

Abstract
The combined actions of natural selection, mutation, and recombination forge the landscape of genetic variation across genomes. One frequently observed manifestation of these processes is a positive association between neutral genetic variation and local recombination rates. Two selective mechanisms and/or recombination-associated mutation (RAM) could generate this pattern, and the relative importance of these alternative possibilities remains unresolved generally. Here we quantify nucleotide differences within populations, between populations, and between species to test for genome-wide effects of selection and RAM in the partially selfing nematode Caenorhabditis briggsae. We find that nearly half of genome-wide variation in nucleotide polymorphism is explained by differences in local recombination rates. By quantifying divergence between several reproductively isolated lineages, we demonstrate that ancestral polymorphism generates a spurious signal of RAM for closely related lineages, with implications for analyses of humans and primates; RAM is, at most, a minor factor in C. briggsae. We conclude that the positive relation between nucleotide polymorphism and the rate of crossover represents the footprint of natural selection across the C. briggsae genome and demonstrate that background selection against deleterious mutations is sufficient to explain this pattern. Hill-Robertson interference also leaves a signature of more effective purifying selection in high-recombination regions of the genome. Finally, we identify an emerging contrast between widespread adaptive hitchhiking effects in species with large outcrossing populations (e.g., Drosophila) versus pervasive background selection effects on the genomes of organisms with self-fertilizing lifestyles and/or small population sizes (e.g., Caenorhabditis elegans, C. briggsae, Arabidopsis thaliana, Lycopersicon, human). These results illustrate how recombination, mutation, selection, and population history interact in important ways to shape molecular heterogeneity within and between genomes.
Central to the aims of evolutionary genomics is the characterization of the relative roles of positive and negative selective forces, mutation, recombination, and population history as determinants of the distribution of genetic variants across the genome. The landmark study of Begun and Aquadro (1992) laid bare the power of natural selection in combination with linkage, in particular, to shape genome patterns of genetic variation in Drosophila melanogaster. It is now clear that adaptation via positive selection plays a critical role in Drosophila genome evolution (Begun et al. 2007; Hahn 2008; Sella et al. 2009), but the generality of this result across taxa remains tenebrous, with mounting evidence for important roles of recombination-associated mutation (RAM) and purifying selection for humans and other organisms (Lercher and Hurst 2002; Cutter and Payseur 2003; Hellmann et al. 2003; Innan and Stephan 2003; Nordborg et al. 2005; Cai et al. 2009; Kaiser and Charlesworth 2009; McVicker et al. 2009). In the most sophisticated analysis of selection at linked sites for humans to date, McVicker et al. (2009) found that ongoing and ancestral background selection effects can account for genome-wide patterns of polymorphism, although this does not preclude a contributing role of recurrent genetic hitchhiking. Here we use patterns of nucleotide differences within populations, between isolated populations, and between species to investigate the relative importance of mutation and of selection in current and ancestral populations as drivers of genome sequence polymorphism and divergence for the nematode Caenorhabditis briggsae.
Linkage of neutral genetic variants to targets of natural selection is predicted to result in depressed levels of neutral polymorphism, and disproportionately so in regions of the genome that experience little recombination. Theory details how this effect can result from recurrent selective sweeps, due to adaptive evolution causing stronger genetic hitchhiking effects in low-recombination portions of the genome (Maynard Smith and Haigh 1974; Wiehe and Stephan 1993; Andolfatto 2001). But the action of pervasive purifying selection that drives extinct deleterious mutations and the variants linked to them (“background selection”) will generate a similar pattern (Charlesworth et al. 1993; Hudson and Kaplan 1995). Despite some unique predictions (Braverman et al. 1995; Charlesworth et al. 1995; Innan and Stephan 2003), it often is difficult to distinguish these two models of selection at linked sites, and both may operate simultaneously (Kim and Stephan 2000). Moreover, should the process of mutation be tied to recombination (i.e., RAM), a similar disparity in polymorphism levels between high- and low-recombination regions would result (Lercher and Hurst 2002; Hellmann et al. 2003). Experiments in yeast implicate RAM as a real phenomenon (Strathern et al. 1995; Rattray et al. 2002; but see Noor 2008b). If prevalent, this selectively neutral process could undermine selective explanations for patterns of higher population variation in high-recombination regions.
For closely related species, the added complication arises of ancestral polymorphism possibly contributing to genomic heterogeneity in measures of divergence (Li 1977; Charlesworth et al. 2005). Specifically, selection at linked sites in the common ancestor could generate a spurious signal of RAM because genealogies will be deeper for genomic regions with more recombination, with a correspondingly elevated contribution of ancestral polymorphism to naive measures of divergence (Hellmann et al. 2003; Begun et al. 2007; Kulathinal et al. 2008; Noor 2008a; McVicker et al. 2009). Indeed, ancestral polymorphism is implicated as the source of correlations between divergence and recombination rate for human–chimpanzee comparisons (McVicker et al. 2009) and at least in part for Drosophila pseudoobscura–Drosophila persimilis (Kulathinal et al. 2008; Noor 2008a). A potential solution to this difficulty in distinguishing neutral from selective processes is to quantify divergence with respect to multiple reproductively isolated lineages that span a range of times to common ancestry. When ancestral polymorphism drives the correlation between divergence and recombination, the association will become weaker with increasing evolutionary distance, provided that the overall recombination environment is conserved. We take this approach here by quantifying sequence differences among individuals within a population, between several reproductively isolated populations, and between distantly related species (Fig. 1).
Figure 1.

Relationships among populations of C. briggsae and species of Caenorhabditis.
Populations of the nematode C. briggsae are separated into phylogeographically distinct entities, in which hermaphrodites reproduce predominantly by self-fertilization (Cutter et al. 2006). Self-fertilization has caused extensive linkage disequilibrium across the genome, by reducing the effective recombination rate (Nordborg 2000), potentially resulting in more striking consequences of linked selection than in outcrossing taxa (Baudry et al. 2001). Indeed, compared to outcrossing relatives, overall levels of genetic variation in C. briggsae are roughly 10-fold lower than would be expected from the standard twofold predicted effects of selfing (Graustein et al. 2002; Cutter et al. 2009). Chromosomal regions with low crossover rates also exhibit higher gene density in C. briggsae (Hillier et al. 2007), which may further facilitate linked selection effects in the genome (Payseur and Nachman 2002)—and the lack of defined centromeres for Caenorhabditis chromosomes precludes potential complicating centromeric effects on low-recombination regions (Henikoff et al. 2001). The genomic distribution of single nucleotide differences between two strains of the related species, Caenorhabditis elegans, is consistent with selection having shaped genome patterns (Cutter and Payseur 2003). However, the lack of a close outgroup prevents a strong test of selective and neutral mutational models for C. elegans, whereas C. briggsae presents itself as exceptionally tractable for understanding the forces that govern the distribution of genetic variants along chromosomes. Here we demonstrate how the C. briggsae genome reveals that natural selection provides the dominant force shaping heterogeneity in patterns of neutral polymorphism among genomic regions with disparate crossover rates and that ancestral polymorphism generates spurious signals of RAM in closely related lineages.
