

Abstract
Using high-throughput sequencing, we devised a technique to determine the insertion sites of virtually all members of the human-specific L1 retrotransposon family in any human genome. Using diagnostic nucleotides, we were able to locate the approximately 800 L1Hs copies corresponding specifically to the pre-Ta, Ta-0, and Ta-1 L1Hs subfamilies, with over 90% of sequenced reads corresponding to human-specific elements. We find that any two individual genomes differ at an average of 285 sites with respect to L1 insertion presence or absence. In total, we assayed 25 individuals, 15 of which are unrelated, at 1139 sites, including 772 shared with the reference genome and 367 nonreference L1 insertions. We show that L1Hs profiles recapitulate genetic ancestry, and determine the chromosomal distribution of these elements. Using these data, we estimate that the rate of L1 retrotransposition in humans is between 1/95 and 1/270 births, and the number of dimorphic L1 elements in the human population with gene frequencies greater than 0.05 is between 3000 and 10,000.
Retrotransposon insertion polymorphisms (RIPs) are an often-overlooked source of inter- and intra-individual genomic variation that, like other genomic variants such as SNPs and CNVs, can influence phenotype and predisposition to disease. Every studied mammalian genome contains retrotransposons whose past activity accounts for a substantial fraction of the genome. Roughly one-third of the human genome (Lander et al. 2001), 27% of the mouse genome (Mouse Genome Sequencing Consortium 2002), 30% of the rat genome (Gibbs et al. 2004), and 30% of the domestic dog genome (Lindblad-Toh et al. 2005) are composed of autonomous and nonautonomous non-LTR retroelements. Here, we focus on RIPs in the human genome caused by the human-specific subfamily of autonomous long interspersed element-1 (LINE-1 or L1). In humans, three classes of retrotransposons have active members: LINEs, SINEs, and SVAs. LINE-1 (L1) is transcribed as a bicistronic mRNA encoding two proteins, ORF1p and ORF2p (Scott et al. 1987). Because these proteins are critical for mobilization of the L1 RNA from which they are derived (Moran et al. 1996), termed cis-preference (Esnault et al. 2000; Wei et al. 2001), L1s are called autonomous retrotransposons. These proteins can also act in trans to mobilize noncoding human retroelements, such as Alu (Dewannieux et al. 2003) and SVA (Ostertag et al. 2003; Wang et al. 2005), and processed pseudogenes (Esnault et al. 2000).
Throughout the evolutionary history of the human genome, there has been a succession of active L1 subfamilies, distinguishable from one another by sequence differences. Over the last ∼40 million years (Myr) of primate evolution, one active proliferating subfamily of L1s has been replaced by another, such that only one subfamily is active at any time (Boissinot and Furano 2001; Khan et al. 2006). The currently active subfamily in the human genome is L1Hs (for human specific) and can be subdivided into pre-Ta and Ta (for transcribed group a) subfamilies (Kazazian et al. 1988; Skowronski et al. 1988; Boissinot et al. 2000; Salem et al. 2003). Ta elements are further subdivided into Ta-0 and Ta-1 based on diagnostic nucleotides scattered throughout the otherwise almost identical nucleotide sequences (Boissinot et al. 2000; Ovchinnikov et al. 2002; Brouha et al. 2003). Aside from the Ta subfamily, and the active subfamilies of Alu and SVA elements driven by L1, all other retroelement subfamilies in the human genome are for the most part inactive fossils, decaying due to random mutation over time. An active retroelement is capable of being transcribed into an RNA (Skowronski and Singer 1985) that is reverse-transcribed and integrated into another genomic site via target-primed reverse transcription (Luan et al. 1993; George et al. 2006). Because this activity is ongoing, some fraction of an active subfamily is dimorphic with respect to presence or absence at a specified locus. In this article, the term dimorphic refers to simple presence or absence of an insertion at a given site. The term polymorphic refers to instances where the homo- or heterozygosity of an insertion at a specified locus is of interest.
Whereas polymorphic insertions are present in more than one individual of a species, some insertions must be present in only one individual. These insertions are de novo events that occurred either in a parental genome of the individual (present at one copy per genome), or in some cell of the individual (present at less than one copy per genome). Both types of de novo events have been detected in rodent models with somatic insertions being more prevalent (Kano et al. 2009). However, these rodent models contained transgenically introduced L1s and so may not recapitulate true endogenous L1 biology. In mice and humans, there is evidence of somatic L1 retrotransposition in neural progenitor cells (Muotri et al. 2005; Coufal et al. 2009), and somatic insertion leading to disease (Miki et al. 1992; van den Hurk et al. 2007).
Prior studies characterizing human L1 RIPs have focused on determining the allele frequencies of known L1Hs polymorphisms using PCR (Myers et al. 2002; Brouha et al. 2003; Salem et al. 2003) and comparative bioinformatics approaches (Bennett et al. 2004; Konkel et al. 2007; Xing et al. 2009). Previous methods to identify nonreference L1 insertions in human genomic DNA include L1 display (Sheen et al. 2000; Ovchinnikov et al. 2001), ATLAS (Badge et al. 2003), and others (Buzdin et al. 2003; Boissinot et al. 2004). In all, about 400 human L1 RIPs have been cataloged in dbRIP (Wang et al. 2006), 131 of which are not present in the reference genome assembly. There have also been recent studies on endogenous retroelement-induced polymorphism in the mouse genome. In one, an analysis of whole-genome shotgun sequences of mouse strains led to a database of thousands of polymorphic mouse retroelement insertions (Akagi et al. 2008). A high level of retroelement polymorphism in the mouse genome was supported by another analysis of this data set, along with data generated from mate-paired sequencing of mouse strain genomes on the Illumina platform, in which 43% of all structural variations caused by transposable elements were due to L1 (Quinlan et al. 2010). Here, we report the development of a robust, generalizable, deep-sequencing approach to identify essentially all members of a retroelement subfamily in any genome, and double the number of currently known human L1 RIPs.
Results
Sequencing and validation
Briefly, our method for acquiring the genomic coordinates of L1 insertions consists of a hemi-specific nested PCR scheme (Fig. 1). The technique results in a library of L1 3′ flanking DNA that is sequenced on an Illumina Genome Analyzer and analyzed with an in-house computational pipeline (see Methods).
