation of InsP3R channel activity, we have focused on channel activity observed in saturating concentrations of InsP3. We now examine the InsP3 dependence of InsP3R gating by looking at the channel activity in subsaturating InsP3. Although spontaneous InsP3R activity of very low Po (approximately a few percent) has been observed in the absence of InsP3 (286, 382), both Ca2+ (in appropriate concentrations) and InsP3 must be present on the cytoplasmic side of the channel to activate it to appreciable activity levels. However, InsP3 activates the InsP3R in a radically different manner from Ca2+, so InsP3 and Ca2+ are not simply equivalent coagonists of the channel. Systematic studies of the gating properties of the Xenopus type 1 (X-InsP3R-1), rat type 3 (r-InsP3R-3), and Sf9 InsP3R channels under a broad range of concentrations of both InsP3 and Ca2+ in steady-state conditions revealed that the InsP3R becomes more sensitive to inhibition by high cytoplasmic Ca2+ concentrations in the presence of subsaturating concentrations of InsP3, i.e., at lower InsP3, the channel is inhibited by Ca2+ at lower concentrations (Fig. 7, A–C). Importantly, all other aspects of Ca2+ regulation of channel activity: its sensitivity to Ca2+ activation, the level of cooperativity of Ca2+ activation, and even the level of cooperativity of Ca2+ inhibition, are not significantly affected by InsP3. At very low concentrations of InsP3, the maximum channel Po observed, and the range of cytoplasmic Ca2+ concentrations over which the channel is active, were both substantially reduced (Fig. 7, A–C). Both effects can be fully accounted for by the increase in the sensitivity of the channel to Ca2+ inhibition. At very low InsP3 concentrations, the channel is so sensitive to Ca2+ inhibition that it begins to be inhibited by Ca2+ before it is fully activated. This pattern of InsP3 regulation of channel activity by solely modulating the sensitivity of the channel to inhibition by cytoplasmic Ca2+ is consistently observed for InsP3R channels of different isoforms (type 1 and 3) from very different species (Xenopus, rat, and insect) (196, 282, 283). This suggests that this is probably the common mechanism underlying ligand regulation of all InsP3R.
Similar effects of InsP3 on channel Po were also observed for endogenous canine cerebellar InsP3R-1 reconstituted in planar lipid bilayers (219), namely, increases in InsP3 concentrations within a subsaturating range reduced the sensitivity of the channel to Ca2+ inhibition, thus broadening the channel versus Po curve from a narrow, bell shape to a plateau shape. Although the concentrations of InsP3 necessary to observe this effect were quite high (180 μM), the channel nevertheless retained a significant level of activity even when Ca2+ concentration was 20 μM. Similar to the observations in nuclear patch-clamp studies, InsP3 had little effect on Ca2+ activation properties of the channel.
Because InsP3 affects only the sensitivity of InsP3R channels to Ca2+ inhibition, the sensitivity of the channels to InsP3 can be defined by changes caused by InsP3 in the sensitivity of the channel to Ca2+ inhibition. Accordingly, a half-maximal effect was observed at ~50 nM InsP3 for both X-InsP3R-1 and r-InsP3R-3 (Fig. 7, A and B). The response of these channels to InsP3 is fully saturated by 100 nM InsP3 (282, 283). In general, this functional sensitivity to InsP3 is in reasonable agreement with the dissociation constant Kd derived from InsP3 binding assays, and the EC50 for InsP3 stimulation of Ca2+ release (229, 300, 307, 319, 380, 464).
The insect Sf9 InsP3R channel has lower sensitivity to InsP3, with half-maximal effect at ~400 nM, and its response is not saturated until the concentration of InsP3 is ~1 μM (Fig. 7C) (196). Canine cerebellar InsP3R-1 apparently has a very low sensitivity to InsP3 such that the Po versus [Ca2+]i curve of the channel was still substantially changed when InsP3 was increased from 2 to 180 μM (219). This significantly lower InsP3 sensitivity agrees with the low InsP3 sensitivity of InsP3R-mediated Ca2+ release observed in cerebellar neurons (234, 235) and may be due to the differences in InsP3R splice variants: InsP3R examined in Reference 282 was the peripheral SII− form, whereas in Reference 219, the cerebellar SII+ form was examined, or due to the interference of InsP3R channel gating by phosphatidylinositol 4,5-bisphosphate bound to the cerebellar InsP3R (262).
2. Physiological significance of InsP3 modulation of Ca2+ inhibition of InsP3R channel
The modulation of InsP3R channel Kinh by InsP3 provides a possible mechanism intrinsic to the channel to generate graded Ca2+ release in response to different levels of extracellular stimulus intensity, instead of the expected all-or-nothing signal expected for the process of CICR (26, 47). Because InsP3R exposed to higher InsP3 concentrations has lower susceptibility to Ca2+ inhibition, cytoplasmic Ca2+ concentrations that can inhibit channel activity at low InsP3 concentrations will be insufficient to inhibit the channel when InsP3 concentration is increased. Higher Ca2+ concentrations at the vicinity of an open channel can therefore be achieved before reaching levels that will inhibit other closed channels in the same InsP3R channel cluster before they can open (26). This enables coordinated release of channels in the same local cluster, whereas in lower InsP3 concentrations, Ca2+ released by the stochastic opening of one channel suppresses gating of the rest of the channels in the cluster. By enabling higher Ca2+ concentrations to be achieved at the local level by coordinated Ca2+ release, higher InsP3 concentrations associated with more intense stimuli would promote greater diffusive spread of the local Ca2+ signal to other InsP3R channel clusters, thereby transforming highly localized signals at low levels of stimulation to more global coordinated Ca2+ release signals as the intensity of the stimulus is increased. Thus the mechanistic insights derived from single-channel patch-clamp studies of the InsP3R response to InsP3 suggest that graded Ca2+ response can be generated without the need to invoke channels with different InsP3 sensitivities or clusters of different channel densities (26, 47, 71, 192), although these or other mechanisms are in no way excluded.
In addition, modulation by InsP3 of Ca2+ inhibition of InsP3R enables channel activity to be exquisitely sensitive to small changes in InsP3 concentration within the subsaturating range. For X-InsP3R-1 in 10 nM InsP3, the channel is appreciably active only within a very narrow range of cytoplasmic Ca2+ concentrations (50–300 nM) with very low Po (Pmax ~ 0.1). In contrast, in 100 nM InsP3, the channel is active over a very wide range of Ca2+ concentrations (50 nM to 100 μM). It gates robustly with high Po (Pmax ~0.8) over Ca2+ concentrations from 1 to 20 μM (Fig. 1B). Thus the InsP3R-mediated Ca2+ signal can be finely controlled by small differences in the InsP3 concentration that the channel is exposed to.
3. Inadequate characterization of the InsP3 dependence of InsP3R channel
InsP3R channel activity is regulated by its ligands InsP3 and Ca2+ in a complicated manner, with InsP3 affecting gating through modulation of Ca2+ inhibition of the channel. This complex relationship between channel Po and the concentrations of InsP3 and cytoplasmic Ca2+, as described in References 196, 219, 282, 283, cannot be adequately characterized by determining the channel Po in various concentrations of InsP3 at just one Ca2+ concentration. It was pointed out (282) that depending on the Ca2+ concentrations used, different apparent functional affinities for InsP3 with different degrees of cooperativity would be observed.
Unfortunately, only a minority (196, 219, 282, 283, 294) of the single-channel studies of InsP3 regulation investigated channel Po over different combinations of cytoplasmic Ca2+ and InsP3 concentrations to characterize the channel behaviors adequately. The majority of studies have only investigated channel Po dependence on InsP3 at a single, arbitrarily selected Ca2+ concentration (164, 262, 331, 380–382, 434, 456, 482, 505). The Po of the channel in any subsaturating concentration of InsP3 at cytoplasmic Ca2+ concentrations other than the selected one cannot be estimated based on these studies. Thus it is impossible to properly compare results obtained in different studies if the dependence of channel Po on InsP3 concentration was determined at different selected single cytoplasmic Ca2+ concentrations.
4. A different kind of InsP3 dependence
A radically different type of InsP3 dependence of channel Po was recently reported for neuronal InsP3R-1 observed by nuclear patch-clamp experiments of the inner membrane of nuclei isolated from Purkinje neurons (294). Instead of modulating the sensitivity to Ca2+ inhibition, increases in InsP3 concentrations in the subsaturating range activated the channel by increasing only the maximum Po of the channel, with very little impact on the shape of the channel Po versus [Ca2+]i curve (294). Thus InsP3 regulation and the Ca2+ regulation of channel Po are totally independent, i.e., Ca2+ regulation is not affected by InsP3 and, conversely, InsP3 regulation is not affected by Ca2+. In this surprising situation, ligand regulation of channel Po can be adequately characterized by determining the Ca2+ dependence of channel Po at saturating InsP3 concentrations and the InsP3 dependence of channel Po at one selected Ca2+ concentration. However, the independence of Ca2+ regulation from InsP3 can only be ascertained by observing the Ca2+ dependence of channel Po at at least two different InsP3 concentrations: a saturating and a subsaturating level.
D. InsP3 and Ca2+ Regulate InsP3R Channel Activities Through Multiple Ca2+ Sensors
The most comprehensive studies of ligand regulation of InsP3R channel activity, investigating channel Po in various combinations of saturating and subsaturating concentrations of InsP3, and activating, optimal and inhibitory concentrations of cytoplasmic Ca2+ (196, 219, 282, 283), indicate that InsP3 regulates gating mainly by modulating its sensitivity to Ca2+ inhibition. However, InsP3R channels with either no discernable inhibition by Ca2+ (382), or with Ca2+ inhibition abolished by preexposure to very low levels of Ca2+ (285), are nevertheless still sensitive to InsP3 (Fig. 7D). These InsP3R channels that were not inhibited by high Ca2+ exhibited no appreciable activity in the absence of InsP3. Increases in InsP3 through subsaturating levels increased the observed Pmax, with little effect on the sensitivity of the channel to Ca2+ activation, or on the level of cooperativity of Ca2+ activation. These results suggest that there are at least two distinct kinds of Ca2+ sensors responsible for ligand regulation of InsP3R channel Po. One kind of Ca2+ sensor is responsible for Ca2+ inhibition in saturating InsP3 concentrations. This sensor has an apparent Ca2+ affinity of ~20–50 μM in most nuclear patch-clamp studies (42, 196, 282, 283) and accounts for the descending phase of the Po versus [Ca2+]i curve in saturating InsP3. This Ca2+ binding site is nonfunctional in the InsP3R channels that are not inhibited by high Ca2+. The second kind of Ca2+ sensor is InsP3 dependent. It is responsible for the InsP3 sensitivity of the channel regardless of the functionality of the other inhibitory Ca2+ sensor. In the absence of InsP3, this site has very high Ca2+ affinity and inhibits channel opening. Thus channels that lack high Ca2+ inhibition still require InsP3 for activation. In addition, comparisons of the sensitivities and levels of cooperativity for Ca2+ and InsP3 activation of different InsP3R isoform channels argue for the existence of a third Ca2+ sensor that is activating yet InsP3 independent. The following describes the properties of each of these sensors separately.
1. The InsP3-independent inhibitory Ca2+ sensor
As noted earlier, single-channel studies of InsP3R have revealed a vast diversity of properties for Ca2+ inhibition of the channel, with very different sensitivity to Ca2+ observed even for the same InsP3R isoform and splice variant in different studies (Fig. 8). As a result, the Po versus [Ca2+]i curves observed in some studies are narrow and bell-shaped, whereas in other studies, they are broad and plateau-shaped. In some InsP3R channels, Ca2+ inhibition actually appeared to be totally absent in some, but not all, studies. It has also been demonstrated that this inhibitory Ca2+ sensor can be reversibly rendered completely nonfunctional, thus abolishing high Ca2+ inhibition of the channel, by treating the channel with nanomolar cytoplasmic Ca2+ for a few minutes before exposure of the channel to InsP3 (285). Remarkably, channels that were not sensitive to Ca2+ inhibition were nevertheless fully InsP3 dependent (285, 380). Together, these observations indicate that this inhibitory Ca2+ sensor is not responsible for InsP3 dependence of the channel and therefore likely InsP3 independent. Furthermore, the functional affinity for Ca2+ for this InsP3-independent inhibitory Ca2+ sensor is malleable, suggesting that it could possibly be under physiological regulation. Besides establishing the malleability of the inhibitory Ca2+ sensor, abolition of Ca2+ inhibition by preexposure of the channel to nanomolar Ca2+ further indicates that its functionality is controlled by another, different type of Ca2+ binding site(s). This latter site(s) is likely to operate with a high level of cooperativity (285).
2. The InsP3-dependent Ca2+ sensor
Another type of Ca2+ sensor regulating InsP3R channel activity is regulated by the InsP3 binding sites in the channel. Nuclear patch-clamp single-channel studies indicated that this InsP3-dependent Ca2+ sensor is extremely sensitive to small changes in InsP3 concentration within the subsaturating range. In particular, the sensitivity of the X-InsP3R-1 channel to Ca2+ inhibition was reduced dramatically as InsP3 concentration was raised from 10 to 100 nM. The channel was significantly inhibited by 160 nM Ca2+ in 10 nM InsP3, but it was not inhibited until cytoplasmic Ca2+ concentration was >30 μM when the channel was exposed to 100 nM InsP3 (Fig. 7A). To reconcile this exquisite sensitivity of the channel to subsaturating levels of InsP3 with the tetrameric structure of the channel consisting of four InsP3R molecules each with a single InsP3-binding site (discussed in sect. IIIC1), an allosteric model was proposed in which the InsP3-dependent Ca2+ sensors in the channel (1 per InsP3R molecule, total of 4 in each channel) act as inhibitory Ca2+-binding sites to inhibit channel gating when bound to Ca2+ in the absence of InsP3 (286). However, as InsP3 concentration is raised and InsP3 binds to the channel, Ca2+ binding to the InsP3-dependent Ca2+ sensors starts to favor opening of the channel. In effect, this Ca2+ sensor becomes an activating Ca2+-binding site. Thus InsP3 regulates InsP3R channel activity with very high effectiveness by modifying not only the functional affinity of the InsP3-dependent Ca2+ sensors, but also their functional nature, changing them from inhibitory to activating sites.
The interplay of the two different types of Ca2+ sensors, one InsP3 sensitive and the other InsP3 insensitive, enables the response of InsP3R channel to InsP3 to saturate very abruptly despite its high sensitivity to subsaturating concentrations of InsP3. Once InsP3 exceeds the saturating level of 100 nM, the Po versus [Ca2+]i curve of the X-InsP3R-1 channel exhibits no discernable change even as InsP3 is further increased by over three orders of magnitude from 100 nM to 180 μM (Fig. 7A). This behavior results from the influence of the inhibitory Ca2+ sensors that are InsP3 independent. Thus, at InsP3 > 100 nM, Ca2+ inhibition of the channel is caused by the InsP3-independent, purely inhibitory Ca2+ sensor. The abruptness in the saturation of the response of the channel to changes in InsP3 concentration is due to the insensitivity to InsP3 of these inhibitory Ca2+ sensors.
3. The InsP3-independent activating Ca2+ sensor
In nuclear patch-clamp experiments, X-InsP3R-1 and r-InsP3R-3 channels exhibited similar sensitivities to activation by InsP3, even though the sensitivity and degree of cooperativity for Ca2+ activation of the two types of channels were very different (Fig. 7, A and B) (282, 283). On the other hand, the sensitivity and level of cooperativity for InsP3 activation of X-InsP3R-1 and Sf9 InsP3R channels are very different, even though the two types of channels have similar sensitivities and levels of cooperativity for Ca2+ activation. It is difficult to account for these different characteristics of Ca2+ and InsP3 activation of these channels if they have the single type of InsP3-dependent Ca2+ sensor discussed above that is transformed into an activating Ca2+ binding site by InsP3. To quantitatively account for Ca2+ activation of channel gating, a third type of Ca2+ sensor must also play a role. This Ca2+ site is InsP3 independent and responsible for the consistent sensitivity to Ca2+ activation observed in various InsP3R channels despite their differences in InsP3 sensitivity or the presence or absence of Ca2+ inhibition.
Numerical calculations (285) indicate that an allosteric model postulating the three types of Ca2+ binding sites as described above can account for all single-channel behaviors of various InsP3R channels studied by nuclear patch-clamp experiments while taking into consideration the homotetrameric structure of the channels (144, 196, 285).
E. Regulation of InsP3R Gating by Luminal Divalent Cations
There have been several reports indicating that besides affecting the channel conductance properties, the concentration and identity of divalent cations present on the luminal side of the InsP3R can also regulate channel gating. The study by Bezprozvanny and Ehrlich (33) remains the most detailed investigation of this aspect of gating regulation. They found that in the presence of the same level of cytoplasmic Ca2+ (0.2 μM), the mean channel open duration to depends on the identity of the divalent cation acting as the charge carrier (using 55 mM divalent ion on the luminal side of the channel). to with Ba2+ as charge carrier is approximately equal to that with Sr2+ is greater than that with Mg2+ is approximately equal to that with Mn2+ (438) is greater than that with Ca2+. Recently, it was reported that channel Po of rat cerebellar InsP3R-1 in the inner nuclear membrane of Purkinje neurons with Ba2+ as charge carrier (100 mM) was nearly 10 times that observed with K+ (50 mM) under the same cytoplasmic ionic conditions (294). However, it is not clear whether this effect has any physiological significance since Ca2+ and Mg2+ are the only physiologically relevant divalent cations that can occur in substantial concentrations in the ER lumen.
A possibly important observation is that InsP3R activity is inhibited by luminal Ca2+, with channel Po elicited, by optimal cytoplasmic concentrations of InsP3 and Ca2+, decreasing 66% as luminal Ca2+ concentration was raised from 3 μM to 10 mM (33). High luminal Ca2+ concentration was also reported to cause rapid inactivation (approximately seconds) of the InsP3R channel after InsP3 activation, whereas the channel remained active for extensive periods (~100 s) in the presence of lower luminal Ca2+ concentrations (469). Functionally, it has been suggested that luminal Ca2+ regulation of channel activity could possibly play a role in quantal Ca2+ release (197). However, generating quantal Ca2+ release by luminal Ca2+ requires inhibition of InsP3R channel activity by low luminal [Ca2+], not the inhibition of channel activity by high luminal [Ca2+] as reported (33, 469). Structurally, such sensitivity of the channel to luminal Ca2+ may be related to a putative Ca2+ binding site located in a luminal loop of the InsP3R (421). On the other hand, because of the important regulation of the channel by cytoplasmic Ca2+, it is possible that Ca2+ permeating through the channel, expected to be considerable in the face of tens of millimolar Ca2+ on the luminal side of the channel, acts on cytoplasmic binding sites to exert the observed effects. At lower luminal Ca2+ concentrations (between 0.2 and 1.5 μM), no significant effects of luminal Ca2+ on channel Po have been observed (282). Beyond these studies, there is no systematic study of regulation of InsP3R channel activity by luminal Ca2+ under various cytoplasmic conditions.
F. Regulation of InsP3R Channel Gating by ATP
1. ATP potentiation of Ca2+ activation of InsP3R channel activity
Besides being activated by InsP3 and suitable concentrations of cytoplasmic Ca2+, InsP3R channel activity is also potentiated by ATP, although ATP is not necessary for channel gating (307, 429, 463). A systematic investigation of the effects of ATP on both endogenous X-InsP3R-1 (281) and recombinant r-InsP3R-3 (280) revealed that ATP regulation of channel activity is both complex and isoform dependent (Fig. 10, A and B). For the type 1 InsP3R, increases in cytoplasmic free ATP concentrations ([ATP]free, the concentration of ATP not bound to divalent cations) increased channel Po primarily by allosterically enhancing the sensitivity of the channel to Ca2+ activation (Fig. 10 A). [ATP]free had no significant effect on the degree of cooperativity of Ca2+ activation (Fig. 10A), nor did it affect the Pmax, although in the absence of ATP, higher [Ca2+]i was needed to activate the channel to Pmax (Figs. 10 A and 11A).
FIG. 11.

Typical single-channel current traces of X-InsP3R-1 in various [Mg2+] and [ATP]free. A: current trace was recorded at optimal (6.2 μM) [Ca2+]i and saturating (10 μM) [InsP3] in the absence of ATP and Mg2+.[Modified from Mak et al. (279).] The remaining current traces were recorded at 0.25 μM [Ca2+]i and saturating [InsP3] (10 μM). [Modified from Mak et al. (281).] B: [ATP]free = 0.5 mM, [Mg2+] = 0 mM. C: [ATP]free = 0 mM, [Mg2+] = 0 mM. D: [ATP]free = 0 mM, [Mg2+] = 3 mM. E: total [ATP] = 0.5 mM, [Mg2+] = 3 mM, [ATP]free = 12 μM calculated by MaxChelator software (29). F: total [ATP] = 4.8 mM, [Mg2+] = 3 mM, [ATP]free = 1.9 mM calculated by MaxChelator. Arrows indicate closed-channel current level in the traces. Channel Po evaluated for the single-channel patch-clamp experiments yielding the current traces shown in A–F are 0.79, 0.46, 0.10, 0.10, 0.10, and 0.76, respectively.
Since ATP potentiates the activity of the X-InsP3R-1 by modulating only the sensitivity of the channel to Ca2+ activation, the functional affinity of the channel for ATP can be determined from the ATP concentration dependence of this effect. Accordingly, the half-maximal [ATP]free was 270 μM (Fig. 10A). Furthermore, ATP modulation of the channel was found to be noncooperative (281), so increasing [ATP]free up to several millimolar continued to increase the sensitivity of the type 1 channel to Ca2+ activation (Fig. 10A).
Similar to X-InsP3R-1, the maximum observed channel Po of the r-InsP3R-3 channel was not affected by [ATP]free, and the channel could be fully activated to Po of 0.8 even in the absence of ATP. However, other aspects of the regulation by [ATP]free of the r-InsP3R-3, observed under identical circumstances were dramatically different. Both its sensitivity to Ca2+ activation as well as the degree of cooperativity for Ca2+ activation were continuous functions of ATP concentration (Fig. 10B). Increases in [ATP]free increased the functional Ca2+ affinity and reduced the level of cooperativity of Ca2+ activation for r-InsP3R-3 channel. As a result, the channel had higher Po at suboptimal cytoplasmic Ca2+ concentrations in the presence of ATP. Furthermore, the r-InsP3R-3 channel was sensitive to submillimolar levels of ATP, but the effects of ATP on Ca2+ activation were saturated by 0.5 mM ATP (Fig. 10B).
Of interest is that the Ca2+ activation responses of the two isoforms are essentially the same in the absence of ATP (Fig. 10B, blue curves). Remarkably, therefore, the major feature distinguishing the types 1 and 3 channel isoforms, their Ca2+ activation properties, is only observable in the presence of ATP. In the absence of ATP, the gating behaviors of the two isoforms are indistinguishable in nuclear patch-clamp studies (279). This intricate regulation of InsP3R channel activity by ATP may have important consequences in cells that express both isoforms.
The effect of ATP to increase channel sensitivity to Ca2+ activation can be accounted for qualitatively by an allosteric model in which ATP and Ca2+ act as heterotropic activating ligands for an InsP3-bound InsP3R channel. Binding of either of the two agonists to the channel stabilizes the channel in its active form (329). However, ATP and Ca2+ are not equivalent agonists, since Ca2+ binding to an InsP3-bound channel can maximally activate it in the absence of free ATP, whereas channel Po remains low at low cytoplasmic Ca2+ concentrations even in the presence of saturating [ATP]free. This suggests that at least one Ca2+ must bind to an activating site (the InsP3-independent activating Ca2+ sensor discussed in sect. VID3) before an InsP3-liganded channel can gate open robustly, while there is no similar requirement for ATP binding (280). Depending on the relative efficacies of ATP and Ca2+ binding to stabilize the active state of the channel, ATP can either modify mainly the sensitivity of the channel to Ca2+ activation, with little effect on the level of cooperativity for Ca2+ activation (as in the case for X-InsP3R-1), or modify both the sensitivity of the channel to Ca2+ activation and the level of cooperativity for Ca2+ activation observed (as in the case for r-InsP3R-3) (280).
The observations that changes in Ca2+ activation of r-InsP3R-3 channels were saturated by 0.5 mM ATP whereas Ca2+ activation of X-InsP3R-1 continued to be affected by changes in ATP in the millimolar range (280, 281) are superficially consistent with the observation in Reference 318 that Ca2+ release was enhanced by ATP to a lesser extent in DT40 B cells expressing only InsP3R-3 than in cells expressing only InsP3R-1, although trying to account for Ca2+ signal characteristics observed at the whole cell level by single-channel InsP3R behaviors is tenuous at best (see discussion in sect. IIIB4E). The single-channel observations that both InsP3R-1 and InsP3R-3 have high sensitivity to ATP potentiation apparently contradict the conclusion derived from measurements of competitive binding of ATP to InsP3R (272) and ATP-stimulated Ca2+ release from permeabilized cells that supposedly express mostly InsP3R-1 or InsP3R-3 (316). There, InsP3R-3 channels were found to be substantially less sensitive to ATP than InsP3R-1 channels. However, it should be noted that Ca2+ release from permeabilized cells can be affected by factors in addition to the intrinsic sensitivity of the InsP3R to ATP, including the presence of different isoforms and feedback regulation of channel activity by released Ca2+. In addition, the ability of ATP to compete against ATP-derived label to bind to InsP3R may not reflect the functional affinity of InsP3R channels to ATP.
2. Significance of ATP potentiation of InsP3R channel activity
Regulation of the Ca2+ activation properties of the InsP3R by ATP complements the effects of InsP3. InsP3 activates the InsP3R mostly by reducing the sensitivity of the channel to Ca2+ inhibition, with little effect on Ca2+ activation properties (see discussion above). In contrast, physiological levels of free ATP activate the channel by potentiating Ca2+ activation. Together, cytoplasmic free ATP and InsP3 act as allosteric regulators to tune the activation and inhibition, respectively, of the InsP3R by cytoplasmic Ca2+.
The interplay between free ATP and Ca2+ concentrations in the control of InsP3R channel activities likely has important physiological significance. Whereas the MgATP concentration in the cytoplasm is in the range of 3–8 mM, the cytoplasmic free ATP concentration is in the range of 400–600 μM. The apparent affinity of the ATP sensors of types 1 and 3 InsP3R (~300 μM) coincides with the normal cytoplasmic free ATP concentrations (280, 281). InsP3R channels are therefore poised in vivo to respond to changes in the free ATP concentration. Thus the nucleotide sensitivity may enable Ca2+ release by the InsP3R to be tuned to the metabolic state of the cell. Furthermore, mitochondria and the ER have been observed to form a tightly coupled, complex signaling unit with the mitochondria in close physical proximity to the ER (393), especially to sites of Ca2+ release with high densities of InsP3R (354, 408, 424). This structural arrangement enables Ca2+ released during agonist-stimulated InsP3R activity to be effectively transmitted into the mitochondrial matrix due to the locally high Ca2+ concentrations in the microdomain of the release channels and rapid uptake of released Ca2+ by the mitochondria (16, 94, 391, 392, 402). Conversely, it also may enable local changes in ATP concentration, due to release from mitochondria into the microdomains of close ER-mitochondria apposition, to rapidly effect local InsP3R-mediated Ca2+ release. Of significance, the ATP released by mitochondria is free ATP, the InsP3R ligand, not MgATP (238). Thus communication between these two organelles may be two way, with local Ca2+ release as the means of communication from ER to mitochondria, and local ATP release providing the crosstalk from mitochondria to ER.
3. Inadequate characterization of ATP potentiation of InsP3R channel
ATP potentiates InsP3R channel activity chiefly by modulating Ca2+ activation of the channel. Even with the relatively simple [ATP]free dependence of the X-InsP3R-1, in which [ATP]free affects only the sensitivity of the channel to Ca2+ activation, the channel can exhibit different apparent sensitivity and extent of ATP potentiation (the difference between channel Po in saturating [ATP]free and 0 ATP) depending on the cytoplasmic Ca2+ concentration (Fig. 10D). Thus ATP regulation of InsP3R channel activity is intricately related to the channel’s Ca2+ dependence. Describing the ATP dependence of channel Po in a saturating concentration of InsP3 at only one cytoplasmic Ca2+ concentration, as done in most single-channel studies of the effects of ATP on InsP3R channel activities (31, 164, 478, 481, 482), is insufficient to characterize the complex allosteric effects of [ATP]free. ATP dependence of channel Po in other cytoplasmic Ca2+ concentrations cannot be deduced from the information provided in these studies. At best, only general, qualitative properties of the channel, such as whether the channel exhibits functional ATP dependence or not, can be derived from such studies. Beyond that, quantitative conclusions drawn from such investigations are unreliable. Most importantly, the functional affinity of the channel for ATP as determined from response of channel Po to ATP in a particular cytoplasmic Ca2+ concentration does not necessarily reflect the actual sensitivity of the channel to ATP. This may be a reason for the diverse values for functional ATP affinity for InsP3R observed in single-channel studies (31, 164, 478, 481, 482) and Ca2+ flux and fluorescence imaging measurements (135, 195, 317, 318).
4. Identification of functional ATP sensors regulating InsP3R channel activity
Three putative ATP binding sites were identified in InsP3R by sequence homology because they contain the glycine-rich sequence GxGxxG (274, 511), which is also found in sequences involved in nucleotide binding, including the Walker A motif (406) and the ADP-binding β-α-β-fold (512). An acidic residue (Asp or Glu) found 19–22 residues downstream from the GxGxxG sequence is conserved for all putative sites in InsP3R and may also be important for ATP binding (512). Of these three putative sites, one [termed ATPB (274), residues 2016–2021; Fig. 2B] is present in all three InsP3R isoforms, including the Drosophila InsP3R; another [termed ATPA (274), residues 1773–1778; Fig. 2B] is found only in InsP3R-1; and the third [termed ATPC (478), residues 1687–1732; Fig. 2B] is unique to SII− splice variant of InsP3R-1 (363, 365, 478). Only ATP binding to ATPA and ATPB sites has been demonstrated biochemically (273, 274). Nonneuronal X-InsP3R-1 has all three sites (241). The opisthotonos (opt) mutant InsP3R-1 has only the ATPB site (437).
With the assumption that the study by Maes et al. (273) had successfully identified all ATP binding sites in InsP3R-1 (SII+ variant, with ATPA and ATPB sites) and InsP3R-3 (with ATPB sites only) homotetrameric channels, the nuclear patch-clamp single-channel observations of the ATP dependence of X-InsP3R-1 and r-InsP3R-3 channels (280, 281) suggest that the lone ATPB site in r-InsP3R-3 must be functional with high ATP affinity. The effects of ATP binding to this sensitive ATP site were saturated by 0.5 mM ATP. In contrast, the X-InsP3R-1 studied was the nonneuronal SII− variant containing all three ATP sites. The fact that it was sensitive to ATP over a much broader range of concentrations is probably due to ATP binding to site(s) (ATPA or ATPC) either functioning in addition to and independently of the ATPB site with lower ATP affinity, or affecting the function of the ATPB site allosterically.
In another set of studies, single-channel activities of various InsP3R isoforms (481), splice variants (SII±), and opt mutant (478) were investigated under identical experimental conditions and characterized for various [ATP]free at the same cytoplasmic Ca2+ concentration. However, no simple pattern emerged from those studies to clearly relate the presence of specific ATP binding sites in the primary sequence of various InsP3R isoforms and splice variants to the functional ATP dependence of single-channel Po in those channels. InsP3R-1 SII+ channels with ATPA and ATPB sites had higher apparent ATP affinity than the InsP3R-1 SII− channel which has three sites. Whereas InsP3R-3, InsP3R-2, and InsP3R-1 opt mutant all contain the ATPB site only, the InsP3R-2 channel was not sensitive to ATP, but InsP3R-3 and InsP3R-1 opt mutant channels were, with InsP3R-3 more sensitive. Of the channels sensitive to ATP, InsP3R-1 opt mutant with only the ATPB site exhibited the greatest extent of ATP potentiation (Po in saturating [ATP]/Po in 0 ATP), whereas the InsP3R-1 SII− variant with three ATP sites had the least extent of ATP potentiation. These confusing results may be partly caused by the inadequate characterization of the ATP effect on channel Po at only one single Ca2+ concentration in these investigations. However, the results may also indicate that these ATP sites are not independent of each other, but instead function cooperatively. There may be other structural elements besides the three ATP sites that are different in the isoforms and splice variants that affect ATP regulation of InsP3R channels.
5. Potentiation of InsP3R channel activity by other nucleotides
In single-channel studies of X-InsP3R-1 channel Po at 250 nM Ca2+ and 10 μM InsP3 in the presence of various concentrations of ATP free acid and MgATP, channel Po in the presence of MgATP alone (Fig. 11E) was similar to that in total absence of ATP (Fig. 11C), which was significantly lower than that in the presence of free ATP, whether Mg2+ was present (Fig. 11F) or not (Fig. 11B). The presence of Mg2+ by itself did not affect InsP3R channel Po (Fig. 11D). Similar results were observed in extensive studies of r-InsP3R-3 (Fig. 10B) as well as X-InsP3R-1 (Fig. 10A), in experiments using different cytoplasmic Ca2+ concentrations. Together, there is a large body of evidence showing convincingly that ATP hydrolysis is not involved, and that ATP free acid (ATP3− or ATP4−) is the relevant ionic species regulating InsP3R channel Po (281, 283). In contrast, in only one set of experiments using reconstituted InsP3R channels was channel Po reportedly potentiated by MgATP to a similar extent as free ATP (31). Potentiating effects of MgATP on InsP3R channel activity were also reported in some Ca2+ flux and fluorescence imaging studies (135, 195, 272), but such studies are complicated by multiple cellular effects of MgATP.
Other nucleotides have also been reported to potentiate InsP3R channel activity, including ADP (135, 195, 272, 316, 317), AMP (135, 195, 272, 281, 316, 317), and GTP (31, 195, 272, 281, 316, 317). Adenine and adenosine were also reported to be active (317). However, the degree of potentiation by these agents reported varies considerably, in part probably because each of the studies was limited to determining channel activity at one arbitrarily selected Ca2+ concentration. Furthermore, the existence of potentiating effects has been disputed for ADP (245, 281), AMP (31), GTP (164), adenine (195), and adenosine (281). Some of this diversity may reflect differences in various isoforms studied (31, 164, 272).
6. Effects of ATP on Ca2+ inhibition of InsP3R channel
Besides modifying the sensitivity of InsP3R channel to Ca2+ activation, nuclear patch-clamp single-channel studies also revealed that increases in [ATP]free increase the sensitivities of both types 1 and 3 InsP3R channels to Ca2+ inhibition in the presence of saturating InsP3 (Fig. 10, A and B). This increase in sensitivity to Ca2+ inhibition by ATP is not due to possible displacement of bound InsP3 by ATP (195, 270) because the reduction is not reversed by increasing InsP3 (even to 180 μM) (Fig. 7A). Since type 3 InsP3R has only one putative ATP binding site according to its primary sequence (32), this observation raises the possibility that binding of ATP to a single site in the InsP3R channel can allosterically modify the properties of both Ca2+ activation and inhibition sites of the channel.
Although the presence of 0.5 mM ATP increased the sensitivity to Ca2+ inhibition of the channel, it also increased the apparent efficacy of InsP3 to reduce the sensitivity of the channel to Ca2+ inhibition so that X-InsP3R-1 in 33 nM InsP3 was half-maximally inhibited by 11 μM Ca2+ in the presence of ATP (0.5 mM, Fig. 7A) but is half-maximally inhibited by 2.3 μM Ca2+ in the absence of ATP (Fig. 10C).
A radically different effect of ATP on Ca2+ inhibition was observed for recombinant r-InsP3R-3 reconstituted in planar bilayers (481, 482). As [ATP]free was raised from 0.5 to 5 mM, the sensitivity of the channel to Ca2+ inhibition was dramatically decreased so that the channel Po versus [Ca2+]i curve was broadened from bell-shaped to plateau-shaped, resembling the curves observed in nuclear patch-clamp experiments (Fig. 8). The channel Pmax was also increased by more than eightfold. It is possible that the high [ATP]free greatly enhanced the ability of InsP3 to relieve Ca2+ inhibition of the reconstituted r-InsP3R-3 channel. However, such effects of ATP have not been observed for any other InsP3R isoform or splice variant, in particular r-InsP3R-3 channels in native membrane environment.
