

Abstract
Nitric -oxide reductase (NOR) from Paracoccus denitrificans catalyzes the reduction of nitric oxide (NO) to nitrous oxide (N2O) (2NO + 2H+ + 2e− →N2O + H2O) by a poorly understood mechanism. NOR contains two low spin hemes c and b, one high spin heme b3, and a non-heme iron FeB. Here, we have studied the reaction between fully reduced NOR and NO using the “flow-flash” technique. Fully (four-electron) reduced NOR is capable of two turnovers with NO. Initial binding of NO to reduced heme b3 occurs with a time constant of ∼1 μs at 1.5 mm NO, in agreement with earlier studies. This reaction is [NO]-dependent, ruling out an obligatory binding of NO to FeB before ligation to heme b3. Oxidation of hemes b and c occurs in a biphasic reaction with rate constants of 50 s−1 and 3 s−1 at 1.5 mm NO and pH 7.5. Interestingly, this oxidation is accelerated as [NO] is lowered; the rate constants are 120 s−1 and 12 s−1 at 75 μm NO. Protons are taken up from solution concomitantly with oxidation of the low spin hemes, leading to an acceleration at low pH. This effect is, however, counteracted by a larger degree of substrate inhibition at low pH. Our data thus show that substrate inhibition in NOR, previously observed during multiple turnovers, already occurs during a single oxidative cycle. Thus, NO must bind to its inhibitory site before electrons redistribute to the active site. The further implications of our data for the mechanism of NO reduction by NOR are discussed.
Keywords: Enzyme Catalysis, Enzyme Mechanisms, Nitric Oxide, Oxidation-Reduction, Proton Transport, Flow-flash, Non-heme Iron, Substrate Inhibition
Introduction
Bacterial nitric-oxide reductases (NOR)3 are integral membrane proteins that reduce NO to N2O (Reaction 1) as part of the sequential reduction of nitrate to dinitrogen, a process termed denitrification (for recent reviews, see Refs. 1, 2).
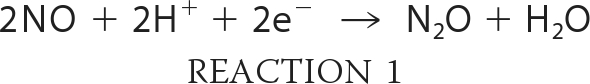 |
The largest subunit in the NORs is a divergent member of the superfamily of O2-reducing heme-copper oxidases (HCuOs), which are characterized by having a catalytic subunit with six invariant histidines at the same predicted positions in 12 trans-membrane helices (3, 4). Two of these histidines coordinate a low spin heme, one a high spin heme, and in the O2-reducing heme-copper oxidases, the remaining three histidines coordinate a copper ion. In NOR, this copper is presumed to be replaced by a non-heme iron (5, 6).
When purified, the NOR from Paracoccus denitrificans contains two subunits; NorB and NorC. NorB is the catalytic subunit harboring a low spin heme b, a high spin heme b3, and a non-heme iron, FeB. The heme b3 and FeB form a magnetically coupled binuclear center, which is the site of NO reduction. This binuclear center is connected via a μ-oxo bridge in the resting oxidized state of the enzyme (7, 8). NorC is a membrane-anchored protein harboring a low spin heme c, which is believed to be the site of electron entry from the water-soluble electron source, either cytochrome c551 or pseudoazurin (1, 9).
The purified P. denitrificans NOR catalyzes NO reduction with a maximum turnover number of ∼40 NO s−1 (6, 10). The mitochondrial aa3-type HCuOs, like bovine cytochrome c oxidase, are not capable of NO reduction at significant turnover rates (11, 12). Some properties of NOR that differ from other HCuOs and thus might be involved in conferring the high NO-reducing capability include: (i) the iron instead of copper in the catalytic site and the binding characteristics of ligands such as CO (13); (ii) a very low midpoint potential (∼60 mV) of high spin heme b3, suggested to avoid formation of a stable heme b32+-NO “dead-end” complex during turnover (14); (iii) a reaction decoupled from the generation of an electrochemical gradient, NO reduction by NOR is nonelectrogenic and protons as well as electrons are derived from the periplasmic side of the membrane (15–17).
The detailed mechanism for NO reduction by NOR is not known, but models have been suggested that involve either a “trans” mechanism (16, 18, 19), where one NO binds to each of the metals in the binuclear site, or “cis” mechanisms, where two NOs bind either to the non-heme FeB (20; see also 14), or consecutively to heme b3 such that the second NO binds to the intermediate formed upon binding of the first (21).
A detailed catalytic model needs to be compatible with the observation that the maximum turnover activity is obtained at ∼5 μm NO, whereas higher NO concentrations lead to a decrease in activity. This substrate inhibition has been suggested to be due to NO binding also to oxidized NOR (6, 22).
