

Abstract
As one of the most widespread drugs of abuse, nicotine has long been known to impact the brain, particularly with respect to addiction. However, the regional effects of nicotine on the concentrations and kinetics of amino acid neurotransmitters and some energetically related neurochemicals have been little studied. In this investigation, acute effects of nicotine were measured by 1H-observed/13C-edited nuclear magnetic resonance spectroscopy method in extracts obtained from nicotine-naïve, freely moving rats given 0.7 mg/kg nicotine or saline, with [1-13C] glucose to track metabolism. Nicotine was observed to exert significant effects on the concentrations of N-acetylaspartate (NAA), and γ-aminobutyric acid (GABA), particularly in the striatum. Nicotine decreased brain glucose oxidation, glutamate-glutamine neurotransmitter cycling, and GABA synthesis regionally, including in the parietal and occipital cortices and the striatum. The olfactory bulb showed kinetics that differed markedly from those observed in the rest of the brain. Independently of nicotine, the concentration of glutamate was found to be correlated significantly with levels of NAA and GABA, suggesting a potential interplay of energetics and excitatory and inhibitory neurotransmission. In summary, the study revealed that the neurochemicals were most affected in the cortex and striatum of the rat brain after acute nicotine treatment.
Keywords: Nicotine, Brain, Carbon-13, Magnetic resonance spectroscopy, Linear discriminant analysis
Introduction
It has long been known that cigarette smoking and other forms of tobacco consumption are addicting, and that addiction is at least in part caused by nicotine (Benowitz 1999). Nicotine, believed to be the substance primarily responsible for addiction to tobacco smoking, is a simple molecule that affects many parts of the brain through binding sites on nicotinic acetylcholine receptors (nAChRs). When acetylcholinergic neurons release acetylcholine, it binds to muscarinic and nicotinic acetylcholine receptors and generally stimulates the release of glutamate and GABA. Nicotine modifies the behavior of the nAChRs and produces many behavioral and physiological effects, including influences on learning, memory, analgesia, anxiety, and seizures (Stapleton et al. 2003, Matta et al. 2007). The acute effects of nicotine have been investigated by imaging methods, including as positron emission tomography (PET) (Stapleton et al. 2003, Nagata et al. 1995), and autoradiography (Weissman et al. 1987, London et al. 1988, London et al. 1990, Marenco et al. 2000).
Many drugs of abuse decrease regional cerebral metabolic rates of glucose (CMRgl) in human subjects chronically (Weissman et al. 1987, London et al. 1990) and acutely; nicotine might, as well, as reported previously (Marenco et al. 2000, Stapleton et al. 2003), but nicotine is known to stimulate glutamatergic and dopaminergic activity and so might increase CMRgl as reported previously (Weissman et al. 1987, London et al. 1988, London et al. 1990). To investigate this and several other neurochemicals aspects of nicotine, we implemented a model to study effects of acute nicotine in awake, freely moving rats.
The purpose of the present study was to evaluate the effect of nicotine on regional concentrations and rates of turnover of glutamate, glutamine, and GABA and other metabolites in the rat brain, in addition to the determination of regional rates of glucose oxidation. Nuclear magnetic resonance spectroscopy (MRS) was used to quantify a number of compounds in vivo and in vitro (de Graaf 2007). We hypothesized that acute nicotine would stimulate glutamate release, thereby increasing glutamate-glutamine neurotransmitter cycling and glucose oxidation, and that the increased glutamate turnover and nicotine together would stimulate GABA synthesis, likely in a region-specific manner. We also hypothesized that GABA levels would rise in the occipital cortex, according to a small study in humans (Mason et al. 2007) and, potentially, in the striatum if synthesis rose, which might occur as the circuits attempt to balance stimulated glutamate release.
Materials and Methods
Animal preparation
All experiments were carried out in accordance with protocols approved by the Yale Animal Care and Use Committee. Male Sprague-Dawley rats (186.8 ± 4.0g, mean ± SD) were maintained on a 12h/12h light-dark cycle, and food and water were available ad libitum. To accustom the rats to human interaction and minimize stress on the day of the experiment, they were handled and weighed twice daily for one week. In this study metabolic kinetics were assessed with infusions of [1-13C]glucose, and that approach yields greater sensitivity when the fractional enrichment of the glucose is higher in the blood. Therefore, to lower the endogenous, unlabeled glucose level, the rats were fasted 17–19 hours overnight before the experiment. Rats were anesthetized with 1.5–2.5% isoflurane mixed with 30% O2 and 70% N2O; an adequate level of anesthesia was verified by lack of withdrawal response to a foot pinch. One sample of 50 μL of blood was collected from one lateral tail vein, and the other lateral tail vein was catheterized with PE50 tubing for the infusion of [1-13C]glucose. After catheterization, the rat recovered, and within 10 minutes was moving normally and grooming, with the catheterization site sealed with labeling tape. The infusion line was passed through flexible plastic tubing and suspended from the center of the cage cover to avoid entanglement of the line with the rat. At this point each rat was housed individually to avoid entanglement of multiple lines and to avoid the risk of chewing each other’s catheterization sites and lines, and allowed to recover for one hour. The rats were divided into two groups: saline control (S) and acute nicotine-treated (N). Nicotine tartrate (Sigma N-5260) was dissolved in saline to achieve a concentration of 0.7 mg/mL of nicotine. After the rats recovered, a volume of solution calculated to contain 7 mg/kg nicotine or an equivalent volume of saline was injected subcutaneously. Ten minutes after administration of the solution, [1-13C]glucose was infused (Fitzpatrick et al. 1990) through the catheterized lateral tail vein while the rat was moving freely around the cage. Rats were sacrificed in a different room by focused microwave irradiation using the commercially available 10 kW Muromachi Microwave Fixation System (Stoelting Co, Wood Dale, IL, USA) (10kW, 1.25sec) in accordance with guidelines established by the Yale Animal Care and Use Committee, at t=0, 10, 20, 60 and 120 minutes after the start of the glucose infusion. Four animals were measured per time point, except for the olfactory bulb, in which three animals were studied per time point. For the measurement at t = 0, the animal was prepared the same as the others, but no glucose infusion was performed. After microwave irradiation, the brain, now approximately the texture of a boiled egg, was removed and dissected into 12 regions: olfactory bulb (OB), cerebellum (CE); medulla (MEA), midbrain (MID), thalamus (THA), hypothalamus (HYP), hippocampus (HP), striatum (ST), frontal cortex (FC), occipital cortex (OC), parietal cortex (PC), and temporal cortex (TC). A second blood sample of 1 mL was drawn from the caudal vena cava.
One measurement in a nicotine-treated rat failed due to very low glucose enrichment, caused by failure of the tail vein catheterization, so the data from that rat were not used. Therefore, experiments were successfully completed for 20 rats with saline and 19 rats with nicotine.
Preparation of plasma and tissue extracts
Blood samples were treated with centrifugation at 10000g for 1min immediately after collection, and 30–40 μL plasma was used to measure the plasma glucose concentration, with the rest of the plasma frozen for subsequent measurement of [1-13C] glucose enrichment.
The brain samples were ground with 0.1M HCl/Methanol (2:1 vol/wt) cooled with dry ice and then transferred to 900 μl of 90% ice-cold ethanol and ground until pulverized (Patel et al. 2005, Patel et al. 2001). Then the mixture was left for 15 minutes on wet ice and diluted with 450 μl deionized water. The homogenate underwent centrifugation at 15000g for 30min at 4°C, was filtered with a 0.2μm Acrodisc syringe filter (Gelman sciences, Ann Arbor, MI, USA), and passed through a 1-mL bed volume column of chelex-100 resin (200–400 mesh, sodium form, pH=7.0, Bio-Rad Laboratories, Hercules, CA, USA). The sample was neutralized, lyophilized, and dissolved in a D2O buffer (0.4ml D2O with 0.2ml buffer (K2HPO4 and KH2PO4, pH=7.0)) for 1H NMR analysis.
