

Abstract
Genetic variation in the human serotonin system has long-been studied because of its functional consequences and links to various behavior-related disorders and it being routinely targeted in research and development for drug therapy. However, aside from clinical studies, little is known about this genetic diversity and how it differs within and between human populations with respect to haplotype structure, which can greatly impact phenotype association studies. In addition, no evolutionary approach among humans and other primates has examined how long- and short-term selective pressures explain existing serotonin variation. Here, we examine DNA sequence variation in natural population samples of 192 human and 40 chimpanzee chromosome sequences for the most commonly implicated ∼38-kb serotonin transporter (SLC6A4) and ∼63-kb serotonin 2A receptor (HTR2A) genes. Our comparative population genetic analyses find significant linkage disequilibrium associated with functionally relevant variants in humans, as well as geographic variation for these haplotypes, at both loci. In addition, although amino acid divergence is consistent with purifying selection, promoter and untranslated regions exhibit significantly high divergence in both species lineages. These evolutionary analyses imply that the serotonin system may have accumulated significant regulatory variation over both recent and ancient periods of time in both humans and chimpanzees. We discuss the implications of this variation for disease association studies and for the evolution of behavior-related phenotypes during the divergence of humans and our closest primate relatives.
Keywords: HTR2A, SLC6A4, depression
Introduction
The serotonin system that includes a number of ligands and their receptors affects almost every physiological aspect of the body such as the cardiovascular, gastrointestinal, and nervous systems. Meta-analyses have shown that variation in these molecules has been linked to various human behavior-related conditions, such as bipolar (Lasky-Su et al. 2005), anxiety–depression (Sen et al. 2004), and personality (Savitz and Ramesar 2004) disorders, as well as clinical diseases such as schizophrenia (Abdolmaleky et al. 2004) and Alzheimer's (Holmes et al. 1998). Because of this strong association with so many common behavioral phenotypes, the serotonin system is the focus of the most popularly used antidepressant drugs, the serotonin-selective reuptake inhibitors or “SSRIs” (Butler and Meegan 2008). However, the inconsistent success of these SSRIs in treating the population as a whole is not because the genes are unknown, but because we lack a general knowledge about the level and impact of serotonin functional variation within these genes in natural populations. As pharmacological research leans toward “personalized medicine” (Malhotra et al. 2007; Sjöqvist and Eliasson 2007; Nebert et al. 2008; Laje et al. 2009), successful treatment of these diseases will come from understanding the evolutionary change within the genes that code for these neurotransmitters and receptors.
Several genic variants have been associated with behavior-related disorders and diseases at two major loci, the serotonin transporter (SLC6A4 or 5-HTT) gene, which is the target for the most important class of SSRIs and is the most studied of the serotonin system molecules (Murphy et al. 2008) and the serotonin receptor 2A (HTR2A) gene (e.g., Serretti et al. 2007). Following the release of serotonin in the brain, and the central and peripheral nervous systems where it binds to several receptors like HTR2A, serotonin is then bound to SLC6A4 for recycling and transportation back to the neurotransmitter pool, and thus, protein expression variation in this cycling is expected to have large interacting consequences (Jonnakuty and Gragnoli 2008). The most examined SLC6A4 variants are a promoter “linked polymorphic region” (LPR) 44-bp insertion/deletion variant with “short” (S) and “long” (L) alleles (Heils et al. 1996) and an intron 2 variable number tandem repeat (VNTR) ∼17-bp variant with 9-, 10-, and 12-repeat alleles (Ogilvie et al. 1996). Two single nucleotide polymorphisms (SNPs) have been commonly studied at HTR2A, the A/G SNP at site −1438 in the promoter region (Spurlock et al. 1998) and the T/C SNP at synonymous site 102 in exon 1 (Warren et al. 1993).
Both of the SLC6A4 length variants have been linked to serotonin transporter expression, with the LPR-S allele exhibiting as much as a 2-fold decrease in promoter activity and mRNA transcription (Lesch et al. 1996; Heinz et al. 2000), and differential development in core brain regions (Hariri et al. 2002; Heinz et al. 2005; Pezawas et al. 2005). However, others have shown significant conflicting results, and even complex interactions among the two variants when combined (Kaiser et al. 2002; Sakai et al. 2002; Ali et al. 2009). The HTR2A −1438A and 102T alleles have been associated with a 10%–20% increased brain and central nervous system serotonin receptor expression and serotonin binding (Polesskaya and Sokolov 2002; Parsons et al. 2004; Fukuda et al. 2006), which downregulates brain-derived neurotrophic factor that is linked to increased depression (Vaidya et al. 1997; Manji et al. 2001). Yet again, several have shown that this genotype–expression association is not consistent (e.g., Bray et al. 2004), which may be explained by other functional promoter variants not well characterized (Myers et al. 2007). These different functional associations have translated to inconsistent results in genotype–phenotype association analyses for the most common behavioral traits, such as depression disorders (e.g., Langley et al. 2003; Munafò et al. 2003; Willeit et al. 2003; Cho et al. 2005). For example, a highly recognized study by Caspi et al. (2003) revealed that subjects who possess the SLC6A4 LPR-S low-expression allele, and thus predicted low serotonin reuptake, had a significantly greater probability of a major depression episode, but only when they experienced four or more stressful life events, implying a complex temporal component. In addition, a large-scaled study of individuals with depressive disorders who were treated with the SSRI drug citalopram found that the HTR2A −1438A allele experienced an 18% increase in response to the drug, but only when ethnicity was taken into consideration (McMahon et al. 2006), suggesting underlying variation across populations.
The SLC6A4 intron 2 VNTR is actually in the intron after the first translated exon (the first exon is only transcribed, i.e., untranslated region [UTR], fig. 1). These “first” introns are typically enriched by transcription factor–binding domains (Bornstein et al. 1987; Majewski and Ott 2002), and thus, their impact on expression may be predicted. However, although the HTR2A −1438 SNP is in close proximity to the functionally identified promoter sequence (Zhu et al. 1995), the synonymous 102 SNP in exon 1 has no obvious predicted explanation. Thus, it is likely the case that these variants are in strong linkage disequilibrium (LD) with other variants and even with each other. As human populations have a demographic history of substructure that has resulted in differential levels of nucleotide diversity and LD, this structure alone could explain failures to resolve consistent genotype–phenotype associations in disease studies (e.g., Tishkoff and Verrelli 2003b; Wall and Pritchard 2003). In addition, although many studies have genotyped these functional SLC6A4 and HTR2A variants in clinical samples for behavior association analyses, as exemplified above, the interactions are clearly complex and results are often inconsistent owing to the use of small sample sizes, single ethnic groups, or single variants (Lohmueller et al. 2003; Smits et al. 2004; Fan and Sklar 2005). In fact, given the intimate relationship between the two proteins, if there are strong epistatic interactions between variants, genotyping single loci or SNPs could also explain failures to identify significant associations.
FIG. 1.

Gene diagrams for the (a) ∼63-kb HTR2A locus and (b) ∼38-kb SLC6A4 locus (with promoter “LPR” and intron 2 “VNTR” variants). Black boxes denote amino acid coding exons, white boxes denote UTRs, and striped boxes denote functionally identified promoter regions. SNP numbering is with respect to the beginning of amino acid translation. Genes are to scale (except where noted with collapsed distances in kb), and only regions for which data were collected are shown. Significant pairwise associations (filled boxes) among informative variants in 192 human (>5% in frequency) and 40 chimpanzee (>12% in frequency) chromosome sequences are above and below gene diagrams, respectively (see Materials and Methods and Results for more details).
A molecular evolutionary genetic approach that examines the historical impact of these functional variants would circumvent these problems that previous genotyping studies have encountered. For example, characterizing an unbiased sample of haplotypes, and not simply genotyping ascertained SNPs, for coding and noncoding regions across multiple human and primate populations would finally address questions concerning the age and origin of these variants (i.e., ancestral vs. derived states), the LD among them, and how this structure differs across ethnically diverse groups, all of which lend significant statistical power to genotype–phenotype association studies (Pritchard, Stephens, Rosenberg, et al. 2000). In addition, although few have examined evolutionary patterns among mammalian serotonin proteins (e.g., Andrés et al. 2007), functional work suggests that serotonin variation is based in expression regulatory regions, yet no studies of divergence and long-term selective constraints in these latter regions have been conducted.
Here, we conduct the first comparative population genetic analysis and tests of neutrality of the two most commonly studied serotonin system genes, SLC6A4 and HTR2A, in natural population samples of both humans and chimpanzees. Characterizing the evolutionary history of coding, noncoding, and regulatory regions both within and between humans and other primates can shed light on the observed functional variation observed across human populations, whether it is unique to the human lineage, and if it reflects a signature of adaptive or demographic processes. As many studies target these two genes and the common variants within them for behavioral studies, these analyses are necessary to determine how the evolutionary history of these genes and the interactions among them may explain inconsistencies in association studies and the variation in both prevalence and treatment response of common behavioral phenotypes found in human populations today.
