

Abstract
Prion diseases are a group of transmissible, invariably fatal neurodegenerative diseases that affect both humans and animals. According to the protein-only hypothesis, the infectious agent is a prion (proteinaceous infectious particle) that is composed primarily of PrPSc, the disease-associated isoform of the cellular prion protein, PrP. PrPSc arises from the conformational change of the normal, glycosylphosphatidylinositol (GPI)-anchored protein, PrPC. The mechanism by which this process occurs, however, remains enigmatic. Rabbits are one of a small number of mammalian species reported to be resistant to prion infection. Sequence analysis of rabbit PrP revealed that its C-terminal amino acids differ from those of PrP from other mammals and may affect the anchoring of rabbit PrP through its GPI anchor. Using a cell culture model, this study investigated the effect of the rabbit PrP-specific C-terminal amino acids on the addition of the GPI anchor to PrPC, PrPC localization, and PrPSc formation. The incorporation of rabbit-specific C-terminal PrP residues into mouse PrP did not affect the addition of a GPI anchor or the localization of PrP. However, these residues did inhibit PrPSc formation, suggesting that these rabbit-specific residues interfere with a C-terminal PrPSc interaction site.
Prion diseases, traditionally known as transmissible spongiform encephalopathies (TSE), are a group of invariably fatal neurodegenerative diseases that affect both humans and animals. According to the protein-only hypothesis, an abnormal isoform of the host-encoded prion protein (PrPC), referred to as PrPSc, is the sole or major component of the infectious agent causing these diseases (33). These disorders affect a wide range of mammals and include diseases such as Creutzfeldt-Jakob disease (CJD), variant CJD, Gerstmann-Straüssler-Scheinker (GSS) syndrome, kuru, and fatal familial insomnia (FFI) in humans, scrapie in sheep and goats, chronic wasting disease (CWD) in cervids, and bovine spongiform encephalopathy (BSE) in cattle. The term “prion” was first used to describe the unique infectious agent and was derived from “proteinaceous infectious particle” to distinguish it from conventional pathogens such as bacteria and viruses (33).
To date, rabbits are one of the few mammalian species reported to be resistant to prion infection. Rabbits do not develop clinical disease after inoculation with brain tissue from individuals affected by the human prion diseases CJD and kuru, or by a number of animal forms of the disease, including scrapie and transmissible mink encephalopathy (TME) (12). In addition, mouse neuroblastoma (MNB) cells overexpressing rabbit PrP are also resistant to prion infection (45). Evidence that rabbit cells per se have the correct cellular machinery to support prion propagation has come from studies using the rabbit kidney epithelial cell line RK13. Upon transfection with appropriate PrP-expressing transgenes, these cells are a highly efficient and robust model of prion infection (6, 25, 41, 43). RK13 cells do not have detectable levels of endogenous rabbit PrPC and are therefore ideal for studying exogenous PrPC and the propagation of prions from different species (6). Originally, it was shown that RK13 cells overexpressing ovine PrP became susceptible to infection with scrapie (43), and more recently, RK13 cells expressing rodent PrPC, from either the mouse or the bank vole, were readily infected by prions adapted to and propagated in these two species (6, 41). RK13 cells expressing human PrPC, however, were resistant to infection with human prions derived directly from a patient with sporadic CJD (25). Since RK13 cells overexpressing PrP are a well-established model of prion propagation, we can therefore conclude that while these cells apparently have the appropriate cellular machinery to support prion propagation, it may be a characteristic of the rabbit prion protein itself that results in the resistance of this species to prion infection. However, the loss of a cellular cofactor may also be a contributing factor.
Analysis of the rabbit PrP amino acid sequence shows that it has all the features previously described for members of the PrP protein family, including an N-terminal signal peptide, an octapeptide repeat region, and a C-terminal signal sequence (26). While amino acid sequence comparison of both mouse and rabbit PrP species reveals 87% sequence homology, there are 22 amino acid differences between the two, and several of these reside in regions of PrP known to be important in PrPSc formation. In scrapie-infected MNB cells, the residues Gly99 and Met108 within the N terminus, Ser173 within the central region, and Ile214 within the C terminus of rabbit PrP were shown to inhibit PrPSc generation when incorporated into mouse PrP, suggesting that multiple amino acid residues in rabbit PrP inhibit PrPSc formation (45). Approximately one-third (9/33 residues in the immature sequence) of the amino acid difference between mouse and rabbit PrPs was shown to occur at the glycosylphosphatidylinositol (GPI) anchor attachment site (see Fig. S1 in the supplemental material). As yet, studies involving this region of rabbit PrP have not been performed. Therefore, this region of rabbit PrP may provide further insight into the resistance of rabbits to prion infection.
GPI anchor addition occurs via a transamination reaction in the endoplasmic reticulum (ER) following cleavage of the C-terminal signal sequence (39). There is no consensus sequence with which to identify the C-terminal cleavage site, but there are three key C-terminal elements: (i) the cleavage site, or ω site, where the GPI anchor attaches to the COOH group of the ω amino acid; (ii) a hydrophilic spacer region of 8 to 12 amino acids (ω + 1 up to ω + 10); and (iii) a hydrophobic region of 10 to 20 amino acids (ω + 11 onwards) (9). Analysis of known GPI-anchored proteins has given rise to sequence motifs in the C-terminal signal peptide allowing the prediction of the ω site of proteins. Due to the complexity of experimentally determining the ω site of GPI-anchored proteins, relatively few of the many known GPI-anchored proteins have had their ω sites determined (36 of 340 proteins in 2008) (32) The ω site of hamster PrP was determined experimentally to be at amino acid 231 (34) and is predicted to be at the same site for PrPs from all mammals, based on amino acid sequence comparison. Amino acid substitutions near the ω site of mouse PrP revealed that mouse PrP has an ω site at residue 230 (17). It was also shown that single amino acid substitutions at and near the ω site of mouse PrP affect the anchoring and conversion efficiency of PrP (17). It is therefore possible that the amino acids at the C terminus and within the GPI anchor signal sequence of rabbit PrP lead to the resistance to prion infection.
To date, no protein structures containing a GPI anchor have been determined by X-ray crystallography, and although the nuclear magnetic resonance (NMR) structures of mouse and rabbit PrP have been solved, they do not contain any structural information for the residues immediately preceding the GPI anchor. We therefore created a mutant mouse PrP model containing rabbit PrP-specific amino acids at the ω site to investigate whether these residues are involved in rabbit resistance to prion infection. Here we demonstrate that the GPI anchor attachment site is an important site that controls the ability of PrP to be converted into PrPSc and that residues ω and ω + 1 of PrP are important modulators of this pathogenic process.
MATERIALS AND METHODS
Antibodies.
The antibodies used in this study were the anti-prion monoclonal antibodies ICSM-18 (residues 143 to 153; D-Gen) and 8F9 (residues 221 to 231; a gift from Man Su Sy), the anti-prion polyclonal antibody 03R22 (residues 218 to 232), and anti-flotillin 1 (BD Biosciences). The secondary antibodies were horseradish peroxidase (HRP)-conjugated anti-mouse and anti-rabbit (GE Healthcare).
