

Abstract
The DA strain of Theiler's murine encephalomyelitis virus (TMEV), a member of the Cardiovirus genus of the family Picornaviridae, causes persistent infection in susceptible mice, associated with restricted expression of viral proteins, and induces a demyelinating disease of the central nervous system. DA-induced demyelinating disease serves as a model of human multiple sclerosis because of similarities in pathology and because host immune responses contribute to pathogenesis in both disorders. In contrast, the GDVII strain of TMEV causes acute lethal encephalitis with no virus persistence. Cardiovirus L is a multifunctional protein that blocks beta interferon (IFN-β) gene transcription. We show that both DA L and GDVII L disrupt IFN-β gene transcription induction by IFN regulatory factor 3 (IRF-3) but do so at different points in the signaling pathway. DA L blocks IFN-β gene transcription downstream of mitochondrial antiviral signaling protein (MAVS) but upstream of IRF-3 activation, while GDVII L acts downstream of IRF-3 activation. Both DA L and GDVII L block IFN-β gene transcription in infected mice; however, IFN-β mRNA is expressed at low levels in the central nervous systems of mice persistently infected with DA. The particular level of IFN-β mRNA expression set by DA L as well as other factors in the IRF-3 pathway may play a role in virus persistence, inflammation, and the restricted expression of viral proteins during the late stage of demyelinating disease.
The Cardiovirus genus of the Picornaviridae family comprises two species, Encephalomyocarditis virus, which includes encephalomyocarditis virus (EMCV) and mengovirus, and Theilovirus, which includes Theiler's murine encephalomyelitis virus (TMEV) and Saffold virus. TMEV strains fall into two subgroups based on their different pathogenicities (reviewed in reference 45). Members of the GDVII subgroup of TMEV cause an acute lytic neuronal disease and do not persist. In contrast, the DA strain and other members of the TO subgroup cause a subclinical neuronal disease that is followed by a chronic inflammatory demyelinating process that is thought to be immune mediated. The TO subgroup strains persist in the central nervous system (CNS) for the life of the mouse, with restricted expression of viral proteins (6), and cause a disease that is remarkably different from that seen with other picornaviruses. DA virus-induced chronic demyelinating disease serves as a model of multiple sclerosis (MS) because of similarities in the demyelinating pathology and because the host immune response appears to contribute to the pathogenesis of both disorders.
Because of its role in the inflammatory demyelinating disease induced by TO subgroup strains, the host immune response has been a focus of investigations of TMEV. However, little is known of the innate immune response to infections with either GDVIII or DA subgroup viruses, despite the importance of early innate immune responses for the acquired immune response and for the pathogenesis of virus infections and autoimmune disease.
Type I interferons (IFNs) are produced following recognition of viral pathogens by cellular receptors, including Toll-like receptors and cytoplasmic DEX(D/H) box helicases, such as those encoded by retinoic acid-inducible gene I (RIG-I) and melanoma-differentiation-associated gene 5 (MDA-5). Among cytosolic receptors, MDA-5 is principally responsible for recognition of picornaviruses, including cardioviruses, and TMEV in particular (21, 27). Binding of viral RNA by MDA-5 results in its association with mitochondrial antiviral signaling adaptor protein (MAVS, also known as IPS-1, VISA, and CARDIF) on the outer mitochondrial membrane. MAVS associates with tumor necrosis factor (TNF) receptor-associated factor 3 (TRAF-3). TRAF-3 recruits and activates two IKK-related kinases, TANK-binding kinase 1 (TBK1) and inducible IkB kinase (IKKɛ), leading to phosphorylation of interferon regulatory factor 3 (IRF-3) and resulting in IRF-3 dimerization and subsequent translocation to the nucleus. IRF-3 activation leads to the transcription of the IFN-β gene, which requires the formation of an enhanceosome on the IFN-β gene promoter involving IRF-3 as well as the activation transcription factor 2/c-jun complex (ATF-2/c-Jun) and NF-κB. This IFN-β gene promoter-enhancer region contains four positive regulatory domains (PRDI to -IV) and one negative regulatory domain (NRDI). PRDI and -III contain the binding sites for IRF-3, as well as for other IRF members; PRDII is recognized by NF-κB and PRDIV by ATF-2/c-Jun heterodimers. Virus infection leads to the recruitment of histone acetyltransferase coactivators (GCN5 and CBP/p300), as well as a high mobility group protein [HMG 1(Y)], which bind to the IFN-β gene enhancer and contribute to the stability of the enhanceosome. Interference with the activation of any of the coactivators of the IFN-β gene enhanceosome abrogates IFN-β gene transcription completely (38). IFN-β subsequently acts in an autocrine/paracrine fashion via Jak-STAT signaling to induce other type I IFNs (e.g., IFN-α) as well as a large number of IFN-stimulated genes (ISGs). Transcription of a small subset of ISGs may also be stimulated directly by IRF-3.
The importance of the type I IFN response in virus infections is demonstrated by increases in mortality and level of infectious virus in mice deficient in the IFN-α/β receptor in the case of many infections, including those by DA (16, 42), and the number of viral gene products, including picornaviral gene products, that have been found to antagonize this response. For example, hepatitis A virus targets its viral 3ABC protease precursor to the mitochondria, where it cleaves MAVS (55) and poliovirus cleaves MDA-5 by an apoptotic activity triggered by the infection (2).
L designates a picornavirus protein located between the start of the polyprotein and the P1 capsid proteins. Not all picornavirus genera have an L protein. The cardiovirus L has a unique sequence compared to those of other picornaviruses, suggesting that it has a special function. The fact that the L coding region is located at the beginning of the long open reading frame of the gene for the polyprotein ensures that it will be robustly synthesized, even in the event of the ribosome falling off the viral genome. Interestingly, there are prominent differences in sequences of L proteins even among cardioviruses: the nucleotide sequences of the L protein genes of EMCV and mengovirus differ from that of the TMEV L gene at ∼42% of base positions, while the amino acid sequences of the proteins differ by ∼23%. Part of this difference results from the presence of a Ser-Thr domain in TMEV L that is absent in EMCV and mengovirus L proteins (Fig. 1A). Furthermore, comparison of DA viral proteins with GDVII viral proteins shows that L is the protein with the greatest difference in amino acid sequences (36).
FIG. 1.

Cardiovirus L protein sequence and interference with IFN-β mRNA. (A) Sequence of L proteins of different cardioviruses. The zinc finger, acidic domain, and serine threonine domain are noted, and the locations of amino acids that vary among TMEV strains are highlighted. (B) IFN-β mRNA levels determined by qRT-PCR in cell lysates harvested 9 h following cardiovirus and SV infection. The difference between the levels following infection with TMEV L mutants versus wt cardioviruses was significant (*, P < 0.001). HeLa cell cytoplasmic lysates used for the qRT-PCR were probed in a Western blot for TMEV VP1 and β-actin, as shown. The bars (B to G) represent means ± standard errors (SE). (C) pISG56-luc activation following infection. Cells were transfected with pISG56-Luc and pRL-TK1 (to normalize transfection) and then infected 16 h later with various viruses. Luciferase levels following infection with TMEV L mutants were significantly higher than those following infection with wt TMEV and EMCV (*, P < 0.001). The same lysates in which the RLUs were determined were Western blotted and probed with anti-TMEV VP1 antibody and anti-β-actin antibody. M corresponds to uninfected cells. (D to F) Experiments in mice infected with TMEV. Levels of IFN-β mRNA (D and F) and virus genome (E and G) in brain 1 day (D and E) and 5 days (F and G) after infection with TMEV wt or ΔL viruses. The levels of IFN-β mRNA and virus genome were different following infection with DA versus DAΔL virus (P < 0.001) as well as GDVII versus GDVIIΔL virus (P < 0.001). The data are from 4 mice in each group. Note the different scales in the different panels. M corresponds to uninfected cells (B and C) or mice (D to G).
