

Abstract
Autism is currently considered a multigene disorder with epigenetic influences. To investigate the contribution of DNA methylation to autism spectrum disorders, we have recently completed large-scale methylation profiling by CpG island microarray analysis of lymphoblastoid cell lines derived from monozygotic twins discordant for diagnosis of autism and their nonautistic siblings. Methylation profiling revealed many candidate genes differentially methylated between discordant MZ twins as well as between both twins and nonautistic siblings. Bioinformatics analysis of the differentially methylated genes demonstrated enrichment for high-level functions including gene transcription, nervous system development, cell death/survival, and other biological processes implicated in autism. The methylation status of 2 of these candidate genes, BCL-2 and retinoic acid-related orphan receptor alpha (RORA), was further confirmed by bisulfite sequencing and methylation-specific PCR, respectively. Immunohistochemical analyses of tissue arrays containing slices of the cerebellum and frontal cortex of autistic and age- and sex-matched control subjects revealed decreased expression of RORA and BCL-2 proteins in the autistic brain. Our data thus confirm the role of epigenetic regulation of gene expression via differential DNA methylation in idiopathic autism, and furthermore link molecular changes in a peripheral cell model with brain pathobiology in autism.—Nguyen, A., Rauch, T. A., Pfeifer, G. P., Hu, V. W. Global methylation profiling of lymphoblastoid cell lines reveals epigenetic contributions to autism spectrum disorders and a novel autism candidate gene, RORA, whose protein product is reduced in autistic brain.
Keywords: DNA methylation, brain expression, BLC-2 gene expression, CpG island microarrays, immunohistochemistry
Autism spectrum disorders (ASDs) collectively represent a heterogeneous range of pervasive neurodevelopmental disorders that are characterized by deficits in social interactions and understanding, aberrant communication and/or language development, and restricted interests and stereotyped behaviors (1, 2). It is believed that multiple etiologies contribute to the heterogeneity in clinical presentation of ASDs and further complicate efforts to identify genetic/epigenetic or molecular markers of the disease.
Although autism is largely considered genetic in origin because of its high heritability observed in twin and family studies (3, 4), recent evidence supports the involvement of epigenetic regulatory mechanisms in the pathogenesis of ASDs (5,6,7). These regulatory mechanisms include DNA methylation at CpG sites, genomic imprinting, chromatin modifications, and noncoding RNA (8,9,10,11,12,13). Two single-gene disorders that may be associated with autistic symptoms, fragile X syndrome and Rett syndrome, arise from epigenetic dysregulation. Fragile X syndrome results from an expansion of the trinucleotide CGG repeat in the 5′UTR region of the FMR1 gene, leading to increased susceptibility to methylation and epigenetic silencing of FMR1(14, 15), and Rett syndrome arises from a mutation in the methyl CpG binding protein 2 (MeCP2) gene, whose protein product is responsible for recognizing methylated genes and is one of the key mediators of epigenetic regulation (16, 17). Our laboratory has recently demonstrated differential gene expression in lymphoblastoid cell lines (LCLs) from monozygotic twins discordant for diagnosis of autism (18), which strongly suggests that epigenetic factors are also involved in idiopathic autism. In addition, recent interest in MeCP2 as a global epigenetic regulator has led to the discovery of increased methylation of the MeCP2 promoter and correlated gene silencing in the frontal cortex of autistic brain (19), and certain MeCP2 polymorphisms that confer increased risk of autism have been identified (20). Other studies have suggested that “epigenetic hotspots” or regions susceptible to genomic imprinting are located in chromosomal regions (e.g., 15q and 7q) identified in genetic linkage analysis of autism (5, 21). Hogart et al.(22) argues that genes located close to these hotspots (such as genes encoding for GABAA-receptor subunits, GABRB3, GABRA5, and GABRG3), while not necessarily subject to imprinting, can still convey an ASD risk on disrupted epigenetic regulation. Recently, increased methylation of the promoter of the oxytocin receptor gene was associated with autism (23). Collectively, these studies provide both indirect as well as direct support for the role of DNA methylation in autism.
Methylation of cytosine residues of CpG sites is the most characterized of the epigenetic mechanisms and involves the addition of a methyl group onto the 5′ position of a cytosine residue. Although 60–90% of CpGs are found methylated throughout the genome (24), unmethylated CpG sites can be found clustered in CpG islands most often associated with 5′ promoter regions of genes. Methylation of CpG islands is most often associated with transcriptional silencing and has been found to be a significant contributor to altered gene expression. The mechanism of methylation as a silencing signal is thought to occur by either recruitment of repressive transcriptional silencing machinery, or by steric hindrance preventing the binding of transcription factors necessary for transcriptional activation.
In this study, we use global methylation profiling of discordantly diagnosed monozygotic twins and their nonautistic siblings on CpG island arrays to test the hypothesis that differential gene expression in idiopathic autism is, at least in part, the result of aberrant methylation. Our study reveals distinct methylation differences in multiple genes between the discordant MZ twins as well as common epigenetic differences distinguishing the twins (the undiagnosed twin exhibiting milder autistic traits that are below the threshold for diagnosis) from nonautistic sibling controls. Overlaying methylation profiles with gene expression profiles previously obtained for these individuals (18) revealed specific differentially expressed ASD candidate genes that are potential targets of aberrant methylation. Immunohistochemical staining of the protein products of 2 of the candidate genes that showed increased methylation in LCLs revealed protein reduction in autistic cerebellum and frontal cortex as postulated, demonstrating the usefulness of LCLs as a surrogate model with which to explore mechanisms of dysregulation of gene expression in ASDs.
MATERIALS AND METHODS
Cell culture and 5-Aza-2-deoxycytidine treatment
LCLs, which were obtained from the Autism Genetic Research Exchange (AGRE; Los Angeles, CA, USA), were derived from lymphocytes of autistic and normal siblings and cultured as described previously (18). Demethylating treatments were carried out by incubating cells in culture with 5 μM 5-Aza-2-deoxycytidine (Fluka, St. Louis, MO, USA) or vehicle control, DMSO (Fisher Bio Tech, Pittsburgh, PA, USA), for 48 h before harvesting cells.
DNA and RNA isolation procedures
Genomic DNA was isolated using the Qiagen DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA, USA) and used for bisulfite modification for methylation analyses. Total RNA was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol and used for gene expression analyses by quantitative RT-PCR. The RNA samples were further purified using the RNeasy Mini kit (Qiagen) and tested for integrity on RNA 6000 NanoChips using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA).
Selection of samples
Three pairs of male monozygotic twins discordant for diagnosis of autism as determined by the Autism Diagnostic Interview–Revised (ADI-R) assessment instrument were selected for this study. The undiagnosed co-twin from each set, although not clinically autistic, exhibited some autistic traits and were described as either “broad spectrum” or “not quite autistic” (NQA) by the AGRE repository based on their stated criteria (http://www.agre.org). Cell lines derived from respective nonautistic siblings of 2 pairs of twins were also included in the analyses, in addition to cell lines derived from a set of monozygotic twins unaffected by autism. For confirmation experiments (qRT-PCR, bisulfite sequencing, and methylation-specific PCR), 2 additional case-control pairs of siblings (affected autistic individual and respective unaffected male siblings) were analyzed. LCLs from individuals with specific genetic and chromosomal abnormalities, diagnosed comorbid disorders, or those born prematurely (<35 wk of gestation) were excluded from this study. Additional demographic and diagnostic information for these subjects is included in Supplemental Table 1.
Global methylation profiling using CpG island microarrays
Global methylation profiling on CpG island arrays was performed as described previously by Rauch et al.(25) to screen for differentially methylated autism candidate genes. Briefly, 20 μg of genomic DNA was digested with the frequent cutter, MseI (5′-TTAA). Two unidirectional linkers, oligo-Long (5′-GCGGTGACCCGGGAGATCTGAATTC-3′) and oligo-MseI (5′-TAGAATTCAGATCTCCCG-3′), were annealed and ligated to the MseI digested genomic DNA by incubation at 16°C overnight. Enrichment of the methylated fraction was accomplished by methyl-CpG-island recovery assay (MIRA). Briefly, 1 μg of purified GST-tagged MBD2b protein and 1 μg of purified His-tagged MBD3L1 protein were preincubated with glutathione beads in the presence of SC110 genomic DNA at 4°C on a rocking platform. The beads were then incubated with 1 μg of the digested and linker-ligated genomic DNA in a binding reaction mixture [10 mM Tris-HCl, pH 7.5; 50 mM NaCl; 1 mM EDTA; 1 mM DTT; 3 mM MgCl2; 0.1% Triton-X100; 5% glycerol; 25 μg/ml bovine serum albumin; and 1.25 μg/ml (dcm minus) SC110 bacterial DNA]. The methylation-enriched MIRA fraction was washed 3 times with wash buffer (10 mM Tris-HCl, pH 7.5; 700 mM NaCl; 1 mM EDTA; 3 mM MgCl2; and 0.1% Triton-X-100), eluted from the glutathione beads using the Qiagen QIAquick PCR purification kit, and PCR-amplified. Enriched and unenriched fractions derived from each individual were then indirectly labeled with aminoallyl-dUTP, conjugated with either Alexa 647 or Alexa 555 (Invitrogen), and hybridized onto 8.1 K CpG island microarrays (UHN Microarray Centre, University of Toronto, Toronto, ON, Canada). For all paired analyses, a direct comparison was performed in which the methylation-enriched fractions from 2 individuals were pooled and hybridized onto the same microarray. In addition, indirect comparisons were performed by cohybridizing the methylation-enriched (MIRA) fraction with the respective unenriched DNA fraction obtained from the same individual. For each paired analysis (between autistic MZ twins and/or between autistic co-twin and unaffected sibling), a total number of 4 replicates were performed, including direct and indirect comparisons.