Figure 1.

Hemi-specific PCR scheme to amplify 3′ flanking regions of human-specific LINE-1 insertion sites. The first five cycles of PCR enrich for sequences containing human-specific L1 sequences via primer extension with the single primer pictured above. The AC and G nucleotides in the primers for L1 are diagnostic for the human-specific subfamily for this element. After enrichment for human-specific L1 flanks, a degenerate primer is added that has a specified 5-mer at the 3′ end preceded by five degenerate bases (NNNNN) and a sequencing primer used for the Illumina Genome Analyzer. Eight different reactions are performed, each with a different specified 5mer. The next round of PCR enriches for human-specific L1 3′ flanks with another primer complementary to the L1 and adds the necessary adapter sequences via primer overhangs. The resulting products from each 5-mer are mixed and sequenced on the Illumina Genome Analyzer platform. Following sequencing and initial processing, tags representing the 3′ flanks of human-specific L1 insertions are aligned to the human reference genome (hg18).
On average, 12.32 million reads were sequenced per individual (25 individuals studied), for an average of 8539 peaks per individual (Supplemental Table S2). The majority of these peaks were very small, consisting of only a few reads, and were insignificant. The vast majority of uniquely alignable reads, an average of 90% per individual, correspond to peaks that represent the genomic locations of either reference L1Hs elements or verified nonreference L1 insertions. In this application, a peak refers to the collection of reads uniquely aligning within a 500-bp window 3′ of the expected insertion site (Fig. 2A).
Figure 2.

Validation of peaks resulting from the clustering of alignments. A typical sequence peak is indicated in A. The genome is represented as the colored band spanning the bottom of the figure, and the bases are represented as colored squares (T, red; A, yellow; G, blue; C, green). Stacks of reads are represented on top of the genome as aligned, with a maximum of five unique reads per alignment shown. Evidence for the presence of a polyadenylated sequence absent from the reference genome is indicated by the red outline, which corresponds to the 3′ polyA sequence associated with L1 insertions. The step-like appearance of the sequence peak is due to multiple binding sites for degenerate primers. (B) Genotyping PCR scheme used for the validation of insertions indicated by sequencing peaks. Primers FP and EP flank the expected insertion, indicated by the schematic L1 of unknown length. PCR using these two primers yields an empty site band E of a predetermined size in the cases where the L1 is heterozygous (+/−) for presence or absent entirely (false-positive, −/−). PCR using the AC-specific primer in the L1 3′ UTR (L1P) along with the FP primer yields a band corresponding to the presence of an L1 insertion F. Presence of the filled site, F, and empty site, E, indicates a heterozygous insertion, while presence of only the filled site band indicates homozygous insertion at the specific site. Bands shown on the gel are for three different sites.
Sequenced individuals share an average of 628 L1Hs insertions with the reference genome out of a possible 797. We find that an average individual genome contains 152 L1 insertions that are absent from the human reference sequence. Many of these nonreference insertions are shared between two or more individuals, yielding a total of 367 nonreference insertions that were validated by PCR. For each novel insertion locus identified from a given genome, validation PCR was performed (see Methods) (Fig. 2B) using that genome. Once a given locus was validated by site-specific PCR for one individual genome, further genomes with or without evidence for a given insertion were considered to have or not have that insertion according to the sequence-based evidence, but not further PCR validation in most cases. Thus, we are usually detecting dimorphism of a given insertion but not its copy number when present. These results are summarized for 15 unrelated individuals in Figure 3.
Figure 3.

L1Hs insertions found in various human genomes. L1Hs insertions found for each individual are categorized based on whether or not they are in the reference genome. Reference insertions are subcategorized into pre-Ta, Ta-0, and Ta-1 based on the presence of diagnostic nucleotides. Uncategorized Ta elements (green) are missing one or both characters necessary for placement into either group, often because the nucleotides are not present due to 3′ truncation of the elements. Bars marked with an asterisk (*) indicate samples that did not yield the expected number of insertions, likely due to poor genomic DNA quality or errors in sample preparation.
Genomic distribution
Relative to L1 elements present in the reference genome, nonreference insertions are not significantly enriched in any particular chromosomal region (Fig. 4), suggesting a nonbiased sampling was achieved. Significant enrichment of nonreference insertions relative to reference insertions was tested by Fisher's exact test for insertion counts in 10-Mb windows across the genome.
Figure 4.

Genomic distribution of reference and nonreference L1 insertions. Reference L1 insertions are shown below the genome; nonreference L1 insertions are shown above the genome. The width of each vertical bar corresponds to a 10-Mb window of a chromosome, represented by the alternating dark and light regions as indicated. The heights of the bins are normalized to be comparable across reference and nonreference bins.
Classification of reference and nonreference L1Hs insertions based on genic and nongenic regions yielded an interesting difference: Nonreference insertions were significantly depleted in introns compared with reference insertions. Of 772 insertions present in the reference genome sequence, 243 are intronic and 529 are intergenic. For the 367 nonreference insertions, the numbers are 87 and 280, respectively. (P = 0.0039, Fisher's exact test). Upon further investigation, this depletion appears to be associated with whether or not an insertion is present at an allele frequency close to one, or nearly fixed in the human population. A total of 369 insertions, including both reference and nonreference insertions, were present in every individual we tested, and a total of 770 were absent in at least one individual. Comparing these two groups of insertions with respect to presence in introns yielded 129 intronic elements and 240 intergenic elements in the “fixed” group. In the “dimorphic” group, 201 intronic and 569 intergenic elements were found, a significant depletion of the dimorphic class from intronic regions (P = 0.0027, Fisher's exact test). The correlation still holds when considering only autosomal insertions (P < 0.01). In our data, 131 (39.7%) of the total 330 genic insertions are in the same orientation as the gene, a significant difference from the expected 50% (P = 0.00022, exact binomial test), but consistent with the distribution in the reference genome. When we break this down by allele frequency, insertions present in all 15 individuals examined are significantly biased away from same-sense orientation (44/129 same-sense, P = 0.00039, exact binomial test), but dimorphic insertions do not show a significant bias, although there is a trend (87/201 same-sense, P = 0.066, exact binomial test).
