

Abstract
MicroRNAs (miRNA) play a key role in cellular regulation and, if deregulated, in the development of neoplastic disorders including chronic lymphocytic leukemia (CLL). RNAs from primary cells of 50 treatment-naive CLL patients and peripheral B cells of 14 healthy donors were applied to miRNA expression profiling using bead chip technology. In CLL cells, a set of 7 up- and 19 down-regulated miRNAs was identified. Among the miRNAs down-regulated in CLL cells, 6 of 10 miRNA promoters examined showed gain of methylation compared with normal B-cell controls. Subsequent target prediction of deregulated miRNAs revealed a highly significant binding prediction at the 3′ untranslated region of the pleomorphic adenoma gene 1 (PLAG1) oncogene. Luciferase reporter assays including site-directed mutagenesis of binding sites revealed a significant regulation of PLAG1 by miR-181a, miR-181b, miR-107, and miR-424. Although expression of PLAG1 mRNA was not affected, PLAG1 protein expression was shown to be significantly elevated in CLL cells compared with the levels in healthy donor B cells. In summary, we could demonstrate disruption of miRNA-mediated translational control, partly due to epigenetic transcriptional silencing of miRNAs, with subsequent overexpression of the oncogenic transcription factor PLAG1 as a putative novel mechanism of CLL pathogenesis.
Introduction
B-cell chronic lymphocytic leukemia (CLL) is the most common leukemic disorder in the western hemisphere and is characterized by an accumulation of mature B cells in the blood, bone marrow, and secondary lymphoid organs. The clinical heterogeneity in CLL is reflected by various chromosomal aberrations, for example, deletion of chromosome 17p indicating highly unfavorable prognosis due to loss of p53, and, on the other hand deletion of chromosome 13q14 being associated with favorable prognosis.1
In previous work, the localization of microRNA (miRNA) and noncoding RNA genes on chromosome 13q14 led to the assumption that down-regulation of miR-15 and -16 is mediated by deletion of the locus and subsequently leads to overexpression of BCL2. In independent approaches, miR-155 was identified to be overexpressed in basically all CLL patients, whereas down-regulation of miR-15/-16 appeared to be critical only in a subset of patients.2–5
miRNA-mediated control of messenger-RNA stability and protein translation plays a pivotal role in the fine regulation of central cellular pathways,6 particularly in embryonic development and cell differentiation. In the multistep development of immune cells, miRNAs have been shown to be key players in differentiation; in particular, the multistep development of B cells shows stage-specific miRNA signatures.7,8 With regard to pathogenesis of neoplastic diseases, miRNA control of putative oncogenic target genes has been previously shown to be directly involved in malignant transformation.9,10 There is increasing evidence for extensive miRNA deregulations in multiple neoplasias. However, both deregulations of miRNAs as well as the identification of their functional relevant targets and regulatory circuits in CLL pathogenesis are only partly understood and remain to be elucidated. Namely, the underlying mechanisms of predominant miRNA down-regulation cannot be generally explained by genetic alterations or defects in the miRNA processing machinery.
By applying miRNA profiling to CLL B cells derived from a large cohort of previously untreated CLL patients, we identified a novel group of deregulated miRNA besides previously identified alterations in CLL. Based on the predominant down-regulation of miRNAs in this cohort, we investigated genetic and epigenetic alterations as potential underlying mechanisms of the observed miRNA deregulations. As a downstream result of miRNA deregulation, we attempted to identify miRNA target genes affected by loss of miRNA control. Surprisingly, a strong oncogenic transcription factor, pleomorphic adenoma gene 1 (PLAG1), was predicted to be a target of several deregulated miRNAs. Indeed, overexpression of PLAG1, primarily identified to be involved in tumorigenesis by promoter swapping in pleomorphic salivary gland adenomas, could be verified in CLL. Alterations in the axis of miRNA-mediated oncogene control by epigenetic transcriptional silencing of miRNAs might be a novel oncogenic pathway in CLL pathogenesis.
Methods
Patients and cells
After informed consent, blood was obtained from patients fulfilling diagnostic criteria for CLL. Only patients without prior therapy were included in this study. Fresh CLL samples were enriched by applying B-RosetteSep (Stem Cell Technologies) and Ficoll-Hypaque (Seromed) density gradient purification resulting in purity of more than 98% of CD19+/CD5+ CLL cells. CLL cells were characterized for CD19, CD5, CD23, FMC7, CD38, ZAP70, and sIgM expression on a FACSCanto flow cytometer (BD PharMingen). IgVH-hypermutational status of CLL patients was analyzed as previously published. Control cells were isolated by positive selection with anti-CD19-MACS beads (Miltenyi Biotec). All cells were immediately lysed by TRIzol and cryopreserved at −80°C. This study was approved by the ethics committee of the University of Cologne (approval no. 01-163). Blood samples were given with informed consent according to the Declaration of Helsinki protocol.
miRNA expression analysis
BeadCHip miRNA expression assay (PAS; Illumina) was carried out as previously described according to the manufacturer's protocol.11 Briefly, 200 ng total RNA extracted from TRIzol-lysed samples was first applied by polyadenylation agent (PAS; Illumina). The attached poly(A) tail was used for further priming of cDNA synthesis (CSS; Illumina) using biotinylated oligo-dT primers containing an additional 5′-universal polymerase chain reaction (PCR) primer sequence. Biotinylated cDNA was captured to a solid phase by streptavidin binding and further hybridized with miRNA-specific assay oligonucleotides. Assembled oligonucleotides were extended by DNA polymerase and subsequently eluted for universal PCR-based amplification with fluorescently labeled universal primers. Single-stranded PCR products were hybridized to a 96 Sentrix Array Matrix (Illumina). Arrays were scanned using a BeadArray reader and data were exported to BeadStudio Version 3.2 (Illumina). Real-time (RT)–PCR expression analysis for further validation was carried out using mirCURY LNA (Exiquon). SNORD48 and U6 RNA served as endogenous housekeeping controls. Microarray data are available online from the University of Cologne.12
Methylation analysis
Isolation of genomic DNA was carried out by lysis of cells for 15 minutes with lysis buffer (155 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA, pH 7.4) at 4°C. Cell nuclei were sedimented by centrifugation, subsequently resuspended by nuclear lysis buffer (10 mM Tris-HCl, 400 mM NaCl, 2 mM Na2-EDTA, pH 8.2), and lysed by adding sodium dodecyl sulfate. Proteinase K digestion was carried out overnight at 50°C. Saturated NaCl solution was added for precipitation of proteins; after centrifugation for 30 minutes at 3500g, solved genomic DNA was isolated from the supernatant, precipitated with isopropanol, washed with 70% ethanol, and dissolved in Tris EDTA (TE) buffer for storage. Genomic DNA (1-2 μg) was chemically modified with sodium bisulfite using the EZ methylation kit (Zymo Research) according to the manufacturer's instructions. Quantitative DNA methylation analysis at single CpG units (containing one or more CpG dinucleotides) was performed by MassARRAY technique, as previously described, targeting amplicons of up to 600 bp in size.13 Regions of analysis were chosen by putative promoter regions of expressed sequence tags and/or protein coding genes harboring pre-miRNAs.14,15 The supposed promoter activity in some of the selected regions was confirmed by genome-wide miRNA promoter analyses (7 of 17 analyzed loci).16 In addition, arbitrarily chosen genomic regions approximately 1-kb upstream of the pre-miRNAs were analyzed to test whether changes in DNA methylation were specific for promoter regions.