Plasma and brain tissue extract analysis
Plasma glucose concentrations were determined using a Beckman Glucose Analyzer II (Beckman Instruments, Fullerton, CA, USA). The 13C enrichment of plasma glucose and metabolites were measured using 1H-[13C] NMR (Fitzpatrick et al. 1990) at 11.75T on a Bruker Avance vertical bore spectrometer. Briefly, the heteronuclear editing method consists of the acquisition of two spin-echo measurements, one with a broad-banded inversion pulse applied at the 13C frequency, and the other without the inversion pulse. The measurement without the inversion pulse yields the total metabolite concentrations, and the difference between the two yields only the 13C-labeled components of the spectra. An echo time of 8 ms, repetition time of 20s, sweep width of 12ppm, and 16k acquisition data points were used.
The spectra were processed with 64k zero filling and 1Hz line broadening. The quantification of total and 13C-labeled metabolite levels was achieved by integration of the resonances of the sum and difference spectra using XWINNMR software (Bruker Instruments, Billerica, MA). The metabolites and carbon positions measured were aspartate C3, glutamate C4, glutamine C4, GABA C2, and the combined glutamate and glutamine C3 labeling. Also measured were the combined concentrations of the methyl groups of NAA and N-acetylaspartyl glutamate (NAA), as well as the sum of creatine and phosphocreatine (Cr). 13C enrichments were normalized by the fractional [1-13C] enrichment of glucose in the brain, which was designed to rise within seconds to a value that remained constant over time (Fitzpatrick et al. 1990)and whose level was confirmed to be constant after the start of the infusion.
Metabolic modeling of 13C time-course data
The metabolic rates were calculated by fitting the three-compartment metabolic model (astrocytes, glutamatergic neurons, and GABAergic neurons) depicted in Fig. 1 to the time courses of 13C enrichment of glutamate C4, glutamine C4, glutamate + glutamine C3, and GABA C2 obtained during the infusions of [1-13C] glucose. CWave software (Mason et al. 2003) was used to simulate the metabolic model as a series of differential equations that represented mass balance and 13C isotope balance in each metabolite pool (Patel et al. 2005). The equations, which are shown in Table 1 of the supplemental material, were solved using a first-/second-order Runge-Kutta algorithm, and the fitting was done with a Levenburg-Marquardt algorithm hybridized with simulated annealing (Alcolea et al. 1999).
Fig. 1.

Schematic diagram of the labeling of cerebral metabolites from [1-13C] glucose. Whole filed circle: 100% 13C enrichment in this position; Half filled circle: 50% 13C enrichment in this position; Red: labeled position after the first TCA circle, Blue: labeled position of the second TCA circle. The number “1” denotes the position of Carbon 1 in each molecule.
Statistical Analysis
First, the metabolite levels were evaluated using MANOVA with a Holm- Bonferroni correction for multiple comparisons (Holm 1979), testing for effects of group (nicotine or saline) and time after nicotine or glucose on metabolite levels. Time was found to have an insignificant effect except for a small extent on striatal GABA and was ignored for all metabolites and regions except for striatal GABA. ANOVA was then performed considering only group and not time.
The primary hypotheses, that GABA would increase in the occipital cortex and the striatum (ST), and that metabolic rates would rise in response to nicotine in the same regions, were treated individually, using a Bonferroni adjustment of 2 in each case to account for the two brain regions that corresponded to the primary hypotheses. The remaining metabolites and associated regions were evaluated as one group, and the remaining regional metabolic rates were evaluated as another group, in each case using the Holm-Bonferroni method (Holm 1979) to account for multiple comparisons.
To further explore the effects of nicotine on the brain, considering all measurements in all regions, linear discriminant analysis (LDA) was used. Because fewer samples were obtained from the olfactory bulb, measurements from that region were not included in the LDA. Therefore, the LDA evaluated 66 descriptors (11 regions × 6 metabolites. The LDA classification was done with all 39 successfully completed rats. The predicted ability of the linear discriminant analysis (LDA) model was estimated by the Leave One Out (LOO) method and by the predictability of the test set.
Results
Within one minute of the subcutaneous injection of nicotine, the rats began to stumble, losing coordination. The effect disappeared by 8 to 10 minutes, just before the infusion of glucose began, and the rats were recovered by 10 minutes, though still appearing a little weak.
After each rat was sacrificed, the brain was removed and dissected into 12 different regions. Most of the regions were completely separated (Supplemental Table 2S), with variability of tissue mass below 15%, except for the FC (15.1%), ST (20.5%) and HYP (23.7%). The greater variability of the FC and ST were caused by physical operation. For the first half of the animals, both nicotine- and saline-treated, the brains were frozen for subsequent dissection and processing. However, once the brains were frozen and thawed, the FC and ST were stuck together, making it difficult to separate them completely. The rest of the brains were therefore dissected just after sacrifice. To ensure the purity of the ST and FC, slightly less mass was collected for those two regions. Thus their variability comprised higher percentage than for the other regions. The largest percentage variability was seen in the HYP, because it had the least mass of any brain region collected, and also because of its tight connection with thalamus. Thus, we can say the rat brain was completely separated into 12 different regions.
Effects of Nicotine on Regional Metabolite Levels
Table 1 shows the concentration of the metabolites. The MANOVA analysis showed that the effect of time was insignificant except for on striatal GABA, which rose from 1.86 to 2.10 μmol/g over the two-hour period after injection of nicotine (Supplemental Fig. 1S). Therefore, time was dropped as a covariant and the concentration measurements for all metabolites and regions were evaluated considering only group, with the exception of striatal GABA. For the consideration of multiple comparisons, using the primary hypothesis that GABA would be affected, GABA was treated separately, and the other metabolites were evaluated together.
Table 1.
Regional concentrations of metabolites in the rat brain, averaged across all animals and all time points.
| GABA | Glutamine | Glutamate | NAA | Creatine | Asp | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Brain | Saline | Nicotine | Saline | Nicotine | Saline | Nicotine | Saline | Nicotine | Saline | Nicotine | Saline | Nicotine |
| CE | 1.15±0.17 | 1.12±0.09 | 6.17±0.56 | 6.29±0.53 | 8.12±0.62 | 7.98±0.58 | 6.00±0.58 | 5.84±0.64 | 12.19±0.85 | 12.11±0.63 | 2.11±0.27 | 2.14±0.22 |
| FC | 1.48±0.17 | 1.50±0.14 | 6.84±0.74 | 6.69±0.47 | 10.85±0.80 | 10.47±0.65 | 7.65±0.87 | 7.03±0.52* | 9.57±0.87 | 9.51±0.61 | 2.94±0.30 | 3.11±0.60 |
| TC | 1.56±0.31 | 1.65±0.22 | 6.40±0.80 | 6.72±0.90 | 9.89±1.07 | 10.27±0.77 | 6.64±1.00 | 6.67±0.90 | 9.71±0.67 | 10.54±1.67 | 2.78±0.40 | 3.02±0.31 |
| HP | 1.72±0.18 | 1.73±0.21 | 6.58±0.51 | 6.59±0.62 | 10.19±0.54 | 9.90±0.63 | 7.50±0.65 | 7.39±0.72 | 10.92±1.09 | 10.50±0.63 | 2.61±0.42 | 2.67±0.54 |
| HYP | 2.81±0.46 | 2.73±0.34 | 6.29±0.78 | 6.37±0.44 | 8.50±0.76 | 8.17±0.56 | 7.16±0.87 | 6.81±0.54 | 10.05±0.97 | 9.99±0.60 | 2.68±0.32 | 2.60±0.32 |
| MEA | 1.41±0.19 | 1.33±0.10 | 4.63±0.40 | 4.64±0.27 | 6.04±0.36 | 5.76±0.36* | 6.17±0.66 | 5.87±0.65 | 10.03±1.13 | 9.52±0.72 | 2.51±0.34 | 2.24±0.34 |
| MID | 2.32±0.22 | 2.24±0.26 | 6.03±0.48 | 6.18±0.47 | 8.39±0.44 | 8.45±0.40 | 7.47±0.56 | 7.58±0.78 | 11.21±0.94 | 11.13±0.67 | 2.95±0.31 | 2.88±0.35 |
| OB1 | 2.88±0.40 | 2.76±0.32 | 6.45±0.53 | 6.55±0.54 | 8.68±0.64 | 8.79±0.45 | 6.58±0.52 | 5.86±0.85* | 8.71±0.93 | 8.77±0.34 | 2.51±0.27 | 2.64±0.37 |
| OC | 1.38±0.20 | 1.36±0.11 | 6.67±0.71 | 6.72±0.43 | 10.29±0.97 | 9.97±0.53 | 7.70±0.92 | 7.23±0.89 | 10.25±0.83 | 10.61±1.23 | 2.97±0.31 | 3.00±0.33 |
| PC | 1.29±0.17 | 1.24±0.15 | 6.52±0.61 | 6.30±0.83 | 10.48±0.74 | 9.76±0.83* | 8.35±0.72 | 7.69±1.03* | 9.98±0.61 | 9.62±1.67 | 3.16±0.26 | 2.98±0.39 |
| ST | 2.19±0.19 | 2.00±0.16* | 6.88±0.59 | 6.68±0.47 | 9.93±0.79 | 9.28±0.96* | 7.18±0.79 | 6.38±0.52* | 10.13±0.87 | 9.92±0.64 | 2.69±0.31 | 2.61±0.33 |
| THA | 2.19±0.18 | 2.20±0.15 | 6.41±0.59 | 6.58±0.40 | 9.66±0.74 | 9.65±0.50 | 7.69±0.51 | 8.01±0.93 | 10.85±0.93 | 10.79±0.56 | 2.79±0.28 | 2.85±0.31 |
15 rats were measured in this region, and 20 rats were used in other regions.
