

Abstract
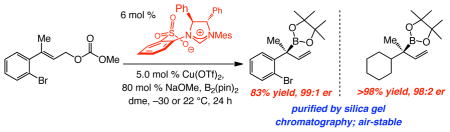
Allylic substitutions that afford α-substituted allylboronates bearing B-substituted tertiary or quaternary carbon stereogenic centers are presented. C–B bond forming reactions, catalyzed by chiral bidentate Cu–NHC complexes, are performed in the presence of commercially available bis(pinacolato)diboron. Transformations proceed in high yield (up to >98%), site selectivity (>98% SN2′) and in up to >99:1 enantiomer ratio (er). Trans as well as cis disubstituted alkenes can be used; alkyl- (linear as well as branched) and aryl-substituted trisubstituted allylic carbonates serve as effective substrates. Allylboronates that bear a quaternary carbon center are air-stable and can be easily purified by silica gel chromatography; in contrast, secondary allylboronates cannot be purified in the same manner and are significantly less stable. Oxidation of the enantiomerically enriched products furnishes secondary or tertiary allylic alcohols, valuable small molecules that cannot be easily obtained in high enantiomeric purity by alternative synthesis or kinetic resolution approaches.
Allylboronates are used as nucleophiles for additions to carbonyls or imines and can be converted to valuable allylic alcohols or amines.1 Design of efficient catalytic protocols for enantioselective preparation of such B-containing unsaturated molecules thus represents a compelling objective in chemical synthesis. In their notable disclosure, Ito and Sawamura outlined an enantioselective protocol for synthesis of α-substituted allylboronates that contain a B-substituted tertiary carbon; this was accomplished by reactions of allylic carbonates with bis(pinacolato)diboron (1), catalyzed by a chiral Cu–phosphine complex.2,3 Use of Z-allylic carbonates bearing a linear alkyl substituent was necessary, however, since enantioselectivity diminished substantially with E isomers; furthermore, boronate substitution was not observed with substrates containing a sterically hindered alkyl (e.g., cyclohexyl) or an aryl unit. Herein, we report methods for synthesis of an assortment of α-substituted allylboronates through reactions promoted by bidentate NHC–Cu complexes developed in these laboratories (eq 1).4 Transformations proceed with 1 and various allylic carbonates, delivering allylboronates carrying a tertiary or – for the first time – a quaternary α carbon stereogenic center,5 in >98% site selectivity and up to 99:1 enantiomer ratio (er). E-Disubstituted allylic carbonates as well as trisubstituted alkyl (linear or branched) or aryl alkenes are effective substrates. Subsequent oxidation furnishes enantiomerically enriched carbinols,6 including tertiary allylic alcohols, valuable entities not easily accessible by other synthesis7 or kinetic resolution strategies.8
 |
(1) |
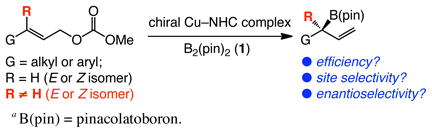
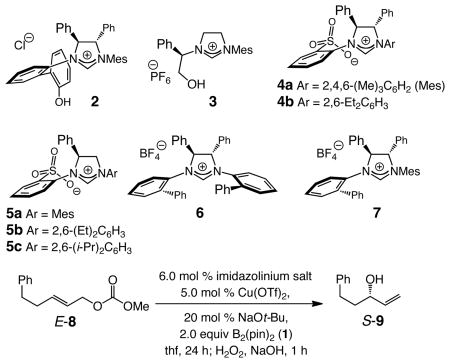
We began by examining the reaction of an E-disubstituted alkene in the presence of a representative set of chiral NHC–Cu complexes, generated in situ from treatment of an imidazolinium salt with a sodium alkoxide and Cu(OTf)2; initial screening had indicated that, somewhat surprisingly, high reactivity and selectivity can be obtained with the Cu(II) salt.9 Representative findings emerging from initial catalyst screening are summarized in Table 1. Reactions with complexes derived from bidentate hydroxyl-containing 2 or 3 are inefficient (≤22% conv). In contrast, sulfonates 4a–b and 5a–c give rise to significantly more effective catalysts: substantial conversion (34–81%) is observed in spite of reactions being performed at −30 °C (vs 22 °C for entries 1–2). Among the bidentate sulfonate–NHC–Cu complexes probed, 5c, bearing a di-i-propylphenyl moiety, promotes the most enantioselective transformation (94:6 er). It should be noted that, although the reaction in the presence of 5c proceeds to 69% conversion, the desired allylic alcohol S-9 is isolated in 68% yield after purification (including the follow-up oxidation procedure); the allylic substitution thus proceeds without generating significant amounts of byproducts. As indicated in entries 8–9 of Table 1, C1- or C2-symmetric monodentate Cu complexes of 6 and 710 are relatively ineffective catalysts (63:37 and 61:39 er, respectively).