Generation of PrP constructs.
The mouse PrP coding sequence, MoPrP, and the rabbit PrP coding sequence (Codon Devices), RbPrP, were cloned into the pIRESpuro2 (Clontech) expression vector. The S230G/S231V double mutant was constructed by site-directed mutagenesis (Stratagene) using the following primers: primer 1, 5′-GACGGGAGAAGAGGCGTCAGCACCGTG-3′; and primer 2, 5′-CACGGTGCTGACGCCTCTTCTCCCGTC-3′. This construct was termed MoPrP-RbGPI. The S231V single mutant was constructed using the following primers: primer 1, 5′-GACGGGAGAAGATCCGTCAGCACCGTG-3′; and primer 2, 5′-CACGGTGCTGACGGATCTTCTCCCGTC-3′. This construct was termed MoPrPS231V.
Generation of cell lines.
Constructs were transfected using Lipofectamine 2000 (Invitrogen) into rabbit kidney epithelial (RK13) cells (41, 43). Cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal calf serum and 1 mM glutamine (Invitrogen) at 37°C in 5% CO2. Transfected cells were bulk selected and maintained using 2.5 μg/ml puromycin (Sigma-Aldrich).
Cell lysis and Western immunoblotting.
Cells were lysed in lysis buffer (0.5% deoxycholate, 0.5% Triton X-100, 150 mM NaCl, 50 mM Tris [pH 7.5]) and centrifuged at 16,100 × g to remove cell debris. Lysates were diluted in SDS sample buffer (125 mM Tris-HCl, pH 6.8, at 4% [wt/vol], 10% [vol/vol] glycerol, 0.02% [wt/vol] bromophenol blue) containing 10% 2-mercaptoethanol (BME) and boiled for 5 min at 100°C. Samples were electrophoresed using 4 to 12% Bis-Tris acrylamide gels (Invitrogen). Proteins were transferred to a polyvinylidene difluoride (PVDF) (Millipore) membrane and blocked for 1 h in 5% nonfat milk powder in PBST (phosphate-buffered saline [PBS] plus 0.5% Tween 20). Membranes were washed in PBST and incubated with the appropriate antibody diluted in PBST overnight at 4°C. Membranes were then washed four times over 0.5 h in PBST and probed with a secondary antibody diluted in PBST for 1 h at room temperature. The membranes were washed four times over 0.5 h in PBST, and specific binding was determined using enhanced chemiluminescence (ECL; GE Healthcare). PrP expression was quantified using Image J software.
Immunofluorescence.
Cells at 40% confluence were washed once with 1× PBS and then fixed with 3.2% paraformaldehyde at room temperature for 20 min. Fixed cells were washed three times with 1× PBS and then blocked with 10% goat serum and 2% bovine serum albumin (BSA) in PBS for 30 min at 37°C. Cells were labeled with the anti-PrP antibody ICSM-18 (1:250) for 45 min at 37°C and then incubated with Alexa 488-conjugated goat anti-mouse IgG (1:500) and DAPI (4′,6-diamidino-2-phenylindole; 1:1,000) for 45 min at 37°C. Samples were observed using a Leica DMIRE 2 confocal microscope with SP2 control software.
PI-PLC treatment of cell lines.
Cells at confluence in a six-well plate were incubated in Hanks buffered salt solution (HBSS) with or without 0.2 U phosphatidylinositol-specific phospholipase C (PI-PLC) at 37°C while shaking for 3 h. The supernatant was collected and spun down at 2,300 × g to remove any cell debris. The supernatant was collected, and the proteins were precipitated with trichloroacetic acid (TCA). Protein pellets were resuspended in 2× SDS sample buffer before SDS-PAGE and immunoblotting.
Sucrose density gradient centrifugation.
Cells at confluence in a 175-cm2 flask were scraped into PBS and pelleted by centrifugation at 600 × g for 3 min. Cells were resuspended in 1 ml of morpholineethanesulfonic acid (MES)-buffered saline (MBS; 25 mM MES, 150 mM NaCl, pH 6.5) containing 1% (vol/vol) Triton X-100 and were homogenized by 20 passages through an 18-gauge and a 24-gauge needle. The homogenate was mixed with 1 ml 80% (wt/vol) sucrose in MBS to make a 40% (wt/vol) sucrose solution and was loaded onto the bottom of a 5%-30% sucrose gradient. Gradients were centrifuged at 100,000 × g for 18 h in an SW30 rotor and separated into 12 1-ml fractions, with fraction 1 containing the lowest density of sucrose. PrP was concentrated from each fraction by TCA precipitation, and pellets were resuspended in PBS and analyzed by SDS-PAGE and immunoblotting.
Deglycosylation.
Cell lysates were deglycosylated by peptide N-glycosidase F (PNGase F) (Roche) treatment. Lysates were incubated with 10% (vol/vol) denaturing buffer (5% SDS, 10% BME) at 100°C for 10 min and then centrifuged at 16,100 × g for 1 min. The supernatant was collected, and deglycosylation was performed using 160 U/μl PNGase F in the presence of 10% (vol/vol) NP-40 and 50 mM sodium phosphate, pH 7.5, at 37°C overnight with shaking at 300 rpm.
Polarization of RK13 cells.
Cells were grown in a Transwell system (pore size, 0.4 μm; Nunc), and the electrical resistance (TER) was measured daily with a Millicell-ERS instrument (Millipore) until a stable TER (100 Ω·cm2) was reached (approximately 4 days).
Fluorescence labeling of polarized cells.
Polarized cells were washed once with 1× PBS and then fixed with 3.2% paraformaldehyde (wt/vol) at room temperature for 20 min. Following fixation, the cells were washed three times with 1× PBS, and then the membrane was cut out of the Transwell and placed into a single well of a 12-well dish (Nunc). Fixed cells were permeabilized with 0.2% (vol/vol) Triton X-100 for 2 min and then blocked with 2% (wt/vol) BSA in 1× PBS for 30 min at 37°C. The blocking reagent was removed, and cells were labeled with the anti-PrP antibody ICSM-18 (1:500) made up in blocking reagent for 1 h at 4°C. Cells were washed 10 times with 1× PBS and then incubated for 45 min at 4°C with Alexa 488-conjugated goat anti-mouse IgG (1:1,000), DAPI (1:1,000), and phalloidin (1:2,000), to detect filamentous actin, in blocking reagent. The cells were washed 10 times with 1× PBS, and then the membrane was mounted onto a glass slide (76 × 22 mm; Sail brand) with Mowiol mounting buffer (Calbiochem). Cells were visualized using a Leica DMI6000 microscope with LAS AF software, using a 63× objective.
Biotinylation of polarized cells.