Comparison of L1Hs profiles
Each individual genome has a profile based on the presence or absence of insertions at sites where reference or nonreference insertions have been previously defined in the genome of one or more individuals. This profile corresponds to a binary string where a 1 indicates the presence of an insertion at a given location and a 0 indicates the absence of an insertion (Fig. 5A). Here, we consider the relationship between these profiles in terms of maximum parsimony, which recapitulates the relationships between individuals at the population and family levels (Fig. 5B), albeit for the limited number of individuals analyzed. The number of locations that differ between two individuals because of an L1 insertion in either genome is normally distributed (P = 0.15, Shapiro-Wilk test), with a mean of 285 insertions. Since the number of differences is normally distributed, we can state a 95% confidence interval of 148–422 insertion sites differing between any two individuals. It is important to clarify that this is the total number of differences between two individuals in terms of their L1 profiles; that is, if individual A has 60 insertions not present in the genome of individual B and if B has 100 insertions not present in A, then A and B differ at a total of 160 sites.
Figure 5.

LINE-1 profiles recapitulate genetic ancestry. (A) Depiction of an L1 profile. Each row of squares corresponds to a different individual, and each column corresponds to an L1 insertion that exists in one or more individuals analyzed. A black square indicates the presence of an insertion at the corresponding site in the corresponding individual's genome. (B) Dendrogram representing the maximum parsimony relationship between 19 individuals (three pairs of Mz twins are excluded). Family trios are as follows: SB4Mo/Fa/Ch, SB3Mo/Fa/Ch, GM12891/92/78, JapnIMo/Fa/Ch, JapnYMo/Fa/Ch, GM19238/39/40. These individuals are members of Caucasian, Japanese, and Yoruba ethnic groups as indicated. Individuals prefixed with “GM” are from the Utah CEPH population.
Presence/absence dimorphism
In this report, we have cataloged 367 L1 insertions not present in the reference genome. Of these, at least 78 are also present in dbRIP, an online database of RIPs (Wang et al. 2006). Our data share 42 of 52 nonreference L1 insertion sites identified by comparing the HuRef genome sequence to the human genome assembly hg18 (Xing et al. 2009). Our assay does not directly reflect whether a given L1 insertion dimorphism is homo- or heterozygous; thus allele frequencies are not directly measured. However, if we permit the assumption that most L1 RIPs are in Hardy-Weinberg equilibrium, then the proportion of individuals in which a dimorphic insertion is detected correlates directly with its allele frequency.
Each individual genome assayed has more than 100 insertions not present in the reference genome assembly hg18 (Fig. 3). On average, these nonreference insertions are present in fewer individuals, 6.6 on average, than those present in the reference genome, which are present in an average of 12.1 individual genomes, indicating the lower average allele frequency of the former (Wilcoxon test P < 2 × 10−16). This difference is expected simply as a function of element age: Older insertions are more likely to have been fixed in human populations and consequently present in the reference sequence than newer ones, which are more likely to be absent from the reference due to lower allele frequencies. In Figure 6, we compare the distribution of allele frequencies for reference and nonreference insertions as a function of the number of diploid genomes in which a given insertion is found. A large number of reference insertions are present in all 15 unrelated genomes (Fig. 6A). The distribution is more random for nonreference insertions but skewed toward presence in only one to two genomes (Fig. 6B). The combined distribution for the 1139 L1Hs in this study is more uniform, except for elements that are likely fixed in human populations (Fig. 6C).
Figure 6.

Insertions shared between various numbers of individuals. Histograms for reference (A), nonreference (B), and combined reference and nonreference (C) L1 insertions are shown. The height of each bar represents the number of reference or nonreference insertions shared between the corresponding number of unrelated individuals (genomes). The y-axis (number of shared insertions) is scaled differently for reference and nonreference insertions.
We examined the length distribution of nonreference L1 insertions by taking advantage of available mate-paired whole-genome sequencing data from three individuals: two Africans (ABT and KB1) (Schuster et al. 2010) and one Korean (SJK) (Ahn et al. 2009). We have developed an algorithm to identify novel L1 insertions from datasets such as these (AD Ewing and HH Kazazian Jr., in prep.) and have cross-referenced insertion sites from these three individuals with the data from the 15 unrelated individuals presented here, yielding an overlap of 165 elements. Using information about both the 5′ and 3′ junctions of these 165 elements, we find that the distribution of L1 element lengths for nonreference insertions is similar to the distribution for reference insertions (Supplemental Fig. S5) and agrees with data of previous studies (Grimaldi et al. 1984; Pavlicek et al. 2002).
Estimating the rate of retrotransposition
Using our data, we estimate the rate of retrotransposition for the human-specific L1 subfamily by applying the population genetics theories of Watterson and Tajima (Supplemental Methods; Watterson 1975; Tajima 1989). In these analyses, we estimate the parameter θ using segregating L1 insertion sites and then use this to estimate the rate of L1Hs retrotransposition per live birth. Assuming an effective population size of 10,000 (Harpending et al. 1998), we estimate this rate as 1/140 live births per generation, with upper and lower bounds of 1/95 and 1/270 live births. While this method has caveats (see Discussion), our mean rate is only slightly higher than the rate of 1/212 events per meiosis derived from comparison of the HuRef genome to hg18 (Xing et al. 2009).
Estimating the number of polymorphic L1Hs elements in the human population
Based on our data, we can make an informed estimate of the number of polymorphic L1Hs elements in the global population with the obvious caveat of our small sample size covering only a few populations. Despite this caveat, a reasonable estimate is possible, in part due to the observation that the majority of human variation, perhaps up to 85%, is present within any population, and the remaining minority of variants are distributed among populations (Lewontin 1972; Barbujani et al. 1997; Jorde et al. 2000; Witherspoon et al. 2007). We use two parallel methods to obtain an estimate: One is based on logistic regression over the change in the number of polymorphic insertions as new individuals are successively added in permuted order, and the other is based on the expected number of segregating sites E(K 2N) ≈ 0.5θ(2log2 N) (Watterson 1975). We made this estimate with and without the two Yoruban (YRI) individuals (GM19238 and GM19239) to assess the robustness of the methods. To build a curve for regression, unrelated individuals were added in succession and the number of polymorphic elements after each addition was recorded (Supplemental Fig. S4). Because the order in which individuals are added affects the shape of this curve, this process was repeated on 1000 unique permutations, each with a different order of addition with respect to the 15 unrelated individuals. This permutation results in a distribution for the number of segregating sites for each number of individuals in the pool, the averages of which form the curve used for regression. Logistic regression on this curve yielded an equation fitting the curve with an R 2 of ∼0.94 (Supplemental Fig. S4). For example, n = 6 × 109 individuals yields 3177 insertions (with the caveat that these insertions were detectable in a sample of 15 diploid individuals) (Table 1). Excluding the two YRI individuals yields a very similar equation.