7. Inhibition of InsP3R channel by millimolar ATP?
ATP at high concentrations (mM) was reported to completely inhibit InsP3R channel activity observed by Ca2+ flux (135, 272) or fluorescence imaging (195, 317) measurements. Displacement of InsP3 bound to the channel by ATP was suggested to be the cause (195, 270). However, such inhibition was not observed in other studies (245, 318). With better control of ligand conditions and direct observation of channel activity, single-channel recordings of InsP3R channel currents should provide a clearer characterization of the effects of high [ATP]free. Complete inhibition of single-channel activity for channels reconstituted into planar bilayers was reported at [ATP]free~5–20 mM for InsP3R-1 (31) and at [ATP]free ~7–10 mM for InsP3R-3 (164). However, a significant fraction of the inhibition observed in these experiments may have been caused by insufficient Ca2+ buffering of the experimental solutions. In these experiments, free Ca2+ concentration in the cytoplasmic solution was buffered using chelator EGTA, and free Ca2+ concentration was calculated (127) without direct measurement. During the experiments, addition of Na2ATP to the cytoplasmic solution lowered its pH (31). This would significantly reduce the Ca2+ affinity of EGTA (29). Our calculations using MaxChelator software (29) indicated that the reported change in pH would raise the free Ca2+ concentration from 0.2 to 0.4 μM at pH 7.2 and 1.0 μM at pH 6.9. The increase in ionic strength of the experimental solutions due to addition of Na2ATP would further reduce the Ca2+ affinity of EGTA, increasing free Ca2+ concentration even more. Given that reconstituted type 1 InsP3R channels have high sensitivity to Ca2+ inhibition (34), the increase in free Ca2+ concentration due to solution acidification would cause substantial inhibition of type 1 InsP3R channel activity, independent of any presumed effects of ATP. In nuclear patch-clamp studies with Ca2+ controlled more rigorously, no reduction in channel Po in activating concentrations of cytoplasmic Ca2+ was observed as [ATP]free was raised from 0.5 to 9.5 mM for either X-InsP3R-1 (281) or r-InsP3R-3 (280).
8. Other effects of ATP on InsP3R channel activity
In addition to affecting the steady-state channel Po, ATP binding to the InsP3R also regulates its phosphorylation by protein kinase A (496) (see sect. VIL1). It also modifies the functional affinity of the channel for AdA and affects the Pmax and gating kinetics of AdA-activated channel activity (279) (see sect. VIH).
G. Ligand Regulation of InsP3R Channel Mean Open and Closed Durations
A feature of InsP3R channel gating, the mean channel open duration (to), is relatively independent of cytoplasmic Ca2+ concentration over a wide range (196, 282, 283). The channel to also shows remarkably little dependence on InsP3 concentration (164, 196, 282, 283) or [ATP] (31, 164, 280, 281). In nuclear patch-clamp experiments, to remained within a narrow range (3–15 ms for vertebrate InsP3R and 10–40 ms for insect InsP3R) in all ligand conditions applied (196, 280–283), except when the channel was activated by AdA in the absence of ATP (see later discussion). Thus the changes in channel Po in response to cytoplasmic concentrations of Ca2+, InsP3, or ATP are mostly due to changes in the mean channel closed duration tc (196, 281, 282). Thus ligand activation of InsP3R channel gating is caused primarily by increasing the channel opening rate. This suggests that an open InsP3R channel may not be sensitive to the ambient concentrations of ligands like Ca2+, InsP3, and ATP. Thus once an InsP3R channel has opened, it will remain open for a duration of approximately to, regardless of the ligand concentrations the channel is then exposed to. Specifically, although the Ca2+ released by one InsP3R channel can activate or inhibit surrounding closed InsP3R channels, the Ca2+-releasing channel itself may not be affected by the Ca2+ it releases (279).
H. Activation of InsP3R Channel by Adenophostin and Its Analogs
AdA, a fungal glyconucleotide metabolite (448), and its many analogs (1, 23, 51, 102, 290, 388, 397, 398, 420, 444, 465) were discovered as agonists of the InsP3R. Although their molecular structures are significantly different from those of InsP3 and its analogs (198), they activate the channel by interacting with the InsP3 binding site (157). AdA binds InsP3R with substantially higher affinity and is significantly more potent in stimulating InsP3R-mediated Ca2+ release than its natural agonist InsP3. Furthermore, AdA is metabolically stable (190, 334, 448). Thus AdA has been applied as a metabolically stable InsP3 substitute in studies of the InsP3R and its regulation (5, 157, 179, 203, 223, 316, 445, 487), Ca2+ release mediated by InsP3R (37, 292) and Ca2+ entry due to depletion of intracellular Ca2+ stores (60, 107, 160, 172, 193, 265). Investigations into the InsP3R binding affinity and biological activity of AdA and its analogs have also provided insights into the structural determinants for ligand interactions with the InsP3 binding site of the channel (51, 93, 332).
In extensive investigations (279), it was revealed that AdA activated the endogenous X-InsP3R-1 channel in the presence of free cytoplasmic ATP (0.5 mM) by exactly the same mechanism as InsP3, alleviating Ca2+ inhibition of the channel. Gating properties of AdA-activated channels were indistinguishable from those of InsP3-activated channels (cf. Figs. 6C and 12C). The potency of AdA to activate channels in 0.5 mM ATP was ~50 times higher than that of InsP3 (cf. Figs. 7A and 12A), which agreed well with observations that AdA binds to the InsP3R and induces Ca2+ release from Ca2+ stores with 8–100 times higher efficacy than InsP3 (190, 332, 334, 397, 420, 448).
In contrast, a very different behavior was observed when similar experiments were performed in the absence of free ATP. Even supra-saturating levels (500 nM) of AdA (Fig. 12B) could not activate the InsP3R channel to the normal Pmax of ~0.8 exhibited by InsP3-liganded channel in either the presence or absence of ATP (Figs. 7A and 10A), or AdA-liganded channels in the presence of ATP (Fig. 12A). The maximum Po achieved by the AdA-liganded channel under optimal ligand conditions was only 0.4 (Fig. 12B). Thus AdA in the absence of ATP was less efficacious than InsP3 in activating channel gating, acting instead as a partial agonist. Gating kinetics of AdA-activated InsP3R channels in optimal cytoplasmic Ca2+ concentrations were also radically different in the absence of ATP. The channels stayed open most of the time with only brief closings when they were optimally activated by InsP3, either in the presence (Fig. 6C) or absence (Fig. 11A) of ATP, as well as when they were AdA-liganded in presence of ATP (Fig. 12C). In contrast, AdA-liganded channels in the absence of ATP had substantially shorter channel openings (Fig. 12D). As noted earlier, in all experimental ligand conditions used in nuclear patch-clamp studies, the mean open duration to of the InsP3R channel was remarkably constant (196, 279–283). The one exception is when the channel was activated by AdA in the absence of free ATP, when the channel was observed to gate with significantly shorter to (279).
The cytoplasmic Ca2+ concentration dependence of the AdA-liganded channel Po in the absence of ATP (Fig. 12B) was comparable to that for channels activated by InsP3. Other than the lower optimal Pmax for the AdA-activated channels, they exhibited similar Ca2+ sensitivities and similar levels of cooperativity for both activation and inhibition as the InsP3-activated channels in the absence of free ATP. Interestingly, the gating kinetics and Pmax of AdA-liganded channels in the absence of ATP are remarkably similar to those observed in many studies of InsP3-activated channels reconstituted in planar lipid bilayer (381, 382, 478, 479, 481, 482). This suggests that the InsP3R may have two distinct activated states when it is liganded at the InsP3 binding site: a fully activated state with higher Pmax (~0.8) and longer to (5–15 ms) achieved when the channel is liganded by InsP3 or AdA in the presence of ATP (observed in outer nuclear membrane) and another partially activated state with lower Pmax (~0.4) and shorter to (~2–5 ms) achieved when the channel is liganded by AdA in the absence of ATP (in outer nuclear membrane patches), or liganded by InsP3 (reconstituted channels in planar bilayers).
Another unique aspect regarding the effect of ATP on the efficacy of AdA as a ligand concerns the potency of AdA. In the absence of ATP, AdA is only ~1.5 times more potent than InsP3 (Figs. 10C and 12B), whereas it is ~50 times more potent than InsP3 in the presence of 0.5 mM ATP (cf. Figs. 10B and 12A). This effect of ATP is not observed when InsP3 is the activating ligand (cf. Figs. 7A and 10C) and indicates that ATP allosterically regulates the affinity of AdA binding to the InsP3R channel (279).
The reduced efficacy of AdA under certain conditions (insufficient free ATP, for instance), producing channel activities with lower Po and shorter to, may explain how, despite being a more potent agonist of InsP3R channel, AdA elicits a slower rate of Ca2+ release than InsP3, thereby generating Ca2+-dependent Cl− currents different from those generated by InsP3 (172, 265), and activates Ca2+ entry with an apparent lack of Ca2+ release from stores (107). More importantly, this may be the reason for the spatial and temporal differences between Ca2+ signals activated by AdA and InsP3 (37, 265, 292). This provides insights into the relationships between the characteristics of Ca2+ signals (duration of Ca2+ puffs and rate of propagation of Ca2+ waves) generated by the coordinated activities of InsP3R channels observed in vivo and the single-channel kinetic properties (channel to) of single InsP3R channels (279).
I. Ligand-Dependent, InsP3-Induced InsP3R Channel Inactivation
A fundamental yet surprising aspect of InsP3R-mediated intracellular signaling is the phenomenon of “quantal release,” defined (308, 333) as the ability of cells to have graded release of Ca2+ from intracellular stores in response to incremental levels of extracellular agonist or InsP3 (reviewed in Refs. 46, 315, 362, 458). This entails two different processes: 1) an initial Ca2+ release whose rate is proportional to InsP3 concentration followed by 2) a substantial reduction in rate or termination of Ca2+ release despite the presence of constant InsP3. Consequently, sustained exposure to submaximal levels of agonists, even over extensive periods, only mobilizes a fraction of total releasable Ca2+ in a cell. This is surprising because, with the steady-state ligand regulation of InsP3R channel Po discussed so far, it might have been expected that all InsP3R channels should become activated in response to sufficient agonist stimulation, releasing all of the InsP3-sensitive Ca2+ stores, albeit at different rates depending on the agonist concentration. Time-dependent reduction in the rate of InsP3-mediated Ca2+ release has been well-documented with flux assays (120, 167, 168, 293) and fast perfusion protocols (5, 82, 91, 120, 140, 293, 513). Furthermore, the InsP3R has been observed to transform from a low-affinity, active state to a high-affinity, desensitized state (92, 293). Moreover, observations of refractory periods following either global (79, 234, 350) or more focal (303, 359) InsP3-mediated Ca2+ release in intact cells are also consistent with channel inactivation in intact cells. However, the ability of maintaining constant conditions in these relatively macroscopic studies of InsP3R channel activity has been questioned (458), and other studies have disputed channel inactivation as a mechanism for release termination (24, 90, 187, 351, 361, 462) and invoked instead other types of mechanisms, including the presence of discrete Ca2+ stores with different densities of InsP3R or sensitivities to InsP3, or the presence of heterogeneous InsP3R channels in a continuous store (different channel isoforms with alternatively spliced variants and variable posttranslational modifications have been proposed), or different proposed mechanisms of release termination, including regulation of InsP3R activity by ER luminal Ca2+ or by desensitization (reviewed in Refs. 46, 315, 362, 458).
In nuclear patch-clamp single-channel studies of the InsP3R, abrupt termination of channel activities despite the constant presence of agonist has been observed for all InsP3R investigated: various isoforms from various species, endogenous or recombinant, InsP3 or AdA stimulated (42, 196, 277–279, 284). The mean duration of channel activity observed from initial activation of the channel by InsP3 until the termination of activity in the presence of constant InsP3 (Ta) for vertebrate InsP3R is typically ~30 s (278, 284). Even though a cytoplasmic Ca2+ concentration dependence of Ta was qualitatively described for recombinant r-InsP3R-1 expressed in COS-7 cell outer nuclear membrane (42), it was impossible, due to technical difficulties, to rule out the possibility that such abrupt termination of channel activity was a nonphysiological artefact associated with patching, for example, collisions of the channels with the walls of the glass pipette. In a recent nuclear patch-clamp study of the Sf9 InsP3R channel gating in which channel Ta under various concentration of Ca2+ and InsP3 was investigated systematically, it was demonstrated that Ta was dependent on the concentrations of both ligands (Fig. 13A) (196). In optimal ligand conditions (InsP3 = 10 μM, Ca2+ = 1 μM), Ta was ~120 s, substantially longer than the vertebrate channels. In 10 μM InsP3, Ta was reduced in [Ca2+]i > 1 μM, with reduction by over 10-fold at 89 μM Ca2+. In subsaturating (33 nM) InsP3, Ta already began to decrease in [Ca2+]i ~300 nM, substantially lower than that observed in saturating InsP3. Furthermore, it was demonstrated that the InsP3-induced termination of InsP3R channel activity was fully reversible upon ligand removal (196). These results suggest that the observed inevitable termination of channel activity is not an experimental artefact, and may be due to the entry of InsP3-liganded channels into a true inactivated state, driven by binding of Ca2+ to the channel at a relatively slow rate (196).
FIG. 13.

Dependencies on [Ca2+]i and [InsP3] of InsP3R channel activity duration and recruitment. A: channel activity duration. Data points are averages of channel activity durations in ligand conditions as tabulated. Smooth curves in graphs in this figure were drawn by hand for clarity. B: ligand-dependent recruitment of InsP3R. Data points are average number of active channels in membrane patches (NA) in ligand concentrations as tabulated. C: ligand-dependent relative magnitude of InsP3R-mediated Ca2+ release. The product NAPo, determined using data shown in Fig. 6B and Fig. 1D, in ligand concentrations as tabulated. [Modified from Ionescu et al. (196).]
The inactivation kinetics observed in nuclear patch-clamp experiments, Ta ~10–100 s in Sf9 InsP3R (196) and ~20–30 s in vertebrate InsP3R (42, 278, 284), were slower than those observed in superfusion experiments (91, 120, 141) and in intact cells in response to photorelease of InsP3 (234), but were comparable to those estimated by ER permeability measurements in permeabilized hepatocytes (167), as well as to the kinetics of InsP3-induced increases in InsP3 affinity of an apparently desensitized InsP3R in cerebellar microsomes (92) (half-life of channel activity ~15–45 s) and the kinetics of the transient fast phase of Ca2+ release in response to initial exposure to InsP3 in permeabilized and intact cells (see Refs. 308, 333, 462). Of note, InsP3R inactivation observed in permeabilized hepatocytes (167), which had kinetics similar to those observed in the single-channel studies, was shown to account for release termination associated with [Ca2+]i oscillations (168). Together these results suggest that the kinetics of inactivation observed in single-channel studies are of physiological relevance for [Ca2+]i signaling in cells. However, it remains unclear whether distinct inactivation kinetics observed in different studies reflect methodological differences, distinct types of inactivation or inhibition, or a physiologically relevant range of kinetics of a common inactivation mechanism.
J. Ligand-Dependent InsP3R Channel Recruitment
In addition to termination of Ca2+ release in the presence of constant InsP3, quantal Ca2+ release requires that the initial rate of Ca2+ release from intracellular stores be proportional to InsP3 concentration. One mechanism to achieve this is by InsP3-tuning of Ca2+ inhibition of channel activity, as discussed in section VIC1. Additional mechanisms were recently discovered to also play a role in recruiting channels into activity as a function of ligand stimulation.
A consistently high rate of detection of InsP3R channel activity in nuclear patch-clamp experiments using isolated Sf9 nuclei (60–80% of nuclear membrane patches obtained exhibited InsP3R channel activity in optimal ligand conditions) enabled detailed quantification of the average number of InsP3R channels detected in a nuclear patch-clamp experiment (NA) under various concentrations of InsP3 and Ca2+ (Fig. 13B) (196). In saturating InsP3, each patch contained 1.3 active InsP3R channels on average, i.e., NA = 1.3 in 50 nM Ca2+. At higher Ca2+ concentrations, more channels were detected in each patch, with maximum NA of 3.0 at 500 nM Ca2+. Above ~8 μM Ca2+, NA was reduced. In subsaturating InsP3 (33 nM), consistently lower NA was observed over all Ca2+ concentrations.
The observation that NA was a function of stimulus strength is unexpected since it was anticipated that the entire channel population in a membrane patch would always become activated, albeit to different levels of activity (Po) depending on the strength of ligand activation. Instead, these results indicate that suboptimal ligand concentrations are insufficient to activate all available InsP3R channels in a membrane patch that can be activated by optimal ligand concentrations. To account for these observations, a model was proposed in which Ca2+ binding at a very fast rate to a “sequestration” site, before these channels could actively gate open and be observed, sequestered some of the available InsP3R channels into a nonactive state when ligand conditions were suboptimal (196).
Thus two mechanisms revealed by single-channel patch-clamp electrophysiology exist to grade Ca2+ release through a population of InsP3R channels: regulation of channel activity level (Po) as well as recruitment of additional channels. Both mechanisms coexist and can occur even in the absence of cross-talk among channels by released Ca2+ (CICR). The importance of this novel channel recruitment mechanism can be appreciated by considering that the rate of Ca2+ flux through a population of InsP3R channels (J) is given by
| (3) |
where γ is the single-channel conductance, N is the number of activated channels, and Po is the average single-channel open probability. Single-channel studies determined that the InsP3R channel conductance γ is largely InsP3 independent, as discussed in section V. Since both channel Po and NA are regulated by Ca2+ and InsP3, the ligand dependence of Ca2+ flux released through InsP3R channels is more accurately estimated in terms of NAPo (Fig. 13C). In saturating concentrations of InsP3, the dependence of NAPo on cytoplasmic Ca2+ concentration is biphasic: NAPo increases by over 10-fold as Ca2+ is increased from 50 to 500 nM and then gradually decreases as Ca2+ is further increased. NAPo is also strongly dependent on InsP3 concentration. With InsP3 reduced to 33 nM, the dependence of NAPo on Ca2+ remains biphasic with peak NAPo observed at Ca2+ ~0.5–1 μM Ca2+, but maximum NAPo is an order of magnitude lower than that observed in saturating InsP3 (196).
K. Ca2+ Sensors Regulating InsP3R Channel Activity
Single-channel studies of ligand regulation of InsP3R channel activity have revealed that Ca2+ is intimately involved in regulating many aspects of InsP3R channel activity, including activation (increasing steady-state channel Po), inhibition (decreasing steady-state channel Po), inactivation (termination of channel activity in the presence of constant ligand concentrations), and sequestration (changing the fraction of channels in a population that is activated). Moreover, other ligands exert their effect on InsP3R channel activity by altering the functional Ca2+ affinities of the channel: the main effect of InsP3 is modulating sensitivity of the channel to Ca2+ inhibition, and ATP potentiates channel activity by increasing its sensitivity to Ca2+ activation. However, despite the identification of multiple putative Ca2+ binding sites in the InsP3R primary sequences (421, 422), relatively little progress has been made in determining the amino acid sequences that constitute the functional Ca2+ sensors in the InsP3R.
A point mutation of a single glutamate residue in the RyR was found to greatly reduce the sensitivity of the channel to Ca2+ activation (85). This amino acid residue is conserved in all RyR and InsP3R isoform sequences by homology analysis (319). Although the region around this residue (~20 residues upstream and 18 downstream) is also highly conserved among the RyR isoforms and among the InsP3R isoforms, the RyR sequence bears little resemblance to the corresponding InsP3R sequence (319). Point mutations of the equivalent Glu residue in the InsP3R (residue 2100; Fig. 2B) substantially reduced the capability of InsP3R to release Ca2+ (319) and increased the dissociation constant Kd for Ca2+ binding to a peptide fragment containing the highly conserved region (479). Single-channel studies of the effect of point mutations at this residue in the r-InsP3R-1 (479) showed that the mutations seemed to decrease the sensitivities of the InsP3R channels to both Ca2+ activation and inhibition. Furthermore, the Kd for Ca2+ binding to the peptide fragment containing the conserved region was consistent with the Ca2+ activation and inhibition sensitivities of the channels derived from fitting the bell-shaped single-channel Po versus [Ca2+]i curves with a biphasic Hill equation (Eq. 2) (479, 481). On the basis of these results and the observation that the Ca2+ dependence of various isoforms and splice variants of InsP3R reconstituted in planar lipid bilayers could be described with a biphasic Hill equation (Eq. 2) giving apparently similar functional affinities for Ca2+ activation and inhibition (34, 229, 434, 438, 478, 479, 481), it was concluded that the conserved region around the glutamate residue is the Ca2+ sensor for the InsP3R (30, 319, 479).
However, because of the narrow shape of the Ca2+ dependence curve of channel Po observed in the planar lipid bilayer experiments, the set of parameters for the biphasic Hill equation that fit the experimental data is not unique (see sect. VIB5, especially Fig. 3). Thus finding a set of Hill equation parameters with Kact ~ Kinh that provides a good fit to the Po versus [Ca2+]i data for the channels does not mean that the channel has similar Ca2+ affinities for activation and inhibition. Similarly, the fact that Po versus [Ca2+]i data can be fitted by a parameter set with Kact and Kinh similar to the Kd for Ca2+ binding to the conserved peptide region does not imply that the functional affinities of the InsP3R channel for Ca2+ activation and inhibition are similar to the Ca2+ binding affinity of the conserved peptide region. In addition, while the narrow bell-shaped Ca2+ dependence of some InsP3R can be fitted by the biphasic Hill equation with Kact ~ Kinh, narrow bell-shaped Ca2+ dependencies are far from universal for all InsP3R studied. In fact, the functional affinity for Ca2+ activation was over two orders of magnitude greater than that for Ca2+ inhibition for InsP3Rs studied in nuclear patch-clamp studies (42, 196, 282, 283). It is difficult to see how the same Ca2+ sensor can regulate Ca2+ activation and inhibition over such different [Ca2+]i ranges. Moreover, in some studies, InsP3R channels found to be insensitive to Ca2+ inhibition were still activated by Ca2+ (285, 380, 382), suggesting that Ca2+ inhibition is regulated independently from Ca2+ activation for InsP3R channels. Furthermore, it is possible that Ca2+ binding to the conserved region modulates both Ca2+ activation and inhibition of the channel allosterically without being the Ca2+ sensor that regulates channel activity directly, similar to ATP binding affecting both the affinities of the channel for Ca2+ activation and inhibition (Fig. 10, A and B).
Detailed single-channel studies of the effect of [Ca2+]i on InsP3R channel activity revealed that Ca2+ regulates different aspects of InsP3R channel activity with very different functional affinities, binding kinetics, and ligand dependence. Therefore, multiple different distinct Ca2+ sensors are likely to be involved. There is one Ca2+ sensor responsible for InsP3 modulation of Ca2+ inhibition of the channel. It changes from an inhibitory Ca2+ binding site to an activating binding site depending on the concentration of InsP3 (see discussion in sect. VID2). Another InsP3-independent Ca2+ sensor is responsible for the consistent Ca2+ activation observed in a wide variety of InsP3R channels (see discussion in sect. VID3). Yet another Ca2+ sensor is InsP3 independent but inhibitory, with variable functional Ca2+ affinity that can be changed by factors extrinsic to the InsP3R channel like membrane lipid composition and exposure to low [Ca2+]i (see discussion in sect. VID1). Elimination of the functionality of this purely inhibitory Ca2+ binding site by exposure to very low bath Ca2+ concentrations (see discussion in sect. VIB5) implies that the channel possesses a fourth Ca2+ binding site with very high Ca2+ affinity that must be occupied by Ca2+ for the Ca2+ inhibitory site to be functional. There is a fifth Ca2+ sensor that binds Ca2+ at a low rate (~0.01 s−1) but only when the channel is liganded with InsP3. This sensor is responsible for InsP3-induced inactivation of InsP3R channels. Ca2+ binding to this site causes the channel to enter into an inactive state from which it cannot emerge in the continuous presence of InsP3 (see discussion in sect. VII). A sixth Ca2+ sensor is responsible for sequestration of InsP3R channels before they can become active. Ca2+ binding to this Ca2+ sensor also causes the channel to enter into a nonactive state, but at a rate (~1 s−1) orders of magnitude higher than that for the inactivation Ca2+ sensor (see discussion in sect. VIJ).
Thus, instead of the simple picture with one single identified Ca2+ sensor being responsible for Ca2+ regulation of InsP3R channel activity (30), evidence points to a much more complex picture with multiple functional Ca2+ sensors in the channel, and the molecular identities of these sensors remain to be properly elucidated. Regulation of InsP3R channel activity by these Ca2+ sensors is probably complicated, with various degrees of cooperativity and hierarchical organization so that Ca2+ binding to one sensor can regulate functionality of another Ca2+ sensor. Structurally, these Ca2+ sensors can either be intrinsic to the channel or reside in a protein(s) associated with the InsP3R channel complex.
L. Channel Regulation by Phosphorylation
The most important regulators of InsP3R channel function are InsP3 and Ca2+. Nevertheless, the channels are importantly regulated by phosphorylation by numerous kinases, including cAMP-dependent protein kinase (PKA) (63, 110, 133, 456, 498, 518), cGMP-dependent protein kinase (PKG) (174, 239, 395, 410), calmodulin-dependent protein kinase II (CaMKII) (17, 19, 180), protein kinase C (PKC) (134, 299, 492), and various protein tyrosine kinases (PTK) (95, 202).
1. Phosphorylation by PKA
Although PKA phosphorylation of the InsP3R-1 was recognized soon after it was cloned (133), the functional consequences of InsP3R phosphorylation were not clear, with discrepancies existing regarding whether PKA phosphorylation of InsP3Rs increased (110, 166, 338, 456, 494, 518) or decreased (73, 442) Ca2+ release activity. With hindsight, some of the discrepancies were likely caused by use of different methods (e.g., 45Ca2+ flux versus single-channel patch-clamping), presence of confounding contributions of increased ER Ca2+ uptake activity (98), and the presence or absence of accessory proteins or different channel isoforms. It now appears to be more generally agreed that PKA phosphorylation of InsP3Rs augments Ca2+ release (371).
The InsP3R-1 contains two PKA consensus sequences (RRXS/T) at Ser-1589 and Ser-1755 (Fig. 2B), which become phosphorylated upon the elevation of cAMP levels (133). These sites are conserved from Drosophila to human, but this canonical motif is not present in either InsP3R-2 or InsP3R-3. The 39-amino acid region that is spliced out in the peripheral type 1 receptor variant (SII− variant) is located between the two phosphorylation sites (Fig. 2B). The neuronal type 1 variant (SII+ variant) is more heavily phosphorylated at Ser-1755, whereas the peripheral SII− variant is preferentially phosphorylated at Ser-1589 (133). As discussed earlier, splicing of this region creates a glycine-rich motif (GXGXXG) at residues 1688–1732 in the SII− variant (137, 363) that binds ATP (496) (called the ATPC site in Ref. 478). Agonist-induced InsP3-mediated Ca2+ release was dramatically potentiated following elevation of cAMP in DT40 InsP3R triple-knock-out (DT40-TKO) cells expressing the wild-type SII− InsP3R or channels with mutations in the ATPA and ATPB sites, but phosphorylation of the receptor and potentiation of Ca2+ release were absent in cells expressing a channel with a mutation in the ATPC site (496). These results suggest that ATP binding specifically to the ATPC site in SII− InsP3R-1 controls the susceptibility of the receptor to PKA-mediated phosphorylation, which in turn contributes to the functional sensitivity of the SII− InsP3R-1 to InsP3.
The functional roles of phosphorylation at either PKA site in InsP3R-1 have been examined by studies of DT40-TKO cells engineered to express InsP3R-1 channels with mutations at either site designed to either mimic or prevent phosphorylation (497, 498). Interestingly, the functionally important phosphorylation sites were different in the two splice variants. Despite the equal susceptibility of phosphorylation of either site in the SII+ variant (431), enhancement of InsP3-induced Ca2+ release by PKA was mediated specifically by phosphorylation of Ser-1755. In contrast, both phosphorylation sites were functionally relevant in the SII− variant (497, 498). However, phosphorylation of both sites could not be observed, suggesting that phosphorylation of one site may preclude phosphorylation of the other site (497).
Effects of PKA-mediated phosphorylation of wild-type InsP3R-1 have been studied at the single-channel level (110, 123, 456). Phosphorylation by PKA of reconstituted recombinant rat InsP3R-1 enhanced single channel Po approximately fivefold from ~0.07 to ~0.35 without affecting the narrow bell-shaped [Ca2+] dependence (456). It was suggested that PKA phosphorylation enhanced the apparent sensitivity to InsP3.
By analogy with the RyR, it was suggested that phosphorylation of InsP3R-1 might be accomplished within a macromolecular signaling complex containing PKA, phosphatases, and the InsP3R itself (110). Biochemical pull-down experiments indicated that PKA, protein phosphatase (PP)1 and PP2A and InsP3R-1 formed intermolecular complexes in rat brain (110). An association of PKA and PP1α with the InsP3R-1 was shown to be mediated by AKAP9 (Yotiao), a multifunctional PKA-anchoring protein, via a leucine/isoleucine zipper (LIZ) motif (residues 1251–1287; Fig. 2B) (480). In addition, PP1α was also shown to bind directly to the COOH terminus of InsP3R-1 specifically (Fig. 2B) (456). PKA catalytic subunit-mediated enhancement of reconstituted recombinant rat InsP3R-1 channel Po was reversed by addition of recombinant PP1α (456). Taken together, these data indicate that phosphorylation of InsP3R-1 by cAMP in situ may be mediated by a complex of proteins associated with the channel, in which PKA and PP1α work antagonistically to modulate InsP3R phosphorylation status. However, it remains to be established whether these in vitro interactions and functional effects are recapitulated in vivo.
Although the canonical motifs expressed in the type 1 receptor are not conserved in the type 2 and type 3 InsP3Rs, it has nevertheless been demonstrated that both InsP3R isoforms can also be phosphorylated by PKA (518). The type 1 channel appears to be more susceptible to PKA-mediated phosphorylation than the other two isoforms (432, 518). The specific sites in the type 3 channel have been localized to Ser-916, Ser-934, and Ser-1832, with Ser-934 most susceptible (432). The functional consequences of specific phosphorylation of either of the types 2 or 3 isoforms are still unknown however.
2. Phosphorylation by PKG
PKG phosphorylates InsP3R-1 at the identical sites as PKA (239, 431). Using S1589A and S1755A mutated InsP3R-1-transduced DT40-TKO cells, Wagner et al. (498) showed that, as with PKA, PKG preferentially phosphorylated S1755 in the SII+ InsP3R-1 variant, which similarly increased the sensitivity of InsP3R to InsP3 and enhanced agonist-induced Ca2+ release. Cells expressing S1755A SII+ InsP3R-1 were insensitive to cGMP, suggesting that PKG does not phosphorylate Ser-1589 or that its phosphorylation has no effect. The latter seems unlikely though, since PKA phosphorylation of Ser-1589 enhances InsP3-mediated Ca2+ release. In contrast, similar studies in 293 cells indicated that Ser-1589 is the preferred phosphorylation site (431). The SII− InsP3R-1 variant is insensitive to cGMP, suggesting that it may not be capable of being directly phosphorylated by PKG (498). Nevertheless, PKG appears to inhibit InsP3-mediated Ca2+ release in peripheral tissues that express predominately the SII− splice variant (335). PKG inhibition of InsP3-mediated Ca2+ signals in smooth muscle is associated with the phosphorylation of a widely expressed accessory protein termed IRAG (InsP3R-associated cGMP kinase substrate)(11, 410). cGMP-dependent protein kinase I (cGKI) forms a trimeric complex with the InsP3R and IRAG (11, 410), and heterologous coexpression of the three proteins in COS-7 cells conferred cGMP inhibition of bradykinin-stimulated Ca2+ release (410). The molecular mechanisms by which the complex inhibits InsP3R-1 channel activity have yet to be defined.
3. Phosphorylation by PKC and CaMKII
PKC and CaMKII phosphorylate purified reconstituted InsP3R-1 in liposomes at distinct sites (134). PKC enhanced InsP3-mediated Ca2+ release from isolated nuclei (299). PKA enhanced, whereas Ca2+ inhibited, PKC phosphorylation of InsP3R-1 (492). However, the influence of PKC on InsP3R channel properties has not been investigated.
The InsP3R contains several CaMKII consensus phosphorylation sequences. Phosphorylation of InsP3R by CaMKII has been implicated in neurotransmitter release (180) and InsP3-dependent Ca2+ oscillations in some systems (296, 549). Specific pharmacological inhibition of CaMKII in HeLa cells (549) and Xenopus oocytes (296) potentiated InsP3-dependent Ca2+ release, whereas introduction into the cytoplasm of a constitutively active CaMKII catalytic subunit inhibited Ca2+ release in HeLa cells (549). The predominant InsP3R isoform in cardiac ventricular myocytes, the InsP3R-2, localizes with CaMKIIδB in the nuclear envelope (19). CaMK-IIδB interacts with and phosphorylates InsP3R-2 in ventricular myocytes, with the interacting and phosphorylated region within the InsP3R localized to the NH2-terminal 1078 residues (19). In single-channel studies using microsomes from InsP3R-2 transfected COS-1 cells, Po of reconstituted InsP3R-2 channels in planar bilayer membranes activated by 2 μM InsP3 (Cs+ was the current carrier with 250 nM Ca2+ present on the cytoplasmic solution) was profoundly reduced from ~0.43 to 0.04 by pretreatment of the microsomes with CaMKII (19). CaMKII-mediated inhibition of channel Po was associated with a 12-fold increase of the channel mean closed time and 2-fold decrease of the channel mean open time. Inhibition of channel Po was not observed when the microsomes were pretreated with CaMKII together with the kinase inhibitor KN-39, suggesting that CaMKII-mediated phosphorylation was responsible for the reduced Po. Although limited, the published results taken together suggest that the activity of InsP3R-2, and possibly other isoforms, is inhibited by CaMKII-mediated phosphorylation. However, it has still not been firmly established that CaMKII-mediated inhibition of channel activity is a direct consequence of channel phosphorylation. Furthermore, additional single-channel experiments using a range of InsP3 and cytoplasmic Ca2+ concentrations are needed to establish the mechanism of channel inhibition. It has been proposed that InsP3R-mediated released Ca2+ activates CaMKII to enable it to modulate Ca2+ oscillations (549) or drive transcription factor translocation between the cytoplasm and nucleus (521). As such, inhibition of InsP3R activity by CaMKII may provide a negative-feedback mechanism that is important in these processes.
4. Phosphorylation by Akt kinase
The serine/threonine protein kinase Akt/protein kinase B is activated by various growth factors and cytokines and phosphorylates a number of key substrates of intermediary metabolism and promotes cell survival, proliferation, and growth (510). A consensus sequence for Akt phosphorylation [RXRXX(S/T)] (246) is present in the COOH-terminal tail of all three InsP3R isoforms, and is conserved in InsP3Rs from most species (233). Akt phosphorylates InsP3R in vivo and in vitro at Ser-2681 (Fig. 2B), but mutagenesis of that residue to either mimic phosphorylation or inhibit it was without effect on InsP3-induced 45Ca2+ release from microsomes or agonist-induced Ca2+ transients in transfected DT40-TKO cells (233). On the other hand, Akt phosphorylation of InsP3R-1 modulated caspase-3 activation in response to apoptotic stress (233), but the link between these events was not established. It was suggested that Akt phosphorylation of InsP3R-1 may modulate protein interactions with the channel that impinge on apoptosis progression (233), such as cytochrome c (44) and Bcl-XL (508), which also bind to the COOH terminus.