Results
First, we tested for evidence of higher mutation rates in regions of high recombination in the C. briggsae genome using interspecific sequence divergence information. Neutrally evolving nucleotide sites should diverge between reproductively isolated lineages at a rate that is directly proportional to the mutation rate at such sites (Kimura 1968). We used lineage-specific rates of synonymous-site divergence (dS) for C. briggsae from 7025 C. briggsae–Caenorhabditis remanei–C. elegans ortholog trios (Cutter 2008) and estimated crossover rates from the C. briggsae genetic map (Fig. 2; Supplemental Fig. 1; Hillier et al. 2007). In a multiple regression model, we found that variation in divergence (dS) was explained significantly by variation in crossover rate, independently of background base composition (introns), levels of codon bias, and chromosome of origin (F26,6991 = 51.7, P < 0.0001; crossover-rate effect test, P < 0.0001) (Supplemental Table 1). Specifically, dS is slightly, but significantly, higher on average in regions of high recombination (Fig. 3A), consistent with previous findings in Caenorhabditis (Cutter and Payseur 2003). And yet, local rates of crossover explain <1% of the variation in synonymous-site divergence (Supplemental Table 1). Lineage-specific rates of amino acid substitution (dN) also correlate positively with crossover rate (Fig. 3B), independently of these other factors (F35,6989 = 65.2, P < 0.0001; crossover-rate effect test, P < 0.0001) (Supplemental Table 2). Because of saturation or near saturation of divergence at synonymous sites for most loci and possible changes in recombination environment of loci in the long history since the common ancestor with C. remanei, contrasts with closer relatives of C. briggsae in the future will be valuable. Overall, these data implicate a potential role, albeit very weak, for mutation (via elevating dS) in having mediated patterns of molecular evolution across the C. briggsae genome.
Figure 2.

Crossover rate estimates for C. briggsae chromosome 1 with each point-estimate based on the slope of the least-squares regression line through five mapped loci. Other chromosomes have similar marker density and general recombination profiles (Supplemental Fig. 2). Finer- and coarser-scale estimation of crossover rates are qualitatively similar, as well (Supplemental Fig. 1B). Note that the low-recombination chromosome centers do not correspond to centromeres, as centromeres are not distinct features of Caenorhabditis chromosomes.
Figure 3.

Lineage-specific synonymous-site (A) and replacement-site divergence (B) for quartiles of recombination rate in C. briggsae, in genome comparisons with C. remanei and C. elegans as outgroups. ANOVA models that include recombination rate, codon bias, background nucleotide composition, chromosome of origin, divergence, and their first-order interactions provide statistical support for the positive dS-recombination and dN-recombination associations. Error bars indicate ±1 standard error.
We next explored the potential for mutation, selection, and recombination to shape population nucleotide polymorphism. We quantified nucleotide differences in 24 intron loci (19 kb total, including two loci from Cutter et al. 2006) for each of 16 C. briggsae strains from populations representing two phylogeographic groups (“Tropical” and “Temperate”), plus two additional strains from more-divergent populations (Nairobi and Kerala) (Fig. 1; Cutter et al. 2010). These loci derive from all five autosomes and span the distribution of crossover rates in the genome. The strains from tropical belts of the globe harbor greater than twofold more genetic diversity than the strains found throughout temperate regions (mean per site πsi = 0.18% vs. 0.074%) (Supplemental Table 3), with nearly half of all polymorphisms in the combined sample of Temperate and Tropical strains representing fixed differences between them (113 of 245 SNPs; mean divergence Dxy = 0.89%; average FST = 0.78) and no shared polymorphisms between the groups. We focus on polymorphism within the Tropical sample, due to its greater variation overall. Within populations, effective recombination is low, with only two or three recombination events detected with a four-gamete test across these 24 loci on five chromosomes. Linkage disequilibrium therefore is pervasive across the genome. As noted previously (Cutter et al. 2006), this evinces extensive, but partial, selfing. Consistent with previous findings (Cutter et al. 2006), the patterns of polymorphism indicate that gene flow is undetectable between the different phylogeographic groups. Consequently, we treat strains from each group as reproductively isolated, meaning that fixed, neutral differences between them should accumulate by genetic drift at a rate equal to the mutation rate (Kimura 1968). We quantified interpopulation divergence between the Tropical-strain samples and each of the other C. briggsae lineages (Temperate, Nairobi, and Kerala) (Fig. 1; Dolgin et al. 2008; Cutter et al. 2010), because recent separation of populations could cause artifacts of ancestral polymorphism in patterns of sequence divergence between the populations. For the six loci in regions of highest recombination, which will be least affected by linked selection, we estimate that the Tropical strains diverged from Temperate, Nairobi, and Kerala populations ∼3.2 Ne, ∼4.9 Ne, and ∼7.3 Ne generations ago, respectively.
Nucleotide polymorphism for Tropical strains is significantly higher among loci in highly recombining portions of the genome (Fig. 4A;  , P = 0.0003). Polymorphism in the Tropical sample also correlates with divergence between Temperate and Tropical populations (
, P = 0.0003). Polymorphism in the Tropical sample also correlates with divergence between Temperate and Tropical populations ( , P = 0.008). The relatively dense genetic map for C. briggsae (Hillier et al. 2007), with median marker spacing of ∼230 kb, also permitted us to explore how the scale of crossover rate estimation influences the relationship of recombination with polymorphism and divergence (local rates estimated with three, five, or seven neighboring mapped loci) (Supplemental Fig. 1B). Bivariate analysis using finer- and coarser-scale estimates of crossover rates indicates stronger associations between polymorphism and recombination at coarser scales (
, P = 0.008). The relatively dense genetic map for C. briggsae (Hillier et al. 2007), with median marker spacing of ∼230 kb, also permitted us to explore how the scale of crossover rate estimation influences the relationship of recombination with polymorphism and divergence (local rates estimated with three, five, or seven neighboring mapped loci) (Supplemental Fig. 1B). Bivariate analysis using finer- and coarser-scale estimates of crossover rates indicates stronger associations between polymorphism and recombination at coarser scales (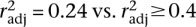 , all P ≤ 0.006), consistent with C. elegans (Cutter and Payseur 2003) but in contrast to D. pseudoobscura (Kulathinal et al. 2008). Fine-scale crossover-rate estimates identify a significant association between interpopulation divergence and crossover rate (Fig. 5), but only for more closely related populations (Temperate–Tropical, , P = 0.007; Nairobi–Tropical,
, all P ≤ 0.006), consistent with C. elegans (Cutter and Payseur 2003) but in contrast to D. pseudoobscura (Kulathinal et al. 2008). Fine-scale crossover-rate estimates identify a significant association between interpopulation divergence and crossover rate (Fig. 5), but only for more closely related populations (Temperate–Tropical, , P = 0.007; Nairobi–Tropical,  , P = 0.021; Kerala–Tropical,
, P = 0.021; Kerala–Tropical, 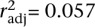 , P = 0.14).