Materials and Methods
Population Samples
Our human population data set includes 96 DNA samples (192 chromosomes) publicly available from the Coriell Institute for Medical Research (Camden, NJ). The 10 sample populations were chosen to reflect a global estimate of nucleotide diversity for geographic regions within and outside of sub-Saharan Africa, the latter typically having the most nucleotide and haplotype diversity owing to its “older” history and larger estimated effective population size (Ne) compared with more recently colonized regions (e.g., Tishkoff and Verrelli 2003a; Campbell and Tishkoff 2008). These samples have been previously used in similar population analyses and, thus, enable direct comparisons of diversity across loci. Samples include (with abbreviation, catalog number, and number of sampled chromosomes): Northern European (NE, HD01, 20), Italian (IT, HD21, 20), Russian (RU, HD23, 20), Chinese (CH, HD32, 20), Japanese (JA, HD07, 20), Southeast Asian (SA, HD13, 20), Mexican (MX, HD08, 20), Middle Eastern (ME, HD05, 20), North African (NA, HD11, 14), and sub-Saharan African (SS, HD12, 18).
Whole-blood samples were collected from wild-born chimpanzees housed at research facilities or zoological institutions (in accordance with the Convention on International Trade in Endangered Species of Wild Fauna and Flora under permit 99US013176/9), and DNA was isolated using a standard phenol–chloroform extraction method. Chimpanzee subspecies type was determined from analyses of mitochondrial DNA and Y-chromosome nucleotide sequences as described by Stone et al. (2002). The sample of 20 western Africa Pan troglodytes verus chimpanzees (40 chromosomes) included in this study represents wild-born, documented unrelated individuals from our database of a collection of over 400 chimpanzees, which we have examined in previous population genetic analyses (Stone et al. 2002; Wooding et al. 2005, 2006; Verrelli et al. 2006, 2008; Perry et al. 2006, 2008; Becquet et al. 2007). Although several subspecies are recognized, population samples of P. t. verus show no evidence of an unusual historical demographic history (e.g., population expansion or subdivision) and have nuclear genetic diversity similar to that of humans and, thus, are most suitable for direct comparative analyses (Kaessmann et al. 1999; Gilad et al. 2003; Fischer et al. 2004; Wooding et al. 2005, 2006; Verrelli et al. 2006, 2008). For inferences on ancestral and derived states, as well as polarizing fixed differences between humans and chimpanzees into their respective lineages for statistical analyses, trace nucleotide sequence files with at least 4× overlap from orangutan (Pongo pygmaeus) and macaque (Macaca mulatta) genome projects were obtained from the National Center for Biotechnology Information (NCBI) database.
Polymerase Chain Reaction and DNA Sequencing
HTR2A spans ∼63 kb on chromosome 13q14–q21 in humans and chimpanzees and codes for a 471-amino acid protein. Figure 1a shows the 7,596 bp sampled including the 740-bp functionally identified promoter (Zhu et al. 1995), 5′ and 3′ UTR (638 bp), and amino acid coding exons (1,413 bp). Intron 2, which is ∼55 kb and includes many Alu elements, was largely avoided here. SLC6A4 spans ∼38 kb on chromosome 17q11–q12 in humans and chimpanzees and codes for a 630-amino acid protein. Figure 1b shows the 11,482 bp sampled including 5′ and 3′ UTR (788 bp) and amino acid coding exons (1,890 bp). Polymerase chain reaction (PCR) and sequencing primers were designed using the GenBank human reference sequence (NCBI Build 36.1) and are available upon request. PCR products were prepared using shrimp alkaline phosphate and exonuclease I (US Biochemicals, Cleveland, OH). Nucleotide sequences were collected with an Applied Biosystems (Foster City, CA) 3730 capillary sequencer, and trace files were aligned in the Sequencher v. 4.5 program (Gene Codes, Ann Arbor, MI).
At SLC6A4, we also genotyped the promoter LPR (which is ∼15 kb upstream of the first amino acid coding exon 2; fig. 1b) and the intron 2 VNTR in the human and chimpanzee samples. For the promoter LPR, primers from Heils et al. (1996) were used to PCR amplify a 484- to 528-bp fragment (depending on the S/L-allelic genotype). The primers for the intron 2 VNTR were designed from the human reference sequence (NCBI Build 36.1) to amplify a ∼338- to 389-bp fragment (depending on the 9-/10-/12-repeat genotype). PCR products for the length variants were electrophoresed on a 2% sodium borate agarose gel, stained with ethidium bromide, and visualized under UV transillumination. Homozygous and heterozygous genotypes could easily be detected for all samples. Although the SLC6A4 LPR was genotyped, nucleotide sequence population diversity was not collected for this promoter. However, for a comparable divergence data set across regions as in HTR2A, nucleotide sequence for the functionally identified promoter region (1,319 bp; Ramamoorthy et al. 1993) was obtained from the human, chimpanzee, orangutan, and macaque genome projects (as above).
Statistical Analyses
As previously mentioned, specific geographic populations such as those from sub-Saharan Africa may have markedly different evolutionary histories, which can influence estimates of diversity, SNP frequency distributions, and LD. We calculated statistics for the overall human sample as a global estimate in comparison to our chimpanzee sample at both genes, as well as within our 10 human population samples for comparison. Unless otherwise noted, we used the Rozas et al. (2003) DnaSP v. 5.1 program for summary statistic estimates. Nonsynonymous sites in exons may be expected to be most functionally conserved given their potential impact on the protein, whereas, although intron and synonymous sites are not completely void of function, compared with amino acid sequences, these “silent” sites typically show less evolutionary constraint in human data sets and are often used to reflect estimates of neutrality. On the other hand, there is very little a priori expectation for which sites are “functional” for promoter and UTRs, yet it is clear that these regions greatly influence expression regulation and exhibit patterns of nonneutral evolution (Wray et al. 2003; Haygood et al. 2007; Cheung and Spielman 2009). Thus, we also conducted all intra- and interspecific comparative analyses with these latter gene regions to evaluate hypotheses of functional constraint.
Haplotypes were reconstructed from our sequence data with the PHASE program v2.1.1 (Stephens et al. 2001; Stephens and Scheet 2005) using 250, 500, and 1,000 iterations to examine consistency among runs. Haplotypes with the highest average goodness-of-fit were chosen for each individual (as in Scheinfeldt et al. 2009), and iterations >250 resulted in no difference in haplotype estimation. Locus-specific estimates of the population parameter θ = 4Neμ were calculated using the number of SNPs (S) from Watterson's (1975) θW as well as the average number of pairwise differences among all sequences (θπ). These two estimates, θW and θπ, can be compared using Tajima's (1989) D test, which identifies deviations in the SNP frequency spectrum that may be the result of nonneutral or demographic influences. To evaluate the impact that different historical demographic forces have had on human nucleotide sequence variation, we performed coalescent simulations with the Hudson (2002) MS program and compared our observed summary statistics (e.g., θπ and Tajima's D) with those compiled from the simulated data sets. We modeled various human demographic scenarios typically employed by others (e.g., Schaffner et al. 2005; Auton et al. 2009; Scheinfeldt et al. 2009) as well as by us (e.g., Verrelli and Tishkoff 2004; Verrelli et al. 2008). Specifically, we generated 10,000 genealogical trees simulating exponential population expansion at periods between 10 and 50 thousand years ago (kya) from a historical Ne of 5,000–50,000, with generation times of 20 years; these analyses were performed for the global population data set as a whole as well as for hypotheses consistent with human population structure (i.e., African vs. non-African population split times). Recombination was factored into these simulations by varying the parameter ρ = 4Ner (where r is the recombination rate per nucleotide site) at intervals of 0 (no recombination) to 100 for each run.
We used the Hudson (2000) Snn statistic to describe the haplotype structure across populations at both genes. In contrast to a standard FST analysis that examines differentiation among individuals and populations on a site-by-site basis, the Snn statistic examines SNPs as haplotypes and has been shown to be a more statistically powerful way to detect genetic differentiation among samples, even with sample sizes as small as 10 sequences per group (Hudson 2000). Pairwise comparisons among the 10 human population samples were conducted and a permutation test that resamples haplotypes from a pooled data set and reconstructs the populations with 1,000 replicates was performed to assess statistical significance (implemented in DnaSP, with Bonferroni corrections applied for multiple-comparisons among populations).
We used this Snn analysis along with analyses of LD to detect SNPs significantly correlated as haplotypes and that can explain differentiation among population samples. Many clinical studies of these genes are typically interested in the strict question of whether SNPs are simply “correlated”; thus, we first calculated LD using a standard model, that is, correlations as r2 and significance by chi-squared tests with Bonferroni correction, computed in DnaSP for both species. However, given that correlations among SNPs are not rare in the human genome, conventional null hypotheses now consider a background model of recombination and distance among variants to identify the LD that is significantly unusual (i.e., Hudson 2001). Thus, second, we used the LDhat program of McVean et al. (2002), which applies the approximate-likelihood method of Hudson (2001) and uses a permutation analysis to determine if pairwise comparisons among SNPs exhibit significant LD given a locus-specific estimate of θ. SNPs sufficiently rare in frequency and are uninformative are determined by LDhat and omitted prior to the analysis.