The L proteins of cardioviruses have recently been shown to have multiple functions of interest, including inhibition of type I IFN gene transcription (23, 42), regulation of apoptosis (15, 44), and interference with nucleocytoplasmic transport and host cell translation (13, 40). L is critically important in TMEV-induced demyelinating disease (TMEV-IDD) since TMEV with a mutation in L is cleared (5, 39), at least partly because of a lack of inhibition of the type I IFN response. Here, we describe studies of the type I IFN response to TMEV infection. We demonstrate that there are important differences in this early innate immune response between DA and GDVIII viruses. The different mechanisms by which the L proteins of these viruses disrupt the induction of IFN-β synthesis through IRF-3 and the level of IFN-β in the CNS may play an important role in TMEV-IDD.
MATERIALS AND METHODS
Cells and viruses.
BHK-21 cells (baby hamster kidney cells) were used for plaque assays and the growth of virus stocks, as previously described (8). Studies examining IRF-3 and the IFN-β pathway were performed in L-929 cells and HeLa cells.
Viruses.
Wild-type (wt) DA was derived from a full-length infectious cDNA clone known as pDAFL3 (46). DAΔL, which has a deletion of amino acids 2 to 67, was prepared by introducing MluI restriction sites in pDAFL3 at the site of the 2nd and the 67th codons of the L coding region using the QuikChange XL site-directed mutagenesis kit (Stratagene, La Jolla, CA). For generation of the MluI site at the second codon, the forward primer was CCTTTTTATTACTATTGACACTATGACGCGTGCTTGCAAACATGGATAC and the reverse primer was GTATCCATGTTTGCAAGCACGCGTCATAGTGTCAATAGTAATAAAAAGG. For the MluI site insertion at the 67th codon, the forward primer was GGACTGACTTACCGCTCACGGCTGTACGCGATATTGTGATG and the reverse primer was CATCACAATATCGCGTACACGCGTGAGCGGTAAGTCAGTCC. The mutated pDAFL3 was then digested with MluI and religated with T4 DNA ligase. The resulting MluI site was eliminated using the QuikChange XL site-directed mutagenesis kit with forward primer CCTTTTTATTACTATTGACACTATGGTACGCGATATTGTCATG and reverse primer CATGACAATATCGCGTACCATAGTGTCAATAGTAATAAAAAGG. DALZn has a mutated Zn finger without a change in the sequence of L* (8). It was prepared by mutating nucleotides (nt) 63, 1096, 1099, and 1105 in the L gene coding region using the QuikChange XL site-directed mutagenesis kit with forward primer CATGGATACCCAGATGTGCGCACTATTCGCACAGCCGTTGACG and reverse primer CGTCAACGGCTGTGCGAATAGTGCGCACATCTGGGTATCCATG. wt GDVII was derived from pGDFL2 (20). GDVIIΔL, in which L has a deletion of amino acids 2 to 71, was a gift from M. K. Rundell and was previously called dl-L (5). EMCV was a gift from Ann Palmenberg. DA/LGDVII and DA/LEMCV, in which DA L in the DA backbone was replaced by GDVII L and EMCV L, respectively, have been previously described (28). Sendai virus (SV), Cantell strain (Charles River Laboratory), was used as a control; 160 hemagglutination units was usually used to infect a 60-mm plate.
The viruses were generated from transfection of in vitro-derived transcripts of their respective infectious cDNA clone, as previously described (46). The L coding regions of the plasmids and viruses were sequenced in order to make certain that the anticipated sequence was present. Viruses were infected at a multiplicity of infection (MOI) of 10.
Plasmids.
pDAL, which is a eukaryotic expression construct of DA L with myc/His epitope tags at the carboxyl terminus, was prepared by amplifying the L sequence with a Superscript PCR mix (Invitrogen, Carlsbad, CA) using pDAFL3 as a template with forward primer GGGAATTCATGGCTTGCAAACATGGATACCCAG and reverse primer CCGGATCCCTGGGGTTCCATGACAATATC. The amplified fragment was ligated into EcoRI/BamHI-digested pcDNA3.1/myc-His A (Invitrogen). pGDVIIL, a eukaryotic expression construct of GDVII L myc/His, was prepared similarly to pDAL except that the following primers were used: forward, GGGAATTCATGGCTTGCAAACACGGATACCCAGACG; reverse, CCGGATCCCTGGGGTTCCATGACAGTATC. pRL-TK1 (Promega, Madison, WI), which has a Renilla luciferase reporter driven by the thymidine kinase promoter, was used as an internal control for transfection efficiency. pISG56-Luc expresses firefly luciferase as a means of quantitating the level of activation of IFN-β response gene 56 (ISG56). p4xPRDI/III-Luc expresses firefly luciferase as a means of quantitating IRF-3-dependent binding to the PRDI/III elements of the IFN-β promoter (14). pIRF-3(5D) expresses a constitutively active phosphomimetic form of IRF-3 because of mutations in five clustered residues among amino acids 396 to 405 of IRF-3 to glutamic acid (29, 30). pTBK-1 expresses TBK-1. pIKKɛ expresses IKKɛ.
Quantitative studies.
TMEV RNA and IFN-β RNA in homogenates of HeLa cells or CNS tissues were assayed by quantitative reverse transcription-PCR (qRT-PCR). RNA was extracted from HeLa cells using the RNeasy Plus minikit (Qiagen, Valencia, CA) and from homogenates of mouse brain and spinal cord using TRIzol (Invitrogen). The RNA was DNase treated (Qiagen, Valencia, CA). qRT-PCR was performed using a Superscript III Platinum two-step qRT-PCR kit with SYBR green (Invitrogen). cDNA was prepared from TMEV RNA and IFN-β RNA according to the protocol of the qRT-PCR kit. A region of DA cDNA between nt 1485 and nt 1684 was amplified using forward primer: TACTATGGCACCTCTCCTCTTGGA and reverse primer CAGCCGCAAGAACTTTATCCGTTG. A region of GDVII cDNA between nt 1946 and nt 2171 was amplified using forward primer GCCTTCAGACCCATTTACCA and reverse primer TGCATGTTGAGTCCAAGAGC. Serial dilutions of pDAFL3 and pGDVIIFL2 plasmids were used to extrapolate the amount of virus genome present in the spinal cords and brains of infected mice. The human IFN-β gene was amplified from HeLa cell cDNA using forward primer CGACACTGTTCGTGTTGTCA and reverse primer GAAGCACAACAGGAGAGCAA; this primer pair and those that follow did not amplify genomic DNA. The murine IFN-β gene was amplified using forward primer ATGAACAACAGGTGGATCCTCC and reverse primer AGGAGCTCCTGACATTTCCGAA. The β-actin gene was used as a housekeeping gene for normalization and for determination of the quality of the total mRNA. A region between nt 294 and nt 1131 of the human β-actin gene was amplified using forward primer ATCTGGCACCACACCTTCTACAATGAGCTGCG and reverse primer CGTCATACTCCTGCTTGCTGATCCACATCTGC. A region between nt 182 and nt 721 of the murine β-actin gene was amplified using forward primer GTGGGCCGCTCTAGGCACCAA and the reverse primer CTCTTTGATGTCACGCACGATTTC. qRT-PCR was conducted on iCycler or MyIQ real-time detection systems (Bio-Rad) using a hot start (95°C for 10 min), 40 amplification cycles (95°C for 15 s, 60°C for 1 min), and a melt curve analysis. The ΔΔCT method of relative quantitation was used to calculate fold change of IFN-β, with β-actin serving as the endogenous control for normalization.
Immunofluorescence studies of IRF-3 nuclear entry.
L-929 cells on coverslips were infected at an MOI of 10, harvested at various times, fixed with 4% paraformaldehyde for 10 min at room temperature (RT), and then permeabilized with 100% ice-cold methanol for 30 min at 4°C. The coverslips were then incubated overnight at 4°C with anti-rabbit IRF-3 antibody (Invitrogen) to localize IRF-3 and anti-VP1 mouse monoclonal antibody (also called GDVII mAb2), which reacts against VP1 capsid proteins of all TMEV strains (34). The cells were then washed 3 times for 5 min, incubated with Alexa Fluor 488-conjugated goat anti-rabbit IgG (Invitrogen) or Cy5-conjugated goat anti-mouse IgG (Chemicon, Bullerica, MA) in phosphate-buffered saline (PBS)-5% bovine serum albumin (BSA) for 1 h at RT. The cells were washed 3 times for 5 min and then incubated for 15 min with POPRO-3 iodide (Invitrogen) for nuclear staining. Coverslips were washed for 5 min and mounted in antifade mounting solution (Fisher, Pittsburgh, PA) prior to imaging. Images were captured using an SP2 A OBS laser scanning confocal microscope (Leica, Bannockburn, IL). ImageJ (National Institutes of Health) and Adobe Photoshop 7.0 (Adobe Systems, Inc.) software was used to process the images, add pseudocolors, and merge the images.