Table 1.
Estimated numbers of segregating L1Hs elements for a various numbers of individuals

The column labels correspond to the labels in Figure 7.
Using the lower and upper bounds of θ (see Supplemental Methods), Watterson's formula yields about 3000 and about 10,000 insertions, respectively, for N = 6 × 109 individuals (Table 1). Estimates for 6 × 109 individuals based on the mean value of θ for the two different estimation methods (regression vs. Watterson) are within a factor of ∼2.5 of each other, while estimates for the lower bound θ of 148 are almost identical to those obtained for the regression curve (Fig. 7).
Figure 7.

Estimation of the number of L1Hs elements in humans. The various estimates discussed are plotted as log number of individuals versus number of L1Hs insertions predicted by the given model. The logistic regression model is plotted as gray circles, and the estimate based on segregating sites is plotted as gray triangles. The dotted lines indicate the upper (open diamonds) and lower (open squares) bounds for the estimate based on segregating sites calculated as described.
Discussion
The evolutionary history of the human genome is replete with retrotransposition events, all of which start out as private insertions, are passed on to subsequent generations as polymorphic insertions, and are eventually lost or fixed through either genetic drift or selective (dis)advantage. Until now, we have only been able to glean hints about the genomics of retrotransposition through the tiny window of the available reference genome. With the advent of next-generation sequencing and associated cost-reductions in sequencing additional human genomes, projects such as the 1000 Genomes will reveal much more about human genetic variation than was previously possible. These new technologies, however, are still not capable of cataloging the variation introduced by the activity of transposable elements due to our currently limited ability to assemble the human genome de novo from short read sequencing data. Although there has been progress (Li et al. 2010), this is especially true for repetitive sequences.
Here, we present a solution to this problem of identifying novel dimorphisms associated with mobile elements. While our technique could be applied to any repeat class in any genome, we have focused on human-specific L1 insertions. The number, length, and accuracy of sequence reads obtained from high-throughput sequencing continues to improve and, correspondingly, so does our ability to perform this assay. As the read length improvement continues, we will likely reach a point where the majority of reads span the 3′ poly-A tail, which can exceed 100–150 bp for new insertions, making subsequent PCR validation unnecessary.
In this article, we have studied more L1Hs insertion loci than in any previous study, a total of 772 shared with the reference genome and 367 not present in the human genome assembly (hg18), an approximately twofold increase in the number of nonreference L1 elements known to exist after subtracting those already present in dbRIP and HuRef (Xing et al. 2009). We have shown that this collection of RIPs can be used to (1) recapitulate genetic ancestry (Fig. 5B), (2) examine the genomic distribution of low and high allele frequency insertions relative to coding regions, and (3) form population-based estimates of retrotransposition frequency and the extent of L1 dimorphism in the human species. We discuss each of these topics.
RIPs, along with simple tandem repeats (STRs), permit the inference of genetic ancestry on a much wider scale than presented here. In one study, 100 Alu and 60 STR polymorphisms were used to assign de-identified individuals to the correct geographical continent of origin (Bamshad et al. 2003). In previous studies, LINE-1 dimorphisms were validated as useful markers of genetic ancestry (Sheen et al. 2000; Witherspoon et al. 2006). In this article, we have presented further evidence that L1 markers are well suited for this purpose by examining the maximum parsimony between L1 insertion profiles for 25 individuals. Since we have a larger number of markers with which to work and since RIPs have the property of being identical-by-descent (Ho et al. 2005), we can build trees that reflect the genetic ancestry of diverse humans on a larger scale with fine detail (Fig. 5), similar to the population-level relationships previously examined using L1 and Alu RIP markers (Witherspoon et al. 2006).
It has long been proposed that the insertion of L1 elements into or nearby genes could influence gene expression by affecting transcription through various mechanisms. Direct evidence for this phenomenon has been elusive. Although one study reported a decrease in L1-containing hnRNAs versus L1-lacking hnRNAs, the effect was only present for certain genes in certain cell types (Ustyugova et al. 2006). An in vitro study suggested that the addition of L1 sequence proximal to a reporter decreased the expression of the reporter in cell culture (Han et al. 2004). An interesting observation, possibly related to this effect, is that L1 insertions have a noted orientation bias with respect to the introns in which they reside. We speculate that an L1 in the sense orientation with respect to an intron is more likely to have a negative fitness effect than an L1 in the antisense orientation. This effect is indeed observable over a long time scale, such that insertions that have proliferated to high allele frequency, and are likely to be very old, are well-tolerated in their given orientation with respect to introns, resulting in a bias toward L1s having an antisense orientation. On the other hand, newer insertions are more random with respect to orientation because evolution has not had as much time to act. These data, taken together with our observation that newer insertions are generally less frequent in genes than older ones, indicate that L1 is perhaps not as neutral a player in genome evolution as expected—specifically, a new intronic insertion in the sense orientation relative to a gene is often not tolerated by the host.
We estimated the number of segregating L1Hs insertion sites between two individuals, and since this quantity corresponds to the parameter θ (see Supplemental Methods), we made a straightforward estimate of the retrotransposition rate. One caveat of our method is the uncertainty of the effective population size Ne, for which we used the published value of 10,000 (Harpending et al. 1998). A second consideration is the simplicity of the model used here. More sophisticated models such as those employing maximum likelihood estimation based on the population genealogy (Felsenstein 1992; Fu and Li 1993) exist and may be applicable to take advantage of the genealogical information we can derive from our data.