Polarized cells were washed once with Dulbecco's PBS (DPBS) (Gibco, Invitrogen), and then either the apical or basolateral side of the cells was labeled with 6 mg/ml biotin [sulfosuccinimidyl 2-(biotinamido) ethyl-1,3′-dithiopropionale] (Thermo Scientific) diluted in 12.5 ml cold DPBS for 20 min on ice. Cells were then washed once with a quenching buffer (25 mM Tris, pH 8.0), followed by three DPBS washes. Cells were then incubated with lysis buffer (0.5% [wt/vol] deoxycholate, 0.5% [vol/vol] Triton X-100, 150 mM NaCl, 50 mM Tris [pH 7.5]) for 10 min on ice. Following centrifugation at 16,100 × g for 5 min, the lysates were incubated with 20 μl immobilized streptavidin (Pierce) for 2 h at 4°C on a spinning wheel. The beads were washed three times with DPBS and centrifuged at 4,000 rpm for 3 min. The final pellet was resuspended in 2× SDS sample buffer, and samples were analyzed by SDS-PAGE and immunoblotting.
Infection of cell lines.
Cells were plated in a six-well plate (Nunc) to ensure approximately 40% confluence at the time of infection. Seeded cells were incubated with 1% (wt/vol) homogenate prepared from the brain tissue of mice with terminal prion disease following infection with the M1000 or MU-02 strain of mouse-adapted human prion homogenate in PBS, as described previously (41). After 5 h, additional Optimem was added, and cells were incubated for 3 days. Infected cells were maintained in Optimem supplemented with 10% heat-inactivated fetal calf serum and 1 mM glutamine (Invitrogen).
Cell blotting immunoassay.
Cells were cultured on Thermanox plastic coverslips (ProSciTech; Nunc) until confluent, washed with PBS, and placed cell side up on lysis buffer-soaked filter paper. A nitrocellulose membrane presoaked in lysis buffer was pressed down on the cells and processed as described previously (22a). The membrane was incubated with the monoclonal antibody ICSM-18 (D-Gen) diluted in TBST (0.1% Tween 20, 100 mM NaCl, 10 mM Tris-HCl [pH 7.8]) and developed by ECL as described above.
RESULTS
The GPI anchor ω site of rabbit PrP is predicted to be Ser221.
Since small changes within the GPI anchor signal sequence are known to dramatically affect the anchoring of a protein, we sought to investigate whether RbPrP was predicted to be GPI anchored and, if so, to determine its predicted ω site by using the GPI modification site predictor DGPI (8). DGPI uses the C-terminal amino acid composition to predict the ω site of proteins. Using MoPrP numbering, DGPI revealed the most likely ω site of RbPrP to be at Ser221, with a second potential ω site at Ala229 (Fig. 1). The major site at Ser 221 in rabbit PrP is nine residues N-terminal to the predicted ω site of MoPrP. Despite the PrP amino acid sequence Ser221-Gln222-Ala223 being highly conserved among mammals, we found that none of the other mammals we investigated had a potential ω site of PrP at Ser221, with all other mammals investigated having a predicted ω site at either residue 230 or 231 (data not shown). This suggests that RbPrP may have a unique GPI anchor attachment site approximately nine residues N-terminal to that of PrPs from all other mammals. Upon processing and addition of the GPI anchor, RbPrP would lose the C-terminal residues 222 to 230 that typically remain after processing of PrPs from other mammals. The far C terminus of PrP has been implicated in playing a role in the conversion of PrPC to PrPSc (18, 19, 24, 44), which raises the possibility that RbPrP may lose a critical PrPSc interaction site, which may prevent its conversion to PrPSc.
FIG. 1.

The GPI anchor modification site predictor DGPI was used to predict whether a GPI anchor would attach to RbPrP and, if so, the site of attachment (ω site) (red). The numbering of the amino acid residues is according to the MoPrP sequence. The predicted ω site of MoPrP is at Ser230, whereas RbPrP contains a possible cleavage site at Ser221. DGPI was used to predict the ω site of MoPrP with substitutions of RbPrP-specific amino acids (*). Replacement of Ser230 and Ser231 of MoPrP with the rabbit-specific residues Gly230 and Val231 (MoPrPS230G/S231V) is predicted to shift the ω site of MoPrP to Ser221. The predicted ω site of MoPrPS230G/S231V models the potential ω site of RbPrP.
Alterations in the C terminus of RbPrP shift the predicted GPI anchor attachment site.
Since rabbits are resistant to prion infection, we used a mutant MoPrP model to observe whether the rabbit-specific amino acids at the C terminus are involved in the inability of RbPrPC to form RbPrPSc. To construct the MoPrP model, DGPI was used to determine the minimal amino acid changes needed to alter the predicted ω site of MoPrP so that it resembled the predicted ω site of RbPrP. The single MoPrP amino acid substitutions D226Q, G227R, R228A, R229A, S230G, and S231V were not predicted to alter the ω site of MoPrP, suggesting that multiple amino acid residues must be substituted to shift the predicted ω site N-terminally. Of all the sequential combinations, D226Q/G227R, G227R/R228A, R228A/R229A, R229A/S230G, and S230G/S231V, replacement of amino acids Ser230 and Ser231 of MoPrP with the RbPrP-specific amino acids, i.e., glycine and valine, respectively (MoPrPS230G/S231V), resulted in a shift of the predicted ω site N-terminally to resemble that observed with RbPrP (Fig. 1). Using MoPrP numbering, DGPI predicted MoPrPS230G/S231V to have a primary ω site at Ser221, with a second potential ω site at Gly230.
MoPrP-RbGPI-expressing cells are resistant to infection.
Using site-directed mutagenesis, the Ser230Gly and Ser231Val double MoPrP mutation was created, termed MoPrP-RbGPI, and stably transfected into RK13 cells. As controls, vector only and wild-type MoPrP and RbPrP plasmid DNAs were also stably transfected into wild-type RK13 cells. Expression of the transfected constructs was confirmed by lysing the cells and conducting an immunoblot assay (Fig. 2 A). MoPrP-RbGPI underwent glycosylation to produce un-, mono-, and diglycosylated forms of PrP, similar to MoPrP, indicating that these mutations did not affect the glycosylation of MoPrP-RbGPI.
FIG. 2.

The vector only, MoPrP, RbPrP, and MoPrP-RbGPI were transfected into RK13 cells. To determine expression, cell lysates were immunoblotted using the PrP-specific antibody ICSM-18 (A), and expression levels were quantified using Image J (n = 5) (B). (C) RK13 cells were incubated in the presence of 1% M1000-infected brain homogenate. After removal of the original inoculum, the cells were passaged for several weeks, and then a cell immunoblot was conducted to determine PrPres expression. MoPrP-expressing RK13 cells were susceptible to infection, whereas the RbPrP- or MoPrP-RbGPI-expressing RK13 cells were not susceptible to infection. (D) Nonpermeabilized and permeabilized vector-, MoPrP-, MoPrP-RbGPI-, and RbPrP-expressing RK13 cells were labeled with the PrP-specific antibody ICSM-18 (green) and with DAPI (blue) and were visualized using confocal microscopy.