, P = 0.14).
Figure 4.

Silent-site polymorphism for Tropical strains of C. briggsae (A) and divergence between Tropical and Kerala populations (B) as a function of crossover rate (intermediate scale, five-loci estimates). Only the polymorphism-crossover rate association is statistically significant (see text).
Figure 5.

The correlation between crossover rate and interpopulation divergence is stronger for comparisons of more closely related populations and in analyses using finer-scale estimates of crossover rate. P-values are indicated above the bars.
A positive relation between divergence and crossover rate typically would be interpreted as evidence of RAM. However, we observed a decay in the divergence–recombination correlation as a function of the time separating lineages, indicating that residual ancestral polymorphism largely explains the divergence–recombination relationship between closely related populations. It is unlikely that rates of crossover for orthologous regions differ much between populations of C. briggsae, as the general character of crossover rates along chromosomes is very similar even in the distant relative C. elegans (Hillier et al. 2007). In addition, C. elegans mutation accumulation lines show no consistent heterogeneity in mutation rates as a function of recombinational environment (Denver et al. 2009). However, our observations of elevated interspecific synonymous-site divergence (see above) and previous observations for C. elegans (Cutter and Payseur 2003) suggest a possible weak role of RAM. In light of these results, we focus on the divergence values inferred for the contrast of distantly related Tropical and Kerala populations as least impacted by ancestral polymorphism. Testing for independent effects of recombination and divergence on the level of population polymorphism, multiple regression analysis recovered a significant independent effect only of crossover rate, to explain nearly half of the variation in polymorphism levels across loci (F3,20 = 7.5, 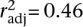 , P = 0.0015). Thus, these polymorphism and interpopulation divergence data (1) identify a spurious signal of ancestral polymorphism that could be mistaken for mutagenic recombination and (2) implicate selection at linked sites as a major force that acts to reduce polymorphism disproportionately in regions of low recombination.
, P = 0.0015). Thus, these polymorphism and interpopulation divergence data (1) identify a spurious signal of ancestral polymorphism that could be mistaken for mutagenic recombination and (2) implicate selection at linked sites as a major force that acts to reduce polymorphism disproportionately in regions of low recombination.
Given the robust signature of selection at linked sites, we attempted to infer whether recurrent selective sweeps or background selection might provide a better general explanation for the patterns of polymorphism in the C. briggsae genome. In an equilibrium population, genetic hitchhiking due to positive selection predicts that low-recombination regions will tend to harbor an excess of low-frequency variants (Braverman et al. 1995; Gillespie 2000), whereas background selection against deleterious mutations predicts no strong association (Charlesworth et al. 1995). A measure of the variant frequency spectrum, Tajima's D (Tajima 1989b), for the 16 loci for which D can be computed for the Tropical sample, does not correlate overall with crossover rate (Spearman ρ = 0.47, P = 0.06). However, Schaeffer's (2002) scaled D/Dmin, which is less biased for loci with few segregating sites, yields a significant positive correlation (Spearman ρ = 0.54, P = 0.03). These patterns are not strong evidence specifically favoring either selective model, however, due to a bimodal distribution of D (and D/Dmin) across loci that suggests deviation from demographic equilibrium (Fig. 6). Plausibly, there could be additional population structure within the Tropical phylogeographic group of C. briggsae that is considered here to represent a single population. It also is conceivable that a species-wide population bottleneck followed the evolutionary origin of high self-fertilization in C. briggsae, which also could generate an excess of intermediate-frequency variants (positive Tajima's D) as well as high variance among loci (Tajima 1989a), if the event was sufficiently recent. If present, structure could generate the elevated Tajima's D values observed for many loci (Pannell 2003), and it also would mean that some “polymorphism” might represent residual ancestral variation or divergence between subpopulations. Because ancestral polymorphism would be expected to predominate for any cryptic structure, our inference that natural selection shapes patterns of polymorphism across the genome would not be compromised.
Figure 6.

Bi-plot of summaries of the variant frequency spectrum as a function of crossover rate. The bimodal distribution of Tajima's D does not correlate significantly with crossover rate, whereas Schaeffer's D/Dmin does (Spearman's ρ = 0.54, P = 0.03).
Recurrent genetic hitchhiking and background selection also predict different curvatures of the polymorphism–recombination association in genomic regions with relatively little crossover (Innan and Stephan 2003). Reduced effective recombination due to selfing places much of the genome in the appropriate parameter space, suggesting that C. briggsae is particularly amenable to this kind of test. Applying the method of Innan and Stephan (2003) to distinguish between these selective forces, we find that a background selection process conforms well to the observed correlation between polymorphism and the polymorphism:recombination ratio (Rp-p:r; P = 0.347; Fig. 7). Fitting of the background selection model to the observed data, modified to accommodate self-fertilization (Nordborg 2000; Innan and Stephan 2003), also yields estimates of the outcrossing rate (c ∼ 0.0001) that are consistent with previous estimates derived from patterns of linkage disequilibrium (Supplemental Fig. 2; Cutter et al. 2006). In contrast, a process of recurrent genetic hitchhiking fit poorly with the observed data, using either of two approaches to capture the effects of selection at linked sites (Rp-p:r neutral simulations scaled by analytical model of recurrent hitchhiking: P < 0.001; Rp-p:r explicit recurrent hitchhiking simulations: P < 0.001; Fig. 7). Simulations that explicitly model recurrent hitchhiking generally fail to recapitulate the observed Rp-p:r for a broad range of assumed background neutral diversity, selection coefficients, and sweep rates, despite otherwise matching the observed average levels of polymorphism and positive correlation between polymorphism and recombination (Supplemental Fig. 3; Supplemental Table 5). Additionally, nonlinear fits of hitchhiking and background selection models that incorporate self-fertilization yield slightly lower mean-square errors for background selection (Supplemental Fig. 2). The simulation procedures we used likely oversimplify the demographic history of the Tropical population, given the unusual distribution of Tajima's D (Fig. 6). Regardless, these data indicate that a process of background selection is sufficient to explain the pattern of polymorphism in C. briggsae. Given the widespread action of purifying selection on functional genome features, we conclude that background selection is the most plausible selective driver of reduced polymorphism in low-recombination regions of the C. briggsae genome, although this does not rule out a contribution from genetic hitchhiking effects of recurrent positive selection. Patterns of polymorphism in some self-fertilizing plants also tend to support a greater role for background selection than for genetic hitchhiking (Innan and Stephan 2003; Nordborg et al. 2005), in contrast to Drosophila (Andolfatto 2001; Innan and Stephan 2003; Sella et al. 2009).