We attempted to extrapolate these results to estimate the number of dimorphic L1Hs elements across the entire human population with similar results for two different methods. The permuted addition of individuals is an intuitive model for the diminishing return with respect to the total number of L1 sites cataloged as the number of individuals in the sample increases. Supplemental Figure S4 illustrates this diminishing return curve, the shape of which suggests a logistic relationship between the number of individual samples and the number of L1 insertion sites. Logistic regression on this curve enables us to estimate the number of insertion sites for a given number of individuals. Another extrapolation method was suggested by G.A. Watterson's 1975 paper (Watterson 1975) on segregating sites. While there is some discrepancy in the results of these estimations, between a one and threefold difference when extrapolating to 6 billion individuals, this deviation is relatively small compared to the scale of the extrapolation. Taken together, these two estimates indicate a relatively small number of polymorphic L1Hs insertion loci, on the order of thousands, with the qualification that they are present with a high enough frequency to be detected in a sample of 15 unrelated individuals. This result is not entirely unexpected because a previous study (Bennett et al. 2004) estimated that on the order of approximately 2000 common RIPs exist across L1, Alu, and SVA elements. Many more insertions should exist at lower allele frequencies.
We and others (Xing et al. 2009) estimate the rate of L1 retrotransposition in humans at one in every approximately 150–200 births. Thus, there must be on the order of 30 million private insertions for the roughly 6 billion persons alive today, some fraction of which should be inherited by the next generation. This approximation is much higher than our estimate, but this discrepancy may be due to one or more of the following possibilities. First, it may take a very long time for an insertion to propagate to a readily detectable allele frequency. A second consideration is the appropriateness of our estimators: Although the regression approach seems intuitive and corroborates the theta-based approximation, the correlation of the diminishing return curve to the logistic regression could decrease as more individuals are added. Finally, there is the possibility that most insertions are maintained at low frequencies because they are not selectively neutral. While the insertion loci we used for estimating the rate of retrotransposition appeared selectively neutral based on the agreement of 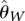 and
and 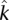 (see Supplemental Methods), many of the lower frequency insertions may not be neutral alleles as suggested by the bias of newer insertions away from introns. Previous studies have provided evidence for significant selection against full-length L1 insertions in the primate lineage (Boissinot et al. 2001).
(see Supplemental Methods), many of the lower frequency insertions may not be neutral alleles as suggested by the bias of newer insertions away from introns. Previous studies have provided evidence for significant selection against full-length L1 insertions in the primate lineage (Boissinot et al. 2001).
In summary, we have developed a technique that allows us to interrogate genomic locations of repeated sequences for which a common 3′ sequence is known based on the reference genome sequence. We applied this method to L1 elements in the human genome and present the largest number of nonreference L1 elements catalogued and analyzed to date. In doing so, we have furthered our understanding of human RIPs and their role in genetic variation.
Methods
Library construction
Because the human genome contains more than 500,000 L1 insertions from the expansions of many previously active subfamilies (Boissinot et al. 2000), specific recognition of the human L1 subfamilies (L1Hs elements) is of the utmost importance. All L1Hs elements from pre-Ta to Ta-1 contain the nucleotide G at position 6015 and the dinucleotide AC at positions 5930-5931 relative to LRE-1 (Dombroski et al. 1991; Boissinot et al. 2000; Ovchinnikov et al. 2002). The human genome assembly hg18 contains 797 L1s with all three of these subfamily-specific characters. We devised a hemi-specific PCR scheme to amplify specifically the 3′ flanking regions of L1Hs in such a way that the resulting library can be sequenced on the Illumina Genome Analyzer platform (Fig. 1). Briefly, the AC-specific primer is used for primer-extension on genomic DNA (for DNA sources, see Supplemental Methods) for five rounds at 58°C to enrich for L1Hs-containing fragments. This is followed by 15 cycles of PCR in eight separate reactions each with a different anchored degenerate primer (for primer sequences, see Supplemental Table 1). These anchored degenerate primers have 5′ overhangs that correspond to the Illumina genomic DNA sequencing primer. Following purification on a Qiagen PCR cleanup column, 2 μL of each reaction is subjected to another 15 cycles of PCR, where the 5′ primer has the “G” diagnostic character at its 3′ end and a 5′ extension corresponding to an Illumina adapter sequence. The 3′ primer corresponds to the Illumina genomic DNA sequencing primer followed by the Illumina adapter sequence. It is important to use a polymerase lacking 3′ → 5′ exonuclease, as this activity removes the 3′ nucleotides of the primers required for subfamily specificity. The supplementary methods provide specific details of the PCR reactions and cycling conditions.
The final PCR products were run on a 1.5% TAE gel stained with EtBr (Invitrogen) or Gel Green (Biotium), and fragments between 200 and 500 bp were excised. DNA from the eight gel sections for each sample was purified using the Qiagen gel extraction kit protocol. Pooled column elutions were further purified on a Qiagen column using the PCR cleanup protocol and eluted in 55 μL of DEPC-treated sterile ddH2O. The products were incubated with Pfu polymerase (Invitrogen) in the supplied buffer at 1× concentration with 0.5 mM dNTPs for 30 min at 72°C to remove any 3′ adenine overhangs. The reaction was then purified on a Qiagen MinElute column following the manufacturer's protocol and eluted in 10 μL of buffer EB. Samples were analyzed on an Agilent 2100 BioAnalyzer to determine DNA concentration prior to dilution and sequencing on the Illumina Genome Analyzer. Determination of DNA concentration and Illumina sequencing were carried out at the University of Pennsylvania IDOM Functional Genomics Core.