Table 1.
Evaluation of Chiral NHC Complexesa
 | |||||
|---|---|---|---|---|---|
| entry | imidazolinium salt | temp (°C) | conv (%)b | yield (%)c | erd |
| 1 | 2 | 22 | 10 | 9 | 61:39 |
| 2 | 3 | 22 | 22 | 19 | 62:38 |
| 3 | 4a | −30 | 81 | 76 | 85:15 |
| 4 | 4b | −30 | 54 | 50 | 84.5:15.5 |
| 5 | 5a | −30 | 70 | 70 | 81:19 |
| 6 | 5b | −30 | 34 | 33 | 85:15 |
| 7 | 5c | −30 | 69 | 68 | 94:6 |
| 8 | 6 | 22 | 53 | 52 | 63:37 |
| 9 | 7 | 22 | 42 | 41 | 61:39 |
Reactions performed under N2 atm; >98% site selectivity in all cases (e.g., <2% SN2 product).
Values determined by analysis of 400 MHz 1H NMR spectra of unpurified mixtures.
Yields of purified products.
By GLC analysis; see the Supporting Information for details.
Subsequent optimization studies indicated that use of dme (vs thf used in Table 1) and excess amounts of NaOMe (80 mol % vs 20 mol % NaOt-Bu) leads to significant improvement in reaction efficiency.11 As shown in Scheme 1, under the new conditions, allylic alcohol S-9 is isolated in 90% yield (>98% conv) and in 95:5 er from reaction of E-8; the related transformation involving Z-8 furnishes R-9 in 94% yield and with identical enantioselectivity (95:5 er). Cu-catalyzed allylic boronate substitutions, involving substrates that bear a β- or an α-branched alkyl substituent proceed readily; allylic carbonate precursor to 11 is unreactive with the previously reported chiral Cu–phosphine catalysts.2 Allylic alcohols 10 and 11 (Scheme 1) are obtained from reactions of the corresponding E-disubstituted allylic carbonates in 71% and 93% yield and 91.5:8.5 and 97:3 er, respectively. In all cases, none of the achiral products derived from SN2 mode of C–B bond formation are detected (see below for more details regarding this selectivity issue).
Scheme 1.

NHC–Cu-Catalyzed Boronate Additions to Disubstituted Allylic Carbonatesa
a All conv >98% by analysis of 400 MHz 1H NMR spectra of unpurified mixtures; >98% SN2′ in all cases. Er determined by GLC analysis (see the SI for details).