values significantly different after correction for multiple comparisons (p<0.05).
Before addressing multiple comparisons with the metabolites other than GABA, it was necessary to evaluate whether the measurements were correlated. The relationships among the five metabolites in the twelve regions were evaluated, and the concentration of glutamate was found to be strongly correlated with NAA (p<0.05 in all the regions), and with aspartate (p<0.05 in 11 regions), and with glutamine and GABA (p<0.05 in 10 regions) (Fig. 2). Other metabolites showed little or no correlation with one another. Because glutamate was strongly correlated with other metabolites, it was not independent and was not counted for adjustment for multiple comparisons, leaving 4 metabolites for which to adjust: aspartate, glutamine, NAA, and creatine. Over 12 regions, the total number of adjustments at the start of the procedure was 48 (4 metabolites by 12 regions). After the Holm-Bonferroni adjustment, striatal NAA remained as significantly affected by nicotine (p=0.0009) after nicotine subcutaneously injection.
Fig. 2.
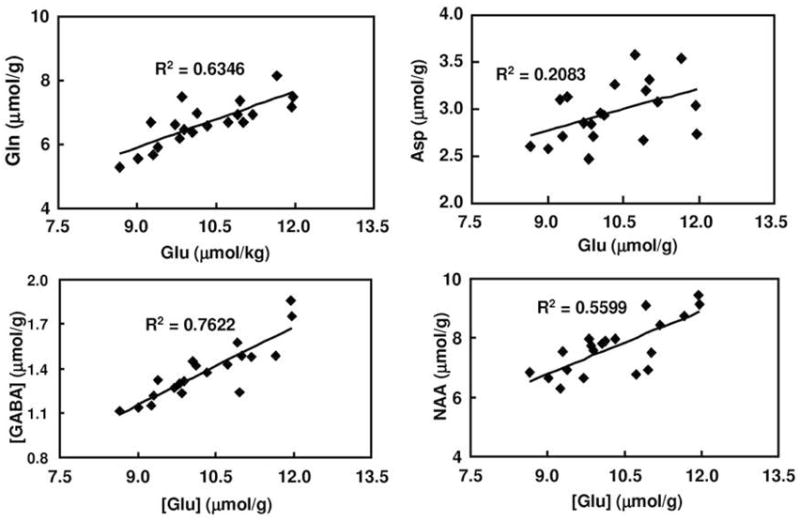
The correlation of Glu with Gln, Asp, GABA, and NAA, using the example of the occipital cortex in the saline group.
For GABA, the Holm-Bonferroni method was also used to account for the GABA in the 12 regions and found that the GABA in ST was significantly affected by nicotine (p=0.0018). Thus, from the perspective of the metabolite concentrations, the ST was the region most affected by acute nicotine.
Results of LDA
Using Wilks’ lambda method (Johnson & Wichern 1992), a stepwise procedure was utilized to select the major descriptors. At first, the entire group of 39 rats was randomly divided into two sets: a training set of 31 rats and a test set of 8 rats. The training set was used to identify the largest differences through the comparison between the saline and nicotine groups and construct a classification model; the test set was used to verify the predictability of the model.
Supplemental Table 3S lists the selected descriptors and their corresponding statistical parameters. This model gave promising predictability, 100.0% for the saline group (16 rats), 93.3% for the nicotine group (15 rats), and 96.7% for the entire group. To verify the reliability of the model, a LOO cross-validated method was also used (Forina et al. 2008), and it yielded 87.1% for the whole training set (81.3% for the saline group, and 93.3% for the nicotine group). Thus, this method shows that the levels of regional metabolites differentiate the rats on the basis of receiving nicotine or saline. Although there is no need to use metabolites to determine which rats have had nicotine and which have not, this method may be useful to highlight patterns of changes that are caused by acute doses of nicotine. Of note is the identification of levels of striatal GABA and NAA and thalamic GABA levels as distinguishing features between the saline- and nicotine-treated groups.
Enrichment of plasma glucose
Plasma glucose levels were similar after the overnight fast between the two groups (5.3 ± 0.8 mM and 5.1 ± 0.7 mM in the groups that were to receive saline or nicotine, respectively. The plasma glucose [1-13C] fractional enrichment increased and remained steady for the duration of the experiment. During the infusion, the plasma glucose concentration increased to 10.8 ± 1.4mM/L (saline group) and 11.9 ± 1.9mM/L (nicotine group), a difference that was not quite significant statistically (p = 0.06). The plasma [1-13C] glucose enrichment was 46.1% ± 6.6% (Nicotine group) and 46.6% ± 8.0% (Saline group), also an insignificant difference (p = 0.84).
Effects of Nicotine on metabolic kinetics of different regions of rat brain
The values of the principal metabolic rates are shown in Table 2, with additional values and mathematical relationships given in Supplemental Tables 4S and 1S. Fig. 3 shows the curves of Glu-C4, Glu-C3, Gln-C4, and GABA-C2 in the ST for the saline and nicotine groups. The values of CMRgl(ox), Vcycle, and Vgad were evaluated individually in the ST and occipital cortex using a Holm-Bonferroni adjustment for 6 parameters. The rates in all other regions were tested using the Holm-Bonferroni procedure to account for multiple comparisons, adjusting for 30 comparisons (3 parameters × 10 regions). Acute nicotine administration was associated with a significant decrease of CMRgl(ox) for almost all parts of the rat brain, especially for parietal cortex (PC) and occipital cortex (OC) (p < 0.05). The rate of Vcycle did not change significantly, except for decreases of 19% in the PC (p<0.0004) and 23% in the OC (p=0.0004). Acute nicotine decreased the rate of GABA formation (Vgad) by 27% in the ST (p = 0.025). Vgad was also lower by 27% in OC (p=0.037) and 21% PC (p=0.039), but not significantly after consideration of multiple comparisons. Relative to the rest of the brain, the PC, OC, and ST were the most affected regions. Supplemental Table 5S shows the rates by region and p-values, indicating which rates survived adjustments for multiple comparisons.
Table 2.