Computational analysis
It was necessary to develop a novel computational pipeline since our technique does not exactly correspond to chromatin immunoprecipitation with massively parallel sequencing (ChIP-seq), whole-genome resequencing, or any other existing application of next-generation sequencing. First, 76-bp sequence reads are trimmed to 40 bp by removing 10 bp from the 5′ end and 26 bp from the 3′ end. For 36-bp reads from earlier runs, only the 5′-most 6 bp were removed, yielding a 30-bp read. The 5′ trimming is necessary because of the degeneracy of the primers after the sequencing primer-binding site. The degenerate primers can bind without the entire degenerate sequence (N 5) matching the reference genome, introducing an excessive number of mismatches on this end in some cases, despite high average base quality scores. For the longer 76-bp read length, the 3′ end is trimmed conservatively due to the drop-off in average sequence quality and the presence of nonreference polyadenylation corresponding to L1 insertions absent from the reference genome assembly. This nonreference polyadenylation can be used to our advantage as discussed. Following trimming, the reads are aligned to the reference genome, allowing two mismatches using bowtie (Langmead et al. 2009) with the option string -n 2 -m 0–best–strata–un. The alignments are sorted and clustered into peaks based on proximity in a 600-bp window using a Perl script. These peaks are somewhat analogous to those derived from ChIP-seq experiments and have the following properties: number of reads per peak, number of unique reads per peak, peak width, average read quality, average number of mismatches per read. The “peaks” may actually consist of multiple disjointed stacks of sequence reads due to multiple binding sites of the degenerate primers (Fig. 2A).
Information about the exact location of some insertions can be obtained by analyzing polyadenylated sequence reads. Starting with reads that are 76 bp in length, the 5′-most 10 bp are trimmed off for reasons discussed above. Following this, reads are aligned to the reference genome using bowtie with the options -n 3 -m 0–best–strata–un. The unalignable reads are then analyzed for the presence of 3′ poly-T tracts of 6 bp or longer. The homopolymers are polythymidine rather than polyadenosine because of the direction in which the sequencing-by-synthesis reaction occurs relative to the L1 sequence. These tracts are trimmed off, and the trimmed reads are realigned to the reference genome again using bowtie. The resulting alignments are sorted and clustered and compared against the peaks created from the previous alignment using 40-bp reads. Those that correspond to previously identified peaks may contain information about the sequence of the junction between the L1 and the genomic site of insertion, if the peak corresponds to a nonreference insertion (Fig. 2A).
After clustering the peaks, another script checks to see whether each peak can be “explained” by a L1 element in the reference genome. For a reference L1 to explain the presence of a peak, it must be oriented opposite to the peak, and the center of the peak must be within 600 bp of the L1 3′ untranslated region (UTR) in the 3′ flanking sequence. Proximity to primate-specific (L1PA*) LINEs is considered as well. Those peaks not corresponding to known locations of L1 elements indicate the possible presence of nonreference L1Hs insertions.
Site-specific PCR
The presence of nonreference insertions is verified via site-specific PCR (Fig. 2B). The 3′ ends and flanking regions of nonreference L1s are amplified using the same AC dinucleotide–specific primer used for the first-round PCR (Fig. 1) and another primer selected from the 3′ flanking region based on the reference genome sequence. The “empty” site, that is, the allele that does not contain an L1 insertion, is also amplified from the genome using primers flanking the suspected site of insertion on the 5′ and 3′ ends (Fig. 2B). PCR reactions were carried out in 1X GoTaq Green master mix (Promega), with 10 pmol for “ES” and “FS” primers and 20 pmol for the L1Hs “AC” primers as indicated in Figure 2B. Reactions were incubated for 3 min at 95°C followed by 30 cycles of 30 sec at 94°C, 30 sec at 57°C, and 1 min at 72°C, followed by final extension of 10 min at 72°C on a DNA engine Dyad (Bio-Rad). Successful primer sequences for validated nonreference insertion sites are included in the supplemental material (Supplemental Table S1). Information about peaks, their locations, and their validation is stored in a relational database in a manner that facilitates the comparison of individuals with respect to their genomic retrotransposon content.
Further bioinformatics and statistical analysis
Reference human-specific L1 sequences were obtained from the RepeatMasker track for the UCSC Genome Browser assembly hg18 (http://genome.ucsc.edu). These were then classified into pre-Ta, Ta-0, and Ta-1 using a Perl script. The maximum parsimony phylogram was created from 1000 bootstrap replicates using the seqboot, dnapars, and consense tools in the PHYLIP package (Felsenstein 1989) and drawn using Dendroscope (Huson et al. 2007). Statistical tests were carried out as indicated using the R language for statistical computing (http://www.r-project.org). We considered a P-value of <0.05 as marginally significant and P < 0.01 as significant.
Data availability
The genomic locations of the 367 verified PCR insertions are provided in Supplemental Table S1. The source code for the computational pipeline is available in the Supplemental material.
Acknowledgments
We thank Warren Ewens for assistance with statistical methods and review of the manuscript, as well as Dustin Hancks, John Goodier, and Sanjida Rangwala for review of the manuscript. We also thank Vivian Cheung, Paul Sniegowski, Frederic Bushman, and Sridhar Hannenhalli for helpful discussions and conceptual contributions. This study was funded by the NIH and the Penn Genome Frontiers Institute (PGFI).
Footnotes
[Supplemental material is available online at http://www.genome.org. The sequence data from this study have been submitted to the NCBI dbGaP (http://www.ncbi.nlm.nih.gov/gap/) under accession no. phs000273.v1.p1.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.106419.110.