Microarray data analysis
Following hybridization and washing according to established protocols, the CpG island microarray slides were scanned using an Axon GenePix 4000B laser scanner (Axon Instruments, Foster City, CA, USA) to elicit dye intensities for each element on the array. Analysis of raw intensity data was performed using the TIGR TM4 Software Suite (http://www.tm4.org) (26). Raw intensity data were normalized and filtered using LOWESS normalization and sd (variance) regularization programs contained within the MIDAS module of TM4. The normalized intensities were then uploaded into the MeV module, and a 70% data filter was applied to remove CpG island regions for which log2 intensity values are missing in >30% of the samples before statistical analyses of the data. Differentially methylated CpG islands were identified using the Significance Analysis of Microarrays (SAM) (27) module within MeV, which also provided false discovery rates (FDRs) for each analysis. A 1-class SAM analysis identified differential methylation between discordant monozygotic twins as well as across both twins in comparison to unaffected control siblings.
To identify genes that were differentially expressed in these samples, gene expression data from our earlier expression profiling of LCLs from these subjects (18) were downloaded from the Gene Expression Omnibus (GEO) database (GEO accession no. GSE4187) and analyzed using a 70% data filter and a 1-class SAM across all sibling pairs. This analysis resulted in the identification of a total of 3043 differentially expressed genes with an FDR of 10%. Microsoft Access software (Microsoft, Redmond, WA, USA) was used to identify the overlap between differentially methylated and differentially expressed genes between the twins and normal siblings, using gene symbol as the common identifier between datasets. The log2 values indicating relative expression or methylation for the twins vs. the normal siblings were obtained by averaging the values across all replicates.
Quantitative real-time PCR (qRT-PCR) analyses
Two candidate genes, BCL-2 and RORA, were chosen for qRT-PCR analysis to examine the correlation between expression of mRNA transcript and methylation status as predicted by microarray analysis, as well as to examine the effect of 5-Aza-2-deoxycytidine treatment on transcript levels. Total RNA was reverse transcribed into cDNA using the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA) following the manufacturer’s protocol, and qPCR was accomplished using Invitrogen’s Platinum SYBR Green qPCR SuperMix-UDG with ROX. The qPCR reactions (in triplicate) were analyzed on an ABI Prism 7300 Sequence Detection System (Applied Biosystems, Foster City, CA, USA), and transcript levels of BCL-2 and RORA were quantified and normalized to standard curves obtained using universal 18S rRNA primers. Sequences of primers used for qRT-PCR are given in Table 1.
TABLE 1.
Primer sequences
| Gene symbol | Application | Forward primer, 5′→3′ | Reverse primer, 5′→3′ |
|---|---|---|---|
| BCL2 | qPCR | TATTGTGGCTGCACTTGCTC | TGTTGCCCAACTGCAAAATA |
| RORA | qPCR | ACACCAGCATCAGGCTTCTT | GGTCTGCCACGTTATCTGCT |
| BCL2 | Bisulfite sequencing Nested PCR-outer | GGTGGTTTAGAGGAGGGTTTTT | ACAAAAATCCTCTTCTAATTAAACTC |
| BCL2 | Bisulfite sequencing Nested PCR-inner | GAGAATGAAGTAAGAGGATAGGT | CACCCTTTCTCCTCCTCCTAATC |
| RORA | MSP: methylated | GGTTGGAGAAGTTTTCGTTAGC | GACGAACGAACAAACAAAAACG |
| MSP: unmethylated | TTGGTTGGAGAAGTTTTTGTTAGT | CAAACAAAAACACAAAAAAACACA |
Bisulfite modification, bisulfite sequencing, and methylation-specific PCR (MSP)
Two micrograms of genomic DNA isolated from LCLs was treated with sodium bisulfite using the Active Motif MethylDetector kit (Active Motif, Carlsbad, CA, USA) according to the manufacturer’s specifications. Successful bisulfite treatment and conversion was verified using positive control PCR primers for the p16 (CDKN2A) locus provided in the MethylDetector kit.
For direct bisulfite sequencing of the BCL-2 promoter region, bisulfite-modified DNA was used for a nested PCR amplifying the P1 promoter region of BCL-2, which overlapped with the CpG island region (UHNhscpg0002696; chr18:59137439–59137855) contained on the 8.1 K CpG island microarray. Successful PCR amplification was verified by gel electrophoresis. PCR products were extracted from the gel and purified using the Qiagen Qiaquick Gel Extraction kit according to the manufacturer’s specifications. Purified PCR products were directly sequenced using the Applied Biosystems Automated 3730xl DNA Analyzer (Cornell DNA Sequencing Facility, Ithaca, NY, USA) with the reverse primer to obtain methylation status of CpG dinucleotides.
For MSP, 2 sets of primers spanning a region ∼300 bp upstream of the first exon of RORA (UHNhscpg0014103; chr15:59309160-59309428) were designed according to assumed differences in methylation status (i.e., fully methylated or fully unmethylated). These primers were previously successful in identifying methylation-specific silencing of RORA in several gastric cancer cell lines (28). Bisulfite-modified genomic DNA was used as a template for PCR, and PCR products were resolved by agarose gel electrophoresis. Sequences of primers used for bisulfite sequencing analysis of BCL-2 and MSP of RORA are given in Table 1.
Immunohistochemistry
Two different sets of tissue arrays were obtained through the Autism Tissue Program (San Diego, CA, USA). One set of arrays was prepared by Dr. Charles Eberhart (Johns Hopkins Pathology Department Tissue Microarray Core Facility, Baltimore, MD, USA), as described previously (29). The samples included thin sections from the cerebellar cortex of 5 autistic, 1 Rett individual, and 5 age-matched control individuals. Additional clinical data on each case have been previously reported by Eberhart et al.(29). The second set of postmortem tissue arrays are from Dr. Janine LaSalle’s laboratory (University of California, Davis, CA, USA) and contained slices from the BA9 region of the frontal cortex from control and age- and sex-matched autistic individuals as well as individuals with a variety of developmental disorders as described by Nagarajan et al.(30).
Immunohistochemistry for RORA and BCL-2 protein was performed using a standard avidin-biotin complex method of detection described by Eberhart et al.(29). Briefly, tissue array slides were deparaffinized in xylene and rehydrated using a dilution series of 100, 95, and 75% ethanol. Slides were subjected to heat-induced antigen retrieval by incubation in Tris-EDTA buffer (10 nM Tris Base, 1 mM EDTA, 0.05% Tween 20, pH 9.0) at 95°C for 25 min. Slides were then incubated overnight at 4°C with a rabbit polyclonal primary antibody (1:50 dilution) against RORA (4 μg/ml, sc-28612; Santa Cruz Biotechnology, Santa Cruz, CA, USA) or against BCL-2 (4 μg/ml, sc-492; Santa Cruz Biotechnology). Biotinylated secondary goat anti-rabbit antibody (1:200 dilution) was added to slides for 30 min at room temperature. All incubations were conducted in a humidified chamber. Antibodies were detected using the Santa Cruz rabbit ABC Staining System using an avidin-biotin complex method and diaminobenzadine as a chromagen. Fixed slides were imaged using an Olympus BX-60 upright light microscope (Olympus, Tokyo, Japan).
Pathway and functional analyses of differentially methylated/expressed genes
Differentially methylated (or expressed) genes were uploaded into either Ingenuity Pathway Analysis or Pathway Studio 5 (PS5) to discover relationships between the genes as well as their association with various processes and disorders. The Fisher’s exact test was used to identify significant functions and pathways represented within the respective datasets, with the P value indicating the probability that the stated function or process is not associated with the genes in the dataset.
RESULTS
Global methylation profiling of LCLs from discordant monozygotic twins and unaffected siblings reveals differential methylation of genes relevant to neurological functions and disorders
CpG island arrays were used to identify differentially methylated genes from LCLs of monozygotic twins discordant in diagnosis of autism. Methylation-enriched DNA was isolated from genomic DNA using the MIRA pulldown method previously established by Rauch et al.(25). Methylation-enriched and unenriched DNA fractions were then hybridized onto CpG island microarrays to identify CpG islands with differential methylation patterns between individuals. The CpG island microarray analysis identified 73 CpG islands as differentially methylated between discordant monozygotic twins (Supplemental Table 2). Network analyses of the differentially methylated genes immediately at the 3′ end, or overlapping, the CpG islands revealed enrichment of high-level functions biologically relevant to the autism phenotype and included numerous genes involved in neurological disorders as well as nervous system development and function (Table 2). In fact, although only 1 twin of each pair is diagnosed autistic, his co-twin still has autistic traits (which we call “mild”), but falls below the criteria for being diagnosed autistic. Thus, if we compare the combined CpG island data from both co-twins against their respective normal siblings, there are a larger number (201) of differentially methylated CpG islands because the phenotypes involved in the comparisons are distinctly different, even though the genotypes are similar (but not identical) because the twins and their respective nonautistic sibling are genetically related (Supplemental Table 3). Network analysis of this set of differentially methylated genes revealed enrichment for high-level functions shown in Table 3. Interestingly, the function with the highest statistical significance (P=9.02×10−8 by Fisher’s exact test) includes genes involved in transcriptional control of gene expression, many of which are required for proper brain development. Other significant high-level functions represented within the gene dataset are nervous system development and function, neurological disease, and cell death, all of which have been implicated in numerous studies on autism.
