

Abstract
It is thought that the pathological cascade in Alzheimer's disease is initiated by the formation of amyloid-β (Aβ) peptide complexes on cell membranes. However, there is considerable debate about the nature of these complexes and the type of solution-phase Aβ aggregates that may contribute to their formation. Also, it is yet to be shown that Aβ attaches strongly to living cell membranes, and that this can happen at low, physiologically relevant Aβ concentrations. Here, we simultaneously measure the aggregate size and fluorescence lifetime of fluorescently labeled Aβ1-40 on and above the membrane of cultured PC12 cells at near-physiological concentrations. We find that at 350 nM Aβ concentration, large (>>10 nm average hydrodynamic radius) assemblies of codiffusing, membrane-attached Aβ molecules appear on the cell membrane together with a near-monomeric species. When the extracellular concentration is 150 nM, the membrane contains only the smaller species, but with a similar degree of attachment. At both concentrations, the extracellular solution contains only small (∼2.3 nm average hydrodynamic radius) Aβ oligomers or monomers. We conclude that at near-physiological concentrations only the small oligomeric Aβ species are relevant, they are capable of attaching to the cell membrane, and they assemble in situ to form much larger complexes.
Introduction
Abnormal aggregation of misfolded proteins and their deposition in the brain are common molecular features of many neurodegenerative diseases, including Alzheimer's and Parkinson's diseases (1,2). A hallmark of Alzheimer's disease is the extracellular aggregation of amyloid-β (Aβ) protein into insoluble β-sheet-rich amyloid fibrils (1,3). Consequently, considerable efforts have been made to understand the aggregation of this peptide in solution (4,5). However, major unresolved questions remain regarding the molecular mechanism underlying Aβ toxicity, including the nature of the interaction of the Aβ with living cell membranes, the type of Aβ aggregates that take part in this interaction, and the size of structures they form on the membrane. Also, although Aβ aggregation is a hallmark of the disease, Aβ does not appear to form any large aggregates in vitro at concentrations that are supposed to exist in vivo.
Because Aβ is an extracellular protein that is produced by proteolytic processing of the membrane-bound amyloid precursor protein (6), interaction with the membrane appears to be an obligatory initial step in the pathway to toxicity (7). A dominant hypothesis for toxicity is that Aβ molecules assemble into ion-permeable structures on the cell membrane. There is considerable evidence for the formation of ion-channel-like Aβ structures on the cell membrane (8,9). Atomic force microscopy imaging has shown ∼16 nm large channel-like structures in artificial membranes incubated with Aβ (10,11). Separate electrophysiological measurements have shown that living cell membranes increase their Ca2+ ion conductivity in the presence of Aβ aggregates, supporting the channel hypothesis (12,13). Molecular modeling results suggest that Aβ molecules can assemble into a small (<5 nm diameter) membrane-spanning, ion-conducting pore (14). On the other hand, other studies indicate that Aβ associates with the plasma membrane outer leaflet and forms nonspecific structures, leading to membrane thinning and deformation (15,16). Binding studies using surface plasmon resonance and fluorescence anisotropy suggest that Aβ binds to membrane lipids (17). However, to date there is no direct proof of the formation of any type of Aβ assemblies on living cell membranes.
Another major issue is the identity of the Aβ aggregate that gives rise to the putative membrane structures. In the aqueous phase, Aβ aggregation into the final fibrillar form occurs via several concentration-dependent steps (18,19), and many of the intermediate species are quasi-stable (20). It is now believed that one or more of the soluble intermediates (species ranging from small soluble oligomers to the larger protofibrils), rather than the mature fibrils, are key to the interaction with the cell membrane (21–24). Zn2+ reduces the population of these soluble Aβ aggregates in solution (22) and it also inhibits the Aβ induced Ca2+ currents and toxicity (9,13), suggesting a link between these soluble aggregates and membrane assemblies. However, other experiments have pointed to different types of soluble aggregates as the key bioactive species. Some studies have shown that Aβ can aggregate in solution into structures that appear to have an annular morphology similar to that of membrane ion channels (14). It is has been hypothesized that these annular structures, formed in the solution phase (25–27), somehow incorporate into the membranes and function as unregulated ion channels, although the thermodynamics of membrane insertion for a structure formed in a hydrophilic environment remains unclear. Some authors have suggested that spherical amyloid aggregates (ranging from 5 nm (27) to 20 nm (28) in diameter) are the most bioactive, and very small oligomers (near-monomeric species) are unable to interact with the membrane (29). A competing hypothesis is that very small oligomers (especially dimers) are bioactive, and a dimer-rich solution of Aβ strongly affects the long-term potentiation of synaptic connections (30,31). Other findings suggest that octamers or similar species may be the basic units that interact with the cell membrane (11). It is possible that these small oligomers initially associate with the membrane as separate entities and then assemble in situ to form larger Aβ assemblies that compromise the cell membrane permeability (11). Some studies suggest that 56 kD oligomers may be the most toxic species (32), and also there is some evidence that larger structures (“protofibrils” ranging in size from tens to hundreds of nanometers) are responsible for neuronal death (33). An alternative hypothesis states that Aβ enters the cytoplasm of the neuronal cells and interacts with some vital cellular processes, such as neurotransmitter packaging (34). Even for this hypothesis, Aβ must first interact with the membrane in a way that facilitates its passage through the membrane. At present, it is fair to say that a multitude of possibilities exist, and no measurement has directly and unambiguously linked any particular type of solution phase aggregate with the formation of the membrane assemblies.