To investigate whether the MoPrP-RbGPI-expressing cells were susceptible to infection, the cells were incubated with 1% (vol/vol) prion-infected mouse brain homogenate. At passage 12 postinfection, a cell blot assay was conducted to determine the presence of protease-resistant PrP (Fig. 2B). The wild-type MoPrP-expressing cells were infected, as demonstrated by the presence of protease-resistant PrP in the cell blot assay. Similar to the RbPrP-expressing cells, however, the MoPrP-RbGPI-expressing cells were not infected, as determined by the absence of protease-resistant PrP. This was confirmed using an animal bioassay in which MoPrP-overexpressing Tga20 mice (10) inoculated with MoPrP-RbGPI-expressing RK13 cell lysate did not show any signs of clinical disease for up to 244 days postinoculation (data not shown). This is in contrast to the case for Tga20 mice inoculated with material from infected MoPrP-expressing RK13 cells, which showed signs of prion disease at approximately 95 days postinoculation (see Fig. S2 in the supplemental material). The brains of mice inoculated with MoPrP-RbGPI-expressing RK13 cell lysates showed no spongiform change and, by immunohistochemistry, did not reveal PrP-positive plaques or astrocytosis (see Fig. S2 in the supplemental material).
To rule out the possibility that the resistance of RbPrP- and MoPrP-RbGPI-expressing RK13 cells to infection was due to the strain of prions used, we also incubated the RK13 cell lines with the MU-02 mouse-adapted human strain of prions. Consistent with the infections with M1000, only the RK13 cells expressing MoPrP were capable of being infected with MU-02 (see Fig. S3 in the supplemental material). Therefore, we concluded that RK-13 cells expressing MoPrP-RbGPI do not propagate infectious prions or generate protease-resistant PrP. This suggests that the rabbit-specific amino acids at the C terminus are involved in prion pathogenesis.
Since the single amino acid substitution mutant MoPrPS231V was not predicted to alter the ω site of MoPrP, its infectibility was determined to further investigate whether a possible shift in the ω site is responsible for the resistance of MoPrP-RbGPI-expressing cells to infection. After infection, MoPrPS231V-expressing RK13 cells were capable of being infected and propagating protease-resistant PrP (see Fig. S4 in the supplemental material), suggesting that the double mutation may alter the ω site of MoPrP and that this change is responsible for the resistance to infection.
The localization and anchoring of MoPrP-RbGPI and RbPrP are similar.
PrPC is predominately localized to the plasma membrane, where it is anchored via a GPI anchor (36). It is believed that PrPC must reach the plasma membrane before it can undergo conversion (4, 13, 21, 37). To investigate whether RbPrP and MoPrP-RbGPI are localized to the plasma membrane, vector- and PrP-expressing cells were fixed and stained with the PrP-specific antibody ICSM-18. Labeled cells were then visualized using confocal microscopy (Fig. 2C). Since the vector-expressing cells did not contain detectable levels of endogenous PrP, no PrP was detected using immunofluorescence. Similar to MoPrP, RbPrP and MoPrP-RbGPI were detected on the surfaces of nonpermeabilized cells, indicating that they are localized to the plasma membrane. After permeabilization, MoPrP, RbPrP, and MoPrP-RbGPI all exhibited predominant plasma membrane localization as well as intracellular localization, consistent with normal PrP trafficking. It was observed that MoPrP-RbGPI-expressing RK13 cells exhibited increased intracellular staining compared to MoPrP-expressing RK13 cells, reminiscent of misfolded PrP. We have shown, however, that MoPrP-RbGPI is localized at the cell surface, suggesting that it does undergo trafficking similar to that of MoPrP. Since plasma membrane localization of PrPC is required for cell infectivity and MoPrP-RbGPI and RbPrP are localized to the cell surface, we can conclude that the localization of RbPrP does not provide further insight into the resistance of rabbits to prion infection.
RbPrP is GPI anchored.
To determine if the double mutation had an effect on the GPI anchoring of MoPrP and whether the C-terminal residues of RbPrP affect its GPI anchoring, the PrP-expressing cells were treated with PI-PLC.
PI-PLC releases GPI-anchored proteins from the cell membrane and catalyzes the release of PrPC from cultured mammalian cells into the medium (36). PrP-expressing cells were incubated in the presence or absence of PI-PLC, and then the media and cell lysates were analyzed using immunoblotting (Fig. 3). After treatment of the PrP-expressing cells with PI-PLC, an increase in MoPrP, RbPrP, and MoPrP-RbGPI was observed in the media and a subsequent decrease in expression was observed in the cell lysates, consistent with PrP being released from the plasma membrane. This indicates that both RbPrP and MoPrP-RbGPI are GPI anchored and that the residues at the C terminus do not affect the anchoring of RbPrP to the cell surface with a GPI moiety.
FIG. 3.

MoPrP-, MoPrP-RbGPI-, and RbPrP-expressing RK13 cells were incubated in the absence (−) or presence (+) of PI-PLC. Cell lysates and media were immunoblotted with the antibody ICSM-18, which revealed that MoPrP, RbPrP, and MoPrP-RbGPI were all able to be released from the surfaces of RK13 cells, consistent with their containing a GPI anchor.
RbPrP is localized to lipid rafts.
PrP is localized to cholesterol-rich microdomains, known as lipid rafts, within the plasma membrane, which are believed to be implicated in prion infection (38, 42). We therefore investigated whether RbPrP and MoPrP-RbGPI were localized to lipid rafts at the plasma membrane. Due to the resistance of rafts to low concentrations of nonionic detergent, cells were lysed in Triton X-100 and then subjected to sucrose density gradient ultracentrifugation. After ultracentrifugation, rafts were shown to float at the 5%-30% sucrose interface due to their buoyancy associated with their high cholesterol content. Flotillin-1 is known to be enriched in lipid rafts and is therefore used as a marker of lipid raft purification. Immunoblotting of MoPrP fractions showed that MoPrP was localized to fractions 4 to 6 and fractions 10 to 12 (Fig. 4). Since flotillin-1 was localized to fractions 4 and 5, which contained the 5%-30% sucrose interface, it is evident that MoPrP is localized to lipid rafts. At the plasma membrane, PrP is believed to move out of lipid rafts prior to internalization. Therefore, localization within fractions 10 to 12 is consistent with internalized PrP that is present in the endocytic compartments. Similar to MoPrP, RbPrP and MoPrP-RbGPI were observed in fractions 4 and 5, indicating that they are also localized to lipid rafts (Fig. 4). The localization of both RbPrP and MoPrP-RbGPI within fractions 10 to 12 is also consistent with that of MoPrP. The distribution of RbPrP and MoPrP-RbGPI between lipid rafts and the endocytic compartments is similar to that of MoPrP and suggests that RbPrP and MoPrP-RbGPI are internalized at a similar rate to that for MoPrP and that they both follow similar trafficking pathways to that for MoPrP.
FIG. 4.

PrP-expressing RK13 cells were lysed in Triton X-100 and subjected to sucrose density gradient ultracentrifugation. Fractions were TCA precipitated and immunoblotted using the PrP-specific antibody ICSM-18 and anti-flotillin antibody. MoPrP, RbPrP, and MoPrP-RbGPI were all localized to flotillin-positive fractions, indicating that C-terminal changes do not affect sorting to lipid rafts.