For the HCuOs, the detailed mechanism of oxygen reduction and its coupled proton pumping have been investigated extensively using the so-called “flow-flash” technique (23). In this technique, fully reduced enzyme with the inhibitor carbon monoxide (CO) bound to the high spin heme in the active site is mixed in a stopped-flow apparatus with an O2-containing solution. Because O2 cannot bind to the high spin heme while CO is bound, the reaction is limited by dissociation of CO, which is slow. However, if a short laser flash (∼10 ns) is applied when mixing is complete, the photolabile Fe-CO bond is broken, and the binding of dioxygen and its subsequent stepwise reduction can be followed using time-resolved spectroscopy. This technique has been used in combination with different detection techniques and has led to insights into the catalytic cycle of the HCuOs, e.g. the conversion rates and chemical structure of different intermediates, the sequence of electron transfer reactions, and the mechanisms and pathways for proton transfer (see e.g. 24–26).
Using the flow-flash technique, we have previously studied the reaction between fully reduced NOR and O2 rather than NO because of the experimental advantages, e.g. in studying the mechanism of proton transfer (27–29), because the chemical reactivity of NO in aqueous solutions hampers direct pH measurements. Our results showed oxygen binding to heme b3 followed by electron transfer from the low spin hemes b and c to the bound O2 with a time constant of 25 ms (at pH 7.5). The 25-ms phase is coupled to proton uptake from the bulk solution, limited by proton transfer from a group with pKa = 6.6 (27). This reaction was impaired or specifically altered in mutant NORs where residues, suggested (17) to form a proton transfer pathway leading from the periplasm to the active site, were exchanged by site-directed mutagenesis (28, 29).
The flow-flash technique has also been used in one study of reduction of NO by NOR (16). The reductive coupling of two NO molecules to form N2O requires two electrons, whereas the fully reduced NOR has four redox-active groups. Hence, two full turnovers are required to reoxidize the enzyme. Moreover, because the oxidized NOR can bind NO (see above and Ref 22), there are multiple possible reaction paths in the reaction of fully reduced NOR with NO.
To elucidate the details of the mechanism of reduction of the physiological substrate NO, here we have studied the reaction between fully reduced NOR and NO, with special emphasis on the effect of the concentration of the substrates NO and protons. Our results show that whereas initial binding of NO to the heme b3 shows an acceleration at higher [NO], subsequent oxidation of the low spin hemes is decelerated. These results indicate that NO does not necessarily have to bind transiently to FeB for producing the first N2O at the active site. Furthermore, NO binds to an inhibitory site, possibly the oxidized heme b3 (or FeB), faster than electrons are transferred to the active site for the second turnover. This slows oxidation already during a single oxidative cycle. We also studied proton uptake from solution, which occurred simultaneously with oxidation of the low spin hemes. This is consistent with the effect of lowering the pH on these reactions, where binding of NO to heme b3 is not dependent on pH, whereas subsequent oxidation of the low spin hemes is. The results further show that at low pH, this increase in rate is counteracted by a more pronounced substrate inhibition, possibly associated with a larger degree of NO binding to the oxidized b3 (or FeB) at low pH.
EXPERIMENTAL PROCEDURES
Growth of Bacteria and Purification of NOR
The NOR used in this study was from P. denitrificans, expressed in Escherichia coli JM109 using the expression system described in Ref. 10. Growth of bacteria, preparation of membranes, and purification of NOR were performed as previously described (10), with the same changes as in (28). NOR was then rapidly frozen in N2(l) and stored at −80 °C until needed.
Sample Preparation for Flow-flash Studies
NOR was diluted to 5–10 μm in a modified Thunberg cuvette, air was exchanged for nitrogen on a vacuum line, and the enzyme was reduced by adding 1–2 mm ascorbate and 0.2 μm 5-methylphenazinium methosulfate. Nitrogen was then exchanged for ∼15% CO as in Ref. 27; low CO concentrations were used to avoid CO recombination interfering with NO binding. Before transferring the sample to the flow-flash apparatus, 10–20 μm dithionite was added to ensure that the sample was completely reduced and to reduce any residual O2.
Flow-flash Measurements
The setup for flow-flash measurements was described in Ref. 30. Briefly, fully reduced CO-bound NOR was mixed 1:5 with NO-containing buffers in a modified stopped-flow apparatus (Applied Photophysics), where both syringes had been made anaerobic by incubation in 50 mm dithionite overnight. After a 200-ms delay, a 10-ns laser flash from the second harmonic (532 nm) of a Nd-YAG laser (Quantel) was applied, dissociating CO and allowing NO to bind and initiate the reaction. For measurements of NO binding at high (millimolar) NO concentrations, the rise time of the instrument was improved, and the xenon lamp of the measuring light was pulsed, generating a bright light pulse, stable for ∼500 μs, as described in Ref. 31.