We used the method implemented by Thomson et al. (2000) and previously adapted by us as well as others for human gene data sets (e.g., Verrelli et al. 2002; Scheinfeldt et al. 2009), to estimate the age of several mutations of relevance to our hypotheses (e.g., SLC6A4 S/L variant). This estimator under a neutral evolutionary substitution model is typically preferred when assumptions of population equilibrium and no recombination are not met (Thomson et al. 2000; Scheinfeldt et al. 2009). The age estimate (t) involves the relationship
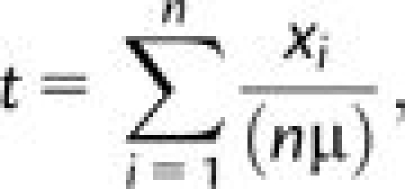 |
where xi is the number of mutational differences between the ith sequence and the estimated most recent common ancestor (MRCA) of all sequences, n is the total number of sequences in the sample, and μ is the mutation rate. The latter is also taken from Thomson et al. (2000) and is a “neutral” estimate for each gene based on the number of substitutions between human and chimpanzee sequences divided by twice the estimated divergence time between these two species with a confidence interval spanning a 4–6 million year split time (e.g., Kumar et al. 2005).
Finally, we conducted several interspecific tests of neutrality. First, we used the McDonald and Kreitman (1991) neutrality test to contrast patterns of diversity within and between humans and chimpanzees. We examined lineage-specific ratios of polymorphism and divergence at nonsynonymous, UTR, and promoter sites compared with silent sites for both HTR2A and SLC6A4, with the orangutan and macaque sequences used as outgroups to estimate polarity within lineages using the maximum composite-likelihood method in the software program MEGA4 (Tamura et al. 2007). Second, we used the software program HYPHY (Kosakovsky Pond and Frost 2005; Kosakovsky Pond et al. 2005), which uses a phylogenetic approach and maximum likelihood to fit evolutionary models to nucleotide sequence alignments in statistically evaluating substitution rates between gene regions. This analysis enables flexibility in “model-free” testing to determine whether rates vary when standard codon-based models do not apply (e.g., promoter and UTR sites). Similar to the hypothesis-driven approach using HYPHY in Haygood et al. (2007), we evaluated how well substitution rates at each of HTR2A and SLC6A4 promoter, UTR, and nonsynonymous sites are consistent with estimates of neutrality within the respective genes in each of the chimpanzee and human lineages (using orangutan and macaque as outgroups). Specifically, an initial likelihood was derived from a null model where the three classes of promoter, UTR, and nonsynonymous sites each evolved independently, whereas a second likelihood model constrained substitution rates at these three classes to fit the silent site divergence estimates at their respective genes. For each of these three classes, the ratio of these two likelihoods was examined using the chi-squared test implemented in HYPHY (i.e., a standard likelihood ratio test).
Results
Human and Chimpanzee Polymorphism and Divergence
A total of 7,596 bp from the HTR2A gene was collected from each of the human and chimpanzee samples (fig. 1a), with 5,264 reflecting silent site variation as described above. A total of 11,482 bp from the SLC6A4 gene was collected for both species samples (fig. 1b), with 9,378 bp reflecting silent sites as above. Although SLC6A4 human silent site diversity (θπ) appears quite low compared with HTR2A (table 1), neither are unusual from that typically seen for gene regions and across populations (e.g., Sachidanandam et al. 2001; Garrigan and Hammer 2006). As expected, sub-Saharan Africans possess a higher level of “neutral” silent diversity both in magnitude (S) and frequency (θπ) for both HTR2A and SLC6A4 compared with other populations here. In addition, although there is some variance in Tajima's D values across populations, neither gene exhibits an unusually skewed frequency distribution compared with that simulated under our demographic models nor compared with other human genes on average for these populations (Garrigan and Hammer 2006). Finally, the P. t. verus sample shows no unusual pattern of silent diversity compared with other gene studies (Gilad et al. 2003; Fischer et al. 2004; Wooding et al. 2005; Verrelli et al. 2006, 2008). Estimates of human–chimpanzee divergence at sites in each of synonymous and intron site classes for both genes are ∼1% and evenly split within lineages (table 2), which is consistent with other single gene (Gilad et al. 2003; Fischer et al. 2004; Wooding et al. 2005; Verrelli et al. 2006, 2008) and genome-wide estimates of nucleotide site divergence (Chimpanzee Sequencing and Analysis Consortium 2005). Thus, synonymous and intron sites at both genes reflect typical polymorphism and fixation rates of evolution for these species and are suitable proxies of neutrality here.
Table 1.
Population Diversity Estimates for Human and Chimpanzee Serotonin Genes.
|
HTR2A |
SLC6A4 |
||||||
| Sample | na | Sb | θπc | TDd | S | θπ | TD |
| Northern European | 20 | 22 | 0.129 | 0.37 | 14 | 0.042 | 0.02 |
| Italian | 20 | 17 | 0.066 | −1.02 | 20 | 0.056 | −0.24 |
| Russian | 20 | 21 | 0.115 | 0.09 | 10 | 0.037 | 0.80 |
| Chinese | 20 | 14 | 0.101 | 1.29 | 11 | 0.036 | 0.36 |
| Japanese | 20 | 11 | 0.084 | 1.53 | 11 | 0.027 | −0.69 |
| Southeast Asian | 20 | 16 | 0.080 | −0.25 | 10 | 0.023 | −0.82 |
| Mexican | 20 | 23 | 0.126 | 0.09 | 13 | 0.042 | 0.29 |
| Middle Eastern | 20 | 14 | 0.096 | 1.05 | 13 | 0.039 | −0.03 |
| North African | 14 | 12 | 0.113 | 2.27 | 15 | 0.059 | 0.68 |
| Sub-Saharan African | 18 | 31 | 0.143 | −0.67 | 26 | 0.081 | 0.00 |
| Global | 192 | 47 | 0.113 | −0.78 | 38 | 0.047 | −1.00 |
| Chimpanzee | 40 | 26 | 0.109 | −0.20 | 17 | 0.045 | 0.19 |
Number of chromosomes sampled.
Refers to number of silent site SNPs (synonymous and intron sites; see Materials and Methods).
Average pairwise sequence differences (%).
Tajima's D analysis of SNP frequency spectrum.
Table 2.
Human and Chimpanzee Divergence in Serotonin Gene Domains.
| Divergencea | Ratios | |||||||
| Gene | Lineage | dSil | dN | dP | dU | dN/dSil | dP/dSil | dU/dSil |
| HTR2A | Human | 0.56 | 0 | 0.54 | 0.80 | 0 | 0.94 | 1.43 |
| Chimpanzee | 0.49 | 0.09 | 0.67 | 0 | 0.18 | 1.37 | 0 | |
| SLC6A4 | Human | 0.53 | 0.21 | 0.23 | 0.63 | 0.40 | 0.43 | 1.19 |
| Chimpanzee | 0.50 | 0.07 | 0.23 | 1.14 | 0.14 | 0.46 | 2.29 | |
Estimates (%) at silent (Sil), nonsynonymous (N), promoter (P), and UTR (U) sites.
McDonald–Kreitman contrasts of polymorphism at nonsynonymous, promoter, and UTR sites with silent sites are not unusual compared with divergence comparisons of these sites in both human and chimpanzee lineages (supplementary table 1, Supplementary Material online). In fact, although there are several amino acid replacement polymorphisms found in both species, they are rare in frequency (<5%) and are most consistent with weak purifying selection (supplementary table 2, Supplementary Material online). On the other hand, patterns of divergence across domains are significantly unusual when modeled under neutrality in our HYPHY analyses (table 2; see Materials and Methods). Specifically, human–chimpanzee divergence at promoter sites appears elevated at HTR2A but with divergence only in the chimpanzee lineage barely significant (P = 0.05). Divergence at UTR sites appears to be elevated at both genes; however, lineage-specific analyses show significantly high and low divergence within human and chimpanzee HTR2A, respectively (P < 0.05) and a significantly high divergence within only chimpanzee SLC6A4 (P < 0.01).