TABLE 2.
Four high-level functions associated with genes differentially methylated between discordant monozygotic twins and identified by CpG island microarray
| Category and no. genes | Function annotation | Molecules | P value |
|---|---|---|---|
| Neurological disease; 13 | Huntington disease | ELMO1, EMX2, FOXN3, GABRA4, MEIS2, NDUFB5, XPA | 2.62E-04 |
| Neurodegeneration of CA3 neurons | GRIA2 | 2.23E-03 | |
| Congenital hydrocephalus of mice | E2F5 | 4.46E-03 | |
| Neurological disorder | E2F5, ELMO1, EMX2, FOXN3, GABRA4, GRIA2, MEIS2, NDUFB5, TIAM1, TOP2A, XPA | 6.24E-03 | |
| Hypoplasia of cerebellum | XPA | 8.91E-03 | |
| Encephalopathy | E2F5, EMX2 | 1.61E-02 | |
| Major depression | GABRA4, GRIA2 | 2.01E-02 | |
| Epilepsy, seizures | GABRA4, GRIA2 | 2.09E-02 | |
| Cell death of pyramidal neurons | GRIA2 | 2.87E-02 | |
| Dyskinesia | GRIA2 | 2.87E-02 | |
| Progressive motor neuropathy | GABRA4, GRIA2, TOP2A | 2.95E-02 | |
| Nervous system development and function; 6 | Calcium permeability of neurons, innervation of Purkinje cells | GRIA2 | 2.23E-03 |
| Secretion of cerebrospinal fluid | E2F5 | 2.23E-03 | |
| Dedepression and depotentiation of CA1 neuron | GRIA2 | 4.46E-03 | |
| Formation of cerebellar folia, size of dendrites, neurogenesis | XPA | 4.46E-03 | |
| Size of hippocampus | EMX2 | 6.69E-03 | |
| Formation of neurite branches | TIAM1 | 8.91E-03 | |
| Size of cerebral cortex | EMX2 | 8.91E-03 | |
| Activation of synapse, LTD of CA1 neurons, Purkinje cells, and mossy fibers | GRIA2 | 1.33E-02 | |
| Development of dentate gyrus | EMX2 | 1.77E-02 | |
| Synaptic transmission of pyramidal neurons | GRIA2 | 1.77E-02 | |
| Memory | GRIA2, ITGA5 | 2.62E-02 | |
| Migration of cortical neurons and Schwann cells | TIAM1 | 3.52E-02 | |
| Morphology of neurons | TIAM1 | 4.38E-02 | |
| Cellular assembly and organization; 5 | Retraction of glial cell projections | GRIA2 | 2.23E-03 |
| Structural integrity of nucleosomes | TOP2A | 4.46E-03 | |
| Retraction of plasma membrane projections | GRIA2, TIAM1 | 8.93E-03 | |
| Size of growth cone | TIAM1 | 1.99E-02 | |
| Formation of actin stress fibers | ITGA5, TIAM1 | 4.83E-02 | |
| Embryonic development; 4 | Degeneration of notochord | ITGA5 | 2.23E-03 |
| Formation of gonadal ridge | CBX2 | 2.23E-03 | |
| Neurogenesis of embryonic tissue | XPA | 6.69E-03 | |
| Cell death of neural crest cells | ITGA5 | 1.77E-02 | |
| Cleavage of embryo | TOP2A | 1.77E-02 |
P values represent significance by Fisher’s exact test and indicate the probability that the indicated function is not associated with the dataset of differentially methylated genes (Ingenuity Pathway Analysis software).
TABLE 3.
Four high-level functions associated with genes differentially methylated between monozygotic twins and unaffected siblings and identified by CpG island microarray
| Category and no. genes | Function annotation | Molecules | P value |
|---|---|---|---|
| Gene expression; 30 | Transcription | ATF1, CBX2, CCNB1, CHAF1A, FOXN3, FRAT1, ILK, IRF1, ITGA5, MED7 (includes EG:9443), MEIS1, MLLT1, MTPN, MYF6, NFIA, NR2C1, PAX7, PWP1, RBM39, RORA, RREB1, RRN3, SET, TFDP2, TG, TGFBR2, VDR, ZNF234 | 9.02E-08 |
| Transactivation | ATF1, FRAT1, IRF1, NFIA, PAX7, RBM39, RORA, TFDP2, TGFBR2, VDR | 1.92E-03 | |
| Activation of TGF beta response element | ITGA5, TGFBR2 | 4.93E-03 | |
| Transactivation of response element | VDR | 5.29E-03 | |
| Transactivation of RORE binding site | RORA | 5.29E-03 | |
| Expression of DNA endogenous promoter | TG, VDR | 7.06E-03 | |
| Activation of Atf-1 binding site | ATF1 | 1.05E-02 | |
| Activation of NFI binding site | NFIA | 1.05E-02 | |
| Binding of TREpal motif | VDR | 1.05E-02 | |
| Transcription of beta catenin response element | FRAT1 | 1.05E-02 | |
| Activation of HAF1/HAF1a binding site | IRF1 | 1.58E-02 | |
| Activation of IRF1/IRF2 binding site | IRF1 | 1.58E-02 | |
| Binding of NFAT response element | VDR | 1.58E-02 | |
| Cell death; 24 | Apoptosis | ATF1, BAK1, CCNB1, CDC73, FAS, FRAT1, FXR1, GRIA2, GULP1, HPN, ILK, IRF1, ITGA5, MCF2L, MED7, MTPN, PAX7, PLA2G6, RRN3, TGFBR2, TIAM1, VDR | 4.30E-03 |
| Delay in cell death of cortical neurons | BAK1 | 5.29E-03 | |
| Killing of oligodendrocytes | MBP | 5.29E-03 | |
| Cell death of microglia | FAS, IRF1 | 5.43E-03 | |
| Apoptosis of embryonic cells | ATF1, FAS, ITGA5 | 6.02E-03 | |
| Cell death | ATF1, BAK1, CCNB1, CDC73, FAS, FRAT1, FXR1, GRIA2, GULP1, HPN, ILK, IRF1, ITGA5, MBP, MCF2L, MED7, MEIS1, MTPN, PAX7, PLA2G6, RRN3, TGFBR2, TIAM1, VDR | 6.61E-03 | |
| Apoptosis of embryonic stem cells | FAS, ITGA5 | 7.65E-03 | |
| Cell death of brain cells | BAK1, FAS, FRAT1, GRIA2 | 2.54E-02 | |
| Cell death of cortical neurons | BAK1, FAS, GRIA2 | 2.56E-02 | |
| Neurological disease (12) | Delay in cell death of cortical neurons | BAK1 | 5.29E-03 |
| Killing of oligodendrocytes | MBP | 5.29E-03 | |
| Neurodegeneration of CA3 neurons | GRIA2 | 5.29E-03 | |
| Cell death of microglia | FAS, IRF1 | 5.43E-03 | |
| Disease of brain cells | GRIA2, TG | 1.02E-02 | |
| Depletion of neurons | DLD | 1.05E-02 | |
| Gracile axonal dystrophy | PLA2G6 | 1.05E-02 | |
| Mobility of brain cancer cell lines | ITGA5 | 1.05E-02 | |
| Multiple sclerosis of humans | MBP | 1.58E-02 | |
| Deurodegeneration | GRIA2, PLA2G6, TGFBR2 | 1.76E-02 | |
| Apoptosis of granule cells | FAS, FRAT1 | 1.89E-02 | |
| Cell death of brain cells, cortical neurons | BAK1, FAS, FRAT1, GRIA2 | 2.54E-02 | |
| Apoptosis of neural precursor cells | FAS | 2.62E-02 | |
| Hypomyelination of axons | MBP | 2.62E-02 | |
| Nervous system development and function; 6 | Calcium permeability of neurons, innervation of Purkinje cells, retraction of glial cell projections | GRIA2 | 5.29E-03 |
| Formation of hippocampal commissure, presence/development of corpus callosum, formation of sling cells | NFIA | 5.29E-03 | |
| Dedepression and depotentiation of CA1 neurons | GRIA2 | 1.05E-02 | |
| Organization of axons, organization of central nervous system | MBP | 1.05E-02 | |
| Quantity of satellite cells | PAX7 | 1.05E-02 | |
| Cilation of cerebral ventricles | NFIA | 1.58E-02 | |
| Function of brain | RORA | 2.10E-02 | |
| Formation of neurite branches | TIAM1 | 2.10E-02 | |
| Migration of neuroglia | NFIA, TIAM1 | 2.26E-02 |
P values represent significance by Fisher’s exact test and indicate the probability that the indicated function is not associated with the dataset of differentially methylated genes (Ingenuity Pathway Analysis software).
Overlap of differentially methylated genes with differentially expressed genes
To prioritize ASD candidate genes for further analysis, differentially methylated genes from the analysis of twins vs. normal siblings were compared to differentially expressed genes derived from prior gene expression profiling of these same samples on DNA microarrays (18). Supplemental Table 4 lists the genes that are both differentially expressed and methylated, and Fig. 1A displays the inverse relationship between expression level and methylation level for all but one of these genes. Network analysis was then performed to examine the relationship between this set of genes and biological processes. As shown in Fig. 1B, many of the associated processes within the network, including synaptic regulation, fetal development, morphogenesis, apoptosis, inflammation, digestion, steroid biosynthesis, and mental deficiency, have been associated with autism. Two genes from this network, BCL-2 and RORA, were selected for further study because of their respective roles in apoptosis and morphogenesis/inflammation. Interestingly, BCL-2 protein has been previously demonstrated to be reduced in the cerebellum and frontal cortex of autistic subjects relative to control subjects (31, 32), but RORA, a nuclear steroid hormone receptor and transcriptional activator that is involved in Purkinje cell differentiation (33) and cerebellar development (34), has never before been implicated in autism. In addition, RORA, a regulator of circadian rhythm (35), is also neuroprotective against inflammation and oxidative stress (36), both of which are increased in autism (37, 38).