The time course of the reaction was studied at different wavelengths in the Soret and α regions. Typically, at each wavelength, 5 · 104 data points were collected, and the dataset was then reduced to ∼1000 points by averaging on a logarithmic time scale.
For the pH-dependence measurements, the buffer concentration in the NOR solution before mixing (in the same 1:5 ratio) was decreased to 10 mm (Hepes at pH 7.5). The NO buffer contained either 200 mm Mes (pH 6.0–7.0) or 200 mm Hepes (pH 7.0–8.0).
Buffers with saturated [NO] were prepared by flushing 100% NO gas through anaerobic buffers as in Ref. 5. The lower [NO] buffers were prepared by flushing 5% NO gas (mixed with 95% N2) in the same way. The solubility of NO was taken to be 1.8 mm at all pH values (32), so that after mixing NOR with NO in the flow-flash setup in the 1:5 ratio, the [NO] was ∼1.5 mm (from 100% NO) or 75 μm (from 5% NO). Preliminary experiments varying the final NO concentrations were made by varying the mixing ratio in the flow-flash setup.
Proton Uptake Measurements
Proton uptake during oxidation of fully reduced NOR by NO was measured essentially as in Ref. 33. Briefly, the protein sample was concentrated and passed through a PD-10 column (Amersham Biosciences) equilibrated with 100 mm KCl, 50 μm EDTA, and 0.05% DDM. Phenol red was added to 40 μm and the pH adjusted to ∼7.6. The protein sample was then reduced and CO-bound as described before. The NO solution also contained 100 mm KCl, 50 μm EDTA, 0.05% DDM, and 40 μm phenol red at pH ∼7.6. The measurements were performed at 567 nm (rather than at the absorbance maximum of the dye at 560 nm) to minimize contributions from oxidation of the low spin heme b. To minimize pH drifts in the unbuffered solutions, due to NO reacting with any residual O2 (producing HNO3), the glucose/glucose oxidase/catalase system (30 mm glucose, 0.1 unit/ml glucose oxidase, and 20 units/ml catalase) was included in both the protein sample and the unbuffered NO mixing solution. Furthermore, only the lower (75 μm) NO concentration was used. The pH of the solution was monitored by the absorbance of phenol red at 570 nm and adjusted immediately before transferring it to the mixing syringes. The buffer control solution contained 50 mm Hepes (pH 7.6), 50 mm KCl, 50 μm EDTA, 0.05% DDM, and 40 μm phenol red. As a further comparison, O2 was instead used as the oxidant in the same experiment (as in Ref. 27) to compare the amplitudes of the observed proton uptake signals.
Data Handling and Analysis
To extract the rate constants and kinetic difference spectra from the traces obtained in the flow-flash experiment, the time-resolved changes in absorbance from different wavelengths were fitted either separately or globally to a model of consecutive irreversible reactions using the software package Pro-K (Applied Photophysics). Alternatively, rate constants were fitted as a sum of exponentials in SigmaPlot (Jandel Scientific).
RESULTS
NO Reduction by the Fully Reduced NOR
After mixing of CO-bound fully reduced NOR with NO-containing buffers, CO is flashed off from heme b3 allowing NO to bind and initiate the reaction. The static optical spectra of oxidized, reduced, and CO-bound NOR are shown in supplemental Fig. 1. Fully reduced NOR has four preloaded electrons, whereas reduction of NO to N2O requires two. Therefore, we will assume that the reaction comprises two turnovers, where the first uses electrons already present at the binuclear site, whereas the second turnover has to involve electron transfer from the low spin hemes b and c.
Initial Binding of NO to Reduced Heme b3 and the First Turnover
After the unresolved flash-induced dissociation of CO from heme b3, the first observed phase in the presence of 1.5 mm NO has a time constant of ∼1 μs and is clearly seen as an absorbance decrease at 430 nm (Fig. 1A). This phase shows a kinetic difference spectrum (data not shown) consistent with NO binding to the heme b3, similar to the spectrum obtained by Hendriks et al. (16). There is a second rapid phase with a time constant of ∼20 μs and a different kinetic difference spectrum with small amplitudes, that we cannot assign to a specific process at this point. The rate constant and spectrum of this phase are similar to the 1.9·104 s−1 (τ = 50 μs) phase obtained by Hendriks et al. (16). However, the amplitude of this phase varies at 430 nm between protein preparations, which we assume is due to an overlapping second phase of NO binding, similar to the biphasic CO binding which is more pronounced in the P. denitrificans NOR when expressed in E. coli (see 13 and 27). After binding of NO during the first 100 μs, there are no significant absorbance changes until oxidation of the low spin hemes takes place on the millisecond time scale (see below and Fig. 1).