5. Phosphorylation by tyrosine kinases
In addition to the serine/threonine residues, phosphorylation of InsP3R also occurs at tyrosine residues. The human SII− InsP3R-1 is tyrosine phosphorylated during T-cell activation (171, 202). Both Src as well as Fyn non-receptor tyrosine kinases directly phosphorylated InsP3R-1 in vitro, and InsP3R-1 and Fyn could be coimmunoprecipitated upon T-cell activation (202). Fyn phosphorylated the InsP3R-1 in vitro and in vivo at Tyr-353 (Fig. 2B) within the β-trefoil domain of the core InsP3-binding domain immediately adjacent to the SI splice site (95). This residue is conserved among all three InsP3R isoforms. However, mutagenesis of this site did not abolish tyrosine phosphorylation, suggesting that other phosphorylation sites are also present. In agreement, tyrosine phosphorylation of InsP3R-1 was diminished but not eliminated during T-cell activation in Fyn knockout mice (202). Nevertheless, [Ca2+]i transients associated with T-cell receptor engagement were diminished in Fyn knockout mice (202), suggesting that tyrosine phosphorylation has functional relevance. Fyn phosphorylation of Tyr-353 increased the InsP3 binding affinity (95). In single-channel planar bilayer studies of reconstituted rat cerebellar microsomal or purified InsP3R (presumably type 1), with Ca2+ as the charge carrier, Fyn enhanced channel activity induced by 2 μM InsP3 as a consequence of a reduction in the channel mean closed time (202). Of note, this effect was observed in the presence of 750 nM cytoplasmic Ca2+, a Ca2+ concentration that normally inhibits InsP3R channel activity in planar bilayers, suggesting that Fyn-mediated phosphorylation may function by relieving high-[Ca2+] inhibition of the channel. Such a functional effect is consistent with a primary effect of Fyn to enhance InsP3 binding affinity, as observed.
6. Phosphorylation by cdks/CyB
Phosphorylation of InsP3R can be modulated by the cyclin-dependent kinases (cdks) that regulate eukaryotic cell cycle progression (288). The cyclin-dependent kinase 1/cyclin B (cdc2/CyB) complex phosphorylates InsP3R-1 at Ser-421 and Thr-799 and InsP3R-3 at Ser-795 (252, 288). Mutagenesis of three residues together (Arg-391, Arg-441, and Arg-871) inhibited binding of the cdc2/CyB complex with the InsP3R-1 (252), suggesting they may be involved in interactions between the proteins that enable the phosphorylation of the channel. Functionally, cdc2/CyB-mediated phosphorylation enhanced the affinity of the InsP3-binding region for InsP3 and enhanced InsP3-mediated Ca2+ release from microsomes (252, 288).
M. Regulation by Redox Status
The redox status of the InsP3R may also play a role in regulating the function of the InsP3R channel. RyR channels are highly sensitive to changes in thiol redox state (118, 183). RyR1 contains up to 100 cysteine residues per monomer, with ~20–50 of these residues free for modification by oxidation, nitrosylation, or alkylation (126, 441). However, much less is known regarding of the roles of thiol redox status of InsP3R. Nearly 70% of the 60 thiol groups in the InsP3R-1 can be modified by small lipophilic thiol-specific probes (213). InsP3R-1 can be activated by oxidative reagents, with thimerosal the best documented (50, 186, 221, 323, 360, 468) for causing increased [Ca2+]i in treated cells. Thimerosal appears to act by sensitizing the InsP3R to subthreshold levels of InsP3 in the cell (50, 314), although it only moderately enhances InsP3 binding (314). Interaction of the first 225 amino acids (suppressor domain) with the InsP3-binding core domain of InsP3R-1 was enhanced by thimerosal (67). It is possible that thimerosal facilitates conformational changes involved in channel activation.
The InsP3R-1 was also shown to interact biochemically and functionally with the ER luminal chaperone ERp44, a member of the thioredoxin family that may link InsP3R function to ER redox status (184). This interaction is discussed further in section VIN6.
N. Regulation by Interacting Proteins
To date, a large number of protein interactions with the InsP3R have been described (reviewed in Ref. 365). In this section, we discuss how various protein-protein interactions contribute to the impressive diversity of spatially and temporally distinct InsP3R signaling behaviors. Focus will be applied to interactions that have been more extensively characterized, in particular, those involved in allosteric modulation of channel gating.
1. Calmodulin
The ubiquitous and highly conserved Ca2+-binding protein calmodulin (CaM) confers Ca2+-dependent regulation on many proteins, including ion channels (404). Binding of Ca2+ to CaM (Ca2+-CaM) triggers a conformational change that promotes or modulates its interaction with binding partners. Ca2+-free CaM (apoCaM) can also interact with target proteins, enabling CaM to function, in essence, as a Ca2+-regulated subunit of the protein. In this way, CaM mediates Ca2+-dependent and -independent regulation of many ion channels, including the RyR (18). Although it is generally accepted in the literature that CaM interacts with the InsP3R, the in vivo association between the two proteins as well as the functional implications are far from unequivocal (recently reviewed in Refs. 336, 401, 461).
The first CaM binding site to be identified in the InsP3R encompassed residues 1564–1585 (Fig. 2B) of the coupling domain of the mouse type 1 receptor (270). The interaction was found to be Ca2+ dependent in CaM-Sepharose binding assays (270, 525). Whereas purified InsP3R-1 channels reconstituted into lipid bilayers lacked inhibition by high [Ca2+], channels reconstituted from cerebellar microsomes displayed high [Ca2+] inhibition (310). This result was interpreted as indicating that a component of Ca2+-dependent inhibition was lost during purification, and implied that Ca2+ inhibition of steady-state InsP3R channel gating was not mediated by Ca2+ interaction with a site intrinsic to the InsP3R itself (310). Addition of CaM to the bath solution restored Ca2+-dependent inhibition to the purified receptors, suggesting that high [Ca2+] inhibition of channel gating was mediated by CaM (310). Addition of the CaM antagonists either W-7 or the CaM binding domain of CaMKII activated reconstituted cerebellar microsomal InsP3R, which was interpreted as indicating that microsomal InsP3R is constitutively associated with and inhibited by CaM. The conclusion that high [Ca2+] inhibition of InsP3R gating is mediated by CaM was reinforced by observations that the type 3 InsP3R did not bind CaM (76, 254, 525) and its steady-state gating was not inhibited by high cytoplasmic Ca2+ concentrations (163).
The study of Michikawa et al. (310) has been repeatedly cited as evidence that Ca2+ inhibition of steady-state InsP3R channel gating is mediated by CaM. However, many experimental results suggest that this is not accurate. First, surface plasmon resonance analysis indicated that the Kd for Ca2+-CaM binding to the channel was 27 μM (191), suggesting a low-affinity interaction, inconsistent with constitutive association with reconstituted channels. In agreement, high concentrations (10 μM) of CaM were required for inhibition of Ca2+ release from purified InsP3R-1 to be observed in the presence of ~200 μM Ca2+ (191). Second, the type 3 InsP3R channel is not insensitive to Ca2+ inhibition as originally claimed. Biphasic Ca2+ concentration-dependent regulation of type 3 channels has been demonstrated in electrophysiological recordings of the rat channel by nuclear membrane patching (283) and in planar bilayer reconstitutions (482). Because the type 3 channel does not bind CaM, Ca2+ inhibition of steady-state channel gating can therefore occur in the absence of CaM binding (76). Third, in nuclear patch-clamp recordings of the InsP3R-1 in Xenopus oocytes, inhibition of channel activity by high concentrations of cytoplasmic Ca2+ was observed in the presence of the CaM antagonist W-7 and despite the overexpression of a dominant-negative, Ca2+-insensitive CaM (286). Fourth, mutation of the CaM-binding site eliminated CaM interaction with the InsP3R (525), but it did not affect Ca2+ inhibition of channel activity (347, 545). Finally, it should be noted that the channel recorded in the original study that claimed that CaM-mediated Ca2+ inhibition had a single-channel conductance of ~60 pS in 250 mM K+ (310), whereas subsequent studies have revealed that the conductance of the InsP3R is ~360 pS in only 140 mM K+ (see sect. V). The channel observed in Reference 310 was therefore likely a dysfunctional InsP3R or not the InsP3R at all. In either case, the published data taken together provide little support for the proposition that high cytoplasmic Ca2+ concentration inhibition of InsP3R gating is mediated by CaM.
In addition to a putative role in Ca2+ inhibition of channel gating, CaM has also been invoked to regulate other properties of the InsP3R. Based on CaM-Sepharose binding studies and surface plasmon resonance (SPR) assays, the interaction between InsP3R and CaM revealed only a single Ca2+-dependent binding site in the regulatory domain (191, 525). Nevertheless, because the affinity of the interaction was dependent on the phosphorylation status of the channel, and the SII− splice variant bound more CaM, it was suggested that an additional CaM binding site is created by deletion of the SII splice region (254). Furthermore, an additional NH2-terminal site was proposed that bound both Ca2+-CaM and apoCaM equally well, with a Kd of ~1 μM in scintillation proximity assays (4, 76, 364). Analysis of CaM binding to InsP3R NH2-terminal peptides indicated that two sites within this region, residues 49–81 and 106–128 (Fig. 2B), were involved in the interaction (423). In [3H]InsP3 competition assays, CaM dose-dependently (1–10 μM) inhibited InsP3 binding to the NH2-terminal fragment of the type 1 and type 3 InsP3R only, an effect that was Ca2+ independent (428, 487). However, in similar assays of full-length InsP3R, CaM inhibited InsP3 binding only to InsP3R-1 (76). Consistent with these observations, CaM inhibited InsP3-mediated 45Ca2+ fluxes from cerebellar microsomes (enriched with InsP3R-1) independently of Ca2+ (364). However, in flux studies of permeabilized cells that predominantly expressed either types 1, 2, or 3 InsP3R, CaM inhibited InsP3-dependent Ca2+ release in all cell lines (4). Furthermore, the effects of CaM were Ca2+ dependent in a cell line predominantly expressing type 1 InsP3R (4). The role of CaM is further complicated by the observation that overexpression of a non-Ca2+-binding CaM mutant only affected InsP3-dependent Ca2+ release at higher Ca2+ concentrations, suggesting that this effect of CaM is Ca2+ dependent but that CaM is not the Ca2+ sensor (224).
Thus the history of the putative involvement of CaM in the regulation of the InsP3R has evolved from an interaction that was strictly Ca2+ dependent and had no effect on either InsP3 binding or Ca2+ release (270), to a role in high [Ca2+]i inhibition of channel gating mediated by binding to the coupling domain, to the postulation of additional binding sites that mediate Ca2+-dependent and -independent regulation of InsP3 binding that modulate channel function. Most recently, it has also been suggested that CaM may be constitutively bound to the channel, acting as a distinct subunit, as established for some other ion channels (404). However, this conclusion was based solely on the observation that a CaM-binding peptide disrupted InsP3R-mediated Ca2+ fluxes, which was overcome by exogenously applied CaM (227). Although these data are intriguing, there is at present no molecular evidence.
There are few biochemical data to suggest that CaM and InsP3R interact in vivo. The evidence of interactions relies almost exclusively on the various in vitro binding and pharmacological studies outlined above; meanwhile, successful coimmunoprecipitation of the two proteins has not been demonstrated. Therefore, despite considerable effort to place CaM in the InsP3R signaling complex, there is little consensus on its functional role.
2. CaBP and CIB1
The InsP3R interacts with a family of CaM-related Ca2+-binding proteins termed CaBPs, a subset of the neuronal Ca2+ sensor (NCS) family of EF-hand-containing proteins (527). Eight CaBP genes have been identified (CaBP1–8) with alternate splicing generating long and short forms of CaBP1 and CaBP2 (161, 162). An interaction between the CaBP gene family and the InsP3R was first identified using a yeast two-hybrid screen and further characterized using coimmunoprecipitation and pull-down assays (527). CaBP1 binds with high affinity (apparent Kd <50 nM) in a Ca2+-dependent manner (apparent affinity ~1 μM) to the NH2-terminal 600 residues of all three mammalian InsP3R isoforms (527), a region that encompasses the InsP3-binding domain. The Ca2+ dependence of the interaction was mediated by the EF-hands of CaBP (509, 527). Truncation mutagenesis of GST-fusion proteins of the InsP3R ligand-binding domain localized CaBP1 binding to the NH2-terminal 225 residues (225) in a region that overlaps with the most NH2-terminal putative CaM binding site (423). However, binding of CaBP1 was Ca2+ independent in these studies, whereas it was demonstrated that high-affinity interaction of CaBP1 with the InsP3R is strongly Ca2+ dependent (509, 527). Thus the significance of CaBP1 binding to this region is unclear.
The functional consequences of this interaction were studied at the single-channel level by patch-clamp recordings of the endogenous Xenopus InsP3R-1 in the outer membrane of isolated oocyte nuclei. Robust channel activity with high Po and gating kinetics similar to those elicited by InsP3 was observed in the absence of InsP3 when recombinant CaBP1 was included in the pipette solution at optimal cytoplasmic Ca2+ concentrations. In contrast, the triple-EF-hand mutant CaBP1 failed to activate the channel (527). Furthermore, purified bovine CaBP2 and mouse CaBP5 each had similar effects on the channel (527). These results identified the CaBP Ca2+ sensors as a family of protein ligands of the InsP3R channel. It was proposed that this regulation might enable the channel to become activated in the absence of phospho-inositide metabolism and that it may modulate the sensitivity of the channel to InsP3, which may play a role in spatially restricting Ca2+ release (527). Because InsP3R-mediated Ca2+ signals are shaped by messenger diffusion, degradation and removal, processes that will have distinct kinetics for InsP3 compared with CaBPs, the identification of novel ligands for the InsP3R provided new insights into the dimensions and versatility of this ubiquitous signaling pathway.
Although members of the CaBP family are exclusively expressed in the brain and retina (162), this novel mode of InsP3R regulation likely extends more widely to nonneuronal cell types as well. The ubiquitously expressed protein CIB1 (calcium- and integrin-binding protein; also called calmyrin or KIP) was subsequently shown to interact with the InsP3R (509). Structural analysis has placed CIB1 in a subfamily of NCS proteins that is distinct from other NCS proteins, including CaBPs (156). Nevertheless, CIB1 shares many of the biochemical and functional properties of CaBP1 with respect to its interactions with the InsP3R (509). Like CaBP1, CIB1 binds to the first 600-residue ligand-binding region of all isoforms of the InsP3R in a Ca2+-sensitive fashion, with an affinity in the micromolar range, and has a similar dependence on functional EF-hands. The functional consequences of the CIB1-InsP3R interaction were determined by recording single InsP3R channel activities in nuclei isolated from either Xenopus oocytes or Sf9 cells. Under conditions of optimal cytoplasmic Ca2+ concentration and in the absence of InsP3, CIB1 activated channel gating from both species, establishing it as a novel protein agonist. Compared with optimal concentrations of InsP3, however, CIB1 appears to be less efficacious as it activated channels to a lesser extent in both the oocyte and Sf9 systems; therefore, CIB1 appears to behave as a partial agonist of the channel. These results extend the concept of protein ligands of the InsP3R to peripheral tissues, and suggest that regulation of the InsP3R-mediated [Ca2+]i signaling may be under complex regulation by protein ligands in many cell types throughout the body.
At the molecular level, acute exposure of the InsP3R to purified recombinant CaBP/CIB1 protein activates channel gating, whereas on the other hand, overexpression of either CaBP or CIB1 was found to inhibit agonist-induced InsP3R-dependent Ca2+ release in intact cells (178, 225, 509). This apparent conflict was resolved by the demonstration that CaBP/CIB1 initially activates InsP3R channel activity, but then subsequently drives the channel into an inactivated state that cannot be activated by InsP3 (509). In CaBP1/CIB1 overexpression studies, much of the InsP3R channel population would therefore be in an inactivated state, accounting for the muted InsP3-mediated responses observed in CaBP1 or CIB1-expressing cells (178, 225, 509). These results suggest that prolonged exposure to CIB1 can effectively remove functional channels from the total InsP3R population by a process of ligand-induced channel inactivation.
Phosphorylation of CaM decreases its affinity for target substrates (378), and mutation of the conserved phosphorylation site in CaBP1 resulted in even greater [Ca2+]i signaling inhibition (225). Although this site is not conserved in CIB1, it remains possible that phosphorylation, other covalent modifications, and other protein interactions could regulate the affinities of the interactions of CaBP1 and CIB1 with the InsP3R. Unbinding of the protein ligands could enable the channel to escape from inactivation, allowing the channel to again be activated by protein ligand rebinding, in a mechanism of repeated channel activation that is completely independent of InsP3.
3. RACK1 and Gβγ
Several other proteins have been identified that also bind to the InsP3R NH2 terminus and modulate the channel activity. An interaction between the adapter protein RACK1 and the InsP3R was detected in a yeast two-hybrid screen (366). RACK1 binds to two sites within the NH2 terminus (residues 90–110 and 580–600; Fig. 2B) that effectively bracket the InsP3-binding domain (Fig. 2B). The third and fourth WD40 repeat in RACK1 were important for the interaction. It was proposed that each molecule of a RACK1 dimer binds to the separate sites (366). RACK1 increased the affinity of InsP3R for InsP3 by approximately twofold and potentiated Ca2+ release from microsomes in response to submaximal but not saturating concentration of InsP3. Thus binding of RACK1 to the ligand-binding region appears to increase the sensitivity of the channel to InsP3. Overexpression of RACK1 enhanced agonist-induced Ca2+ signals in several cell types. siRNA downregulation of RACK1 expression had surprisingly major inhibitory effects on Ca2+ signals elicited by InsP3-mobilizing agonists, suggesting that the interaction constitutively enhances channel responses to stimulation. However, it is not certain that this inhibition was not a secondary effect mediated through, for example, other proteins, such as PKC, with which RACK1 also interacts.
RACK1 has a high degree of structural homology with the G protein β-subunit and interacts with the Gβγ-complex (84). Interestingly, Gβγ could also be coimmunoprecipitated with InsP3R-1, but this association decreased InsP3 binding (542). Notably, recombinant Gβγ evoked robust gating of the Xenopus InsP3R in the absence of InsP3 in single-channel nuclear patch-clamp studies (542). Furthermore, Gβγ stimulated Ca2+ oscillations in whole cell experiments, despite blockade of PLC activity (542). These data demonstrate that Gβγ can activate the InsP3R independently of PLC and InsP3. Although nothing is known about the molecular determinants of the Gβγ-InsP3R interaction, the structural similarity between RACK1 and Gβγ may suggest that they bind to similar NH2-terminal sites to modulate InsP3R function in distinct ways.
4. IRBIT
IRBIT (InsP3R binding protein released with inositol 1,4,5-trisphosphate) was identified in a proteomics approach as a protein that eluted from the InsP3R in the presence of InsP3 (13). IRBIT contains an acidic serine/threonine-rich NH2 terminus that mediates its interaction with all three isoforms of the InsP3R (12, 111). Phosphorylated, but not nonphosphorylated, IRBIT competes with InsP3 for binding to the NH2 terminus of the InsP3R (12, 13, 111). There is some dispute whether the suppressor domain is necessary for the interaction (111), or just the core InsP3 binding domain is sufficient (12). The InsP3 binding pocket contributes to the interaction, since 10 of 12 residues shown to be important for InsP3 binding are also important for IRBIT binding (12). Physiologically, IRBIT binding may reduce the sensitivity of the channel to InsP3. Recombinant IRBIT suppressed Ca2+ release in response to low, but not saturating, doses of InsP3 in 45Ca2+ flux assays (12, 111), and siRNA knockdown of IRBIT enhanced Ca2+ signals in response to low agonist concentrations (12). Unlike CaBP1, CIB1, and Gβγ, IRBIT does not appear to function as a protein ligand of the channel, since application of recombinant IRBIT alone failed to activate reconstituted mouse cerebellar InsP3R channel activity in planar bilayer experiments (12). Taken together, these data suggest a model in which IRBIT binding to the ligand-binding domain functions to reduce the apparent InsP3 sensitivity by masking the InsP3 binding site.
5. Chromogranins
Chromogranins are high-capacity, low-affinity Ca2+ binding proteins that are enriched in secretory granules but are also found in ER and nuclear compartments (15). Both chromogranins A and B (CGA/CGB) interact within the third luminal loop of InsP3R-1 (532, 535). With the use of synthetic peptides, the interacting region was narrowed to the stretch of amino acids that link the luminal end of the pore selectivity filter to the beginning of TM6 (Fig. 2B) (533). In vitro binding studies showed that the CGA-InsP3R interaction was promoted by acidic pH, as would be experienced in the lumen of a mature secretory vesicle, while the CGB-InsP3R interaction was pH independent (532–534). Incorporation of CGA into liposomes containing purified InsP3R enhanced InsP3-mediated Ca2+ release (532). In functional studies of reconstituted InsP3R-1 incorporated into bilayers, both CGA and CGB dramatically increased channel Po from ~5%, the typical maximum Po observed for the reconstituted type 1 channel (Fig. 8), to 20–80% in different studies (87, 467, 470). The effect of CGA was manifested at luminal pH 5.5 but not 7.5, while CGB showed much less pH dependence, consistent with biochemical binding assays (467, 470). This remarkable level of stimulation was associated with the appearance of a new, long open state (467, 470). Because ligand modulation of InsP3R activity is primarily through modulation of the channel opening rate (196, 278, 283), i.e., the channel closed time, this effect of chromogranin is notable not only for the magnitude of its effect on Po but also for its distinct mode of action. Remarkably, the chromogranin-activated channel lacked all cytoplasmic Ca2+ sensitivity, being equally active at 10 nM, 1 μM, and 100 μM Ca2+ (470).
It has been proposed that as the luminal pH decreases during secretory granule development, enhanced chromogranin-InsP3R interaction might prime the vesicle for a Ca2+ release associated with exocytosis (531). It is not clear that the InsP3R-CGA interaction would have physiological significance at the ER, where the pH is expected to be ~7.2. However, the observation that CGB is heterogeneously distributed in ER and enriched in neurites has suggested that an interaction of CGB with the InsP3R could possibly regulate InsP3R-dependent Ca2+ release in local cellular regions (207). Interference of the interaction between chromogranins and InsP3R with an expressed chromogranin peptide targeted to the ER lumen in neuronally differentiated PC12 cells, inhibited Ca2+ release response to the muscarinic agonist carbachol (87). CGB is also found in the nucleus in a complex with phospholipids and the InsP3R, and it has been suggested that the interaction could modulate InsP3-dependent Ca2+ signaling within the nucleoplasm (194).
6. ERp44
The ER lumen-specific protein ERp44 was also shown to interact with the third luminal loop of the InsP3R (184), upstream of the region identified to interact with chromogranins (Fig. 2B). In pull-down and coimmunoprecipitation assays, ERp44 bound only to the type 1 InsP3R isoform, dependent on Ca2+ and redox state (184). The biochemical interaction was inhibited by high Ca2+ concentrations normally found in the ER lumen (>100 μM) and was promoted in a reducing environment, although the ER lumen is considered to be an oxidizing environment. Mutation of InsP3R cysteines within the interacting region decreased the in vitro interaction. Addition of recombinant ERp44 to the luminal aspect of cerebellar microsomal InsP3R reconstituted into planar bilayers dose-dependently decreased channel Po by up to 60% at ~7 μM ERp44 in the presence of saturating InsP3 (the cytoplasmic Ca2+ concentration was not specified). However, this inhibition required the presence of 3 mM luminal dithiothreitol. Whereas the in vitro conditions required to demonstrate binding (low Ca2+, reducing conditions) were distinct from those associated with the ER lumen, physiological relevance of the interaction was strongly suggested by the observation that siRNA knockdown of Erp44 caused enhanced Ca2+ signaling, and only in cell lines predominantly expressing the type 1 InsP3R (184). Reciprocal overexpression studies were consistent with these results (184). These data suggest that binding of ERp44 to the luminal loop of the InsP3R, possibly regulated by changes in luminal Ca2+ concentration and redox state, inhibits InsP3R activity. Thus two luminal proteins, chromogranins and ERp44, appear to bind to regions of the luminal loop that connect TM5 with TM6 and have opposite effects on channel activity, activating and inhibitory, respectively.
7. FKBP12
FK506 and rapamycin mediate their immunosuppressant effects by interacting with the immunophilin family of proteins. A physiological interaction between the FK506 binding protein, FKBP12, and the RyR has been well established (249). Binding of FKBP12 to RyR modulates channel gating by stabilizing the full conductance state and facilitating coupled activation within receptor clusters (59, 295). Binding of FK506 to FKBP12 dissociates it from the RyR, resulting in persistent subconductance gating (59). A role for FKBP12 in the regulation of InsP3R-dependent Ca2+ signaling, however, is somewhat controversial. A biochemical interaction between InsP3R and FKBP12, first demonstrated using copurification, coimmunoprecipitation, and yeast two-hybrid assays, localized an interaction to a leucine-proline (1400–1401; Fig. 2B) dipeptide sequence that is conserved across channel isoforms (72–74). However, other studies failed to replicate these findings, using either coimmunoprecipitation or GST-fusion protein pull-down strategies (64, 66, 78). The functional data are also conflicting. Displacement of FKBP12 by FK506 was reported to increase the sensitivity to InsP3 of the InsP3R in 45Ca2+ flux experiments (73, 74), whereas enhanced channel activity was observed after addition of FKBP12 in planar bilayer studies (100). However, other studies failed to observe an effect of FKBP12 on InsP3-dependent Ca2+ signaling (41, 65). Some of these discrepancies may be reconciled if FKBP12 modifies channel behavior indirectly. For example, it was proposed that FKBP12 binding to the phosphatase calcineurin effectively recruited it to the InsP3R, where it could prevent phosphorylation, and thereby depress InsP3R activity (73). Indeed, calcineurin was shown to copurify with the InsP3R-FKBP12 complex (73). Because the FK506-FKBP12 complex inhibits calcineurin activity, FK506 may relieve FKBP12-calcineurin inhibition, presenting as an increase in apparent InsP3 sensitivity (72, 73).
Rapamycin also binds to FKBP12 and reportedly displaces it from the InsP3R (74); however, the rapamycin-FKBP12 complex does not inhibit calcineurin (2). Physiologically, the rapamycin-FKBP12 complex inhibits mTOR (mammalian target of rapamycin), a protein kinase that regulates growth and cell cycle progression (247). Rapamycin was shown to inhibit Ca2+ release from cerebral microsomes (100) and in intact cells (268). It has been proposed that the InsP3R is a target for mTOR and that rapamycin-FKBP12 inhibition of mTOR reduces channel phosphorylation to inhibit Ca2+ release (268).
In summary, whereas it appears that FKBP12 may not directly modulate the InsP3R, it may influence channel activity by interacting with effector proteins involved in regulating the phosphorylation status of the InsP3R. However, the inconsistency among the data and the reliance on pharmacological manipulations suggest that at present such models must be regarded as highly speculative.
8. Glyceraldehyde 3-phosphate dehydrogenase
The glycolytic enzyme glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was found to directly bind to rat InsP3R-1 within residues 981–1000 (Fig. 2B) (367). Mutagenesis of Cys-992 and Cys-995 abolished the interaction. GAPDH catalyzes the oxidative phosphorylation of its aldehyde substrate with the reduction of NAD+ to NADH. NADH was previously shown to stimulate InsP3R-mediated Ca2+ release, whereas NAD+ was ineffective (222). Addition of purified GAPDH to liposomes containing purified cerebellar InsP3R in the presence of NAD+ enhanced InsP3-mediated Ca2+ release, dependent on the two cysteine residues important for GAPDH binding (367). It was proposed that local generation of NADH by GAPDH bound to the InsP3R may couple channel activity to cell metabolic state (367), for example, during hypoxia-induced stimulation of glycolysis (222). It had previously been suggested that NADH binds to the same site(s) as ATP, because the effects of ATP and NADH were not additive (222). However, the detailed effects on gating of single InsP3R channels have not been investigated.
9. Bcl-2 proteins
Ca2+ signaling is a key regulatory process in the progression of necrotic and apoptotic cell death mechanisms (reviewed in Refs. 353, 374). Ca2+ released by InsP3R activation can be taken up by mitochondria to stimulate oxidative phosphorylation and enhance ATP production (119). However, if Ca2+ uptake is coincident with certain apoptotic stimuli, it can trigger the swelling and rupture of the outer mitochondrial membrane, releasing an array of apoptotic factors, such as cytochrome c (165). A central feature of molecular models of apoptosis is the control of outer mitochondrial membrane permeability by pro- and antiapoptotic Bcl-2-related proteins (385). Nevertheless, Bcl-2 proteins also localize to the ER (240, 551). Furthermore, apoptosis protection conferred by overexpression of the antiapoptotic proteins Bcl-2 and Bcl-XL has been correlated with a decrease in the ER luminal Ca2+ concentration (145, 372), whereas upregulation of the proapoptotic molecules Bax and Bak has the opposite effect (81, 349). There is now a growing body of evidence that links ER Ca2+ homeostasis with a direct interaction between Bcl-2/Bcl-XL and the InsP3R (83, 349, 508).
Bcl-XL was shown to interact with the COOH terminus of all three isoforms of the InsP3R (508). In patch-clamp studies of isolated Sf9 cell nuclei, application of recombinant antiapoptotic Bcl-XL was shown to increase both the number and activity of InsP3R channels evoked by a subsaturating (10 nM) InsP3, to levels comparable to those observed with saturating (10 μM) InsP3. Furthermore, the InsP3R behavior was similarly affected by transient overexpression of Bcl-XL, demonstrating an in vivo physical and functional interaction that was sufficiently strong to survive the nuclear isolation protocol. Proapoptotic Bax and tBid each disrupted binding of the InsP3R to GST-Bcl-XL, and in patch-clamp studies from cells transiently transfected with Bcl-XL, inclusion of either tBid or Bax in the pipette solution abolished the Bcl-XL-dependent effects on the InsP3R (508).
In the absence of Bcl-XL expression, the expression of InsP3R in DT40 cells enhances apoptosis in response to B-cell receptor ligation, suggesting that InsP3R-mediated Ca2+ release may be toxic to mitochondria (508). Paradoxically, expression of the InsP3R was necessary for Bcl-XL to exert its full antiapoptotic effects (508). Bcl-XL expression resulted in enhanced Ca2+ signaling in vivo in quiescent and submaximally stimulated cells (508), consistent with the activating effects of Bcl-XL on channel gating observed in the patch-clamp studies. Activation of low-level InsP3-dependent Ca2+ signaling by Bcl-2 has also been observed in other cell types (355, 547). When expressed in DT40 cells, Bcl-XL reduced ER Ca2+ concentration in wild-type but not in InsP3R triple-knockout cells (508). Thus Bcl-XL interaction with the InsP3R sensitizes the channel to low levels of InsP3 that may exist in un-stimulated cells, enhancing InsP3-dependent channel activity and lowering ER Ca2+ concentration. The reduced ER Ca2+concentration does not, however, account for the antiapoptotic function of Bcl-XL/Bcl-2 at the ER, because the triple knockout cells should have had maximum protection from ER-mediated Ca2+ insults, because they lacked a mechanism (InsP3R expression) to convey any Ca2+ signal to the mitochondria, yet expression of InsP3R with Bcl-XL conferred more protection. The InsP3R-mediated, Bcl-XL-dependent low-level Ca2+ signaling associated with enhanced apoptosis resistance was correlated with enhanced mitochondrial bioenergetics (508). This suggests that the effects of Bcl-2/Bcl-XL at the ER are antiapoptotic due to a specific modulation by these proteins of an exquisitely regulated Ca2+ permeability, the InsP3R, providing enhanced low-level Ca2+ signaling to mitochondria, improving cellular energetics that enables or adapts the cell to better withstand apoptotic insults.
10. Cytochrome c
While the InsP3R-Bcl-2/Bcl-XL interaction primes cells to better withstand a potential apoptotic hit, there is also compelling evidence that the InsP3R can function as a proapoptotic mediator in cells undergoing apoptosis. With the use of the COOH terminus of the rat InsP3R-1 as bait in a yeast two-hybrid screen, the key apoptotic signaling molecule cytochrome c (CytC) was identified as an interacting protein (44). CytC bound to the COOH terminus of type 1 and type 3 InsP3R (type 2 was not determined), mapped to residues 2621–2636 (Fig. 2B) (44, 45). In in vitro flux assays, high Ca2+ concentration inhibition of 45Ca2+ release in the presence of a saturating InsP3 concentration was relieved by addition of nanomolar CytC. However, single-channel studies have not been undertaken, so the details of how CytC regulates the channel remain to be determined. Disruption of the interaction in vivo by intracellular delivery of a cell-permeant peptide based on the 16-residue InsP3R-binding region, was protective against a range of apoptotic stimuli (45), possibly suggesting that CytC interaction with the InsP3R modulated Ca2+ signals that impinged on apoptotic pathways.
11. Huntingtin
The neurodegenerative disorder Huntington’s disease is caused by a polyglutamine expansion in the Huntingtin (Htt) protein. Huntingtin-associated protein 1 (HAP1) interacts with Htt, an association promoted by the polyglutamine expansion of Htt (Httexp) (130). A yeast two-hybrid screen identified an interaction between the COOH terminus of InsP3R-1 and HAP1 (454). The biochemical interaction was confirmed by coimmunoprecipitation and pull-down of HAP1 by a GST-InsP3R COOH-terminal construct (residues 2627–2736; Fig. 2B). This construct also directly bound Httexp, and to a lesser extent Htt. In both cases, the interaction was strengthened by the presence of HAP1, suggesting that all three proteins exist in a complex (454).
Addition of recombinant HAP1 to the cytoplasmic aspect of the reconstituted InsP3R-1 in planar bilayers was without effect on Po evoked by a subsaturating concentration of InsP3. However, subsequent sequential additions of recombinant NH2-terminal fragments of Htt or Httexp, or application of premixed HAP1-Htt/Httexp, increased channel activity, with Httexp producing the more dramatic change (454, 455). Furthermore, recombinant full-length Httexp but not Htt increased Po in response to subsaturating but not saturating InsP3 without HAP1 pre-exposure (454). Consistent with these observations, overexpression of full-length Httexp, but not Htt, increased InsP3-dependent Ca2+ release in medium spiny neurons in response to threshold levels of agonist concentration. The increased sensitivity caused by Httexp was partially dependent on HAP1, as the effect was less robust in HAP1−/− cells (455). It was suggested that enhanced InsP3 sensitivity of the Htt/HAP1-bound InsP3R may contribute to neuronal apoptosis associated with progression of Huntington’s disease (453). As discussed above, binding of Bcl-XL to the COOH terminus of the InsP3R also increases the apparent sensitivity of the channel to subsaturating concentrations of InsP3 (508). Furthermore, an increase in the apparent sensitivity to InsP3 may also be the mechanism by which CytC binding to the COOH terminus relieves high [Ca2+] inhibition of Ca2+ release (45). It is interesting that three different proteins that have been shown to interact with the COOH terminus of the InsP3R all enhance the apparent InsP3 sensitivity of channel activity. Furthermore, all these studies have implicated this enhancement in apoptosis progression, yet the conclusions differ regarding whether the interactions are pro- or antiapoptotic. Further studies are necessary to understand how apparent functional effects on the channel elicit these different physiological outcomes.