Bisulfite-treated DNA was PCR-amplified (primer sequences given in supplemental Table 4, available on the Blood website; see the Supplemental Materials link at the top of the online article), in vitro transcribed, base-specifically cleaved by RNase A, and subjected to matrix-assisted laser desorption ionization–time-of-flight mass spectrometry. DNA methylation standards (0%, 20%, 40%, 60%, 80%, and 100% methylated genomic DNA) and correction algorithms based on R statistical computing environment were used for data correction and normalization.
PLAG1 mRNA expression analysis
Quantification of PLAG1 mRNA was performed by LightCycler TaqMan Master (Roche). Universal probe no. 70 (Roche) with forward primer 5′-GTCCAGCCCGAAATATGAGA-3′ and reverse primer 5′-CAGCACCAAGAGGCAACC-3′ was used for PLAG1 amplification. Beta-2-microglobulin was applied as housekeeping gene standard by universal probe no. 42 (Roche) with forward primer 5′-TTCTGGCCTGGAGGCTATC-3′ and reverse primer 5′-TCAGGAAATTTGACTTTCCATTC-3′. All experiments were performed in replicates and crossing points were determined by second derivative maximum method. Relative quantification analysis was done by Exor3 software package (Roche).
PLAG1 protein expression analysis
A PLAG1-specific mouse monoclonal antibody (clone 3B7; Abnova) was used for detection of PLAG1 protein. For control of antibody specificity, PLAG1-GFP was transfected by calcium phosphate transfection to HEK293 cells with PLAG1-specific 50 nM small interfering RNA (siRNA). Cellular lysates were processed with radioimmunoprecipitation assay buffer, sonicated, and further blotted onto nitrocellulose membranes. Western blot detection and density measurements were performed on an Odyssey infrared imaging system (Licor).
miRNA binding assays
For luciferase reporter experiments, a 2-kb 3′ untranslated region (UTR) segment containing predicted microRNA interaction sites was amplified from human PLAG1 cDNA (ImaGenes) using forward primer 5′-gactagttgcctatttgttgcttgtgc-3′ and reverse primer 5′-ttttccttttgcggccgcaagcccactttccattctga-3′ and cloned into pTrcHis2-TOPO (CR972286; Invitrogen). From this construct, a SpeI/NotI fragment was cleaved out and inserted into pIS15 adjacent to the stop codon of the firefly luciferase gene. Site-directed mutagenesis at predicted miRNA binding sites within the PLAG1 3′-UTR was performed using the QuikChange XL mutagenesis kit (Stratagene). The variants (supplemental Table 3) were confirmed by sequencing.
HeLa cells at a density of 2 × 104 cells/well were cotransfected in 96-well plates using Lipofectamine 2000 (Invitrogen) with 45 ng pIS1-PLAG1 constructs containing the Renilla luciferase gene, 5 ng pISO expressing firefly luciferase as transfection control, and 1 pg microRNA duplex (miRIDIAN; Dharmacon). The miRNAs used for cotransfection were miR-181a, -181b, -107, -424, and -141, and a miRNA mimic-negative control. Light emission by firefly and Renilla luciferase activities was measured consecutively using Dual-Luciferase assays (Promega) 24 hours after transfection using a Glomax luminometer (Promega). Ratios of Renilla versus firefly signals served as a measure for reporter activity normalized for transfection efficiency.
Bioinformatic and statistical analysis
The array intensity data were imported into BeadStudio Version 3.2 (Illumina) and a quantile normalization procedure was applied. Further analysis of differential expression was carried out using R software package and dCHIP (Windows; Microsoft). DNA methylation data were normalized and corrected by R software package and analyzed with Wilcoxon test by JMP software (Version 8.0; SAS Inc).
For target prediction TargetScan Version 4.1 (http://www.targetscan.org), PicTar (http://pictar.mdc-berlin.de),17 MiRDB (http://mirdb.org),18,19 and miRanda (http://www.microrna.org)20,21 algorithms were applied. GeneTrail database (http://genetrail.bioinf.uni-sb.de)22 was accessed for functional annotation of predicted target genes.23 SPSS (SPSS Inc), Excel (Microsoft), and SigmaPlot (Systat Software Inc) were applied for statistical analysis of data.
Results
Identification of a novel set of miRNAs reduced in CLL
To assess deregulation of global miRNA expression, we profiled human miRNAs using a bead chip–based miRNA expression profiling platform. Purified B cells from either CLL patients (n = 50) or healthy controls (n = 14) were assessed. All CLL patients were treatment naive. The majority of patients presented with Binet stage A disease and showed a favorable risk profile as assessed by clinical and molecular features (Tables 1–2). The most frequent aberration was a deletion 13q14, although only 6 of 45 patients were documented to have a homozygous deletion in this area. Comparing the total number of miRNAs expressed, a significantly lower number of miRNAs were detected in CLL compared with normal B cells (Figure 1A). The predominance of down-regulated miRNAs in CLL cells was accompanied by a highly significantly lower total number of miRNAs expressed above the detection threshold in CLL patients (19.8% vs 23.5%; P < .001).
Table 1.
Patients' characteristics of miRNA screening cohort
| Negative for feature, n/% | Positive for feature, n/% | |
|---|---|---|
| Thymidine kinase (> 10 U/L) | 34/50 (68.0%) | 16/50 (32.0%) |
| CD38 (> 10% positive CLL cells) | 34/50 (68.0%) | 16/50 (32.0%) |
| ZAP70 (> 20% positive CLL cells) | 25/50 (50.0%) | 25/50 (50.0%) |
| IgVH hypermutational status (homology > 98%) | 20/31 (64.5%) | 11/31 (35.4%) |
Molecular features of 50 patients being diagnosed with CLL were assessed before miRNA profiling was performed.
Table 2.
Patients' prognostic factors of miRNA screening cohort
| n/% | |
|---|---|
| Binet stage | |
| A | 33/50 (66.0 %) |
| B | 11/50 (22.0%) |
| C | 6/50 (12.0%) |
| Cytogenetics | |
| Normal | 12/45 (26.6%) |
| del13q14 | 25/45 (55.5%) |
| Homozygous del13q14 | 6/45 (13.2%) |
| Trisomy 12 | 3/45 (6.6%) |
| del11q | 3/45 (6.6%) |
| del17p | 1/45 (2.2%) |
Clinical and molecular features including cytogenetic analysis by FISH of 50 patients being diagnosed with CLL were assessed before miRNA profiling was performed.
Figure 1.