FIGURE 1.

Absorbance changes during the reaction between fully reduced NOR and NO (1.5 mm). Traces are shown for 430 nm (A, reporting mainly on the heme b3), 420 nm (B, all hemes contributing), 550 nm (C, heme c) and 560 nm (D, heme b). The initial rise in absorbance at t = 0 at 430 nm is due to the unresolved dissociation of CO. Experimental conditions: 200 mm Hepes at pH 7.5, 50 mm KCl, 0.1 mm EDTA, 0.05% DDM, ∼2 μm reacting NOR, [NO] = 1.5 mm, T = 295 K. The short time scales in A and B were recorded using a brighter light pulse (see “Experimental Procedures”). The red line in the left, shorter panel for 430 nm is a fit to the two time constants 1 μs and 20 μs (see “Experimental Procedures”). The red lines on the longer time scales are two-exponential fits to the two rate constants 50 s−1 and 3 s−1 for oxidation of hemes c and b.
Oxidation of the Low Spin Hemes; Second Turnover
At 1.5 mm NO and pH 7.5, oxidation of the low spin hemes occurs with rate constants of 50 s−1 and 3 s−1 (τ = 20 ms and 300 ms, respectively; see fits in Fig. 1). As seen in Fig. 1, the c-type heme has a larger contribution to the fast phase (about 50% of absorbance change at 550 nm) than the b-type hemes (6–20% contribution at the other wavelengths). Heme c thus oxidizes faster than heme b, which is consistent with previously obtained data (16).
NO Concentration Dependence
The rapid phase(s) of NO-binding clearly displays NO concentration-dependent kinetics. At 75 μm NO (Fig. 2), the trace on the rapid time scale was fitted with two time constants of 6 μs and 100 μs, each contributing ∼50% to the observed amplitude. This gives a difference of a factor of ∼5 to the rates for binding at 1.5 mm NO (for both components).
FIGURE 2.

Comparison of the reaction between fully reduced NOR and NO at high (1.5 mm) and lower (75 μm) NO concentrations. Other conditions are as in Fig. 1. The amplitudes of the signals have been normalized to the same CO step. For the traces on the short time scale, a linear slope originating from the pulsed lamp has been subtracted.
Oxidation of the low spin hemes is also clearly dependent on the NO concentration, as the process speeds up at lower [NO] (see Fig. 2). A global fit to data at 420, 430, and 550 nm (data at 420 and 550 nm not shown) for 75 μm NO shown in Fig. 2 gave major rate constants of 120 s−1 and 12 s−1. Also, at this [NO], the fast process is mainly heme c oxidation (60% contribution at 550 nm, below 35% at 420 and 430). This gives a difference of a factor of ∼3–4 compared with the rates observed at 1.5 mm NO (50 s−1 and 3 s−1).
pH Dependence
Whereas rapid binding of NO on the microsecond time scale at 1.5 mm is not dependent on pH (Fig. 3A), oxidation of the low spin hemes is, as seen in Fig. 3. The slower heme oxidation phase at pH 6.0 with 1.5 mm NO had a rate constant of ∼6 s−1 (Fig. 3A), a factor of 2 faster than at pH 7.5. The pH effect is more pronounced at lower [NO] (75 μm) (Fig. 3B), where the slower (major) rate constants differ by a factor of 8 (∼100 s−1 at pH 6.0 compared with 12 s−1 at pH 7.5).
FIGURE 3.

Comparison of the oxidation of fully reduced NOR by NO at pH 7.5 and 6.0. Shown are also the differences at high (1.5 mm, A) and low (75 μm, B) [NO]. Other conditions are as in Fig. 1.
The same data also show that the effect of lowering the [NO] on the kinetics is greater at pH 6.0, the rate of the major (slower) heme oxidation phase increases from ∼6 s−1 (1.5 mm) to ∼100 s−1 (75 μm), a factor of 17, which is linear with the 20-fold decrease in [NO]. Table 1 summarizes the kinetic data for oxidation of the low spin hemes.
TABLE 1.
Rate constants observed for the oxidation of the low spin hemes during oxidation of fully reduced NOR by NO
| NO concentration | Rate constants | % of Total amplitude |
|
|---|---|---|---|
| 560 nm (heme b) | 550 nm (heme c) | ||
| s−1 | |||
| 1.5 mm NO, pH 7.5 | 50 | 10 | 50 |
| 3 | 90 | 50 | |
| 75 μm NO, pH 7.5 | 120 | 10 | 55 |
| 12 | 90 | 45 | |
| 1.5 mm NO, pH 6.0 | 150 | ∼10a | 40 |
| 6 | ∼90a | 60 | |
| 75 μm NO, pH 6.0 | ∼500 | 35 | 60 |
| 100 | 65 | 40 | |
a Not detected at 560 nm, amount of heme b oxidized estimated from other wavelengths.