The ω site of RbPrP is similar to that of MoPrP.
Due to the difficulty associated with experimentally determining the ω sites of GPI-anchored proteins and the absence of RbPrP-specific C-terminal antibodies to investigate whether RbPrP does have an ω site N-terminal to the ω site of MoPrP, we labeled MoPrP-RbGPI-expressing cell lysates with the monoclonal antibody 8F9 (MoPrP220-231) (29) and the polyclonal antibody 03R22 (MoPrP218-232) (Fig. 5). Following processing and addition of the GPI anchor, MoPrP-RbGPI is predicted to lose amino acids 222 to 230, which would normally remain during processing of MoPrP. Therefore, one would expect 03R22 and 8F9 to detect MoPrP but not MoPrP-RbGPI. For the purpose of this experiment, cell lysates were treated with PNGase F to deglycosylate PrP and simplify the banding patterns detected by 8F9 and 03R22. After PNGase F treatment, the N-glycans are removed and PrP is detected as a single band of approximately 28 kDa on an immunoblot. As shown in previous immunoblots, ICSM-18, which has an epitope at residues 143 to 153 of MoPrP, was capable of detecting MoPrP (Fig. 5B). As expected, both 03R22 and 8F9 were also capable of detecting MoPrP (Fig. 5B). The C-terminal fragment C1, which is produced by proteolytic cleavage between residues 110 and 111, was also detected by ICSM-18 and 03R22, at approximately 16 kDa. It is unclear why C1 was not detected by 8F9, but it may have been due to masking of this epitope, as previously reported (22). Immunoblotting of MoPrP-RbGPI with ICSM-18 and the C-terminal antibodies revealed that ICSM-18, as well as 03R22 and 8F9, detected MoPrP-RbGPI (Fig. 5B). Consistent with the case for MoPrP, ICSM-18 and 03R22 also detected the C1 fragment produced by MoPrP-RbGPI-expressing RK13 cells (Fig. 5B). Therefore, this suggests that MoPrP-RbGPI had not lost residues 222 to 230 during processing and addition of the GPI anchor and that the ω site of MoPrP-RbGPI is at Gly230, the second potential cleavage site predicted by DGPI. Since MoPrP-RbGPI was created to model RbPrP and contained RbPrP-specific residues at the two most critical sites within the GPI anchor signal sequence to determine the ω site, this suggests that the ω site of RbPrP is consistent with the ω site of PrPs from mammals capable of being infected with prions. Using MoPrP numbering, DGPI predicted a second potential cleavage site of RbPrP at Ala229, the equivalent of Ala231 in RbPrP (Fig. 5C). It is therefore likely that Ala231 is the ω site of RbPrP. Interestingly, this is the same amino acid that another GPI anchor predictor, Big-PI, predicted to be the most probable ω site of RbPrP.
FIG. 5.

(A) Antibody epitopes. The monoclonal MoPrP antibody ICSM-18 binds to amino acids 143 to 153. The monoclonal C-terminal antibody 8F9 binds to residues 220 to 231, and the polyclonal antibody 03R22 (22.2) binds to residues 218 to 232. (B) Glycosylated (−) and deglycosylated (+) vector-, MoPrP- and MoPrP-RbGPI-expressing RK13 cell lysates were immunoblotted with the antibodies ICSM-18, 8F9, and 22.2. (C) C-terminal antibody detection suggests that MoPrP-RbGPI is anchored at Ala229, the equivalent of Ala232 in RbPrP.
RbPrP is sorted to the apical side of polarized cells.
GPI-anchored proteins are sorted to either the apical or basolateral side of polarized cells, with the majority of GPI-anchored proteins being sorted to the apical side. This is observed for PrP in polarized RK13 cells, where it is sorted to the apical side of the cell (30). This is relevant to prion infection, as PrP-expressing RK13 cells were capable of being infected when the inoculum was added to the apical side of polarized cells but not when the inoculum was added to the basolateral side of polarized cells (30). Since RbPrP exhibits a difference in amino acid sequence at the GPI anchor attachment site compared to PrPs from other mammals, it is possible that this results in the differential sorting of RbPrP in polarized cells, thereby preventing cellular infection. Since we determined that the ω site of RbPrP is similar to that of MoPrP, we compared only the sorting of MoPrP and RbPrP in polarized RK13 cells. To investigate the sorting of RbPrP, PrP-expressing cells were polarized by growing the cells to confluence on porous filters. A stable electrical resistance reading across the epithelial monolayer (TER) of approximately 100 Ω·cm−2 indicated that tight junctions had formed between the cells and that the cells were polarized. Polarized cells were fixed, permeabilized, and labeled with the anti-PrP antibody ICSM-18 and the actin filament stain phalloidin. Confocal microscopy revealed that both MoPrP and RbPrP were predominately sorted to the apical side of polarized RK13 cells (Fig. 6 A). Polarized cells were also labeled with biotin, a non-membrane-permeating agent that has previously been shown to successfully label PrP at the cell surface (3). The apical or basolateral side of the polarized cells was labeled with biotin, and the cell lysates were incubated with streptavidin-coated beads to capture the biotin-labeled proteins. Immunoblotting of the captured proteins revealed that both MoPrP and RbPrP were localized predominately at the apical side of the cell (Fig. 6B). This indicates that the resistance of RbPrP-expressing cells to infection is not due to RbPrP undergoing an alternate sorting pathway.
FIG. 6.
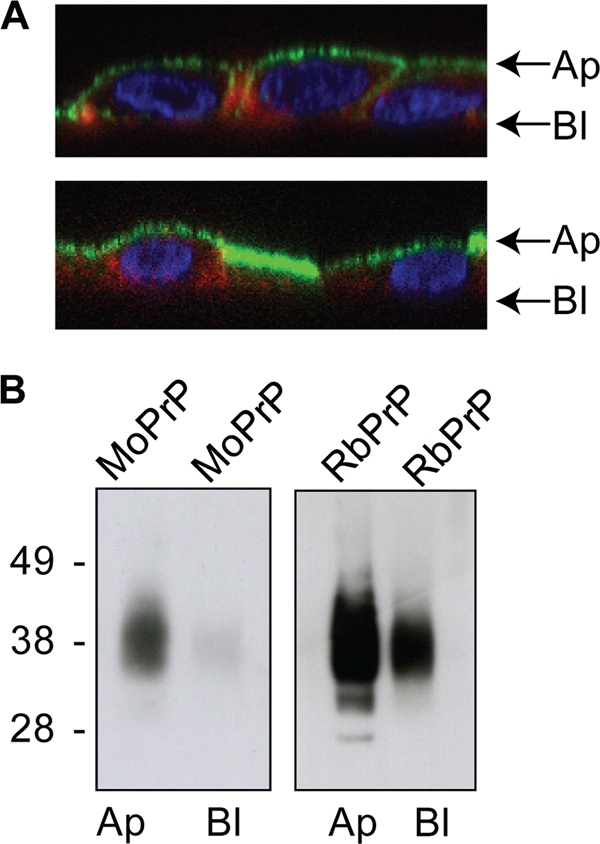
MoPrP- and RbPrP-expressing RK13 cells were polarized on Transwell membranes. (A) Polarized cells were fixed, permeabilized, and labeled with DAPI (blue), phalloidin (red), and the PrP-specific antibody ICSM-18 (green). Images taken are z sections through confocal images. In addition, the apical (Ap) or basolateral (Bl) side of the polarized cells was labeled with biotin. (B) Lysates were incubated with streptavidin and immunoblotted with the PrP-specific antibody SAF-23, which shows the enrichment of both MoPrP and RbPrP at the apical surface, with a minor fraction observed in the basolateral side of the cell.