With an effective procedure for reactions of E- and Z-disubstituted allylic carbonates established, we turned to the more challenging issue of enantioselective synthesis of allylboronates with an α B-substituted quaternary carbon stereogenic center. The findings summarized in Table 2 illustrate that in the presence of 6.0 mol % chiral imidazolinium salt 4a, alkyl-bearing trisubstituted allylic carbonates, including those that contain a sterically hindered substituent (e.g., entry 7), efficiently undergo reaction to afford the derived allylboronates in up to 98:2 er (86 to >98% conv, 81–97% yield of tertiary allylic alcohol).12 Preliminary studies indicated that the reaction in entry 1 of Table 2 in the presence of 5c (22 °C), used in the case of disubstituted alkenes,13 affords the desired product with somewhat lower enantioselectivity (89:11 er vs 95:5 er with 4a).
Table 2.
NHC–Cu-Catalyzed Enantioselective Synthesis of Alkyl-Substituted Tertiary Allylboronates and Alcoholsa
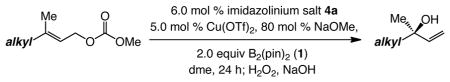 | |||||
|---|---|---|---|---|---|
| entry | substrate (alkyl) | temp (°C) | conv (%)b | yield (%)c | erd |
| 1 | 22 | >98 | 97 | 95:5 | |
| 2 | −30 | 86 | 82 | 97:3 | |
| 3 | 22 | >98 | 96 | 96.5:3.5 | |
| 4 | −30 | >98 | 83 | 95:5 | |
| 5 | −30 | >98 | 81 | 95:5 | |
| 6 | 22 | >98 | 89 | 81:19e | |
| 7 | −30 | >98 | 95 | 98:2 | |
See Table 1.
Enantioselectivity does not improve at −30 °C.
Reaction rates and enantioselectivities can be sensitive to substrate structure. The more facile processes can be performed at −30 °C so that higher er values can be achieved; the representative comparison in entries 1–2, where (−)-linalool can be obtained in 82% yield and 97:3 er, illustrate this point. The lowest er value (81:19 er in entry 6, Table 2) corresponds to a substrate that contains a β-branched alkyl, a trend observed with reactions of disubstituted alkenes as well (see S-9, 10 and 11, Scheme 1). In further contrast to the less substituted alkenes, reactions of Z-trisubstituted allylic carbonates are less efficient and relatively non-selective. As an example, when the Z isomer of substrate in entry 1 of Table 2 is used with the Cu complex of 4a under the conditions shown in Table 2, (+)-linalool is isolated in 88% yield (>98% conv at 22 °C) and 76:24 er (vs >98% conv, 97% yield and 95:5 er with the E isomer in entry 1 of Table 2).
Transformations of aryl-substituted alkenes, which pose a noteworthy mechanistic question, were subsequently investigated. We have reported that NHC–Cu complexes in the presence of B2(pin)2 promote boron–copper additions with disubstituted aryl olefins (vs those in Table 1 and Scheme 1) with high site selectivity, affording benzylic C–Cu bonds exclusively (I, Figure 1). Reaction of an allylic carbonate (R = CH2OCO2Me in I), bearing a disubstituted olefin, was found to afford only a homobenzylic C–B bond (>98% α to C–O).14 Such preferences were attributed to the stabilization of the developing electron density at the carbon of the benzylic C–Cu bond (I). The above considerations offer a rationale for the site selectivity in NHC–Cu-catalyzed reactions of alkyl-substituted carbonates discussed above: the intermediate C–Cu is likely stabilized by the adjacent C–O. With an aryl-containing trisubstituted allylic carbonate, C–Cu bond formation may be electronically favored at either the benzylic or the homobenzylic position (stabilization by aryl group or C–O, respectively). Partial positive charge developed at the site of the C–B bond formation, however, should be better accommodated at the more substituted benzylic carbon (II), and disfavored adjacent to the electron withdrawing carbonate, thus culminating in site selective allylic substitution.15
Figure 1.

Effect of alkene substitution on selectivity of Cu–B addition to an aryl alkene.