Proton Uptake during Oxidation of Fully Reduced NOR by NO
Proton uptake by NOR during oxidation by 75 μm NO is shown in Fig. 4A. The traces obtained were fitted (globally, including also data at 420 and 550 nm) to two phases with rate constants ∼200 s−1 and 20 s−1 with approximately equal amplitudes (ΔA∼1.5·10−3 for each phase) at 567 nm. These rate constants are slightly faster than those obtained in the buffered solutions at pH 7.5 (see above), indicating that the pH has drifted to a somewhat lower pH. The trace acquired at 430 nm the same experiment is shown in Fig. 4B, and as seen in the figure, proton uptake does not occur on the time scale of NO binding, but only coupled to the slower phases of oxidation of the low spin hemes.
FIGURE 4.

A, proton uptake from solution, measured as the change in absorbance of phenol red at 567 nm, during the reaction between fully reduced NOR and NO (75 μm). B, absorbance changes at 430 nm in the same experiment as in A. Experimental conditions: 100 mm KCl (50 mm Hepes/50 mm KCl for the buffered trace), 40 μm phenol red, 50 μm EDTA, 0.05% DDM, pH ∼7.5 (see “Experimental Procedures”). A laser artifact around t = 0 at 567 nm has been truncated for clarity. Other conditions are as in Fig. 1.
For the control using O2 as the oxidant, the rate constants obtained correlated well with those obtained earlier (27) with a total amplitude of 5–6 milliabsorbance units. We assume that this difference in total amplitudes (∼3 milliabsorbance units when NO is the oxidant) is due to a higher buffering capacity with NO and that approximately ∼1H+/reacting NOR is taken up in both cases (see 27 and 33 for a discussion on proton uptake amplitudes).
DISCUSSION
In this work, we have studied the reaction between fully reduced bacterial NO reductase and NO time-resolved using the flow-flash technique. The fully reduced NOR contains four electrons available in the reaction with the substrate, whereas the conversion of NO to N2O is a two-electron reduction (see Reaction 1). Therefore the enzyme is capable of completing two full turnovers, involving the binding of four NO molecules. Two electrons reside in the binuclear site, so we expect the first turnover to use these electrons and involve no oxidation of the low spin hemes b and c. The second turnover, however, must involve transfer of electrons from the low spin hemes to the active site, as well as binding of two additional NOs.
Initial binding of NO (at 1.5 mm) to reduced heme b3 occurs on the microsecond time scale (Fig. 1A), in agreement with previous measurements (16). When [NO] is decreased to 75 μm, binding is clearly slowed (see Fig. 2), and preliminary data indicate that binding is slowed also when the NO concentration is decreased to intermediate [NO], around 0.5 mm (data not shown). Hendriks et al. (16) suggested that the initial binding of NO to reduced heme b3 is independent of [NO], possibly because of limiting internal transfer from FeB to the heme b3, which is not in agreement with our data. However, at this stage we cannot rule out the possibility that the rate of binding saturates at [NO]<1.5 mm because the deceleration of NO binding is not linear with [NO] between 75 μm and 1.5 mm (the rate constant(s) is ∼6-fold slower for 20 times lower [NO]). Hence, internal transfer of NO from FeB to heme b3 might limit the rate at very high [NO]. It should be noted that we do not expect the binding of NO to FeB to give rise to any significant absorbance changes at the wavelengths used, so FeB might bind NO faster than the heme b3, or it could bind NO during the 200-ms mixing time, with CO still bound to the heme b3. Such simultaneous binding of CO to heme b3 and NO to FeB has been suggested previously (19). The second NO molecule could also bind to the intermediate formed at heme b3 upon binding of the first (21). Our data do not allow discrimination between these scenarios.
Further, we report here the first studies of how the oxidation of the low spin hemes in the reaction between NO and the fully reduced NOR depends on the concentration of NO. Interestingly, this process accelerates as [NO] is lowered. Bacterial NOR is known to show substrate inhibition on steady-state turnover (6), where the maximum activity occurs at a few micromolar NO. The molecular mechanism for this inhibition is not known, but it was suggested to be due to NO binding also to the oxidized NOR, with an obtained Ki of 13.5 μm at pH 6.0 (6). There are several possible mechanisms for how NO binding to the oxidized NOR could inhibit the catalytic rate; e.g. binding to ferric heme b3 lowering its midpoint potential, slowing the rate of electron transfer from the low spin hemes, conformational changes affecting the input of electrons into the protein, binding of NO to ferric FeB, affecting its electron donation rate, or binding to some other “allosteric” site. In the reaction between O2 and fully reduced NOR, the rate of oxidation of the low spin hemes is independent of [O2] (27), showing that regardless of where the inhibitory NO site is, it is specific for NO.