miRNA expression profiling of CLL patient samples and healthy donor B cells. (A) Significant decrease of expressed miRNA in CLL cells (n = 50) versus healthy donor B cells (n = 14; P < .001) shown as percentage of present calls. (B) Heat map of microRNA expression patterns in CLL cells versus healthy donor B cells: 26 miRNAs are significantly deregulated in CLL versus healthy control, 19 miRNAs show down-regulation in CLL and 7 miRNAs were demonstrated to be up-regulated.
Applying a threshold of a median fold change larger than 2, a minimal absolute difference of signal intensity defined by the detection threshold level, and a P value lower than .001 in CLL patient cells, 7 miRNAs appeared to be up-regulated, whereas 19 miRNAs were shown to be significantly down-regulated (Table 3, Figure 1B, and supplemental Table 1). Applying Bonferroni correction due to multiple testing of miRNA probe sets underlined significant deregulation of all identified miRNAs except miR-449 and miR-565.
Table 3.
Deregulated miRNAs in CLL versus healthy donor B cells
| miRNA | Mean expression of CLL cells | Mean expression of healthy B cells | Fold change | P | Absolute difference of means |
|---|---|---|---|---|---|
| hsa-miR-107 | 692.53 | 1764.03 | −2.55 | < .001* | 1071.5 |
| hsa-miR-125a | 418.88 | 2039.2 | −4.87 | < .001* | 1620.32 |
| hsa-miR-126 | 4540.85 | 15 815.12 | −3.48 | < .001* | 11 274.27 |
| hsa-miR-126* | 1983.83 | 21 761.79 | −10.97 | < .001* | 19 777.95 |
| hsa-miR-130a | 1272.19 | 10 530.37 | −8.28 | < .001* | 9258.18 |
| hsa-miR-139 | 1001.53 | 3095.88 | −3.09 | < .001* | 2094.35 |
| hsa-miR-141 | 3871.12 | 1623.27 | 2.38 | < .001* | 2247.85 |
| hsa-miR-143 | 299.48 | 1506.67 | −5.03 | < .001* | 1207.19 |
| hsa-miR-148a | 14 042.43 | 4724.08 | 2.97 | < .001* | 9318.35 |
| hsa-miR-155 | 26 073.76 | 9956.07 | 2.62 | < .001* | 16 117.69 |
| hsa-miR-181a | 1093.34 | 9251.04 | −8.46 | < .001* | 8157.7 |
| hsa-miR-181b | 316.23 | 1364.86 | −4.32 | < .001* | 1048.63 |
| hsa-miR-199a | 386.26 | 4393.66 | −11.37 | < .001* | 4007.4 |
| hsa-miR-199a* | 1511.02 | 8537.04 | −5.65 | < .001* | 7026.02 |
| hsa-miR-326 | 222.65 | 3376.34 | −15.16 | < .001* | 3153.69 |
| hsa-miR-34a | 1876.1 | 444.46 | 4.22 | < .001* | 1431.64 |
| hsa-miR-368 | 270.28 | 2118.85 | −7.84 | < .001* | 1848.57 |
| hsa-miR-369-3p | 294.96 | 1307.95 | −4.43 | < .005* | 1012.99 |
| hsa-miR-424 | 902.62 | 2536.33 | −2.81 | < .002* | 1633.71 |
| hsa-miR-449 | 1713.84 | 5159.03 | −3.01 | .002 | 3445.2 |
| hsa-miR-451 | 16 872.23 | 5770.64 | 2.92 | < .001* | 11 101.59 |
| hsa-miR-565 | 2861.11 | 8192.31 | −2.86 | .003 | 5331.2 |
| hsa-miR-582 | 333.08 | 1793.82 | −5.39 | < .001* | 1460.74 |
| hsa-miR-584 | 344.23 | 2809.91 | −8.16 | < .001* | 2465.68 |
| hsa-miR-598 | 2010.3 | 910.88 | 2.21 | < .001* | 1099.42 |
| hsa-miR-660 | 2000.38 | 896.36 | 2.23 | < .001* | 1104.02 |
BeadChip microarray–based miRNA expression values of quantile normalized data comparing purified CLL cells (n = 50) to healthy donor peripheral B cells (n = 14). A t test was carried out for determination of differential expression. For rigid correction of multiple testing, Bonferroni correction was applied and miRNAs remaining significant by this test are indicated by asterisks (*).
In addition, for miR-21 previously demonstrated to be up-regulated in CLL,24 we could detect the highest absolute expression values of all assessed miRNAs. However, in healthy controls the miR-21 expression was not significantly lower.
Due to the clinical heterogeneity of CLL, we analyzed for differential expression of miRNAs in CLL patient subsets defined by cytogenetics and prognostic markers. We could not identify significantly differentially expressed miRNAs in cytogenetically defined subgroups; in particular, we could not detect significant deregulation of miRNAs in patients harboring del13q14. However in CD38+ versus CD38− CLL cells, we could detect significantly reduced expression of miR-660. In IgVH unmutated cases, a significantly reduced expression of miR-146b could be observed (supplemental Table 1).
With regard to previous miRNA screening approaches assessing either spotted miRNA arrays, Solexa sequencing, miRNA cloning, or RT-PCR–based assays, we confirmed up-regulation of miR-155 and down-regulation of miR-181a/b. We also detected heterogeneous up-regulation of miR-34a, previously identified as heterogeneously expressed in CLL and down-regulated in CLL patients harboring del17p, facilitating chemotherapy resistance.25 We could not identify significant down-regulation of miR-15 and miR-16 except in one patient harboring a homozygous deletion of chromosome 13q14. However, the previous up-regulation of miR-155, a key regulator of B-cell ontogenesis,24 appeared to be the most prominent up-regulated miRNA in our cohort.
Here we identified a previously unknown down-regulation of a set of miRNAs in CLL, including miR-107, -424, -125a, -126, and -326.
For validation of the microarray-based dataset, we performed quantitative RT-PCR–based relative expression of selected miRNAs. No down-regulation of miR-16 was seen in CLL cells (n = 13) compared with healthy donor B cells (n = 10), whereas miR-107 and -181b were proven to be significantly down-regulated in CLL (P = .009 and P = .001, respectively). In summary, we identified a set of predominantly down-regulated miRNAs in CLL including several hitherto unknown deregulated miRNAs.
Deregulation of miRNAs is not associated with genomic alterations but with epigenetic transcriptional silencing
With respect to genomic localization of miRNA genes deregulated in CLL, no obvious association with known breakpoints in CLL, frequent deletions, or other cytogenetic alterations was seen. At chromosome 11q, 4 miRNA loci (miR-130, -139a, -143, -326) were identified, however, genomic localizations are widely distributed apart from ATM locus. In addition, only 3 of the CLL patients in this cohort harbored a del11q deletion, thus rendering major cytogenetic breaks unlikely as the underlying mechanism in general miRNA suppression in CLL cells.