Figure 7.

Distribution of correlation coefficients between neutral polymorphism and the polymorphism-recombination rate ratio (Rp-p:r). The observed correlation (Spearman rank correlation = 0.930; vertical line) falls within the distribution simulated for background selection (A), but not for the genetic hitchhiking model (B,C). The hitchhiking model in B uses scaled output of neutral coalescent simulations, whereas C uses direct simulation of a recurrent sweep process (Jensen et al. 2008). Simulations for A assume π0 = 0.0138 and for B assume π0 = 0.0486 (similar distributions result when π0 are identical), as well as c = 0.000158, a = 7.67 × 10−9, 800-bp loci, μ = 1.7 × 10−9 (see Supplemental Fig. 2), and the observed fine-scale crossover rates for 22 loci. Parameters from both A and B are based on least-squares fitting of analytical selection models. Explicit recurrent hitchhiking simulations for C are based on π0 = 0.035, c = 0.000158, s = 0.001, λ = 1 × 10−6, and fine-scale crossover rates for 22 loci.
Finally, we assessed sequence differences at synonymous sites using shotgun-sequencing reads made for several strains of C. briggsae (Hillier et al. 2007) that happen to span one of the phylogeographic splits that we exploited in the resequencing analysis presented above. We uniquely aligned exonic sequence to the reference genome of Tropical strain AF16, comprising ∼343 kb from strain VT847 (Tropical) and ∼306 kb from strain HK104 (Temperate). From these alignments, we derived the number of synonymous-site pairwise differences for AF16-VT847 (intrapopulation comparison) and AF16-HK104 (interpopulation comparison) and assessed the density of these putatively neutral nucleotide differences among genomic bins corresponding to crossover rate quartiles. Again, we observed a significantly greater density of synonymous-site differences in high- relative to low-recombination regions in the intrapopulation comparison (VT847–AF16 G-test, P << 0.0001), along with greater synonymous-site divergence between recently isolated populations for regions of high recombination (HK104–AF16 G-test, P << 0.0001; Fig. 8A). The interpopulation divergence for these data likely is confounded by ancestral polymorphism, as illustrated above. These observations for exon-sequence data corroborate the population sample analyses, further connecting recombination with the action of selection.
Figure 8.

Sequence differences (S) per site between AF16 and shotgun reads of Tropical strain VT847 (polymorphism, gray squares) and shotgun reads of Temperate strain HK104 (divergence, black diamonds) are greater in high-crossover regions of the genome, for both synonymous sites (A) and replacement sites (B). (C) The ratio of replacement to synonymous site differences within or between populations indicates an excess of replacement differences in regions experiencing little crossover. Black and gray horizontal lines indicate the null expectation for inter- and intrapopulation differences, respectively.
Differences at amino acid changing sites for these shotgun-sequence strain comparisons mimic the synonymous-site pattern: Coding sequences in high-recombination regions contain a greater density of replacement-site differences than do coding sequences in low-recombination regions, both for interpopulation and intrapopulation contrasts (both G-tests, P << 0.0001; Fig. 8B). However, the ratio of replacement- to synonymous-site changes indicates a significant excess of replacement-site differences in low-recombination regions (both G-tests, P << 0.0001; Fig. 8C). This pattern implies that purifying selection on replacement sites is more effective in high-recombination regions, assuming that functional constraints are equivalent among loci, and is indicative of Hill-Robertson interference among simultaneously selected sites (Hill and Robertson 1968). In contrast, replacement-site divergence between species (dN, controlled for dS and other factors) shows the opposite pattern: Genes in regions with frequent crossover exhibit higher sequence divergence at amino acid changing sites, independently of synonymous-site divergence (Fig. 3B; Supplemental Table 2). We hypothesize that this disparity reflects the recent origin of a highly selfing lifestyle in C. briggsae, because most of the divergence between C. briggsae and its common ancestor with C. remanei took place in an obligately outbreeding state (Cutter et al. 2009, 2010). In the present day, recombination may act primarily to facilitate selective elimination of deleterious mutations, in contrast to possibly facilitating positive selection more commonly prior to the onset of selfing in C. briggsae's outbreeding ancestor. The inference that both background selection and Hill-Robertson interference contribute to patterns of polymorphism in the C. briggsae genome indicates that it will be important in this species to apply approaches that can simultaneously incorporate both linkage effects (Loewe and Charlesworth 2007; Kaiser and Charlesworth 2009).
Discussion
Here, we demonstrate that sequences in regions of high recombination in the C. briggsae genome have more differences between species, between isolated populations, and between individuals within a population than do sequences in regions of low recombination. Most of this association is due to the genome-wide effects of natural selection on patterns of polymorphism, and we identify background selection against deleterious mutations as a sufficient mechanism to explain this pattern. Moreover, we observe excess polymorphism at amino acid–encoding sites in genes in low-recombination chromosome centers, indicating that selective interference in the genome permits more effective elimination of deleterious replacement-site mutations in high-recombination regions. The hallmarks of both background selection and Hill-Robertson interference in the C. briggsae genome also highlight the importance of developing and implementing theory that successfully accounts for multiple aspects of linkage in genome evolution (Loewe and Charlesworth 2007; Kaiser and Charlesworth 2009).