There are several reports of NO binding to ferric heme b3 (see e.g. 22, 34), an interaction that was suggested to be pH-dependent, with more NO bound at lower pH values (34). The reason for the pH dependence has been suggested to be linked to the μ-oxo bridged structure between heme b3 and FeB in the resting oxidized NOR. This μ-oxo bridge is presumed to be easier to displace at low pH (35). The larger extent of NO binding to ferric NOR at lower pH is consistent with larger absorbance changes obtained when oxidized P. denitrificans NOR reacts with NO at pH 6.0 compared with 7.5.4 Also, ferric FeB has been suggested to bind NO (34). Catalysis by NOR was suggested by Blomberg et al. (21) to involve binding of NO to ferrous heme b3 followed by the second NO molecule binding to b32+-NO, forming a hyponitrous acid coordinating between heme b3 and FeB. In this scenario, binding of NO to FeB is a site of inhibition, as it would interfere with catalytic binding.
We also note that there are no major absorbance changes occurring between those attributed to the binding of NO and later phases due to oxidation of the low spin hemes (see below). We therefore (in agreement with Hendriks et al. (16)) see no evidence for the suggested displacement of the proximal His ligand to heme b3 upon NO binding (18). We presume that there are several processes occurring during this time; the binuclear center should oxidize, producing the first molecule of N2O, and NO should bind to its inhibitory site affecting the rate of electron transfer from the low spin hemes (see below). None of these processes is thus associated with large absorbance changes (at the wavelengths used), which is in agreement with the relatively small absorbance changes seen for NO binding to the resting oxidized NOR at pH 7.5 (22,34).4 Also, we see no uptake of protons on the time scale of the first NO turnover (Fig. 4), so the protons needed for producing the first N2O (and H2O) must be taken internally from the protein, possibly from the Glu-198 or Glu-267 close to the binuclear site (see e.g. 29).
Here, we have shown that substrate inhibition occurs already when the fully reduced NOR reacts with NO, ruling out that the inhibition involves electron input into the protein, which does not occur in our experiments. Our work also shows that binding of NO to its inhibitory site must occur on the time scale of oxidation of the low spin hemes in our measurements, i.e. on the millisecond time scale or faster. This is because after the first presumed turnover, using electrons from the binuclear site, NO must bind to its inhibitory site (at the oxidized binuclear center) before electrons redistribute from the low spin hemes. If otherwise, the second turnover would be a repetition of the first, showing acceleration instead of deceleration at high [NO]. This means that reaction schemes involving such electron redistribution before NO binding (such as in Ref. 19) are incompatible with our data. A simplified reaction scheme consistent with the data obtained in this study is shown in Fig. 5.
FIGURE 5.

Schematic simplified illustration of the reaction between fully reduced NOR and NO. All redox-active cofactors are shown as filled circles when reduced and open circles when oxidized. The reaction starts with the fully reduced NOR after CO is flashed off from heme b3 (A). NO can now bind, and the b3-NO intermediate (B) is observed in the kinetic difference spectrum of the rapid (microsecond) phase. The second NO molecule is assumed to bind to FeB, although our data are equally consistent with consecutive binding to the NO intermediate at heme b3. The first turnover is assumed to use electrons from the binuclear site and protons from donors inside the NOR (leading to intermediate C) because no protons are taken from solution and no oxidation of the low spin hemes is observed. Before the second turnover involving oxidation of the low spin hemes, NO must bind to its inhibitory site (D) because we observe inhibition by NO on the rate constant of low spin heme oxidation. Here, we have indicated that this site is the oxidized heme b3, although our data are equally consistent with FeB as the site of inhibition (see “Discussion”). Furthermore, NO binding presumably competes with reformation of the μ-oxo bridge (not shown) between ferric heme b3 and FeB after the first oxidation of the active site. We then observe oxidation of the low spin hemes concomitant with the uptake of protons from the external solution, presumably completing the second turnover, and producing the fully oxidized NOR (E).
It should be noted that in the resting state of oxidized NOR, the μ-oxo bridge needs to be broken for NO to bind. In our kinetic measurements, however, it is possible that after the first turnover, NO binds to the oxidized NOR before the μ-oxo bridge is reformed (see Fig. 5 and its legend), hampering a direct correlation to data obtained with the resting enzyme. Whether or not the μ-oxo bridge forms during steady-state turnover is not known.