Figure 1.

Overlap of dysregulated genes identified from CpG island microarray and global gene expression profiling previously performed using the same samples. A) Scatter plot shows inverse correlation of log2 ratios of gene expression and methylation of genes with differentially methylated CpG islands directly overlapping with its 5′ end. Inset: plot of log2 ratios of genes with differentially methylated CpG islands located either upstream or downstream of the CpG island. Interestingly, the majority of genes identified from these analyses were hypermethylated in the twins relative to their respective nonautistic siblings, with a corresponding decrease in gene expression. B) Network analysis of 25 genes (circumscribed in blue) that were both differentially methylated and expressed. The network was generated using Pathway Studio 5 network prediction software and identified common biological themes, including apoptosis, cellular differentiation, and inflammation. The analysis also revealed neurologically relevant functions and disorders including synaptic regulation, development, and mental deficiency.
Confirmation of differential methylation of specific CpG sites in upstream promoter regions of BCL-2 and RORA
The CpG islands immediately upstream of BCL-2 and RORA were chosen for further confirmation of methylation status in LCLs of autistic and control individuals. Specifically, a 417 bp CpG island overlapping the P1 promoter region of the BCL-2 gene (chr18:59137439-59137855) was identified as hypermethylated in all co-twins exhibiting autistic traits compared to unaffected siblings in the CpG island microarray, and a 269-bp CpG island associated with the 5′ end of the RORA1 isoform (chr15:59309160-59309428) was identified as hypermethylated in the same analysis. To obtain high-resolution information regarding which specific CpG sites may be hypermethylated in autistic individuals, 2 separate experimental approaches taking advantage of bisulfite modification of genomic DNA were employed: bisulfite sequencing and methylation-specific PCR. Bisulfite modification of genomic DNA results in deamination of all unmethylated cytosine residues to uracils, while methylated cytosine residues are protected against conversion.
Figure 2 displays the results from bisulfite sequencing of the BCL-2 promoter region after bisulfite treatment of genomic DNA isolated from LCLs derived from the monozygotic twins, their unaffected siblings, an additional autistic/unaffected sibling pair (A_2020/C_2019), and a pair of unaffected monozygotic twins (C_2744/C_2745). Following bisulfite conversion, nested primers were designed to amplify across the P1 promoter region of BCL-2 (Fig. 2A). This is the same region interrogated by the CpG island microarrays and spans 38 total CpG sites. As shown in Fig. 2B, an overall increase in percentage methylation across the 38 CpG sites (∼25% on average) was observed in the diagnosed autistic samples in comparison to an average of 11.4% for the undiagnosed co-twins and 9.5% for the unaffected siblings. Increased methylation was especially noted on CpG sites 3–13 for autistic individuals only. Also, there appears to be a decrease in methylation at site 34 in the autistic samples that is found methylated in 4 of 5 control individuals. Interestingly, the same methylated CpG sites are not observed across all autistic subjects (with the exception of CpG site no. 12), suggesting that regulation is not dependent on specific sites, but instead may involve the entire region encompassed by CpG sites 3–13.
Figure 2.

A) Bisulfite sequencing of the BCL-2 P1 promoter region. B). Bisulfite sequencing was performed across 38 CpG sites from genomic DNA isolated from LCLs of discordant MZ twins. Autistic co-twins are designated by A_ preceding the blood identifier number (e.g., A_809); undiagnosed co-twins are designated by M_ preceding the blood number (e.g., M_810). Control (C) individuals include unaffected siblings (e.g., C_813), one normal monozygotic twin pair (C_2744 and C_2745). A pair of siblings, one autistic (A_2020) and one unaffected (C_2019), was also included in this analysis. Inset: average percentage methylation with sd across all 38 CpG sites for each group of samples.
MSP was utilized to analyze methylation in a region upstream of RORA because of the presence of homopolymer sequences between the CpG sites of interest that would complicate sequencing analyses. This region overlaps with the region interrogated by the CpG island microarray and is located ∼300 bp before the first exon of the RORα1 isoform (Fig. 3A). Two sets of primers encompassing several CpG sites were designed according to assumed differences in methylation status to amplify the same region upstream of RORA. These particular primers were previously successful in identifying epigenetic silencing of RORA in several gastric cancer cell lines (28). The presence of a PCR product corresponding to methylated DNA only in the vehicle-treated samples of autistic individuals in contrast to those of the unaffected controls (Fig. 3B) confirms methylation of the RORA promoter region only in the autistic individuals. The absence of methylated product in the autistic samples following treatment with 5 μM 5-Aza-2-deoxycytidine further confirms methylation in the untreated autistic samples and specificity of the methylated PCR primers.
Figure 3.

A) Representative MSP image of the upstream CpG island region of RORA in LCL from autistic (A_737 and A_2020) and unaffected control (C_735 and C_2019) siblings. LCLs were treated with DMSO (vehicle control) or 5 uM of 5-Aza-2-deoxycytidine for 48 h. Genomic DNA was isolated and bisulfite modified. B) Modified DNA was used for PCR containing primers specific for unmethylated (U) and methylated (M) CpG sites of RORA.
Confirmation of methylation-dependent silencing of candidate genes identified by CpG island arrays
Quantitative RT-PCR was used to confirm decreased expression of BCL-2 and RORA in autistic samples and to evaluate the effect of a global methylation inhibitor, 5-Aza-2-deoxycytidine, on gene expression. For both BCL-2 and RORA, gene expression was significantly higher (P<0.05) in the unaffected control than autistic co-twins (Fig. 4A). Generally, the diagnosed autistic co-twin (_A) had the lowest level of expression of BCL-2 and RORA, while the milder undiagnosed co-twin (_M) exhibited transcript levels between that observed for unaffected sibling controls and autistic co-twins. This suggests a quantitative relationship between phenotype and gene expression of these 2 genes, although additional studies are required to confirm this observation. BCL-2 and RORA gene expression was found to be methylation-dependent because both transcripts were up-regulated by 1.5- and 1.8-fold, respectively, following global inhibition of methylation with 5-Aza-2-deoxycytidine (Fig. 4B). By contrast, there were no statistically significant increases in the expression levels of BCL-2 and RORA in the LCLs from the “mild” co-twins with 5-Aza-2-deoxycytidine treatment.
Figure 4.

qRT-PCR of BCL-2 and RORA transcripts from LCLs of 3 pairs of discordant twins and respective sibling controls for 2 pairs of twins. A) Percentage of gene expression of autistic co-twin (#_A). B) RNA isolated from LCLs following 48 h treatment with 5 μM 5-Aza-2-deoxycytidine was also analyzed by qRT-PCR. Results are average ± se fold change in expression following treatment.
RORA and BCL-2 proteins are also reduced in post mortem brain tissues of autistic subjects
To explore the relevance of our findings of enhanced methylation and reduced expression of RORA and BCL-2 in LCLs to the expression of these proteins in the brain, the levels of RORA and BCL-2 protein were investigated in postmortem tissues from autistic subjects and age- and sex-matched controls, using tissue arrays containing sections from both the cerebellum and frontal cortex (29, 30). Immunohistochemical staining of the tissue arrays confirmed reduced levels of RORA protein in the cerebellum of autistic individuals, including the molecular cell, granule cell, and Purkinje cell layers, where expression of RORA has been previously demonstrated (34, 39, 40). Figure 5 illustrates representative immunohistochemical staining results for RORA protein in cerebellum of autistic subjects in comparison to that of respective age- and sex-matched controls. Immunoreactivity was decreased in most autistic samples (male: Fig. 5A, C, I; female: Fig. 5E, G) as indicated by the decreased extent of RORA staining. As reduced expression of RORA (as well as other regulators of the circadian rhythm) was observed only in the subtype of ASD exhibiting severe language impairment (41), we do not expect to see reduced RORA protein in all autistic brain samples.
Figure 5.