The highly selfing lifestyle of this organism and the tendency for low-recombination regions to be gene-dense (Hillier et al. 2007) likely contribute to the widespread effects of natural selection on the landscape of genetic variation across its genome, as also reported for some self-compatible plants like Arabidopsis thaliana and tomato species (Innan and Stephan 2003; Nordborg et al. 2005; Roselius et al. 2005). This suggests that the genomes of species with small and/or selfing populations—like C. briggsae and A. thaliana—might generally reveal background selection effects more readily than species with large outbreeding populations, in which the hitchhiking effects of selective sweeps might be relatively more apparent. Inferences from Drosophila that recurrent selective sweeps generate widespread genetic hitchhiking throughout the genome (Hahn 2008; Sella et al. 2009)—despite an important role for background selection in portions of the genome (Charlesworth 1996; Kaiser and Charlesworth 2009)—might reflect a combination of their large, outbreeding populations and ecological circumstances that facilitate recurrent adaptation. Anopheles gambiae mosquitoes exhibit some similar patterns to Drosophila, but not the close relative Anopheles arabiensis, which has a smaller effective population size (Stump et al. 2005). Such dramatic effects of adaptive evolution on genomes are not evident in many other organisms, however, including humans (Lercher and Hurst 2002; Hellmann et al. 2003; Reed et al. 2005; McVicker et al. 2009), many plants (Innan and Stephan 2003; Nordborg et al. 2005; Roselius et al. 2005; Wright et al. 2006), and C. briggsae. Of course, some contribution of hitchhiking with selective sweeps must also be important in these organisms, as there are many examples and candidate regions with evidence of recent adaptive evolution (Nielsen et al. 2007; Kelley and Swanson 2008). Given the disparate modes of reproduction and effective population size among Caenorhabditis species (Cutter et al. 2009), this group represents a useful genus for future exploration of variation among species in the relative roles of positive and negative selection shaping genomic patterns of polymorphism.
Finally, our contrasts between multiple reproductively isolated lineages demonstrate explicitly the confounding effect of ancestral polymorphism on an inference of RAM. Fine-scale recombination rate estimates showed the greatest sensitivity to this ancestral polymorphism, suggesting that denser genetic maps that would permit even finer resolution of crossover rates will be beneficial for understanding selection at linked sites in C. briggsae. High-diversity portions of genomes for recently diverged lineages will be most affected by ancestral polymorphism, suggesting that a multiple-outgroup approach, as used here, might generally help elucidate the potential for and relative contribution of this spurious effect to have influenced inferences of recombination-associated mutation in Drosophila and primates (Hellmann et al. 2003; Kulathinal et al. 2008; Noor 2008a; McVicker et al. 2009). There is an inherent trade-off in selecting outgroups for analyses of divergence: persistent ancestral polymorphism as a problem for close relatives and the confounding of discordant genetic parameters for distant relatives (e.g., different rates of recombination and mutation for orthologous regions). Application of multiple outgroups should facilitate the identification of a divergence “sweet spot” that minimizes these conflating effects. The observation that the divergence–recombination association diminishes by half when human sequences are compared to baboon rather than chimpanzee (Hellmann et al. 2003), along with similar effects of using human–macaque or human–dog divergence values versus human–chimpanzee (McVicker et al. 2009), indicates the importance of ancestral polymorphism in primates as well as Caenorhabditis for understanding the evolutionary forces governing genome-wide patterns of polymorphism. Indeed, McVicker et al. (2009) recently argued that background selection in human–chimpanzee ancestors plausibly generated the pattern previously inferred to be RAM (Hellmann et al. 2003, 2005; see also Hellmann et al. 2008; Cai et al. 2009). Fine-scale analyses of recombination hotspots in humans also run counter to the expectations of RAM (Spencer et al. 2006). Together with analyses of yeast, Arabidopsis, Anopheles, Drosophila, C. elegans, and now C. briggsae (Betancourt and Presgraves 2002; Stump et al. 2005; Wright et al. 2006; Begun et al. 2007; Noor 2008b; Denver et al. 2009), these studies undermine the notion that recombination, or some correlate, generally exerts mutagenic effects on genomes. Genome-scale comparisons that incorporate gorilla and orangutan should further elucidate the effects of ancestral polymorphism and incomplete lineage sorting, with special relevance to understanding how selection and mutation shape the genomic landscape of polymorphism and divergence.
Methods
Crossover rate estimation
C. briggsae crossover rates were estimated from the genetic map of 248 loci and their physical positions from Hillier et al. (2007); this marker density (median spacing 234 kb) is double that of Kulathinal et al.'s (2008) study of D. pseudoobscura. Specifically, point estimates at each genetically mapped locus were calculated as the slope of the least-squares regression line of genetic on physical position for that locus and two neighboring loci on each side (five loci total). Crossover rate estimates for a given unmapped locus were calculated as the average of the point estimates of the flanking two mapped loci, weighted proportionately by physical proximity to the two flanking loci. Similar crossover rate estimates were calculated at finer (three mapped loci) and coarser (seven mapped loci) scales; analyses use the intermediate scale unless noted otherwise. The windows for these three scales of recombination rate estimation have median sizes of 571 kb, 1.26 Mb, and 2.05 Mb. Quartiles of crossover rate were calculated based on the observed distributions of crossover rates among 16,467 gene annotations that are mapped to C. briggsae chromosomes, with quartile boundaries at 1.07 cM/Mb, 2.25 cM/Mb, and 4.29 cM/Mb.
Interspecific divergence analysis
Calculations of synonymous-site substitution (dS) and replacement-site substitution (dN) for the C. briggsae lineage, based on three-way comparisons of 7025 C. elegans–remanei–briggsae orthologs from Cutter (2008), were cross-referenced with crossover rate estimates. Calculations of codon bias (Fop, frequency of optimal codons) also were obtained from this previous study. Intron base composition was calculated as the combined G+C content across all introns for each gene, averaged between C. briggsae and C. elegans (WormBase WS190). Log10-transformed values of dS, dN, Fop, and recombination rate (“5-locus” estimates) were used in multiple regression models of divergence (intron G+C content was left untransformed); transformed distributions were bell-shaped with little skew.
Intron polymorphism analysis
Twenty-two autosomal introns were sequenced in eight strains from Temperate isolates of C. briggsae (strains BW287, EG4181, HK104, HK105, PB800, PB826, JU439, JU516) and seven Tropical isolates (ED3032, ED3033, ED3034, ED3036, ED3083, JU725, VT847), as well as for ED3101 (Nairobi) and JU1341 (Kerala). GenBank accessions for these sequences are GU953293–GU953666. The corresponding sequences from the genome of the Tropics-derived AF16 strain were included (Stein et al. 2003), and published sequence for two autosomal intron loci for which we could infer crossover rates also were included in the analysis (Cutter et al. 2006, 2010). Locus locations, primers, and summary information are provided in Supplemental Tables 3 and 4. Forward- and reverse-strand sequence quality was confirmed manually in Sequencher v. 4.9 and BioEdit, and resulting intra- and interpopulation polymorphism statistics (π, θ, Dxy, Rmin, Tajima's D, Fst) were computed in dnaSP v.5.0 (Rozas et al. 2003). We then manually calculated Schaeffer's (2002) D/Dmin.