Haplotype Structure and Age Estimates
The Snn analysis of human HTR2A haplotypes revealed two significant patterns (supplementary fig. 1, Supplementary Material online). First, pairwise comparisons involving sub-Saharan Africans are statistically different, which as previously noted, is not unusual owing to their different demographic history. Second, Chinese and Japanese samples were significantly different compared with others, except the Southeast Asian sample. Correlations among variants are shown in figure 1 for both genes, where SNPs >5% in frequency were informative for human analyses (see Materials and Methods); although many SNPs are unsurprisingly correlated across both genes, our LDhat analysis was used to further identify significant LD. Under this latter model, as expected, the sub-Saharan sample exhibited less LD at HTR2A, yet the overall relationship among SNPs as core haplotypes was similar across each of the 10 samples, that is, common SNPs were similarly associated within and across populations (data not shown). Thus, it is important to note that the significant HTR2A differences found with the Snn tests (supplementary fig. 1, Supplementary Material online) are primarily the result of haplotype frequency differences among populations and less the result of different associations among these SNPs (i.e., different core haplotypes).
The strongest evidence of LD at HTR2A was found in the haplotype group bearing the derived alleles at the two commonly genotyped SNPs −1438 and 102 within all samples but with frequencies differing from 20% to 70% outside of sub-Saharan Africa alone (fig. 2). Our estimate of the MRCA of this −1438A/102T derived haplotype is 326 ± 65 kya (see Materials and Methods), which was similar across all samples, with the estimate within sub-Saharan Africa being older but not significantly different. Interestingly, our MS coalescent simulations under various demographic models (see Materials and Methods) found that this derived haplotype had significantly little diversity (θπ = 0.015%) given its frequency (40%, n = 77) and diversity at HTR2A (P < 0.0001). When simulations independently incorporated different levels of diversity inside and outside of the sub-Saharan Africa sample observed here (table 1) and contrasted models of population change in these different regions (see Materials and Methods), results were similar, suggesting that the pattern associated with this haplotype is not explained by demography alone. Within the −1438A/102T group, our LDhat analysis independently revealed a specific haplotype that uniquely bears the derived 3092C allele in intron 1 (i.e., cannot explain LD in the overall group and vice versa), which reaches 35% in frequency in Japanese and Chinese samples, yet is otherwise virtually absent. In fact, this SNP alone appears to explain the significant HTR2A Snn tests with these two samples (supplementary fig. 1, Supplementary Material online) as its removal results in no significant haplotype structure other than that generally seen with sub-Saharan Africa. Other than the −1438A/102T haplotype group, no other HTR2A haplotypes (of similar frequency or SNP diversity) had an unusual pattern associated with them in our MS coalescent analyses.
FIG. 2.

Histogram plot for HTR2A −1438A/102T, SLC6A4 promoter LPR-S, SLC6A4 intron 2 VNTR 12-repeat, and SLC6A4 LPR-S/12-repeat/23966G haplotype frequencies, for each of the 10 human population samples (see Materials and Methods for abbreviations), as well as the overall global sample of 192 human chromosome sequences.
Our similar analysis of the chimpanzee HTR2A haplotypes (fig. 1a) also shows high correlations, and of these, a few are significantly unusual in our LDhat analysis incorporating background recombination estimates (SNPs > 12% in frequency were informative for both genes). Interestingly, chimpanzees have a similar common mutation at the −1438 upstream promoter site. This variant involves a G to A change at a CpG site, which are generally hypermutable (e.g., Subramanian and Kumar 2003); together with the orangutan and macaque outgroup sequences and the fact that shared SNPs (i.e., identical by descent) in humans and chimpanzees are very rare suggests that it is a “multiple hit” occurring independently within the two lineages. Our LDhat analysis finds that the derived −1438A allele exhibits significant LD with several SNPs in spite of its frequency of 85%. Nonetheless, although −1438A haplotypes have a much lower diversity (θπ = 0.047%) compared with that seen for HTR2A in general (table 1), our MS coalescent simulations find nothing significantly unusual (P = 0.13).
Our Snn analysis of human SLC6A4 haplotypes found that only the sub-Saharan African sample was significantly different from others (supplementary fig. 1, Supplementary Material online). We included the promoter LPR and intron 2 VNTR variants (fig. 1b), which when compared with chimpanzees suggest that the derived states are the S and 12-repeat alleles, respectively. As with HTR2A, although there is strong LD across SLC6A4 (fig. 1b), our LDhat analysis finds that this structure is similar across samples for common variants (data not shown). Of note is that although the LPR and VNTR variants are correlated, there is significant geographic variation for both, with the S allele differing by 25%–85% (fig. 2; chi-squared tests, P < 0.01). Our estimates of the MRCA of the LPR-S (223 ± 43 kya) and VNTR 12-repeat (270 ± 50 kya) alleles were similar to each other and like the −1438 and 102 SNPs are not very recent in age. Our LDhat analysis revealed one haplotype group composed of the LPR-S, intron 2 VNTR 12-repeat, and exon 14 (3′ UTR) site 23966G alleles that showed the strongest LD, with frequencies from 10% to 75% outside of sub-Saharan Africa alone (“S/12/G”; fig. 2). This haplotype had an estimated MRCA of 19 ± 4 kya, and our MS coalescent simulations found it had significantly little associated SNP diversity (θπ = 0.005%) given its frequency (36%, n = 70) and the overall diversity at SLC6A4 (P < 0.0001). As with analyses at HTR2A, this result was not sensitive to MS-simulated differences in diversity and demographic histories inside and outside of Africa. Finally, our similar SLC6A4 LDhat analysis of the chimpanzee SNP associations (fig. 1b) revealed a multi-SNP haplotype group with significantly high LD and which appears to be driven by linkage alone with the 5′ UTR −97 SNP. Our MS simulations found that only the haplotype group defined by the derived −97T allele is unusual in the chimpanzee sample in that it possesses significantly little diversity (θπ = 0.005%) given its frequency (33%, n = 13; P < 0.01).
Discussion
The serotonin-related genes HTR2A and SLC6A4 have been implicated in common behavior-related disorders, and functionally relevant variants have been identified at both. Nonetheless, although studies of ethnically diverse groups show inconsistencies in association studies and failures in treatment of these disorders, the current study is surprisingly the first to use an evolutionary approach to address these problems. To this extent, we find unusual patterns of haplotype structure across human populations for both genes as well as historical patterns of nonneutral evolution across gene domains in both the human and chimpanzee lineages. Below, we discuss these population and species observations in light of the clinical, functional, and evolutionary implications for these two well-studied serotonin pathway genes.
SNP versus Haplotype Studies
Although we find that there are many sites across the human HTR2A gene that exhibit significant LD, the derived −1438A and 102T alleles are found in complete linkage >97% of the time in our global sample. On the other hand, although the LPR and VNTR variants are correlated alone, they do not show the strongest LD at SLC6A4. Thus, although clinical studies genotype these variants and analyze them independently, it is clear that this is unnecessary for the −1438/102 SNPs, and in fact, we find that other variants and combinations may be better candidates. For example, the HTR2A intron 3092C allele is very common in the Chinese and Japanese samples and exhibits the most geographic variation other than that seen for sub-Saharan Africans. Interestingly, our inspection of larger samples (each of 88–120 sequences) in the NCBI HapMap database found that the 3092C allele is similarly high in frequency in their Chinese and Japanese samples, but very rare elsewhere, confirming that our results are not due to sample size alone. However, as this SNP is one of many common alleles preferentially genotyped in these studies, without haplotype analyses considering unbiased samples of SNPs and populations as done here, the pattern associated with 3092C goes unfounded. Additionally, at SLC6A4, we find that incorporating the 23966 SNP into the LPR-S and VNTR 12-repeat haplotype results in the most significant LD and reveals significant geographic variation at the locus. It is interesting to note that this common 23966 SNP is found in the 3′ UTR, which may reflect undocumented functional variation at this gene given the potential regulatory properties of these regions.
Our population sampling of HTR2A and SLC6A4 clearly shows that examining only single variants, and not haplotypes, misses a considerable amount of genetic and population diversity. Thus, one reason why association studies find a significant association between functional SNPs and behavioral phenotypes in one population but not in another could be explained by the frequency differences of these SNPs/haplotypes (i.e., sample size and reduced statistical power) and not actually a lack of an association between genotype and phenotype in different groups. One simple solution is not only to examine haplotypes instead of single variants but also to account for population stratification (i.e., Pritchard, Stephens, and Donnelly 2000) to statistically correct for this haplotypic geographic variation in disease association studies with HTR2A and SLC6A4 (Pritchard, Stephens, Rosenberg, et al. 2000; Tishkoff and Verrelli 2003b; Wakeley and Lessard 2003).
Signatures of Demography and Selection
Another interesting observation at both loci is that specific core haplotypes show low SNP diversity associated with them. It is possible that a demographic scenario, such as recent population expansion (i.e., “Out of Africa” colonization events), could generate this pattern associated with haplotypes high in frequency (e.g., Tishkoff and Verrelli 2003a; Campbell and Tishkoff 2008), yet our coalescent simulations are not consistent with this hypothesis alone. In addition, such a demographic scenario would need to explain why only a specific haplotype group shows this pattern, which is inconsistent with patterns seen at other human genes in general (e.g., Garrigan and Hammer 2006). It may be the case that individual population samples are insufficiently small to detect other haplotype patterns or that only specific populations (e.g., non-African samples) are the cause of the pattern overall. Although this is possible, these simulations consider haplotype frequencies, population sample sizes, and SNP diversity, and in fact, although geographic variation exists at both loci, patterns of LD are similar for these specific haplotypes across samples.