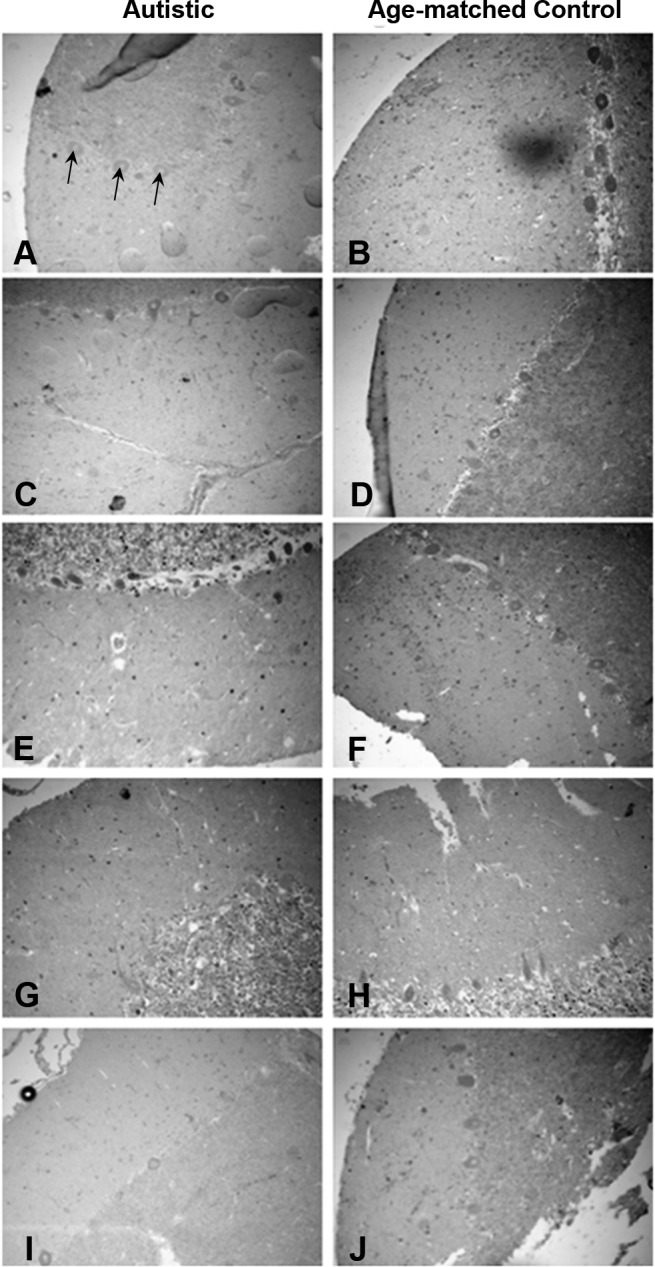
Immunohistochemical staining for RORA protein in the cerebellum. Immunohistochemistry for RORA protein was performed using a standard avidin-biotin complex method of detection, as described in Materials and Methods. Representative images (×20) were taken from the autism tissue array (prepared by Dr. Charles Eberhart, Johns Hopkins University, Baltimore, MD, USA), which contained tissue cores from 5 autistic brains (A, C, E, G, I) and age-matched controls (B, D, F, H, J). Samples E, F, G, H were from female donors. It should be noted that not all autistic samples exhibit reduced RORA staining. This is expected because we observed that only the autistic phenotype associated with severe language impairment exhibited reduced RORA expression (41). Arrows indicate Purkinje cells, which are the largest cells in the sections.
Immunohistochemistry of post mortem brain tissue also revealed decreased BCL-2 protein in the granule cell and Purkinje cell layers of the cerebellum, with 3 of 5 autistic samples showing reduced immunoreactivity for BCL-2 protein (Fig. 6). This reduction in BCL-2 in the post mortem cerebellum of autistic subjects is consistent with that reported previously by Fatemi et al.(31), who performed Western analyses on whole tissue extracts from the cerebellum. Higher-magnification representative images of the cerebellar sections show that both RORA and BCL-2 protein are specifically reduced in Purkinje cells (Fig. 7). RORA protein was observed in both the cytoplasm and nucleus, as reported previously for different isoforms of RORA (42), while counterstaining with hematoxylin revealed that BCL-2 is confined to the cytoplasm. Figure 8 shows that RORA is also reduced in the majority autistic tissue sections vs. age- and sex-matched control sections from the frontal cortex (BA9 region), suggesting that RORA deficiency is not limited to the cerebellum. This deficiency may contribute to reduced protection against both oxidative stress and neural inflammation, which have both been demonstrated in autism (37, 38).
Figure 6.

Immunohistochemical staining for BCL-2 protein in the cerebellum. Immunohistochemistry for BCL-2 protein was performed as described in Materials and Methods for RORA protein with the substitution of a primary antibody for BCL-2 instead of for RORA. In addition, the tissue was counterstained with hematoxylin to reveal the nuclei. Tissue samples from 5 autistic brains (A, C, E, G, I) and age-matched controls (B, D, F, H, J); samples E, F, G, H were from female donors. Images were taken at ×20. Arrows indicate Purkinje cells.
Figure 7.

Higher magnification (×40) images of immunohistochemical staining for RORA and BCL-2 on cerebellar sections from autistic and age-matched control subjects. ML, molecular layer; PL, Purkinje layer; GL, granule layer. Arrows indicate Purkinje cells.
Figure 8.
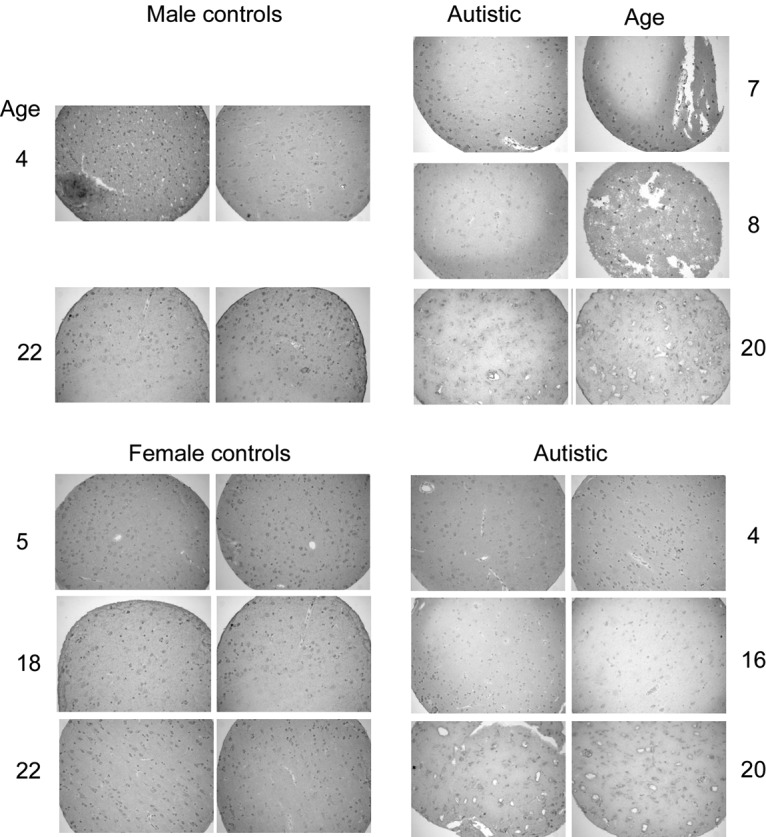
RORA protein also appears to be reduced in the frontal cortex (BA9) in the majority of tissue sections from age- and sex-matched autistic and control subjects. This tissue array, obtained through the Autism Tissue Program, was prepared as described by Nagarajan et al.(30) in the laboratory of Dr. Janine LaSalle (University of California, Davis, CA, USA). Immunohistochemical staining for RORA was accomplished as in Fig. 5.
DISCUSSION
In this study, we used microarray-based epigenomic profiling of LCLs from discordant monozygotic autistic twins and unaffected siblings to test the hypothesis that DNA methylation is involved in global dysregulation of gene expression in ASDs and to screen for candidate genes whose expression is potentially linked to aberrant DNA methylation. We observed DNA-methylation differences in numerous loci containing genes potentially important to the pathobiology of autism. In particular, functional and pathway analyses of the differentially methylated/expressed genes showed enrichment of genes involved in inflammation and apoptosis, cellular differentiation, brain morphogenesis, growth rate, cytokine production, myelination, synaptic regulation, learning, and steroid biosynthesis, all of which have been shown to be altered in ASDs. The candidate genes were prioritized for further analyses by identifying the overlap between the differentially methylated genes and those that had been shown to be differentially expressed in the same set of samples in previous gene expression analyses (18). Pathway analyses of this filtered set of genes thus focused our attention on 2 genes, BCL-2 and RORA, as potential candidate genes for ASDs whose expression may be dysregulated by aberrant methylation.
BCL-2 is a candidate gene for autism
BCL-2 is an antiapoptotic protein located in the outer mitochondrial membrane that is important for cell survival under a variety of stressful conditions. Fatemi et al.(31) measured levels of BCL-2 in the cerebellar cortex of 5 autistic males and found a 34–51% reduction in levels of BCL-2 protein by Western analyses. Subsequent studies also found an ∼30% reduction of BCL-2 protein in the parietal and superior frontal cortex of adult autistic males (32). This marked decrease in BCL-2 protein was concomitant with an increase in p53 expression in the same brain regions. Interestingly, abnormalities in BCL-2 and p53 correlated with the presence of severe mental retardation in these patients. From these studies, it was postulated that dysfunctional apoptotic regulation may lead to altered development of these brain regions, thereby resulting in decreased cognitive function. Our data provide further evidence of the involvement of reduced BCL-2 in ASDs. Moreover, our study provides new evidence that increased DNA methylation of the P1 promoter region of BCL-2 is a possible mechanism for increased gene silencing and decreased protein abundance of BCL-2 in the autistic brain. In contrast to the Western analyses, these studies also localized the changes in BCL-2 to Purkinje cells in the cerebellum.
RORA is a novel candidate gene for autism
RORA is a member of the NR1 subfamily of nuclear hormone receptors and is involved in transcriptional regulation of many genes (34, 43, 44). Among its many relevant functions are regulation of the circadian clock by activation of BMAL1 (35), neuroprotection in the face of oxidative stress and inflammation (36), survival and differentiation of Purkinje cells (33), and cerebellar development (34). All of these associated functions implicate RORA as a potential autism susceptibility gene. In particular, deficiency of Purkinje cells is one of the most consistently reported neuroanatomical abnormalities in post mortem brain as reviewed by Palmen et al.(45) and Polleux and Lauder (46). Moreover, neuroinflammation and oxidative stress have both been shown to be associated with neuropathology in autism (37, 38). Recently, the role of circadian rhythm in ASDs has been highlighted both by genetic studies (47,48,49) as well as our own findings that show that multiple regulators of circadian rhythm, including RORA, are aberrantly expressed in LCLs from autistic individuals with severe language impairment (41). For these reasons, we decided to confirm the methylation and expression differences of RORA in LCLs, and to further explore the involvement of RORA in autistic brain pathology.