DISCUSSION
Gaining insights into how rabbit-specific residues in PrP inhibit the formation of protease-resistant PrP will allow regions involved in the conversion process to be identified, which may aid in the development of future therapeutics that interfere with PrPSc formation. We therefore wanted to determine whether rabbit-specific amino acid residues at the putative ω site and immediately surrounding the ω site of RbPrP are involved in the resistance of RbPrP-expressing cells to PrP conversion. Previous studies identified the rabbit-specific residues Gly99 and Met108 within the N terminus, Ser173 in the central region, and Ile214 within the C terminus of RbPrP as residues capable of inhibiting PrPSc formation in scrapie-infected cells (45). Here we show that the combination of another two rabbit-specific amino acids, Gly230 and Val231, prevents infection in our RK13 cell model. A recent study by Hizume et al. showed that the single mutations in MoPrP230G and MoPrP231V had no effect on the generation of PrPres, suggesting that the double mutation is required to inhibit conversion (17). Since these residues occupy the ω and ω + 1 sites when incorporated into MoPrP, this implicates the ω site of PrP in the infection process.
The C-terminal amino acid sequence determines whether a protein is GPI anchored and, if so, the site at which the GPI anchor attaches. Furthermore, the presence of a GPI anchor determines the cellular sorting and the environment that the protein occupies at the plasma membrane. Since RbPrP exhibits a large amino acid sequence difference around the ω site of PrP compared with those from other mammalian species, this may have a significant impact on the GPI anchoring of RbPrP. Moreover, many pathogenic mutations within PrP have been shown to alter the localization and membrane topology of PrP expressed in cultured cells (14, 20, 40). In this study, we have shown that despite exhibiting a significant amino acid difference around the ω site compared to PrPs from other mammals, RbPrP is anchored to the plasma membrane through a GPI anchor and is localized to lipid rafts. The C-terminal residues of RbPrP do not affect the sorting of PrP to the apical surface of the cell. Moreover, rabbit-specific amino acids at the ω and ω + 1 sites of MoPrP do not shift the ω site of MoPrP N-terminally, suggesting that RbPrP has an ω site at alanine 231, similar to PrPs from other mammals. Therefore, the resistance of RbPrP to conversion is not likely due to its anchoring or cellular localization.
As well as being a GPI anchor determinant, the GPI anchor signal sequence can also affect the properties of GPI-anchored proteins. Studies of Saccharomyces cerevisiae indicate that the residues preceding ω are key determinants of whether the protein will be localized to the plasma membrane or the cell wall (11, 15, 16). In addition, studies with green fluorescent protein (GFP) showed that it was capable of oligomerizing when fused with the GPI anchor signal sequence of PrP but not when fused with the GPI anchor signal sequence of the folate receptor (28). Since the GPI anchor signal sequence is removed upon addition of the GPI anchor, it is thought that the signal sequence promotes the addition of specific anchors which may be involved in the function of the protein to which they are attached. Therefore, the unique amino acids at the ω site and C-terminal of the ω site of RbPrP may result in the attachment of a modified GPI anchor that does not support the propagation of prions.
GPI anchors contain a conserved core of ethanolamine phosphate in an amide linkage to the C terminus of the protein, three mannose residues, glucosamine, and phosphatidylinositol (2, 27). When the protein migrates to the plasma membrane, the GPI anchors are modified so that many variations on this core structure arise. The GPI anchor of PrPC isolated from hamster brains contains large amounts of galactose, mannose, and sialic acid (35). Interestingly, the PrP GPI anchor is one of the only GPI anchors known to contain sialic acid. The role of sialic acid in prion infection remains to be investigated. There is evidence to suggest, however, that modifications specific to the GPI anchor of PrP support prion infection and pathogenesis. The localization of PrP to lipid rafts seems to be critical for prion infection. However, Thy-1, a GPI-anchored protein also localized to lipid rafts, occupies slightly different domains on the plasma membrane compared to those occupied by PrP (5). In addition, the GPI anchor of PrP, but not the GPI anchor of Thy-1, can activate phospholipase 2, a protein involved in apoptosis (2). Therefore, the difference in amino acid sequence around the ω site may cause the GPI anchor of RbPrP to be modified so that it does not support prion propagation.
The GPI anchor can also influence the conformation and structure of the protein to which it is attached. The OX7 antibody that recognizes Thy-1 fails to bind to Thy-1 after treatment with PI-PLC (1), and the circular dichroism spectrum of GPI-anchored human Thy-1 differs from that of soluble Thy-1 (1, 31). If the GPI anchor of RbPrP is modified compared to the GPI anchor of PrPs from other mammals, this may influence the overall structure of RbPrP, rendering it more stable and less susceptible to conversion. Future studies involving the purification and characterization of the RbPrP GPI anchor are therefore essential and will not only provide further insight into the role of the GPI anchor in prion diseases but also enhance our knowledge of the structure and function of GPI anchors.
Alternatively, the amino acids at the ω site and N-terminal of the ω site of RbPrP may disrupt an interaction site between PrPC and PrPSc. RbPrP is the only PrP that is expected to have a GPI anchor attached to an alanine, and the alanine at the ω − 1 site is not observed in PrP from any other mammal. Alanine is a small amino acid that could be hidden in the folded protein and prevent an interaction with PrPSc. Several studies have implicated the C terminus in the infection process. Quinacrine, an acridine analogue that inhibits PrPSc formation in cultured cells (7, 23), binds to recombinant human PrP at residues Tyr225, Tyr226, and Gln227 (44). The monoclonal anti-PrP antibody anti-219-232 inhibited the binding of PrPsen to PrPres when bound to PrPsen in vitro (18). The amino acids 218 and 214 are critical in the interaction with the putative protein X, a molecule other than PrPC that is postulated to be involved in PrPSc formation (19). The human PrP226-237 peptide can bind to PrPSc in infected brain homogenates (24), indicating that the far-C-terminal residues of mature RbPrP could potentially inhibit the interaction of PrPC with PrPSc. This would prevent its conversion and thereby render rabbits resistant to prion infection.