Effect of nicotine on cerebral cortical metabolic fluxes (μmol·g·min−1), including cerebral metabolic rates for glucose (CMRgl(ox)), rates of glutamate-glutamine cycling (Vcycle) and rates of GABA formation (Vgad).
| CMRgl(ox) | Vcycle | Vgad | ||||
|---|---|---|---|---|---|---|
| Brain | Saline | Nicotine | Saline | Nicotine | Saline | Nicotine |
| CE | 0.44±0.02 | 0.43±0.01 | 0.49±0.05 | 0.44±0.04 | 0.16±0.01 | 0.16±0.01 |
| FC | 0.50±0.03 | 0.41±0.02 | 0.51±0.07 | 0.45±0.03 | 0.12±0.01 | 0.10±0.00 |
| TC | 0.45±0.02 | 0.40±0.02 | 0.52±0.05 | 0.47±0.04 | 0.12±0.01 | 0.11±0.01 |
| HP | 0.43±0.02 | 0.36±0.01 | 0.47±0.05 | 0.44±0.04 | 0.12±0.01 | 0.11±0.02 |
| HYP | 0.36±0.02 | 0.32±0.01 | 0.48±0.05 | 0.48±0.06 | 0.12±0.01 | 0.10±0.01 |
| MEA | 0.36±0.02 | 0.32±0.01 | 0.44±0.05 | 0.47±0.04 | 0.13±0.01 | 0.11±0.01 |
| MID | 0.43±0.02 | 0.37±0.01 | 0.50±0.06 | 0.43±0.04 | 0.16±0.02 | 0.15±0.01 |
| OB | 0.36±0.02 | 0.33±0.01 | 0.56±0.07 | 0.55±0.06 | 0.10±0.01 | 0.11±0.01 |
| OC | 0.48±0.03 | 0.42±0.02 | 0.43±0.02 | 0.39±0.01 | 0.13±0.01 | 0.12±0.01 |
| PC | 0.54±0.03 | 0.44±0.01 | 0.49±0.06 | 0.37±0.03 | 0.14±0.01 | 0.11±0.01 |
| ST | 0.45±0.02 | 0.37±0.01 | 0.45±0.05 | 0.43±0.04 | 0.12±0.01 | 0.08±0.01 |
| THA | 0.43±0.02 | 0.42±0.02 | 0.51±0.05 | 0.51±0.04 | 0.13±0.01 | 0.13±0.00 |
Fig. 3.
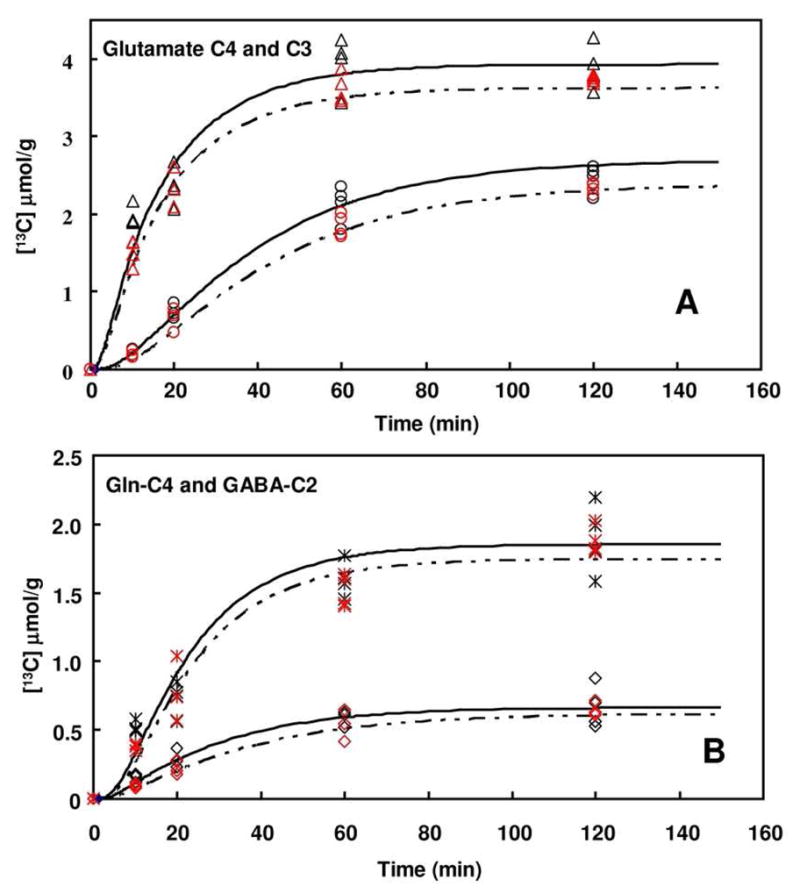
Fit of the metabolic model to experimental data. The time courses of 13C labeling of Glu-C4 (△) and Glu-C3 (○) (A) and Gln-C4 (*) and GABA-C2 (◇) (B) in ST from [1-13C]-glucose are depicted. Note: black: saline; Red: nicotine.
Discussion
In this study of acute nicotine administration, nicotine appears to influence the ST more than other regions, in that it changed two major metabolites in that region, NAA and GABA, as well as the rate of GABA synthesis. Nicotine also exhibited significant effects on metabolite levels and rates of turnover in other cortical regions, including PC and OC. The most regionally consistent effects of nicotine were decreased metabolic rates and concentrations of NAA.
Effects of nicotine on metabolite levels in the rat brain
Nicotine decreased the levels of most metabolites in most regions, although only the most major reductions survived the large adjustment for multiple comparisons. Both with and without nicotine, the concentrations of the metabolites differed among regions of the brain. Of the measured compounds, GABA showed the greatest regional variations. The olfactory bulb had high levels of GABA (Heyden & Korf 1978), as did the hypothalamus (Balcom et al. 1975, Krzalic et al. 1962), consistent with previous findings. These results are consistent with the density of GABAergic neurons in the rat brain, as HYP (Obrietan & van den Pol 1995) and OB (Poirier et al. 2004) have higher densities of GABAergic neurons, compared to other regions. The cerebellum had high creatine but the smallest concentration of GABA. The higher concentration of creatine was consistent with what has been reported in humans (Michaelis et al. 1993) and may be related to the energetics of the cerebellum. In the brain, creatine kinase is very active, and the creatine kinase activity with comparably high levels in the cerebellum (Manos et al. 1991), and the low levels of cerebellar GABA are consistent with a previous report (Palmi et al. 1991). Glutamine was the most homogeneously distributed metabolite across regions, except for in the medulla (Pogodaev & Logunov 1968), which also had the lowest level of glutamate, as reported previously in cat brain (Berl & Frigyesi 1969).
Across brain regions, glutamate levels showed strong correlations with concentrations of glutamine, NAA, GABA, and aspartate. The concentration of glutamine could be related to that of glutamate because of their roles in glutamate-glutamine neurotransmitter cycling, such that lower glutamate levels might be associated with less glutamine, although the situation likely includes many other factors, given that the rate of glutamate-glutamine cycling did not change with nicotine. Another possibility is that the astrocytes, where glutamine is made, were affected in the minutes following nicotine administration in a way that dropped the glutamine to a new, lower steady state from which the glutamine did not increase during the two-hour experiment.
NAA is found mainly in glutamatergic neurons (Moffett et al. 1991, Urenjak et al. 1992), which could explain the correlation between NAA and glutamate. Nicotine reduced levels of NAA in most regions of the rat brain, especially in ST, so it must either have decreased the formation or increased the degradation of NAA. NAA is synthesized by N-acetyl aspartyl transferase, an enzyme associated with mitochondria. It is possible that decreased mitochondrial activity, as would be expected with decreased CMRgl(ox), could decrease levels of NAA, so perhaps this is an energetic effect. Acute nicotine influenced the ST more than other regions, with respect to metabolite levels and kinetics. The metabolites NAA and GABA were the most affected compounds after nicotine injection, especially in the ST. Because the ST has densities of α4β2 and α7 nAChRs that are similar to what is seen in the rest of the brain, except for the thalamus, which has much higher levels (Staley et al. 2006, Whiteaker et al. 1999, Mugnaini et al. 2002), the stronger effects on metabolite levels and kinetic rates must be related to other factors. One possible explanation is action of the α6 subunit of the nAChR, which in monkeys was more highly expressed in the ST than in cortex (Quik et al. 2005) and modulates GABA release onto dopaminergic neurons (Yang et al. 2009).