As shown in Figs. 3 and 4, respectively, RORA was confirmed to be inversely differentially methylated and expressed in LCLs from autistic vs. nonautistic siblings, with expression dependent on methylation, as demonstrated by the absence of methylation in the presence of 5-Aza-2-deoxycytidine. Notably, we also show by immunohistochemical staining of cerebellar and frontal cortex regions of autistic vs. normal brain (Figs. 5, 8), that RORA protein is noticeably reduced in the majority of the autistic samples relative to age- and sex-matched controls. This reduction is also specifically demonstrated in Purkinje cells, which are dependent on RORA for both survival and differentiation (Fig. 7). These findings thus link molecular changes identified in a peripheral cell model of ASDs to actual pathological changes in the autistic brain, suggesting that LCLs is an appropriate surrogate for studies on autism.
An animal model for RORA deficiency: Possible relationship between staggerer (RORA-null) mouse phenotype and autism
Although RORA deficiency has not been previously reported in the context of autism, a natural mutant mouse strain called staggerer (RORAsg/sg) was found to contain a 6.4-kb intragenic deletion in the fifth exon of RORA and exhibits an identical phenotype to that of RORA-null mice (50). Both RORA-deficient mouse strains display ataxia and severe cerebellar atrophy, characterized by reduction in size and number of Purkinje and granule cells that display disordered cellular architecture with underdeveloped dendrites and immature synaptic connections (34). In addition to ataxia, hypotonia, and cerebellar atrophy, these animal models of RORA deficiency replicate some of the characteristics often associated with autism. Although much attention has been focused on the motor and muscle phenotypes of the staggerer mouse, other phenotypes have been reported for the heterozygous RORA-knockout mouse, particularly on aging. These additional phenotypes include atherosclerosis, elevated expression of inflammatory cytokines, abnormalities in bone tissue, as well as defects in muscle differentiation (51). Although there are few descriptions of behavioral phenotypes of RORA-knockout or staggerer mice, there is some evidence that RORA-deficient animals exhibit autistic-like restricted behaviors such as limited maze patrolling (52), anomalous spatial learning, reduced exploration, and perseverative behavior in comparison to wild-type mice (53,54,55,56). Because of RORA’s potential relevance to ASDs, it is clear that the behavioral phenotype of RORA-deficient (staggerer) mice requires additional study, particularly with respect to social behaviors that are prominently affected in autism.
Gene expression profiling of the staggerer mouse vs. the wild-type animal revealed a set of 36 genes that were differentially expressed (57), including 6 genes that overlapped with the set of genes that we identified as differentially expressed in autistic individuals with severe language impairment relative to controls (41). Thus, we undertook a pathway analysis of the differentially expressed genes resulting from the full staggerer mutation to identify other potential functions and disorders that might be associated with this mouse model. Supplemental Table 5 shows a partial list of the cellular processes and disorders that were revealed by our Pathway Studio 5 analysis of the differentially expressed genes identified in the staggerer mouse (57). The network of genes, cellular processes, and disorders specifically related to RORA is shown in Fig. 9. It is notable that many of the processes and associated disorders are reminiscent of those related to autism spectrum disorders, with dysregulated genes converging on the glutamatergic synapse (34). Coupled with the known involvement of RORA in Purkinje cell survival and cerebellar development, defects also noted in autism, these findings strongly implicate RORA as a candidate gene for autism susceptibility.
Figure 9.

Genes, cellular processes (yellow rectangles), disorders (purple), metabolites (green), and functional complexes (orange) associated with RORA by pathway analyses using Pathway Studio. Genes highlighted in blue were found to be differentially expressed between autistic individuals with severe language impairment and nonautistic controls in our recent study profiling gene expression in several autistic subtypes (41); genes highlighted in yellow were associated with autism in other studies.
Role of DNA methylation as an epigenetic mechanism for dysregulation of gene expression in autism
The broad functions of the methylation-regulated differentially expressed genes identified in this study provide support for our hypothesis that the pathological symptoms of autism may, at least in part, be the result of systemic dysregulation of gene expression. Bioinformatic analysis of the differentially methylated genes from the CpG island assays (Tables 2 and 3) revealed enrichment for many high-level functions previously implicated in autism, including aberrant response to inflammation (38, 58), decreased cell survival (31), and abnormal cellular differentiation leading to abnormal brain development (59). In addition, these processes reiterate common biological themes in autism that our laboratory has identified from global gene expression profiling of LCLs in discordant monozygotic twins (18), sibling pairs (60), and large-scale case-control studies of unrelated subjects (41).
Coupling methylation profiles with global gene expression profiles of the same autistic and control individuals provides a means of identifying high-priority candidate genes for autism. Confirmation of the postulated decrease in protein products of 2 of the candidate genes in post mortem brain tissue of autistic individuals highlights the relevance of our findings in LCLs to the autistic brain. This ability to translate findings from peripheral tissues to the brain is consistent with reports of individuals with neuropsychiatric disorders having detectable epigenetic programming disturbances, both in the brain and in secondary tissues (61). Recently, Gregory et al.(23) reported that the methylation status of the oxytocin receptor in the brain replicated that found in LCLs, providing further support for the ability to link molecular alterations in blood to that in the brain of autistic individuals. Thus, LCLs and primary blood lymphocytes have been demonstrated to be reasonable surrogate experimental models of autism to identify both expressed and epigenetic alterations that may also have diagnostic potential as biomarkers for autism (62,63,64,65).
CONCLUSIONS
Global methylation profiling of LCLs from phenotypically discordant monozygotic twins and nonautistic siblings highlights the role of epigenetic regulation in idiopathic autism and reveals DNA methylation as a mechanism through which this regulation may occur. Two candidate genes, BCL-2 and RORA, which were identified from our coupling of methylation with gene expression data, exhibited increased methylation of specific CpG sites in respective upstream regulatory CpG islands that coincided with methylation-specific gene silencing. Translation of these findings from LCLs to the detection of decreased BCL-2 and RORA protein in post mortem brain tissues of autistic individuals further confirms the feasibility of using LCLs as a surrogate model for autism, particularly when investigating dysregulated genes with systemic functions, such as apoptosis and circadian rhythm. In addition to identifying key autism candidate genes, these studies also yield further insight into the pathobiology of this complex disorder by elucidating global epigenomic modifications relevant to the autistic phenotype. Aside from shedding light on higher-order regulation of gene expression, another compelling reason to investigate epigenetic mechanisms in idiopathic autism is that such modifications can be influenced by exposure to biological modulators and environmental factors. Epigenetics may thus mediate the interaction between genotype and intrinsic (biological) or extrinsic (environmental) factors contributing to ASDs.
Supplementary Material
Acknowledgments
This work was supported by a grant from Autistic Speaks (2381 to V.W.H.) and, in part, by U.S. National Institutes of Health grant R21 MH073393 (V.W.H.). The authors are grateful to Dr. Anastas Popratiloff (Director, Center for Microscopy and Image Analysis at George Washington University Medical Center) for his assistance with microscopy. The authors also thank Dr. Charles Eberhart (Johns Hopkins University, Baltimore, MD, USA) and Dr. Janine LaSalle (University of California, Davis, CA, USA) for making the brain tissue arrays available to us through the Autism Tissue Program, and the families of the donors for their invaluable gift to autism research. The authors gratefully acknowledge the resources (both LCL and phenotypic data) provided by the Autism Genetic Resource Exchange (AGRE) Consortium and the participating AGRE families. AGRE is a program of Autism Speaks and is supported, in part, by grant 1U24MH081810 from the National Institute of Mental Health to Clara M. Lajonchere (PI). The AGRE Consortium: Dan Geschwind, M.D., Ph.D., University of California–Los Angeles (UCLA), Los Angeles, CA, USA; Maja Bucan, Ph.D., University of Pennsylvania, Philadelphia, PA, USA; W. Ted Brown, M.D., Ph.D., F.A.C.M.G., New York State Institute for Basic Research in Developmental Disabilities, Long Island, NY, USA; Rita M. Cantor, Ph.D., UCLA School of Medicine, Los Angeles, CA, USA; John N. Constantino, M.D., Washington University School of Medicine, St. Louis, MO, USA; T. Conrad Gilliam, Ph.D., University of Chicago, Chicago, IL, USA; Martha Herbert, M.D., Ph.D., Harvard Medical School, Boston, MA, USA; Clara Lajonchere, Ph.D., Cure Autism Now, Los Angeles, CA, USA; David H. Ledbetter, Ph.D., Emory University, Atlanta, GA, USA; Christa Lese-Martin, Ph.D., Emory University, Atlanta, GA, USA; Janet Miller, J.D., Ph.D., Cure Autism Now, Los Angeles, CA, USA; Stanley F. Nelson, M.D., UCLA School of Medicine, Los Angeles, CA, USA; Gerard D. Schellenberg, Ph.D., University of Washington, Seattle, WA, USA; Carol A. Samango-Sprouse, Ed.D., George Washington University, Washington, DC, USA; Sarah Spence, M.D., Ph.D., UCLA, Los Angeles, CA, USA; Matthew State, M.D., Ph.D., Yale University, New Haven, CT, USA; Rudolph E. Tanzi, Ph.D., Massachusetts General Hospital, Boston, MA, USA. The authors’ contributions are as follows: A.N. performed all of the experiments in this study and cowrote the manuscript in partial fulfillment of the requirements for the master’s degree in biochemistry at George Washington University; T.A.R. and G.P.P. generously provided the purified GST-tagged MBD2b and His-tagged MBD3L1 proteins as well as advice on the MIRA procedures; V.W.H. conceived and designed the study, provided overall supervision, and wrote the manuscript.