As the data in Table 3 indicate, in sharp contrast to their disubstituted counterparts,13 aryl-containing carbonates containing a trisubstituted alkene undergo Cu-catalyzed boronate substitution with completely opposite sense of site-selectivity, in favor of benzylic C–B bond. Reactions afford the corresponding tertiary allylic alcohols (after oxidative workup) with >98% site selectivity and 72–96% yield. The desired allylboronates can be obtained in 90:10–99:1 er, with the highest selectivities being observed with the more sterically demanding aryl units; lower temperature can lead to improved enantioselectivity (entries 6–7; >98% conv and 89:11 er for both cases at 22 °C). The lack of reactivity with p-trifluoromethyl substrate (entry 8) supports the aforementioned scenario (cf. II, Figure 1); reaction via I could be energetically inaccessible, or MeOH may be required for effective turnover with such a mode of addition.13
Table 3.
NHC–Cu-Catalyzed Enantioselective Synthesis of Aryl-Substituted Tertiary Allylboronates and Alcohols a
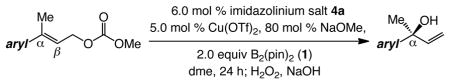 | ||||||
|---|---|---|---|---|---|---|
| entry | aryl | temp (°C) | conv (%)b | α:βb | yield (%)c | erd |
| 1 | Ph | 22 | >98 | >98:2 | 95 | 90:10e |
| 2 | 1-naphthyl | 22 | >98 | >98:2 | 91 | 94.5:5.5e |
| 3 | o-ClC6H4 | 22 | >98 | >98:2 | 96 | 99:1 |
| 4 | o-BrC6H4 | 22 | 86 | >98:2 | 84 | 99:1 |
| 5 | o-MeC6H4 | 22 | >98 | >98:2 | 88 | 98:2 |
| 6 | m-MeC6H4 | −30 | 86 | >98:2 | 77 | 93:7 |
| 7 | p-MeC6H4 | −30 | 74 | >98:2 | 72 | 92:8 |
| 8 | p-CF3C6H4 | 22 | <2 | – | – | – |
See Table 1.
Enantioselectivity does not improve at −30 °C.
A limitation of the present class of reactions is worthy of note. Cu-catalyzed reaction of Et-substituted alkene 12 (eq 2) delivers 13 in 62% yield and 80:20 er. Particularly noteworthy is that the reaction leads to – unlike all previous cases – the formation of the primary C–B bond (20% SN2 addition). Generation of the achiral byproduct suggests the intermediacy of π-allylcopper complexes in reactions promoted by boronate-containing NHC–cuprates (vs neutral copper boronates), and, at least in certain instances, the intermediacy of Cu(III) complex (oxidative addition), followed by B–alkyl reductive elimination (vs Cu–B addition to the alkene proposed for neutral Cu complexes2). The above findings point to the need for still more effective chiral catalysts for transformations of highly congested alkenes.
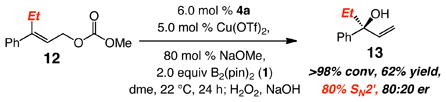 |
(2) |
Unlike the less substituted tertiary derivatives, allylic carbonates with a B-substituted quaternary carbon are sufficiently robust to be purified by silica gel chromatography. Alkyl- or aryl-substituted quaternary allylboronates can be obtained in high yields and er (Scheme 2). Such stability is likely the result of steric congestion at the B center, rendering it less susceptible to nucleophilic attack. Nonetheless, quaternary carbon-containing allyl boronates obtained by the present protocol do serve as nucleophilic reagents. Treatment of 14 with benzaldehyde (22 °C) affords the desired homoallylic alcohol in 98% yield, 97:3 er but as an equal mixture of alkene isomers (improvement not observed at −78 °C).
Scheme 2.