As an alternative mechanism of miRNA down-regulation, we hypothesized that epigenetic transcriptional silencing could be involved. DNA hypermethylation at promoter regions is a well known transcriptional silencing mechanism. To investigate whether altered DNA methylation leads to aberrant expression of miRNAs in CLL, we analyzed the DNA methylation status at known or predicted transcriptional start sites (TSSs) of pri-miRNAs. DNA methylation was measured in 70 CLL samples (including 40 of 50 patient samples applied for miRNA expression profiling) and was compared with peripheral blood CD19+ B cells and tonsillar CD19+ B cells from healthy donors. Among 23 miRNA loci covering all deregulated miRNAs, 3 miRNAs either not overlapping with known ESTs (miR-199a-1 and miR-369-3p) or situated in an EST, harboring a CpG-poor TSS sequence (miR-181a1/b1) were not further analyzed. miR-424 and miR-660 promoters were excluded from analysis, because their DNA methylation patterns showed bimordial distribution in a sex-dependent manner due to X-chromosome inactivation.
In most of the miRNAs down-regulated in CLL (10 of 12 loci), promoter regions were unmethylated in normal B cells consistent with transcriptionally active TSSs in general. Intriguingly, in miRNA promoters associated with reduced miRNA expression, many CLL samples showed gain of methylation, compared with normal B cells. Especially, promoters of miR-139 and miR-582 showed significant gain of methylation in CLL, whereas promoters of the other miRNAs did not show a statistically significant difference (Figure 2). In contrast, up-regulated miRNAs showed either loss of methylation or no change in their promoters that were hypermethylated or hypomethylated in normal B cells, respectively (supplemental Figure 2A). As evidence for specificity of changes of DNA methylation within miR promoter regions, we also analyzed DNA methylation in arbitrarily chosen regions approximately 1 kb upstream of pre-miRNAs. Most of these loci showed significant loss of methylation in CLL B cells compared with normal B cells, irrespective of down- or up-regulated aberrant expression of miRNAs (supplemental Figure 2B). This is likely to reflect a consequence of global loss of methylation in CLL, as previously shown.26 Taken together, the gain of methylation was specific to the promoter regions for down-regulated miRNAs, indicating that transcriptional silencing of pri-miRNA by DNA methylation, at least in part, leads to down-regulation of miRNAs during CLL pathogenesis.
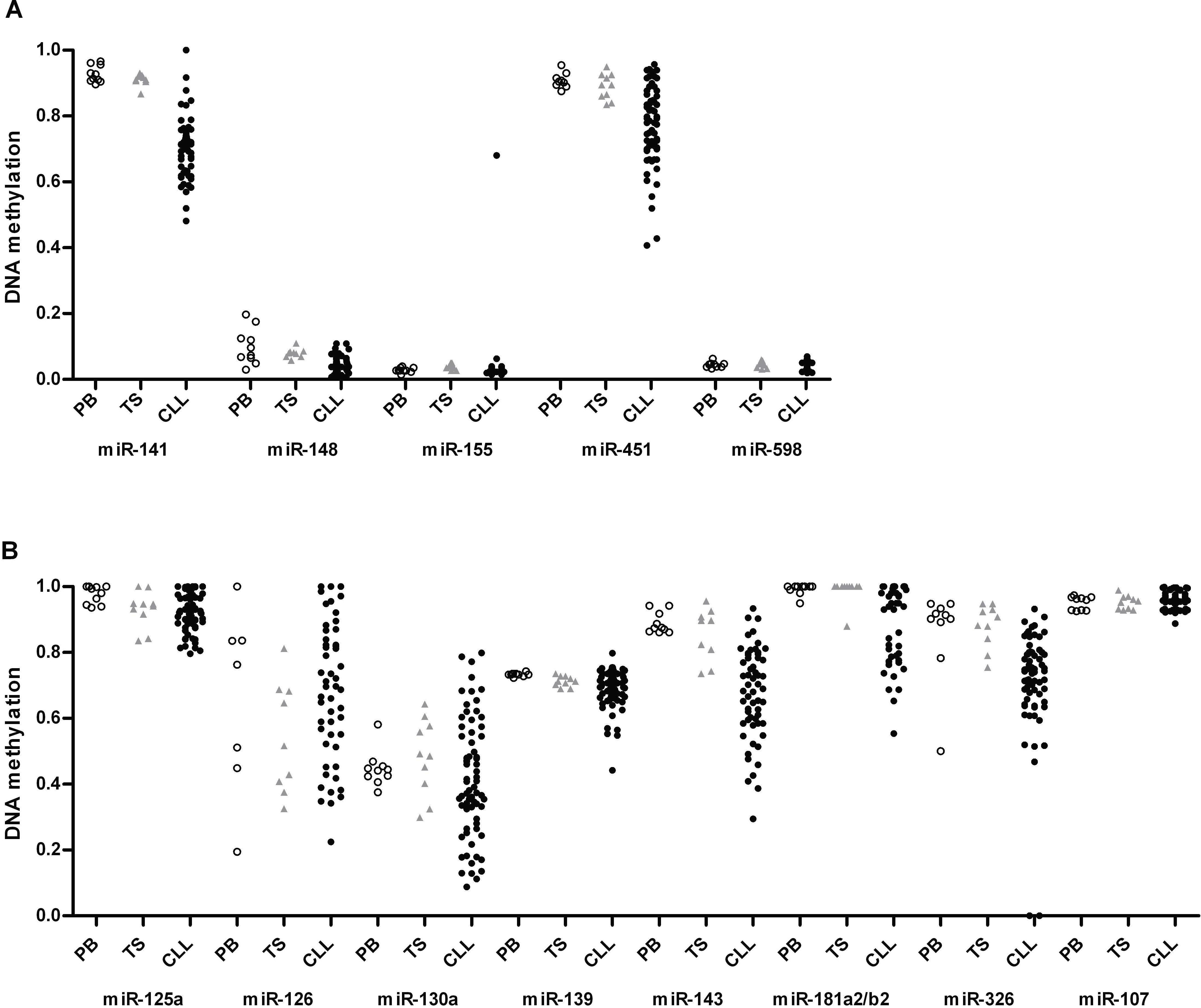
Figure 2.

DNA methylation status at promoters of miRNAs down-regulated in CLL. DNA methylation status of promoter regions associated with down-regulated miRNAs (n = 10) was examined in CLL samples (●; n = 70) and compared with peripheral blood CD19+ B cells (PB, ○) and tonsillar CD19+ B cells (TS,  ) from healthy donors. The statistical analysis was done using the Wilcoxon test between CLL and PB or CLL and TS. *P < .05; **P < .001. For miR-181a2b2, the mean value of the CLL samples showed lower methylation compared with PB and TS samples because the majority of CLL samples had very low methylation levels, although some patients showed highly methylated promoters.
) from healthy donors. The statistical analysis was done using the Wilcoxon test between CLL and PB or CLL and TS. *P < .05; **P < .001. For miR-181a2b2, the mean value of the CLL samples showed lower methylation compared with PB and TS samples because the majority of CLL samples had very low methylation levels, although some patients showed highly methylated promoters.