References
- Volkmar F. R., Klin A., Siegel B., Szatmari P., Lord C., Campbell M., Freeman B. J., Cicchetti D. V., Rutter M., Kline W. Field trial for autistic disorder in DSM-IV. Am J Psychiatry. 1994;151:1361–1367. doi: 10.1176/ajp.151.9.1361. [DOI] [PubMed] [Google Scholar]
- Siegel B. Toward DSM-IV: a developmental approach to autistic disorder. Psychiatr Clin N Am. 1991;14:53–68. [PubMed] [Google Scholar]
- Bailey A., Le Couteur A., Gottesman I., Bolton P., Simonoff E., Yuzda E., Rutter M. Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med. 1995;25:63–77. doi: 10.1017/s0033291700028099. [DOI] [PubMed] [Google Scholar]
- Folstein S., Rutter M. Infantile autism: a genetic study of 21 twin pairs. J Child Psychol Psychiatry. 1977;18:297–321. doi: 10.1111/j.1469-7610.1977.tb00443.x. [DOI] [PubMed] [Google Scholar]
- Schanen N. C. Epigenetics of autism spectrum disorders. Hum Mol Genet. 2006;15(2):R138–R150. doi: 10.1093/hmg/ddl213. [DOI] [PubMed] [Google Scholar]
- Beaudet A. L. Autism: highly heritable but not inherited. Nat Med. 2007;13:534–536. doi: 10.1038/nm0507-534. [DOI] [PubMed] [Google Scholar]
- Nakayama A., Masaki S., Aoki E. Genetics and epigenetics in autism. Nihon Shinkei Seishin Yakurigaku Zasshi. 2006;26:209–212. [In Japanese] [PubMed] [Google Scholar]
- Mehler M. F., Mattick J. S. Non-coding RNAs in the nervous system. J Physiol. 2006;575:333–341. doi: 10.1113/jphysiol.2006.113191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champagne F. A. Epigenetic mechanisms and the transgenerational effects of maternal care. Front Neuroendocrinol. 2008;29:386–397. doi: 10.1016/j.yfrne.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang J. C., Jones P. A. Epigenetics and microRNAs. Pediatr Res. 2007;61:24R–29R. doi: 10.1203/pdr.0b013e3180457684. [DOI] [PubMed] [Google Scholar]
- Van Vliet J., Oates N. A., Whitelaw E. Epigenetic mechanisms in the context of complex diseases. Cell Mol Life Sci. 2007;64:1531–1538. doi: 10.1007/s00018-007-6526-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminsky Z., Wang S. C., Petronis A. Complex disease, gender and epigenetics. Ann Med. 2006;38:530–544. doi: 10.1080/07853890600989211. [DOI] [PubMed] [Google Scholar]
- Lopez-Rangel E., Lewis M. E. Loud and clear evidence for gene silencing by epigenetic mechanisms in autism spectrum and related neurodevelopmental disorders. Clin Genet. 2006;69:21–22. doi: 10.1111/j.1399-0004.2006.00543a.x. [DOI] [PubMed] [Google Scholar]
- Feng Y., Zhang F., Lokey L. K., Chastain J. L., Lakkis L., Eberhart D., Warren S. T. Translational suppression by trinucleotide repeat expansion at FMR1. Science. 1995;268:731–734. doi: 10.1126/science.7732383. [DOI] [PubMed] [Google Scholar]
- Li Z., Zhang Y., Ku L., Wilkinson K. D., Warren S. T., Feng Y. The fragile X mental retardation protein inhibits translation via interacting with mRNA. Nucleic Acids Res. 2001;29:2276–2283. doi: 10.1093/nar/29.11.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Den Veyver I. B., Zoghbi H. Y. Mutations in the gene encoding methyl-CpG-binding protein 2 cause Rett syndrome. Brain Dev. 2001;23:S147–S151. doi: 10.1016/s0387-7604(01)00376-x. [DOI] [PubMed] [Google Scholar]
- Amir R. E., Van den Veyver I. B., Wan M., Tran C. Q., Francke U., Zoghbi H. Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Hu V. W., Frank B. C., Heine S., Lee N. H., Quackenbush J. Gene expression profiling of lymphoblastoid cell lines from monozygotic twins discordant in severity of autism reveals differential regulation of neurologically relevant genes. BMC Genomics. 2006;7:118. doi: 10.1186/1471-2164-7-118. doi: 10.1186/1471-2164-7-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan R. P., Hogart A. R., Gwye Y., Martin M. R., LaSalle J. M. Reduced MeCP2 expression is frequent in autism frontal cortex and correlates with aberrant MECP2 promoter methylation. Epigenetics. 2006;1:e1–e11. doi: 10.4161/epi.1.4.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loat C. S., Curran S., Lewis C. M., Duvall J., Geschwind D., Bolton P., Craig I. W. Methyl-CpG-binding protein 2 polymorphisms and vulnerability to autism. Genes Brain Behav. 2008;7:754–760. doi: 10.1111/j.1601-183X.2008.00414.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies W., Isles A. R., Wilkinson L. S. Imprinted genes and mental dysfunction. Ann Med. 2001;33:428–436. doi: 10.3109/07853890108995956. [DOI] [PubMed] [Google Scholar]
- Hogart A., Nagarajan R. P., Patzel K. A., Yasui D. H., Lasalle J. M. 15q11–13 GABAA receptor genes are normally biallelically expressed in brain yet are subject to epigenetic dysregulation in autism-spectrum disorders. Hum Mol Genet. 2007;16:691–703. doi: 10.1093/hmg/ddm014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory S. G., Connelly J. J., Towers A. J., Johnson J., Biscocho D., Markunas C. A., Lintas C., Abramson R. K., Wright H. H., Ellis P., Langford C. R., Worley G., Delong B. R., Murphy S. K., Cuccaro M. L., Persico A., Pericak-Vance M. A. Genomic and epigenetic evidence for oxytocin receptor deficiency in autism. BMC Med. 2009;7:62. doi: 10.1186/1741-7015-7-62. doi: 10.1186/1741-7015-7-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose R. J., Bird A. P. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Rauch T., Li H., Wu X., Pfeifer G. P. MIRA-assisted microarray analysis, a new technology for the determination of DNA methylation patterns, identifies frequent methylation of homeodomain-containing genes in lung cancer cells. Cancer Res. 2006;66:7939–7947. doi: 10.1158/0008-5472.CAN-06-1888. [DOI] [PubMed] [Google Scholar]
- Quackenbush J. Microarray data normalization and transformation. Nat Genet. 2002;32:496–501. doi: 10.1038/ng1032. [DOI] [PubMed] [Google Scholar]
- Tusher V. G., Tibshirani R., Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–51121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita S., Tsujino Y., Moriguchi K., Tatematsu M., Ushijima T. Chemical genomic screening for methylation-silenced genes in gastric cancer cell lines using 5-aza-2′-deoxycytidine treatment and oligonucleotide microarray. Cancer Sci. 2006;97:64–71. doi: 10.1111/j.1349-7006.2006.00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhart C. G., Copeland J., Abel T. W. Brief report: S6 ribosomal protein phosphorylation in autistic frontal cortex and cerebellum: a tissue array analysis. J Autism Dev Disord. 2006;36:1131–1135. doi: 10.1007/s10803-006-0135-9. [DOI] [PubMed] [Google Scholar]
- Nagarajan R. P., Hogart A. R., Gwye Y., Martin M. R., Lasalle J. M. Reduced MeCP2 expression is frequent in autism frontal cortex and correlates with aberrant MECP2 promoter methylation. Epigenetics. 2006;1:172–182. doi: 10.4161/epi.1.4.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi S. H., Stary J. M., Halt A. R., Realmuto G. R. Dysregulation of reelin and bcl-2 proteins in autistic cerebellum. J Autism Dev Disord. 2001;31:529–535. doi: 10.1023/a:1013234708757. [DOI] [PubMed] [Google Scholar]
- Fatemi S. H., Halt A. R. Altered levels of Bcl2 and p53 proteins in parietal cortex reflect deranged apoptotic regulation in autism. Synapse. 2001;42:281–284. doi: 10.1002/syn.10002. [DOI] [PubMed] [Google Scholar]
- Boukhtouche F., Doulazmi M., Frederic F., Dusart I., Brugg B., Mariani J. RORα, a pivotal nuclear receptor for Purkinje neuron survival and differentiation: from development to ageing. Cerebellum. 2006;5:97–104. doi: 10.1080/14734220600750184. [DOI] [PubMed] [Google Scholar]
- Gold D. A., Gent P. M., Hamilton B. A. RORα in genetic control of cerebellum development: 50 staggering years. Brain Res. 2007;1140:19–25. doi: 10.1016/j.brainres.2005.11.080. [DOI] [PubMed] [Google Scholar]
- Akashi M., Takumi T. The orphan nuclear receptor RORα regulates circadian transcription of the mammalian core-clock Bmal1. Nat Struct Mol Biol. 2005;12:441–448. doi: 10.1038/nsmb925. [DOI] [PubMed] [Google Scholar]