We also report here the first measurements of proton uptake from solution coupled to oxidation of NOR by NO, which showed that transfer of electrons from the low spin hemes into the active site is coupled to proton uptake, whereas initial binding of NO is not (Fig. 4). This is consistent with the finding that oxidation of hemes b and c is pH-dependent, whereas binding of NO to reduced heme b3 (at 1.5 mm) is not (Fig. 3). It should be noted that the kinetic effect of lowering the pH presumably is underestimated because more NO seems to bind to the oxidized NOR at lower pH (see above). Thereby, the slowing of oxidation due to NO bound to oxidized NOR could be larger at low pH, counteracting the acceleration of electron transfer due to the coupled proton uptake in the oxidation process. This is also supported by our observation that the effect of lowering [NO] on the rates of oxidation is larger at pH 6 than at pH 7.5.
Using the binding and inhibition parameters derived by Girsch and deVries from steady-state NO reduction at pH 6 (6), the kcat at 1.5 mm NO compared with 75 μm should differ by a factor of 18. Interestingly, this is the same number as that obtained for the ratio (a factor of 17) of the rate constants in the oxidation of the fully reduced NOR at the same [NO] and pH (see Fig. 3 and Table 1). At pH 7.5, however, this ratio is only 3–4, presumably due to a smaller degree of NO binding to its inhibitory site.
Electron transfer from the low spin hemes, coupled to the transfer of protons into the active site, is observed also upon oxidation of fully reduced NOR by O2 (27). In that study, the rate of oxidation of the low spin hemes displayed pH-dependent kinetics with a pKa of 6.6. The time constants were in the same range (25 ms at pH 7.5) as those found in this work using NO as the electron acceptor (time constants 20 ms and 300 ms at 1.5 mm NO). Additionally, the rates were pH-dependent in the same range, indicating that proton transfer contributes to the rate-limiting step of the reactions.
In summary, in this study we have presented results from studies of the interrelated pH- and [NO] dependence during oxidation of the fully reduced NOR by NO. Although NO binding to heme b3 accelerates with increasing NO concentration, the following oxidation of the low spin hemes, which is coupled to proton uptake from solution, decelerates. Substrate inhibition by NO in NOR has previously been observed during steady-state turnover, but here we have shown that it occurs already upon oxidation of fully reduced NOR. Binding to the inhibitory site must therefore occur before redistribution of electrons to the active site, offering mechanistic insights into how NO reduction is catalyzed by NOR.
Supplementary Material
Acknowledgments
We thank Peter Brzezinski (University of Stockholm) and Nicholas Watmough (University of East Anglia) for stimulating discussions. We also thank N. Watmough for sending us the strains expressing NOR.
This work was supported by a grant from the Swedish Research Council.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. 1.
P. Lachmann, unpublished data.
- NOR
- nitric-oxide reductase
- DDM
- β-d-dodecyl maltoside
- HCuO
- heme-copper oxidase
- Mes
- 4-morpholineethanesulfonic acid.
REFERENCES
- 1.Zumft W. G. (2005) J. Inorg. Biochem. 99, 194–215 [DOI] [PubMed] [Google Scholar]
- 2.Watmough N. J., Field S. J., Hughes R. J., Richardson D. J. (2009) Biochem. Soc. Trans. 37, 392–399 [DOI] [PubMed] [Google Scholar]
- 3.Saraste M., Castresana J. (1994) FEBS Lett. 341, 1–4 [DOI] [PubMed] [Google Scholar]
- 4.van der Oost J., de Boer A. P., de Gier J. W., Zumft W. G., Stouthamer A. H., van Spanning R. J. (1994) FEMS Microbiol. Lett. 121, 1–9 [DOI] [PubMed] [Google Scholar]
- 5.Hendriks J., Warne A., Gohlke U., Haltia T., Ludovici C., Lübben M., Saraste M. (1998) Biochemistry 37, 13102–13109 [DOI] [PubMed] [Google Scholar]
- 6.Girsch P., de Vries S. (1997) Biochim. Biophys. Acta 1318, 202–216 [DOI] [PubMed] [Google Scholar]