It is possible that recent directional selection explains the increase in frequency of specific haplotypes while increasing LD and reducing associated SNP diversity. The estimates of the age of the SLC6A4 LPR and VNTR and the HTR2A −1438 and 102 mutations are all consistent with an origin in Africa and predate estimates of the emergence of modern humans out of Africa. However, coalescent age estimates of some of the specific derived combinations such as the “S/12/G” haplotype appear to be very recent, which altogether may argue an ancient origin, but a recent expansion. As this haplotype is high in Japanese and Southeast Asian populations, yet lower in samples from both sub-Saharan Africa and Northern Europe, again, a simple demographic explanation is difficult to apply, especially because other haplotypes shared among these regions show no such pattern. Instead, a complex model that incorporates both selection and demography may be necessary, and larger sample sizes and estimates of LD in greater distances from these tagged SNPs should be examined to address these scenarios. All adaptive arguments aside, the observation from our coalescent analyses that LD can be haplotype specific provides one of the larger consequences for association analyses. Specifically, because only certain haplotypes bearing the LPR-S allele exhibit unusual SNP diversity and LD and that they differ in frequency across samples, this is compelling evidence for why this functional variant is correlated with phenotypic traits in some studies but not in others.
As previously noted, general patterns of genetic diversity within P. t. verus, including our samples, show no evidence of an unusual demographic history (e.g., Stone et al. 2002; Wooding et al. 2005; Verrelli et al. 2006, 2008). Thus, the find that this species also has an unusually low SNP diversity associated with a putative SLC6A4 promoter variant (−97T haplotypes) may also imply recent positive selection. In addition, the pattern associated with the same −1438 mutation associated with functional variation in humans, although not significant, is also suggestive. Thus, as in our human samples, further long-range LD analyses focused on these tagged SNPs in larger chimpanzee samples, in addition to functional assays of the −97 and −1438 variants will reveal their adaptive potential (Verrelli BC, unpublished data).
Although both humans and chimpanzees possess amino acid variants, their rare frequencies are consistent with purifying selection, albeit relatively weak (e.g., Ohta 1992). On the other hand, the estimated elevated divergence for regions that putatively regulate serotonin receptor and transporter expression appears to reflect historical positive selection. These recent and ancient patterns imply that adaptive changes in serotonin functioning are highly unlikely to come about by altering the structural proteins themselves, but that altering the expression of these proteins via changes to their transcriptional control may be favorable. These results also imply that some level of regulation, the best evidence here coming from the UTRs, was independently adaptive in both lineages. Interestingly, the significant evidence from the population analyses also point to regulatory regions as being putative targets of recent positive selection. To what extent this divergence actually reflects adaptive cis-regulatory variation is unknown; nonetheless, it does provide targets for testing hypotheses in functional assays (Verrelli BC, unpublished data). Specifically, if we determine that these fixed differences represent potential transcriptional variation, it implies serotonin functional diversity and potentially associated behavior, adaptive or not, that separates us from chimpanzees and other primates.
Serotonin Evolutionary Functional Genomics
Our analyses of the HTR2A −1438 and 102 SNPs would imply that several populations, including those of East Asian descent, would experience increased serotonin receptor expression and thus increased depression as well (Vaidya et al. 1997; Manji et al. 2001). In addition, if we conservatively consider only SLC6A4 LPR-S/S homozygotes (and not S-heterozygotes who also show an increased tendency for depression; Caspi et al. 2003), we would predict that over 60% of the Southeast Asian population, but only 1% of the Northern European population, would experience a major and clinical depression episode (based on allele frequencies in fig. 2). Although the highest rates of depression and social anxiety in populations of East Asian descent are well documented, with the rates for those of European descent being lower (e.g., Okazaki 1997; Schoen et al. 1997; Abright 2002; Ozer and McDonald 2006), our estimates are not consistent with those actually observed in clinical studies, thus demonstrating the clear problems in using single variants in association studies at these loci. Furthermore, even when combinations of variants have been used, haplotype analyses are not conducted in samples of varying ethnic backgrounds. For example, Myers et al. (2007) suggested that the −1483A variant increased HTR2A expression only when paired with the −783G promoter allele, but this study included >86% “Caucasians” and would appear biased. In fact, we found that the −783G allele (estimated ancestral state) reaches the highest frequencies in our Northern European (15%) and African samples (22%), yet it is found only four times in the rest of the global sample (∼2%; supplementary fig. 2, Supplementary Material online) and cannot explain the LD and haplotype structure here. Additionally, in a promoter assay, Ali et al. (2009) showed that the LPR-S/VNTR 12-repeat combination actually showed the highest SLC6A4 expression compared with the lowest expression for the LPR-L/VNTR 10-repeat combination. This observation would appear to conflict with large behavioral studies like that of Caspi et al. (2003); however, the latter used exclusively subjects of “Caucasian” background.
A final consideration is not only to use multiple variants as haplotypes across ethnically diverse populations but also a population genomic approach to explain the problem of inconsistent associations. For example, a model that at least considers the interaction of both SLC6A4 and HTR2A proteins would seem to be more suitable. If we simply examine the five best candidates for functional variation at both loci, the SLC6A4 LPR and VNTR and the HTR2A −1483, −783, and 102 variants, we can estimate all possible haplotypes (supplementary fig. 2, Supplementary Material online). Although LD is apparent at both HTR2A and SLC6A4 from our previous analyses, there is no significant haplotype LD between loci either in the overall sample or within any of the population samples. In fact, if we collapse these single-locus haplotypes into predicted “high” and “low” expression phenotypes, the frequencies of di-locus “expression alleles” follow no one pattern and are highly variable across populations (supplementary fig. 2, Supplementary Material online). These analyses not only show that the two loci are statistically independent but also that there is geographic diversity associated with these haplotypes as well.
Our haplotype analyses combined with previous functional analyses suggest that individuals with “low” SLC6A4 expression haplotypes are just as likely to have “low” or “high” HTR2A haplotypes and vice versa; and thus, although these individuals may have reduced capacity for serotonin uptake that would otherwise result in increased risk for depression, they may also have downregulated receptor expression that results in an equilibrium and reduces this risk. Our study is not only the first to explore the haplotype structure of ethnically diverse samples but to provide a potential multilocus haplotype model of serotonin functioning from these major candidate loci. Although our model is simple in its additive assumptions, given the strong LD shared across these samples for core haplotypes and the lack of LD across loci, we would conclude that haplotype frequency differences, and not population-specific combinations of the SNPs themselves, best explain why expression variation appears so variable across samples, and thus, why association studies continue to show inconsistent results.
If it is the case that “low” and “high” expression alleles at these loci interact as hypothesized, then one may speculate that the unusual patterns of diversity associated with these common haplotypes at both loci could be one of balancing selection. That is, overall “low” transporter expression is only adaptive as it counterbalances “high” receptor expression and vice versa. It is also possible that balanced serotonin functioning at the level of expression found today is consistent with the observed historical trend found in primates in general. For example, this balance may be a temporal trade-off as different serotonin expression levels appear to be correlated with behavior at different life stages in rhesus macaques who also possess SLC6A4 promoter length variants similar to that found in humans and chimpanzees (Kinnally et al. 2009).
From an applied perspective, given the vast research on drugs for serotonin regulation, our observations provide direct input into the specific HTR2A and SLC6A4 haplotypes that should be targeted for genotyping across populations. The hypothesis that transcriptional regions were important in the divergence of humans from other primates has long-been supported (e.g., King and Wilson 1975; Gilad et al. 2006), yet we are just beginning to connect this primate variation with complex phenotypes using an adaptive evolutionary genomics model (Stone and Verrelli 2006). In fact, together with similar studies on neurotransmitters related to dopamine expression and catabolism in humans and other primates (Gilad et al. 2002; Miller-Butterworth et al. 2007, 2008), our current study sheds light on the genetic change associated with cognition and behavior, the various disorders linked to them, and how they have coevolved during the divergence of humans and our closest primate relatives.
Supplementary Material
Supplementary tables 1 and 2 and figures 1 and 2 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Supplementary Material
Acknowledgments
The authors thank R. Haygood for advice on the HYPHY analyses and three anonymous reviewers for critical review of the manuscript. Chimpanzee samples were provided by H. Vasken Aposhian, the Detroit Zoological Institute (Detroit, MI), the Lincoln Park Zoo (Chicago, IL), the Primate Foundation of Arizona (Mesa, AZ), the Riverside Zoo (Scottsbluff, NE), the Southwest Foundation for Biomedical Research (San Antonio, TX), the Sunset Zoo (Manhattan, KS), the Welsh Mountain Zoo (North Wales, UK), the Yerkes National Primate Research Center (Atlanta, GA), and the New Iberia Research Center (Lafayette, LA), supported by National Institutes of Health-National Center for Research Resources grant (U42-RR015087) to the University of Louisiana at Lafayette. K.G.C. was supported by National Institutes of Health Post-baccalaureate Research Education Program for Biomedical Research grant (R25-GM071798) to Arizona State University.