- Boukhtouche F., Vodjdani G., Jarvis C. I., Bakouche J., Staels B., Mallet J., Mariani J., Lemaigre-Dubreuil Y., Brugg B. Human retinoic acid receptor-related orphan receptor α1 overexpression protects neurones against oxidative stress-induced apoptosis. J Neurochem. 2006;96:1778–1789. doi: 10.1111/j.1471-4159.2006.03708.x. [DOI] [PubMed] [Google Scholar]
- Pardo C. A., Vargas D. L., Zimmerman A. W. Immunity, neuroglia and neuroinflammation in autism. Int Rev Psychiatry. 2005;17:485–495. doi: 10.1080/02646830500381930. [DOI] [PubMed] [Google Scholar]
- Chauhan A., Chauhan V. Oxidative stress in autism. Pathophysiology. 2006;13:171–181. doi: 10.1016/j.pathophys.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Hadj-Sahraoui N., Frederic F., Zanjani H., Delhaye-Bouchaud N., Herrup K., Mariani J. Progressive atrophy of cerebellar purkinje cell dendrites during aging of the heterozygous staggerer mouse (Rora+/sg) Dev Brain Res. 2001;126:201–209. doi: 10.1016/s0165-3806(01)00095-5. [DOI] [PubMed] [Google Scholar]
- Doulazmi M., Frédéric F., Lemaigre-Dubreuil Y., Hadj-Sahraoui N., Delhaye-Bouchaud N., Mariani J. Cerebellar purkinje cell loss during life span of the heterozygous staggerer mouse (Rora+/Rora(sg)) is gender-related. J Comp Neurol. 1999;411:267–273. [PubMed] [Google Scholar]
- Hu V. W., Sarachana T., Kim K. S., Nguyen A., Kulkarni S., Steinberg M. E., Luu T., Lai Y., Lee N. H. Gene expression profiling differentiates autism case-controls and phenotypic variants of autism spectrum disorders: evidence for circadian rhythm dysfunction in severe autism. Autism Res. 2009;2:78–97. doi: 10.1002/aur.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giguere V., Tini M., Flock G., Ong E., Evans R. M., Otulakowski G. Isoform-specific amino-terminal domains dictate DNA-binding properties of RORα, a novel family of orphan hormone nuclear receptors. Genes Dev. 1994;8:538–553. doi: 10.1101/gad.8.5.538. [DOI] [PubMed] [Google Scholar]
- Giguere V., Beatty B., Squire J., Copeland N. G., Jenkins N. A. The orphan nuclear receptor RORα (RORA) maps to a conserved region of homology on human chromosome 15q21–q22 and mouse chromosome 9. Genomics. 1995;28:596–598. doi: 10.1006/geno.1995.1197. [DOI] [PubMed] [Google Scholar]
- Hamilton B. A., Frankel W. N., Kerrebrock A. W., Hawkins T. L., FitzHugh W., Kusumi K., Russell L. B., Mueller K. L., Van Berkel V., Birren B. W., Kruglyak L., Lander E. S. Disruption of the nuclear hormone receptor RORα in staggerer mice. Nature. 1996;379:736–739. doi: 10.1038/379736a0. [DOI] [PubMed] [Google Scholar]
- Palmen S. J., van Engeland H., Hof P. R., Schmitz C. Neuropathological findings in autism. Brain. 2004;127:2572–2583. doi: 10.1093/brain/awh287. [DOI] [PubMed] [Google Scholar]
- Polleux F., Lauder J. M. Toward a developmental neurobiology of autism. Ment Retard Dev Disabil Res Rev. 2004;10:303–317. doi: 10.1002/mrdd.20044. [DOI] [PubMed] [Google Scholar]
- Nicholas B., Rudrasingham V., Nash S., Kirov G., Owen M. J., Wimpory D. C. Association of Per1 and Npas2 with autistic disorder: support for the clock genes/social timing hypothesis. Mol Psychiatry. 2007;12:581–592. doi: 10.1038/sj.mp.4001953. [DOI] [PubMed] [Google Scholar]
- Melke J., Goubran Botros H., Chaste P., Betancur C., Nygren G., Anckarsäter H., Rastam M., Ståhlberg O., Gillberg I. C., Delorme R., Chabane N., Mouren-Simeoni M.-C., Fauchereau F., Durand C. M., Chevalier F., Drouot X., Collet C., Launay J.-M., Leboyer M., Gillberg C., Bourgeron T., the PARIS study Abnormal melatonin synthesis in autism spectrum disorders. Mol Psychiatry. 2008;13:90–98. doi: 10.1038/sj.mp.4002016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeron T. The possible interplay of synaptic and clock genes in autism spectrum disorders. Cold Spring Harbor Symp Quant Biol. 2007;72:645–654. doi: 10.1101/sqb.2007.72.020. [DOI] [PubMed] [Google Scholar]
- Dussault I., Fawcett D., Matthyssen A., Bader J.-A., Giguère V. Orphan nuclear receptor RORα-deficient mice display the cerebellar defects of staggerer. Mech Dev. 1998;70:147–153. doi: 10.1016/s0925-4773(97)00187-1. [DOI] [PubMed] [Google Scholar]
- Jarvis C. I., Staels B., Brugg B., Lemaigre-Dubreuil Y., Tedgui A., Mariani J. Age-related phenotypes in the staggerer mouse expand the RORα nuclear receptor’s role beyond the cerebellum. Mol Cell Endocrinol. 2002;186:1–5. doi: 10.1016/s0303-7207(01)00668-2. [DOI] [PubMed] [Google Scholar]
- Goodall G., Gheusi G. Abnormal patterns of maze patrolling in the mutant mouse staggerer. Behav Neural Biol. 1987;47:307–320. doi: 10.1016/s0163-1047(87)90422-5. [DOI] [PubMed] [Google Scholar]
- Lalonde R. Exploration and spatial learning in staggerer mutant mice. J Neurogenet. 1987;4:285–292. [PubMed] [Google Scholar]
- Lalonde R., Manseau M., Botez M. I. Spontaneous alternation and exploration in staggerer mutant mice. Behav Brain Res. 1988;27:273–276. doi: 10.1016/0166-4328(88)90124-6. [DOI] [PubMed] [Google Scholar]
- Lalonde R., Strazielle C. Discrimination learning in rorasg and Grid2ho mutant mice. Neurobiol Learn Mem. 2008;90:472–474. doi: 10.1016/j.nlm.2008.05.004. [DOI] [PubMed] [Google Scholar]
- Lalonde R., Botez M. I., Boivin D. Object exploration in staggerer mutant mice. Physiol Behav. 1987;41:115–117. doi: 10.1016/0031-9384(87)90139-9. [DOI] [PubMed] [Google Scholar]
- Gold D. A., Baek S. H., Schork N. J., Rose D. W., Larsen D. D., Sachs B. D., Rosenfeld M. G., Hamilton B. A. RORα coordinates reciprocal signaling in cerebellar development through sonic hedgehog and calcium-dependent pathways. Neuron. 2003;40:1119–1131. doi: 10.1016/s0896-6273(03)00769-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohly H. H., Panja A. Immunological findings in autism. Int Rev Neurobiol. 2005;71:317–341. doi: 10.1016/s0074-7742(05)71013-8. [DOI] [PubMed] [Google Scholar]
- Casanova M. F. Neuropathological and genetic findings in autism: the significance of a putative minicolumnopathy. Neuroscientist. 2006;12:435–441. doi: 10.1177/1073858406290375. [DOI] [PubMed] [Google Scholar]
- Hu V. W., Nguyen A., Kim K. S., Steinberg M. E., Sarachana T., Scully M. A., Soldin S. J., Luu T., Lee N. H. Gene expression profiling of lymphoblasts from autistic and nonaffected sib pairs: altered pathways in neuronal development and steroid biosynthesis. PLoS ONE. 2009;4:e5775. doi: 10.1371/journal.pone.0005775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdolmaleky H. M., Zhou J. R., Thiagalingam S., Smith C. L. Epigenetic and pharmacoepigenomic studies of major psychoses and potentials for therapeutics. Pharmacogenomics. 2008;9:1809–1823. doi: 10.2217/14622416.9.12.1809. [DOI] [PubMed] [Google Scholar]
- Baron C. A., Liu S. Y., Hicks C., Gregg J. P. Utilization of lymphoblastoid cell lines as a system for the molecular modeling of autism. J Autism Dev Disord. 2006;36:973–982. doi: 10.1007/s10803-006-0134-x. [DOI] [PubMed] [Google Scholar]
- Nishimura Y., Martin C. L., Vazquez-Lopez A., Spence S. J., Alvarez-Retuerto A. I., Sigman M., Steindler C., Pellegrini S., Schanen N. C., Warren S. T., Geschwind D. H. Genome-wide expression profiling of lymphoblastoid cell lines distinguishes different forms of autism and reveals shared pathways. Hum Mol Genet. 2007;16:1682–1698. doi: 10.1093/hmg/ddm116. [DOI] [PubMed] [Google Scholar]
- Gregg J. P., Lit L., Baron C. A., Hertz-Picciotto I., Walker W., Davis R. A., Croen L. A., Ozonoff S., Hansen R., Pessah I. N., Sharp F. R. Gene expression changes in children with autism. Genomics. 2008;91:22–29. doi: 10.1016/j.ygeno.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Enstrom A. M., Lit L., Onore C. E., Gregg J. P., Hansen R. L., Pessah I. N., Hertz-Picciotto I., Van de Water J. A., Sharp F. R., Ashwood P. Altered gene expression and function of peripheral blood natural killer cells in children with autism. Brain Behav Immun. 2009;23:124–133. doi: 10.1016/j.bbi.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.