Glucose utilization after acute nicotine treatment
In this experiment, nicotine significantly decreased glucose oxidation in most brain regions, especially in the ST, OC, and PC. In early studies that pioneered the question of nicotine effects on energy metabolism in vivo (Weissman et al. 1987, London et al. 1988, London et al. 1990) evaluated regional glucose utilization in rats after femoral vein and artery catheterization and did the study after 3–5 hours of recovery, with the rats partially immobilized in plaster casts that covered their hindquarters. Although their report that nicotine stimulated glucose utilization in many regions of the rat brain was consistent with our original hypothesis, it differs from the present results. One difference with the present study is the dose of nicotine. In the two studies, dosages of 0.1mg/kg, 0.3mg/kg, and 1.0mg/kg were used in one publication (London et al. 1988) and 1.75mg/kg in another (Weissman et al. 1987), but the current dose of 0.7 mg/kg lies in the midst of that published dose range, so dose differences probably do not explain the different outcome. Another difference is that in the earlier studies Fisher 344 rats were used, whereas this study used Sprague-Dawley rats. However, in another study the same surgical preparation but intravenous dosing was used to study Sprague-Dawley rats (Grünwald et al. 1987) and replicated the increased glucose utilization by nicotine, so the finding appeared to be independent of strain of rat.
In a later study, nicotine’s effects were measured in both partially immobilized and freely moving rats (Marenco et al. 2000). In their freely moving rats given a dose of 0.4 mg/kg of nicotine, and the results were similar to those reported here both regionally and with respect to the reductions of glucose utilization. In the partially immobilized group, the surgical preparation was the same as that used in the previously discussed studies, but lidocaine was added to reduce post-operative discomfort. Nicotine significantly increased glucose utilization in only two brain regions: the anteroventral thalamic nucleus, and the superior colliculus. In other regions, nicotine appeared to have little or no effect. A study of nicotine effects on glucose uptake in humans (Stapleton et al. 2003) showed decreased glucose utilization, which is in agreement with the current results, although the human dose of 1.5 mg via a 10-second intravenous infusion was much lower than any of the doses used in this or any of the other studies of rats. Also similarly to the present results, the thalamus was among the regions least affected by nicotine. Having the thalamus least affected seems contrary to expectation, because of the high density of nAChRs in the thalamus, An explanation proposed previously (Stapleton et al. 2003) was that nicotine has effects on metabolism apart from its actions through the nAChRs, such that areas with a higher density of receptors like the thalamus (Horti et al. 1997) have equal and opposite effects that sum to zero. Perhaps the richness of dopamine receptors in the thalamus contributes to the balance. Indeed, acute nicotine dosing has been reported to reduce glycolysis (Dewar et al. 2002), though those findings were in the liver.
What is in common is that the studies of Marenco, Stapleton, and the present data all imposed less stress on the subjects than did the earlier projects. Based on the comparisons, stress may influence brain metabolism strongly. In fact, a study of the effect of immobilization stress on glucose uptake showed that stress caused an increase of 50% to 90% in glucose utilization in rats (Carlsson et al. 1977). It appears that minimization of stress is an important factor in the study of metabolism with nicotine effects and probably in many other conditions.
Effects of nicotine on turnover of GABA and glutamate
The 13C-labeling of glutamate in the brain during an infusion of [1-13C] glucose reflects primarily glucose oxidation. Supplemental Table 4S shows that that most parts of the brain were affected, especially the PC and OC (p<0.05). Acute nicotine injection also decreased the rate of GABA formation in some parts of the brain. However, the change in GABA was primarily in the OC, PC, and ST. Across the metabolites, the OC, PC and ST were also the most affected regions, as shown by the metabolite levels, as well The decreased GABA synthesis in those regions might result from the time-dependent desensitization of the α4β2 GABA receptors a few minutes after the administration of nicotine (Mansvelder et al. 2002). For regions in which GABA synthesis was unchanged by nicotine, another possibility is that GABA synthesis was subject to two opposite influences and so was not affected by nicotine, as was the case for glucose oxidation. If nicotine tends to reduce GABA synthesis while nAChRs stimulate GABA synthesis, then the opposite effects may cancel. Indeed, nicotine has been shown to reduce GABA levels and synthesis in pancreatic tumors (Al-Wadei et al. 2009), although no effects on brain GABA release or glutamate decarboxylase were seen after a single sub-convulsant dose of nicotine (Pérez de la Mora et al. 1991). There also remains the possibility that in the first few minutes following nicotine administration, GABA synthesis and perhaps glutamate-glutamine cycling and glucose oxidation were elevated but only acutely, such that the effect disappeared before most of the data were acquired.
Relationship between glutamate/glutamine cycling and glucose oxidation
Our previous finding of a near 1:1 proportionality between incremental changes in Vcycle and CMRgl(ox) in non-activated cortex of anesthetized rats is consistent with glucose oxidative support of glutamatergic activity and associated functions of the brain (Sibson et al. 1998, Rothman et al. 1999, Magistretti et al. 1999). Combined with the results from previous studies (Sibson et al. 1998, Patel et al. 2004), the incremental changes were consistent with the idea that incremental changes in cycling are associated with energetic changes and altered glucose oxidation (Supplemental Fig. 2S).
After acute nicotine injection, changes in Vcycle and CMRgl(px) were similar in the cortex (FC: 11.5% and 14.3%; PC: 24.1% and 18.1%; OC: 10.0% and 12.1%), so the glutamatergic neurotransmission and glucose oxidation appear to follow the previously observed coupling in the cortex of the freely moving rat. However, the relationship appears not to be the same for all brain regions, especially for the OB. Accordingly, the distribution of glutamine synthetase is inhomogeneous in the rat brain, with twice the level in the OB compared to other regions (Patel et al. 1985). Table 2 and Supplemental Fig. 3S show that the OB has a much higher value of Vcycle compared with CMRgl(px), and the ratios Vcycle/CMRgl(px) were 1.50 and 1.58 for the saline and nicotine groups, separately.
Glutamine synthetase, required for glutamine synthesis, must contribute a significant component to Vcycle. Glutamine synthetase is predominantly astrocytic, as is pyruvate carboxylase (Yu et al. 1983), and is implicated in glutamatergic and GABAergic function (Okere & Kaba 2000) The synthetase activity can directly influenced the formation of glutamine in astrocytes and, through neurotransmitter cycling can potentially regulated the metabolism of glutamate and GABA. The OB has the highest value activity of glutamate synthetase compared to the other regions. If in the olfactory bulb a greater fraction of glutamine synthesis is dedicated to other than uptake of glutamate and conversion to glutamine, then the associated energy requirements may be lower and the values of Vcycle/CMRgl(ox) disproportionately higher in this region, with a different relationship predicted across a range of functional states. To evaluate part of this hypothesis, the region-specific activities of glutamine synthetase (Patel et al. 1985)were expressed as ratios to the activity in the cortex. The value of Vcycle in the same regions was also expressed as a ratio to what was measured in the parietal cortex. The enzyme activity ratios and Vcycle ratios were expressed relative to one another (Fig. 6), and the OB was clearly separated from the other regions. Thus, the distribution of glutamine synthetase may be related to the different value of Vcycle/CMRgl(ox) in the OB.
Implications for addiction
In the acute nicotine study, the positive and negative effects of glucose oxidation and GABA turnover in the thalamus might be important for the process of nicotine addiction and could perhaps be altered by GABAergic treatments. Studies in humans showed that topiramate, which raises brain GABA levels (Kuzniecky et al. 1998), affects subjective responses to nicotine in ways that may enhance smoking cession (Sofuoglu et al. 2006), and that another GABAergic drug, tiagabine, decreases nicotine craving (Sofuoglu et al. 2005). Studies in animals showed that nicotine alters GABAB receptor expression and that nicotinic modulation of GABAergic function contributes acutely to inhibition of dopamine release and chronically to stimulation of dopamine release (Mansvelder et al. 2002). The GABAB receptor agonists baclofen and CGP44532 each decreased administration of nicotine in rats without affecting food intake (Paterson et al. 2004).