Western blots.
For detection of phosphorylation of IRF-3 at Ser396, cell lysates were subjected to electrophoresis on 10% SDS-polyacrylamide gels and transferred to polyvinylidene difluoride (PVDF) membranes (Whatman, Stanford, ME). Membranes were first blocked overnight at 4°C in 5% (wt/vol) skim milk in Tris-buffered saline (TBS)-0.1% Tween 20, followed by three washings with TBS-0.1% Tween 20, and then incubated for 1 h at RT with rabbit anti-phospho-IRF-3(Ser396) antibody (Cell Signaling Technology, Danvers, MA) in TBS-0.1% Tween 20 with 5% BSA. Membranes were then washed three times, followed by 1 h of incubation at RT with horseradish peroxidase-conjugated anti-rabbit IgG antibody (Cell Signaling Technology) in 5% (wt/vol) skim milk in TBS-0.1% Tween 20.
For detection of monomers and dimers of IRF-3 as well as phosphorylation of IRF-3 at Ser386, lysates were diluted with native sample buffer (125 mM Tris-HCl [pH 6.8], 30% glycerol, 0.01% bromophenol blue) and then separated on Ready Gels J (7.5%; Bio-Rad, Hercules, CA). After electrophoresis and transfer, the membranes were processed as described above except that rabbit anti-IRF-3 antibody (FL-425 clone; Santa Cruz, CA) or rabbit anti-phospho-IRF-3(Ser386) antibody (IBL Biosciences, Gunma, Japan) was used as the primary antibody.
Promoter reporter assays.
HeLa cells were transfected with various combinations of a firefly luciferase reporter plasmid (p4xPRDI/III-Luc or pISG56-Luc) along with the control Renilla luciferase plasmid pRL-TK1 using Lipofectamine 2000 (Invitrogen). In some cases, cells were also transfected with pTBK-1, pIKKɛ, or pIRF-3(5D). In some experiments, pDAL or pGDVIIL was also transfected, and the transfected cells were harvested 16 h later. In other experiments, cells were infected 16 h after transfection with various viruses and then harvested 9 h after infection. Luciferase activities of cell lysates were determined using the Dual-Glo luciferase assay system (Promega, Madison, WI) with the firefly luciferase activity normalized to Renilla luciferase activity in order to control for transfection efficiency. The RLU, which represents the firefly luciferase activity relative to Renilla luciferase activity, was calculated (55). Each experiment was carried out in triplicate.
Animal studies.
Weanling (3- to 4-week-old) SJL/J mice (Jackson Laboratory, Bar Harbor, ME) were inoculated intracerebrally with 0.03 ml (2 × 106 PFU) of DA or GDVII wt or ΔL viruses. Mice were sacrificed at 1 and 5 days postinfection (dpi) or 6 weeks pi. CNS tissue was harvested and processed for quantitation of the amounts of IFN-β mRNA and virus genome.
RESULTS
TMEV L interferes with IFN-β gene transcription.
We measured IFN-β mRNA levels in HeLa cells by quantitative reverse transcription-PCR (qRT-PCR) following infection (MOI, 10) with DA, GDVII, DAΔL, DALZn (7), GDVIIΔL, EMCV, and a control virus, SV (Fig. 1B). There were low levels of IFN-β mRNA 9 h after infection with DA, GDVII, and EMCV, compared to levels seen following infection with TMEV L mutant viruses and SV (P < 0.001). Similar results were obtained at time points prior to 9 h; cell death became prominent subsequent to this time. As shown, there was no evidence of VP1 in a Western blot of GDVIIΔL-infected HeLa cells. This failure in growth of GDVIIΔL in IFN-competent cells was noted before (5) and at that time attributed to a virus assembly defect. The absence of VP1 staining in this Western blot (and subsequent ones that follow) of EMCV-infected cells is due to the lack of immunoreactivity of the anti-TMEV VP1 monoclonal antibody for EMCV VP1. These results suggest that cardiovirus L interferes with IFN-β gene transcription, as previously published (23, 53).
We next examined production of IFN-stimulated gene 56 (ISG56), which is induced by IFN-β or directly activated by IRF-3 (22), by using a pISG56-Luc construct, which has a firefly luciferase reporter gene driven by the ISG56 promoter (17) (Fig. 1C). After cotransfection with pISG56-Luc and pRL-TK1, cells were infected with cardioviruses that were wt or had L mutations. The results showed that infection with either DA or GDVIII viruses, like EMCV, failed to activate the ISG56 promoter, with no significant difference compared to mock-infected cells, while TMEV L mutant viruses had a luciferase activation that was similar to that seen with SV and significantly higher than that seen with the respective wt parental strains (P < 0.001) (Fig. 1C). Since the ISG56 promoter can be directly activated by IRF-3 (22), these results suggest that L proteins of both TMEV strains either block IRF-3 activation or restrict its ability to stimulate IFN-β gene transcription.
We next investigated whether there was evidence that L interferes with IFN-β gene transcription following TMEV infection of mice. IFN-β mRNA and viral RNA levels were measured in total RNA extracted from brains by qRT-PCR following infection with DA, GDVII, DAΔL, and GDVIIΔL viruses. IFN-β mRNA levels in the brains of mice 1 day after infection with DAΔL and GDVIIΔL viruses were significantly higher than those following infection with wt DA and GDVII viruses (Fig. 1D), supporting a role for TMEV L in blocking IFN-β gene transcription. Viral RNA levels were inversely correlated with IFN-β mRNA abundance and significantly lower with DAΔL and GDVIIΔL viruses than with wt DA and GDVII viruses (Fig. 1E), presumably because the high-level IFN response restricts replication of the L mutants. At 5 dpi, high levels of viral RNA continued in the brains of mice infected with wt DA virus (Fig. 1G), with continuing low levels of IFN-β mRNA (Fig. 1F; note the different scales in panels D and F); previous studies have shown that a high level of virus is maintained for 5 dpi (43). In contrast the DAΔL virus had been cleared from the brain. The mice infected with GDVII virus succumbed before 5 dpi, consistent with acute lytic neuronal disease caused by this virus (19).
There is a block in IRF-3 nuclear entry following infection with wt DA virus and EMCV, but not wt GDVII or TMEV L mutant viruses.
The above studies demonstrate a block in IFN-β gene transcription following cardiovirus infection and suggest that this occurs at the level of IRF-3. Cardiovirus infections are sensed by most cell types via the cytosolic pathogen recognition receptor MDA-5 (21, 27), leading to IRF-3 phosphorylation, dimerization, and subsequent nuclear entry, culminating in IFN-β gene transcription. We initially examined this pathway by determining whether IRF-3 enters the nucleus following infection with wt or L mutant virus.
The localization of IRF-3 after infections with various viruses is shown in Fig. 2. IRF-3 was localized to the cytoplasm in mock-infected cells but was present in the nucleus by 9 h after SV infection. In contrast, there was no nuclear translocation of IRF-3 after DA virus infection; however, infection with DAΔL virus led to IRF-3 nuclear entry. The L zinc finger was necessary for blocking IRF-3 nuclear entry since DALZn virus infection also led to IRF-3 nuclear entry. Surprisingly, and in sharp contrast to results for DA, substantial nuclear translocation of IRF-3 occurred after GDVII virus infection, similar to that seen following SV infection, demonstrating that GDVII infection does not prevent IRF-3 nuclear entry. IRF-3 also entered the nucleus after GDVIIΔL virus infection. EMCV infection led to some translocation of IRF-3 to the nucleus, as previously reported for mengovirus (23), but not as much as seen with GDVII virus infection.
FIG. 2.