This study has indicated that careful sequence analysis can identify potentially important amino acids and motifs. Sequence comparison of all known PrP sequences revealed that the brush-tailed possum is the only other species which exhibits a difference in amino acid sequence around the GPI anchor attachment site. Since the brush-tailed possum has never been infected experimentally with prions, we cannot comment on its susceptibility to infection. It would be useful to determine if the brush-tailed possum, along with the rabbit, is resistant to prion infection, which would further implicate the C terminus of PrP in prion propagation.
Supplementary Material
Acknowledgments
We thank Charles Weissmann for the gift of Tga20 mice, John Collinge for the gift of monoclonal antibody ICSM-18, and Man Sun Sy for monoclonal antibody 8F9. We thank the animal facility staff of the Department of Pathology for their technical assistance.
This work was supported by NHMRC program grant 400202. R.M.N. is the recipient of a University of Melbourne research scholarship, C.F.H. is the recipient of an Australian postgraduate award scholarship, A.F.H. is the recipient of an NHMRC career development award (level 2), R.C. is the recipient of an NHMRC senior research fellowship, and V.A.L. is the recipient of the University of Melbourne C. R. Roper Fellowship.
Footnotes
Published ahead of print on 28 April 2010.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Barboni, E., B. P. Rivero, A. J. George, S. R. Martin, D. V. Renoup, E. F. Hounsell, P. C. Barber, and R. J. Morris. 1995. The glycophosphatidylinositol anchor affects the conformation of Thy-1 protein. J. Cell Sci. 108:487-497. [DOI] [PubMed] [Google Scholar]
- 2.Bate, C., S. Reid, and A. Williams. 2004. Phospholipase A2 inhibitors or platelet-activating factor antagonists prevent prion replication. J. Biol. Chem. 279:36405-36411. [DOI] [PubMed] [Google Scholar]
- 3.Borchelt, D. R., M. Scott, A. Taraboulos, N. Stahl, and S. B. Prusiner. 1990. Scrapie and cellular prion proteins differ in their kinetics of synthesis and topology in cultured cells. J. Cell Biol. 110:743-752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borchelt, D. R., A. Taraboulos, and S. B. Prusiner. 1992. Evidence for synthesis of scrapie prion proteins in the endocytic pathway. J. Biol. Chem. 267:16188-16199. [PubMed] [Google Scholar]
- 5.Brugger, B., C. Graham, I. Leibrecht, E. Mombelli, A. Jen, F. Wieland, and R. Morris. 2004. The membrane domains occupied by glycosylphosphatidylinositol-anchored prion protein and Thy-1 differ in lipid composition. J. Biol. Chem. 279:7530-7536. [DOI] [PubMed] [Google Scholar]
- 6.Courageot, M. P., N. Daude, R. Nonno, S. Paquet, M. A. Di Bari, A. Le Dur, J. Chapuis, A. F. Hill, U. Agrimi, H. Laude, and D. Vilette. 2008. A cell line infectible by prion strains from different species. J. Gen. Virol. 89:341-347. [DOI] [PubMed] [Google Scholar]
- 7.Doh-Ura, K., T. Iwaki, and B. Caughey. 2000. Lysosomotropic agents and cysteine protease inhibitors inhibit scrapie-associated prion protein accumulation. J. Virol. 74:4894-4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eisenhaber, B., P. Bork, and F. Eisenhaber. 1999. Prediction of potential GPI-modification sites in proprotein sequences. J. Mol. Biol. 292:741-758. [DOI] [PubMed] [Google Scholar]
- 9.Eisenhaber, B., P. Bork, and F. Eisenhaber. 1998. Sequence properties of GPI-anchored proteins near the omega-site: constraints for the polypeptide binding site of the putative transamidase. Protein Eng. 11:1155-1161. [DOI] [PubMed] [Google Scholar]
- 10.Fischer, M., T. Rulicke, A. Raeber, A. Sailer, M. Moser, B. Oesch, S. Brandner, A. Aguzzi, and C. Weissmann. 1996. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J. 15:1255-1264. [PMC free article] [PubMed] [Google Scholar]
- 11.Frieman, M. B., and B. P. Cormack. 2003. The omega-site sequence of glycosylphosphatidylinositol-anchored proteins in Saccharomyces cerevisiae can determine distribution between the membrane and the cell wall. Mol. Microbiol. 50:883-896. [DOI] [PubMed] [Google Scholar]
- 12.Gibbs, C. J., Jr., and D. C. Gajdusek. 1973. Experimental subacute spongiform virus encephalopathies in primates and other laboratory animals. Science 182:67-68. [DOI] [PubMed] [Google Scholar]
- 13.Gilch, S., K. F. Winklhofer, M. H. Groschup, M. Nunziante, R. Lucassen, C. Spielhaupter, W. Muranyi, D. Riesner, J. Tatzelt, and H. M. Schatzl. 2001. Intracellular re-routing of prion protein prevents propagation of PrP(Sc) and delays onset of prion disease. EMBO J. 20:3957-3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gu, Y., A. Singh, S. Bose, and N. Singh. 2008. Pathogenic mutations in the glycosylphosphatidylinositol signal peptide of PrP modulate its topology in neuroblastoma cells. Mol. Cell. Neurosci. 37:647-656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hamada, K., H. Terashima, M. Arisawa, and K. Kitada. 1998. Amino acid sequence requirement for efficient incorporation of glycosylphosphatidylinositol-associated proteins into the cell wall of Saccharomyces cerevisiae. J. Biol. Chem. 273:26946-26953. [DOI] [PubMed] [Google Scholar]
- 16.Hamada, K., H. Terashima, M. Arisawa, N. Yabuki, and K. Kitada. 1999. Amino acid residues in the omega-minus region participate in cellular localization of yeast glycosylphosphatidylinositol-attached proteins. J. Bacteriol. 181:3886-3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hizume, M., A. Kobayashi, H. Mizusawa, and T. Kitamoto. 2009. Amino acid conditions near the GPI anchor attachment site of prion protein for the conversion and the GPI anchoring. Biochem. Biophys. Res. Commun. 391:1681-1686. [DOI] [PubMed] [Google Scholar]
- 18.Horiuchi, M., and B. Caughey. 1999. Specific binding of normal prion protein to the scrapie form via a localized domain initiates its conversion to the protease-resistant state. EMBO J. 18:3193-3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaneko, K., M. Vey, M. Scott, S. Pilkuhn, F. E. Cohen, and S. B. Prusiner. 1997. COOH-terminal sequence of the cellular prion protein directs subcellular trafficking and controls conversion into the scrapie isoform. Proc. Natl. Acad. Sci. U. S. A. 94:2333-2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kiachopoulos, S., A. Bracher, K. F. Winklhofer, and J. Tatzelt. 2005. Pathogenic mutations located in the hydrophobic core of the prion protein interfere with folding and attachment of the glycosylphosphatidylinositol anchor. J. Biol. Chem. 280:9320-9329. [DOI] [PubMed] [Google Scholar]