Identification of the transcription factor PLAG1 as a prime target gene for deregulated miRNAs in CLL
miRNAs are hypothesized as a major control mechanism in cell differentiation and control of oncogenes. To identify novel oncogenes evading miRNA control due to the tremendous deregulations of miRNAs in CLL, we assessed target binding prediction algorithms for deregulated miRNAs. All miRNAs classified here as specifically deregulated in CLL were subjected to prediction by the TargetScan algorithm (http://www.targetscan.org, Version 4.1).27–29 All candidate genes predicted by TargetScan were extracted. The initial screen of putative miRNA targets in CLL contained 7760 putative interactions and 3700 candidate genes. Due to the extensive amount of candidate genes, eliminating false-positive predictions is required to further focus on highly probable targets. Binding scores calculated by TargetScan to validate the predicted stringency of the miRNA-mRNA binding were assessed to an additive quantification of cumulative putative binding. First, each binding score of predicted binding sites was added to a cumulative binding score of each candidate gene. Second, the total number of predicted binding sites for the CLL-specific miRNAs was assessed by additive counting. Third, conservation of binding sites was included in ranking of target candidates. Forth, we focused on genes affected by down-regulated miRNAs. Applying these filter criteria, we ranked all genes for single filter criteria and a total rank was calculated. We focused on the 167 genes ranked in the group of 5% highest total rank for further analysis of functional association. GeneTrail database for functional annotation was assessed to the high-rank group of 167 genes revealing a highly significant overrepresentation of transcription factors and DNA-binding proteins (supplemental Table 2).30 To functionally focus on genes involved in malignant transformation, we evaluated the functional context of the 167 predicted target genes regarding oncogenesis, proliferation, antiapoptotic properties, and cell-cycle control, revealing a list of 48 genes (supplemental Table 1).
Here, the strong oncogenic transcription factor pleomorphic adenoma gene 1 (PLAG1) was identified as the top-ranked most significant gene. Nine putative evolutionarily highly conserved (including chicken) and an additional 8 less conserved putative binding sites for deregulated miRNAs in CLL were identified in the 3′UTR of the PLAG1 mRNA (Figure 3). Here we primarily assessed the TargetScan algorithm for first prediction target genes. To verify our hypotheses with an independent target prediction algorithm, we performed miRNA-binding prediction of PLAG1 3′UTR in several other bioinformatic approaches. By PicTar approach, miR-107, -141, -181a, and -181b as for the miR-15/16/195/424/497 family were analogously predicted.17 MiRDB also predicted miR-141, -424, -181a, and -181b.19 Finally, miRanda algorithms identified miR-107, -141, -424, -181a, and -181b.21
Figure 3.

Schematic representation of predicted miRNA-binding sites at the PLAG1 3′UTR. CLL-specific deregulated miRNAs were applied to TargetScan 4.1–based prediction of miRNA-binding sites for identification of putative targets genes. PLAG1 gene was identified to harbor multiple binding sites of CLL-specific deregulated miRNAs. Evolutionarily highly conserved (including chicken) miRNA-binding sites of 3′UTR are indicated in black; less conserved binding sites, in gray.
In conclusion, the oncogene PLAG1 showed up as a key candidate to evade miRNA control especially of the underexpressed miRNAs miR-181a, -181b, -107, and -424 in CLL, leading to disturbed oncogene control in leukemia cells.
PLAG1 is under the control of miRNAs deregulated in CLL
To experimentally address miRNA target interaction identified above, we cotransfected the individual miRNAs (miRNA-181a, -181b, -107, -424, and -141) together with a Renilla luciferase reporter construct containing a PLAG1 3′-UTR mRNA fragment into HeLa cells. After 24 hours, luciferase activities in transfected HeLa cells with and without the specific miRNAs were compared. Relative to unspecific control miRNA, cotransfection with miR-181a significantly suppressed activity of the PLAG1-3′UTR-luciferase construct by 43.6% (P = .03, n = 5) and in miR-181b by 48.4% (P = .03, n = 5; Figure 4A). PLAG1-3′UTR-reporter construct was also significantly suppressed, with 28.6% activity reduction by miR-107 (P = .01, n = 5) and 42.7% activity reduction by miR-424 (P = .005, n = 5; Figure 4B). In contrast, by transfection of miR-141, up-regulated in CLL, no significant activity reduction was seen. When combining all miRNAs simultaneously to reporter assays, no additive effect outranging the sole application of miR-181a/b was seen (data not shown).
Figure 4.

miRNA reporter assay of PLAG1-miRNA interaction. PLAG1-3′UTR was cloned to pIS1-vector containing a Renilla luciferase construct. PLAG1-3′UTR construct was cotransfected with pIS0 firefly luciferase control vector into HEK-293 cells (n = 5). For detection of miRNA binding, synthetic miRNAs were cotransfected as indicated. miR-181a, -181b, -424, and -107 are demonstrated to significantly reduce luciferase activity via PLAG1-3′UTR interaction. Specificity of miRNA-PLAG1 interaction was proven by site-directed mutagenesis of miRNA-binding sites in PLAG1 3′UTR. Concerning the miR-181 family, 2 binding sites were mutated, revealing abrogated inhibitory effects of miR-181a and -181b.
To examine whether the observed repression of a reporter gene by miRNAs was specifically due to the presence of predicted binding sites in the PLAG1 3′-UTR fragment, these sites were disrupted by site-directed mutagenesis (supplemental Table 3). For interrupting a perfect seed pairing, 2 nucleotides were substituted in each predicted binding site. Because there are 2 predicted highly conserved binding sites for miR-181a/b in the PLAG1 3′UTR, 3 variants were generated, in which one respectively both of these sites were mutated. For assessing the role of the predicted binding sites, the PLAG1 3′-UTR constructs with wild-type or mutated binding sites were cotransfected with the corresponding miRNAs and subjected to reporter assays. Significantly reversed binding of miRNA-181a was demonstrated in miR-181–binding site mutants PLAG1-3′UTR-mut4390 (P = .04), -mut3500 (P = .02), and -mut4390/3500 (P = .001). Similarly, for miR-181b significantly reversed binding by site-directed mutagenesis of predicted binding sites PLAG1-3′UTR-mut4390 (P = .039), -mut3500 (P = .011), and -mut4390/3500 (P = .011) was shown (Figure 4A). Moreover, significantly reversed miRNA-mediated suppression of activity was also seen by miR-107–binding site mutant PLAG1-3′UTR-mut4437 (P = .006) and miR-424–binding site mutant PLAG1-3′UTR-mut4437 (P = .018; Figure 4B). Altogether miR-181a, -181b, -107, and -424 significantly bind PLAG1-3′UTR, and mutating predicted binding sites completely abrogates miRNA-mediated suppression.