IRF-3 enters the nucleus after infection with wt GDVII, GDVIIΔL, DALZn, DAΔL, or SV, but not wt DA. Following infection with the indicated virus or mock infection, cells were immunostained with anti-IRF-3 antibody for IRF-3 (green) and anti-TMEV VP1 antibody for virus capsid (blue) and POPRO-3 was used for nuclear staining (red). The panels in the bottom row show merged images.
IRF-3 phosphorylation and dimerization are blocked following infection with wt DA virus and EMCV, but not wt GDVII or TMEV L mutant viruses.
The above studies demonstrate that infection with wt DA virus, like EMCV, does not induce IRF-3 nuclear translocation and that this is due specifically to the L protein and dependent upon the Zn finger of L. In contrast, GDVII infection leads to IRF-3 nuclear entry but fails to stimulate either the IFN-β gene or ISG56 promoter. This suggests that GDVII L blocks IFN-β gene transcription downstream of IRF-3 phosphorylation, which leads typically to its nuclear translocation. In contrast, the DA virus appears to block the activation of IRF-3. To confirm this difference in the two viruses, we examined whether infection with the wt or L mutant viruses induces IRF-3 phosphorylation of IRF-3 at Ser396 and S386, amino acids that are critical targets for activation of IRF-3 following infection (48) and dimerization.
As expected, SV infection induced IRF-3 phosphorylation at S386 and S396 and its dimerization (Fig. 3A to H). In contrast, DA virus infection failed to induce IRF-3 phosphorylation of IRF-3 at S386 or S396 and dimerization (Fig. 3A to C), while infection with the DALZn and DAΔL mutants induced both IRF-3 phosphorylation and dimerization (Fig. 3A to C). Since the wt DA virus replicates robustly in these HeLa cells, as indicated by VP1 protein expression (Fig. 3C), these results suggest that the DA L protein interferes with the IFN-β induction pathway upstream of IRF-3 phosphorylation. In contrast, robust IRF-3 S386 and S396 phosphorylation and dimerization occurred as early as 6 h after GDVII virus infection (Fig. 3D to F). Phosphorylation and dimerization of IRF-3 were also induced by GDVIIΔL virus infection (Fig. 3D to F), although the amount of phosphorylated and dimerized IRF-3 was less than that seen with wt GDVII, possibly because of different virus replication kinetics. Interestingly, there was evidence of an additional less-well-defined band above the main phosphorylated band in the case of infection with GDVII (Fig. 3F) and DAΔL virus (Fig. 3A), presumably corresponding to IRF-3 hyperphosphorylation, as previously described (48). Similar to DA virus, EMCV failed to induce IRF-3 phosphorylation or dimerization (Fig. 3D to F); however, the block did not seem as complete as that seen following DA virus infection.
FIG. 3.

DA and GDVII L proteins target different sites in the IFN-β pathway. Western blots of cell lysates harvested 6 and 9 h pi and immunostained with anti-IRF-3 antibody (A, D, and G), anti-phospho-IRF-3(Ser386) antibody (B and E), and anti-phospho-IRF-3(Ser396) antibody (C, F, and H) are shown. The same lysates were probed with anti-TMEV VP1 antibody and anti-β-actin antibody. IRF-3 was dimerized and phosphorylated after infection with DAΔL, wt GDVII, GDVIIΔL, DA/LGDVII, and SV, but not wt DA. The arrowheads show the locations of dimers, while the asterisks show monomers. In panel A, less protein was loaded in the SV lane than in other lanes. Note that DA/LGDVII and DA/LEMCV viruses (G) were not probed with anti-phospho-IRF-3(Ser386) antibody. M corresponds to uninfected cells.
These findings suggested that DA L disrupts the IFN-β induction pathway at a different step than GDVII L. We confirmed this finding by constructing mutant DA viruses in which the DA L coding region was replaced by that of GDVII L or EMCV L to make DA/LGDVII virus and DA/LEMCV virus, respectively. DA/LGDVII and DA/LEMCV viruses, like wild-type DA virus, grew to >107 PFU/ml in HeLa cells at 9 h postinfection; however, there was a lag in DA/LEMCV virus growth at 7 h compared to that of wild-type DA virus (data not shown). Infection with DA/LGDVII virus, but not infection with wt DA virus, led to IRF-3 phosphorylation at S396 and dimerization (Fig. 3G and H). Thus, the GDVII L protein imposes a block at a different point within the IFN-β pathway than DA L, even when placed in the backbone of the DA genome: DA L interferes with IRF-3 phosphorylation and dimerization, while GDVII L does not. These results also demonstrate that the difference in the site at which the pathway is disrupted is due entirely to L and not an interaction of L with another coding region. The results for DA/LEMCV virus infection were more difficult to interpret, since infection led to a greater amount of IRF-3 phosphorylation at S386 and dimerization than that seen following wt DA virus infection, but less than that observed with DA/LGDVII virus (Fig. 3G and H). This may reflect the slowed growth of this virus as well as an impairment in processing at the L-P1 junction (since there is a change in the −4 position at this junction), as demonstrated by a decreased abundance of VP1 in DA/LEMCV chimeric virus-infected cells (Fig. 3H, VP1).
DA L blocks IFN-β gene transcription downstream of MAVS but upstream of IRF-3 phosphorylation.
As shown above, IRF-3 is not phosphorylated or dimerized and does not enter the nucleus following DA virus infection. We continued to refine the site of the block by DA L by examining the effect of overexpression of the IRF-3-activating kinases, TBK-1 and IKKɛ. DA and GDVII virus (but not the related ΔL viruses) blocked activation of the IFN-β gene promoter caused by TBK-1 (Fig. 4A) and IKKɛ (Fig. 4B) overexpression, suggesting that both the DA and GDVII L proteins block the activation of the IFN-β gene promoter downstream of TBK-1 and IKKɛ. These results are consistent with the data presented in Fig. 3C to F that demonstrate that GDVIII L does not inhibit IRF-3 phosphorylation or nuclear entry. In contrast, since DA L blocks IRF-3 activation (Fig. 3A to C), it appears to impose a block at the level of IRF-3 phosphorylation by TBK1 and IKKɛ.
FIG. 4.