References
- Abdolmaleky HM, Faraone SV, Glatt SJ, Tsuang MT. Meta-analysis of association between the T102C polymorphism of the 5HT2a receptor gene and schizophrenia. Schizophr Res. 2004;67:53–62. doi: 10.1016/s0920-9964(03)00183-x. [DOI] [PubMed] [Google Scholar]
- Abright AR. Depression in Asian American children. West J Med. 2002;176:244–248. [PMC free article] [PubMed] [Google Scholar]
- Ali FR, Vasiliou SA, Haddley K, Paredes UM, Roberts JC, Miyajima F, Klenova E, Bubb VJ, Quinn JP. Combinatorial interaction between two human serotonin transporter gene variable number tandem repeats and their regulation by CTCF. J Neurochem. 2009;112:296–306. doi: 10.1111/j.1471-4159.2009.06453.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrés AM, de Hemptinne C, Bertranpetit J. Heterogeneous rate of protein evolution in serotonin genes. Mol Biol Evol. 2007;24:2707–2715. doi: 10.1093/molbev/msm202. [DOI] [PubMed] [Google Scholar]
- Auton A, Bryc K, Boyko AR, et al. (13 co-authors) Global distribution of genomic diversity underscores rich complex history of continental human populations. Genome Res. 2009;19:795–803. doi: 10.1101/gr.088898.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becquet C, Patterson N, Stone AC, Przeworski M, Reich D. Genetic structure of chimpanzee populations. PLoS Genet. 2007;3:e66. doi: 10.1371/journal.pgen.0030066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornstein P, McKay J, Morishima JK, Devarayalu S, Gelinas RE. Regulatory elements in the first intron contribute to transcriptional control of the human alpha 1(I) collagen gene. Proc Natl Acad Sci USA. 1987;84:8869–8873. doi: 10.1073/pnas.84.24.8869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray NJ, Buckland PR, Hall H, Owen MJ, O'Donovan MC. The serotonin-2A receptor gene locus does not contain common polymorphism affecting mRNA levels in adult brain. Mol Psychiatry. 2004;9:109–114. doi: 10.1038/sj.mp.4001366. [DOI] [PubMed] [Google Scholar]
- Butler SG, Meegan MJ. Recent developments in the design of anti-depressive therapies: targeting the serotonin transporter. Curr Med Chem. 2008;15:1737–1761. doi: 10.2174/092986708784872357. [DOI] [PubMed] [Google Scholar]
- Campbell MC, Tishkoff SA. African genetic diversity: implications for human demographic history, modern human origins, and complex disease mapping. Annu Rev Genomics Hum Genet. 2008;9:403–433. doi: 10.1146/annurev.genom.9.081307.164258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi A, Sugden K, Moffitt TE, et al. (11 co-authors) Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. 2003;301:386–389. doi: 10.1126/science.1083968. [DOI] [PubMed] [Google Scholar]
- Cheung VG, Spielman RS. Genetics of human gene expression: mapping DNA variants that influence gene expression. Nat Rev Genet. 2009;10:595–604. doi: 10.1038/nrg2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chimpanzee Sequencing and Analysis Consortium. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature. 2005;437:69–87. doi: 10.1038/nature04072. [DOI] [PubMed] [Google Scholar]
- Cho HJ, Meira-Lima I, Cordeiro Q, Michelon L, Sham P, Vallada H, Collier DA. Population-based and family-based studies on the serotonin transporter gene polymorphisms and bipolar disorder: a systematic review and meta-analysis. Mol Psychiatry. 2005;10:771–781. doi: 10.1038/sj.mp.4001663. [DOI] [PubMed] [Google Scholar]
- Fan JB, Sklar P. Meta-analysis reveals association between serotonin transporter gene STin2 VNTR polymorphism and schizophrenia. Mol Psychiatry. 2005;10:928–938. doi: 10.1038/sj.mp.4001690. [DOI] [PubMed] [Google Scholar]
- Fischer A, Wiebe V, Paabo S, Przeworski M. Evidence for a complex demographic history of chimpanzees. Mol Biol Evol. 2004;21:799–808. doi: 10.1093/molbev/msh083. [DOI] [PubMed] [Google Scholar]
- Fukuda Y, Koga M, Arai M, et al. (11-co-authors) Monoallelic and unequal allelic expression of the HTR2A gene in human brain and peripheral lymphocytes. Biol Psychiatry. 2006;60:1331–1335. doi: 10.1016/j.biopsych.2006.06.024. [DOI] [PubMed] [Google Scholar]
- Garrigan D, Hammer MF. Reconstructing human origins in the genomic era. Nat Rev Genet. 2006;7:669–680. doi: 10.1038/nrg1941. [DOI] [PubMed] [Google Scholar]
- Gilad Y, Bustamante CD, Lancet D, Paabo S. Natural selection on the olfactory receptor gene family in humans and chimpanzees. Am J Hum Genet. 2003;73:489–501. doi: 10.1086/378132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilad Y, Oshlack A, Smyth GK, Speed TP, White KP. Expression profiling in primates reveals a rapid evolution of human transcription factors. Nature. 2006;440:242–245. doi: 10.1038/nature04559. [DOI] [PubMed] [Google Scholar]
- Gilad Y, Rosenberg S, Przeworski M, Lancet D, Skorecki K. Evidence for positive selection and population structure at the human MAO-A gene. Proc Natl Acad Sci USA. 2002;99:862–867. doi: 10.1073/pnas.022614799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hariri AR, Mattay VS, Tessitore A, Kolachana B, Fera F, Goldman D, Egan MF, Weinberger DR. Serotonin transporter genetic variation and the response of the human amygdala. Science. 2002;297:400–403. doi: 10.1126/science.1071829. [DOI] [PubMed] [Google Scholar]
- Haygood R, Fedrigo O, Hanson B, Yokoyama KD, Wray GA. Promoter regions of many neural- and nutrition-related genes have experienced positive selection during human evolution. Nat Genet. 2007;39:1140–1144. doi: 10.1038/ng2104. [DOI] [PubMed] [Google Scholar]
- Heinz A, Braus DF, Smolka MN, et al. (12 co-authors) Amygdala-prefrontal coupling depends on a genetic variation of the serotonin transporter. Nat Neurosci. 2005;8:20–21. doi: 10.1038/nn1366. [DOI] [PubMed] [Google Scholar]
- Heinz A, Jones DW, Mazzanti C, Goldman D, Ragan P, Hommer D, Linnoila M, Weinberger DR. A relationship between serotonin transporter genotype and in vivo protein expression and alcohol neurotoxicity. Biol Psychiatry. 2000;47:643–649. doi: 10.1016/s0006-3223(99)00171-7. [DOI] [PubMed] [Google Scholar]
- Heils A, Teufel A, Petri S, Stober G, Riederer P, Bengel D, Lesch KP. Allelic variation of human serotonin transporter gene expression. J Neurochem. 1996;66:2621–2624. doi: 10.1046/j.1471-4159.1996.66062621.x. [DOI] [PubMed] [Google Scholar]
- Holmes C, Arranz MJ, Powell JF, Collier DA, Lovestone S. 5-HT2A and 5-HT2C receptor polymorphisms and psychopathology in late onset Alzheimer's disease. Hum Mol Genet. 1998;7:1507–1509. doi: 10.1093/hmg/7.9.1507. [DOI] [PubMed] [Google Scholar]
- Hudson RR. A new statistic for detecting genetic differentiation. Genetics. 2000;155:2011–2014. doi: 10.1093/genetics/155.4.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR. Two-locus sampling distributions and their application. Genetics. 2001;159:1805–1817. doi: 10.1093/genetics/159.4.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics. 2002;18:337–338. doi: 10.1093/bioinformatics/18.2.337. [DOI] [PubMed] [Google Scholar]
- Jonnakuty C, Gragnoli C. What do we know about serotonin? J Cell Physiol. 2008;217:301–306. doi: 10.1002/jcp.21533. [DOI] [PubMed] [Google Scholar]
- Kaessmann H, Wiebe V, Paabo S. Extensive nuclear DNA sequence diversity among chimpanzees. Science. 1999;286:1159–1162. doi: 10.1126/science.286.5442.1159. [DOI] [PubMed] [Google Scholar]
- Kaiser R, Muller-Oerlinghausen B, Filler D, Tremblay PB, Berghofer A, Roots I, Brockmoller J. Correlation between serotonin uptake in human blood platelets with the 44-bp polymorphism and the 17-bp variable number of tandem repeat of the serotonin transporter. Am J Med Genet. 2002;114:323–328. doi: 10.1002/ajmg.10119. [DOI] [PubMed] [Google Scholar]
- King MC, Wilson AC. Evolution at two levels in humans and chimpanzees. Science. 1975;188:107–116. doi: 10.1126/science.1090005. [DOI] [PubMed] [Google Scholar]