Reduced miRNA-based repression is associated with PLAG1 overexpression in CLL
Based on the reduced expression of multiple miRNAs in CLL that bind to the 3′UTR of the PLAG1 mRNA, one would postulate that PLAG1 mRNA and/or protein expression is enhanced in CLL cells. We therefore assessed PLAG1 expression in CLL patients compared with healthy controls. Quantification of PLAG1 mRNA by RT-PCR revealed a slightly elevated level of PLAG1 mRNA in CLL cells (n = 11) versus healthy donor control B cells (n = 8), although the difference did not reach statistical significance (Figure 5A). These findings are in line with the current concept that most mammalian miRNA target genes are regulated mainly by repression of protein translation rather than mRNA destruction. We therefore addressed PLAG1 protein expression by immunoblotting of lysates derived from purified CLL cells and healthy donor peripheral B cells, respectively. Specificity of the applied antibody was confirmed by controls including recombinantly overexpressed PLAG1 and cotransfection controls with PLAG1-specific siRNA, respectively (Figure 5D). Immunoblotting of primary CLL cells (n = 29) versus healthy donor controls (n = 22) demonstrated a significantly elevated PLAG1 protein expression in CLL cells. Almost no detectable expression was seen in healthy donor control B cells. Applying immunohistochemistry, PLAG1 could not be detected in CLL lymph nodes (data not shown). This finding is in line with the low absolute expression of PLAG1 in CLL cells. However, applying Odyssey densitometry of specific immunoblotting, signal intensities revealed a significantly elevated mean of PLAG1-specific fluorescence intensity rate, 10.31 (95% confidence interval, 4.4-16.2; P = .014) in CLL cells compared with 0.85 (95% confidence interval, 0.48-1.22) in healthy donor B cells (Figure 5B). Although PLAG1 expression in some patients (CLL2, CLL4) was indistinguishable compared with healthy controls, 3 patients (CLL1, CLL3, CLL5) showed high expression of PLAG1 protein expression; however no obvious clinical correlation with methylation patterns of miRNA loci, disease stage, or prognostic factors could be identified (Figure 5C).
Figure 5.

PLAG1 expression in CLL and healthy donor B cells. PLAG1 mRNA expression was assessed in CLL cells (n = 11) versus healthy donor control B cells (n = 8) by RT-PCR (A). PLAG1 protein expression was detected by immunoblotting of lysates from CLL cells and healthy donor B cells. Specificity of the PLAG1-specific mAb was proven in PLAG1-transfected HeLa cells (Rec.PLAG1) and cotransfection controls with PLAG1-specific siRNA (D). Protein expression in primary CLL cells (n = 29) and healthy donor B cells (n = 22) was assessed by immunoblotting of lysates with PLAG1-specific mAb and quantified by Odyssey densitometry. Two representative experiments are shown (B-C).
In conclusion, the oncogene PLAG1 shows a strong aberrant expression in CLL that is significantly elevated in comparison with healthy B cells harboring no detectable PLAG1.
Discussion
Here we describe a novel set of 26 miRNAs significantly deregulated in CLL. In addition to miR-181a and miR-181b, a total set of 19 down-regulated genes was identified. Strikingly, the majority of down-regulated miRNAs show hypermethylation in the respective putative transcriptional starting sites within the promoter sequence, strongly suggesting epigenetic rather than mutational regulation of miRNA in CLL. In fact, except for one patient, we could not confirm wide distribution of miR-15 and mir-16 down-regulation attributed to homozygous deletion of chromosome 13q14. To determine oncogenic events downstream of down-regulated miRNA, a computational approach revealed the novel finding of the oncogene PLAG1 to be a potential target of miRNA deregulation. In fact, we clearly demonstrate regulation of PLAG1 expression by 6 of the down-regulated miRNAs (miR-181a, -181b, -107, -424, -155, and -141). As a consequence of missing miRNA regulation, PLAG1 is overexpressed in CLL.
In total, the number of miRNAs detectable was significantly lower in malignant CLL cells. This phenomenon of decreased miRNA levels in malignantly transformed cells has been previously described.31,32 Comparing our data to recently published data, we could verify the down-regulation of miR-181a and miR-181b in CLL. We also showed significant up-regulation of miR-155 known to be a key regulator of B-cell maturation and associated with lymphoma development.24,33,34 However, miR-21 that was previously reported to be highly up-regulated in CLL was shown to be only slightly overexpressed compared with healthy donor B cells in our screening set.2 This difference could be partially due to different screening approaches: miRNA cloning and quantitative RT-PCR of mature miRNAs versus bead chip array technology as used by us. Furthermore, we could not detect high frequencies of miR-15 and miR-16 down-regulation as previously described.4 Nevertheless, this finding is in line with previous independent observations applying both sequencing and hybridization methods for miRNA detection.2,4 We observed miR-16 down-regulation in only one patient harboring homozygous deletion of chromosome 13q14. Nevertheless, we identified significant down-regulation of miR-424 belonging to the miR-15/16/195/424/497 miRNA family sharing the same 3′UTR-binding seed sequence.
We demonstrate that gain of methylation was observed especially in the promoter regions of down-regulated miRNAs, whereas nonpromoter regions showed loss of methylation irrespective of up- or down-regulation of miRNAs in CLL. This indicates that DNA methylation could be an important mechanism of transcriptional silencing, in this case of pri-miRNA, with subsequent down-regulation of mature miRNAs, although we cannot exclude that loss of methylation in some locations still has biologic significance in the individual locus.
We identified several novel miRNAs being significantly deregulated in CLL. This novel group of miRNAs was assessed to predict relevant downstream targets of miRNA in CLL. Due to the high amount of false positives by in silico prediction of binding sites for a single miRNA, we combined the integrated perspective of a deregulated cluster with rigorous filtering for predicted binding affinity and frequency of predicted binding sites to reveal high probability of miRNA-target interaction. The oncogenic zinc-finger transcription factor PLAG1 was identified by application of our step-wise algorithm to be targeted by miR-181a, -181b, -107, -424, -155, and -141. So far the PLAG1 oncogene has not been described in CLL pathogenesis. However, the well-described oncogenic potential of this transcription factor on salivary gland adenomas, breast cancer, and acute myeloid leukemia pathogenesis indicate the influence of PLAG1 on malignant transformation.35–37 PLAG1 was primarily identified to be involved in tumorigenesis of salivary gland adenomas by translocation and promoter swapping.35 Although we previously identified chromosomal translocations in CLL as prognostic parameter, no significantly enhanced frequency of translocations involving the PLAG1 locus could be identified in CLL.38 In previous gene expression profiling attempts, PLAG1 was not identified as overexpressed gene in CLL. Due to the low overall mRNA expression level and the demonstrated miRNA control mechanisms, the overexpression of this oncogene was undetectable by classical screening attempts. However, we could demonstrate low detectable mRNA expression levels for PLAG1 by modern RT-PCR technique and even aberrant protein expression of PLAG1 in a significant proportion of CLL cases. In contrast, no protein expression of PLAG1 was detectable in healthy donor B cells, although mRNA could be shown to be prevalent in healthy B-cell control cells. We therefore hypothesized that miRNA-mediated control mechanisms and their disruption are crucial for PLAG1 oncogene overexpression in CLL.