The lack of experimental evidence is due to the demanding requirements of sensitivity, size-resolution, and biocompatibility of the candidate technique. Aβ concentrations in the cerebrospinal fluid in the brain of Alzheimer's patients are much less than 1 μM (35,36). Electrophysiological measurements have also established the disruptive effect of such low concentrations of Aβ on neuronal communication (30,37,38). However, most aggregation measurements are carried out at concentrations of many micromolar because techniques employed to measure protein aggregation, such as dynamic light scattering, thio-T binding, and size-exclusion chromatography are typically not sensitive to nanomolar-level protein concentrations and are applicable only in the solution phase. Such high concentrations may allow the formation of large aggregates that may not be relevant at lower physiological concentrations.
A few studies have probed the presence of Aβ aggregates on living cells, mainly using fluorescence-based techniques that are sensitive and biocompatible. Fluorescence imaging studies have shown that Aβ colocalizes with neurons and neuronal processes (39), but these studies did not show whether they physically attach to the membranes. A recent pioneer study of fluorescent Aβ (at 2 μM concentration) using confocal imaging and photobleaching suggested that Aβ associates with PC12 cell membranes, and a fraction of these Aβ molecules are within the Forster radii (i.e., a few nanometers) of each other (40). This raises several important questions: Do the observed Aβ patches represent true membrane aggregates? Can these aggregates form at near-physiological concentrations? How large are these aggregates? Do these aggregates represent the incorporation of similar-sized aggregates that are preformed in the solution? If not, what type of solution aggregates correlate with the formation of the membrane aggregates? Here, we explore these questions by studying the aggregation of Aβ at submicromolar concentrations on the cell membrane of living PC12 cells, as well as in the extracellular medium close to the cell membranes. PC12 cells are mammalian in origin, have been used as neuronal mimics, are easily cultured, and have been used in the past to study the effects of Aβ on living cells (40,41). Our use of these cells allows us to directly compare our results with those of previous studies.
We use confocal fluorescence imaging to locate the fluorescently labeled Aβ on the cell membrane and in the extracellular space. We then measure the diffusion constant (which is related to the size) of Aβ monomers and aggregates using fluorescence correlation spectroscopy (FCS) (42–44). This technique has been shown to be effective for studying Aβ aggregation in the solution phase at nanomolar concentrations (45) and is biocompatible (46,47). We simultaneously determine the fluorescence lifetime with time-correlated single photon counting (TCSPC) to verify the attachment of Aβ with the cell membrane. These experiments are repeated at two different concentrations to determine whether the observed characteristics are concentration-dependent.
Materials and Methods
Materials
Purified synthetic Aβ1-40 and fluorescein-labeled Aβ1-40 (labeled at the N-terminus through a lysine, Fl-Lys-Aβ1-40) was purchased from rPeptide (San Jose, CA). The labeled and unlabeled peptide was used in a ratio of 1:1 without further purification. Buffer salts were purchased from Sigma (St. Louis, MO). The peptide was made in artificial cerebrospinal fluid buffer (146 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 0.8 mM MgSO4, 0.4 mM KH2PO4, 5 mM dextrose, and 20 mM Na2HPO4, pH adjusted to 7.35). All experiments were performed at 25°C.
Cell culture
PC12 cell lines were maintained in Dulbecco's modified Eagle's medium (high glucose; ATCC, Manassas, VA) containing 10% horse serum (Sigma) and 5% fetal bovine serum (Sigma), and then placed in cell culture dishes precoated with 1 mg/mL poly-L-lysine (Sigma). Cells were maintained in an incubator at 37°C with 5% CO2. For living-cell microscopy studies, cells were seeded onto sterilized No. 1 coverslip (VWR International, Batavia, IL) and placed inside a six-well plate.