- Kinnally EL, Tarara ER, Mason WA, Mendoza SP, Abel K, Lyons LA, Capitanio JP. Serotonin transporter expression is predicted by early life stress and is associated with disinhibited behavior in infant rhesus macaques. Genes Brain Behav. 2009;9:45–52. doi: 10.1111/j.1601-183X.2009.00533.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Frost SDW. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol. 2005;22:1208–1222. doi: 10.1093/molbev/msi105. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Frost SDW, Muse SV. HyPhy: hypothesis testing using phylogenies. Bioinformatics. 2005;21:676–679. doi: 10.1093/bioinformatics/bti079. [DOI] [PubMed] [Google Scholar]
- Kumar S, Filipski A, Swarna V, Walker A, Hedges SB. Placing confidence limits on the molecular age of the human-chimpanzee divergence. Proc Natl Acad Sci USA. 2005;102:18842–18847. doi: 10.1073/pnas.0509585102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laje G, Perlis RH, Rush AJ, McMahon FJ. Pharmacogenetics studies in STAR*D: strengths, limitations, and results. Psychiatr Serv. 2009;60:1446–1457. doi: 10.1176/appi.ps.60.11.1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley K, Payton A, Hamshere ML, et al. (11 co-authors) No evidence of association of two 5HT transporter gene polymorphisms and attention deficit hyperactivity disorder. Psychiatr Genet. 2003;13:107–110. doi: 10.1097/01.ypg.0000056177.32550.a5. [DOI] [PubMed] [Google Scholar]
- Lasky-Su JA, Faraone SV, Glatt SJ, Tsuang MT. Meta-analysis of the association between two polymorphisms in the serotonin transporter gene and affective disorders. Am J Med Genet B Neuropsychiat Genet. 2005;133B:110–115. doi: 10.1002/ajmg.b.30104. [DOI] [PubMed] [Google Scholar]
- Lesch KP, Bengel D, Heils A, Sabol SZ, Greenberg BD, Petri S, Benjamin J, Muller CR, Hamer DH, Murphy DL. Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science. 1996;274:1527–1530. doi: 10.1126/science.274.5292.1527. [DOI] [PubMed] [Google Scholar]
- Lohmueller KE, Pearce CL, Pike M, Lander ES, Hirschhorn JN. Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat Genet. 2003;33:177–182. doi: 10.1038/ng1071. [DOI] [PubMed] [Google Scholar]
- Majewski J, Ott J. Distribution and characterization of regulatory elements in the human genome. Genome Res. 2002;12:1827–1836. doi: 10.1101/gr.606402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra AK, Lencz T, Correll CU, Kane JM. Genomics and the future of pharmacotherapy in psychiatry. Int Rev Psychiatry. 2007;19:523–530. doi: 10.1080/09540260701563460. [DOI] [PubMed] [Google Scholar]
- Manji HK, Drevets WC, Charney DS. The cellular neurobiology of depression. Nat Med. 2001;7:541–547. doi: 10.1038/87865. [DOI] [PubMed] [Google Scholar]
- McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991;351:652–654. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- McMahon FJ, Buervenich S, Charney D, et al. (13 co-authors) Variation in the gene encoding the serotonin 2A receptor is associated with outcome of antidepressant treatment. Am J Hum Genet. 2006;78:804–814. doi: 10.1086/503820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean G, Awadalla P, Fearnhead P. A coalescent-based method for detecting and estimating recombination from gene sequences. Genetics. 2002;160:1231–1241. doi: 10.1093/genetics/160.3.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller-Butterworth CM, Kaplan JR, Barmada MM, Manuck SB, Ferrell RE. The serotonin transporter: sequence variation in Macaca fascicularis and its relationship to dominance. Behav Genet. 2007;37:678–696. doi: 10.1007/s10519-007-9162-3. [DOI] [PubMed] [Google Scholar]
- Miller-Butterworth CM, Kaplan JR, Shaffer J, Devlin B, Manuck SB, Ferrell RE. Sequence variation in the primate dopamine transporter gene and its relationship to social dominance. Mol Biol Evol. 2008;25:18–28. doi: 10.1093/molbev/msm219. [DOI] [PubMed] [Google Scholar]
- Munafò MR, Clark TG, Moore LR, Payne E, Walton R, Flint J. Genetic polymorphisms and personality in healthy adults: a systematic review and meta-analysis. Mol Psychiatry. 2003;8:471–484. doi: 10.1038/sj.mp.4001326. [DOI] [PubMed] [Google Scholar]
- Murphy DL, Fox MA, Timpano KR, Moya P, Ren-Patterson R, Andrews AM, Holmes A, Lesch KP, Wendland JR. How the serotonin story is being rewritten by new gene-based discoveries principally related to SLC6A4, the serotonin transporter gene, which functions to influence all cellular serotonin systems. Neuropharmacology. 2008;55:932–960. doi: 10.1016/j.neuropharm.2008.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers RL, Airey DC, Manier DH, Shelton RC, Sanders-Bush E. Polymorphisms in the regulatory region of the human serotonin 5-HT2A receptor gene (HTR2A) influence gene expression. Biol Psychiatry. 2007;61:167–173. doi: 10.1016/j.biopsych.2005.12.018. [DOI] [PubMed] [Google Scholar]
- Nebert DW, Zhang G, Vesell ES. From human genetics and genomics to pharmacogenetics and pharmacogenomics: past lessons, future directions. Drug Metab Rev. 2008;40:187–224. doi: 10.1080/03602530801952864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogilvie AD, Battersby S, Bubb VJ, Fink G, Harmar AJ, Goodwim GM, Smith CA. Polymorphism in serotonin transporter gene associated with susceptibility to major depression. Lancet. 1996;347:731–733. doi: 10.1016/s0140-6736(96)90079-3. [DOI] [PubMed] [Google Scholar]
- Ohta T. The nearly neutral theory of molecular evolution. Annu Rev Ecol Syst. 1992;23:263–286. [Google Scholar]
- Okazaki S. Sources of ethnic differences between Asian American and White American college students on measures of depression and social anxiety. J Abnorm Psychol. 1997;106:52–60. doi: 10.1037//0021-843x.106.1.52. [DOI] [PubMed] [Google Scholar]
- Ozer EJ, McDonald KL. Exposure to violence and mental health among Chinese American urban adolescents. J Adolesc Health. 2006;39:73–79. doi: 10.1016/j.jadohealth.2005.09.015. [DOI] [PubMed] [Google Scholar]
- Parsons MJ, D'Souza UM, Arranz MJ, Kerwin RW, Makoff AJ. The -1438A/G polymorphism in the 5-hydroxytryptamine type 2A receptor gene affects promoter activity. Biol Psychiatry. 2004;56:406–410. doi: 10.1016/j.biopsych.2004.06.020. [DOI] [PubMed] [Google Scholar]
- Perry GH, Tchinda J, McGrath SD, et al. (12 co-authors) Hotspots for copy number variation in chimpanzees and humans. Proc Natl Acad Sci USA. 2006;103:8006–8011. doi: 10.1073/pnas.0602318103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry GH, Yang F, Marques-Bonet T, et al. (14 co-authors) Copy number variation and evolution in humans and chimpanzees. Genome Res. 2008;18:1698–1710. doi: 10.1101/gr.082016.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezawas L, Meyer-Lindenberg A, Drabant EM, Verchinski BA, Munoz KE, Kolachana BS, Egan MF, Mattay VS, Hariri AR, Weinberger DR. 5-HTTLPR polymorphism impacts human cingulate-amygdala interactions: a genetic susceptibility mechanism for depression. Nat Neurosci. 2005;8:828–834. doi: 10.1038/nn1463. [DOI] [PubMed] [Google Scholar]
- Polesskaya OO, Sokolov BP. Differential expression of the “C” and “T” alleles of the 5-HT2A receptor gene in the temporal cortex of normal individuals and schizophrenics. J Neurosci Res. 2002;67:812–822. doi: 10.1002/jnr.10173. [DOI] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Rosenberg NA, Donnelly P. Association mapping in structured populations. Am J Hum Genet. 2000;67:170–181. doi: 10.1086/302959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramamoorthy S, Bauman AL, Moore KR, Han H, Yang-Feng T, Chang AS, Ganapathy V, Blakely RD. Antidepressant- and cocaine-sensitive human serotonin transporter: molecular cloning, expression, and chromosomal localization. Proc Natl Acad Sci USA. 1993;90:2542–2546. doi: 10.1073/pnas.90.6.2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas J, Sánchez-DelBarrio JC, Messeguer X, Rozas R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19:2496–2497. doi: 10.1093/bioinformatics/btg359. [DOI] [PubMed] [Google Scholar]