- 7.Field S. J., Prior L., Roldan M. D., Cheesman M. R., Thomson A. J., Spiro S., Butt J. N., Watmough N. J., Richardson D. J. (2002) J. Biol. Chem. 277, 20146–20150 [DOI] [PubMed] [Google Scholar]
- 8.Moënne-Loccoz P., Richter O. M. H., Huang H. W., Wasser I. M., Ghiladi R. A., Karlin K. D., deVries S. (2000) J. Am. Chem. Soc. 122, 9344–9345 [Google Scholar]
- 9.Thorndycroft F. H., Butland G., Richardson D. J., Watmough N. J. (2007) Biochem. J. 401, 111–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Butland G., Spiro S., Watmough N. J., Richardson D. J. (2001) J. Bacteriol. 183, 189–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giuffrè A., Stubauer G., Sarti P., Brunori M., Zumft W. G., Buse G., Soulimane T. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 14718–14723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stubauer G., Giuffrè A., Brunori M., Sarti P. (1998) Biochem. Biophys. Res. Commun. 245, 459–465 [DOI] [PubMed] [Google Scholar]
- 13.Hendriks J. H., Prior L., Baker A. R., Thomson A. J., Saraste M., Watmough N. J. (2001) Biochemistry 40, 13361–13369 [DOI] [PubMed] [Google Scholar]
- 14.Grönberg K. L., Roldán M. D., Prior L., Butland G., Cheesman M. R., Richardson D. J., Spiro S., Thomson A. J., Watmough N. J. (1999) Biochemistry 38, 13780–13786 [DOI] [PubMed] [Google Scholar]
- 15.Bell L. C., Richardson D. J., Ferguson S. J. (1992) J. Gen. Microbiol. 138, 437–443 [DOI] [PubMed] [Google Scholar]
- 16.Hendriks J. H., Jasaitis A., Saraste M., Verkhovsky M. I. (2002) Biochemistry 41, 2331–2340 [DOI] [PubMed] [Google Scholar]
- 17.Reimann J., Flock U., Lepp H., Honigmann A., Ädelroth P. (2007) Biochim. Biophys. Acta 1767, 362–373 [DOI] [PubMed] [Google Scholar]
- 18.Moënne-Loccoz P., deVries S. (1998) J. Am. Chem. Soc. 120, 5147–5152 [Google Scholar]
- 19.Kumita H., Matsuura K., Hino T., Takahashi S., Hori H., Fukumori Y., Morishima I., Shiro Y. (2004) J. Biol. Chem. 279, 55247–55254 [DOI] [PubMed] [Google Scholar]
- 20.Watmough N. J., Cheesman M. R., Butler C. S., Little R. H., Greenwood C., Thomson A. J. (1998) J. Bioenerg. Biomembr. 30, 55–62 [DOI] [PubMed] [Google Scholar]
- 21.Blomberg L. M., Blomberg M. R., Siegbahn P. E. (2006) Biochim. Biophys. Acta 1757, 240–252 [DOI] [PubMed] [Google Scholar]
- 22.Pinakoulaki E., Gemeinhardt S., Saraste M., Varotsis C. (2002) J. Biol. Chem. 277, 23407–23413 [DOI] [PubMed] [Google Scholar]
- 23.Gibson Q. H., Greenwood C. (1963) Biochem. J. 86, 541–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Proshlyakov D. A., Pressler M. A., Babcock G. T. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 8020–8025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brzezinski P., Ädelroth P. (1998) J. Bioenerg. Biomembr. 30, 99–107 [DOI] [PubMed] [Google Scholar]
- 26.Verkhovsky M. I., Morgan J. E., Verkhovskaya M. L., Wikström M. (1997) Biochim. Biophys. Acta 1318, 6–10 [Google Scholar]
- 27.Flock U., Watmough N. J., Ädelroth P. (2005) Biochemistry 44, 10711–10719 [DOI] [PubMed] [Google Scholar]
- 28.Flock U., Thorndycroft F. H., Matorin A. D., Richardson D. J., Watmough N. J., Ädelroth P. (2008) J. Biol. Chem. 283, 3839–3845 [DOI] [PubMed] [Google Scholar]
- 29.Flock U., Lachmann P., Reimann J., Watmough N. J., Ädelroth P. (2009) J. Inorg. Biochem. 103, 845–850 [DOI] [PubMed] [Google Scholar]
- 30.Brändén M., Sigurdson H., Namslauer A., Gennis R. B., Ädelroth P., Brzezinski P. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 5013–5018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Namslauer A., Brändén M., Brzezinski P. (2002) Biochemistry 41, 10369–10374 [DOI] [PubMed] [Google Scholar]
- 32.Armor J. N. (1974) J. Chem. Eng. Data 19, 82–84 [Google Scholar]
- 33.Huang Y., Reimann J., Lepp H., Drici N., Ädelroth P. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 20257–20262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sakurai T., Nakashima S., Kataoka K., Seo D., Sakurai N. (2005) Biochem. Biophys. Res. Commun. 333, 483–487 [DOI] [PubMed] [Google Scholar]
- 35.Moënne-Loccoz P. (2007) Nat. Prod. Rep. 24, 610–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.