Stability of Enantiomerically Enriched Allylboronates
We put forth protocols for enantioselective synthesis of allyl boronates and allylic alcohols that were not easily attainable previously,16 and further underline the special utility of sulfonate-bearing NHC–Cu catalysts.17 These studies, and a small number of other disclosures,18 underline the need for certain future developments. Identification of effective catalysts that promote reactions of sterically congested allylboronates (e.g., 14–15) to electrophilic substrates, introduction of effective procedures leading to efficient and stereoselective conversion of hindered C–B to C–N19 and C–C bonds are among such challenges. Investigations along these lines, mechanistic studies, development of more effective chiral catalysts and use of the NHC–Cu-catalyzed C–B bond formation to other substrate classes are in progress.
Supplementary Material
Acknowledgments
Support was provided by the NSF (CHE-0715138); A. G.-M. is an NIH Postdoctoral Fellow (GM-47480). We are grateful to B. Maybury-Lewis for experimental assistance and to Frontier, Inc. for gifts of reagent 1. Mass spectrometry facilities at Boston College are supported by the NSF (DBI-0619576).
Footnotes
Supporting Information Available: Experimental procedures and spectral, analytical data for all products (PDF). This material is available on the web: http://www.pubs.acs.org
References
- 1.Hall DG, editor. Boronic Acids. Wiley-VCH; Weinheim, Germany: 2000. [Google Scholar]
- 2.Ito H, Ito S, Sasaki Y, Matsuura K, Sawamura M. J Am Chem Soc. 2007;129:14856. doi: 10.1021/ja076634o. [DOI] [PubMed] [Google Scholar]
- 3.For catalytic enantioselective synthesis of α-substituted allyl boronates (tertiary C–B), see: Gao X, Hall DG. J Am Chem Soc. 2003;125:9308. doi: 10.1021/ja036368o.Pelz NF, Woodward AR, Burks HE, Sieber JD, Morken JP. J Am Chem Soc. 2004;126:16328. doi: 10.1021/ja044167u.Gerdin M, Moberg C. Adv Synth Catal. 2005;347:749.Carosi L, Hall DG. Angew Chem, Int Ed. 2007;46:5913. doi: 10.1002/anie.200700975.Peng F, Hall DG. Tetrahedron Lett. 2007;48:3305.For related auxiliary-based methods, see: Fang GY, Aggarwal VK. Angew Chem, Int Ed. 2007;46:359. doi: 10.1002/anie.200603659. and references cited therein.
- 4.For example, see: Larsen AO, Leu W, Oberhuber CN, Campbell JE, Hoveyda AH. J Am Chem Soc. 2004;126:11130. doi: 10.1021/ja046245j.Van Veldhuizen JJ, Campbell JE, Giudici RE, Hoveyda AH. J Am Chem Soc. 2005;127:6877. doi: 10.1021/ja050179j.Brown MK, May TL, Baxter CA, Hoveyda AH. Angew Chem, Int Ed. 2007;46:1097. doi: 10.1002/anie.200604511.May TL, Brown MK, Hoveyda AH. Angew Chem, Int Ed. 2008;47:7358. doi: 10.1002/anie.200802910.Brown MK, Hoveyda AH. J Am Chem Soc. 2008;130:12904. doi: 10.1021/ja8058414.
- 5.To the best of our knowledge, there is only a limited number of reported examples involving a chiral enantiomerically enriched substrate (non-catalytic) for enantioselective synthesis of an α-substituted allyl boronate with a quaternary carbon stereogenic center. See: Stymiest JL, Bagutski V, French RM, Aggarwal VK. Nature. 2008;456:778. doi: 10.1038/nature07592.Bagutski V, Ros A, Aggarwal VK. Tetrahedron. 2009;65:9956.
- 6.For representative recent catalytic enantioselective protocols that afford secondary allylic alcohols, see: Tomita D, Wada R, Kanai M, Shibasaki M. J Am Chem Soc. 2005;127:4138. doi: 10.1021/ja0507362.Kirsch SF, Overman LE. J Am Chem Soc. 2005;127:2866. doi: 10.1021/ja0425583.Lyothier I, Defieber C, Carreira EM. Angew Chem, Int Ed. 2006;45:6204. doi: 10.1002/anie.200602408.For additional cases, see the Supporting Information.