We showed inhibitory potential of predicted miRNAs on the PLAG1 3′UTR by luciferase reporter assays. Moreover, site-directed mutagenesis of binding sites could demonstrate specificity of miRNA-3′UTR interaction by abrogation of miRNA regulatory effects. As a consequence for deregulation of miRNAs and disrupted control of PLAG1 3′UTR by the down-regulated miR-107, -424, -181a, and 181b, we demonstrated for the first time overexpression of PLAG1 in CLL cells. Assessing mRNA expression of PLAG1, a nonsignificant up-regulation of PLAG1 was seen. However, PLAG1 protein is significantly up-regulated in primary CLL cells compared with healthy donor B cells. As the control of target genes by miRNAs is mediated by multiple mechanisms, including both mRNA degradation as well as translational inhibition, the significant up-regulation of PLAG1 protein accompanied by a nonsignificant increase of PLAG1 mRNA in CLL indicates translational regulation of PLAG1 as the predominant mechanism of miRNA control on the PLAG1 oncogene.39,40 Here we identified for the first time the up-regulation of PLAG1 in CLL cells, whereas previous attempts of mRNA gene expression profiling could not detect modulation of PLAG1 in CLL.
The physiologic function of this zinc-finger protein is characterized by direct DNA binding to a defined sequence motif.41 Expression of PLAG1 is detectable only in early developmental stages but not in differentiated human tissues, indicating that PLAG1 is involved in embryogenesis.42 PLAG1 mediates transcriptional control of the IGF-II mitogenic pathway and on various other target genes such as VEGF, BPGF-II, Bcl-2, MAPK11, TRAF1, and WT1, as a previous study revealed by gene expression profiling of PLAG1-transfected cells.41 We applied this PLAG1-dependent gene cluster to our previously published mRNA gene expression dataset comparing CLL cells versus healthy donor B cells. By supervised hierarchical cluster analysis, the PLAG1-dependent gene cluster separated malignant CLL cells from healthy B cells, thus indicating differential expression of PLAG1-dependent genes in CLL and an effect of the above-described overexpression of PLAG1 in vivo (supplemental Figure 1).43 Aberrant expression of VEGF in CLL leading to autostimulatory loops has been previously described, and elevated bcl-2 is a key phenomenon of apoptosis resistance in CLL.44 The overexpression of PLAG1-dependent genes indicates regulatory effects of PLAG1 as oncogenic transcription factor in CLL, however ongoing research efforts using transgenic mice models will reveal the oncogenic potential of PLAG1 in B cells.
In conclusion, we (1) demonstrated predominant down-regulation of miRNAs in CLL, (2) identified novel deregulated miRNAs in CLL, (3) unraveled underlying epigenetic changes in loci of deregulated miRNA, (4) applied in silico target prediction of miRNA interactions for identification of novel pathogenetic factors, and (5) identified specific interaction of deregulated miRNA with PLAG1 3′UTRs resulting in overexpression of this oncogene in CLL. Therefore, PLAG1 overexpression in CLL cells represents a novel oncogenic mechanism in CLL pathogenesis on the background of deregulation in miRNA-mediated control mechanisms.
Supplementary Material
Acknowledgments
We thank Reinhild Brinker for excellent technical support. We thank Drs Ibrahim Hasan, Stefan Fronhoffs, Franz Josef Heidgen, Friedhelm Breuer, Roland Schnell, Holger Schulz, Helmut Forstbauer, Carsten Ziske, Ruth Reihs, and Björn Schöttker for referral of patient samples.
C.P.P., C.-M.W., and M.H. were supported by grants from the Deutsche Forschungsgemeinschaft (DFG; Excellence Cluster 229: Cellular Stress Responses in Aging-Associated Diseases), German Cancer Aid (Program for the Development of Interdisciplinary Oncology Centers of Excellence in Germany), CLL Global Research Foundation, and the Marga und Walter Boll-Stiftung. J.L.S. was supported by a research collaboration grant from Illumina. C.P. was supported by National Institutes of Health (NIH) grant CA101956. Y.J.P. was a recipient of a Roman Herzog stipend (Alexander von Humboldt Stiftung). R.C. received a fellowship by the DFG. A.R. was supported by the CLL Global Research Foundation.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: C.P.P. and C.-M.W. conceived and designed the present work; C.P.P., M.P., Y.J.P., S.H., D.E., R.C., S.D.-P., A.S., L.P.F., J.C., and N.K. performed the research; G.K. and A.R. contributed analytical tools; C.P.P., D.E., S.D.-P., Y.J.P., and R.C. conducted statistical analysis; C.P.P., Y.J.P., C.M., C.P., J.L.S., M.H., and C.-M.W. analyzed the data; C.P.P. and C.-M.W. wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Clemens-Martin Wendtner, University of Cologne, Department I of Internal Medicine, Center for Integrated Oncology, Kerpener Strasse 62, D-50924 Cologne, Germany; e-mail: clemens.wendtner@uni-koeln.de.
References
- 1.Döhner H, Stilgenbauer S, Benner A, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343(26):1910–1916. doi: 10.1056/NEJM200012283432602. [DOI] [PubMed] [Google Scholar]
- 2.Fulci V, Chiaretti S, Goldoni M, et al. Quantitative technologies establish a novel microRNA profile of chronic lymphocytic leukemia. Blood. 2007;109(11):4944–4951. doi: 10.1182/blood-2006-12-062398. [DOI] [PubMed] [Google Scholar]
- 3.Calin GA, Dumitru CD, Shimizu M, et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2002;99(24):15524–15529. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marton S, Garcia MR, Robello C, et al. Small RNAs analysis in CLL reveals a deregulation of miRNA expression and novel miRNA candidates of putative relevance in CLL pathogenesis. Leukemia. 2008;22(2):330–338. doi: 10.1038/sj.leu.2405022. [DOI] [PubMed] [Google Scholar]
- 5.Wang M, Tan LP, Dijkstra MK, et al. miRNA analysis in B-cell chronic lymphocytic leukaemia: proliferation centres characterized by low miR-150 and high BIC/miR-155 expression. J Pathol. 2008;215(1):13–20. doi: 10.1002/path.2333. [DOI] [PubMed] [Google Scholar]
- 6.Lau NC, Lim LP, Weinstein EG, Bartel DP. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science. 2001;294(5543):858–862. doi: 10.1126/science.1065062. [DOI] [PubMed] [Google Scholar]
- 7.Basso K, Sumazin P, Morozov P, et al. Identification of the human mature B cell miRNome. Immunity. 2009;30(5):744–752. doi: 10.1016/j.immuni.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tan LP, Wang M, Robertus JL, et al. miRNA profiling of B-cell subsets: specific miRNA profile for germinal center B cells with variation between centroblasts and centrocytes. Lab Invest. 2009;89(6):708–716. doi: 10.1038/labinvest.2009.26. [DOI] [PubMed] [Google Scholar]
- 9.Mayr C, Hemann MT, Bartel DP. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science. 2007;315(5818):1576–1579. doi: 10.1126/science.1137999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ventura A, Jacks T. MicroRNAs and cancer: short RNAs go a long way. Cell. 2009;136(4):586–591. doi: 10.1016/j.cell.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen J, Lozach J, Garcia EW, et al. Highly sensitive and specific microRNA expression profiling using BeadArray technology. Nucleic Acids Res. 2008;36(14):e87. doi: 10.1093/nar/gkn387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.University of Cologne. Microarray data. [Accessed August 2009]. http://www.uk-koeln.de/kliniken/innere1/forschung/Illumina_miRNA_CLL_versus_HD-B_cells_raw_data.txt.