We estimated divergence time between C. briggsae populations with the method of Hudson et al. (1987), restricting analysis to the six loci in the highest-recombination regions (>5 cM/Mb measured at all scales) that will be least impacted by selection at linked sites. For these same loci, we estimate the effective population size for the Tropical sample to be ∼1.9 × 105, assuming neutral equilibrium and a mutation rate in C. briggsae (5.4 × 10−9/site/generation; mean πsi = 0.41% per site) double that of C. elegans (Denver et al. 2009; Phillips et al. 2009). Divergence between Tropical and Kerala populations (∼7.3 Ne gen) implies a common ancestor ∼140,000 yr ago, assuming 10 gen/yr.
Comparison of background selection and recurrent hitchhiking models
We used two approaches based on that of Innan and Stephan (2003) to distinguish recurrent genetic hitchhiking and background selection as causes of the observed correlation between polymorphism and crossover rate (Supplemental Fig. 2). In summary, we first fit analytical approximations of background selection and recurrent hitchhiking models, modified to accommodate selfing, to estimate model parameters given the observed polymorphism and recombination rates. We then used coalescent simulations to obtain baseline polymorphism values that were scaled according to the selection models. For each model, we derived a distribution of Spearman rank correlation coefficients between polymorphism and the polymorphism:recombination ratio to contrast with the observed correlation (Innan and Stephan 2003). We refer to this correlation coefficient test statistic as Rp-p:r. In a second implementation, we directly simulated recurrent hitchhiking effects rather than scaling the output of neutral coalescent simulations (Jensen et al. 2008), but similarly contrasted simulated distributions of correlation coefficients (Rp-p:r) to the observed values (Innan and Stephan 2003).
First, we fit a background selection model π = π0(1 + F)−1 exp{−μ[r(1 − F)]−1} to the observed data using JMP v.7, where π is the observed silent-site polymorphism, π0 is the baseline neutral polymorphism in the absence of selection and selfing, r is the observed rate of crossover, μ is the deleterious mutation rate, and c is the outcrossing rate (where c = 1 − S and F = S/(2 − S), with selfing rate S and inbreeding coefficient F). This model assumes that recombination is sufficiently constant around a given locus and sufficiently common that the approximation in Equation 9 of Hudson and Kaplan (1995) holds; this likely is reasonable for the C. briggsae loci considered here, except those with r = 0, which we excluded. Assuming the C. briggsae mutation rate as a fixed parameter (1.7 × 10−9/site per generation [gen]; given 32% constrained sites in the genome [Shabalina and Kondrashov 1999] and neutral mutation rate 5.4 × 10−9/site per generation, double that of C. elegans [Denver et al. 2009; Phillips et al. 2009]), we used least-squares fitting to obtain estimates for π0 and c. The estimates of c ∼ 0.0001 from the background selection model match well with previous estimates based on linkage disequilibrium in C. briggsae (Cutter et al. 2006). We then separately fit the recurrent hitchhiking model π = π0(1 + F)−1 × r(1 − F)[r(1 − F) + a(1 + F)]−1, where a is a selection and sweep-rate parameter. In the hitchhiking model, we applied the estimates of c derived from the background selection model as a fixed parameter and then estimated π0 and a. The inclusion of selfing in the models follows Nordborg (2000), such that selfing reduces the effective recombination rate by a factor 1 − F and the effective population size by (1 + F)−1, and increases the effect of selection by a factor of (1 + F).
Using the estimated values of π0 from these models, we then performed coalescent simulations to obtain a distribution of values (rather than apply a normal approximation as in Innan and Stephan 2003), scaled them with the selection models to obtain simulated polymorphism values (π*), and computed Spearman rank correlation coefficients for π* with π*/r (Rp-p:r) using the observed crossover rates (Innan and Stephan 2003). This incorporated custom Perl scripts with Hudson's ms program (Hudson 2002). We then generated 1000 simulated correlation coefficients (Rp-p:r) for each model to compare with the observed Spearman rank correlation coefficients.
Second, we performed coalescent simulations explicitly in a recurrent hitchhiking framework, using the rsweep_stochCG program (Jensen et al. 2008), because the non-equilibrium dynamics of positive selection might compromise inferences based on scaling of neutral coalescent output. These simulations matched the loci in our data set in terms of sample size, length, and recombination rate. We assumed a fixed outcrossing rate c = 0.000158 and effective population size Ne = 1.9 × 105, but varied other parameters (selection coefficient s, rate of sweeps λ, and baseline diversity π0). As described above, the outcrossing rate was used to scale r, s, and π0 (Nordborg 2000). After initially exploring a broad parameter space, we considered in-depth simulations for parameter combinations that generated average π values across loci that matched the mean for the observed data as well as correlations between π and r of similar magnitude as observed (0.0005 ≤ s ≤ 0.01, 10−10 ≤ λ ≤ 10−3, 0.005 ≤ π0 ≤ 0.045). We then used the same approach as above to contrast the distribution of 1000 simulated correlation coefficients (Rp-p:r) between π* with π*/r with the observed coefficient. We used the Spearman correlation coefficients because they had greater power to differentiate background selection and recurrent hitchhiking models than did Pearson correlation coefficients, likely because the underlying distributions of π* and r were somewhat skewed.
Exon polymorphism analysis
High-quality shotgun-sequence reads for C. briggsae strains HK104 (4.9 Mb) and VT847 (6.1 Mb) reported by Hillier et al. (2007) were compared by BLASTX to an exon-derived peptide database based on C. briggsae gene annotations obtained from WormBase release WS190. Sequences for the resulting best hits were trimmed and aligned with ClustalW using customized Perl scripts. Alignment pairs were evaluated for synonymous and replacement differences using a version of the polyMORPHOrama program (Bachtrog and Andolfatto 2006) modified by S.I. Wright. Duplicate reads or exons were removed from further analysis by retaining alignments with the lowest polymorphism; alignments <45 bp also were excluded. This procedure yielded 1232 and 1354 unique exon alignments of AF16 with HK104 and VT847, respectively, representing 343 kb and 306 kb of sequence.