12. Proteases (caspase-3 and calpain)
The primary role of CytC in the apoptotic cascade is its involvement in the activation of the cysteine protease, caspase-3, a key proteolytic enzyme responsible for cellular disassembly. The InsP3R is one of its many substrates (189). The coupling domain of InsP3R-1 possesses a highly conserved DEVD consensus sequence for caspase-3 cleavage (Fig. 2B), and the channel was shown to be specifically cleaved at this site, both in vitro biochemical assays and in vivo during apoptosis (175, 189). As caspase-3 activation is a downstream event in apoptosis, these observations raise the question of whether InsP3R cleavage represents a key regulatory step in the apoptotic pathway, or whether it simply reflects degradation after a commitment to death has already been made. Genetic deletion of all InsP3R isoforms in the DT40 cells provided some protection from apoptotic insults (440), a result that has been recapitulated in several model systems using various InsP3R-knockdown strategies (38, 201, 232). Reintroduction of the type 1 receptor into DT40-TKO cells restored apoptotic sensitivity, whereas expression of the channel with a mutated DEVD sequence, although still functional, rendered the channel resistant to caspase-3 cleavage and the cells less sensitive to apoptosis (14). Accordingly, it was suggested that caspase cleavage of InsP3R-1 has a direct role in apoptosis progression (14). However, it should be noted that the types 2 and 3 channels lack the caspase-3 site but can nevertheless sensitize cells to apoptosis (440). Thus caspase cleavage of the InsP3R-1 cannot account for all the effects of InsP3R in apoptosis. It was suggested that the mechanism by which caspase 3-cleavage of InsP3R-1 sensitized cells to apoptosis was by dysregulating Ca2+ homeostasis (189). Expression of an NH2-terminal truncated InsP3R-1 corresponding to the caspase-3 cleaved channel-only domain (341) caused apparent depletion of ER Ca2+ stores. It was suggested that the channel-only domain is constitutively “leaky” and that the NH2-terminal domain is required to keep the channel closed. It was speculated that this “leakiness” may impinge on apoptosis progression (341). However, these studies assume that the NH2-terminally cleaved domain completely dissociates from the channel domain, but there is no evidence that this occurs. Indeed, in cerebral microsomes exposed to levels of caspase-3 that almost completely degraded the channel, Ca2+ accumulation persisted, although InsP3R-dependent release was attenuated (189). Similarly, trypsinization of the InsP3R digested it into five separate fragments, yet the channel retained a degree of structural and functional integrity (189, 537).
In addition to caspase, there is some evidence that calpain, a Ca2+-dependent cysteine protease activated in some apoptotic paradigms, is involved in InsP3R degradation (112). Calpain was shown to cleave InsP3R-1 at two undefined sites (275) and contributed to InsP3R-1 (519) and InsP3R-3 (112) degradation in vivo. However, at present, there is no information on the functional effects of calpain on InsP3R channel activity.
13. Adapter proteins (protein 4.1N, ankyrin, and Homer)
Protein 4.1N is the neuronal homolog of the 4.1 family of cytoskeleton proteins structurally and functionally characterized by three domains: an NH2-terminal region important for interactions with plasma membrane proteins, a central spectrin-actin binding domain, and a COOH-terminal domain shown to bind nuclear mitotic apparatus protein and the glutamate receptor GluR1 (298, 417, 499). Using yeast two-hybrid screening, two groups independently identified an interaction between the COOH termini of protein 4.1N and InsP3R-1 specifically (301, 544) within the terminal 14 residues of the channel (Fig. 2B) (147). Protein 4.1N was shown to translocate InsP3R-1 to the basolateral membrane of polarized Madin-Darby canine kidney cells (544), suggesting that the interaction may provide a mechanism to spatially restrict InsP3-mediated Ca2+ signals. In neurons, a protein 4.1N-InsP3R complex may be associated with cytoskeletal elements enriched in postsynaptic compartments (301). It has been widely observed that disruption of the actin filament network has profound effects on InsP3-evoked Ca2+ signals (56, 387, 484, 503). Actin plays a role in the translational mobility of InsP3R-1 (148) but not InsP3R-3 (132, 148). As InsP3R-3 does not bind to protein 4.1N, it was postulated that InsP3R-1 is specifically linked to the actin cytoskeleton via an interaction with protein 4.1N (148).
The ankyrin family of adapter proteins also serves to couple target proteins to the cytoskeleton, by spectrin binding (for review, see Ref. 25). An antibody to erythrocyte ankyrin immunoprecipitated InsP3R from brain (215), and ankyrin purified from human erythrocytes bound to the InsP3R with high affinity (Kd = 0.2 nM) and inhibited InsP3 binding and InsP3R-dependent 45Ca2+ fluxes (56). An 11-amino acid stretch of the InsP3R (residues 2548–2558; Fig. 2B) with homology to a known ankyrin-binding domain was identified as the interaction region. However, because this region overlaps with the InsP3R pore sequence, it is unlikely to be the binding site in the full-length receptor (57). Ankyrin B interacts with the sigma-1 receptor (Sig-1R), an integral ER membrane protein that binds neurosteroids and psychotopic drugs, and there is strong evidence for an ankyrin B-Sig-1R-InsP3R complex (177). Ligand-binding to Sig-1R increased intracellular Ca2+ release in response to InsP3-generating agonists concomitantly with ankyrin dissociation from the type 3 channel (176, 177). Taken together, these data suggest a functional role of ankyrin interaction with InsP3R in providing tonic inhibition of Ca2+ release activity through decreased InsP3 sensitivity, with reversibility provided by an ankyrin-Sig-1R interaction.
In addition to directly modulating Ca2+ release, ankyrins are also involved in the localization of InsP3Rs. Ankyrin may recruit InsP3Rs into lipid rafts upon ligand binding of the cell surface receptor CD44 (426). Neonatal cardiomyocytes from ankyrin B−/− mice displayed a Ca2+ dysregulation phenotype, which was correlated with altered spatial distribution of membrane proteins including Na+-K+-ATPase, Na+-Ca2+ exchanger, and the InsP3R. Protein localization was restored by the expression of exogenous ankyrin B but not by expression of the E1425G loss-of-function ankyrin B mutant (325–327, 485). The physiological consequences of this mutation are thought to underlie type 4 long-QT cardiac arrhythmia in humans (327). However, it is difficult to assess the contribution of the InsP3R to the alterations in Ca2+ signaling observed in ankyrin B−/− or ankyrin B−/− cells expressing E1425G-ankyrin, as other Ca2+ regulating proteins are also affected.
Subcellular targeting of the InsP3R is also mediated by indirect coupling to membrane receptors and channels through an interaction with the Homer family of scaffolding proteins (483, 539). The Homer proteins bind to polyproline motifs of mGlu-receptors, TRPC1, InsP3R, and RyR, among others (128). A consensus Homer binding motif is present in the NH2 terminus of the InsP3R, in the suppressor domain (residues 49–54; Fig. 2B). In addition to a proline-binding domain, Homer also possesses a coiled-coil region required for the formation of homo- or heterodimers. These interactions serve to cross-link Homer-bound proteins, forming multimeric complexes. In this way, Homer facilitates an interaction between the InsP3R and mGluR1α (483) and TRPC1 (539). In both cases, disruption of the interaction profoundly affected Ca2+ signaling that was dependent on the cross-linking capacity of Homer. However, a direct interaction between Homer and the InsP3R has not been demonstrated. Planar bilayer studies of the RyR demonstrated enhanced gating in the presence of both full-length recombinant Homer and a protein lacking the coiled-coil region (131). However, it is not known whether Homer can directly modify the behavior of InsP3R channel.
14. TRPC channels and Na+-K+-ATPase
The transient receptor potential channels (TRPC) are a diversely regulated family of plasma membrane weakly voltage-dependent Ca2+-permeable cation channels (384). Biochemical and functional studies suggest a close coupling of some TRPC channels and InsP3R. In cells overexpressing TRPC3, the single-channel activity of TRPC3 that was lost upon patch excision or excessive patch washing was restored upon addition of InsP3, cerebellar microsomes enriched in InsP3R, or purified InsP3R (237). These effects on TRPC gating were dependent on the presence of the InsP3R NH2 terminus (236). TRPC3 was successfully coimmunoprecipitated with InsP3R (55, 236, 539) with GST pull-down experiments, indicating that the interaction was localized to two regions in the InsP3R distal to the ligand-binding domain (residues 669–702 and 755–824; Fig. 2B) (55). With similar experimental approaches, biochemical interactions have also been identified between the InsP3R and TRPC1, TRPC4, and TRPC6 (55, 258, 306, 396, 425, 452, 539). These studies have suggested that the InsP3R is directly coupled to the TRPC family members or that both proteins are contained within a larger protein complex (258, 539). It has been suggested that InsP3 binding to the InsP3R is required to fully gate the TRPC channels (237, 539) and that the interaction can couple InsP3R-dependent Ca2+ release to voltage-independent or store depletion-dependent Ca2+ influx (reviewed in Refs. 356, 384). Nevertheless, these models are controversial, since several studies demonstrated that the InsP3R is not required for TRP channel activation (264, 476, 506). What has not been evaluated is whether direct interactions with TRP channels have reciprocal, allosteric effects on InsP3R function.
As the principle plasma membrane ion exchanger, the Na+-K+-ATPase serves to maintain cellular electrical and chemical gradients. In addition, it functions as a steroid receptor, and thus is involved in signal transduction independent from ion exchange (reviewed in Ref. 523). All three isoforms of the InsP3R were shown to coimmunoprecipitate with the Na+-K+-ATPase, Src kinase, and PLC-γ1 (320, 540), suggesting the Na+-K+-ATP-ase may exist as part of a Ca2+-signaling complex (540). GST-fusion constructs of the 1–604 NH2 terminus of the InsP3R pulled down the Na+-K+-ATPase, suggesting a direct interaction between the proteins (543). Ouabain, an exogenous steroid ligand of the Na+-K+-ATPase, promoted the association of the InsP3R with the Na+-K+-ATPase, and stimulated Src-dependent PLC-γ1 activation and InsP3R phosphorylation (540). At the cellular level, ouabain evoked repetitive Ca2+ oscillations that reflected periodic Ca2+ release from the ER (6, 251, 320). In primary rat proximal tubular cells, the ouabain-evoked Ca2+ oscillations were correlated with increased apoptotic resistance that was blocked by NFκB inhibition (251). The mechanism by which ouabain triggers Ca2+ release is unknown. It has been proposed that ouabain binding causes a conformational change in the Na+-K+-ATPase that allosterically modulates the activity of the InsP3R through a direct interaction of the two proteins (251). If this hypothesis is confirmed experimentally, it will be interesting to consider whether conformational changes in either protein during their normal transport functions couple their activities through mutual allosteric influences.
VII. CONCLUDING REMARKS
It is hoped that this review of the structural and functional aspects of the properties of InsP3R as intracellular Ca2+ release channels has revealed the remarkable richness of its gating behaviors and regulation. The single-channel properties of the InsP3R undoubtedly contribute to the ability of the InsP3 signaling system to regulate diverse cell physiological processes, even within a single cell. However, because of the complexity of the structure, behavior, and regulation of the InsP3R channel, many questions remain regarding the molecular physiology of the channel and its roles in cellular biology. In the future, we should anticipate progress in understanding the relationship between the structure of the channel and its various functional properties, the relationship of various channel functional properties to features of cellular Ca2+ signals, and the functional and physiological implications of the remarkable diversity of InsP3R channel expression. To achieve these goals, ongoing approaches and new strategies will need to be applied to gain further insights into the structure of the InsP3R channel. Protein expression and single-channel recording systems need to be further developed to allow recombinant channels, without contamination from background endogenous channels, to be studied rigorously under a wide range of conditions with a high level of consistency. In addition, experimental systems that enable non-steady-state gating properties of the channel to be recorded in response to rapid alterations of ligands and regulators will facilitate development of quantitative models that relate dynamic changes in cytoplasmic Ca2+ signals to the kinetic properties of InsP3R channels. Development of animal models, both vertebrate and invertebrate, that enable cell- and tissue-specific expression of wild-type and mutant recombinant InsP3R channel function will provide insights into the relationships of channel gating properties and their regulation with cell physiological processes in vivo, providing an integrated physiology of the InsP3R Ca2+ release channel.
Acknowledgments
GRANTS
This work was supported by the National Institutes of Health and American Heart Association.
References
- 1.Abe H, Shuto S, Matsuda A. Synthesis of the C-glycosidic analog of adenophostin A and its uracil congener as potential IP3 receptor ligands. Stereoselective construction of the C-glycosidic structure by a temporary silicon-tethered radical coupling reaction. J Org Chem. 2000;65:4315–4325. doi: 10.1021/jo0001333. [DOI] [PubMed] [Google Scholar]
- 2.Abraham RT, Wiederrecht GJ. Immunopharmacology of rapamycin. Annu Rev Immunol. 1996;14:483–510. doi: 10.1146/annurev.immunol.14.1.483. [DOI] [PubMed] [Google Scholar]
- 3.Acharya JK, Jalink K, Hardy RW, Hartenstein V, Zuker CS. InsP3 receptor is essential for growth and differentiation but not for vision in Drosophila. Neuron. 1997;18:881–887. doi: 10.1016/s0896-6273(00)80328-1. [DOI] [PubMed] [Google Scholar]
- 4.Adkins CE, Morris SA, De Smedt H, Sienaert I, Torok K, Taylor CW. Ca2+-calmodulin inhibits Ca2+ release mediated by type-1, -2 and -3 inositol trisphosphate receptors. Biochem J. 2000;345:357–363. [PMC free article] [PubMed] [Google Scholar]
- 5.Adkins CE, Wissing F, Potter BV, Taylor CW. Rapid activation and partial inactivation of inositol trisphosphate receptors by adenophostin A. Biochem J. 2000;352:929–933. [PMC free article] [PubMed] [Google Scholar]
- 6.Aizman O, Uhlen P, Lal M, Brismar H, Aperia A. Ouabain, a steroid hormone that signals with slow calcium oscillations. Proc Natl Acad Sci USA. 2001;98:13420–13424. doi: 10.1073/pnas.221315298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Allbritton NL, Meyer T, Stryer L. Range of messenger action of calcium ion and inositol 1,4,5-trisphosphate. Science. 1992;258:1812–1815. doi: 10.1126/science.1465619. [DOI] [PubMed] [Google Scholar]
- 8.Alvarez J, Montero M. Measuring [Ca2+] in the endoplasmic reticulum with aequorin. Cell Calcium. 2002;32:251–260. doi: 10.1016/s0143416002001860. [DOI] [PubMed] [Google Scholar]
- 9.Alzayady KJ, Panning MM, Kelley GG, Wojcikiewicz RJ. Involvement of the p97-Ufd1-Npl4 complex in the regulated endoplasmic reticulum-associated degradation of inositol 1,4,5-trisphosphate receptors. J Biol Chem. 2005;280:34530–34537. doi: 10.1074/jbc.M508890200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alzayady KJ, Wojcikiewicz RJ. The role of Ca2+ in triggering inositol 1,4,5-trisphosphate receptor ubiquitination. Biochem J. 2005;392:601–606. doi: 10.1042/BJ20050949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ammendola A, Geiselhoringer A, Hofmann F, Schlossmann J. Molecular determinants of the interaction between the inositol 1,4,5-trisphosphate receptor-associated cGMP kinase substrate (IRAG) and cGMP kinase Ibeta. J Biol Chem. 2001;276:24153–24159. doi: 10.1074/jbc.M101530200. [DOI] [PubMed] [Google Scholar]
- 12.Ando H, Mizutani A, Kiefer H, Tsuzurugi D, Michikawa T, Mikoshiba K. IRBIT suppresses IP3 receptor activity by competing with IP3 for the common binding site on the IP3 receptor. Mol Cell. 2006;22:795–806. doi: 10.1016/j.molcel.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 13.Ando H, Mizutani A, Matsuura T, Mikoshiba K. IRBIT, a novel inositol 1,4,5-trisphosphate (IP3) receptor-binding protein, is released from the IP3 receptor upon IP3 binding to the receptor. J Biol Chem. 2003;278:10602–10612. doi: 10.1074/jbc.M210119200. [DOI] [PubMed] [Google Scholar]
- 14.Assefa Z, Bultynck G, Szlufcik K, Nadif Kasri N, Vermassen E, Goris J, Missiaen L, Callewaert G, Parys JB, De Smedt H. Caspase-3-induced truncation of type 1 inositol trisphosphate receptor accelerates apoptotic cell death and induces inositol trisphosphate-independent calcium release during apoptosis. J Biol Chem. 2004;279:43227–43236. doi: 10.1074/jbc.M403872200. [DOI] [PubMed] [Google Scholar]
- 15.Aunis D, Metz-Boutigue MH. Chromogranins: current concepts. Structural and functional aspects. Adv Exp Med Biol. 2000;482:21–38. doi: 10.1007/0-306-46837-9_2. [DOI] [PubMed] [Google Scholar]
- 16.Babcock DF, Hille B. Mitochondrial oversight of cellular Ca2+ signaling. Curr Opin Neurobiol. 1998;8:398–404. doi: 10.1016/s0959-4388(98)80067-6. [DOI] [PubMed] [Google Scholar]
- 17.Bagni C, Mannucci L, Dotti CG, Amaldi F. Chemical stimulation of synaptosomes modulates α-Ca2+/calmodulin-dependent protein kinase II mRNA association to polysomes. J Neurosci. 2000;20:RC76. doi: 10.1523/JNEUROSCI.20-10-j0004.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Balshaw DM, Yamaguchi N, Meissner G. Modulation of intracellular calcium-release channels by calmodulin. J Membr Biol. 2002;185:1–8. doi: 10.1007/s00232-001-0111-4. [DOI] [PubMed] [Google Scholar]
- 19.Bare DJ, Kettlun CS, Liang M, Bers DM, Mignery GA. Cardiac type 2 inositol 1,4,5-trisphosphate receptor: interaction and modulation by calcium/calmodulin-dependent protein kinase II. J Biol Chem. 2005;280:15912–15920. doi: 10.1074/jbc.M414212200. [DOI] [PubMed] [Google Scholar]
- 20.Barrans JD, Allen PD, Stamatiou D, Dzau VJ, Liew CC. Global gene expression profiling of end-stage dilated cardiomyopathy using a human cardiovascular-based cDNA microarray. Am J Pathol. 2002;160:2035–2043. doi: 10.1016/S0002-9440(10)61153-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bassik MC, Scorrano L, Oakes SA, Pozzan T, Korsmeyer SJ. Phosphorylation of Bcl-2 regulates ER Ca2+ homeostasis and apoptosis. EMBO J. 2004;23:1207–1216. doi: 10.1038/sj.emboj.7600104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baylis HA, Furuichi T, Yoshikawa F, Mikoshiba K, Sattelle DB. Inositol 1,4,5-trisphosphate receptors are strongly expressed in the nervous system, pharynx, intestine, gonad and excretory cell of Caenorhabditis elegans and are encoded by a single gene (itr-1) J Mol Biol. 1999;294:467–476. doi: 10.1006/jmbi.1999.3229. [DOI] [PubMed] [Google Scholar]
- 23.Beecroft MD, Marchant JS, Riley AM, Van Straten NC, Van der Marel GA, Van Boom JH, Potter BV, Taylor CW. Acyclophostin: a ribose-modified analog of adenophostin A with high affinity for inositol 1,4,5-trisphosphate receptors and pH-dependent efficacy. Mol Pharmacol. 1999;55:109–117. doi: 10.1124/mol.55.1.109. [DOI] [PubMed] [Google Scholar]
- 24.Beecroft MD, Taylor CW. Incremental Ca2+ mobilization by inositol trisphosphate receptors is unlikely to be mediated by their desensitization or regulation by luminal or cytosolic Ca2+ Biochem J. 1997;326:215–220. doi: 10.1042/bj3260215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bennett V, Baines AJ. Spectrin and ankyrin-based pathways: metazoan inventions for integrating cells into tissues. Physiol Rev. 2001;81:1353–1392. doi: 10.1152/physrev.2001.81.3.1353. [DOI] [PubMed] [Google Scholar]
- 26.Berridge MJ. Elementary and global aspects of calcium signalling. J Physiol. 1997;499:291–306. doi: 10.1113/jphysiol.1997.sp021927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- 28.Berridge MJ, Bootman MD, Lipp P. Calcium—a life and death signal. Nature. 1998;395:645–648. doi: 10.1038/27094. [DOI] [PubMed] [Google Scholar]
- 29.Bers DM, Patton CW, Nuccitelli R. A practical guide to the preparation of Ca2+ buffers. Methods Cell Biol. 1994;40:3–29. doi: 10.1016/s0091-679x(08)61108-5. [DOI] [PubMed] [Google Scholar]
- 30.Bezprozvanny I. The inositol 1,4,5-trisphosphate receptors. Cell Calcium. 2005;38:261–272. doi: 10.1016/j.ceca.2005.06.030. [DOI] [PubMed] [Google Scholar]
- 31.Bezprozvanny I, Ehrlich BE. ATP modulates the function of inositol 1,4,5-trisphosphate-gated channels at two sites. Neuron. 1993;10:1175–1184. doi: 10.1016/0896-6273(93)90065-y. [DOI] [PubMed] [Google Scholar]
- 32.Bezprozvanny I, Ehrlich BE. The inositol 1,4,5-trisphosphate (InsP3) receptor. J Membr Biol. 1995;145:205–216. doi: 10.1007/BF00232713. [DOI] [PubMed] [Google Scholar]
- 33.Bezprozvanny I, Ehrlich BE. Inositol (1,4,5)-trisphosphate (InsP3)-gated Ca channels from cerebellum: conduction properties for divalent cations and regulation by intraluminal calcium. J Gen Physiol. 1994;104:821–856. doi: 10.1085/jgp.104.5.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bezprozvanny I, Watras J, Ehrlich BE. Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature. 1991;351:751–754. doi: 10.1038/351751a0. [DOI] [PubMed] [Google Scholar]
- 35.Bhanumathy CD, Nakao SK, Joseph SK. Mechanism of proteasomal degradation of inositol trisphosphate receptors in CHO-K1 cells. J Biol Chem. 2006;281:3722–3730. doi: 10.1074/jbc.M509966200. [DOI] [PubMed] [Google Scholar]
- 36.Bichet D, Haass FA, Jan LY. Merging functional studies with structures of inward-rectifier K+ channels. Nat Rev Neurosci. 2003;4:957–967. doi: 10.1038/nrn1244. [DOI] [PubMed] [Google Scholar]
- 37.Bird GStJ, Takahashi M, Tanzawa K, Putney JW., Jr Adenophostin A induces spatially restricted calcium signaling in Xenopus laevis oocytes. J Biol Chem. 1999;274:20643–20649. doi: 10.1074/jbc.274.29.20643. [DOI] [PubMed] [Google Scholar]
- 38.Blackshaw S, Sawa A, Sharp AH, Ross CA, Snyder SH, Khan AA. Type 3 inositol 1,4,5-trisphosphate receptor modulates cell death. FASEB J. 2000;14:1375–1379. doi: 10.1096/fj.14.10.1375. [DOI] [PubMed] [Google Scholar]
- 39.Blondel O, Takeda J, Janssen H, Seino S, Bell GI. Sequence and functional characterization of a third inositol trisphosphate receptor subtype, IP3R-3, expressed in pancreatic islets, kidney, gastrointestinal tract, other tissues. J Biol Chem. 1993;268:11356–11363. [PubMed] [Google Scholar]
- 40.Boehning D, Joseph SK. Direct association of ligand-binding and pore domains in homo- and heterotetrameric inositol 1,4,5-trisphosphate receptors. EMBO J. 2000;19:5450–5459. doi: 10.1093/emboj/19.20.5450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boehning D, Joseph SK. Functional properties of recombinant type I and type III inositol 1,4,5-trisphosphate receptor isoforms expressed in COS-7 cells. J Biol Chem. 2000;275:21492–21499. doi: 10.1074/jbc.M001724200. [DOI] [PubMed] [Google Scholar]
- 42.Boehning D, Joseph SK, Mak DOD, Foskett JK. Single-channel recordings of recombinant inositol trisphosphate receptors in mammalian nuclear envelope. Biophys J. 2001;81:117–124. doi: 10.1016/s0006-3495(01)75685-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boehning D, Mak DOD, Foskett JK, Joseph SK. Molecular determinants of ion permeation and selectivity in inositol 1,4,5-trisphosphate receptor Ca2+ channels. J Biol Chem. 2001;276:13509–13512. doi: 10.1074/jbc.C100094200. [DOI] [PubMed] [Google Scholar]
- 44.Boehning D, Patterson RL, Sedaghat L, Glebova NO, Kurosaki T, Snyder SH. Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat Cell Biol. 2003;5:1051–1061. doi: 10.1038/ncb1063. [DOI] [PubMed] [Google Scholar]
- 45.Boehning D, van Rossum DB, Patterson RL, Snyder SH. A peptide inhibitor of cytochrome c/inositol 1,4,5-trisphosphate receptor binding blocks intrinsic and extrinsic cell death pathways. Proc Natl Acad Sci USA. 2005;102:1466–1471. doi: 10.1073/pnas.0409650102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bootman MD. Quantal Ca2+ release from InsP3-sensitive intracellular Ca2+ stores. Mol Cell Endocrinol. 1994;98:157–166. doi: 10.1016/0303-7207(94)90134-1. [DOI] [PubMed] [Google Scholar]
- 47.Bootman MD, Berridge MJ. The elemental principles of calcium signaling. Cell. 1995;83:675–678. doi: 10.1016/0092-8674(95)90179-5. [DOI] [PubMed] [Google Scholar]
- 48.Bootman MD, Berridge MJ. Subcellular Ca2+ signals underlying waves and graded responses in HeLa cells. Curr Biol. 1996;6:855–865. doi: 10.1016/s0960-9822(02)00609-7. [DOI] [PubMed] [Google Scholar]
- 49.Bootman MD, Lipp P, Berridge MJ. The organisation and functions of local Ca2+ signals. J Cell Sci. 2001;114:2213–2222. doi: 10.1242/jcs.114.12.2213. [DOI] [PubMed] [Google Scholar]
- 50.Bootman MD, Taylor CW, Berridge MJ. The thiol reagent, thimerosal, evokes Ca2+ spikes in HeLa cells by sensitizing the inositol 1,4,5-trisphosphate receptor. J Biol Chem. 1992;267:25113–25119. [PubMed] [Google Scholar]
- 51.Borissow CN, Black SJ, Paul M, Tovey SC, Dedos SG, Taylor CW, Potter BV. Adenophostin A and analogues modified at the adenine moiety: synthesis, conformational analysis and biological activity. Org Biomol Chem. 2005;3:245–252. doi: 10.1039/b415229h. [DOI] [PubMed] [Google Scholar]
- 52.Bosanac I, Alattia JR, Mal TK, Chan J, Talarico S, Tong FK, Tong KI, Yoshikawa F, Furuichi T, Iwai M, Michikawa T, Mikoshiba K, Ikura M. Structure of the inositol 1,4,5-trisphosphate receptor binding core in complex with its ligand. Nature. 2002;420:696–700. doi: 10.1038/nature01268. [DOI] [PubMed] [Google Scholar]
- 53.Bosanac I, Michikawa T, Mikoshiba K, Ikura M. Structural insights into the regulatory mechanism of IP3 receptor. Biochim Biophys Acta. 2004;1742:89–102. doi: 10.1016/j.bbamcr.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 54.Bosanac I, Yamazaki H, Matsu-Ura T, Michikawa T, Mikoshiba K, Ikura M. Crystal structure of the ligand binding suppressor domain of type 1 inositol 1,4,5-trisphosphate receptor. Mol Cell. 2005;17:193–203. doi: 10.1016/j.molcel.2004.11.047. [DOI] [PubMed] [Google Scholar]
- 55.Boulay G, Brown DM, Qin N, Jiang M, Dietrich A, Zhu MX, Chen Z, Birnbaumer M, Mikoshiba K, Birnbaumer L. Modulation of Ca2+ entry by polypeptides of the inositol 1,4,5-trisphos-phate receptor (IP3R) that bind transient receptor potential (TRP): evidence for roles of TRP and IP3R in store depletion-activated Ca2+ entry. Proc Natl Acad Sci USA. 1999;96:14955–14960. doi: 10.1073/pnas.96.26.14955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bourguignon LY, Iida N, Jin H. The involvement of the cytoskeleton in regulating IP3 receptor-mediated internal Ca2+ release in human blood platelets. Cell Biol Int. 1993;17:751–758. doi: 10.1006/cbir.1993.1136. [DOI] [PubMed] [Google Scholar]
- 57.Bourguignon LY, Jin H. Identification of the ankyrin-binding domain of the mouse T-lymphoma cell inositol 1,4,5-trisphosphate (IP3) receptor and its role in the regulation of IP3-mediated internal Ca2+ release. J Biol Chem. 1995;270:7257–7260. doi: 10.1074/jbc.270.13.7257. [DOI] [PubMed] [Google Scholar]
- 58.Brelidze TI, Niu XW, Magleby KL. A ring of eight conserved negatively charged amino acids doubles the conductance of BK channels and prevents inward rectification. Proc Natl Acad Sci USA. 2003;100:9017–9022. doi: 10.1073/pnas.1532257100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brillantes AB, Ondrias K, Scott A, Kobrinsky E, Ondriasova E, Moschella MC, Jayaraman T, Landers M, Ehrlich BE, Marks AR. Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell. 1994;77:513–523. doi: 10.1016/0092-8674(94)90214-3. [DOI] [PubMed] [Google Scholar]
- 60.Broad LM, Armstrong DL, Putney JW., Jr Role of the inositol 1,4,5-trisphosphate receptor in Ca2+ feedback inhibition of calcium release-activated calcium current (Icrac) J Biol Chem. 1999;274:32881–32888. doi: 10.1074/jbc.274.46.32881. [DOI] [PubMed] [Google Scholar]
- 61.Brown V, Jin P, Ceman S, Darnell JC, O’Donnell WT, Tenenbaum SA, Jin X, Feng Y, Wilkinson KD, Keene JD, Darnell RB, Warren ST. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell. 2001;107:477–487. doi: 10.1016/s0092-8674(01)00568-2. [DOI] [PubMed] [Google Scholar]
- 62.Bruening-Wright A, Schumacher MA, Adelman JP, Maylie J. Localization of the activation gate for small conductance Ca2+ activated K+ channels. J Neurosci. 2002;22:6499–6506. doi: 10.1523/JNEUROSCI.22-15-06499.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bugrim AE. Regulation of Ca2+ release by cAMP-dependent protein kinase. A mechanism for agonist-specific calcium signaling? Cell Calcium. 1999;25:219–226. doi: 10.1054/ceca.1999.0027. [DOI] [PubMed] [Google Scholar]
- 64.Bultynck G, De Smet P, Rossi D, Callewaert G, Missiaen L, Sorrentino V, De Smedt H, Parys JB. Characterization and mapping of the 12 kDa FK506-binding protein (FKBP12)-binding site on different isoforms of the ryanodine receptor and of the inositol 1,4,5-trisphosphate receptor. Biochem J. 2001;354:413–422. doi: 10.1042/0264-6021:3540413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bultynck G, De Smet P, Weidema AF, Ver Heyen M, Maes K, Callewaert G, Missiaen L, Parys JB, De Smedt H. Effects of the immunosuppressant FK506 on intracellular Ca2+ release and Ca2+ accumulation mechanisms. J Physiol. 2000;525:681–693. doi: 10.1111/j.1469-7793.2000.t01-1-00681.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bultynck G, Rossi D, Callewaert G, Missiaen L, Sorrentino V, Parys JB, De Smedt H. The conserved sites for the FK506-binding proteins in ryanodine receptors and inositol 1,4,5-trisphosphate receptors are structurally and functionally different. J Biol Chem. 2001;276:47715–47724. doi: 10.1074/jbc.M106573200. [DOI] [PubMed] [Google Scholar]
- 67.Bultynck G, Szlufcik K, Kasri NN, Assefa Z, Callewaert G, Missiaen L, Parys JB, De Smedt H. Thimerosal stimulates Ca2+ flux through inositol 1,4,5-trisphosphate receptor type 1, but not type 3, via modulation of an isoform-specific Ca2+-dependent intramolecular interaction. Biochem J. 2004;381:87–96. doi: 10.1042/BJ20040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bush KT, Stuart RO, Li SH, Moura LA, Sharp AH, Ross CA, Nigam SK. Epithelial inositol 1,4,5-trisphosphate receptors. Multiplicity of localization, solubility, isoforms. J Biol Chem. 1994;269:23694–23699. [PubMed] [Google Scholar]
- 69.Bygrave FL, Benedetti A. What is the concentration of calcium ions in the endoplasmic reticulum? Cell Calcium. 1996;19:547–551. doi: 10.1016/s0143-4160(96)90064-0. [DOI] [PubMed] [Google Scholar]
- 70.Cai W, Hisatsune C, Nakamura K, Nakamura T, Inoue T, Mikoshiba K. Activity-dependent expression of inositol 1,4,5-trisphosphate receptor type 1 in hippocampal neurons. J Biol Chem. 2004;279:23691–23698. doi: 10.1074/jbc.M313296200. [DOI] [PubMed] [Google Scholar]