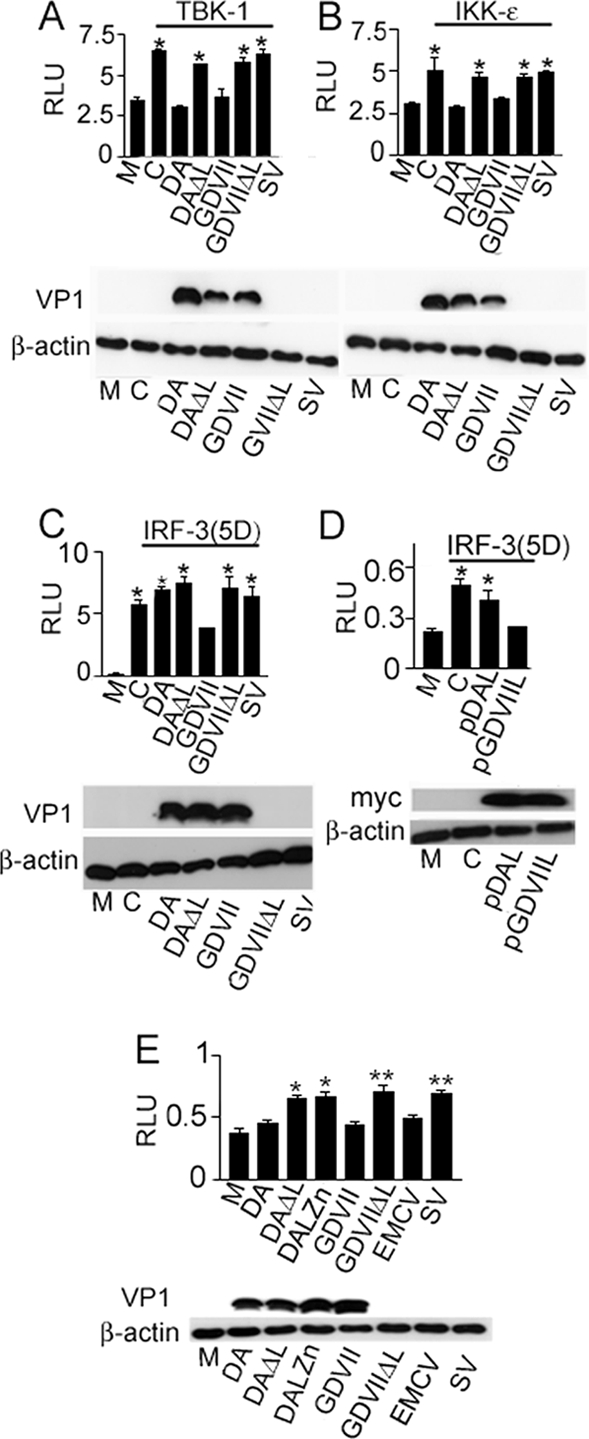
DA and GDVII L proteins target different sites in the IRF-3-dependent IFN-β pathway. IFN-β-dependent luciferase activity in cells infected with wt TMEV or ΔL viruses following transfection with pIFN-β-Luc, pRL-TK1, and either pTBK-1 (A) or pIKKɛ (B) is shown. Following transfection, the cells were infected with various viruses. Cells infected with DA and GDVII virus showed a significantly lower RLU than cells infected with TMEV ΔL viruses or overexpressing TBK-1 or IKKɛ alone (*, P < 0.001). M corresponds to cells that were transfected with the reporter plasmids (pIFN-β-Luc and pRL-TK1) but were not infected or transfected with expression constructs. C corresponds to cells that were transfected with the reporter plasmids and either pTBK-1 (A) or pIKKɛ (B) but were not infected. Bars (all panels) represent means ± SE. (C and D) IFN-β-dependent luciferase activity in cells transfected with an activated form of IRF-3, IRF-3(5D), along with pIFN-β-Luc and pRL-TK1 and then infected with wt TMEV or ΔL viruses (C) or transfected with DA or GDVII L (D). Cells infected with GDVII virus (C) showed a significantly lower RLU than cells infected with DA or TMEV ΔL viruses or cells overexpressing IRF-3(5D) alone (*, P < 0.001). Cells transfected with GDVII L (D) showed a significantly lower RLU than cells transfected with DA L or cells overexpressing IRF-3(5D) alone (*, P < 0.001). M corresponds to cells that were transfected with the reporter plasmids but were not infected or transfected with IRF-3(5D). C corresponds to cells that were transfected with the reporter plasmids and IRF-3(5D) but not infected. (E) Luciferase-dependent binding of IRF-3 to the PRDI/III elements of the IFN-β promoter following transfection with p4XIRF-3-Luc and pRL-TK1 and then infection with various viruses. Significant differences in RLU for DA versus DAΔL infection (**, P < 0.01) and for GDVII versus GDVIIΔL and SV infection (*, P < 0.001) were seen. M corresponds to cells that were transfected with the reporter plasmids but were not infected or transfected with IRF-3(5D). C corresponds to cells that were transfected with the reporter plasmids and p4XIRF-3-Luc. Error bars are not visible in the case of GDVII (C) and pGDVIIL (D) because the standard errors differ so minimally from the mean.
GDVII L blocks IFN-β gene transcription at a site between IRF-3 nuclear entry and IRF-3 binding to the PRDIII/I element of the IFN-β gene promoter.
To further investigate the site of the block in the IFN-β pathway by GDVII L, we determined the effect of virus infection on IFN-β gene promoter activation by an ectopically expressed IRF-3 phosphomimetic mutant, IRF-3(5D) (30). We infected cells with DA or GDVII virus 16 h after transfection with pIRF-3(5D) and the IFN-β-Luc reporter construct. Overexpression of IRF-3(5D) led to activation of the IRF-3-dependent IFN-β construct in uninfected cells, as previously described (29) (Fig. 4C). This activation was inhibited in cells infected with GDVII virus (P < 0.001) (Fig. 4C); the inhibition was a result of GDVII L since no inhibition was seen following infection with GDVIIΔL virus (Fig. 4C). DA (and DAΔL) virus failed to inhibit the activation by IRF-3(5D) (Fig. 4C), presumably because the disruption in the IFN-β pathway caused by DA L is at the level of the kinases responsible for IRF-3 phosphorylation. Consistent with these results, overexpression of GDVII L inhibited IFN-β activation by IRF-3(5D) (P < 0.001), while no inhibition was seen with DA L (Fig. 4D). Thus, while DA L interferes with IFN-β gene transcription upstream of IRF-3 activation, GDVII L interferes with IFN-β gene transcription within the nucleus, downstream of IRF-3 activation.
Since interference with the activation of any of the coactivators of the IFN-β enhanceosome can abrogate IFN-β gene transcription (38), we determined whether GDVII L specifically interferes with IRF-3-dependent IFN-β gene transcription (rather than a non-IRF-3 pathway required for formation of the IFN-β gene enhanceosome) by transfecting cells with p4xPRDI/III-Luc (14), in which the luciferase reporter is driven by multiple copies of the PRDIII and PRDI IRF-3-binding sites. As expected, p4xPRDI/III-Luc was activated after infection with TMEV L mutant viruses and SV, but not DA virus (or EMCV). GDVII virus infection also failed to activate the p4xPRDI/III-Luc reporter (Fig. 4E), indicating that the GDVII L protein inhibits the ability of activated IRF-3 to stimulate the IFN-β gene promoter. These data confirm that GDVII (and DA and EMCV) L interferes with the IRF-3 pathway that leads to activation of IFN-β.
DA virus-infected mice have low but measurable levels of IFN-β mRNA in the CNS during persistent infection.
We questioned whether a particular level of IFN-β might be important during persistent infection and play a role in the restriction in DA virus protein expression. To assess this possibility, we measured IFN-β mRNA and viral RNA levels in CNS tissue homogenates collected from mice 6 weeks after inoculation, when persistent infection with DA virus was well established. Although the level of IFN-β expression was presumably too low to clear virus, IFN-β mRNA levels were significantly elevated compared to those in mock-infected animals (P < 0.001) (Fig. 5A). The high levels of DA virus genome present in the spinal cord at this time (Fig. 5B) are known to be accompanied by low levels of infectious virus and small amounts of viral antigen (i.e., the expression of viral proteins is restricted) (reviewed in reference 45).
FIG. 5.
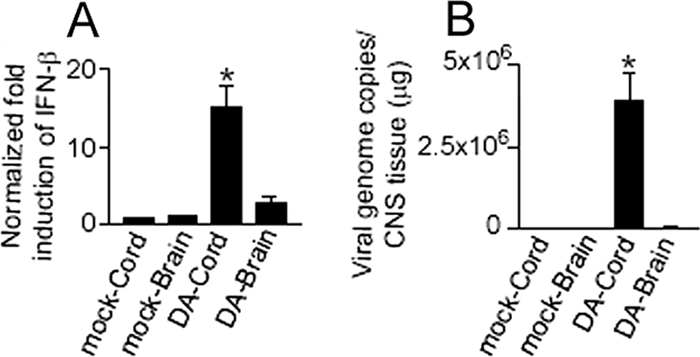
There are low levels of IFN-β mRNA in the spinal cords of DA virus-infected mice that are significantly elevated compared to controls. Levels of IFN-β mRNA (A) and virus genome (B) in spinal cord and brain 6 weeks pi. Low levels of IFN-β mRNA, which are significantly elevated compared to controls, are present in the spinal cord 6 weeks following infection with wt DA virus (*, P < 0.001). The data are from 4 mice in each group. Mock corresponds to uninfected mice. Bars (both panels) represent means ± SE.
DISCUSSION
The DA strain and other members of the TO subgroup of TMEV cause a late inflammatory demyelinating disease in which the immune system contributes to the pathology; virus persists in the CNS for the life of the mouse, with restricted expression of virus proteins (6). In contrast, the GDVII strain and other members of the GDVII subgroup of TMEV cause an acute neuronal infection and neither persist nor cause demyelination.