Acknowledgments
We thank R. Waterston and the WashU Genome Center for making shotgun sequence reads available to us, and S. Wright for discussion and comments on the manuscript. We also thank three anonymous reviewers who provided thoughtful and constructive comments. We thank NSERC for financial support, with a Discovery Grant to A.D.C. and an Undergraduate Summer Research Award to J.Y.C. Open access publication costs were defrayed by the Ontario Genomics Institute Genomics Publication Fund.
Footnotes
[Supplemental material is available online at http://www.genome.org. The sequence data from this study have been submitted to GenBank (http://www.ncbi.nlm.nih.gov/genbank) under accession nos. GU953293–GU953666.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.104331.109.
References
- Andolfatto P 2001. Adaptive hitchhiking effects on genome variability. Curr Opin Genet Dev 11: 635–641 [DOI] [PubMed] [Google Scholar]
- Bachtrog D, Andolfatto P 2006. Selection, recombination and demographic history in Drosophila miranda. Genetics 174: 2045–2059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudry E, Kerdelhue C, Innan H, Stephan W 2001. Species and recombination effects on DNA variability in the tomato genus. Genetics 158: 1725–1735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begun DJ, Aquadro CF 1992. Levels of naturally-occurring DNA polymorphism correlate with recombination rates in Drosophila melanogaster. Nature 356: 519–520 [DOI] [PubMed] [Google Scholar]
- Begun DJ, Holloway AK, Stevens K, Hillier LW, Poh YP, Hahn MW, Nista PM, Jones CD, Kern AD, Dewey CN, et al. 2007. Population genomics: Whole-genome analysis of polymorphism and divergence in Drosophila simulans. PLoS Biol 5: e310 doi: 10.1371/journal.pbio.0050310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betancourt AJ, Presgraves DC 2002. Linkage limits the power of natural selection in Drosophila. Proc Natl Acad Sci 99: 13616–13620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braverman JM, Hudson RR, Kaplan NL, Langley CH, Stephan W 1995. The hitchhiking effect on the site frequency-spectrum of DNA polymorphisms. Genetics 140: 783–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai JJ, Macpherson JM, Sella G, Petrov DA 2009. Pervasive hitchhiking at coding and regulatory sites in humans. PLoS Genet 5: e1000336 doi: 10.1371/journal.pgen.1000336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B 1996. Background selection and patterns of genetic diversity in Drosophila melanogaster. Genet Res 68: 131–149 [DOI] [PubMed] [Google Scholar]
- Charlesworth B, Morgan MT, Charlesworth D 1993. The effect of deleterious mutations on neutral molecular variation. Genetics 134: 1289–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth D, Charlesworth B, Morgan MT 1995. The pattern of neutral molecular variation under the background selection model. Genetics 141: 1619–1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, Bartolome C, Noel V 2005. The detection of shared and ancestral polymorphisms. Genet Res 86: 149–157 [DOI] [PubMed] [Google Scholar]
- Cutter AD 2008. Divergence times in Caenorhabditis and Drosophila inferred from direct estimates of the neutral mutation rate. Mol Biol Evol 25: 778–786 [DOI] [PubMed] [Google Scholar]
- Cutter AD, Payseur BA 2003. Selection at linked sites in the partial selfer Caenorhabditis elegans. Mol Biol Evol 20: 665–673 [DOI] [PubMed] [Google Scholar]
- Cutter AD, Félix M-A, Barriere A, Charlesworth D 2006. Patterns of nucleotide polymorphism distinguish temperate and tropical wild isolates of Caenorhabditis briggsae. Genetics 173: 2021–2031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutter AD, Dey A, Murray RL 2009. Evolution of the Caenorhabditis elegans genome. Mol Biol Evol 26: 1199–1234 [DOI] [PubMed] [Google Scholar]
- Cutter AD, Yan W, Tsvetkov N, Sunil S, Felix MA 2010. Molecular population genetics and phenotypic sensitivity to ethanol for a globally diverse sample of the nematode Caenorhabditis briggsae. Mol Ecol 19: 798–809 [DOI] [PubMed] [Google Scholar]
- Denver DR, Dolan PC, Wilhelm LJ, Sung W, Lucas-Lledo JI, Howe DK, Lewis SC, Okamoto K, Thomas WK, Lynch M, et al. 2009. A genome-wide view of Caenorhabditis elegans base-substitution mutation processes. Proc Natl Acad Sci 106: 16310–16324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolgin ES, Félix M-A, Cutter AD 2008. Hakuna nematoda: Genetic and phenotypic diversity in African isolates of Caenorhabditis elegans and C. briggsae. Heredity 100: 304–315 [DOI] [PubMed] [Google Scholar]
- Gillespie JH 2000. Genetic drift in an infinite population: The pseudohitchhiking model. Genetics 155: 909–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graustein A, Gaspar JM, Walters JR, Palopoli MF 2002. Levels of DNA polymorphism vary with mating system in the nematode genus Caenorhabditis. Genetics 161: 99–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn MW 2008. Toward a selection theory of molecular evolution. Evolution 62: 255–265 [DOI] [PubMed] [Google Scholar]
- Hellmann I, Ebersberger I, Ptak SE, Paabo S, Przeworski M 2003. A neutral explanation for the correlation of diversity with recombination rates in humans. Am J Hum Genet 72: 1527–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmann I, Prufer K, Ji H, Zody MC, Paabo S, Ptak SE 2005. Why do human diversity levels vary at a megabase scale? Genome Res 15: 1222–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmann I, Mang Y, Gu Z, Li P, de la Vega FM, Clark AG, Nielsen R 2008. Population genetic analysis of shotgun assemblies of genomic sequences from multiple individuals. Genome Res 18: 1020–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S, Ahmad K, Malik HS 2001. The centromere paradox: Stable inheritance with rapidly evolving DNA. Science 293: 1098–1102 [DOI] [PubMed] [Google Scholar]
- Hill WG, Robertson A 1968. Linkage disequilibrium in finite populations. Theor Appl Genet 38: 226–231 [DOI] [PubMed] [Google Scholar]
- Hillier LW, Miller RD, Baird SE, Chinwalla A, Fulton LA, Koboldt DC, Waterston RH 2007. Comparison of C. elegans and C. briggsae genome sequences reveals extensive conservation of chromosome organization and synteny. PLoS Biol 5: e167 doi: 10.1371/journal.pbio.0050167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR 2002. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 18: 337–338 [DOI] [PubMed] [Google Scholar]