- 71.Callamaras N, Marchant JS, Sun XP, Parker I. Activation and co-ordination of InsP3-mediated elementary Ca2+ events during global Ca2+ signals in Xenopus oocytes. J Physiol. 1998;509:81–91. doi: 10.1111/j.1469-7793.1998.081bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cameron AM, Nucifora FC, Jr, Fung ET, Livingston DJ, Aldape RA, Ross CA, Snyder SH. FKBP12 binds the inositol 1,4,5-trisphosphate receptor at leucine-proline (1400–1401) and anchors calcineurin to this FK506-like domain. J Biol Chem. 1997;272:27582–27588. doi: 10.1074/jbc.272.44.27582. [DOI] [PubMed] [Google Scholar]
- 73.Cameron AM, Steiner JP, Roskams AJ, Ali SM, Ronnett GV, Snyder SH. Calcineurin associated with the inositol 1,4,5-trisphosphate receptor-FKBP12 complex modulates Ca2+ flux. Cell. 1995;83:463–472. doi: 10.1016/0092-8674(95)90124-8. [DOI] [PubMed] [Google Scholar]
- 74.Cameron AM, Steiner JP, Sabatini DM, Kaplin AI, Walensky LD, Snyder SH. Immunophilin FK506 binding protein associated with inositol 1,4,5-trisphosphate receptor modulates calcium flux. Proc Natl Acad Sci USA. 1995;92:1784–1788. doi: 10.1073/pnas.92.5.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cannon B, Hermansson M, Gyorke S, Somerharju P, Virtanen JA, Cheng KH. Regulation of calcium channel activity by lipid domain formation in planar lipid bilayers. Biophys J. 2003;85:933–942. doi: 10.1016/S0006-3495(03)74532-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cardy TJ, Taylor CW. A novel role for calmodulin: Ca2+-independent inhibition of type-1 inositol trisphosphate receptors. Biochem J. 1998;334:447–455. doi: 10.1042/bj3340447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cardy TJ, Traynor D, Taylor CW. Differential regulation of types-1 and -3 inositol trisphosphate receptors by cytosolic Ca2+ Biochem J. 1997;328:785–793. doi: 10.1042/bj3280785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Carmody M, Mackrill JJ, Sorrentino V, O’Neill C. FKBP12 associates tightly with the skeletal muscle type 1 ryanodine receptor, but not with other intracellular calcium release channels. FEBS Lett. 2001;505:97–102. doi: 10.1016/s0014-5793(01)02787-9. [DOI] [PubMed] [Google Scholar]
- 79.Carter TD, Ogden D. Kinetics of Ca2+ release by InsP3 in pig single aortic endothelial cells: evidence for an inhibitory role of cytosolic Ca2+ in regulating hormonally evoked Ca2+ spikes. J Physiol. 1997;504:17–33. doi: 10.1111/j.1469-7793.1997.00017.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chadwick CC, Saito A, Fleischer S. Isolation and characterization of the inositol trisphosphate receptor from smooth muscle. Proc Natl Acad Sci USA. 1990;87:2132–2136. doi: 10.1073/pnas.87.6.2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chami M, Prandini A, Campanella M, Pinton P, Szabadkai G, Reed JC, Rizzuto R. Bcl-2 and Bax exert opposing effects on Ca2+ signaling, which do not depend on their putative pore-forming region. J Biol Chem. 2004;279:54581–54589. doi: 10.1074/jbc.M409663200. [DOI] [PubMed] [Google Scholar]
- 82.Champeil P, Combettes L, Berthon B, Doucet E, Orlowski S, Claret M. Fast kinetics of calcium release induced by myo-inositol trisphosphate in permeabilized rat hepatocytes. J Biol Chem. 1989;264:17665–17673. [PubMed] [Google Scholar]
- 83.Chen R, Valencia I, Zhong F, McColl KS, Roderick HL, Bootman MD, Berridge MJ, Conway SJ, Holmes AB, Mignery GA, Velez P, Distelhorst CW. Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J Cell Biol. 2004;166:193–203. doi: 10.1083/jcb.200309146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen S, Spiegelberg BD, Lin F, Dell EJ, Hamm HE. Interaction of Gβγ with RACK1 and other WD40 repeat proteins. J Mol Cell Cardiol. 2004;37:399–406. doi: 10.1016/j.yjmcc.2004.04.019. [DOI] [PubMed] [Google Scholar]
- 85.Chen SR, Ebisawa K, Li X, Zhang L. Molecular identification of the ryanodine receptor Ca2+ sensor. J Biol Chem. 1998;273:14675–14678. doi: 10.1074/jbc.273.24.14675. [DOI] [PubMed] [Google Scholar]
- 86.Cheng H, Lederer MR, Lederer WJ, Cannell MB. Calcium sparks and [Ca2+]i waves in cardiac myocytes. Am J Physiol Cell Physiol. 1996;270:C148–C159. doi: 10.1152/ajpcell.1996.270.1.C148. [DOI] [PubMed] [Google Scholar]
- 87.Choe CU, Harrison KD, Grant W, Ehrlich BE. Functional coupling of chromogranin with the inositol 1,4,5-trisphosphate receptor shapes calcium signaling. J Biol Chem. 2004;279:35551–35556. doi: 10.1074/jbc.M311261200. [DOI] [PubMed] [Google Scholar]
- 88.Clandinin TR, DeModena JA, Sternberg PW. Inositol trisphosphate mediates a RAS-independent response to LET-23 receptor tyrosine kinase activation in C. elegans. Cell. 1998;92:523–533. doi: 10.1016/s0092-8674(00)80945-9. [DOI] [PubMed] [Google Scholar]
- 89.Clapham DE. Calcium signaling. Cell. 1995;80:259–268. doi: 10.1016/0092-8674(95)90408-5. [DOI] [PubMed] [Google Scholar]
- 90.Combettes L, Cheek TR, Taylor CW. Regulation of inositol trisphosphate receptors by luminal Ca2+ contributes to quantal Ca2+ mobilization. EMBO J. 1996;15:2086–2093. [PMC free article] [PubMed] [Google Scholar]
- 91.Combettes L, Hannaert-Merah Z, Coquil JF, Rousseau C, Claret M, Swillens S, Champeil P. Rapid filtration studies of the effect of cytosolic Ca2+ on inositol 1,4,5-trisphosphate-induced 45Ca2+ release from cerebellar microsomes. J Biol Chem. 1994;269:17561–17571. [PubMed] [Google Scholar]
- 92.Coquil JF, Mauger JP, Claret M. Inositol 1,4,5-trisphosphate slowly converts its receptor to a state of higher affinity in sheep cerebellum membranes. J Biol Chem. 1996;271:3568–3574. doi: 10.1074/jbc.271.7.3568. [DOI] [PubMed] [Google Scholar]
- 93.Correa V, Riley AM, Shuto S, Horne G, Nerou EP, Marwood RD, Potter BV, Taylor CW. Structural determinants of adenophostin A activity at inositol trisphosphate receptors. Mol Pharmacol. 2001;59:1206–1215. doi: 10.1124/mol.59.5.1206. [DOI] [PubMed] [Google Scholar]
- 94.Csordás G, Thomas AP, Hajnóczky G. Quasi-synaptic calcium signal transmission between endoplasmic reticulum and mitochondria. EMBO J. 1999;18:96–108. doi: 10.1093/emboj/18.1.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cui J, Matkovich SJ, deSouza N, Li S, Rosemblit N, Marks AR. Regulation of the type 1 inositol 1,4,5-trisphosphate receptor by phosphorylation at tyrosine 353. J Biol Chem. 2004;279:16311–16316. doi: 10.1074/jbc.M400206200. [DOI] [PubMed] [Google Scholar]
- 96.Da Fonseca PC, Morris SA, Nerou EP, Taylor CW, Morris EP. Domain organization of the type 1 inositol 1,4,5-trisphosphate receptor as revealed by single-particle analysis. Proc Natl Acad Sci USA. 2003;100:3936–3941. doi: 10.1073/pnas.0536251100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dal Santo P, Logan MA, Chisholm AD, Jorgensen EM. The inositol trisphosphate receptor regulates a 50-second behavioral rhythm in C. elegans. Cell. 1999;98:757–767. doi: 10.1016/s0092-8674(00)81510-x. [DOI] [PubMed] [Google Scholar]
- 98.Danila CI, Hamilton SL. Phosphorylation of ryanodine receptors. Biol Res. 2004;37:521–525. doi: 10.4067/s0716-97602004000400005. [DOI] [PubMed] [Google Scholar]
- 99.Danoff SK, Ferris CD, Donath C, Fischer GA, Munemitsu S, Ullrich A, Snyder SH, Ross CA. Inositol 1,4,5-trisphosphate receptors: distinct neuronal and nonneuronal forms derived by alternative splicing differ in phosphorylation. Proc Natl Acad Sci USA. 1991;88:2951–2955. doi: 10.1073/pnas.88.7.2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dargan SL, Lea EJ, Dawson AP. Modulation of type-1 Ins(1,4,5)P3 receptor channels by the FK506-binding protein, FKBP12. Biochem J. 2002;361:401–407. doi: 10.1042/0264-6021:3610401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dawson SP, Keizer J, Pearson JE. Fire-diffuse-fire model of dynamics of intracellular calcium waves. Proc Natl Acad Sci USA. 1999;96:6060–6063. doi: 10.1073/pnas.96.11.6060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.De Kort M, Correa V, Valentijn AR, van der Marel GA, Potter BV, Taylor CW, van Boom JH. Synthesis of potent agonists of the D-myo-inositol 1,4, 5-trisphosphate receptor based on clustered disaccharide polyphosphate analogues of adenophostin A. J Med Chem. 2000;43:3295–3303. doi: 10.1021/jm000957c. [DOI] [PubMed] [Google Scholar]
- 103.De Smedt F, Verjans B, Mailleux P, Erneux C. Cloning and expression of human brain type I inositol 1,4,5-trisphosphate 5-phosphatase. High levels of mRNA in cerebellar Purkinje cells. FEBS Lett. 1994;347:69–72. doi: 10.1016/0014-5793(94)00509-5. [DOI] [PubMed] [Google Scholar]
- 104.De Smedt H, Missiaen L, Parys JB, Bootman MD, Mertens L, Van Den Bosch L, Casteels R. Determination of relative amounts of inositol trisphosphate receptor mRNA isoforms by ratio polymerase chain reaction. J Biol Chem. 1994;269:21691–21698. [PubMed] [Google Scholar]
- 105.De Smedt H, Missiaen L, Parys JB, Henning RH, Sienaert I, Vanlingen S, Gijsens A, Himpens B, Casteels R. Isoform diversity of the inositol trisphosphate receptor in cell types of mouse origin. Biochem J. 1997;322:575–583. doi: 10.1042/bj3220575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Del Camino D, Yellen G. Tight steric closure at the intracellular activation gate of a voltage-gated K+ channel. Neuron. 2001;32:649–656. doi: 10.1016/s0896-6273(01)00487-1. [DOI] [PubMed] [Google Scholar]
- 107.DeLisle S, Marksberry EW, Bonnett C, Jenkins DJ, Potter BV, Takahashi M, Tanzawa K. Adenophostin A can stimulate Ca2+ influx without depleting the inositol 1,4,5-trisphosphate-sensitive Ca2+ stores in the Xenopus oocyte. J Biol Chem. 1997;272:9956–9961. doi: 10.1074/jbc.272.15.9956. [DOI] [PubMed] [Google Scholar]
- 108.Dellis O, Dedos SG, Tovey SC, Taufiq Ur R, Dubel SJ, Taylor CW. Ca2+ entry through plasma membrane IP3 receptors. Science. 2006;313:229–233. doi: 10.1126/science.1125203. [DOI] [PubMed] [Google Scholar]
- 109.Deshpande M, Venkatesh K, Rodrigues V, Hasan G. The inositol 1,4,5-trisphosphate receptor is required for maintenance of olfactory adaptation in Drosophila antennae. J Neurobiol. 2000;43:282–288. doi: 10.1002/(sici)1097-4695(20000605)43:3<282::aid-neu6>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 110.DeSouza N, Reiken S, Ondrias K, Yang YM, Matkovich S, Marks AR. Protein kinase A and two phosphatases are components of the inositol 1,4,5-trisphosphate receptor macromolecular signaling complex. J Biol Chem. 2002;277:39397–39400. doi: 10.1074/jbc.M207059200. [DOI] [PubMed] [Google Scholar]
- 111.Devogelaere B, Nadif Kasri N, Derua R, Waelkens E, Callewaert G, Missiaen L, Parys JB, De Smedt H. Binding of IRBIT to the IP3 receptor: determinants and functional effects. Biochem Biophys Res Commun. 2006;343:49–56. doi: 10.1016/j.bbrc.2006.02.119. [DOI] [PubMed] [Google Scholar]
- 112.Diaz F, Bourguignon LY. Selective down-regulation of IP3 receptor subtypes by caspases and calpain during TNFα-induced apoptosis of human T-lymphoma cells. Cell Calcium. 2000;27:315–328. doi: 10.1054/ceca.2000.0126. [DOI] [PubMed] [Google Scholar]
- 113.Dingwall C, Laskey R. The nuclear membrane. Science. 1992;258:942–947. doi: 10.1126/science.1439805. [DOI] [PubMed] [Google Scholar]
- 114.Dirksen RT, Avila G. Altered ryanodine receptor function in central core disease: leaky or uncoupled Ca2+ release channels? Trends Cardiovasc Med. 2002;12:189–197. doi: 10.1016/s1050-1738(02)00163-9. [DOI] [PubMed] [Google Scholar]
- 115.Doyle DA, Cabral JM, Pfuetzner RA, Kuo AL, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 116.Du GG, Guo XH, Khanna VK, MacLennan DH. Functional characterization of mutants in the predicted pore region of the rabbit cardiac muscle Ca2+ release channel (ryanodine receptor isoform 2) J Biol Chem. 2001;276:31760–31771. doi: 10.1074/jbc.M102751200. [DOI] [PubMed] [Google Scholar]
- 117.Du GG, MacLennan DH. Functional consequences of mutations of conserved, polar amino acids in transmembrane sequences of the Ca2+ release channel (ryanodine receptor) of rabbit skeletal muscle sarcoplasmic reticulum. J Biol Chem. 1998;273:31867–31872. doi: 10.1074/jbc.273.48.31867. [DOI] [PubMed] [Google Scholar]
- 118.Du W, Frazier M, McMahon TJ, Eu JP. Redox activation of intracellular calcium release channels (ryanodine receptors) in the sustained phase of hypoxia-induced pulmonary vasoconstriction. Chest. 2005;128:556S–558S. doi: 10.1378/chest.128.6_suppl.556S. [DOI] [PubMed] [Google Scholar]
- 119.Duchen MR. Mitochondria and calcium: from cell signalling to cell death. J Physiol. 2000;529:57–68. doi: 10.1111/j.1469-7793.2000.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dufour JF, Arias IM, Turner TJ. Inositol 1,4,5-trisphosphate and calcium regulate the calcium channel function of the hepatic inositol 1,4,5-trisphosphate receptor. J Biol Chem. 1997;272:2675–2681. doi: 10.1074/jbc.272.5.2675. [DOI] [PubMed] [Google Scholar]
- 121.Dutzler R, Campbell EB, MacKinnon R. Gating the selectivity filter in ClC chloride channels. Science. 2003;300:108–112. doi: 10.1126/science.1082708. [DOI] [PubMed] [Google Scholar]
- 122.Dyer JL, Michelangeli F. Inositol 1,4,5-trisphosphate receptor isoforms show similar Ca2+ release kinetics. Cell Calcium. 2001;30:245–250. doi: 10.1054/ceca.2001.0231. [DOI] [PubMed] [Google Scholar]
- 123.Dyer JL, Mobasheri H, Lea EJ, Dawson AP, Michelangeli F. Differential effect of PKA on the Ca2+ release kinetics of the type I and III InsP3 receptors. Biochem Biophys Res Commun. 2003;302:121–126. doi: 10.1016/s0006-291x(03)00120-7. [DOI] [PubMed] [Google Scholar]
- 124.Ehrlich BE, Watras J. Inositol 1,4,5-trisphosphate activates a channel from smooth muscle sarcoplasmic reticulum. Nature. 1988;336:583–586. doi: 10.1038/336583a0. [DOI] [PubMed] [Google Scholar]
- 125.Eismann E, Muller F, Heinemann SH, Kaupp UB. A single negative charge within the pore region of a cGMP-gated channel controls rectification, Ca2+ blockage, ionic selectivity. Proc Natl Acad Sci USA. 1994;91:1109–1113. doi: 10.1073/pnas.91.3.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Eu JP, Sun J, Xu L, Stamler JS, Meissner G. The skeletal muscle calcium release channel: coupled O2 sensor and NO signaling functions. Cell. 2000;102:499–509. doi: 10.1016/s0092-8674(00)00054-4. [DOI] [PubMed] [Google Scholar]
- 127.Fabiato A. Computer programs for calculating total from specified free or free from specified total ionic concentrations in aqueous solutions containing multiple metals and ligands. Methods Enzymol. 1988;157:378–417. doi: 10.1016/0076-6879(88)57093-3. [DOI] [PubMed] [Google Scholar]
- 128.Fagni L, Worley PF, Ango F. Homer as both a scaffold and transduction molecule. Sci STKE. 2002;2002:RE8. doi: 10.1126/stke.2002.137.re8. [DOI] [PubMed] [Google Scholar]
- 129.Faure AV, Grunwald D, Moutin MJ, Hilly M, Mauger JP, Marty I, De Waard M, Villaz M, Albrieux M. Developmental expression of the calcium release channels during early neurogenesis of the mouse cerebral cortex. Eur J Neurosci. 2001;14:1613–1622. doi: 10.1046/j.0953-816x.2001.01786.x. [DOI] [PubMed] [Google Scholar]
- 130.Feigin A, Zgaljardic D. Recent advances in Huntington’s disease: implications for experimental therapeutics. Curr Opin Neurol. 2002;15:483–489. doi: 10.1097/00019052-200208000-00013. [DOI] [PubMed] [Google Scholar]
- 131.Feng W, Tu J, Yang T, Vernon PS, Allen PD, Worley PF, Pessah IN. Homer regulates gain of ryanodine receptor type 1 channel complex. J Biol Chem. 2002;277:44722–44730. doi: 10.1074/jbc.M207675200. [DOI] [PubMed] [Google Scholar]
- 132.Ferreri-Jacobia M, Mak DOD, Foskett JK. Translational mobility of the type 3 inositol 1,4,5-trisphosphate receptor Ca2+ release channel in endoplasmic reticulum membrane. J Biol Chem. 2005;280:3824–3831. doi: 10.1074/jbc.M409462200. [DOI] [PubMed] [Google Scholar]
- 133.Ferris CD, Cameron AM, Bredt DS, Huganir RL, Snyder SH. Inositol 1,4,5-trisphosphate receptor is phosphorylated by cyclic AMP-dependent protein kinase at serines 1755 and 1589. Biochem Biophys Res Commun. 1991;175:192–198. doi: 10.1016/s0006-291x(05)81219-7. [DOI] [PubMed] [Google Scholar]
- 134.Ferris CD, Huganir RL, Bredt DS, Cameron AM, Snyder SH. Inositol trisphosphate receptor: phosphorylation by protein kinase C and calcium calmodulin-dependent protein kinases in reconstituted lipid vesicles. Proc Natl Acad Sci USA. 1991;88:2232–2235. doi: 10.1073/pnas.88.6.2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ferris CD, Huganir RL, Snyder SH. Calcium flux mediated by purified inositol 1,4,5-trisphosphate receptor in reconstituted lipid vesicles is allosterically regulated by adenine nucleotides. Proc Natl Acad Sci USA. 1990;87:2147–2151. doi: 10.1073/pnas.87.6.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ferris CD, Huganir RL, Supattapone S, Snyder SH. Purified inositol 1,4,5-trisphosphate receptor mediates calcium flux in reconstituted lipid vesicles. Nature. 1989;342:87–89. doi: 10.1038/342087a0. [DOI] [PubMed] [Google Scholar]
- 137.Ferris CD, Snyder SH. Inositol 1,4,5-trisphosphate-activated calcium channels. Annu Rev Physiol. 1992;54:469–488. doi: 10.1146/annurev.ph.54.030192.002345. [DOI] [PubMed] [Google Scholar]
- 138.Ferris CD, Snyder SH. IP3 receptors. Ligand-activated calcium channels in multiple forms. Adv Second Messenger Phosphoprotein Res. 1992;26:95–107. [PubMed] [Google Scholar]
- 139.Fill M, Copello JA. Ryanodine receptor calcium release channels. Physiol Rev. 2002;82:893–922. doi: 10.1152/physrev.00013.2002. [DOI] [PubMed] [Google Scholar]
- 140.Finch EA, Turner TJ, Goldin SM. Calcium as a coagonist of inositol 1,4,5-trisphosphate-induced calcium release. Science. 1991;252:443–446. doi: 10.1126/science.2017683. [DOI] [PubMed] [Google Scholar]
- 141.Finch EA, Turner TJ, Goldin SM. Subsecond kinetics of inositol 1,4,5-trisphosphate-induced calcium release reveal rapid potentiation and subsequent inactivation by calcium. Ann NY Acad Sci. 1991;635:400–403. doi: 10.1111/j.1749-6632.1991.tb36509.x. [DOI] [PubMed] [Google Scholar]
- 142.Flynn GE, Johnson JP, Zagotta WN. Cyclic nucleotide-gated channels: shedding light on the opening of a channel pore. Nat Rev Neurosci. 2001;2:643–651. doi: 10.1038/35090015. [DOI] [PubMed] [Google Scholar]
- 143.Flynn GE, Zagotta WN. Conformational changes in S6 coupled to the opening of cyclic nucleotide-gated channels. Neuron. 2001;30:689–698. doi: 10.1016/s0896-6273(01)00324-5. [DOI] [PubMed] [Google Scholar]
- 144.Foskett JK, Mak DOD. Novel model of calcium and inositol 1,4,5-trisphosphate regulation of InsP3 receptor channel gating in native endoplasmic reticulum. Biol Res. 2004;37:513–519. doi: 10.4067/s0716-97602004000400004. [DOI] [PubMed] [Google Scholar]
- 145.Foyouzi-Youssefi R, Arnaudeau S, Borner C, Kelley WL, Tschopp J, Lew DP, Demaurex N, Krause KH. Bcl-2 decreases the free Ca2+ concentration within the endoplasmic reticulum. Proc Natl Acad Sci USA. 2000;97:5723–5728. doi: 10.1073/pnas.97.11.5723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Fujino I, Yamada N, Miyawaki A, Hasegawa M, Furuichi T, Mikoshiba K. Differential expression of type 2 and type 3 inositol 1,4,5-trisphosphate receptor mRNAs in various mouse tissues: in situ hybridization study. Cell Tissue Res. 1995;280:201–210. doi: 10.1007/BF00307790. [DOI] [PubMed] [Google Scholar]
- 147.Fukatsu K, Bannai H, Inoue T, Mikoshiba K. 4.1N binding regions of inositol 1,4,5-trisphosphate receptor type 1. Biochem Biophys Res Commun. 2006;342:573–576. doi: 10.1016/j.bbrc.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 148.Fukatsu K, Bannai H, Zhang S, Nakamura H, Inoue T, Mikoshiba K. Lateral diffusion of inositol 1,4,5-trisphosphate receptor type 1 is regulated by actin filaments and 4.1N in neuronal dendrites. J Biol Chem. 2004;279:48976–48982. doi: 10.1074/jbc.M408364200. [DOI] [PubMed] [Google Scholar]
- 149.Furuichi T, Simon-Chazottes D, Fujino I, Yamada N, Hasegawa M, Miyawaki A, Yoshikawa S, Guenet JL, Mikoshiba K. Widespread expression of inositol 1,4,5-trisphosphate receptor type 1 gene (Insp3r1) in the mouse central nervous system. Receptors Channels. 1993;1:11–24. [PubMed] [Google Scholar]
- 150.Furuichi T, Yoshikawa S, Miyawaki A, Wada K, Maeda N, Mikoshiba K. Primary structure and functional expression of the inositol 1,4,5-trisphosphate-binding protein P400. Nature. 1989;342:32–38. doi: 10.1038/342032a0. [DOI] [PubMed] [Google Scholar]
- 151.Futatsugi A, Kuwajima G, Mikoshiba K. Muscle-specific mRNA isoform encodes a protein composed mainly of the N-terminal 175 residues of type 2 Ins(1,4,5)P3 receptor. Biochem J. 1998;334:559–563. doi: 10.1042/bj3340559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Futatsugi A, Nakamura T, Yamada MK, Ebisui E, Nakamura K, Uchida K, Kitaguchi T, Takahashi-Iwanaga H, Noda T, Aruga J, Mikoshiba K. IP3 receptor types 2 and 3 mediate exocrine secretion underlying energy metabolism. Science. 2005;309:2232–2234. doi: 10.1126/science.1114110. [DOI] [PubMed] [Google Scholar]
- 153.Galvan DL, Borrego-Diaz E, Perez PJ, Mignery GA. Subunit oligomerization, topology of the inositol 1,4,5-trisphosphate receptor. J Biol Chem. 1999;274:29483–29492. doi: 10.1074/jbc.274.41.29483. [DOI] [PubMed] [Google Scholar]
- 154.Galvan DL, Mignery GA. Carboxyl-terminal sequences critical for inositol 1,4,5-trisphosphate receptor subunit assembly. J Biol Chem. 2002;277:48248–48260. doi: 10.1074/jbc.M209990200. [DOI] [PubMed] [Google Scholar]
- 155.Gao L, Balshaw D, Xu L, Tripathy A, Xin CL, Meissner G. Evidence for a role of the luminal M3–M4 loop in skeletal muscle Ca2+ release channel (ryanodine receptor) activity and conductance. Biophys J. 2000;79:828–840. doi: 10.1016/S0006-3495(00)76339-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Gentry HR, Singer AU, Betts L, Yang C, Ferrara JD, Sondek J, Parise LV. Structural and biochemical characterization of CIB1 delineates a new family of EF-hand-containing proteins. J Biol Chem. 2005;280:8407–8415. doi: 10.1074/jbc.M411515200. [DOI] [PubMed] [Google Scholar]
- 157.Glouchankova L, Krishna UM, Potter BV, Falck JR, Bezprozvanny I. Association of the inositol (1,4,5)-trisphosphate receptor ligand binding site with phosphatidylinositol (4,5)-bisphosphate and adenophostin A. Mol Cell Biol Res Commun. 2000;3:153–158. doi: 10.1006/mcbr.2000.0208. [DOI] [PubMed] [Google Scholar]
- 158.Goldberg MW, Allen TD. The nuclear pore complex: three-dimensional surface structure revealed by field emission, in-lens scanning electron microscopy, with underlying structure uncovered by proteolysis. J Cell Sci. 1993;106:261–274. doi: 10.1242/jcs.106.1.261. [DOI] [PubMed] [Google Scholar]
- 159.Gower NJ, Temple GR, Schein JE, Marra M, Walker DS, Baylis HA. Dissection of the promoter region of the inositol 1,4,5-trisphosphate receptor gene, itr-1, in C. elegans: a molecular basis for cell-specific expression of IP3R isoforms. J Mol Biol. 2001;306:145–157. doi: 10.1006/jmbi.2000.4388. [DOI] [PubMed] [Google Scholar]
- 160.Gregory RB, Wilcox RA, Berven LA, van Straten NC, van der Marel GA, van Boom JH, Barritt GJ. Evidence for the involvement of a small subregion of the endoplasmic reticulum in the inositol trisphosphate receptor-induced activation of Ca2+ inflow in rat hepatocytes. Biochem J. 1999;341:401–408. [PMC free article] [PubMed] [Google Scholar]
- 161.Haeseleer F, Imanishi Y, Sokal I, Filipek S, Palczewski K. Calcium-binding proteins: intracellular sensors from the calmodulin superfamily. Biochem Biophys Res Commun. 2002;290:615–623. doi: 10.1006/bbrc.2001.6228. [DOI] [PubMed] [Google Scholar]
- 162.Haeseleer F, Sokal I, Verlinde CL, Erdjument-Bromage H, Tempst P, Pronin AN, Benovic JL, Fariss RN, Palczewski K. Five members of a novel Ca2+-binding protein (CABP) subfamily with similarity to calmodulin. J Biol Chem. 2000;275:1247–1260. doi: 10.1074/jbc.275.2.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Hagar RE, Burgstahler AD, Nathanson MH, Ehrlich BE. Type III InsP3 receptor channel stays open in the presence of increased calcium. Nature. 1998;396:81–84. doi: 10.1038/23954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hagar RE, Ehrlich BE. Regulation of the type III InsP3 receptor and its role in β cell function. Cell Mol Life Sci. 2000;57:1938–1949. doi: 10.1007/PL00000674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hajnóczky G, Davies E, Madesh M. Calcium signaling and apoptosis. Biochem Biophys Res Commun. 2003;304:445–454. doi: 10.1016/s0006-291x(03)00616-8. [DOI] [PubMed] [Google Scholar]
- 166.Hajnóczky G, Gao E, Nomura T, Hoek JB, Thomas AP. Multiple mechanisms by which protein kinase A potentiates inositol 1,4,5-trisphosphate-induced Ca2+ mobilization in permeabilized hepatocytes. Biochem J. 1993;293:413–422. doi: 10.1042/bj2930413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Hajnóczky G, Thomas AP. The inositol trisphosphate calcium channel is inactivated by inositol trisphosphate. Nature. 1994;370:474–477. doi: 10.1038/370474a0. [DOI] [PubMed] [Google Scholar]
- 168.Hajnóczky G, Thomas AP. Minimal requirements for calcium oscillations driven by the IP3 receptor. EMBO J. 1997;16:3533–3543. doi: 10.1093/emboj/16.12.3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Hamada K, Miyata T, Mayanagi K, Hirota J, Mikoshiba K. Two-state conformational changes in inositol 1,4,5-trisphosphate receptor regulated by calcium. J Biol Chem. 2002;277:21115–21118. doi: 10.1074/jbc.C200244200. [DOI] [PubMed] [Google Scholar]
- 170.Hamada K, Terauchi A, Mikoshiba K. Three-dimensional rearrangements within inositol 1,4,5-trisphosphate receptor by calcium. J Biol Chem. 2003;278:52881–52889. doi: 10.1074/jbc.M309743200. [DOI] [PubMed] [Google Scholar]
- 171.Harnick DJ, Jayaraman T, Ma Y, Mulieri P, Go LO, Marks AR. The human type 1 inositol 1,4,5-trisphosphate receptor from T lymphocytes. Structure, localization, tyrosine phosphorylation. J Biol Chem. 1995;270:2833–2840. doi: 10.1074/jbc.270.6.2833. [DOI] [PubMed] [Google Scholar]
- 172.Hartzell HC, Machaca K, Hirayama Y. Effects of adenophostin-A and inositol-1,4,5-trisphosphate on Cl− currents in Xenopus laevis oocytes. Mol Pharmacol. 1997;51:683–692. doi: 10.1124/mol.51.4.683. [DOI] [PubMed] [Google Scholar]
- 173.Hattori M, Suzuki AZ, Higo T, Miyauchi H, Michikawa T, Nakamura T, Inoue T, Mikoshiba K. Distinct roles of inositol 1,4,5-trisphosphate receptor types 1 and 3 in Ca2+ signaling. J Biol Chem. 2004;279:11967–11975. doi: 10.1074/jbc.M311456200. [DOI] [PubMed] [Google Scholar]
- 174.Haug LS, Jensen V, Hvalby O, Walaas SI, Ostvold AC. Phosphorylation of the inositol 1,4,5-trisphosphate receptor by cyclic nucleotide-dependent kinases in vitro and in rat cerebellar slices in situ. J Biol Chem. 1999;274:7467–7473. doi: 10.1074/jbc.274.11.7467. [DOI] [PubMed] [Google Scholar]
- 175.Haug LS, Walaas SI, Ostvold AC. Degradation of the type I inositol 1,4,5-trisphosphate receptor by caspase-3 in SH-SY5Y neuroblastoma cells undergoing apoptosis. J Neurochem. 2000;75:1852–1861. doi: 10.1046/j.1471-4159.2000.0751852.x. [DOI] [PubMed] [Google Scholar]
- 176.Hayashi T, Maurice T, Su TP. Ca2+ signaling via σ1-receptors: novel regulatory mechanism affecting intracellular Ca2+ concentration. J Pharmacol Exp Ther. 2000;293:788–798. [PubMed] [Google Scholar]
- 177.Hayashi T, Su TP. Regulating ankyrin dynamics: roles of σ1 receptors. Proc Natl Acad Sci USA. 2001;98:491–496. doi: 10.1073/pnas.98.2.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Haynes LP, Tepikin AV, Burgoyne RD. Calcium-binding protein 1 is an inhibitor of agonist-evoked, inositol 1,4,5-trisphosphate-mediated calcium signaling. J Biol Chem. 2004;279:547–555. doi: 10.1074/jbc.M309617200. [DOI] [PubMed] [Google Scholar]
- 179.He CL, Damiani P, Ducibella T, Takahashi M, Tanzawa K, Parys JB, Fissore RA. Isoforms of the inositol 1,4,5-trisphosphate receptor are expressed in bovine oocytes and ovaries: the type-1 isoform is down-regulated by fertilization and by injection of adenophostin A. Biol Reprod. 1999;61:935–943. doi: 10.1095/biolreprod61.4.935. [DOI] [PubMed] [Google Scholar]
- 180.He X, Yang F, Xie Z, Lu B. Intracellular Ca2+ and Ca2+/calmodulin-dependent kinase II mediate acute potentiation of neurotransmitter release by neurotrophin-3. J Cell Biol. 2000;149:783–792. doi: 10.1083/jcb.149.4.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Heginbotham L, Abramson T, Mackinnon R. A functional connection between the pores of distantly related ion channels as revealed by mutant K+ channels. Science. 1992;258:1152–1155. doi: 10.1126/science.1279807. [DOI] [PubMed] [Google Scholar]
- 182.Hess P, Tsien RW. Mechanism of ion permeation through calcium channels. Nature. 1984;309:453–456. doi: 10.1038/309453a0. [DOI] [PubMed] [Google Scholar]
- 183.Hidalgo C, Bull R, Behrens MI, Donoso P. Redox regulation of RyR-mediated Ca2+ release in muscle and neurons. Biol Res. 2004;37:539–552. doi: 10.4067/s0716-97602004000400007. [DOI] [PubMed] [Google Scholar]
- 184.Higo T, Hattori M, Nakamura T, Natsume T, Michikawa T, Mikoshiba K. Subtype-specific and ER lumenal environment-dependent regulation of inositol 1,4,5-trisphosphate receptor type 1 by ERp44. Cell. 2005;120:85–98. doi: 10.1016/j.cell.2004.11.048. [DOI] [PubMed] [Google Scholar]
- 185.Hille B. Ionic Channels of Excitable Membranes. Sunderland, MA: Sinauer; 2001. p. 814. [Google Scholar]
- 186.Hilly M, Pietri-Rouxel F, Coquil JF, Guy M, Mauger JP. Thiol reagents increase the affinity of the inositol 1,4,5-trisphosphate receptor. J Biol Chem. 1993;268:16488–16494. [PubMed] [Google Scholar]
- 187.Hirose K, Iino M. Heterogeneity of channel density in inositol-1,4,5-trisphosphate-sensitive Ca2+ stores. Nature. 1994;372:791–794. doi: 10.1038/372791a0. [DOI] [PubMed] [Google Scholar]
- 188.Hirota J, Baba M, Matsumoto M, Furuichi T, Takatsu K, Mikoshiba K. T-cell-receptor signalling in inositol 1,4,5-trisphosphate receptor (IP3R) type-1-deficient mice: is IP3R type 1 essential for T-cell-receptor signalling? Biochem J. 1998;333:615–619. doi: 10.1042/bj3330615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Hirota J, Furuichi T, Mikoshiba K. Inositol 1,4,5-trisphosphate receptor type 1 is a substrate for caspase-3 and is cleaved during apoptosis in a caspase-3-dependent manner. J Biol Chem. 1999;274:34433–34437. doi: 10.1074/jbc.274.48.34433. [DOI] [PubMed] [Google Scholar]
- 190.Hirota J, Michikawa T, Miyawaki A, Takahashi M, Tanzawa K, Okura I, Furuichi T, Mikoshiba K. Adenophostin-medicated quantal Ca2+ release in the purified and reconstituted inositol 1,4,5-trisphosphate receptor type 1. FEBS Lett. 1995;368:248–252. doi: 10.1016/0014-5793(95)00659-w. [DOI] [PubMed] [Google Scholar]
- 191.Hirota J, Michikawa T, Natsume T, Furuichi T, Mikoshiba K. Calmodulin inhibits inositol 1,4,5-trisphosphate-induced calcium release through the purified and reconstituted inositol 1,4,5-trisphosphate receptor type 1. FEBS Lett. 1999;456:322–326. doi: 10.1016/s0014-5793(99)00973-4. [DOI] [PubMed] [Google Scholar]
- 192.Horne JH, Meyer T. Elementary calcium-release units induced by inositol trisphosphate. Science. 1997;276:1690–1693. doi: 10.1126/science.276.5319.1690. [DOI] [PubMed] [Google Scholar]
- 193.Huang Y, Takahashi M, Tanzawa K, Putney JW., Jr Effect of adenophostin A on Ca2+ entry and calcium release-activated calcium current (Icrac) in rat basophilic leukemia cells. J Biol Chem. 1998;273:31815–31821. doi: 10.1074/jbc.273.48.31815. [DOI] [PubMed] [Google Scholar]
- 194.Huh YH, Chu SY, Park SY, Huh SK, Yoo SH. Role of nuclear chromogranin B in inositol 1,4,5-trisphosphate-mediated nuclear Ca2+ mobilization. Biochemistry. 2006;45:1212–1226. doi: 10.1021/bi051594r. [DOI] [PubMed] [Google Scholar]