- 13.Ehrich M, Nelson MR, Stanssens P, et al. Quantitative high-throughput analysis of DNA methylation patterns by base-specific cleavage and mass spectrometry. Proc Natl Acad Sci U S A. 2005;102(44):15785–15790. doi: 10.1073/pnas.0507816102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baskerville S, Bartel DP. Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA. 2005;11(3):241–247. doi: 10.1261/rna.7240905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004;14(10A):1902–1910. doi: 10.1101/gr.2722704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ozsolak F, Poling LL, Wang Z, et al. Chromatin structure analyses identify miRNA promoters. Genes Dev. 2008;22(22):3172–3183. doi: 10.1101/gad.1706508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krek A, Grun D, Poy MN, et al. Combinatorial microRNA target predictions. Nat Genet. 2005;37(5):495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 18.miRDB. [Accessed August 2009]. http://mirdb.org/miRDB.
- 19.Wang X, El Naqa IM. Prediction of both conserved and nonconserved microRNA targets in animals. Bioinformatics. 2008;24(3):325–332. doi: 10.1093/bioinformatics/btm595. [DOI] [PubMed] [Google Scholar]
- 20.Memorial Sloan-Kettering Cancer Center. microRNA.org: a resource for predicted microRNA targets and expression. [Accessed August 2009]. http://www.microrna.org/microrna/home.do.
- 21.John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human MicroRNA targets. PLoS Biol. 2004;2(11):e363. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.GeneTrail. [Accessed August 2009]. http://genetrail.bioinf.uni-sb.de.
- 23.Keller A, Backes C, Lenhof HP. Computation of significance scores of unweighted Gene Set Enrichment Analyses. BMC Bioinformatics. 2007;8:290. doi: 10.1186/1471-2105-8-290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thai TH, Calado DP, Casola S, et al. Regulation of the germinal center response by microRNA-155. Science. 2007;316(5824):604–608. doi: 10.1126/science.1141229. [DOI] [PubMed] [Google Scholar]
- 25.Zenz T, Mohr J, Eldering E, et al. miR-34a as part of the resistance network in chronic lymphocytic leukemia. Blood. 2009;113(16):3801–3808. doi: 10.1182/blood-2008-08-172254. [DOI] [PubMed] [Google Scholar]
- 26.Rush LJ, Raval A, Funchain P, et al. Epigenetic profiling in chronic lymphocytic leukemia reveals novel methylation targets. Cancer Res. 2004;64(7):2424–2433. doi: 10.1158/0008-5472.can-03-2870. [DOI] [PubMed] [Google Scholar]
- 27.Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115(7):787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 28.Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27(1):91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 30.Keller A, Backes C, Al-Awadhi M, et al. GeneTrailExpress: a web-based pipeline for the statistical evaluation of microarray experiments. BMC Bioinformatics. 2008;9:552. doi: 10.1186/1471-2105-9-552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Michael MZ, O'Connor SM, van Holst Pellekaan NG, Young GP, James RJ. Reduced accumulation of specific microRNAs in colorectal neoplasia. Mol Cancer Res. 2003;1(12):882–891. [PubMed] [Google Scholar]
- 32.Lu J, Getz G, Miska EA, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435(7043):834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 33.Eis PS, Tam W, Sun L, et al. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci U S A. 2005;102(10):3627–3632. doi: 10.1073/pnas.0500613102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dorsett Y, McBride KM, Jankovic M, et al. MicroRNA-155 suppresses activation-induced cytidine deaminase-mediated Myc-Igh translocation. Immunity. 2008;28(5):630–638. doi: 10.1016/j.immuni.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kas K, Voz ML, Roijer E, et al. Promoter swapping between the genes for a novel zinc finger protein and beta-catenin in pleiomorphic adenomas with t(3;8)(p21;q12) translocations. Nat Genet. 1997;15(2):170–174. doi: 10.1038/ng0297-170. [DOI] [PubMed] [Google Scholar]
- 36.Declercq J, Skaland I, Van Dyck F, et al. Adenomyoepitheliomatous lesions of the mammary glands in transgenic mice with targeted PLAG1 overexpression. Int J Cancer. 2008;123(7):1593–1600. doi: 10.1002/ijc.23586. [DOI] [PubMed] [Google Scholar]
- 37.Landrette SF, Kuo YH, Hensen K, et al. Plag1 and Plagl2 are oncogenes that induce acute myeloid leukemia in cooperation with Cbfb-MYH11. Blood. 2005;105(7):2900–2907. doi: 10.1182/blood-2004-09-3630. [DOI] [PubMed] [Google Scholar]
- 38.Mayr C, Speicher MR, Kofler DM, et al. Chromosomal translocations are associated with poor prognosis in chronic lymphocytic leukemia. Blood. 2006;107(2):742–751. doi: 10.1182/blood-2005-05-2093. [DOI] [PubMed] [Google Scholar]
- 39.Eulalio A, Huntzinger E, Izaurralde E. Getting to the root of miRNA-mediated gene silencing. Cell. 2008;132(1):9–14. doi: 10.1016/j.cell.2007.12.024. [DOI] [PubMed] [Google Scholar]
- 40.Selbach M, Schwanhausser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by micro-RNAs. Nature. 2008;455(7209):58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- 41.Kas K, Voz ML, Hensen K, Meyen E, Van de Ven WJ. Transcriptional activation capacity of the novel PLAG family of zinc finger proteins. J Biol Chem. 1998;273(36):23026–23032. doi: 10.1074/jbc.273.36.23026. [DOI] [PubMed] [Google Scholar]
- 42.Van Dyck F, Declercq J, Braem CV, Van de Ven WJ. PLAG1, the prototype of the PLAG gene family: versatility in tumour development. Int J Oncol. 2007;30(4):765–774. [PubMed] [Google Scholar]
- 43.Voz ML, Mathys J, Hensen K, et al. Microarray screening for target genes of the proto-oncogene PLAG1. Oncogene. 2004;23(1):179–191. doi: 10.1038/sj.onc.1207013. [DOI] [PubMed] [Google Scholar]
- 44.Chen H, Treweeke AT, West DC, et al. In vitro and in vivo production of vascular endothelial growth factor by chronic lymphocytic leukemia cells. Blood. 2000;96(9):3181–3187. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}