References
- Akagi K, Li J, Stephens RM, Volfovsky N, Symer DE 2008. Extensive variation between inbred mouse strains due to endogenous L1 retrotransposition. Genome Res 18: 869–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn SM, Kim TH, Lee S, Kim D, Ghang H, Kim DS, Kim BC, Kim SY, Kim WY, Kim C 2009. The first Korean genome sequence and analysis: Full genome sequencing for a socio-ethnic group. Genome Res 19: 1622–1629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badge RM, Alisch RS, Moran JV 2003. ATLAS: A system to selectively identify human-specific L1 insertions. Am J Hum Genet 72: 823–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamshad MJ, Wooding S, Watkins WS, Ostler CT, Batzer MA, Jorde LB 2003. Human population genetic structure and inference of group membership. Am J Hum Genet 72: 578–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbujani G, Magagni A, Minch E, Cavalli-Sforza LL 1997. An apportionment of human DNA diversity. Proc Natl Acad Sci 94: 4516–4519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett EA, Coleman LE, Tsui C, Pittard WS, Devine SE 2004. Natural genetic variation caused by transposable elements in humans. Genetics 168: 933–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissinot S, Furano AV 2001. Adaptive evolution in LINE-1 retrotransposons. Mol Biol Evol 18: 2186–2194 [DOI] [PubMed] [Google Scholar]
- Boissinot S, Chevret P, Furano AV 2000. L1 (LINE-1) retrotransposon evolution and amplification in recent human history. Mol Biol Evol 17: 915–928 [DOI] [PubMed] [Google Scholar]
- Boissinot S, Entezam A, Furano AV 2001. Selection against deleterious LINE-1-containing loci in the human lineage. Mol Biol Evol 18: 926–935 [DOI] [PubMed] [Google Scholar]
- Boissinot S, Entezam A, Young L, Munson PJ, Furano AV 2004. The insertional history of an active family of L1 retrotransposons in humans. Genome Res 14: 1221–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouha B, Schustak J, Badge RM, Lutz-Prigge S, Farley AH, Moran JV, Kazazian HH Jr 2003. Hot L1s account for the bulk of retrotransposition in the human population. Proc Natl Acad Sci 100: 5280–5285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzdin A, Ustyugova S, Gogvadze E, Lebedev Y, Hunsmann G, Sverdlov E 2003. Genome-wide targeted search for human specific and polymorphic L1 integrations. Hum Genet 112: 527–533 [DOI] [PubMed] [Google Scholar]
- Coufal NG, Garcia-Perez JL, Peng GE, Yeo GW, Mu Y, Lovci MT, Morell M, O'Shea KS, Moran JV, Gage FH 2009. L1 retrotransposition in human neural progenitor cells. Nature 460: 1127–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewannieux M, Esnault C, Heidmann T 2003. LINE-mediated retrotransposition of marked Alu sequences. Nat Genet 35: 41–48 [DOI] [PubMed] [Google Scholar]
- Dombroski BA, Mathias SL, Nanthakumar E, Scott AF, Kazazian HH Jr 1991. Isolation of an active human transposable element. Science 254: 1805–1808 [DOI] [PubMed] [Google Scholar]
- Esnault C, Maestre J, Heidmann T 2000. Human LINE retrotransposons generate processed pseudogenes. Nat Genet 24: 363–367 [DOI] [PubMed] [Google Scholar]
- Felsenstein J 1989. PHYLIP—Phylogeny Inference Package (version 3.2). Cladistics 5: 164–166 [Google Scholar]
- Felsenstein J 1992. Estimating effective population size from samples of sequences: Inefficiency of pairwise and segregating sites as compared to phylogenetic estimates. Genet Res 59: 139–147 [DOI] [PubMed] [Google Scholar]
- Fu YX, Li WH 1993. Maximum likelihood estimation of population parameters. Genetics 134: 1261–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- George JA, Burke WD, Eickbush TH 2006. Analysis of the 5′ junctions of R2 insertions with the 28S gene: Implications for non-LTR retrotransposition. Genetics 142: 853–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs RA, Weinstock GM, Metzker ML, Muzny DM, Sodergren EJ, Scherer S, Scott G, Steffen D, Worley KC, Burch PE, et al. 2004. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature 428: 493–521 [DOI] [PubMed] [Google Scholar]
- Grimaldi G, Skowronski J, Singer MF 1984. Defining the beginning and end of KpnI family segments. EMBO J 3: 1753–1759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JS, Szak ST, Boeke JD 2004. Transcriptional disruption by the L1 retrotransposon and implications for mammalian transcriptomes. Nature 429: 268–274 [DOI] [PubMed] [Google Scholar]
- Harpending HC, Batzer MA, Gurven M, Jorde LB, Rogers AR, Sherry ST 1998. Genetic traces of ancient demography. Proc Natl Acad Sci 95: 1961–1967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho HJ, Ray DA, Salem AH, Myers JS, Batzer MA 2005. Straightening out the LINEs: LINE-1 orthologous loci. Genomics 85: 201–207 [DOI] [PubMed] [Google Scholar]
- Huson DH, Richter DC, Rausch C, Dezulian T, Franz M, Rupp R 2007. Dendroscope: An interactive viewer for large phylogenetic trees. BMC Bioinformatics 8: 460 doi: 10.1186/1471-2105-8-460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorde LB, Watkins WS, Bamshad MJ, Dixon ME, Ricker CE, Seielstad MT, Batzer MA 2000. The distribution of human genetic diversity: A comparison of mitochondrial, autosomal, and Y-chromosome data. Am J Hum Genet 66: 979–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano H, Godoy I, Courtney C, Vetter MR, Gerton GL, Ostertag EM, Kazazian HH Jr 2009. L1 retrotransposition occurs mainly in embryogenesis and creates somatic mosaicism. Genes Dev 23: 1303–1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazazian HH Jr, Wong C, Youssoufian H, Scott AF, Phillips DG, Antonarakis SE 1988. Haemophilia A resulting from de novo insertion of L1 sequences represents a novel mechanism for mutation in man. Nature 332: 164–166 [DOI] [PubMed] [Google Scholar]
- Khan H, Smit A, Boissinot S 2006. Molecular evolution and tempo of amplification of human LINE-1 retrotransposons since the origin of primates. Genome Res 16: 78–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konkel MK, Wang J, Liang P, Batzer MA 2007. Identification and characterization of novel polymorphic LINE-1 insertions through comparison of two human genome sequence assemblies. Gene 390: 28–38 [DOI] [PubMed] [Google Scholar]
- Lander ES, Heaford A, Sheridan A, Linton LM, Birren B, Subramanian A, Coulson A, Nusbaum C, Zody MC, Dunham A, et al. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860–921 [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25 doi: 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewontin RC 1972. The apportionment of human diversity. Evol Biol 6: 381–398 [Google Scholar]