- 21.Kim, C. L., A. Karino, N. Ishiguro, M. Shinagawa, M. Sato, and M. Horiuchi. 2004. Cell-surface retention of PrPC by anti-PrP antibody prevents protease-resistant PrP formation. J. Gen. Virol. 85:3473-3482. [DOI] [PubMed] [Google Scholar]
- 22.Klingeborn, M., L. Wik, M. Simonsson, L. H. Renstrom, T. Ottinger, and T. Linne. 2006. Characterization of proteinase K-resistant N- and C-terminally truncated PrP in Nor98 atypical scrapie. J. Gen. Virol. 87:1751-1760. [DOI] [PubMed] [Google Scholar]
- 22a.Klöhn, P. C., L. Stoltze, E. Flechsig, M. Enari, and C. Weissmann. 2003. A quantitative, highly sensitive cell-based infectivity assay for mouse scrapie prions. Proc. Natl. Acad. Sci. U. S. A. 100:11666-11671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Korth, C., B. C. May, F. E. Cohen, and S. B. Prusiner. 2001. Acridine and phenothiazine derivatives as pharmacotherapeutics for prion disease. Proc. Natl. Acad. Sci. U. S. A. 98:9836-9841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lau, A. L., A. Y. Yam, M. M. Michelitsch, X. Wang, C. Gao, R. J. Goodson, R. Shimizu, G. Timoteo, J. Hall, A. Medina-Selby, D. Coit, C. McCoin, B. Phelps, P. Wu, C. Hu, D. Chien, and D. Peretz. 2007. Characterization of prion protein (PrP)-derived peptides that discriminate full-length PrPSc from PrPC. Proc. Natl. Acad. Sci. U. S. A. 104:11551-11556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lawson, V. A., L. J. Vella, J. D. Stewart, R. A. Sharples, H. Klemm, D. M. Machalek, C. L. Masters, R. Cappai, S. J. Collins, and A. F. Hill. 2008. Mouse-adapted sporadic human Creutzfeldt-Jakob disease prions propagate in cell culture. Int. J. Biochem. Cell Biol. 40:2793-2801. [DOI] [PubMed] [Google Scholar]
- 26.Loftus, B., and M. Rogers. 1997. Characterization of a prion protein (PrP) gene from rabbit—a species with apparent resistance to infection by prions. Gene 184:215-219. [DOI] [PubMed] [Google Scholar]
- 27.Mayor, S., and H. Riezman. 2004. Sorting GPI-anchored proteins. Nat. Rev. Mol. Cell. Biol. 5:110-120. [DOI] [PubMed] [Google Scholar]
- 28.Paladino, S., S. Lebreton, S. Tivodar, V. Campana, R. Tempre, and C. Zurzolo. 2008. Different GPI-attachment signals affect the oligomerisation of GPI-anchored proteins and their apical sorting. J. Cell Sci. 121:4001-4007. [DOI] [PubMed] [Google Scholar]
- 29.Pan, T., R. Li, S. C. Kang, B. S. Wong, T. Wisniewski, and M. S. Sy. 2004. Epitope scanning reveals gain and loss of strain specific antibody binding epitopes associated with the conversion of normal cellular prion to scrapie prion. J. Neurochem. 90:1205-1217. [DOI] [PubMed] [Google Scholar]
- 30.Paquet, S., E. Sabuncu, J. L. Delaunay, H. Laude, and D. Vilette. 2004. Prion infection of epithelial Rov cells is a polarized event. J. Virol. 78:7148-7152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paulick, M. G., and C. R. Bertozzi. 2008. The glycosylphosphatidylinositol anchor: a complex membrane-anchoring structure for proteins. Biochemistry 47:6991-7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pierleoni, A., P. L. Martelli, and R. Casadio. 2008. PredGPI: a GPI-anchor predictor. BMC Bioinformatics 9:392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prusiner, S. B. 1982. Novel proteinaceous infectious particles cause scrapie. Science 216:136-144. [DOI] [PubMed] [Google Scholar]
- 34.Stahl, N., M. A. Baldwin, A. L. Burlingame, and S. B. Prusiner. 1990. Identification of glycoinositol phospholipid linked and truncated forms of the scrapie prion protein. Biochemistry 29:8879-8884. [DOI] [PubMed] [Google Scholar]
- 35.Stahl, N., M. A. Baldwin, R. Hecker, K. Pan, A. L. Burlingame, and S. B. Prusiner. 1992. Glycosylinositol phospholipid anchors of the scrapie and cellular prion protein contain sialic acid. Biochemistry 31:5043-5053. [DOI] [PubMed] [Google Scholar]
- 36.Stahl, N., D. R. Borchelt, K. Hsiao, and S. B. Prusiner. 1987. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 51:229-240. [DOI] [PubMed] [Google Scholar]
- 37.Taraboulos, A., A. J. Raeber, D. R. Borchelt, D. Serban, and S. B. Prusiner. 1992. Synthesis and trafficking of prion proteins in cultured cells. Mol. Biol. Cell 3:851-863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taraboulos, A., M. Scott, A. Semenov, D. Avraham, L. Laszlo, and S. B. Prusiner. 1995. Cholesterol depletion and modification of COOH-terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J. Cell Biol. 129:121-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Udenfriend, S., and K. Kodukula. 1995. How glycosylphosphatidylinositol-anchored membrane proteins are made. Annu. Rev. Biochem. 64:563-591. [DOI] [PubMed] [Google Scholar]
- 40.Uelhoff, A., J. Tatzelt, A. Aguzzi, K. F. Winklhofer, and C. Haass. 2005. A pathogenic PrP mutation and doppel interfere with polarized sorting of the prion protein. J. Biol. Chem. 280:5137-5140. [DOI] [PubMed] [Google Scholar]
- 41.Vella, L. J., R. A. Sharples, V. A. Lawson, C. L. Masters, R. Cappai, and A. F. Hill. 2007. Packaging of prions into exosomes is associated with a novel pathway of PrP processing. J. Pathol. 211:582-590. [DOI] [PubMed] [Google Scholar]
- 42.Vey, M., S. Pilkuhn, H. Wille, R. Nixon, S. J. DeArmond, E. J. Smart, R. G. Anderson, A. Taraboulos, and S. B. Prusiner. 1996. Subcellular colocalization of the cellular and scrapie prion proteins in caveolae-like membranous domains. Proc. Natl. Acad. Sci. U. S. A. 93:14945-14949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vilette, D., O. Andreoletti, F. Archer, M. F. Madelaine, J. L. Vilotte, S. Lehmann, and H. Laude. 2001. Ex vivo propagation of infectious sheep scrapie agent in heterologous epithelial cells expressing ovine prion protein. Proc. Natl. Acad. Sci. U. S. A. 98:4055-4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vogtherr, M., S. Grimme, B. Elshorst, D. M. Jacobs, K. Fiebig, C. Griesinger, and R. Zahn. 2003. Antimalarial drug quinacrine binds to C-terminal helix of cellular prion protein. J. Med. Chem. 46:3563-3564. [DOI] [PubMed] [Google Scholar]
- 45.Vorberg, I., M. H. Groschup, E. Pfaff, and S. A. Priola. 2003. Multiple amino acid residues within the rabbit prion protein inhibit formation of its abnormal isoform. J. Virol. 77:2003-2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.