- 7.For example, see: Uenishi J, Kubo Y. Tetrahedron Lett. 1994;35:6697.Wu Y-K, Liu H-J, Zhu J-L. Synlett. 2008. p. 621.A tertiary allylic alcohol has been prepared by Cu-catalyzed enantioselective vinyl addition to an α-ketoester. See ref 6a.
- 8.Tertiary allylic alcohols cannot be effectively resolved through Ti-catalyzed epoxidation: Johnson, R. A., Sharpless, K. B. in Catalytic Asymmetric Synthesis; Ojima, I., Ed., VCH: Weinheim, Germany, 1993, p. 103.
- 9.For details of Cu screening, see the Supporting Information. It is noteworthy that a Cu(II) salt is reduced to the catalytic active Cu(I) complex by 1. All previous Cu-catalyzed C–B bond forming reactions involve use of a Cu(I) salt (e.g., ref 2, 13 and 18b). Related mechanistic details will be discussed in the full account of this work.
- 10.For use of C1-symmetric chiral NHC–Cu complexes in enantioselective synthesis, see: Lee K-s, Hoveyda AH. J Org Chem. 2009;74:4455. doi: 10.1021/jo900589x.Lee K-s, Hoveyda AH. J Am Chem Soc. 2010;132:2898. doi: 10.1021/ja910989n.
- 11.The exact reason for improvement in reaction efficiency due to use of larger excess of NaOMe is not clear, but may be partly the result of more efficient imidazolinium salt deprotonation and Cu–NHC complex formation.
- 12.The stereochemical outcome of reactions in Table 2 (determined based on formation of (−)-linalool in entry 1) differs from E disubstituted alkenes (see Scheme 1). The origin of this difference is difficult to establish in the absence of mechanistic data (identity of turnover-limiting step).
- 13.Lee Y, Hoveyda AH. J Am Chem Soc. 2009;131:3160. doi: 10.1021/ja809382c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.When alkyl-containing (vs aryl) disubstituted olefins are subjected to Cu–NHC-catalyzed boron–copper additions (in the presence of MeOH), <2% hydroboration product is generated. See ref 13.
- 15.It is possible that boron–copper addition is reversible and only when mode of addition II occurs, Cu–alkoxide elimination leads to the formation of the allylic substitution product. Studies carried out with products from additions to alkynes suggest that boron–copper addition process is largely irreversible (Lee, Y., Hoveyda, A. H., unpublished results); similar investigations corresponding to alkene substrates are in progress.
- 16.Reactions with the less air sensitive Cu(OAc)2·H2O, although at times less efficient, deliver similar enantioselectivities. For example, 9 and product from reaction in entry 3 of Table 2 are obtained in 52% yield, 94:6 er and 96% yield, 97:3 er, respectively, with the latter Cu salt.
- 17.For another example, see: Lee Y, Akiyama K, Gillingham DG, Brown MK, Hoveyda AH. J Am Chem Soc. 2008;130:446. doi: 10.1021/ja0782192.
- 18.(a) Lee K-s, Zhugralin AR, Hoveyda AH. J Am Chem Soc. 2009;131:7253. doi: 10.1021/ja902889s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chen IH, Yin L, Itano W, Kanai M, Shibasaki M. J Am Chem Soc. 2009;131:11664. doi: 10.1021/ja9045839. [DOI] [PubMed] [Google Scholar]
- 19.Primary or secondary benzylic C–B bonds have been converted to C–N bonds. For representative cases, see: Fernandez E, Maeda K, Hooper MW, Brown JM. Chem Eur J. 2000;6:1840. doi: 10.1002/(sici)1521-3765(20000515)6:10<1840::aid-chem1840>3.0.co;2-6.However, application of such procedures to compounds bearing a B-substituted quaternary carbon has not been disclosed.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.