Attention has recently focused on the cardiovirus L protein in TMEV-IDD pathogenesis because of its multiple important functions, including inhibition of the type I IFN response. Initial studies showed that DAΔL and GDVIIΔL viruses replicate well in BHK-21 cells but had significantly decreased growth in L-929 cells and in mice (5, 28). Subsequent studies demonstrated that DA and GDVII L proteins interfered with transcription of the IFN-β gene (53), providing an explanation for the cell-type-specific differences in growth (since L-929 cells are type I IFN competent, while BHK-21 cells have a known defect in IFN production [10]). L proteins of DA, EMCV, and mengovirus were subsequently found to interfere with nucleocytoplasmic transport, including IRF-3 entry into the nucleus (13, 40). Additional investigations showed that L of the BeAn strain, a TMEV TO subgroup strain that causes persistent infection and demyelinating disease like that seen with DA, is proapoptotic in murine macrophage cell lines and BHK-21 cells (15). DA L expression also leads to oligodendrocyte death in the CNS of transgenic mice (G. Ghadge, G. Baida, and R. P. Roos, unpublished data). On the other hand, infection of BHK-21 cells with wt mengovirus versus mengovirus with L mutations demonstrated that mengovirus L was antiapoptotic (44). Recent investigations probing the mechanism by which the L protein blocks IFN-β expression have found that mengovirus virus infection leads to IRF-3 phosphorylation, but not IRF-3 dimerization, while infection with mengovirus with a deletion of L or mutation in the L protein zinc finger leads to both IRF-3 phosphorylation and dimerization (23) and that IRF-3 fails to dimerize following DA virus infection of L-929 cells while IRF-3 dimerizes following infection with DA virus that has a Zn finger mutation or deletion of L (42). IRF-3 phosphorylation following DA infection has not been investigated.
Although both DA L and GDVII L interfere with IFN-β gene transcription, we have shown that they target different points in the IFN-β induction pathway. The DA L-mediated block in the IFN-β pathway is at the level of phosphorylation of IRF-3 by TBK-1 and IKKɛ. This process, which involves a complex with TRAF3, is an attractive point at which to block signaling and is targeted by a number of viral proteins (see, e.g., reference 49). In contrast to these findings related to DA L, our work shows that the block imposed in the IFN-β pathway by GDVII L is downstream of IRF-3 nuclear entry and upstream of IRF-3 binding to PRDIII/I elements of the IFN-β gene promoter. This could occur by several different mechanisms. L could act directly by binding and sequestering activated IRF-3 or CBP/p300, as reported for herpes simplex virus ICP0 (32). Alternatively, L could induce ubiquitination and proteasomal degradation of IRF-3 without interfering with its activation, as reported for bovine viral diarrhea virus Npro (9); this possibility seems unlikely, however, as IRF-3 expression levels were not reduced by DA virus infection (Fig. 3). Finally, L could bind directly to PRDIII/I sites in the promoter, thereby interfering with binding of IRF-3 and assembly of the enhanceosome complex.
The different mechanisms by which DA and GDVII L proteins block the IFN-β pathway were confirmed in experiments with a chimeric DA virus in which DA L was replaced by GDVII L (Fig. 3G and H). GDVII L and DA L, two proteins 76 amino acids in length, differ by 11 amino acids, with 6 of these changes in the Ser-Thr domain (Fig. 1A). It will be of interest to clarify which of these amino acids are responsible for differences in disrupting IFN-β gene transcription and to determine whether these differences are associated with other L protein activities. It may be that the differences in sites that are targeted by DA versus GDVII L are important in the subgroup-specific disease phenotypes. Studies of intertypic recombinant DA/GDVII viruses suggested that GDVII L can replace DA L with no change in the persistence of virus (39); however, it is possible that the disease phenotype (DA-induced demyelination versus GDVII neurovirulence) varies depending on the presence of DA or GDVII L.
Like DA virus-infected cells, EMCV-infected cells exhibit a block in IRF-3 dimerization and S396 and S386 phosphorylation (Fig. 3D to F). These findings contrast with those reported by Hato et al., who described IRF-3 phosphorylation following mengovirus infection (23). We suspect that the reason underlying this difference is our use of antibodies specific for IRF-3 phosphorylation at S386 and S396 rather than an anti-IRF-3 antibody that does not specifically recognize a phosphorylation site, as used in the previous investigation. Although we did not specifically test EMCV L mutants, we presume that EMCV L is responsible for the block in IFN-β gene transcription, as previously reported for mengovirus (23). Although the disruption of IRF-3 signaling following EMCV infection appears to be similar to that following DA infection, the results we obtained with the chimeric DA/LEMCV virus suggest there may be differences in the activities of EMCV and DA L proteins. Further studies are in progress to clarify this point, as well as to investigate the activity of the L protein of Saffold virus. It is of interest that the mechanism by which the L proteins block IFN-β varies among different cardioviruses, despite close homology in the sequences of these proteins (Fig. 1A). This variation demonstrates how viral proteins with similar structures may evolve distinct functions.
A number of published studies have suggested a role for IFN-β in TMEV-IDD. TMEV-infected dendritic cells, microglia, and astrocytes of SJL/J mice, which are highly susceptible to the late demyelinating disease, produce more type I IFN than those of resistant mice (24, 26). Investigations of DA virus-infected mice showed that pathogenic chemokine expression in the CNS of demyelinated mice correlated with the innate immune response in the CNS rather than the presence of T cells (41). The effect of (parenteral) IFN-β on TMEV-IDD has been assessed in two previous studies (35, 37). Early IFN-β treatment around the time of the initiation of infection ameliorated the late demyelinating disease (37). Later treatment during the demyelinating stage had more complicated effects: a short-term treatment promoted remyelination, while a longer treatment enhanced demyelination (35). One important issue related to the above investigations is that the parenteral route may affect IFN-β levels systemically and in CNS endothelial cells but have little or no effect on levels in neural cells. It is likely that that a critical balance of IFN-β and proinflammatory cytokines is important in TMEV-IDD, as recently proposed for other autoimmune disorders (31). This critical balance may depend on the IFN-β concentration (reviewed in reference 4), the timing of its expression (24, 33) (especially in the case of DA infection, where IFN-β may be protective early but pathogenic late), and the duration of the expression (47).
There has been increasing attention directed to the role of IFN-β, a mainstay of treatment (administered parenterally) of MS, and the innate immune system in the immunopathology of this disease (54). Studies have reported that some patients with relapsing-remitting MS have upregulation of type I IFN genes in peripheral blood cells (11, 50) and that MS patients with this IFN signature are unlikely to benefit from IFN-β treatment (11, 51); an IFN signature in a number of autoimmune diseases has also been described (1, 3). Recent investigations identified the IRF-8 locus (12) and other genes that have a close relationship to type I IFN (reviewed in reference 25) as being important in MS susceptibility.
In order to investigate the importance of the IFN-β response in TMEV-IDD, we measured IFN-β mRNA levels in the CNS following inoculation with DA virus. At 6 weeks pi, a time of prominent demyelination, we found a low abundance of IFN-β mRNA in the mouse CNS (Fig. 5A). Nonetheless, these mRNA levels were significantly increased over those in mock-infected mice. This low level of IFN-β mRNA may be insufficient to clear virus but possibly sufficient to induce chemokines, cytokines, and an acquired immune response that contribute to autoimmune inflammation during the demyelinating disease. The IFN-β response in mice persistently infected with DA virus could also inhibit viral translation, leading to the restriction in virus protein expression that is characteristically seen in the CNS of mice with demyelinating disease. The restricted expression of viral proteins may allow evasion of the host's immune system, thereby fostering virus persistence (which is necessary for demyelination). In addition, the type I IFN response, along with the inhibition by L of host cell mRNA exit from the nucleus (42), could restrict expression of host proteins in oligodendrocytes, the cells that make myelin and sites in which the viral genome persists; this restriction could lead to demyelination since oligodendrocytes are very susceptible to defects in translation (52).
Acknowledgments
This work was supported by grants from the National Institutes of Health (R.P.R.) (1RO1 NS37958-07), the National Multiple Sclerosis Society (R.P.R.), and the Multiple Sclerosis Foundation (S.S.).
Footnotes
Published ahead of print on 7 July 2010.