- 195.Iino M. Effects of adenine nucleotides on inositol 1,4,5-trisphosphate-induced calcium release in vascular smooth muscle cells. J Gen Physiol. 1991;98:681–698. doi: 10.1085/jgp.98.4.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Ionescu L, Cheung KH, Vais H, Mak DOD, White C, Foskett JK. Graded recruitment and inactivation of single InsP3 receptor Ca2+-release channels: implications for quantal Ca2+release. J Physiol. 2006;573:645–662. doi: 10.1113/jphysiol.2006.109504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Irvine RF. “Quantal” Ca2+ release and the control of Ca2+ entry by inositol phosphates—a possible mechanism. FEBS Lett. 1990;263:5–9. doi: 10.1016/0014-5793(90)80692-c. [DOI] [PubMed] [Google Scholar]
- 198.Irvine RF, Brown KD, Berridge MJ. Specificity of inositol trisphosphate-induced calcium release from permeabilized Swiss-mouse 3T3 cells. Biochem J. 1984;222:269–272. doi: 10.1042/bj2220269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Iwai M, Tateishi Y, Hattori M, Mizutani A, Nakamura T, Futatsugi A, Inoue T, Furuichi T, Michikawa T, Mikoshiba K. Molecular cloning of mouse type 2 and type 3 inositol 1,4,5-trisphosphate receptors and identification of a novel type 2 receptor splice variant. J Biol Chem. 2005;280:10305–10317. doi: 10.1074/jbc.M413824200. [DOI] [PubMed] [Google Scholar]
- 200.Iwasaki H, Chiba K, Uchiyama T, Yoshikawa F, Suzuki F, Ikeda M, Furuichi T, Mikoshiba K. Molecular characterization of the starfish inositol 1,4,5-trisphosphate receptor and its role during oocyte maturation and fertilization. J Biol Chem. 2002;277:2763–2772. doi: 10.1074/jbc.M108839200. [DOI] [PubMed] [Google Scholar]
- 201.Jayaraman T, Marks AR. T cells deficient in inositol 1,4,5-trisphosphate receptor are resistant to apoptosis. Mol Cell Biol. 1997;17:3005–3012. doi: 10.1128/mcb.17.6.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Jayaraman T, Ondrias K, Ondriasova E, Marks AR. Regulation of the inositol 1,4,5-trisphosphate receptor by tyrosine phosphorylation. Science. 1996;272:1492–1494. doi: 10.1126/science.272.5267.1492. [DOI] [PubMed] [Google Scholar]
- 203.Jellerette T, He CL, Wu H, Parys JB, Fissore RA. Down-regulation of the inositol 1,4,5-trisphosphate receptor in mouse eggs following fertilization or parthenogenetic activation. Dev Biol. 2000;223:238–250. doi: 10.1006/dbio.2000.9675. [DOI] [PubMed] [Google Scholar]
- 204.Jiang QX, Thrower EC, Chester DW, Ehrlich BE, Sigworth FJ. Three-dimensional structure of the type 1 inositol 1,4,5-trisphosphate receptor at 24 A ° resolution. EMBO J. 2002;21:3575–3581. doi: 10.1093/emboj/cdf380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Jiang YX, Lee A, Chen JY, Cadene M, Chait BT, MacKinnon R. The open pore conformation of potassium channels. Nature. 2002;417:523–526. doi: 10.1038/417523a. [DOI] [PubMed] [Google Scholar]
- 206.Jiang YX, Lee A, Chen JY, Ruta V, Cadene M, Chait BT, MacKinnon R. X-ray structure of a voltage-dependent K+ channel. Nature. 2003;423:33–41. doi: 10.1038/nature01580. [DOI] [PubMed] [Google Scholar]
- 207.Johenning FW, Zochowski M, Conway SJ, Holmes AB, Koulen P, Ehrlich BE. Distinct intracellular calcium transients in neurites and somata integrate neuronal signals. J Neurosci. 2002;22:5344–5353. doi: 10.1523/JNEUROSCI.22-13-05344.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Jonas EA, Knox RJ, Kaczmarek LK. Gigaohm seals on intracellular membranes: a technique for studying intracellular ion channels in intact cells. Neuron. 1997;19:7–13. doi: 10.1016/s0896-6273(00)80343-8. [DOI] [PubMed] [Google Scholar]
- 209.Joseph SK, Boehning D, Pierson S, Nicchitta CV. Membrane insertion, glycosylation, oligomerization of inositol trisphosphate receptors in a cell-free translation system. J Biol Chem. 1997;272:1579–1588. doi: 10.1074/jbc.272.3.1579. [DOI] [PubMed] [Google Scholar]
- 210.Joseph SK, Bokkala S, Boehning D, Zeigler S. Factors determining the composition of inositol trisphosphate receptor heterooligomers expressed in COS cells. J Biol Chem. 2000;275:16084–16090. doi: 10.1074/jbc.M000506200. [DOI] [PubMed] [Google Scholar]
- 211.Joseph SK, Brownell S, Khan MT. Calcium regulation of inositol 1,4,5-trisphosphate receptors. Cell Calcium. 2005;38:539–546. doi: 10.1016/j.ceca.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 212.Joseph SK, Lin C, Pierson S, Thomas AP, Maranto AR. Heteroligomers of type-I and type-III inositol trisphosphate receptors in WB rat liver epithelial cells. J Biol Chem. 1995;270:23310–23316. doi: 10.1074/jbc.270.40.23310. [DOI] [PubMed] [Google Scholar]
- 213.Joseph SK, Nakao SK, Sukumvanich S. Reactivity of free thiol groups in type-I inositol trisphosphate receptors. Biochem J. 2006;393:575–582. doi: 10.1042/BJ20050889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Joseph SK, Pierson S, Samanta S. Trypsin digestion of the inositol trisphosphate receptor: implications for the conformation and domain organization of the protein. Biochem J. 1995;307:859–865. doi: 10.1042/bj3070859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Joseph SK, Samanta S. Detergent solubility of the inositol trisphosphate receptor in rat brain membranes. Evidence for association of the receptor with ankyrin. J Biol Chem. 1993;268:6477–6486. [PubMed] [Google Scholar]
- 216.Joshi R, Venkatesh K, Srinivas R, Nair S, Hasan G. Genetic dissection of itpr gene function reveals a vital requirement in aminergic cells of Drosophila larvae. Genetics. 2004;166:225–236. doi: 10.1534/genetics.166.1.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Jurkat-Rott K, McCarthy T, Lehmann-Horn F. Genetics and pathogenesis of malignant hyperthermia. Muscle Nerve. 2000;23:4–17. doi: 10.1002/(sici)1097-4598(200001)23:1<4::aid-mus3>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 218.Jurkovicova D, Kubovcakova L, Hudecova S, Kvetnansky R, Krizanova O. Adrenergic modulation of the type 1 IP3 receptors in the rat heart. Biochim Biophys Acta. 2006;1763:18–24. doi: 10.1016/j.bbamcr.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 219.Kaftan EJ, Ehrlich BE, Watras J. Inositol 1,4,5-trisphosphate (InsP3) and calcium interact to increase the dynamic range of InsP3 receptor-dependent calcium signaling. J Gen Physiol. 1997;110:529–538. doi: 10.1085/jgp.110.5.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Kao JP. Practical aspects of measuring [Ca2+] with fluorescent indicators. Methods Cell Biol. 1994;40:155–181. doi: 10.1016/s0091-679x(08)61114-0. [DOI] [PubMed] [Google Scholar]
- 221.Kaplin AI, Ferris CD, Voglmaier SM, Snyder SH. Purified reconstituted inositol 1,4,5-trisphosphate receptors. Thiol reagents act directly on receptor protein. J Biol Chem. 1994;269:28972–28978. [PubMed] [Google Scholar]
- 222.Kaplin AI, Snyder SH, Linden DJ. Reduced nicotinamide adenine dinucleotide-selective stimulation of inositol 1,4,5-trisphosphate receptors mediates hypoxic mobilization of calcium. J Neurosci. 1996;16:2002–2011. doi: 10.1523/JNEUROSCI.16-06-02002.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Kashiwayanagi M, Tatani K, Shuto S, Matsuda A. Inositol 1,4,5-trisphosphate and adenophostin analogues induce responses in turtle olfactory sensory neurons. Eur J Neurosci. 2000;12:606–612. doi: 10.1046/j.1460-9568.2000.00948.x. [DOI] [PubMed] [Google Scholar]
- 224.Kasri NN, Bultynck G, Smyth J, Szlufcik K, Parys JB, Callewaert G, Missiaen L, Fissore RA, Mikoshiba K, de Smedt H. The N-terminal Ca2+-independent calmodulin-binding site on the inositol 1,4,5-trisphosphate receptor is responsible for calmodulin inhibition, even though this inhibition requires Ca2+ Mol Pharmacol. 2004;66:276–284. doi: 10.1124/mol.66.2.276. [DOI] [PubMed] [Google Scholar]
- 225.Kasri NN, Holmes AM, Bultynck G, Parys JB, Bootman MD, Rietdorf K, Missiaen L, McDonald F, De Smedt H, Conway SJ, Holmes AB, Berridge MJ, Roderick HL. Regulation of InsP3 receptor activity by neuronal Ca2+-binding proteins. EMBO J. 2004;23:312–321. doi: 10.1038/sj.emboj.7600037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Kasri NN, Kocks SL, Verbert L, Hebert SS, Callewaert G, Parys JB, Missiaen L, De Smedt H. Up-regulation of inositol 1,4,5-trisphosphate receptor type 1 is responsible for a decreased endoplasmic-reticulum Ca2+ content in presenilin double knockout cells. Cell Calcium. 2006;40:41–51. doi: 10.1016/j.ceca.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 227.Kasri NN, Torok K, Galione A, Garnham C, Callewaert G, Missiaen L, Parys JB, De Smedt H. Endogenously bound calmodulin is essential for the function of the inositol 1,4,5-trisphosphate receptor. J Biol Chem. 2006;281:8332–8338. doi: 10.1074/jbc.M510971200. [DOI] [PubMed] [Google Scholar]
- 228.Katayama E, Funahashi H, Michikawa T, Shiraishi T, Ikemoto T, Iino M, Mikoshiba K. Native structure and arrangement of inositol-1,4,5-trisphosphate receptor molecules in bovine cerebellar Purkinje cells as studied by quick-freeze deep-etch electron microscopy. EMBO J. 1996;15:4844–4851. [PMC free article] [PubMed] [Google Scholar]
- 229.Kaznacheyeva E, Lupu VD, Bezprozvanny I. Single-channel properties of inositol (1,4,5)-trisphosphate receptor heterologously expressed in HEK-293 cells. J Gen Physiol. 1998;111:847–856. doi: 10.1085/jgp.111.6.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Kessler DA, Levine H. Fluctuation-induced diffusive instabilities. Nature. 1998;394:556–558. [Google Scholar]
- 231.Kettlun C, Gonzalez A, Rios E, Fill M. Unitary Ca2+ current through mammalian cardiac and amphibian skeletal muscle ryanodine receptor channels under near-physiological ionic conditions. J Gen Physiol. 2003;122:407–417. doi: 10.1085/jgp.200308843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Khan AA, Soloski MJ, Sharp AH, Schilling G, Sabatini DM, Li SH, Ross CA, Snyder SH. Lymphocyte apoptosis: mediation by increased type 3 inositol 1,4,5-trisphosphate receptor. Science. 1996;273:503–507. doi: 10.1126/science.273.5274.503. [DOI] [PubMed] [Google Scholar]
- 233.Khan MT, Wagner L, 2nd, Yule DI, Bhanumathy C, Joseph SK. Akt kinase phosphorylation of inositol 1,4,5-trisphosphate receptors. J Biol Chem. 2006;281:3731–3737. doi: 10.1074/jbc.M509262200. [DOI] [PubMed] [Google Scholar]
- 234.Khodakhah K, Ogden D. Fast activation and inactivation of inositol trisphosphate-evoked Ca2+ release in rat cerebellar Purkinje neurones. J Physiol. 1995;487:343–358. doi: 10.1113/jphysiol.1995.sp020884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Khodakhah K, Ogden D. Functional heterogeneity of calcium release by inositol trisphosphate in single Purkinje neurones, cultured cerebellar astrocytes, peripheral tissues. Proc Natl Acad Sci USA. 1993;90:4976–4980. doi: 10.1073/pnas.90.11.4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Kiselyov K, Mignery GA, Zhu MX, Muallem S. The N-terminal domain of the IP3 receptor gates store-operated hTrp3 channels. Mol Cell. 1999;4:423–429. doi: 10.1016/s1097-2765(00)80344-5. [DOI] [PubMed] [Google Scholar]
- 237.Kiselyov K, Xu X, Mozhayeva G, Kuo T, Pessah I, Mignery G, Zhu X, Birnbaumer L, Muallem S. Functional interaction between InsP3 receptors and store-operated Htrp3 channels. Nature. 1998;396:478–482. doi: 10.1038/24890. [DOI] [PubMed] [Google Scholar]
- 238.Klingenberg M. The ADP-ATP translocation in mitochondria, a membrane potential controlled transport. J Membr Biol. 1980;56:97–105. doi: 10.1007/BF01875961. [DOI] [PubMed] [Google Scholar]
- 239.Komalavilas P, Lincoln TM. Phosphorylation of the inositol 1,4,5-trisphosphate receptor by cyclic GMP-dependent protein kinase. J Biol Chem. 1994;269:8701–8707. [PubMed] [Google Scholar]
- 240.Krajewski S, Tanaka S, Takayama S, Schibler MJ, Fenton W, Reed JC. Investigation of the subcellular-distribution of the Bcl-2 oncoprotein: residence in the nuclear envelope, endoplasmic reticulum, outer mitochondrial membranes. Cancer Res. 1993;53:4701–4714. [PubMed] [Google Scholar]
- 241.Kume S, Muto A, Aruga J, Nakagawa T, Michikawa T, Furuichi T, Nakade S, Okano H, Mikoshiba K. The Xenopus IP3 receptor: structure, function, localization in oocytes and eggs. Cell. 1993;73:555–570. doi: 10.1016/0092-8674(93)90142-d. [DOI] [PubMed] [Google Scholar]
- 242.Kume S, Muto A, Okano H, Mikoshiba K. Developmental expression of the inositol 1,4,5-trisphosphate receptor and localization of inositol 1,4,5-trisphosphate during early embryogenesis in Xenopus laevis. Mech Dev. 1997;66:157–168. doi: 10.1016/s0925-4773(97)00101-9. [DOI] [PubMed] [Google Scholar]
- 243.Kuo AL, Gulbis JM, Antcliff JF, Rahman T, Lowe ED, Zimmer J, Cuthbertson J, Ashcroft FM, Ezaki T, Doyle DA. Crystal structure of the potassium channel KirBac1.1 in the closed state. Science. 2003;300:1922–1926. doi: 10.1126/science.1085028. [DOI] [PubMed] [Google Scholar]
- 244.Labro AJ, Raes AL, Bellens I, Ottschytsch N, Snyders DJ. Gating of Shaker-type channels requires the flexibility of S6 caused by prolines. J Biol Chem. 2003;278:50724–50731. doi: 10.1074/jbc.M306097200. [DOI] [PubMed] [Google Scholar]
- 245.Landolfi B, Curci S, Debellis L, Pozzan T, Hofer AM. Ca2+ homeostasis in the agonist-sensitive internal store: functional interactions between mitochondria and the ER measured in situ in intact cells. J Cell Biol. 1998;142:1235–1243. doi: 10.1083/jcb.142.5.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Lawlor MA, Alessi DR. PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J Cell Sci. 2001;114:2903–2910. doi: 10.1242/jcs.114.16.2903. [DOI] [PubMed] [Google Scholar]
- 247.Lawrence JC, Lin TA, McMahon LP, Choi KM. Modulation of the protein kinase activity of mTOR. Curr Top Microbiol Immunol. 2004;279:199–213. doi: 10.1007/978-3-642-18930-2_12. [DOI] [PubMed] [Google Scholar]
- 248.Lechleiter J, Girard S, Peralta E, Clapham D. Spiral calcium wave propagation and annihilation in Xenopus laevis oocytes. Science. 1991;252:123–126. doi: 10.1126/science.2011747. [DOI] [PubMed] [Google Scholar]
- 249.Lehnart SE, Huang F, Marx SO, Marks AR. Immunophilins and coupled gating of ryanodine receptors. Curr Top Med Chem. 2003;3:1383–1391. doi: 10.2174/1568026033451907. [DOI] [PubMed] [Google Scholar]
- 250.Li C, Fox CJ, Master SR, Bindokas VP, Chodosh LA, Thompson CB. Bcl-XL affects Ca2+ homeostasis by altering expression of inositol 1,4,5-trisphosphate receptors. Proc Natl Acad Sci USA. 2002;99:9830–9835. doi: 10.1073/pnas.152571899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Li J, Zelenin S, Aperia A, Aizman O. Low doses of ouabain protect from serum deprivation-triggered apoptosis and stimulate kidney cell proliferation via activation of NF-κB. J Am Soc Nephrol. 2006;17:1848–1857. doi: 10.1681/ASN.2005080894. [DOI] [PubMed] [Google Scholar]
- 252.Li X, Malathi K, Krizanova O, Ondrias K, Sperber K, Ablamunits V, Jayaraman T. Cdc2/cyclin B1 interacts with and modulates inositol 1,4,5-trisphosphate receptor (type 1) functions. J Immunol. 2005;175:6205–6210. doi: 10.4049/jimmunol.175.9.6205. [DOI] [PubMed] [Google Scholar]
- 253.Li X, Zima AV, Sheikh F, Blatter LA, Chen J. Endothelin-1-induced arrhythmogenic Ca2+ signaling is abolished in atrial myocytes of inositol-1,4,5-trisphosphate(IP3)-receptor type 2-deficient mice. Circ Res. 2005;96:1274–1281. doi: 10.1161/01.RES.0000172556.05576.4c. [DOI] [PubMed] [Google Scholar]
- 254.Lin C, Widjaja J, Joseph SK. The interaction of calmodulin with alternatively spliced isoforms of the type-I inositol trisphosphate receptor. J Biol Chem. 2000;275:2305–2311. doi: 10.1074/jbc.275.4.2305. [DOI] [PubMed] [Google Scholar]
- 255.Lindsay AR, Manning SD, Williams AJ. Monovalent cation conductance in the ryanodine receptor-channel of sheep cardiac muscle sarcoplasmic reticulum. J Physiol. 1991;439:463–480. doi: 10.1113/jphysiol.1991.sp018676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Lipp P, Laine M, Tovey SC, Burrell KM, Berridge MJ, Li W, Bootman MD. Functional InsP3 receptors that may modulate excitation-contraction coupling in the heart. Curr Biol. 2000;10:939–942. doi: 10.1016/s0960-9822(00)00624-2. [DOI] [PubMed] [Google Scholar]
- 257.Liu J, Siegelbaum SA. Change of pore helix conformational state upon opening of cyclic nucleotide-gated channels. Neuron. 2000;28:899–909. doi: 10.1016/s0896-6273(00)00162-8. [DOI] [PubMed] [Google Scholar]
- 258.Lockwich TP, Liu X, Singh BB, Jadlowiec J, Weiland S, Ambudkar IS. Assembly of Trp1 in a signaling complex associated with caveolin-scaffolding lipid raft domains. J Biol Chem. 2000;275:11934–11942. doi: 10.1074/jbc.275.16.11934. [DOI] [PubMed] [Google Scholar]
- 259.Long SB, Campbell EB, Mackinnon R. Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science. 2005;309:903–908. doi: 10.1126/science.1116270. [DOI] [PubMed] [Google Scholar]
- 260.Lu T, Ting AY, Mainland J, Jan LY, Schultz PG, Yang J. Probing ion permeation and gating in a K+ channel with backbone mutations in the selectivity filter. Nature Neurosci. 2001;4:239–246. doi: 10.1038/85080. [DOI] [PubMed] [Google Scholar]
- 261.Ludtke SJ, Serysheva II, Hamilton SL, Chiu W. The pore structure of the closed RyR1 channel. Structure. 2005;13:1203–1211. doi: 10.1016/j.str.2005.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Lupu VD, Kaznacheyeva E, Krishna UM, Falck JR, Bezprozvanny I. Functional coupling of phosphatidylinositol 4,5-bisphosphate to inositol 1,4,5-trisphosphate receptor. J Biol Chem. 1998;273:14067–14070. doi: 10.1074/jbc.273.23.14067. [DOI] [PubMed] [Google Scholar]
- 263.Lynch PJ, Tong JF, Lehane M, Mallet A, Giblin L, Heffron JA, Vaughan P, Zafra G, MacLennan DH, McCarthy TV. A mutation in the transmembrane/luminal domain of the ryanodine receptor is associated with abnormal Ca2+ release channel function and severe central core disease. Proc Natl Acad Sci USA. 1999;96:4164–4169. doi: 10.1073/pnas.96.7.4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Ma HT, Venkatachalam K, Li HS, Montell C, Kurosaki T, Patterson RL, Gill DL. Assessment of the role of the inositol 1,4,5-trisphosphate receptor in the activation of transient receptor potential channels and store-operated Ca2+ entry channels. J Biol Chem. 2001;276:18888–18896. doi: 10.1074/jbc.M100944200. [DOI] [PubMed] [Google Scholar]
- 265.Machaca K, Hartzell HC. Adenophostin A and inositol 1,4,5-trisphosphate differentially activate Cl− currents in Xenopus oocytes because of disparate Ca2+ release kinetics. J Biol Chem. 1999;274:4824–4831. doi: 10.1074/jbc.274.8.4824. [DOI] [PubMed] [Google Scholar]
- 266.MacKinnon R. Potassium channels. FEBS Lett. 2003;555:62–65. doi: 10.1016/s0014-5793(03)01104-9. [DOI] [PubMed] [Google Scholar]
- 267.MacKrill JJ. Protein-protein interactions in intracellular Ca2+-release channel function. Biochem J. 1999;337:345–361. [PMC free article] [PubMed] [Google Scholar]
- 268.MacMillan D, Currie S, Bradley KN, Muir TC, McCarron JG. In smooth muscle, FK506-binding protein modulates IP3 receptor-evoked Ca2+ release by mTOR and calcineurin. J Cell Sci. 2005;118:5443–5451. doi: 10.1242/jcs.02657. [DOI] [PubMed] [Google Scholar]
- 269.Madden DR. The structure and function of glutamate receptor ion channels. Nat Rev Neurosci. 2002;3:91–101. doi: 10.1038/nrn725. [DOI] [PubMed] [Google Scholar]
- 270.Maeda N, Kawasaki T, Nakade S, Yokota N, Taguchi T, Kasai M, Mikoshiba K. Structural and functional characterization of inositol 1,4,5-trisphosphate receptor channel from mouse cerebellum. J Biol Chem. 1991;266:1109–1116. [PubMed] [Google Scholar]
- 271.Maeda N, Niinobe M, Mikoshiba K. A cerebellar Purkinje cell marker P400 protein is an inositol 1,4,5-trisphosphate (InsP3) receptor protein. Purification and characterization of InsP3 receptor complex. EMBO J. 1990;9:61–67. doi: 10.1002/j.1460-2075.1990.tb08080.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Maes K, Missiaen L, De Smet P, Vanlingen S, Callewaert G, Parys JB, De Smedt H. Differential modulation of inositol 1,4,5-trisphosphate receptor type 1 and type 3 by ATP. Cell Calcium. 2000;27:257–267. doi: 10.1054/ceca.2000.0121. [DOI] [PubMed] [Google Scholar]
- 273.Maes K, Missiaen L, Parys JB, De Smet P, Sienaert I, Waelkens E, Callewaert G, De Smedt H. Mapping of the ATP-binding sites on inositol 1,4,5-trisphosphate receptor type 1 and type 3 homotetramers by controlled proteolysis and photoaffinity labeling. J Biol Chem. 2001;276:3492–3497. doi: 10.1074/jbc.M006082200. [DOI] [PubMed] [Google Scholar]
- 274.Maes K, Missiaen L, Parys JB, Sienaert I, Bultynck G, Zizi M, De Smet P, Casteels R, De Smedt H. Adenine-nucleotide binding sites on the inositol 1,4,5-trisphosphate receptor bind caffeine, but not adenophostin A or cyclic ADP-ribose. Cell Calcium. 1999;25:143–152. doi: 10.1054/ceca.1998.0011. [DOI] [PubMed] [Google Scholar]
- 275.Magnusson A, Haug LS, Walaas SI, Ostvold AC. Calcium-induced degradation of the inositol (1,4,5)-trisphosphate receptor/Ca2+-channel. FEBS Lett. 1993;323:229–232. doi: 10.1016/0014-5793(93)81345-z. [DOI] [PubMed] [Google Scholar]
- 276.Mak DOD, Foskett JK. Effects of divalent cations on single-channel conduction properties of Xenopus IP3 receptor. Am J Physiol Cell Physiol. 1998;275:C179–C188. doi: 10.1152/ajpcell.1998.275.1.C179. [DOI] [PubMed] [Google Scholar]
- 277.Mak DOD, Foskett JK. Single-channel inositol 1,4,5-trisphosphate receptor currents revealed by patch clamp of isolated Xenopus oocyte nuclei. J Biol Chem. 1994;269:29375–29378. [PubMed] [Google Scholar]
- 278.Mak DOD, Foskett JK. Single-channel kinetics, inactivation, spatial distribution of inositol trisphosphate (IP3) receptors in Xenopus oocyte nucleus. J Gen Physiol. 1997;109:571–587. doi: 10.1085/jgp.109.5.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Mak DOD, McBride S, Foskett JK. ATP-dependent adenophostin activation of inositol 1,4,5-trisphosphate receptor channel gating: kinetic implications for the durations of calcium puffs in cells. J Gen Physiol. 2001;117:299–314. doi: 10.1085/jgp.117.4.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Mak DOD, McBride S, Foskett JK. ATP regulation of recombinant type 3 inositol 1,4,5-trisphosphate receptor gating. J Gen Physiol. 2001;117:447–456. doi: 10.1085/jgp.117.5.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Mak DOD, McBride S, Foskett JK. ATP regulation of type 1 inositol 1,4,5-trisphosphate receptor channel gating by allosteric tuning of Ca2+ activation. J Biol Chem. 1999;274:22231–22237. doi: 10.1074/jbc.274.32.22231. [DOI] [PubMed] [Google Scholar]
- 282.Mak DOD, McBride S, Foskett JK. Inositol 1,4,5-trisphosphate activation of inositol trisphosphate receptor Ca2+ channel by ligand tuning of Ca2+ inhibition. Proc Natl Acad Sci USA. 1998;95:15821–15825. doi: 10.1073/pnas.95.26.15821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Mak DOD, McBride S, Foskett JK. Regulation by Ca2+ and inositol 1,4,5-trisphosphate (InsP3) of single recombinant type 3 InsP3 receptor channels. Ca2+ activation uniquely distinguishes types 1 and 3 InsP3 receptors. J Gen Physiol. 2001;117:435–446. doi: 10.1085/jgp.117.5.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Mak DOD, McBride S, Raghuram V, Yue Y, Joseph SK, Foskett JK. Single-channel properties in endoplasmic reticulum membrane of recombinant type 3 inositol trisphosphate receptor. J Gen Physiol. 2000;115:241–256. doi: 10.1085/jgp.115.3.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Mak DOD, McBride SM, Foskett JK. Spontaneous channel activity of the inositol 1,4,5-trisphosphate (InsP3) receptor (InsP3R). Application of allosteric modeling to calcium and InsP3 regulation of InsP3R single-channel gating. J Gen Physiol. 2003;122:583–603. doi: 10.1085/jgp.200308809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Mak DOD, McBride SM, Petrenko NB, Foskett JK. Novel regulation of calcium inhibition of the inositol 1,4,5-trisphosphate receptor calcium-release channel. J Gen Physiol. 2003;122:569–581. doi: 10.1085/jgp.200308808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Mak DOD, White C, Ionescu L, Foskett JK. Nuclear patch clamp electrophysiology of inositol trisphosphate receptor Ca2+ release channels. In: Putney JW Jr, editor. Methods in Calcium Signaling Research. Boca Raton, FL: CRC; 2005. pp. 203–229. [Google Scholar]
- 288.Malathi K, Kohyama S, Ho M, Soghoian D, Li X, Silane M, Berenstein A, Jayaraman T. Inositol 1,4,5-trisphosphate receptor (type 1) phosphorylation and modulation by Cdc2. J Cell Biochem. 2003;90:1186–1196. doi: 10.1002/jcb.10720. [DOI] [PubMed] [Google Scholar]
- 289.Maranto AR. Primary structure, ligand binding, localization of the human type 3 inositol 1,4,5-trisphosphate receptor expressed in intestinal epithelium. J Biol Chem. 1994;269:1222–1230. [PubMed] [Google Scholar]
- 290.Marchant JS, Beecroft MD, Riley AM, Jenkins DJ, Marwood RD, Taylor CW, Potter BV. Disaccharide polyphosphates based upon adenophostin A activate hepatic D-myo-inositol 1,4,5-trisphosphate receptors. Biochemistry. 1997;36:12780–12790. doi: 10.1021/bi971397v. [DOI] [PubMed] [Google Scholar]
- 291.Marchant JS, Parker I. Functional interactions in Ca2+ signaling over different time and distance scales. J Gen Physiol. 2000;116:691–696. doi: 10.1085/jgp.116.5.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Marchant JS, Parker I. Kinetics of elementary Ca2+ puffs evoked in Xenopus oocytes by different Ins(1,4,5)P3 receptor agonists. Biochem J. 1998;334:505–509. doi: 10.1042/bj3340505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Marchant JS, Taylor CW. Rapid activation and partial inactivation of inositol trisphosphate receptors by inositol trisphosphate. Biochemistry. 1998;37:11524–11533. doi: 10.1021/bi980808k. [DOI] [PubMed] [Google Scholar]
- 294.Marchenko SM, Yarotskyy VV, Kovalenko TN, Kostyuk PG, Thomas RC. Spontaneously active and InsP3-activated ion channels in cell nuclei from rat cerebellar Purkinje and granule neurones. J Physiol. 2005;565:897–910. doi: 10.1113/jphysiol.2004.081299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Marx SO, Ondrias K, Marks AR. Coupled gating between individual skeletal muscle Ca2+ release channels (ryanodine receptors) Science. 1998;281:818–821. doi: 10.1126/science.281.5378.818. [DOI] [PubMed] [Google Scholar]
- 296.Matifat F, Hague F, Brule G, Collin T. Regulation of InsP3-mediated Ca2+ release by CaMKII in Xenopus oocytes. Pflügers Arch. 2001;441:796–801. doi: 10.1007/s004240000479. [DOI] [PubMed] [Google Scholar]
- 297.Matsumoto M, Nakagawa T, Inoue T, Nagata E, Tanaka K, Takano H, Minowa O, Kuno J, Sakakibara S, Yamada M, Yoneshima H, Miyawaki A, Fukuuchi Y, Furuichi T, Okano H, Mikoshiba K, Noda T. Ataxia and epileptic seizures in mice lacking type 1 inositol 1,4,5-trisphosphate receptor. Nature. 1996;379:168–171. doi: 10.1038/379168a0. [DOI] [PubMed] [Google Scholar]
- 298.Mattagajasingh SN, Huang SC, Hartenstein JS, Snyder M, Marchesi VT, Benz EJ. A nonerythroid isoform of protein 4.1R interacts with the nuclear mitotic apparatus (NuMA) protein. J Cell Biol. 1999;145:29–43. doi: 10.1083/jcb.145.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Matter N, Ritz MF, Freyermuth S, Rogue P, Malviya AN. Stimulation of nuclear protein kinase C leads to phosphorylation of nuclear inositol 1,4,5-trisphosphate receptor and accelerated calcium release by inositol 1,4,5-trisphosphate from isolated rat liver nuclei. J Biol Chem. 1993;268:732–736. [PubMed] [Google Scholar]
- 300.Mauger JP, Lièvremont JP, Piétri-Rouxel F, Hilly M, Coquil JF. The inositol 1,4,5-trisphosphate receptor: kinetic properties and regulation. Mol Cell Endocrinol. 1994;98:133–139. doi: 10.1016/0303-7207(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 301.Maximov A, Tang TS, Bezprozvanny I. Association of the type 1 inositol (1,4,5)-trisphosphate receptor with 4.1N protein in neurons. Mol Cell Neurosci. 2003;22:271–283. doi: 10.1016/s1044-7431(02)00027-1. [DOI] [PubMed] [Google Scholar]
- 302.Mayrleitner M, Chadwick CC, Timerman AP, Fleischer S, Schindler H. Purified IP3 receptor from smooth muscle forms an IP3 gated and heparin sensitive Ca2+ channel in planar bilayers. Cell Calcium. 1991;12:505–514. doi: 10.1016/0143-4160(91)90032-a. [DOI] [PubMed] [Google Scholar]
- 303.McCarron JG, MacMillan D, Bradley KN, Chalmers S, Muir TC. Origin and mechanisms of Ca2+ waves in smooth muscle as revealed by localized photolysis of caged inositol 1,4,5-trisphosphate. J Biol Chem. 2004;279:8417–8427. doi: 10.1074/jbc.M311797200. [DOI] [PubMed] [Google Scholar]
- 304.McCarthy TV, Quane KA, Lynch PJ. Ryanodine receptor mutations in malignant hyperthermia and central core disease. Hum Mutat. 2000;15:410–417. doi: 10.1002/(SICI)1098-1004(200005)15:5<410::AID-HUMU2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 305.McGowan TA, Sharma K. Regulation of inositol 1,4,5-trisphosphate receptors by transforming growth factor-β: implications for vascular dysfunction in diabetes. Kidney Int Suppl. 2000;77:S99–S103. doi: 10.1046/j.1523-1755.2000.07716.x. [DOI] [PubMed] [Google Scholar]
- 306.Mery L, Magnino F, Schmidt K, Krause KH, Dufour JF. Alternative splice variants of hTrp4 differentially interact with the C-terminal portion of the inositol 1,4,5-trisphosphate receptors. FEBS Lett. 2001;487:377–383. doi: 10.1016/s0014-5793(00)02362-0. [DOI] [PubMed] [Google Scholar]
- 307.Meyer T, Holowka D, Stryer L. Highly cooperative opening of calcium channels by inositol 1,4,5-trisphosphate. Science. 1988;240:653–656. doi: 10.1126/science.2452482. [DOI] [PubMed] [Google Scholar]
- 308.Meyer T, Stryer L. Transient calcium release induced by successive increments of inositol 1,4,5-trisphosphate. Proc Natl Acad Sci USA. 1990;87:3841–3845. doi: 10.1073/pnas.87.10.3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Michikawa T, Hamanaka H, Otsu H, Yamamoto A, Miyawaki A, Furuichi T, Tashiro Y, Mikoshiba K. Transmembrane topology and sites of N-glycosylation of inositol 1,4,5 trisphosphate receptor. J Biol Chem. 1994;269:9184–9189. [PubMed] [Google Scholar]
- 310.Michikawa T, Hirota J, Kawano S, Hiraoka M, Yamada M, Furuichi T, Mikoshiba K. Calmodulin mediates calcium-dependent inactivation of the cerebellar type 1 inositol 1,4,5-trisphosphate receptor. Neuron. 1999;23:799–808. doi: 10.1016/s0896-6273(01)80037-4. [DOI] [PubMed] [Google Scholar]
- 311.Mignery GA, Newton CL, Archer BT, 3rd, Sudhof TC. Structure and expression of the rat inositol 1,4,5-trisphosphate receptor. J Biol Chem. 1990;265:12679–12685. [PubMed] [Google Scholar]
- 312.Mignery GA, Sudhof TC. The ligand binding site and transduction mechanism in the inositol 1,4,5-triphosphate receptor. EMBO J. 1990;9:3893–3898. doi: 10.1002/j.1460-2075.1990.tb07609.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Mignery GA, Sudhof TC, Takei K, De Camilli P. Putative receptor for inositol 1,4,5-trisphosphate similar to ryanodine receptor. Nature. 1989;342:192–195. doi: 10.1038/342192a0. [DOI] [PubMed] [Google Scholar]
- 314.Missiaen L, De Smedt H, Parys JB, Sienaert I, Valingen S, Casteels R. Threshold for inositol 1,4,5-trisphosphate action. J Biol Chem. 1996;271:12287–12293. doi: 10.1074/jbc.271.21.12287. [DOI] [PubMed] [Google Scholar]
- 315.Missiaen L, Parys JB, De Smedt H, Oike M, Casteels R. Partial calcium release in response to submaximal inositol 1,4,5-trisphosphate receptor activation. Mol Cell Endocrinol. 1994;98:147–156. doi: 10.1016/0303-7207(94)90133-3. [DOI] [PubMed] [Google Scholar]