Limitations
The study has some limitations: 1) The rats could not be scanned in vivo in the MRI machine, because of the need for long-term, general anesthesia, so the measurements had to be made in isolated tissue. Therefore, individual rats had to be used for each time point, rather than evaluating entire time courses with individual rats. 2) The current study used a large, single acute dose of nicotine, so the results may differ from what one would find after chronic repetition of nicotine administration. 3) The rats were awake and moving freely during the nicotine administration and the infusion of [1-13C]glucose, and seconds before placement in the microwave, the rats were anesthetized. The turnover of the metabolites measured are processes that require tens of minutes and can only be minimally affected by the brief anesthesia before euthanasia.
Another limitation is that the techniques used in this study only provide information on the total concentrations of the neurochemicals in the tissue, not their fractionation within the tissue, so it was not possible to differentiate glutamate released from glutamate remaining within the neurons. At present, the relationships between neurotransmitter activity and function remain unknown but are the subject of investigation. The reviewer is correct that the techniques used in this study can only address the total amount of neurochemical in the tissue, not its distribution. However, it is possible to relate the level of glutamate with its rate of release, using 13C-glucose and 13C-acetate (Patel et al. 2005, Lebon et al. 2002). Also, some regions were inaccessible with the present methods. For example, the ventral tegmental area and nucleus accumbens were too small to remove and measure, and the prefrontal cortex was difficult to differentiate from the frontal cortex, problems made more difficult by the texture of the brain after microwaving. However, it would certainly have been interesting and important to examine those regions. The brain dissection was limited by size and the ability to discriminate among regions in the microwaved brain..
A limitation is the issue of species differences between rats and humans, including potential discrepancies in the distribution of nAChRs. Though there are some differences, such as a subunit distribution in the thalamus, reviewed previously (Spurden et al. 1997), they are in general similarly distributed in rats and humans (Turner & Kellar 2005, Staley et al. 2006, Spurden et al. 1997). The metabolism of nicotine in rats is much faster than in humans. In rats, CYP1B1/2 is responsible for nicotine metabolism and CYP2A6 is inactive, but CYP2A6 is responsible for nicotine in humans. The plasma nicotine half-time is 45 minutes in rats but two hours in humans (Matta et al. 2007). This means that rats must be given a higher dose of nicotine, and the concentration of nicotine will drop after injection, just as plasma nicotine levels fall after a human smokes. In a previous study (Rossi et al. 2005), 0.15mg/kg nicotine was used, and concentration of nicotine was 140, 125, 92 and 44.9ng/g at 10, 30, 60, and 120 minutes. In spite of the falling plasma nicotine levels, the current results is showed little time-dependence of metabolite levels. For the kinetic portion of the study, the majority of the kinetic information is contained in the first 30 minutes of the 13C-labeling time courses, so changes after that that have less impact on the metabolic rates. Although the level of nicotine in the circulation certainly dropped during the present time courses, which is also true for human study, nicotine remains tightly bound in the brain for several days (Matta et al. 2007), which might tend to keep neurochemical conditions more stable than one would predict based on the plasma nicotine levels.
A methodological limitation was the use of the three-compartment model of neurotransmitter and energy metabolism for all brain regions. The model has been evaluated for cortical tissue using [1-13C] and [1,6-13C2]glucose, [2-13C]glucose, and [2-13C]acetate and so far behaved with internal consistency and in agreement with measurements by other techniques. However, the model remains to be evaluated for the olfactory bulb and other brain regions that have contain different fractions of glutamatergic, GABAergic, and other types of neurons. If the model is incorrect in the present case, one could argue that the same error will exist with nicotine treatment, so directional changes should remain valid even if the absolute rates are biased in one direction or another. This study is just the first step in evaluating the model throughout the brain. Other isotopic precursors are to test and, if necessary, change the model appropriately for non-cortical brain regions, and these experiments are in progress.
On a related point, the non-cortical regions required assumptions for some values that can only be determine using isotopic precursors other than [1-13C] or [1,6-13C2]glucose. For example, the study could not independently evaluate the ratio of GABA-glutamine neurotransmitter cycling as a fraction of the GABAergic TCA cycle rate as was done previous for the cortex (Patel et al. 2005), but measurements with 13C-labeled acetate will yield that value. In this case, the cortical value of 0.46 was used, as reported in cortex in the absence of nicotine(Patel et al. 2005). The value has the potential to alter the determination of the rates of glutamate-glutamine cycling, glucose oxidation, and GABA synthesis, so a sensitivity analysis was done. The sensitivity analysis showed that if the ratio is as low as 0.2, the values of Vgad, CMRgl(ox), and Vcycle can be reduced by 24, 4%, and 7%, respectively. If the ratio is as high as 0.7, then the respective values are 26%, 13%, and 8% higher. Therefore, if nicotine affects the ratio Vcycle/VtcaGABA, such an effect could potentially appear as a change in Vgad, and 13C-acetate provides a means to establish the value of that ratio.
Summary
The main findings of this study are the following: (a) acute nicotine administration decreased total concentrations of NAA and GABA in the striatum; (b) the main effect of nicotine on the measured parameters was to decrease glucose utilization and GABA synthesis, particularly in the striatum and occipital and parietal cortices; (c) nicotine effects on the neurotransmitter cycle (glutamate-glutamine) cycle did not reach significance, except in the parietal and occipital cortices, although by using 13C-labeled acetate the precision might be improved sufficiently to detect such effects (Patel et al. 2010, Lebon et al. 2002, Deelchand et al. 2009); (e) incremental changes in glutamatergic neurotransmission and glucose oxidation follow previously observed coupling in the cortex of the wake rats, but the coupling was not the same for the olfactory bulb or hypothalamus; (f) the concentration of glutamate was strongly correlated with those of glutamine, NAA, GABA and aspartate throughout most of the brain.
Supplementary Material
Acknowledgments
The authors thank Prof. Marina R. Picciotto, who gave us suggestion on the dose of nicotine for acute injection on rats, Prof. Ralitza Gueorguieva, who provided guidance on the statistical procedures, Golam Chowdhury for advice on dissection of the brain, and Peter Brown and Scott McIntyre for technical support. The research was supported by R01 DA021785, R21 AA018210, and P30 NS052519.