REFERENCES
- 1.Baechler, E. C., J. W. Bauer, C. A. Slattery, W. A. Ortmann, K. J. Espe, J. Novitzke, S. R. Ytterberg, P. K. Gregersen, T. W. Behrens, and A. M. Reed. 2007. An interferon signature in the peripheral blood of dermatomyositis patients is associated with disease activity. Mol. Med. 13:59-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barral, P. M., J. M. Morrison, J. Drahos, P. Gupta, D. Sarkar, P. B. Fisher, and V. R. Racaniello. 2007. MDA-5 is cleaved in poliovirus-infected cells. J. Virol. 81:3677-3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bennett, L., A. K. Palucka, E. Arce, V. Cantrell, J. Borvak, J. Banchereau, and V. Pascual. 2003. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med. 197:711-723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Biron, C. A. 2001. Interferons alpha and beta as immune regulators—a new look. Immunity 14:661-664. [DOI] [PubMed] [Google Scholar]
- 5.Calenoff, M. A., C. S. Badshah, M. C. Dal Canto, H. L. Lipton, and M. K. Rundell. 1995. The leader polypeptide of Theiler's virus is essential for neurovirulence but not for virus growth in BHK cells. J. Virol. 69:5544-5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cash, E., M. Chamorro, and M. Brahic. 1985. Theiler's virus RNA and protein synthesis in the central nervous system of demyelinating mice. Virology 144:290-294. [DOI] [PubMed] [Google Scholar]
- 7.Chen, H. H., W. P. Kong, and R. P. Roos. 1995. The leader peptide of Theiler's murine encephalomyelitis virus is a zinc-binding protein. J. Virol. 69:8076-8078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen, H. H., W. P. Kong, L. Zhang, P. L. Ward, and R. P. Roos. 1995. A picornaviral protein synthesized out of frame with the polyprotein plays a key role in a virus-induced immune-mediated demyelinating disease. Nat. Med. 1:927-931. [DOI] [PubMed] [Google Scholar]
- 9.Chen, Z., R. Rijnbrand, R. K. Jangra, S. G. Devaraj, L. Qu, Y. Ma, S. M. Lemon, and K. Li. 2007. Ubiquitination and proteasomal degradation of interferon regulatory factor-3 induced by Npro from a cytopathic bovine viral diarrhea virus. Virology 366:277-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chinsangaram, J., M. E. Piccone, and M. J. Grubman. 1999. Ability of foot-and-mouth disease virus to form plaques in cell culture is associated with suppression of alpha/beta interferon. J. Virol. 73:9891-9898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Comabella, M., J. D. Lunemann, J. Rio, A. Sanchez, C. Lopez, E. Julia, M. Fernandez, L. Nonell, M. Camina-Tato, F. Deisenhammer, E. Caballero, M. T. Tortola, M. Prinz, X. Montalban, and R. Martin. 2009. A type I interferon signature in monocytes is associated with poor response to interferon-beta in multiple sclerosis. Brain 132:3353-3365. [DOI] [PubMed] [Google Scholar]
- 12.De Jager, P. L., X. Jia, J. Wang, P. I. de Bakker, L. Ottoboni, N. T. Aggarwal, L. Piccio, S. Raychaudhuri, D. Tran, C. Aubin, R. Briskin, S. Romano, S. E. Baranzini, J. L. McCauley, M. A. Pericak-Vance, J. L. Haines, R. A. Gibson, Y. Naeglin, B. Uitdehaag, P. M. Matthews, L. Kappos, C. Polman, W. L. McArdle, D. P. Strachan, D. Evans, A. H. Cross, M. J. Daly, A. Compston, S. J. Sawcer, H. L. Weiner, S. L. Hauser, D. A. Hafler, and J. R. Oksenberg. 2009. Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat. Genet. 41:776-782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delhaye, S., V. van Pesch, and T. Michiels. 2004. The leader protein of Theiler's virus interferes with nucleocytoplasmic trafficking of cellular proteins. J. Virol. 78:4357-4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ehrhardt, C., C. Kardinal, W. J. Wurzer, T. Wolff, C. von Eichel-Streiber, S. Pleschka, O. Planz, and S. Ludwig. 2004. Rac1 and PAK1 are upstream of IKK-epsilon and TBK-1 in the viral activation of interferon regulatory factor-3. FEBS Lett. 567:230-238. [DOI] [PubMed] [Google Scholar]
- 15.Fan, J., K. N. Son, S. Y. Arslan, Z. Liang, and H. L. Lipton. 2009. Theiler's murine encephalomyelitis virus leader protein is the only nonstructural protein tested that induces apoptosis when transfected into mammalian cells. J. Virol. 83:6546-6553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fiette, L., C. Aubert, U. Muller, S. Huang, M. Aguet, M. Brahic, and J. F. Bureau. 1995. Theiler's virus infection of 129Sv mice that lack the interferon alpha/beta or interferon gamma receptors. J. Exp. Med. 181:2069-2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fitzgerald, K. A., S. M. McWhirter, K. L. Faia, D. C. Rowe, E. Latz, D. T. Golenbock, A. J. Coyle, S. M. Liao, and T. Maniatis. 2003. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4:491-496. [DOI] [PubMed] [Google Scholar]
- 18.Reference deleted.
- 19.Fu, J., M. Rodriguez, and R. P. Roos. 1990. Strains from both Theiler's virus subgroups encode a determinant for demyelination. J. Virol. 64:6345-6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fu, J. L., S. Stein, L. Rosenstein, T. Bodwell, M. Routbort, B. L. Semler, and R. P. Roos. 1990. Neurovirulence determinants of genetically engineered Theiler viruses. Proc. Natl. Acad. Sci. U. S. A. 87:4125-4129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gitlin, L., W. Barchet, S. Gilfillan, M. Cella, B. Beutler, R. A. Flavell, M. S. Diamond, and M. Colonna. 2006. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc. Natl. Acad. Sci. U. S. A. 103:8459-8464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grandvaux, N., M. J. Servant, B. tenOever, G. C. Sen, S. Balachandran, G. N. Barber, R. Lin, and J. Hiscott. 2002. Transcriptional profiling of interferon regulatory factor 3 target genes: direct involvement in the regulation of interferon-stimulated genes. J. Virol. 76:5532-5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hato, S. V., C. Ricour, B. M. Schulte, K. H. Lanke, M. de Bruijni, J. Zoll, W. J. Melchers, T. Michiels, and F. J. van Kuppeveld. 2007. The mengovirus leader protein blocks interferon-alpha/beta gene transcription and inhibits activation of interferon regulatory factor 3. Cell. Microbiol. 9:2921-2930. [DOI] [PubMed] [Google Scholar]
- 24.Hou, W., E. Y. So, and B. S. Kim. 2007. Role of dendritic cells in differential susceptibility to viral demyelinating disease. PLoS Pathog. 3:e124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jakkula, E., V. Leppa, A. M. Sulonen, T. Varilo, S. Kallio, A. Kemppinen, S. Purcell, K. Koivisto, P. Tienari, M. L. Sumelahti, I. Elovaara, T. Pirttila, M. Reunanen, A. Aromaa, A. B. Oturai, H. B. Sondergaard, H. F. Harbo, I. L. Mero, S. B. Gabriel, D. B. Mirel, S. L. Hauser, L. Kappos, C. Polman, P. L. De Jager, D. A. Hafler, M. J. Daly, A. Palotie, J. Saarela, and L. Peltonen. 2010. Genome-wide association study in a high-risk isolate for multiple sclerosis reveals associated variants in STAT3 gene. Am. J. Hum. Genet. 86:285-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jin, Y. H., M. Mohindru, M. H. Kang, A. C. Fuller, B. Kang, D. Gallo, and B. S. Kim. 2007. Differential virus replication, cytokine production, and antigen-presenting function by microglia from susceptible and resistant mice infected with Theiler's virus. J. Virol. 81:11690-11702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kato, H., O. Takeuchi, S. Sato, M. Yoneyama, M. Yamamoto, K. Matsui, S. Uematsu, A. Jung, T. Kawai, K. J. Ishii, O. Yamaguchi, K. Otsu, T. Tsujimura, C. S. Koh, C. Reis e Sousa, Y. Matsuura, T. Fujita, and S. Akira. 2006. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441:101-105. [DOI] [PubMed] [Google Scholar]