- Sachidanandam R, Weissman D, Schmidt SC, et al. (42 co-authors) A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 2001;409:928–933. doi: 10.1038/35057149. [DOI] [PubMed] [Google Scholar]
- Sakai K, Nakamura M, Ueno S, Sano A, Sakai N, Shirai Y, Saito N. The silencer activity of the novel human serotonin transporter linked polymorphic regions. Neurosci Lett. 2002;327:13–16. doi: 10.1016/s0304-3940(02)00348-8. [DOI] [PubMed] [Google Scholar]
- Savitz JB, Ramesar RS. Genetic variants implicated in personality: a review of the more promising candidates. Am J Med Genet B Neuropsychiat Genet. 2004;131B:20–32. doi: 10.1002/ajmg.b.20155. [DOI] [PubMed] [Google Scholar]
- Schaffner SF, Foo C, Gabriel S, Reich D, Daly MJ, Altshuler D. Calibrating a coalescent simulation of human genome sequence variation. Genome Res. 2005;15:1576–1583. doi: 10.1101/gr.3709305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheinfeldt LB, Biswas S, Madeoy J, Connelly CF, Schadt EE, Akey JM. Population genomic analysis of ALMS1 in humans reveals a surprisingly complex evolutionary history. Mol Biol Evol. 2009;26:1357–1367. doi: 10.1093/molbev/msp045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoen C, Davis K, Collins KS, Greenberg L, Des Roches C, Abrams M. New York: Louis Harris and Associates, Inc; 1997. The Commonwealth Fund survey of the health of adolescent girls. [Google Scholar]
- Sen S, Burmeister M, Ghosh D. Meta-analysis of the association between a serotonin transporter promoter polymorphism (5-HTTLPR) and anxiety-related personality traits. Am J Med Genet B Neuropsychiat Genet. 2004;127B:85–89. doi: 10.1002/ajmg.b.20158. [DOI] [PubMed] [Google Scholar]
- Serretti A, Drago A, De Ronchi D. HTR2A gene variants and psychiatric disorders: a review of current literature and selection of SNPs for future studies. Curr Med Chem. 2007;14:2053–2069. doi: 10.2174/092986707781368450. [DOI] [PubMed] [Google Scholar]
- Sjöqvist F, Eliasson E. The convergence of conventional therapeutic drug monitoring and pharmacogenetic testing in personalized medicine: focus on antidepressants. Clin Pharmacol Ther. 2007;81:899–902. doi: 10.1038/sj.clpt.6100188. [DOI] [PubMed] [Google Scholar]
- Smits KM, Smits LJM, Schouten JSAG, Stelma FF, Nelemans P, Prins MH. Influence of SERTPR and STin2 in the serotonin transporter gene on the effect of selective serotonin reuptake inhibitors in depression: a systematic review. Mol Psychiat. 2004;9:433–441. doi: 10.1038/sj.mp.4001488. [DOI] [PubMed] [Google Scholar]
- Spurlock G, Heils A, Holmans P, et al. (11 co-authors) A family based association study of T102C polymorphism in 5HT2A and schizophrenia plus identification of new polymorphisms in the promoter. Mol Psychiatry. 1998;3:42–49. doi: 10.1038/sj.mp.4000342. [DOI] [PubMed] [Google Scholar]
- Stephens M, Scheet P. Accounting for decay of linkage disequilibrium in haplotype inference and missing-data imputation. Am J Hum Genet. 2005;76:449–462. doi: 10.1086/428594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone AC, Griffiths RC, Zegura SL, Hammer MF. High levels of Y-chromosome nucleotide diversity in the genus Pan. Proc Natl Acad Sci USA. 2002;99:43–48. doi: 10.1073/pnas.012364999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone AC, Verrelli BC. Focusing on comparative ape population genetics in the post-genomic age. Curr Opin Genet Dev. 2006;16:586–591. doi: 10.1016/j.gde.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Subramanian S, Kumar S. Neutral substitutions occur at a faster rate in exons than in noncoding DNA in primate genomes. Genome Res. 2003;13:838–844. doi: 10.1101/gr.1152803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Thomson R, Pritchard JK, Shen P, Oefner PJ, Feldman MW. Recent common ancestry of human Y chromosomes: evidence from DNA sequence data. Proc Natl Acad Sci USA. 2000;97:7360–7365. doi: 10.1073/pnas.97.13.7360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff SA, Verrelli BC. Patterns of human genetic diversity: implications for human evolutionary history and disease. Annu Rev Genomics Hum Genet. 2003a;4:293–340. doi: 10.1146/annurev.genom.4.070802.110226. [DOI] [PubMed] [Google Scholar]
- Tishkoff SA, Verrelli BC. Role of evolutionary history on haplotype block structure in the human genome: implications for disease mapping. Curr Opin Genet Dev. 2003b;13:569–575. doi: 10.1016/j.gde.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Vaidya VA, Marek GJ, Aghajanian GK, Duman RS. 5-HT2A receptor-mediated regulation of brain-derived neurotrophic factor mRNA in the hippocampus and the neocortex. J Neurosci. 1997;17:2785–2795. doi: 10.1523/JNEUROSCI.17-08-02785.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verrelli BC, Lewis CM, Jr., Stone AC, Perry GH. Different selective pressures shape the molecular evolution of color vision in chimpanzee and human populations. Mol Biol Evol. 2008;25:2735–2743. doi: 10.1093/molbev/msn220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verrelli BC, McDonald JH, Argyropoulos G, Destro-Bisol G, Froment A, Drousiotou A, Lefranc G, Helal AN, Loiselet J, Tishkoff SA. Evidence for balancing selection from nucleotide sequence analyses of human G6PD. Am J Hum Genet. 2002;71:1112–1128. doi: 10.1086/344345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verrelli BC, Tishkoff SA. Signatures of selection and gene conversion associated with human color vision variation. Am J Hum Genet. 2004;75:363–375. doi: 10.1086/423287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verrelli BC, Tishkoff SA, Stone AC, Touchman JW. Contrasting histories of G6PD molecular evolution and malarial resistance in humans and chimpanzees. Mol Biol Evol. 2006;23:1592–1601. doi: 10.1093/molbev/msl024. [DOI] [PubMed] [Google Scholar]
- Wakeley J, Lessard S. Theory of the effects of population structure and sampling on patterns of linkage disequilibrium applied to genomic data from humans. Genetics. 2003;164:1043–1053. doi: 10.1093/genetics/164.3.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall JD, Pritchard JK. Haplotype blocks and linkage disequilibrium in the human genome. Nat Rev Genet. 2003;4:587–597. doi: 10.1038/nrg1123. [DOI] [PubMed] [Google Scholar]
- Warren JT, Jr., Peacock ML, Rodriguez LC, Fink JK. An MspI polymorphism in the human serotonin receptor gene (HTR2): detection by DGGE and RFLP analysis. Hum Mol Genet. 1993;2:338. doi: 10.1093/hmg/2.3.338. [DOI] [PubMed] [Google Scholar]
- Watterson GA. On the number of segregating sites in genetic models without recombination. Theor Popul Biol. 1975;7:256–276. doi: 10.1016/0040-5809(75)90020-9. [DOI] [PubMed] [Google Scholar]
- Willeit M, Praschak-Rieder N, Neumeister A, et al. (16 co-authors) A polymorphism (5-HTTLPR) in the serotonin transporter promoter gene is associated with DSM-IV depression subtypes in seasonal affective disorder. Mol Psychiatry. 2003;8:942–946. doi: 10.1038/sj.mp.4001392. [DOI] [PubMed] [Google Scholar]
- Wooding S, Bufe B, Grassi C, Howard MT, Stone AC, Vazquez M, Dunn DM, Meyerhof W, Weiss RB, Bamshad MJ. Independent evolution of bitter-taste sensitivity in humans and chimpanzees. Nature. 2006;440:930–934. doi: 10.1038/nature04655. [DOI] [PubMed] [Google Scholar]
- Wooding S, Stone AC, Dunn DM, Mummidi S, Jorde LB, Weiss RK, Ahuja S, Bamshad MJ. Contrasting effects of natural selection on human and chimpanzee CC chemokine receptor 5. Am J Hum Genet. 2005;76:291–301. doi: 10.1086/427927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray GA, Hahn MW, Abouheif E, Balhoff JP, Pizer M, Rockman MV, Romano LA. The evolution of transcriptional regulation in eukaryotes. Mol Biol Evol. 2003;20:1377–1419. doi: 10.1093/molbev/msg140. [DOI] [PubMed] [Google Scholar]
- Zhu QS, Chen K, Shih JC. Characterization of the human 5-HT2A receptor gene promoter. J Neurosci. 1995;15:4885–4895. doi: 10.1523/JNEUROSCI.15-07-04885.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.