- 316.Missiaen L, Parys JB, Sienaert I, Maes K, Kunzelmann K, Takahashi M, Tanzawa K, De Smedt H. Functional properties of the type-3 InsP3 receptor in 16HBE14o- bronchial mucosal cells. J Biol Chem. 1998;273:8983–8986. doi: 10.1074/jbc.273.15.8983. [DOI] [PubMed] [Google Scholar]
- 317.Missiaen L, Parys JB, Smedt HD, Sienaert I, Sipma H, Vanlingen S, Maes K, Casteels R. Effect of adenine nucleotides on myo-inositol-1,4,5-trisphosphate-induced calcium release. Biochem J. 1997;325:661–666. doi: 10.1042/bj3250661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Miyakawa T, Maeda A, Yamazawa T, Hirose K, Kurosaki T, Iino M. Encoding of Ca2+ signals by differential expression of IP3 receptor subtypes. EMBO J. 1999;18:1303–1308. doi: 10.1093/emboj/18.5.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Miyakawa T, Mizushima A, Hirose K, Yamazawa T, Bezprozvanny I, Kurosaki T, Iino M. Ca2+-sensor region of IP3 receptor controls intracellular Ca2+ signaling. EMBO J. 2001;20:1674–1680. doi: 10.1093/emboj/20.7.1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Miyakawa-Naito A, Uhlen P, Lal M, Aizman O, Mikoshiba K, Brismar H, Zelenin S, Aperia A. Cell signaling microdomain with Na,K-ATPase and inositol 1,4,5-trisphosphate receptor generates calcium oscillations. J Biol Chem. 2003;278:50355–50361. doi: 10.1074/jbc.M305378200. [DOI] [PubMed] [Google Scholar]
- 321.Miyawaki A, Furuichi T, Maeda N, Mikoshiba K. Expressed cerebellar-type inositol 1,4,5-trisphosphate receptor, P400, has calcium release activity in a fibroblast L cell line. Neuron. 1990;5:11–18. doi: 10.1016/0896-6273(90)90029-f. [DOI] [PubMed] [Google Scholar]
- 322.Miyawaki A, Furuichi T, Ryou Y, Yoshikawa S, Nakagawa T, Saitoh T, Mikoshiba K. Structure-function relationships of the mouse inositol 1,4,5-trisphosphate receptor. Proc Natl Acad Sci USA. 1991;88:4911–4915. doi: 10.1073/pnas.88.11.4911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Miyazaki S, Shirakawa H, Nakada K, Honda Y, Yuzaki M, Nakade S, Mikoshiba K. Antibody to the inositol trisphosphate receptor blocks thimerosal-enhanced Ca2+-induced Ca2+ release and Ca2+ oscillations in hamster eggs. FEBS Lett. 1992;309:180–184. doi: 10.1016/0014-5793(92)81090-9. [DOI] [PubMed] [Google Scholar]
- 324.Miyazawa A, Fujiyoshi Y, Unwin N. Structure and gating mechanism of the acetylcholine receptor pore. Nature. 2003;423:949–955. doi: 10.1038/nature01748. [DOI] [PubMed] [Google Scholar]
- 325.Mohler PJ, Davis JQ, Davis LH, Hoffman JA, Michaely P, Bennett V. Inositol 1,4,5-trisphosphate receptor localization and stability in neonatal cardiomyocytes requires interaction with ankyrin-B. J Biol Chem. 2004;279:12980–12987. doi: 10.1074/jbc.M313979200. [DOI] [PubMed] [Google Scholar]
- 326.Mohler PJ, Gramolini AO, Bennett V. The ankyrin-B C-terminal domain determines activity of ankyrin-B/G chimeras in rescue of abnormal inositol 1,4,5-trisphosphate and ryanodine receptor distribution in ankyrin-B (−/−) neonatal cardiomyocytes. J Biol Chem. 2002;277:10599–10607. doi: 10.1074/jbc.M110958200. [DOI] [PubMed] [Google Scholar]
- 327.Mohler PJ, Schott JJ, Gramolini AO, Dilly KW, Guatimosim S, duBell WH, Song LS, Haurogne K, Kyndt F, Ali ME, Rogers TB, Lederer WJ, Escande D, Le Marec H, Bennett V. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003;421:634–639. doi: 10.1038/nature01335. [DOI] [PubMed] [Google Scholar]
- 328.Monkawa T, Miyawaki A, Sugiyama T, Yoneshima H, Yamamoto-Hino M, Furuichi T, Saruta T, Hasegawa M, Mikoshiba K. Heterotetrameric complex formation of inositol 1,4,5-trisphosphate receptor subunits. J Biol Chem. 1995;270:14700–14704. doi: 10.1074/jbc.270.24.14700. [DOI] [PubMed] [Google Scholar]
- 329.Monod J, Wyman J, Changeux JP. On the nature of allosteric transitions: a plausible model. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 330.Montero M, Brini M, Marsault R, Alvarez J, Sitia R, Pozzan T, Rizzuto R. Monitoring dynamic changes in free Ca2+ concentration in the endoplasmic reticulum of intact cells. EMBO J. 1995;14:5467–5475. doi: 10.1002/j.1460-2075.1995.tb00233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Moraru II, Kaftan EJ, Ehrlich BE, Watras J. Regulation of type 1 inositol 1,4,5-trisphosphate-gated calcium channels by InsP3 and calcium: simulation of single channel kinetics based on ligand binding and electrophysiological analysis. J Gen Physiol. 1999;113:837–849. doi: 10.1085/jgp.113.6.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Morris SA, Nerou EP, Riley AM, Potter BV, Taylor CW. Determinants of adenophostin A binding to inositol trisphosphate receptors. Biochem J. 2002;367:113–120. doi: 10.1042/BJ20020675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Muallem S, Pandol SJ, Beeker TG. Hormone-evoked calcium release from intracellular stores is a quantal process. J Biol Chem. 1989;264:205–212. [PubMed] [Google Scholar]
- 334.Murphy CT, Riley AM, Lindley CJ, Jenkins DJ, Westwick J, Potter BV. Structural analogues of D-myo-inositol-1,4,5-trisphosphate and adenophostin A: recognition by cerebellar and platelet inositol-1,4,5-trisphosphate receptors. Mol Pharmacol. 1997;52:741–748. doi: 10.1124/mol.52.4.741. [DOI] [PubMed] [Google Scholar]
- 335.Murthy KS, Zhou H. Selective phosphorylation of the IP3R-I in vivo by cGMP-dependent protein kinase in smooth muscle. Am J Physiol Gastrointest Liver Physiol. 2003;284:G221–G230. doi: 10.1152/ajpgi.00401.2002. [DOI] [PubMed] [Google Scholar]
- 336.Nadif Kasri N, Bultynck G, Sienaert I, Callewaert G, Erneux C, Missiaen L, Parys JB, De Smedt H. The role of calmodulin for inositol 1,4,5-trisphosphate receptor function. Biochim Biophys Acta. 2002;1600:19–31. doi: 10.1016/s1570-9639(02)00440-5. [DOI] [PubMed] [Google Scholar]
- 337.Nakade S, Maeda N, Mikoshiba K. Involvement of the C-terminus of the inositol 1,4,5-trisphosphate receptor in Ca2+ release analysed using region-specific monoclonal antibodies. Biochem J. 1991;277:125–131. doi: 10.1042/bj2770125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Nakade S, Rhee SK, Hamanaka H, Mikoshiba K. Cyclic AMP-dependent phosphorylation of an immunoaffinity-purified homotetrameric inositol 1,4,5-trisphosphate receptor (type I) increases Ca2+ flux in reconstituted lipid vesicles. J Biol Chem. 1994;269:6735–6742. [PubMed] [Google Scholar]
- 339.Nakagawa T, Okano H, Furuichi T, Aruga J, Mikoshiba K. The subtypes of the mouse inositol 1,4,5-trisphosphate receptor are expressed in a tissue-specific and developmentally specific manner. Proc Natl Acad Sci USA. 1991;88:6244–6248. doi: 10.1073/pnas.88.14.6244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Nakagawa T, Shiota C, Okano H, Mikoshiba K. Differential localization of alternative spliced transcripts encoding inositol 1,4,5-trisphosphate receptors in mouse cerebellum and hippocampus: in situ hybridization study. J Neurochem. 1991;57:1807–1810. doi: 10.1111/j.1471-4159.1991.tb06385.x. [DOI] [PubMed] [Google Scholar]
- 341.Nakayama T, Hattori M, Uchida K, Nakamura T, Tateishi Y, Bannai H, Iwai M, Michikawa T, Inoue T, Mikoshiba K. The regulatory domain of the inositol 1,4,5-trisphosphate receptor is necessary to keep the channel domain closed: possible physiological significance of specific cleavage by caspase 3. Biochem J. 2004;377:299–307. doi: 10.1042/BJ20030599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Naraghi M, Neher E. Linearized buffered Ca2+ diffusion in microdomains and its implications for calculation of [Ca2+] at the mouth of a calcium channel. J Neurosci. 1997;17:6961–6973. doi: 10.1523/JNEUROSCI.17-18-06961.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Neher E. Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron. 1998;20:389–399. doi: 10.1016/s0896-6273(00)80983-6. [DOI] [PubMed] [Google Scholar]
- 344.Nerou EP, Riley AM, Potter BV, Taylor CW. Selective recognition of inositol phosphates by subtypes of the inositol trisphosphate receptor. Biochem J. 2001;355:59–69. doi: 10.1042/0264-6021:3550059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Newton CL, Mignery GA, Sudhof TC. Co-expression in vertebrate tissues and cell lines of multiple inositol 1,4,5-trisphosphate (InsP3) receptors with distinct affinities for InsP3. J Biol Chem. 1994;269:28613–28619. [PubMed] [Google Scholar]
- 346.Nimigean CM, Chappie JS, Miller C. Electrostatic tuning of ion conductance in potassium channels. Biochemistry. 2003;42:9263–9268. doi: 10.1021/bi0348720. [DOI] [PubMed] [Google Scholar]
- 347.Nosyreva E, Miyakawa T, Wang Z, Glouchankova L, Mizushima A, Iino M, Bezprozvanny I. The high-affinity calcium-calmodulin-binding site does not play a role in the modulation of type 1 inositol 1,4,5-trisphosphate receptor function by calcium and calmodulin. Biochem J. 2002;365:659–667. doi: 10.1042/BJ20011789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Nucifora FC, Jr, Li SH, Danoff S, Ullrich A, Ross CA. Molecular cloning of a cDNA for the human inositol 1,4,5-trisphosphate receptor type 1, the identification of a third alternatively spliced variant. Brain Res. 1995;32:291–296. doi: 10.1016/0169-328x(95)00089-b. [DOI] [PubMed] [Google Scholar]
- 349.Oakes SA, Scorrano L, Opferman JT, Bassik MC, Nishino M, Pozzan T, Korsmeyer SJ. Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum. Proc Natl Acad Sci USA. 2005;102:105–110. doi: 10.1073/pnas.0408352102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Ogden D, Capiod T. Regulation of Ca2+ release by InsP3 in single guinea pig hepatocytes and rat Purkinje neurons. J Gen Physiol. 1997;109:741–756. doi: 10.1085/jgp.109.6.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Oldershaw KA, Richardson A, Taylor CW. Prolonged exposure to inositol 1,4,5-trisphosphate does not cause intrinsic desensitization of the intracellular Ca2+-mobilizing receptor. J Biol Chem. 1992;267:16312–16316. [PubMed] [Google Scholar]
- 352.Onoue H, Tanaka H, Tanaka K, Doira N, Ito Y. Heterooligomer of type 1 and type 2 inositol 1,4,5-trisphosphate receptor expressed in rat liver membrane fraction exists as tetrameric complex. Biochem Biophys Res Commun. 2000;267:928–933. doi: 10.1006/bbrc.1999.2065. [DOI] [PubMed] [Google Scholar]
- 353.Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- 354.Otsu H, Yamamoto A, Maeda N, Mikoshiba K, Tashiro Y. Immunogold localization of inositol 1, 4, 5-trisphosphate (InsP3) receptor in mouse cerebellar Purkinje cells using three monoclonal antibodies. Cell Struct Funct. 1990;15:163–173. doi: 10.1247/csf.15.163. [DOI] [PubMed] [Google Scholar]
- 355.Palmer AE, Jin C, Reed JC, Tsien RY. Bcl-2-mediated alterations in endoplasmic reticulum Ca2+ analyzed with an improved genetically encoded fluorescent sensor. Proc Natl Acad Sci USA. 2004;101:17404–17409. doi: 10.1073/pnas.0408030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Parekh AB, Putney JW., Jr Store-operated calcium channels. Physiol Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- 357.Parker AK, Gergely FV, Taylor CW. Targeting of inositol 1,4,5-trisphosphate receptors to the endoplasmic reticulum by multiple signals within their transmembrane domains. J Biol Chem. 2004;279:23797–23805. doi: 10.1074/jbc.M402098200. [DOI] [PubMed] [Google Scholar]
- 358.Parker I, Choi J, Yao Y. Elementary events of InsP3-induced Ca2+ liberation in Xenopus oocytes: hot spots, puffs and blips. Cell Calcium. 1996;20:105–121. doi: 10.1016/s0143-4160(96)90100-1. [DOI] [PubMed] [Google Scholar]
- 359.Parker I, Ivorra I. Inhibition by Ca2+ of inositol trisphosphate-mediated Ca2+ liberation: a possible mechanism for oscillatory release of Ca2+ Proc Natl Acad Sci USA. 1990;87:260–264. doi: 10.1073/pnas.87.1.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Parys JB, Missiaen L, De Smedt H, Droogmans G, Casteels R. Bell-shaped activation of inositol-1,4,5-trisphosphate-induced Ca2+ release by thimerosal in permeabilized A7r5 smooth-muscle cells. Pflügers Arch. 1993;424:516–522. doi: 10.1007/BF00374916. [DOI] [PubMed] [Google Scholar]
- 361.Parys JB, Missiaen L, De Smedt H, Sienaert I, Henning RH, Casteels R. Quantal release of calcium in permeabilized A7r5 cells is not caused by intrinsic inactivation of the inositol trisphosphate receptor. Biochem Biophys Res Commun. 1995;209:451–456. doi: 10.1006/bbrc.1995.1523. [DOI] [PubMed] [Google Scholar]
- 362.Parys JB, Missiaen L, Smedt HD, Sienaert I, Casteels R. Mechanisms responsible for quantal Ca2+ release from inositol trisphosphate-sensitive calcium stores. Pflügers Arch. 1996;432:359–367. doi: 10.1007/s004240050145. [DOI] [PubMed] [Google Scholar]
- 363.Patel S, Joseph SK, Thomas AP. Molecular properties of inositol 1,4,5-trisphosphate receptors. Cell Calcium. 1999;25:247–264. doi: 10.1054/ceca.1999.0021. [DOI] [PubMed] [Google Scholar]
- 364.Patel S, Morris SA, Adkins CE, O’Beirne G, Taylor CW. Ca2+-independent inhibition of inositol trisphosphate receptors by calmodulin: redistribution of calmodulin as a possible means of regulating Ca2+ mobilization. Proc Natl Acad Sci USA. 1997;94:11627–11632. doi: 10.1073/pnas.94.21.11627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Patterson RL, Boehning D, Snyder SH. Inositol 1,4,5-trisphosphate receptors as signal integrators. Annu Rev Biochem. 2004;73:437–465. doi: 10.1146/annurev.biochem.73.071403.161303. [DOI] [PubMed] [Google Scholar]
- 366.Patterson RL, van Rossum DB, Barrow RK, Snyder SH. RACK1 binds to inositol 1,4,5-trisphosphate receptors and mediates Ca2+ release. Proc Natl Acad Sci USA. 2004;101:2328–2332. doi: 10.1073/pnas.0308567100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Patterson RL, van Rossum DB, Kaplin AI, Barrow RK, Snyder SH. Inositol 1,4,5-trisphosphate receptor/GAPDH complex augments Ca2+ release via locally derived NADH. Proc Natl Acad Sci USA. 2005;102:1357–1359. doi: 10.1073/pnas.0409657102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Perez PJ, Ramos-Franco J, Fill M, Mignery GA. Identification and functional reconstitution of the type 2 inositol 1,4,5-trisphosphate receptor from ventricular cardiac myocytes. J Biol Chem. 1997;272:23961–23969. doi: 10.1074/jbc.272.38.23961. [DOI] [PubMed] [Google Scholar]
- 369.Perozo E. New structural perspectives on K+ channel gating. Structure. 2002;10:1027–1029. doi: 10.1016/s0969-2126(02)00812-2. [DOI] [PubMed] [Google Scholar]
- 370.Perozo E, Cortes DM, Cuello LG. Structural rearrangements underlying K+-channel activation gating. Science. 1999;285:73–78. doi: 10.1126/science.285.5424.73. [DOI] [PubMed] [Google Scholar]
- 371.Pieper AA, Brat DJ, O’Hearn E, Krug DK, Kaplin AI, Takahashi K, Greenberg JH, Ginty D, Molliver ME, Snyder SH. Differential neuronal localizations and dynamics of phosphorylated and unphosphorylated type 1 inositol 1,4,5-trisphosphate receptors. Neuroscience. 2001;102:433–444. doi: 10.1016/s0306-4522(00)00470-x. [DOI] [PubMed] [Google Scholar]
- 372.Pinton P, Ferrari D, Magalhaes P, Schulze-Osthoff K, Di Virgilio F, Pozzan T, Rizzuto R. Reduced loading of intracellular Ca2+ stores and downregulation of capacitative Ca2+ influx in Bcl-2-overexpressing cells. J Cell Biol. 2000;148:857–862. doi: 10.1083/jcb.148.5.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Pinton P, Pozzan T, Rizzuto R. The Golgi apparatus is an inositol 1,4,5-trisphosphate-sensitive Ca2+ store, with functional properties distinct from those of the endoplasmic reticulum. EMBO J. 1998;17:5298–5308. doi: 10.1093/emboj/17.18.5298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Pinton P, Rizzuto R. Bcl-2 and Ca2+ homeostasis in the endoplasmic reticulum. Cell Death Differ. 2006;13:1409–1418. doi: 10.1038/sj.cdd.4401960. [DOI] [PubMed] [Google Scholar]
- 375.Ponting CP. Novel repeats in ryanodine and IP3 receptors and protein O-mannosyltransferases. Trends Biochem Sci. 2000;25:48–50. doi: 10.1016/s0968-0004(99)01513-3. [DOI] [PubMed] [Google Scholar]
- 376.Priori SG, Napolitano C. Cardiac and skeletal muscle disorders caused by mutations in the intracellular Ca2+ release channels. J Clin Invest. 2005;115:2033–2038. doi: 10.1172/JCI25664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Putney JW, Jr, Bird GStJ. The inositol phosphate-calcium signaling system in nonexcitable cells. Endocr Rev. 1993;14:610–631. doi: 10.1210/edrv-14-5-610. [DOI] [PubMed] [Google Scholar]
- 378.Quadroni M, L’Hostis EL, Corti C, Myagkikh I, Durussel I, Cox J, James P, Carafoli E. Phosphorylation of calmodulin alters its potency as an activator of target enzymes. Biochemistry. 1998;37:6523–6532. doi: 10.1021/bi972930+. [DOI] [PubMed] [Google Scholar]
- 379.Ramos J, Jung W, Ramos-Franco J, Mignery GA, Fill M. Single channel function of inositol 1,4,5-trisphosphate receptor type-1 and -2 isoform domain-swap chimeras. J Gen Physiol. 2003;121:399–411. doi: 10.1085/jgp.200208718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Ramos-Franco J, Bare D, Caenepeel S, Nani A, Fill M, Mignery G. Single-channel function of recombinant type 2 inositol 1,4,5-trisphosphate receptor. Biophys J. 2000;79:1388–1399. doi: 10.1016/S0006-3495(00)76391-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Ramos-Franco J, Caenepeel S, Fill M, Mignery G. Single channel function of recombinant type-1 inositol 1,4,5-trisphosphate receptor ligand binding domain splice variants. Biophys J. 1998;75:2783–2793. doi: 10.1016/S0006-3495(98)77721-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Ramos-Franco J, Fill M, Mignery GA. Isoform-specific function of single inositol 1,4,5-trisphosphate receptor channels. Biophys J. 1998;75:834–839. doi: 10.1016/S0006-3495(98)77572-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Ramos-Franco J, Galvan D, Mignery GA, Fill M. Location of the permeation pathway in the recombinant type 1 inositol 1,4,5-trisphosphate receptor. J Gen Physiol. 1999;114:243–250. doi: 10.1085/jgp.114.2.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Ramsey IS, Delling M, Clapham DE. An introduction to TRP channels. Annu Rev Physiol. 2006;68:619–647. doi: 10.1146/annurev.physiol.68.040204.100431. [DOI] [PubMed] [Google Scholar]
- 385.Reed JC. Proapoptotic multidomain Bcl-2/Bax-family proteins: mechanisms, physiological roles, therapeutic opportunities. Cell Death Differ. 2006;13:1378–1386. doi: 10.1038/sj.cdd.4401975. [DOI] [PubMed] [Google Scholar]
- 386.Regan MR, Lin DD, Emerick MC, Agnew WS. The effect of higher order RNA processes on changing patterns of protein domain selection: a developmentally regulated transcriptome of type 1 inositol 1,4,5-trisphosphate receptors. Proteins. 2005;59:312–331. doi: 10.1002/prot.20225. [DOI] [PubMed] [Google Scholar]
- 387.Ribeiro CM, Reece J, Putney JW., Jr Role of the cytoskeleton in calcium signaling in NIH 3T3 cells. An intact cytoskeleton is required for agonist-induced [Ca2+]i signaling, but not for capacitative calcium entry. J Biol Chem. 1997;272:26555–26561. doi: 10.1074/jbc.272.42.26555. [DOI] [PubMed] [Google Scholar]
- 388.Riley AM, Correa V, Mahon MF, Taylor CW, Potter BV. Bicyclic analogues of D-myo-inositol 1,4,5-trisphosphate related to adenophostin A: synthesis and biological activity. J Med Chem. 2001;44:2108–2117. doi: 10.1021/jm0005499. [DOI] [PubMed] [Google Scholar]
- 389.Riley AM, Morris SA, Nerou EP, Correa V, Potter BV, Taylor CW. Interactions of inositol 1,4,5-trisphosphate (IP3) receptors with synthetic poly(ethylene glycol)-linked dimers of IP3 suggest close spacing of the IP3-binding sites. J Biol Chem. 2002;277:40290–40295. doi: 10.1074/jbc.M206925200. [DOI] [PubMed] [Google Scholar]
- 390.Rios E, Stern MD. Calcium in close quarters: microdomain feedback in excitation-contraction coupling and other cell biological phenomena. Annu Rev Biophys Biomol Struct. 1997;26:47–82. doi: 10.1146/annurev.biophys.26.1.47. [DOI] [PubMed] [Google Scholar]
- 391.Rizzuto R, Bastianutto C, Brini M, Murgia M, Pozzan T. Mitochondrial Ca2+ homeostasis in intact cells. J Cell Biol. 1994;126:1183–1194. doi: 10.1083/jcb.126.5.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Rizzuto R, Brini M, Murgia M, Pozzan T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science. 1993;262:744–747. doi: 10.1126/science.8235595. [DOI] [PubMed] [Google Scholar]
- 393.Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998;280:1763–1766. doi: 10.1126/science.280.5370.1763. [DOI] [PubMed] [Google Scholar]
- 394.Roes J, Choi BK, Cazac BB. Redirection of B cell responsiveness by transforming growth factor β receptor. Proc Natl Acad Sci USA. 2003;100:7241–7246. doi: 10.1073/pnas.0731875100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Rooney TA, Joseph SK, Queen C, Thomas AP. Cyclic GMP induces oscillatory calcium signals in rat hepatocytes. J Biol Chem. 1996;271:19817–19825. doi: 10.1074/jbc.271.33.19817. [DOI] [PubMed] [Google Scholar]
- 396.Rosado JA, Sage SO. Coupling between inositol 1,4,5-trisphosphate receptors and human transient receptor potential channel 1 when intracellular Ca2+ stores are depleted. Biochem J. 2000;350:631–635. [PMC free article] [PubMed] [Google Scholar]
- 397.Rosenberg HJ, Riley AM, Laude AJ, Taylor CW, Potter BV. Synthesis and Ca2+-mobilizing activity of purine-modified mimics of adenophostin A: a model for the adenophostin-Ins(1,4,5)P3 receptor interaction. J Med Chem. 2003;46:4860–4871. doi: 10.1021/jm030883f. [DOI] [PubMed] [Google Scholar]
- 398.Rosenberg HJ, Riley AM, Marwood RD, Correa V, Taylor CW, Potter BVL. Xylopyranoside-based agonists of D-myo-inositol 1,4,5-trisphosphate receptors: synthesis and effect of stereochemistry on biological activity. Carbohydr Res. 2001;332:53–66. doi: 10.1016/s0008-6215(01)00067-2. [DOI] [PubMed] [Google Scholar]
- 399.Ross CA, Danoff SK, Schell MJ, Snyder SH, Ullrich A. Three additional inositol 1,4,5-trisphosphate receptors: molecular cloning and differential localization in brain and peripheral tissues. Proc Natl Acad Sci USA. 1992;89:4265–4269. doi: 10.1073/pnas.89.10.4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Ross CA, Meldolesi J, Milner TA, Satoh T, Supattapone S, Snyder SH. Inositol 1,4,5-trisphosphate receptor localized to endoplasmic reticulum in cerebellar Purkinje neurons. Nature. 1989;339:468–470. doi: 10.1038/339468a0. [DOI] [PubMed] [Google Scholar]
- 401.Rossi AM, Taylor CW. Ca2+ regulation of inositol 1,4,5-trisphosphate receptors: can Ca2+ function without calmodulin? Mol Pharmacol. 2004;66:199–203. doi: 10.1124/mol.104.002592. [DOI] [PubMed] [Google Scholar]
- 402.Rutter GA, Burnett P, Rizzuto R, Brini M, Murgia M, Pozzan T, Tavare JM, Denton RM. Subcellular imaging of intramitochondrial Ca2+ with recombinant targeted aequorin: significance for the regulation of pyruvate dehydrogenase activity. Proc Natl Acad Sci USA. 1996;93:5489–5494. doi: 10.1073/pnas.93.11.5489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Ryu GR, Sung CH, Kim MJ, Sung JH, Lee KH, Park DW, Sim SS, Min do S, Rhie DJ, Yoon SH, Hahn SJ, Kim MS, Jo AY. Changes in IP3 receptor are associated with altered calcium response to cholecystokinin in diabetic rat pancreatic acini. Pancreas. 2004;29:e106–e112. doi: 10.1097/00006676-200411000-00164. [DOI] [PubMed] [Google Scholar]
- 404.Saimi Y, Kung C. Calmodulin as an ion channel subunit. Annu Rev Physiol. 2002;64:289–311. doi: 10.1146/annurev.physiol.64.100301.111649. [DOI] [PubMed] [Google Scholar]
- 405.Samso M, Wagenknecht T, Allen PD. Internal structure and visualization of transmembrane domains of the RyR1 calcium release channel by cryo-EM. Nat Struct Mol Biol. 2005;12:539–544. doi: 10.1038/nsmb938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Saraste M, Sibbald PR, Wittinghofer A. The P-loop-a common motif in ATP- and GTP-binding proteins. Trends Biochem Sci. 1990;15:430–434. doi: 10.1016/0968-0004(90)90281-f. [DOI] [PubMed] [Google Scholar]
- 407.Sato C, Hamada K, Ogura T, Miyazawa A, Iwasaki K, Hiroaki Y, Tani K, Terauchi A, Fujiyoshi Y, Mikoshiba K. Inositol 1,4,5-trisphosphate receptor contains multiple cavities and L-shaped ligand-binding domains. J Mol Biol. 2004;336:155–164. doi: 10.1016/j.jmb.2003.11.024. [DOI] [PubMed] [Google Scholar]
- 408.Satoh T, Ross CA, Villa A, Supattapone S, Pozzan T, Snyder SH, Meldolesi J. The inositol 1,4,5-trisphosphate receptor in cerebellar Purkinje cells: quantitative immunogold labeling reveals concentration in an ER subcompartment. J Cell Biol. 1990;111:615–624. doi: 10.1083/jcb.111.2.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Sayers LG, Miyawaki A, Muto A, Takeshita H, Yamamoto A, Michikawa T, Furuichi T, Mikoshiba K. Intracellular targeting and homotetramer formation of a truncated inositol 1,4,5-trisphosphate receptor-green fluorescent protein chimera in Xenopus lae-vis oocytes: evidence for the involvement of the transmembrane spanning domain in endoplasmic reticulum targeting and homotetramer complex formation. Biochem J. 1997;323:273–280. doi: 10.1042/bj3230273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Schlossmann J, Ammendola A, Ashman K, Zong X, Huber A, Neubauer G, Wang GX, Allescher HD, Korth M, Wilm M, Hofmann F, Ruth P. Regulation of intracellular calcium by a signalling complex of IRAG, IP3 receptor and cGMP kinase Ibeta. Nature. 2000;404:197–201. doi: 10.1038/35004606. [DOI] [PubMed] [Google Scholar]
- 411.Schug ZT, Joseph SK. The role of the S4–S5 linker and C-terminal tail in inositol 1,4,5-trisphosphate receptor function. J Biol Chem. 2006;281:24431–24440. doi: 10.1074/jbc.M604190200. [DOI] [PubMed] [Google Scholar]
- 412.Serysheva II, Bare DJ, Ludtke SJ, Kettlun CS, Chiu W, Mignery GA. Structure of the type 1 inositol 1,4,5-trisphosphate receptor revealed by electron cryomicroscopy. J Biol Chem. 2003;278:21319–21322. doi: 10.1074/jbc.C300148200. [DOI] [PubMed] [Google Scholar]
- 413.Serysheva II, Hamilton SL, Chiu W, Ludtke SJ. Structure of Ca2+ release channel at 14 Å resolution. J Mol Biol. 2005;345:427–431. doi: 10.1016/j.jmb.2004.10.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Shah PK, Sowdhamini R. Structural understanding of the transmembrane domains of inositol triphosphate receptors and ryanodine receptors towards calcium channeling. Protein Eng. 2001;14:867–874. doi: 10.1093/protein/14.11.867. [DOI] [PubMed] [Google Scholar]
- 415.Sharp AH, Nucifora FC, Jr, Blondel O, Sheppard CA, Zhang C, Snyder SH, Russell JT, Ryugo DK, Ross CA. Differential cellular expression of isoforms of inositol 1,4,5-triphosphate receptors in neurons and glia in brain. J Comp Neurol. 1999;406:207–220. [PubMed] [Google Scholar]
- 416.Shealy RT, Murphy AD, Ramarathnam R, Jakobsson E, Subramaniam S. Sequence-function analysis of the K+-selective family of ion channels using a comprehensive alignment and the KcsA channel structure. Biophys J. 2003;84:2929–2942. doi: 10.1016/S0006-3495(03)70020-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Shen L, Liang F, Walensky LD, Huganir RL. Regulation of AMPA receptor GluR1 subunit surface expression by a 4.1N-linked actin cytoskeletal association. J Neurosci. 2000;20:7932–7940. doi: 10.1523/JNEUROSCI.20-21-07932.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Shibao K, Hirata K, Robert ME, Nathanson MH. Loss of inositol 1,4,5-trisphosphate receptors from bile duct epithelia is a common event in cholestasis. Gastroenterology. 2003;125:1175–1187. doi: 10.1016/s0016-5085(03)01201-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Shiraishi K, Okada A, Shirakawa H, Nakanishi S, Mikoshiba K, Miyazaki S. Developmental changes in the distribution of the endoplasmic reticulum and inositol 1,4,5-trisphosphate receptors and the spatial pattern of Ca2+ release during maturation of hamster oocytes. Dev Biol. 1995;170:594–606. doi: 10.1006/dbio.1995.1239. [DOI] [PubMed] [Google Scholar]
- 420.Shuto S, Tatani K, Ueno Y, Matsuda A. Synthesis of adenophostin analogues lacking the adenine moiety as novel potent IP3 receptor ligands: some structural requirements for the significant activity of adenophostin A. J Org Chem. 1998;63:8815–8824. [Google Scholar]
- 421.Sienaert I, De Smedt H, Parys JB, Missiaen L, Vanlingen S, Sipma H, Casteels R. Characterization of a cytosolic and a luminal Ca2+ binding site in the type I inositol 1,4,5-trisphosphate receptor. J Biol Chem. 1996;271:27005–27012. doi: 10.1074/jbc.271.43.27005. [DOI] [PubMed] [Google Scholar]
- 422.Sienaert I, Missiaen L, De Smedt H, Parys JB, Sipma H, Casteels R. Molecular and functional evidence for multiple Ca2+-binding domains in the type 1 inositol 1,4,5-trisphosphate receptor. J Biol Chem. 1997;272:25899–25906. doi: 10.1074/jbc.272.41.25899. [DOI] [PubMed] [Google Scholar]
- 423.Sienaert I, Nadif Kasri N, Vanlingen S, Parys JB, Callewaert G, Missiaen L, de Smedt H. Localization and function of a calmodulin-apocalmodulin-binding domain in the N-terminal part of the type 1 inositol 1,4,5-trisphosphate receptor. Biochem J. 2002;365:269–277. doi: 10.1042/BJ20020144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Simpson PB, Mehotra S, Lange GD, Russell JT. High density distribution of endoplasmic reticulum proteins and mitochondria at specialized Ca2+ release sites in oligodendrocyte processes. J Biol Chem. 1997;272:22654–22661. doi: 10.1074/jbc.272.36.22654. [DOI] [PubMed] [Google Scholar]
- 425.Singh BB, Liu X, Ambudkar IS. Expression of truncated transient receptor potential protein 1α (Trp1α): evidence that the Trp1 C terminus modulates store-operated Ca2+ entry. J Biol Chem. 2000;275:36483–36486. doi: 10.1074/jbc.C000529200. [DOI] [PubMed] [Google Scholar]
- 426.Singleton PA, Bourguignon LY. CD44 interaction with ankyrin and IP3 receptor in lipid rafts promotes hyaluronan-mediated Ca2+ signaling leading to nitric oxide production and endothelial cell adhesion and proliferation. Exp Cell Res. 2004;295:102–118. doi: 10.1016/j.yexcr.2003.12.025. [DOI] [PubMed] [Google Scholar]
- 427.Sinha M, Hasan G. Sequencing and exon mapping of the inositol 1,4,5-trisphosphate receptor cDNA from Drosophila embryos suggests the presence of differentially regulated forms of RNA and protein. Gene. 1999;233:271–276. doi: 10.1016/s0378-1119(99)00158-4. [DOI] [PubMed] [Google Scholar]
- 428.Sipma H, De Smet P, Sienaert I, Vanlingen S, Missiaen L, Parys JB, De Smedt H. Modulation of inositol 1,4,5-trisphosphate binding to the recombinant ligand-binding site of the type-1 inositol 1,4,5-trisphosphate receptor by Ca2+ and calmodulin. J Biol Chem. 1999;274:12157–12162. doi: 10.1074/jbc.274.17.12157. [DOI] [PubMed] [Google Scholar]
- 429.Smith JB, Smith L, Higgins BL. Temperature and nucleotide dependence of calcium release by myo-inositol 1,4,5-trisphosphate in cultured vascular smooth muscle cells. J Biol Chem. 1985;260:14413–14416. [PubMed] [Google Scholar]
- 430.Sneyd J, Falcke M, Dufour JF, Fox C. A comparison of three models of the inositol trisphosphate receptor. Prog Biophys Mol Biol. 2004;85:121–140. doi: 10.1016/j.pbiomolbio.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 431.Soulsby MD, Alzayady K, Xu Q, Wojcikiewicz RJ. The contribution of serine residues 1588 and 1755 to phosphorylation of the type I inositol 1,4,5-trisphosphate receptor by PKA and PKG. FEBS Lett. 2004;557:181–184. doi: 10.1016/s0014-5793(03)01487-x. [DOI] [PubMed] [Google Scholar]