References
- Al-Wadei HAN, Plummer HK, 3rd, Schuller HM. Nicotine stimulates pancreatic cancer xenografts by systemic increase in stress neurotransmitters and suppression of the inhibitory neurotransmitter gamma-aminobutyric acid. Carcinogenesis. 2009;30:506–511. doi: 10.1093/carcin/bgp010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcolea A, Carrera J, Medina A. ModelCARE. Vol. 99. Zürich; Switzerland: 1999. A hybrid Marquardt-Simulated Annealing method for solving the groundwater inverse problem. [Google Scholar]
- Balcom GJ, Lenox RH, Meyerhoff JL. Regional γ-Aminobutyric acid levels in rat brain determined after microwave fixation. J Neurochem. 1975;24:609–613. [PubMed] [Google Scholar]
- Benowitz NL. Nicotine addiction. Prim Care. 1999;26:611–631. doi: 10.1016/s0095-4543(05)70120-2. [DOI] [PubMed] [Google Scholar]
- Berl S, Frigyesi TL. Comparison of cerebral regional metabolism of [14C]leucine following third ventricle and intravenous administration in the cat. J Neurochem. 1969;16:405–415. doi: 10.1111/j.1471-4159.1969.tb10381.x. [DOI] [PubMed] [Google Scholar]
- Carlsson C, Hägerdal M, Kaasik AE, Siesjö BK. A catecholamine-mediated increase in cerebral oxygen uptake during immobilisation stress in rats. Brain Res. 1977;119:223–231. doi: 10.1016/0006-8993(77)90102-0. [DOI] [PubMed] [Google Scholar]
- de Graaf RA. In vivo NMR Spectroscopy -2nd Edition Principles and Techniques. John Weiley & Sons, Ltd; 2007. [Google Scholar]
- Deelchand DK, Shestov AA, Koski DM, U3urbil K, Henry PG. Acetate transport and utilization in the rat brain. J Neurochem. 2009;109:46–54. doi: 10.1111/j.1471-4159.2009.05895.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewar BJ, Bradford BU, Thurman RG. Nicotine increases hepatic oxygen uptake in the isolated perfused rat liver by inhibiting glycolysis. J Pharmacol Exp Ther. 2002;301:930–937. doi: 10.1124/jpet.301.3.930. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick SM, Hetherington HP, Behar KL, Shulman RG. The Flux from Glucose to Glutamate in the Rat-Brain Invivo as Determined by H-1-Observed, C-13-Edited Nmr-Spectroscopy. Journal of Cerebral Blood Flow and Metabolism. 1990;10:170–179. doi: 10.1038/jcbfm.1990.32. [DOI] [PubMed] [Google Scholar]
- Forina M, Oliveri P, Lanteri S, Casale M. Class-modeling techniques, classic and new, for old and new problems. Chemometrics and Intelligent Laboratory Systems. 2008;93:132–148. [Google Scholar]
- Grünwald F, Schröck H, Kuschinsky W. The effect of an acute nicotine infusion on the local cerebral glucose utilization of the awake rat. Brain Res. 1987;400:232–238. doi: 10.1016/0006-8993(87)90622-6. [DOI] [PubMed] [Google Scholar]
- Heyden JAM, Korf J. Regional Levels of GABA in the brain: rapid semlautomated assay and prevention of postmortem increase by 3-mercapto-propionic acid. J Neurochem. 1978;31:197–203. doi: 10.1111/j.1471-4159.1978.tb12448.x. [DOI] [PubMed] [Google Scholar]
- Holm S. A Simple Sequentially Rejective Multiple Test Procedure. Scand J Stat. 1979;6:65–70. [Google Scholar]
- Horti A, Scheffel U, Stathis M, Finley P, Ravert HT, London ED, Dannals RF. Fluorine-18-FPH for PET imaging of nicotinic acetylcholine receptors. J Nucl Med. 1997;38:1260–1265. [PubMed] [Google Scholar]
- Johnson RA, Wichern DW. Applied Multivariate Statistical Analysis. Prentice-Hall; Englewood Cliffs, NJ: 1992. [Google Scholar]
- Krzalic L, Mandic V, Mihailovic L. On the glutamine and gamma-aminobutyric acid contents of various regions of the cat brain. Experientia. 1962;18:368–369. doi: 10.1007/BF02172255. [DOI] [PubMed] [Google Scholar]
- Kuzniecky R, Hetherington H, Ho S, Pan J, Martin R, Gilliam F, Hugg J, Faught E. Topiramate increases cerebral GABA in healthy humans. Neurology. 1998;51:627–629. doi: 10.1212/wnl.51.2.627. [DOI] [PubMed] [Google Scholar]
- Lebon V, Petersen KF, Cline GW, Shen J, Mason GF, Dufour S, Behar KL, Shulman GI, Rothman DL. Astroglial contribution to brain energy metabolism in humans revealed by 13C nuclear magnetic resonance spectroscopy: Elucidation of the dominant pathway for neurotransmitter glutamate repletion and measurement of astrocytic oxidative metabolism. J Neurosci. 2002;22:1523–1531. doi: 10.1523/JNEUROSCI.22-05-01523.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London ED, Cascella NG, Wong DF, et al. Cocaine-Induced Redoppuction of Glucose Utilization in Human Brain: A Study Using Positron Emission Tomography and [Fluorine 18]-Fluorodeoxyglucose. Arch Gen Psychiatry. 1990;47:567–574. doi: 10.1001/archpsyc.1990.01810180067010. [DOI] [PubMed] [Google Scholar]
- London ED, Connolly RJ, Szikszay M, Wamsley JK, Dam M. Effects of Nicotine on Local Cerebral Glucose-Utilization in the Rat. J Neurosci. 1988;8:3920–3928. doi: 10.1523/JNEUROSCI.08-10-03920.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistretti PJ, Pellerin L, Rothman DL, Shulman RG. NEUROSCIENCE:Energy on Demand. Science. 1999;283:496–497. doi: 10.1126/science.283.5401.496. [DOI] [PubMed] [Google Scholar]
- Manos P, Bryan GK, Edmond J. Creatine kinase activity in postnatal rat brain development and in cultured neurons, astrocytes, and oligodendrocytes. J Neurochem. 1991;56:2101–2107. doi: 10.1111/j.1471-4159.1991.tb03472.x. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, Keath JR, McGehee DS. Synaptic mechanisms underlie Nicotine-Induced excitability of brain reward areas. Neuron. 2002;33:905–919. doi: 10.1016/s0896-6273(02)00625-6. [DOI] [PubMed] [Google Scholar]
- Marenco T, Bernstein S, Cumming P, Clarke PBS. Effects of nicotine and chlorisondamine on cerebral glucose utilization in immobilized and freely-moving rats. British Journal of Pharmacology. 2000;129:147–155. doi: 10.1038/sj.bjp.0703005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason GF, Boumezbeur F, Sanacora G, et al. Acute nicotine stimulates GABA synthesis in human brain. Proc Intern Soc Magn Reson Med. 2007:770. [Google Scholar]
- Mason GF, Falk Petersen K, de Graaf RA, Kanamatsu T, Otsuki T, Rothman DL. A comparison of 13C NMR measurements of the rates of glutamine synthesis and the tricarboxylic acid cycle during oral and intravenous administration of [1–13C]glucose. Brain Res Protoc. 2003;10:181–190. doi: 10.1016/s1385-299x(02)00217-9. [DOI] [PubMed] [Google Scholar]
- Matta SG, Balfour DJ, Benowitz NL, et al. Guidelines on nicotine dose selection for in vivo research. Psychopharmacology. 2007;190:269–319. doi: 10.1007/s00213-006-0441-0. [DOI] [PubMed] [Google Scholar]
- Michaelis T, Merboldt KD, Bruhn H, Hanicke W, Frahm J. Absolute Concentrations of Metabolites in the Adult Human Brain Invivo - Quantification of Localized Proton Mr Spectra. Radiology. 1993;187:219–227. doi: 10.1148/radiology.187.1.8451417. [DOI] [PubMed] [Google Scholar]
- Moffett JR, Namboodiri MAA, Cangro CB, Neale JH. Immunohistochemical Localization of N-Acetylaspartate in Rat-Brain. Neuroreport. 1991;2:131–134. doi: 10.1097/00001756-199103000-00005. [DOI] [PubMed] [Google Scholar]
- Mugnaini M, Tessari M, Tarter G, Merlo Pich E, Chiamulera C, Bunnemann B. Upregulation of [3H]methyllycaconitine binding sites following continuous infusion of nicotine, without changes of alpha7 or alpha6 subunit mRNA: an autoradiography and in situ hybridization study in rat brain. Eur J Neurosci. 2002;16:1633–1646. doi: 10.1046/j.1460-9568.2002.02220.x. [DOI] [PubMed] [Google Scholar]
- Nagata K, Shinohara T, Kanno I, Hatazawa J, Domino E. Effects of tobacco cigarette smoking on cerebral blood flow in normal adults. In: Domino EF, editor. Brain Imaging of Nicotine and Tobacco Smoking. NPP Books; Ann Arbor: 1995. [Google Scholar]