- Li R, Zhu H, Ruan J, Qian W, Fang X, Shi Z, Li Y, Li S, Shan G, Kristiansen K, et al. 2010. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res 20: 265–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindblad-Toh K, Wade CM, Mikkelsen TS, Karlsson EK, Jaffe DB, Kamal M, Clamp M, Chang JL, Kulbokas EJ, Zody MC, et al. 2005. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 438: 803–819 [DOI] [PubMed] [Google Scholar]
- Luan DD, Korman MH, Jakubczak JL, Eickbush TH 1993. Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: A mechanism for non-LTR retrotransposition. Cell 72: 595–605 [DOI] [PubMed] [Google Scholar]
- Miki Y, Nishisho I, Horii A, Miyoshi Y, Utsunomiya J, Kinzler KW, Vogelstein B, Nakamura Y 1992. Disruption of the APC gene by a retrotransposal insertion of L1 sequence in a colon cancer. Cancer Res 52: 643–645 [PubMed] [Google Scholar]
- Moran JV, Holmes SE, Naas TP, DeBerardinis RJ, Boeke JD, Kazazian HH Jr 1996. High frequency retrotransposition in cultured mammalian cells. Cell 87: 917–927 [DOI] [PubMed] [Google Scholar]
- Mouse Genome Sequencing Consortium 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520–562 [DOI] [PubMed] [Google Scholar]
- Muotri AR, Chu VT, Marchetto MC, Deng W, Moran JV, Gage FH 2005. Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature 435: 903–910 [DOI] [PubMed] [Google Scholar]
- Myers JS, Vincent BJ, Udall H, Watkins WS, Morrish TA, Kilroy GE, Swergold GD, Henke J, Henke L, Moran JV, et al. 2002. A comprehensive analysis of recently integrated human Ta L1 elements. Am J Hum Genet 71: 312–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostertag EM, Goodier JL, Zhang Y, Kazazian HH Jr 2003. SVA elements are nonautonomous retrotransposons that cause disease in humans. Am J Hum Genet 73: 1444–1451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovchinnikov I, Troxel AB, Swergold GD 2001. Genomic characterization of recent human LINE-1 insertions: Evidence supporting random insertion. Genome Res 11: 2050–2058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovchinnikov I, Rubin A, Swergold GD 2002. Tracing the LINEs of human evolution. Proc Natl Acad Sci 99: 10522–10527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlicek A, Paces J, Zika R, Hejnar J 2002. Length distribution of long interspersed nucleotide elements (LINEs) and processed pseudogenes of human endogenous retroviruses: Implications for retrotransposition and pseudogene detection. Gene 300: 189–194 [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Clark RA, Sokolova S, Leibowitz ML, Zhang Y, Hurles ME, Mell JC, Hall IM 2010. Genome-wide mapping and assembly of structural variant breakpoints in the mouse genome. Genome Res 20: 623–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salem AH, Myers JS, Otieno AC, Watkins WS, Jorde LB, Batzer MA 2003. LINE-1 preTa elements in the human genome. J Mol Biol 326: 1127–1146 [DOI] [PubMed] [Google Scholar]
- Schuster SC, Miller W, Ratan A, Tomsho LP, Giardine B, Kasson LR, Harris RS, Petersen DC, Zhao F, Qi J, et al. 2010. Complete Khoisan and Bantu genomes from southern Africa. Nature 463: 943–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott AF, Schmeckpeper BJ, Abdelrazik M, Comey CT, O'Hara B, Rossiter JP, Cooley T, Heath P, Smith KD, Margolet L 1987. Origin of the human L1 elements: Proposed progenitor genes deduced from a consensus DNA sequence. Genomics 1: 113–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheen FM, Sherry ST, Risch GM, Robichaux M, Nasidze I, Stoneking M, Batzer MA, Swergold GD 2000. Reading between the LINEs: Human genomic variation induced by LINE-1 retrotransposition. Genome Res 10: 1496–1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skowronski J, Singer MF 1985. Expression of a cytoplasmic LINE-1 transcript is regulated in a human teratocarcinoma cell line. Proc Natl Acad Sci 82: 6050–6054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skowronski J, Fanning TG, Singer MF 1988. Unit-length line-1 transcripts in human teratocarcinoma cells. Mol Cell Biol 8: 1385–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ustyugova SV, Lebedev YB, Sverdlov ED 2006. Long L1 insertions in human gene introns specifically reduce the content of corresponding primary transcripts. Genetica 128: 261–272 [DOI] [PubMed] [Google Scholar]
- van den Hurk JA, Meij IC, Seleme MC, Kano H, Nikopoulos K, Hoefsloot LH, Sistermans EA, de Wijs IJ, Mukhopadhyay A, Plomp AS, et al. 2007. L1 retrotransposition can occur early in human embryonic development. Hum Mol Genet 16: 1587–1592 [DOI] [PubMed] [Google Scholar]
- Wang H, Xing J, Grover D, Hedges DJ, Han K, Walker JA, Batzer MA 2005. SVA elements: A hominid-specific retroposon family. J Mol Biol 354: 994–1007 [DOI] [PubMed] [Google Scholar]
- Wang J, Song L, Grover D, Azrak S, Batzer MA, Liang P 2006. dbRIP: A highly integrated database of retrotransposon insertion polymorphisms in humans. Hum Mutat 27: 323–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson GA 1975. On the number of segregating sites in genetical models without recombination. Theor Popul Biol 7: 256–276 [DOI] [PubMed] [Google Scholar]
- Wei W, Gilbert N, Ooi SL, Lawler JF, Ostertag EM, Kazazian HH Jr, Boeke JD, Moran JV 2001. Human L1 retrotransposition: Cis preference versus trans complementation. Mol Cell Biol 21: 1429–1439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witherspoon DJ, Marchani EE, Watkins WS, Ostler CT, Wooding SP, Anders BA, Fowlkes JD, Boissinot S, Furano AV, Ray DA, et al. 2006. Human population genetic structure and diversity inferred from polymorphic L1(LINE-1) and Alu insertions. Hum Hered 62: 30–46 [DOI] [PubMed] [Google Scholar]
- Witherspoon DJ, Wooding S, Rogers AR, Marchani EE, Watkins WS, Batzer MA, Jorde LB 2007. Genetic similarities within and between human populations. Genetics 176: 351–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing J, Zhang Y, Han K, Salem AH, Sen SK, Huff CD, Zhou Q, Kirkness EF, Levy S, Batzer MA, et al. 2009. Mobile elements create structural variation: Analysis of a complete human genome. Genome Res 19: 1516–1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The genomic locations of the 367 verified PCR insertions are provided in Supplemental Table S1. The source code for the computational pipeline is available in the Supplemental material.