- 432.Soulsby MD, Wojcikiewicz RJ. The type III inositol 1,4,5-trisphosphate receptor is phosphorylated by cAMP-dependent protein kinase at three sites. Biochem J. 2005;392:493–497. doi: 10.1042/BJ20051325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Srikanth S, Banerjee S, Hasan G. Ectopic expression of a Drosophila InsP3R channel mutant has dominant-negative effects in vivo. Cell Calcium. 2006;39:187–196. doi: 10.1016/j.ceca.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 434.Srikanth S, Wang Z, Tu H, Nair S, Mathew MK, Hasan G, Bezprozvanny I. Functional properties of the Drosophila melanogaster inositol 1,4,5-trisphosphate receptor mutants. Biophys J. 2004;86:3634–3646. doi: 10.1529/biophysj.104.040121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435.Stern MD. Theory of excitation-contraction coupling in cardiac muscle. Biophys J. 1992;63:497–517. doi: 10.1016/S0006-3495(92)81615-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Straub SV, Wagner LE, 2nd, Bruce JI, Yule DI. Modulation of cytosolic calcium signaling by protein kinase A-mediated phosphorylation of inositol 1,4,5-trisphosphate receptors. Biol Res. 2004;37:593–602. doi: 10.4067/s0716-97602004000400013. [DOI] [PubMed] [Google Scholar]
- 437.Street VA, Bosma MM, Demas VP, Regan MR, Lin DD, Robinson LC, Agnew WS, Tempel BL. The type 1 inositol 1,4,5-trisphosphate receptor gene is altered in the opisthotonos mouse. J Neurosci. 1997;17:635–645. doi: 10.1523/JNEUROSCI.17-02-00635.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Striggow F, Ehrlich BE. The inositol 1,4,5-trisphosphate receptor of cerebellum. Mn2+ permeability and regulation by cytosolic Mn2+ J Gen Physiol. 1996;108:115–124. doi: 10.1085/jgp.108.2.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Sudhof TC, Newton CL, Archer BT, 3rd, Ushkaryov YA, Mignery GA. Structure of a novel InsP3 receptor. EMBO J. 1991;10:3199–3206. doi: 10.1002/j.1460-2075.1991.tb04882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Sugawara H, Kurosaki M, Takata M, Kurosaki T. Genetic evidence for involvement of type 1, type 2 and type 3 inositol 1,4,5-trisphosphate receptors in signal transduction through the B-cell antigen receptor. EMBO J. 1997;16:3078–3088. doi: 10.1093/emboj/16.11.3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Sun J, Xu L, Eu JP, Stamler JS, Meissner G. Classes of thiols that influence the activity of the skeletal muscle calcium release channel. J Biol Chem. 2001;276:15625–15630. doi: 10.1074/jbc.M100083200. [DOI] [PubMed] [Google Scholar]
- 442.Supattapone S, Danoff SK, Theibert A, Joseph SK, Steiner J, Snyder SH. Cyclic AMP-dependent phosphorylation of a brain inositol trisphosphate receptor decreases its release of calcium. Proc Natl Acad Sci USA. 1988;85:8747–8750. doi: 10.1073/pnas.85.22.8747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Supattapone S, Worley PF, Baraban JM, Snyder SH. Solubilization, purification, characterization of an inositol trisphosphate receptor. J Biol Chem. 1988;263:1530–1534. [PubMed] [Google Scholar]
- 444.Sureshan KM, Trusselle M, Tovey SC, Taylor CW, Potter BV. Guanophostin A: synthesis and evaluation of a high affinity agonist of the D-myo-inositol 1,4,5-trisphosphate receptor. Chem Commun. 2006:2015–2017. doi: 10.1039/b517911d. [DOI] [PubMed] [Google Scholar]
- 445.Swatton JE, Morris SA, Wissing F, Taylor CW. Functional properties of Drosophila inositol trisphosphate receptors. Biochem J. 2001;359:435–441. doi: 10.1042/0264-6021:3590435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Swillens S, Dupont G, Combettes L, Champeil P. From calcium blips to calcium puffs: theoretical analysis of the requirements for interchannel communication. Proc Natl Acad Sci USA. 1999;96:13750–13755. doi: 10.1073/pnas.96.24.13750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Szlufcik K, Missiaen L, Parys JB, Callewaert G, De Smedt H. Uncoupled IP3 receptor can function as a Ca2+-leak channel: cell biological and pathological consequences. Biol Cell. 2006;98:1–14. doi: 10.1042/BC20050031. [DOI] [PubMed] [Google Scholar]
- 448.Takahashi M, Tanzawa K, Takahashi S. Adenophostins, newly discovered metabolites of Penicillium brevicompactum, act as potent agonists of the inositol 1,4,5-trisphosphate receptor. J Biol Chem. 1994;269:369–372. [PubMed] [Google Scholar]
- 449.Takei K, Mignery GA, Mugnaini E, Sudhof TC, De Camilli P. Inositol 1,4,5-trisphosphate receptor causes formation of ER cisternal stacks in transfected fibroblasts and in cerebellar Purkinje cells. Neuron. 1994;12:327–342. doi: 10.1016/0896-6273(94)90275-5. [DOI] [PubMed] [Google Scholar]
- 450.Talon S, Vallot O, Huchet-Cadiou C, Lompre AM, Leoty C. IP3-induced tension and IP3-receptor expression in rat soleus muscle during postnatal development. Am J Physiol Regul Integr Comp Physiol. 2002;282:R1164–R1173. doi: 10.1152/ajpregu.00073.2001. [DOI] [PubMed] [Google Scholar]
- 451.Tamura T, Hashimoto M, Aruga J, Konishi Y, Nakagawa M, Ohbayashi T, Shimada M, Mikoshiba K. Promoter structure and gene expression of the mouse inositol 1,4,5-trisphosphate receptor type 3 gene. Gene. 2001;275:169–176. doi: 10.1016/s0378-1119(01)00658-8. [DOI] [PubMed] [Google Scholar]
- 452.Tang J, Lin Y, Zhang Z, Tikunova S, Birnbaumer L, Zhu MX. Identification of common binding sites for calmodulin and inositol 1,4,5-trisphosphate receptors on the carboxyl termini of trp channels. J Biol Chem. 2001;276:21303–21310. doi: 10.1074/jbc.M102316200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453.Tang TS, Slow E, Lupu V, Stavrovskaya IG, Sugimori M, Llinas R, Kristal BS, Hayden MR, Bezprozvanny I. Disturbed Ca2+ signaling and apoptosis of medium spiny neurons in Huntington’s disease. Proc Natl Acad Sci USA. 2005;102:2602–2607. doi: 10.1073/pnas.0409402102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Tang TS, Tu H, Chan EY, Maximov A, Wang Z, Wellington CL, Hayden MR, Bezprozvanny I. Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1. Neuron. 2003;39:227–239. doi: 10.1016/s0896-6273(03)00366-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455.Tang TS, Tu H, Orban PC, Chan EY, Hayden MR, Bezprozvanny I. HAP1 facilitates effects of mutant huntingtin on inositol 1,4,5-trisphosphate-induced Ca release in primary culture of striatal medium spiny neurons. Eur J Neurosci. 2004;20:1779–1787. doi: 10.1111/j.1460-9568.2004.03633.x. [DOI] [PubMed] [Google Scholar]
- 456.Tang TS, Tu H, Wang Z, Bezprozvanny I. Modulation of type 1 inositol (1,4,5)-trisphosphate receptor function by protein kinase A and protein phosphatase 1α. J Neurosci. 2003;23:403–415. doi: 10.1523/JNEUROSCI.23-02-00403.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Tanimura A, Tojyo Y, Turner RJ. Evidence that type I, II, III inositol 1,4,5-trisphosphate receptors can occur as integral plasma membrane proteins. J Biol Chem. 2000;275:27488–27493. doi: 10.1074/jbc.M004495200. [DOI] [PubMed] [Google Scholar]
- 458.Taylor CW. Inositol trisphosphate receptors: Ca2+-modulated intracellular Ca2+ channels. Biochim Biophys Acta. 1998;1436:19–33. doi: 10.1016/s0005-2760(98)00122-2. [DOI] [PubMed] [Google Scholar]
- 459.Taylor CW, da Fonseca PC, Morris EP. IP3 receptors: the search for structure. Trends Biochem Sci. 2004;29:210–219. doi: 10.1016/j.tibs.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 460.Taylor CW, Genazzani AA, Morris SA. Expression of inositol trisphosphate receptors. Cell Calcium. 1999;26:237–251. doi: 10.1054/ceca.1999.0090. [DOI] [PubMed] [Google Scholar]
- 461.Taylor CW, Laude AJ. IP3 receptors and their regulation by calmodulin and cytosolic Ca2+ Cell Calcium. 2002;32:321–334. doi: 10.1016/s0143416002001859. [DOI] [PubMed] [Google Scholar]
- 462.Taylor CW, Potter BV. The size of inositol 1,4,5-trisphosphate-sensitive Ca2+ stores depends on inositol 1,4,5-trisphosphate concentration. Biochem J. 1990;266:189–194. doi: 10.1042/bj2660189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Taylor CW, Putney JW., Jr Size of the inositol 1,4,5-trisphosphate-sensitive calcium pool in guinea-pig hepatocytes. Biochem J. 1985;232:435–438. doi: 10.1042/bj2320435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Taylor CW, Richardson A. Structure and function of inositol trisphosphate receptors. Pharmacol Ther. 1991;51:97–137. doi: 10.1016/0163-7258(91)90043-l. [DOI] [PubMed] [Google Scholar]
- 465.Terauchi M, Abe H, Tovey SC, Dedos SG, Taylor CW, Paul M, Trusselle M, Potter BV, Matsuda A, Shuto S. A systematic study of C-glucoside trisphosphates as myo-inositol trisphosphate receptor ligands. Synthesis of β-C-glucoside trisphosphates based on the conformational restriction strategy. J Med Chem. 2006;49:1900–1909. doi: 10.1021/jm051039n. [DOI] [PubMed] [Google Scholar]
- 466.Thomas AP, Bird GStJ, Hajnóczky G, Robb-Gaspers LD, Putney JW., Jr Spatial and temporal aspects of cellular calcium signaling. FASEB J. 1996;10:1505–1517. [PubMed] [Google Scholar]
- 467.Thrower EC, Choe CU, So SH, Jeon SH, Ehrlich BE, Yoo SH. A functional interaction between chromogranin B and the inositol 1,4,5-trisphosphate receptor/Ca2+ channel. J Biol Chem. 2003;278:49699–49706. doi: 10.1074/jbc.M309307200. [DOI] [PubMed] [Google Scholar]
- 468.Thrower EC, Duclohier H, Lea EJ, Molle G, Dawson AP. The inositol 1,4,5-trisphosphate-gated Ca2+ channel: effect of the protein thiol reagent thimerosal on channel activity. Biochem J. 1996;318:61–66. doi: 10.1042/bj3180061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Thrower EC, Mobasheri H, Dargan S, Marius P, Lea EJA, Dawson AP. Interaction of luminal calcium and cytosolic ATP in the control of type 1 inositol (1,4,5)-trisphosphate receptor channels. J Biol Chem. 2000;275:36049–36055. doi: 10.1074/jbc.M000970200. [DOI] [PubMed] [Google Scholar]
- 470.Thrower EC, Park HY, So SH, Yoo SH, Ehrlich BE. Activation of the inositol 1,4,5-trisphosphate receptor by the calcium storage protein chromogranin A. J Biol Chem. 2002;277:15801–15806. doi: 10.1074/jbc.M110139200. [DOI] [PubMed] [Google Scholar]
- 471.Tinker A, Lindsay AR, Williams AJ. Cation conduction in the calcium release channel of the cardiac sarcoplasmic reticulum under physiological and pathophysiological conditions. Cardiovasc Res. 1993;27:1820–1825. doi: 10.1093/cvr/27.10.1820. [DOI] [PubMed] [Google Scholar]
- 472.Tinker A, Lindsay AR, Williams AJ. A model for ionic conduction in the ryanodine receptor channel of sheep cardiac muscle sarcoplasmic reticulum. J Gen Physiol. 1992;100:495–517. doi: 10.1085/jgp.100.3.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473.Tinker A, Williams AJ. Divalent cation conduction in the ryanodine receptor channel of sheep cardiac muscle sarcoplasmic reticulum. J Gen Physiol. 1992;100:479–493. doi: 10.1085/jgp.100.3.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Tinker A, Williams AJ. Measuring the length of the pore of the sheep cardiac sarcoplasmic-reticulum calcium-release channel using related trimethylammonium ions as molecular calipers. Biophys J. 1995;68:111–120. doi: 10.1016/S0006-3495(95)80165-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Tinker A, Williams AJ. Probing the structure of the conduction pathway of the sheep cardiac sarcoplasmic-reticulum calcium-release channel with permeant and impermeant organic cations. J Gen Physiol. 1993;102:1107–1129. doi: 10.1085/jgp.102.6.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476.Trebak M, Bird GStJ, McKay RR, Birnbaumer L, Putney JW., Jr Signaling mechanism for receptor-activated canonical transient receptor potential 3 (TRPC3) channels. J Biol Chem. 2003;278:16244–16252. doi: 10.1074/jbc.M300544200. [DOI] [PubMed] [Google Scholar]
- 477.Tsien R, Pozzan T. Measurement of cytosolic free Ca2+ with quin2. Methods Enzymol. 1989;172:230–262. doi: 10.1016/s0076-6879(89)72017-6. [DOI] [PubMed] [Google Scholar]
- 478.Tu H, Miyakawa T, Wang Z, Glouchankova L, Iino M, Bezprozvanny I. Functional characterization of the type 1 inositol 1,4,5-trisphosphate receptor coupling domain SII(+/−) splice variants and the Opisthotonos mutant form. Biophys J. 2002;82:1995–2004. doi: 10.1016/S0006-3495(02)75548-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479.Tu H, Nosyreva E, Miyakawa T, Wang Z, Mizushima A, Iino M, Bezprozvanny I. Functional and biochemical analysis of the type 1 inositol (1,4,5)-trisphosphate receptor calcium sensor. Biophys J. 2003;85:290–299. doi: 10.1016/S0006-3495(03)74474-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Tu H, Tang TS, Wang Z, Bezprozvanny I. Association of type 1 inositol 1,4,5-trisphosphate receptor with AKAP9 (Yotiao) and protein kinase A. J Biol Chem. 2004;279:19375–19382. doi: 10.1074/jbc.M313476200. [DOI] [PubMed] [Google Scholar]
- 481.Tu H, Wang Z, Bezprozvanny I. Modulation of mammalian inositol 1,4,5-trisphosphate receptor isoforms by calcium: a role of calcium sensor region. Biophys J. 2005;88:1056–1069. doi: 10.1529/biophysj.104.049601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482.Tu H, Wang Z, Nosyreva E, De Smedt H, Bezprozvanny I. Functional characterization of mammalian inositol 1,4,5-trisphosphate receptor isoforms. Biophys J. 2005;88:1046–1055. doi: 10.1529/biophysj.104.049593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.Tu JC, Xiao B, Yuan JP, Lanahan AA, Leoffert K, Li M, Linden DJ, Worley PF. Homer binds a novel proline-rich motif and links group 1 metabotropic glutamate receptors with IP3 receptors. Neuron. 1998;21:717–726. doi: 10.1016/s0896-6273(00)80589-9. [DOI] [PubMed] [Google Scholar]
- 484.Turvey MR, Fogarty KE, Thorn P. Inositol (1,4,5)-trisphosphate receptor links to filamentous actin are important for generating local Ca2+ signals in pancreatic acinar cells. J Cell Sci. 2005;118:971–980. doi: 10.1242/jcs.01693. [DOI] [PubMed] [Google Scholar]
- 485.Tuvia S, Buhusi M, Davis L, Reedy M, Bennett V. Ankyrin-B is required for intracellular sorting of structurally diverse Ca2+ homeostasis proteins. J Cell Biol. 1999;147:995–1008. doi: 10.1083/jcb.147.5.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486.Uchida K, Miyauchi H, Furuichi T, Michikawa T, Mikoshiba K. Critical regions for activation gating of the inositol 1,4,5-trisphosphate receptor. J Biol Chem. 2003;278:16551–16560. doi: 10.1074/jbc.M300646200. [DOI] [PubMed] [Google Scholar]
- 487.Vanlingen S, Sipma H, De Smet P, Callewaert G, Missiaen L, De Smedt H, Parys JB. Ca2+ and calmodulin differentially modulate myo-inositol 1,4,5-trisphosphate (IP3)-binding to the recombinant ligand-binding domains of the various IP3 receptor isoforms. Biochem J. 2000;346:275–280. [PMC free article] [PubMed] [Google Scholar]
- 488.Vanlingen S, Sipma H, De Smet P, Callewaert G, Missiaen L, De Smedt H, Parys JB. Modulation of inositol 1,4,5-trisphosphate binding to the various inositol 1,4,5-trisphosphate receptor isoforms by thimerosal and cyclic ADP-ribose. Biochem Pharmacol. 2001;61:803–809. doi: 10.1016/s0006-2952(01)00540-8. [DOI] [PubMed] [Google Scholar]
- 489.Vanlingen S, Sipma H, Missiaen L, De Smedt H, De Smet P, Casteels R, Parys JB. Modulation of type 1, 2 and 3 inositol 1,4,5-trisphosphate receptors by cyclic ADP-ribose and thimerosal. Cell Calcium. 1999;25:107–114. doi: 10.1054/ceca.1998.0010. [DOI] [PubMed] [Google Scholar]
- 490.Venkatesh K, Hasan G. Disruption of the IP3 receptor gene of Drosophila affects larval metamorphosis and ecdysone release. Curr Biol. 1997;7:500–509. doi: 10.1016/s0960-9822(06)00221-1. [DOI] [PubMed] [Google Scholar]
- 491.Verkhratsky A. Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons. Physiol Rev. 2005;85:201–279. doi: 10.1152/physrev.00004.2004. [DOI] [PubMed] [Google Scholar]
- 492.Vermassen E, Fissore RA, Nadif Kasri N, Vanderheyden V, Callewaert G, Missiaen L, Parys JB, De Smedt H. Regulation of the phosphorylation of the inositol 1,4,5-trisphosphate receptor by protein kinase C. Biochem Biophys Res Commun. 2004;319:888–893. doi: 10.1016/j.bbrc.2004.05.071. [DOI] [PubMed] [Google Scholar]
- 493.Vermassen E, Parys JB, Mauger JP. Subcellular distribution of the inositol 1,4,5-trisphosphate receptors: functional relevance and molecular determinants. Biol Cell. 2004;96:3–17. doi: 10.1016/j.biolcel.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 494.Volpe P, Alderson-Lang BH. Regulation of inositol 1,4,5-trisphosphate-induced Ca2+ release II. Effect of cAMP-dependent protein kinase. Am J Physiol Cell Physiol. 1990;258:C1086–C1091. doi: 10.1152/ajpcell.1990.258.6.C1086. [DOI] [PubMed] [Google Scholar]
- 495.Wagenknecht T, Samso M. Three-dimensional reconstruction of ryanodine receptors. Front Biosci. 2002;7:d1464–d1474. doi: 10.2741/A853. [DOI] [PubMed] [Google Scholar]
- 496.Wagner LE, 2nd, Betzenhauser MJ, Yule DI. ATP binding to a unique site in the type-1 S2− inositol 1,4,5-trisphosphate receptor defines susceptibility to phosphorylation by protein kinase A. J Biol Chem. 2006;281:17410–17419. doi: 10.1074/jbc.M601340200. [DOI] [PubMed] [Google Scholar]
- 497.Wagner LE, 2nd, Li WH, Joseph SK, Yule DI. Functional consequences of phosphomimetic mutations at key cAMP-dependent protein kinase phosphorylation sites in the type 1 inositol 1,4,5-trisphosphate receptor. J Biol Chem. 2004;279:46242–46252. doi: 10.1074/jbc.M405849200. [DOI] [PubMed] [Google Scholar]
- 498.Wagner LE, 2nd, Li WH, Yule DI. Phosphorylation of type-1 inositol 1,4,5-trisphosphate receptors by cyclic nucleotide-dependent protein kinases: a mutational analysis of the functionally important sites in the S2+ and S2− splice variants. J Biol Chem. 2003;278:45811–45817. doi: 10.1074/jbc.M306270200. [DOI] [PubMed] [Google Scholar]
- 499.Walensky LD, Blackshaw S, Liao D, Watkins CC, Weier HU, Parra M, Huganir RL, Conboy JG, Mohandas N, Snyder SH. A novel neuron-enriched homolog of the erythrocyte membrane cytoskeletal protein 4.1. J Neurosci. 1999;19:6457–6467. doi: 10.1523/JNEUROSCI.19-15-06457.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500.Wang RW, Bolstad J, Kong HH, Zhang L, Brown C, Chen SRW. The predicted TM10 transmembrane sequence of the cardiac Ca2+ release channel (ryanodine receptor) is crucial for channel activation and gating. J Biol Chem. 2004;279:3635–3642. doi: 10.1074/jbc.M311367200. [DOI] [PubMed] [Google Scholar]
- 501.Wang RW, Zhang L, Bolstad J, Diao N, Brown C, Ruest L, Welch W, Williams AJ, Chen SRW. Residue Gln(4863) within a predicted transmembrane sequence of the Ca2+ release channel (ryanodine receptor) is critical for ryanodine interaction. J Biol Chem. 2003;278:51557–51565. doi: 10.1074/jbc.M306788200. [DOI] [PubMed] [Google Scholar]
- 502.Wang X, Huang G, Luo X, Penninger JM, Muallem S. Role of regulator of G protein signaling 2 (RGS2) in Ca2+ oscillations and adaptation of Ca2+ signaling to reduce excitability of RGS2−/− cells. J Biol Chem. 2004;279:41642–41649. doi: 10.1074/jbc.M406450200. [DOI] [PubMed] [Google Scholar]
- 503.Wang Y, Mattson MP, Furukawa K. Endoplasmic reticulum calcium release is modulated by actin polymerization. J Neurochem. 2002;82:945–952. doi: 10.1046/j.1471-4159.2002.01059.x. [DOI] [PubMed] [Google Scholar]
- 504.Wang Y, Xu L, Pasek DA, Gillespie D, Meissner G. Probing the role of negatively charged amino acid residues in ion permeation of skeletal muscle ryanodine receptor. Biophys J. 2005;89:256–265. doi: 10.1529/biophysj.104.056002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505.Watras J, Bezprozvanny I, Ehrlich BE. Inositol 1,4,5-trisphosphate-gated channels in cerebellum: presence of multiple conductance states. J Neurosci. 1991;11:3239–3245. doi: 10.1523/JNEUROSCI.11-10-03239.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506.Wedel BJ, Vazquez G, McKay RR, Bird GStJ, Putney JW., Jr A calmodulin/inositol 1,4,5-trisphosphate (IP3) receptor-binding region targets TRPC3 to the plasma membrane in a calmodulin/IP3 receptor-independent process. J Biol Chem. 2003;278:25758–25765. doi: 10.1074/jbc.M303890200. [DOI] [PubMed] [Google Scholar]
- 507.Welch W, Rheault S, West DJ, Williams AJ. A model of the putative pore region of the cardiac ryanodine receptor channel. Biophys J. 2004;87:2335–2351. doi: 10.1529/biophysj.104.044180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 508.White C, Li C, Yang J, Petrenko NB, Madesh M, Thompson CB, Foskett JK. The endoplasmic reticulum gateway to apoptosis by Bcl-XL modulation of the InsP3R. Nat Cell Biol. 2005;7:1021–1028. doi: 10.1038/ncb1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509.White C, Yang J, Monteiro MJ, Foskett JK. CIB1, a ubiquitously-expressed Ca2+-binding protein-ligand of the InsP3 receptor Ca2+-release channel. J Biol Chem. 2006;281:20825–20833. doi: 10.1074/jbc.M602175200. [DOI] [PubMed] [Google Scholar]
- 510.Whiteman EL, Cho H, Birnbaum MJ. Role of Akt/protein kinase B in metabolism. Trends Endocrinol Metab. 2002;13:444–451. doi: 10.1016/s1043-2760(02)00662-8. [DOI] [PubMed] [Google Scholar]
- 511.Wierenga RK, Hol WG. Predicted nucleotide-binding properties of p21 protein and its cancer-associated variant. Nature. 1983;302:842–844. doi: 10.1038/302842a0. [DOI] [PubMed] [Google Scholar]
- 512.Wierenga RK, Terpstra P, Hol WG. Prediction of the occurrence of the ADP-binding αβα-fold in proteins, using an amino acid sequence fingerprint. J Mol Biol. 1986;187:101–107. doi: 10.1016/0022-2836(86)90409-2. [DOI] [PubMed] [Google Scholar]
- 513.Wilcox RA, Strupish J, Nahorski SR. Quantal calcium release in electropermeabilized SH-SY5Y neuroblastoma cells perfused with myo-inositol 1,4,5-trisphosphate. Cell Calcium. 1996;20:243–255. doi: 10.1016/s0143-4160(96)90030-5. [DOI] [PubMed] [Google Scholar]
- 514.Williams AJ, West DJ, Sitsapesan R. Light at the end of the Ca2+-release channel tunnel: structures and mechanisms involved in ion translocation in ryanodine receptor channels. Q Rev Biophys. 2001;34:61–104. doi: 10.1017/s0033583501003675. [DOI] [PubMed] [Google Scholar]
- 515.Wojcikiewicz RJ. Regulated ubiquitination of proteins in GPCR-initiated signaling pathways. Trends Pharmacol Sci. 2004;25:35–41. doi: 10.1016/j.tips.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 516.Wojcikiewicz RJ. Type I, II, III inositol 1,4,5-trisphosphate receptors are unequally susceptible to down-regulation and are expressed in markedly different proportions in different cell types. J Biol Chem. 1995;270:11678–11683. doi: 10.1074/jbc.270.19.11678. [DOI] [PubMed] [Google Scholar]
- 517.Wojcikiewicz RJ, Luo SG. Differences among type I, II, III inositol-1,4,5-trisphosphate receptors in ligand-binding affinity influence the sensitivity of calcium stores to inositol-1,4,5-trisphosphate. Mol Pharmacol. 1998;53:656–662. doi: 10.1124/mol.53.4.656. [DOI] [PubMed] [Google Scholar]
- 518.Wojcikiewicz RJ, Luo SG. Phosphorylation of inositol 1,4,5-trisphosphate receptors by cAMP-dependent protein kinase. Type I, II, III receptors are differentially susceptible to phosphorylation and are phosphorylated in intact cells. J Biol Chem. 1998;273:5670–5677. doi: 10.1074/jbc.273.10.5670. [DOI] [PubMed] [Google Scholar]
- 519.Wojcikiewicz RJ, Oberdorf JA. Degradation of inositol 1,4,5-trisphosphate receptors during cell stimulation is a specific process mediated by cysteine protease activity. J Biol Chem. 1996;271:16652–16655. doi: 10.1074/jbc.271.28.16652. [DOI] [PubMed] [Google Scholar]
- 520.Worley PF, Baraban JM, Supattapone S, Wilson VS, Snyder SH. Characterization of inositol trisphosphate receptor binding in brain. Regulation by pH and calcium. J Biol Chem. 1987;262:12132–12136. [PubMed] [Google Scholar]
- 521.Wu X, Zhang T, Bossuyt J, Li X, McKinsey TA, Dedman JR, Olson EN, Chen J, Brown JH, Bers DM. Local InsP3-dependent perinuclear Ca2+ signaling in cardiac myocyte excitation-transcription coupling. J Clin Invest. 2006;116:675–682. doi: 10.1172/JCI27374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 522.Xiao BL, Masumiya H, Jiang DW, Wang RW, Sei Y, Zhang L, Murayama T, Ogawa Y, Lai FA, Wagenknecht T, Chen SRW. Isoform-dependent formation of heteromeric Ca2+ release channels (ryanodine receptors) J Biol Chem. 2002;277:41778–41785. doi: 10.1074/jbc.M208210200. [DOI] [PubMed] [Google Scholar]
- 523.Xie Z, Askari A. Na+/K+-ATPase as a signal transducer. Eur J Biochem. 2002;269:2434–2439. doi: 10.1046/j.1432-1033.2002.02910.x. [DOI] [PubMed] [Google Scholar]
- 524.Xu L, Wang Y, Gillespie D, Meissner G. Two rings of negative charges in the cytosolic vestibule of type-1 ryanodine receptor modulate ion fluxes. Biophys J. 2006;90:443–453. doi: 10.1529/biophysj.105.072538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 525.Yamada M, Miyawaki A, Saito K, Nakajima T, Yamamoto-Hino M, Ryo Y, Furuichi T, Mikoshiba K. The calmodulin-binding domain in the mouse type 1 inositol 1,4,5-trisphosphate receptor. Biochem J. 1995;308:83–88. doi: 10.1042/bj3080083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526.Yamda J, Ohkusa T, Nao T, Ueyama T, Yano M, Kobayashi S, Hamano K, Esato K, Matsuzaki M. Up-regulation of inositol 1,4,5 trisphosphate receptor expression in atrial tissue in patients with chronic atrial fibrillation. J Am Coll Cardiol. 2001;37:1111–1119. doi: 10.1016/s0735-1097(01)01144-5. [DOI] [PubMed] [Google Scholar]
- 527.Yang J, McBride S, Mak DOD, Vardi N, Palczewski K, Haeseleer F, Foskett JK. Identification of a family of calcium sensors as protein ligands of inositol trisphosphate receptor Ca2+ release channels. Proc Natl Acad Sci USA. 2002;99:7711–7716. doi: 10.1073/pnas.102006299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 528.Yang W, Lee HW, Hellinga H, Yang JJ. Structural analysis, identification, design of calcium-binding sites in proteins. Proteins. 2002;47:344–356. doi: 10.1002/prot.10093. [DOI] [PubMed] [Google Scholar]
- 529.Yin X, Gower NJ, Baylis HA, Strange K. Inositol 1,4,5-trisphosphate signaling regulates rhythmic contractile activity of myoepithelial sheath cells in Caenorhabditis elegans. Mol Biol Cell. 2004;15:3938–3949. doi: 10.1091/mbc.E04-03-0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 530.Yoneshima H, Miyawaki A, Michikawa T, Furuichi T, Mikoshiba K. Ca2+ differentially regulates the ligand-affinity states of type 1 and type 3 inositol 1,4,5-trisphosphate receptors. Biochem J. 1997;322:591–596. doi: 10.1042/bj3220591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531.Yoo SH. Coupling of the IP3 receptor/Ca2+ channel with Ca2+ storage proteins chromogranins A and B in secretory granules. Trends Neurosci. 2000;23:424–428. doi: 10.1016/s0166-2236(00)01621-0. [DOI] [PubMed] [Google Scholar]
- 532.Yoo SH, Lewis MS. Interaction of chromogranin B and the near N-terminal region of chromogranin B with an intraluminal loop peptide of the inositol 1,4, 5-trisphosphate receptor. J Biol Chem. 2000;275:30293–30300. doi: 10.1074/jbc.M001204200. [DOI] [PubMed] [Google Scholar]
- 533.Yoo SH, Lewis MS. pH-dependent interaction of an intraluminal loop of inositol 1,4,5-trisphosphate receptor with chromogranin A. FEBS Lett. 1994;341:28–32. doi: 10.1016/0014-5793(94)80234-3. [DOI] [PubMed] [Google Scholar]
- 534.Yoo SH, Lewis MS. Thermodynamic study of the pH-dependent interaction of chromogranin A with an intraluminal loop peptide of the inositol 1,4,5-trisphosphate receptor. Biochemistry. 1995;34:632–638. doi: 10.1021/bi00002a030. [DOI] [PubMed] [Google Scholar]
- 535.Yoo SH, So SH, Kweon HS, Lee JS, Kang MK, Jeon CJ. Coupling of the inositol 1,4,5-trisphosphate receptor and chromogranins A and B in secretory granules. J Biol Chem. 2000;275:12553–12559. doi: 10.1074/jbc.275.17.12553. [DOI] [PubMed] [Google Scholar]
- 536.Yoshida Y, Imai S. Structure and function of inositol 1,4,5-trisphosphate receptor. Jpn J Pharmacol. 1997;74:125–137. doi: 10.1254/jjp.74.125. [DOI] [PubMed] [Google Scholar]
- 537.Yoshikawa F, Iwasaki H, Michikawa T, Furuichi T, Mikoshiba K. Trypsinized cerebellar inositol 1,4,5-trisphosphate receptor. Structural and functional coupling of cleaved ligand binding and channel domains. J Biol Chem. 1999;274:316–327. doi: 10.1074/jbc.274.1.316. [DOI] [PubMed] [Google Scholar]
- 538.Yoshikawa F, Morita M, Monkawa T, Michikawa T, Furuichi T, Mikoshiba K. Mutational analysis of the ligand binding site of the inositol 1,4,5-trisphosphate receptor. J Biol Chem. 1996;271:18277–18284. doi: 10.1074/jbc.271.30.18277. [DOI] [PubMed] [Google Scholar]
- 539.Yuan JP, Kiselyov K, Shin DM, Chen J, Shcheynikov N, Kang SH, Dehoff MH, Schwarz MK, Seeburg PH, Muallem S, Worley PF. Homer binds TRPC family channels and is required for gating of TRPC1 by IP3 receptors. Cell. 2003;114:777–789. doi: 10.1016/s0092-8674(03)00716-5. [DOI] [PubMed] [Google Scholar]
- 540.Yuan Z, Cai T, Tian J, Ivanov AV, Giovannucci DR, Xie Z. Na/K-ATPase tethers phospholipase C and IP3 receptor into a calcium-regulatory complex. Mol Biol Cell. 2005;16:4034–4045. doi: 10.1091/mbc.E05-04-0295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 541.Yule DI, Straub SV, Bruce JI. Modulation of Ca2+ oscillations by phosphorylation of Ins(1,4,5)P3 receptors. Biochem Soc Trans. 2003;31:954–957. doi: 10.1042/bst0310954. [DOI] [PubMed] [Google Scholar]
- 542.Zeng W, Mak DOD, Li Q, Shin DM, Foskett JK, Muallem S. A new mode of Ca2+ signaling by G protein-coupled receptors: gating of IP3 receptor Ca2+ release channels by Gβγ. Curr Biol. 2003;13:872–876. doi: 10.1016/s0960-9822(03)00330-0. [DOI] [PubMed] [Google Scholar]
- 543.Zhang S, Malmersjo S, Li J, Ando H, Aizman O, Uhlen P, Mikoshiba K, Aperia A. Distinct role of the N-terminal tail of the Na,K-ATPase catalytic subunit as a signal transducer. J Biol Chem. 2006;281:21954–21962. doi: 10.1074/jbc.M601578200. [DOI] [PubMed] [Google Scholar]
- 544.Zhang S, Mizutani A, Hisatsune C, Higo T, Bannai H, Nakayama T, Hattori M, Mikoshiba K. Protein 4.1N is required for translocation of inositol 1,4,5-trisphosphate receptor type 1 to the basolateral membrane domain in polarized Madin-Darby canine kidney cells. J Biol Chem. 2003;278:4048–4056. doi: 10.1074/jbc.M209960200. [DOI] [PubMed] [Google Scholar]
- 545.Zhang X, Joseph SK. Effect of mutation of a calmodulin binding site on Ca2+ regulation of inositol trisphosphate receptors. Biochem J. 2001;360:395–400. doi: 10.1042/0264-6021:3600395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 546.Zhao MC, Li P, Li XL, Zhang L, Winkfein RJ, Chen SRW. Molecular identification of the ryanodine receptor pore-forming segment. J Biol Chem. 1999;274:25971–25974. doi: 10.1074/jbc.274.37.25971. [DOI] [PubMed] [Google Scholar]
- 547.Zhong F, Davis MC, McColl KS, Distelhorst CW. Bcl-2 differentially regulates Ca2+ signals according to the strength of T cell receptor activation. J Cell Biol. 2006;172:127–137. doi: 10.1083/jcb.200506189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 548.Zhou YF, MacKinnon R. The occupancy of ions in the K+ selectivity filter: charge balance and coupling of ion binding to a protein conformational change underlie high conduction rates. J Mol Biol. 2003;333:965–975. doi: 10.1016/j.jmb.2003.09.022. [DOI] [PubMed] [Google Scholar]
- 549.Zhu DM, Tekle E, Chock PB, Huang CY. Reversible phosphorylation as a controlling factor for sustaining calcium oscillations in HeLa cells: involvement of calmodulin-dependent kinase II and a calyculin A-inhibitable phosphatase. Biochemistry. 1996;35:7214–7223. doi: 10.1021/bi952471h. [DOI] [PubMed] [Google Scholar]
- 550.Zilberberg N, Ilan N, Goldstein SAN. KCNKO: opening and closing the 2-P-domain potassium leak channel entails “C-type” gating of the outer pore. Neuron. 2001;32:635–648. doi: 10.1016/s0896-6273(01)00503-7. [DOI] [PubMed] [Google Scholar]
- 551.Zong WX, Li C, Hatzivassiliou G, Lindsten T, Yu QC, Yuan JY, Thompson CB. Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis. J Cell Biol. 2003;162:59–69. doi: 10.1083/jcb.200302084. [DOI] [PMC free article] [PubMed] [Google Scholar]