- Obrietan K, van den Pol AN. GABA neurotransmission in the hypothalamus: developmental reversal from Ca2+ elevating to depressing. J Neurosci. 1995;15:5065–5077. doi: 10.1523/JNEUROSCI.15-07-05065.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okere CO, Kaba H. Region-specific localization of glutamine synthetase immunoreactivity in the mouse olfactory bulb: implications for neuron-glia interaction in bulbar synaptic plasticity. Brain Res. 2000;857:308–312. doi: 10.1016/s0006-8993(99)02465-8. [DOI] [PubMed] [Google Scholar]
- Palmi M, Brooke S, Smith AD, Bolam JP. GABA-like immunoreactivity in different cellular populations of cerebellar cortex of rats before and after treatment with amino-oxyacetic acid. Brain Res. 1991;543:277–286. doi: 10.1016/0006-8993(91)90038-w. [DOI] [PubMed] [Google Scholar]
- Patel AB, de Graaf RA, Mason GF, Kanamatsu T, Rothman DL, Shulman RG, Behar KL. Glutamatergic Neurotransmission and Neuronal Glucose Oxidation Are Coupled During Intense Neuronal Activation. J Cereb Blood Flow Metab. 2004;24:972–985. doi: 10.1097/01.WCB.0000126234.16188.71. [DOI] [PubMed] [Google Scholar]
- Patel AB, de Graaf RA, Mason GF, Rothman DL, Shulman RG, Behar KL. The contribution of GABA to glutamate/glutamine cycling and energy metabolism in the rat cortex in vivo. P Nati Cad Sci USA. 2005;102:5588–5593. doi: 10.1073/pnas.0501703102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AB, de Graaf RA, Rothman DL, Behar KL, Mason GF. Evaluation of Cerebral Acetate Transport and Metabolic Rates in the Rat Brain In Vivo using 1H-[13C]NMR. J Cereb Blood Flow Metab. 2010 doi: 10.1038/jcbfm.2010.2. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AB, Rothman DL, Cline GW, Behar KL. Glutamine is the major precursor for GABA synthesis in rat neocortex in vivo following acute GABA-transaminase inhibition. Brain Res. 2001;919:207–220. doi: 10.1016/s0006-8993(01)03015-3. [DOI] [PubMed] [Google Scholar]
- Patel AJ, Weir MD, Hunt A, Tahourdin CSM, Thomas DGT. Distribution of glutamine synthetase and glial fibrillary acidic protein and correlation of glutamine synthetase with glutamate decarboxylase in different regions of the rat central nervous system. Brain Res. 1985;331:1–9. doi: 10.1016/0006-8993(85)90708-5. [DOI] [PubMed] [Google Scholar]
- Paterson NE, Froestl W, Markou A. The GABAB receptor agonists baclofen and CGP44532 decreased nicotine self-administration in the rat. Psychopharmacology. 2004;172:179–186. doi: 10.1007/s00213-003-1637-1. [DOI] [PubMed] [Google Scholar]
- Pérez de la Mora M, ndez-Franco J, Salceda R, Aguirre JA, Fuxe K. Neurochemical effects of nicotine on glutamate and GABA mechanisms in the rat brain. Acta Physiol Scand. 1991;141:241–250. doi: 10.1111/j.1748-1716.1991.tb09074.x. [DOI] [PubMed] [Google Scholar]
- Pogodaev KI, Logunov VV. [Dynamics of the formation and bonding of ammonia in rat brain tissue under the influence of acoustic stimulation inducing convulsions] Biull Eksp Biol Med. 1968;66:46–49. [PubMed] [Google Scholar]
- Poirier K, Van Esch H, Friocourt G, et al. Neuroanatomical distribution of ARX in brain and its localisation in GABAergic neurons. Mol Brain Res. 2004;122:35–46. doi: 10.1016/j.molbrainres.2003.11.021. [DOI] [PubMed] [Google Scholar]
- Quik M, Vailati S, Bordia T, Kulak JM, Fan H, McIntosh JM, Clementi F, Gotti C. Subunit composition of nicotinic receptors in monkey striatum: effect of treatments with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine or L-DOPA. Mol Pharmacol. 2005;67:32–41. doi: 10.1124/mol.104.006015. [DOI] [PubMed] [Google Scholar]
- Rossi S, Singer S, Shearman E, Sershen H, Lajtha A. Regional heterogeneity of nicotine effects on neurotransmitters in rat brains in vivo at low doses. Neurochem Res. 2005;30:91–103. doi: 10.1007/s11064-004-9690-7. [DOI] [PubMed] [Google Scholar]
- Rothman DL, Sibson NR, Hyder F, Shen J, Behar KL, Shulman RG. In vivo Nuclear Magnetic Resonance Spectroscopy Studies of the Relationship between the Glutamate-Glutamine Neurotransmitter Cycle and Functional Neuroenergetics. Philos T Biolog Sci. 1999;354:1165–1177. doi: 10.1098/rstb.1999.0472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibson NR, Dhankhar A, Mason GF, Rothman DL, Behar KL, Shulman RG. Stoichiometric coupling of brain glucose metabolism and glutamatergic neuronal activity. P Nati Cad Sci USA. 1998;95:316–321. doi: 10.1073/pnas.95.1.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofuoglu M, Mouratidis M, Yoo S, Culligan K, Kosten T. Effects of tiagabine in combination with intravenous nicotine in overnight abstinent smokers. Psychopharmacology. 2005;181:504–510. doi: 10.1007/s00213-005-0010-y. [DOI] [PubMed] [Google Scholar]
- Sofuoglu M, Poling J, Mouratidis M, Kosten T. Effects of topiramate in combination with intravenous nicotine in overnight abstinent smokers. Psychopharmacology. 2006;184:645–651. doi: 10.1007/s00213-005-0296-9. [DOI] [PubMed] [Google Scholar]
- Spurden DP, Court JA, Lloyd S, Oakley A, Perry R, Pearson C, Pullen RGL, Perry EK. Nicotinic receptor distribution in the human thalamus: autoradiographical localization of [3H]nicotine and [125I]a-bungarotoxin binding. J Chem Neuroanat. 1997;13:105–113. doi: 10.1016/s0891-0618(97)00038-0. [DOI] [PubMed] [Google Scholar]
- Staley JK, Krishnan-Sarin S, Cosgrove KP, et al. Human tobacco smokers in early abstinence have higher levels of beta2* nicotinic acetylcholine receptors than nonsmokers. J Neurosci. 2006;26:8707–8714. doi: 10.1523/JNEUROSCI.0546-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapleton JM, Gilson SF, Wong DF, Villemagne VL, Dannals RF, Grayson RF, Henningfield JE, London ED. Intravenous Nicotine Reduces Cerebral Glucose Metabolism: A Preliminary Study. Neuropsychopharmacology. 2003;28:765–772. doi: 10.1038/sj.npp.1300106. [DOI] [PubMed] [Google Scholar]
- Turner JR, Kellar KJ. Nicotinic cholinergic receptors in the rat cerebellum: multiple heteromeric subtypes. J Neurosci. 2005;25:9258–9265. doi: 10.1523/JNEUROSCI.2112-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urenjak J, Williams SR, Gadian DG, Noble M. Specific expression of N-acetylaspartate in neurons, oligodendrocyte-type-2 astrocyte progenitors, and immature oligodendrocytes in vitro. J Neurochem. 1992;59:55–61. doi: 10.1111/j.1471-4159.1992.tb08875.x. [DOI] [PubMed] [Google Scholar]
- Weissman AD, Dam M, London ED. Alterations in local cerebral glucose utilization induced by phencyclidine. Brain Res. 1987;435:29–40. doi: 10.1016/0006-8993(87)91583-6. [DOI] [PubMed] [Google Scholar]
- Whiteaker P, Davies AR, Marks MJ, Blagbrough IS, Potter BV, Wolstenholme AJ, Collins AC, Wonnacott S. An autoradiographic study of the distribution of binding sites for the novel alpha7-selective nicotinic radioligand [3H]-methyllycaconitine in the mouse brain. Eur J Neurosci. 1999;11:2689–2696. doi: 10.1046/j.1460-9568.1999.00685.x. [DOI] [PubMed] [Google Scholar]
- Yang K-c, Jin G-z, Wu J. Mysterious alpha6-containing nAChRs: function, pharmacology, and pathophysiology. Chung Kuo Yao Li Hsueh Pao. 2009;30:740–751. doi: 10.1038/aps.2009.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu ACH, Drejer J, Hertz L, Schousboe A. Pyruvate carboxylase activity in primary cultures of astrocytes and neurons. J Neurochem. 1983;41:1484–1487. doi: 10.1111/j.1471-4159.1983.tb00849.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.