- Hudson RR, Kaplan NL 1995. Deleterious background selection with recombination. Genetics 141: 1605–1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR, Kreitman M, Aguade M 1987. A test of neutral molecular evolution based on nucleotide data. Genetics 116: 153–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innan H, Stephan W 2003. Distinguishing the hitchhiking and background selection models. Genetics 165: 2307–2312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen JD, Thornton KR, Andolfatto P 2008. An approximate Bayesian estimator suggests strong, recurrent selective sweeps in Drosophila. PLoS Genet 4: e1000198 doi: 10.1371/journal.pgen.1000198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser VB, Charlesworth B 2009. The effects of deleterious mutations on evolution in non-recombining genomes. Trends Genet 25: 9–12 [DOI] [PubMed] [Google Scholar]
- Kelley JL, Swanson WJ 2008. Positive selection in the human genome: From genome scans to biological significance. Annu Rev Genomics Hum Genet 9: 143–160 [DOI] [PubMed] [Google Scholar]
- Kim Y, Stephan W 2000. Joint effects of genetic hitchhiking and background selection on neutral variation. Genetics 155: 1415–1427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M 1968. Evolutionary rate at molecular level. Nature 217: 624–626 [DOI] [PubMed] [Google Scholar]
- Kulathinal RJ, Bennett SM, Fitzpatrick CL, Noor MA 2008. Fine-scale mapping of recombination rate in Drosophila refines its correlation to diversity and divergence. Proc Natl Acad Sci 105: 10051–10056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lercher MJ, Hurst LD 2002. Human SNP variability and mutation rate are higher in regions of high recombination. Trends Genet 18: 337–340 [DOI] [PubMed] [Google Scholar]
- Li WH 1977. Distribution of nucleotide differences between two randomly chosen cistrons in a finite population. Genetics 85: 331–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewe L, Charlesworth B 2007. Background selection in single genes may explain patterns of codon bias. Genetics 175: 1381–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard Smith J, Haigh J 1974. Hitch-hiking effect of a favorable gene. Genet Res 23: 23–35 [PubMed] [Google Scholar]
- McVicker G, Gordon D, Davis C, Green P 2009. Widespread genomic signatures of natural selection in hominid evolution. PLoS Genet 5: e1000471 doi: 10.1371/journal.pgen.1000471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, Hellmann I, Hubisz M, Bustamante C, Clark AG 2007. Recent and ongoing selection in the human genome. Nat Rev Genet 8: 857–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noor MA 2008a. Connecting recombination, nucleotide diversity and species divergence in Drosophila. Fly 2: 1–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noor MAF 2008b. Mutagenesis from meiotic recombination is not a primary driver of sequence divergence between Saccharomyces species. Mol Biol Evol 25: 2439–2444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordborg M 2000. Linkage disequilibrium, gene trees and selfing: An ancestral recombination graph with partial self-fertilization. Genetics 154: 923–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordborg M, Hu TT, Ishino Y, Jhaveri J, Toomajian C, Zheng H, Bakker E, Calabrese P, Gladstone J, Goyal R, et al. 2005. The pattern of polymorphism in Arabidopsis thaliana. PLoS Biol 3: e196 doi: 10.1371/journal.pbio.0030196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannell JR 2003. Coalescence in a metapopulation with recurrent local extinction and recolonization. Evolution 57: 949–961 [DOI] [PubMed] [Google Scholar]
- Payseur BA, Nachman MW 2002. Gene density and human nucleotide polymorphism. Mol Biol Evol 19: 336–340 [DOI] [PubMed] [Google Scholar]
- Phillips N, Salomon M, Custer A, Ostrow D, Baer CF 2009. Spontaneous mutational and standing genetic (co)variation at dinucleotide microsatellites in Caenorhabditis briggsae and Caenorhabditis elegans. Mol Biol Evol 26: 659–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattray AJ, Shafer BK, McGill CB, Strathern JN 2002. The roles of REV3 and RAD57 in double-strand-break-repair-induced mutagenesis of Saccharomyces cerevisiae. Genetics 162: 1063–1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed FA, Akey JM, Aquadro CF 2005. Fitting background-selection predictions to levels of nucleotide variation and divergence along the human autosomes. Genome Res 15: 1211–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roselius K, Stephan W, Stadler T 2005. The relationship of nucleotide polymorphism, recombination rate and selection in wild tomato species. Genetics 171: 753–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R 2003. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19: 2496–2497 [DOI] [PubMed] [Google Scholar]
- Schaeffer SW 2002. Molecular population genetics of sequence length diversity in the Adh region of Drosophila pseudoobscura. Genet Res 80: 163–175 [DOI] [PubMed] [Google Scholar]
- Sella G, Petrov DA, Przeworski M, Andolfatto P 2009. Pervasive natural selection in the Drosophila genome? PLoS Genet 5: e1000495 doi: 10.1371/journal.pgen.1000495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabalina SA, Kondrashov AS 1999. Pattern of selective constraint in C. elegans and C. briggsae genomes. Genet Res 74: 23–30 [DOI] [PubMed] [Google Scholar]
- Spencer CCA, Deloukas P, Hunt S, Mullikin J, Myers S, Silverman BW, Donnelly P, Bentley D, McVean G 2006. The influence of recombination on human genetic diversity. PLoS Genet 2: 1375–1385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein LD, Bao Z, Blasiar D, Blumenthal T, Brent MR, Chen N, Chinwalla A, Clarke L, Clee C, Coghlan A, et al. 2003. The genome sequence of Caenorhabditis briggsae: A platform for comparative genomics. PLoS Biol 1: 166–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strathern JN, Shafer BK, McGill CB 1995. DNA-synthesis errors associated with double-strand-break repair. Genetics 140: 965–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stump AD, Fitzpatrick MC, Lobo NF, Traore S, Sagnon N, Costantini C, Collins FH, Besansky NJ 2005. Centromere-proximal differentiation and speciation in Anopheles gambiae. Proc Natl Acad Sci 102: 15930–15935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F 1989a. The effect of change in population-size on DNA polymorphism. Genetics 123: 597–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F 1989b. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiehe THE, Stephan W 1993. Analysis of a genetic hitchhiking model, and its application to DNA polymorphism data from Drosophila melanogaster. Mol Biol Evol 10: 842–854 [DOI] [PubMed] [Google Scholar]
- Wright SI, Foxe JP, DeRose-Wilson L, Kawabe A, Looseley M, Gaut BS, Charlesworth D 2006. Testing for effects of recombination rate on nucleotide diversity in natural populations of Arabidopsis lyrata. Genetics 174: 1421–1430 [DOI] [PMC free article] [PubMed] [Google Scholar]