- 28.Kong, W.-P., G. D. Ghadge, and R. P. Roos. 1994. Involvement of cardiovirus leader in host cell-restricted virus expression. Proc. Natl. Acad. Sci. U. S. A. 91:1796-1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin, R., C. Heylbroeck, P. M. Pitha, and J. Hiscott. 1998. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol. Cell. Biol. 18:2986-2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin, R., Y. Mamane, and J. Hiscott. 1999. Structural and functional analysis of interferon regulatory factor 3: localization of the transactivation and autoinhibitory domains. Mol. Cell. Biol. 19:2465-2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsuzawa, A., P. H. Tseng, S. Vallabhapurapu, J. L. Luo, W. Zhang, H. Wang, D. A. Vignali, E. Gallagher, and M. Karin. 2008. Essential cytoplasmic translocation of a cytokine receptor-assembled signaling complex. Science 321:663-668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Melroe, G. T., L. Silva, P. A. Schaffer, and D. M. Knipe. 2007. Recruitment of activated IRF-3 and CBP/p300 to herpes simplex virus ICP0 nuclear foci: potential role in blocking IFN-beta induction. Virology 360:305-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nagai, T., O. Devergne, T. F. Mueller, D. L. Perkins, J. M. van Seventer, and G. A. van Seventer. 2003. Timing of IFN-beta exposure during human dendritic cell maturation and naive Th cell stimulation has contrasting effects on Th1 subset generation: a role for IFN-beta-mediated regulation of IL-12 family cytokines and IL-18 in naive Th cell differentiation. J. Immunol. 171:5233-5243. [DOI] [PubMed] [Google Scholar]
- 34.Nitayaphan, S., M. M. Toth, and R. P. Roos. 1985. Neutralizing monoclonal antibodies to Theiler's murine encephalomyelitis viruses. J. Virol. 53:651-657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Njenga, M. K., M. J. Coenen, N. DeCuir, H. Y. Yeh, and M. Rodriguez. 2000. Short-term treatment with interferon-alpha/beta promotes remyelination, whereas long-term treatment aggravates demyelination in a murine model of multiple sclerosis. J. Neurosci. Res. 59:661-670. [DOI] [PubMed] [Google Scholar]
- 36.Ohara, Y., S. Stein, J. L. Fu, L. Stillman, L. Klaman, and R. P. Roos. 1988. Molecular cloning and sequence determination of DA strain of Theiler's murine encephalomyelitis viruses. Virology 164:245-255. [DOI] [PubMed] [Google Scholar]
- 37.Olson, J. K., and S. D. Miller. 2009. The innate immune response affects the development of the autoimmune response in Theiler's virus-induced demyelinating disease. J. Immunol. 182:5712-5722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Panne, D., S. M. McWhirter, T. Maniatis, and S. C. Harrison. 2007. Interferon regulatory factor 3 is regulated by a dual phosphorylation-dependent switch. J. Biol. Chem. 282:22816-22822. [DOI] [PubMed] [Google Scholar]
- 39.Paul, S., and T. Michiels. 2006. Cardiovirus leader proteins are functionally interchangeable and have evolved to adapt to virus replication fitness. J. Gen. Virol. 87:1237-1246. [DOI] [PubMed] [Google Scholar]
- 40.Porter, F. W., Y. A. Bochkov, A. J. Albee, C. Wiese, and A. C. Palmenberg. 2006. A picornavirus protein interacts with Ran-GTPase and disrupts nucleocytoplasmic transport. Proc. Natl. Acad. Sci. U. S. A. 103:12417-12422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ransohoff, R. M., T. Wei, K. D. Pavelko, J. C. Lee, P. D. Murray, and M. Rodriguez. 2002. Chemokine expression in the central nervous system of mice with a viral disease resembling multiple sclerosis: roles of CD4+ and CD8+ T cells and viral persistence. J. Virol. 76:2217-2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ricour, C., S. Delhaye, S. V. Hato, T. D. Olenyik, B. Michel, F. J. van Kuppeveld, K. E. Gustin, and T. Michiels. 2009. Inhibition of mRNA export and dimerization of interferon regulatory factor 3 by Theiler's virus leader protein. J. Gen. Virol. 90:177-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rodriguez, M., and R. P. Roos. 1992. Pathogenesis of early and late disease in mice infected with Theiler's virus, using intratypic recombinant GDVII/DA viruses. J. Virol. 66:217-225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Romanova, L. I., P. V. Lidsky, M. S. Kolesnikova, K. V. Fominykh, A. P. Gmyl, E. V. Sheval, S. V. Hato, F. J. van Kuppeveld, and V. I. Agol. 2009. Antiapoptotic activity of the cardiovirus leader protein, a viral “security” protein. J. Virol. 83:7273-7284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roos, R. P. 2002. Pathogenesis of Theiler's murine encephalomyelitis virus-induced disease, p. 427-435. In B. L. Semler and E. Wimmer (ed.), Molecular biology of picornaviruses. ASM Press, Washington, DC.
- 46.Roos, R. P., S. Stein, Y. Ohara, J. L. Fu, and B. L. Semler. 1989. Infectious cDNA clones of the DA strain of Theiler's murine encephalomyelitis virus. J. Virol. 63:5492-5496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sarasin-Filipowicz, M., X. Wang, M. Yan, F. H. Duong, V. Poli, D. J. Hilton, D. E. Zhang, and M. H. Heim. 2009. Alpha interferon induces long-lasting refractoriness of JAK-STAT signaling in the mouse liver through induction of USP18/UBP43. Mol. Cell. Biol. 29:4841-4851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Servant, M. J., N. Grandvaux, B. R. tenOever, D. Duguay, R. Lin, and J. Hiscott. 2003. Identification of the minimal phosphoacceptor site required for in vivo activation of interferon regulatory factor 3 in response to virus and double-stranded RNA. J. Biol. Chem. 278:9441-9447. [DOI] [PubMed] [Google Scholar]
- 49.Siu, K. L., K. H. Kok, M. H. Ng, V. K. Poon, K. Y. Yuen, B. J. Zheng, and D. Y. Jin. 2009. Severe acute respiratory syndrome coronavirus M protein inhibits type I interferon production by impeding the formation of TRAF3.TANK.TBK1/IKKepsilon complex. J. Biol. Chem. 284:16202-16209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van Baarsen, L. G., T. C. van der Pouw Kraan, J. J. Kragt, J. M. Baggen, F. Rustenburg, T. Hooper, J. F. Meilof, M. J. Fero, C. D. Dijkstra, C. H. Polman, and C. L. Verweij. 2006. A subtype of multiple sclerosis defined by an activated immune defense program. Genes Immun. 7:522-531. [DOI] [PubMed] [Google Scholar]
- 51.van Baarsen, L. G., S. Vosslamber, M. Tijssen, J. M. Baggen, L. F. van der Voort, J. Killestein, T. C. van der Pouw Kraan, C. H. Polman, and C. L. Verweij. 2008. Pharmacogenomics of interferon-beta therapy in multiple sclerosis: baseline IFN signature determines pharmacological differences between patients. PLoS One 3:e1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van der Knaap, M. S., J. C. Pronk, and G. C. Scheper. 2006. Vanishing white matter disease. Lancet Neurol. 5:413-423. [DOI] [PubMed] [Google Scholar]
- 53.van Pesch, V., O. van Eyll, and T. Michiels. 2001. The leader protein of Theiler's virus inhibits immediate-early alpha/beta interferon production. J. Virol. 75:7811-7817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weiner, H. L. 2009. The challenge of multiple sclerosis: how do we cure a chronic heterogeneous disease? Ann. Neurol. 65:239-248. [DOI] [PubMed] [Google Scholar]
- 55.Yang, Y., Y. Liang, L. Qu, Z. Chen, M. Yi, K. Li, and S. M. Lemon. 2007. Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease precursor. Proc. Natl. Acad. Sci. U. S. A. 104:7253-7258. [DOI] [PMC free article] [PubMed] [Google Scholar]