Method
Measurements were performed with a confocal microscope setup with TCSPC and FCS facility (Time Harp200; PicoQuant GmbH, Berlin, Germany). The system uses a stage scanning confocal setup on an inverted microscope (Olympus IX71, Olympus America Inc., Centre Valley, PA), with two picosecond pulsed diode lasers (467 nm excitation; PicoQuant, GmbH Berlin, Germany). The excitation power and frequencies of the lasers were optimized to avoid photobleaching of the fluorophores. The laser powers used in the experiments were 30 μW and 40 μW at 467 nm and 636 nm excitation, respectively, with a pulse frequency of 40 MHz for both lasers. The laser beams were focused in the sample volume using an apochromatic 60X water immersion objective (Olympus America Inc.) with an NA of 1.2, and the emitted fluorescence was collected using the same objective and separated from the excitation beam by a dichroic mirror. A 50 μm pinhole was used to reject the out-of-focus fluorescence. The overall fluorescence collected was separated using a dichroic cube beam splitter and filtered by a suitable emission filter before it was detected by two single photon avalanche photodiodes (SPCM-AQR; PerkinElmer, Waltham, MA). The fluorescence lifetime was measured using the TCSPC module in time-tagged time-resolved mode. Analyses of the fluorescence lifetime signals were performed with Origin 7.0 Software (Origin Labs, Northampton, MA). An in-house-developed analysis algorithm (45), termed MEMFCS (maximum entropy method-based analysis of FCS data), was used to evaluate the diffusion time distribution.
Results
Fluorescence imaging suggests that Aβ associates with cell membranes
We investigated the association of fluorescently labeled extracellular Aβ with the cell membranes using confocal imaging. We incubated separate sets of PC12 cells with a buffer containing 150 nM and 350 nM fluorescein-labeled Aβ, respectively, for 15 min and imaged the cells with a confocal microscope. Confocal images of the cell membranes show multiple bright patches (Fig. 1 a, arrows), which are in general brighter than the structures observed from cells that were not incubated with fluorescein-labeled Aβ (Fig. 1 b). Images obtained from the cells incubated with the 150 nM solution are similar to those obtained from the 350 nM solution (data not shown). Diffuse brightness is seen near the top and bottom optical sections of the cells, whereas the intermediate sections show a rim-like annular appearance (Fig. 1 b, rightmost cell). These are characteristics of a cell membrane in a confocal image. The appearance of bright patches suggests that Aβ aggregates on the membrane, but the optical resolution of confocal microscopy (∼750 nm in the longitudinal and ∼300 nm in the lateral direction) was not sufficient to establish a molecular-level attachment with the membrane, nor could it provide evidence of a molecular-level association between different Aβ monomers.
Figure 1.
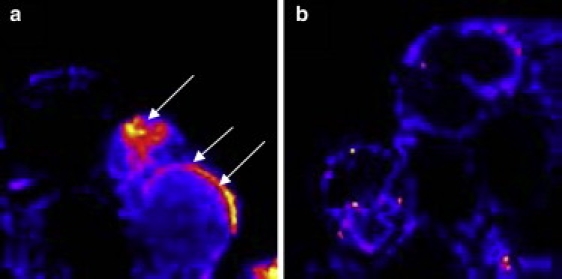
Fluorescence images of labeled Aβ on cell membranes. (a) Representative confocal fluorescence image of a PC12 cell recorded after the addition of 350 nM of fluorescein-labeled Aβ1-40. White arrows show the attachment of Aβ to the membrane. (b) Autofluorescence image from a PC12 cell before Aβ addition.
Fluorescence lifetime data show membrane attachment
Fluorescence lifetimes were measured from the bright membrane-associated patches visible in the confocal images. Lifetime data were obtained from the same confocal image data set (the photon intensity information is used to calculate the confocal image, and the photon arrival information is used to calculate the lifetime and the fluorescence correlation curves). The lifetime of 150 nM Aβ recorded in the solution ∼10 μm above the cell membrane fits well for a single component with a lifetime of 3.9 ± 0.1 ns (Fig. 2, squares). However, the data from the cell membrane (for 150 nM extracellular Aβ) fits well only when a minimum of two components are used (1.3 ± 0.4 and 4.1 ± 0.2 ns at a 1:1.9 amplitude ratio; Fig. 2, circles). The lifetime data obtained from the membrane exposed to 150 nM extracellular Aβ (Fig. 2, circles) is similar to those exposed to the 350 nM solution (see Fig. S1 in the Supporting Material). The long lifetime component is similar to that obtained from the solution. The faster decay component indicates that the fluorescence of a part of the population is quenched. Separate evidence of this quenching can be found from a photon counting histogram (PCH) analysis of the same set of data reported in Fig. S2. The PCH data binned at 10 μs show that the peak emission rate of the particles shifts from a value of 18,700 photons per second (in the extracellular medium) to 5400 photons per second (on the membrane). A decrease in the fluorescence counts and the lifetime indicates dynamic quenching in the system. Together, these data show that a fraction of the Aβ molecules are in an environment different from those in the solution.
Figure 2.
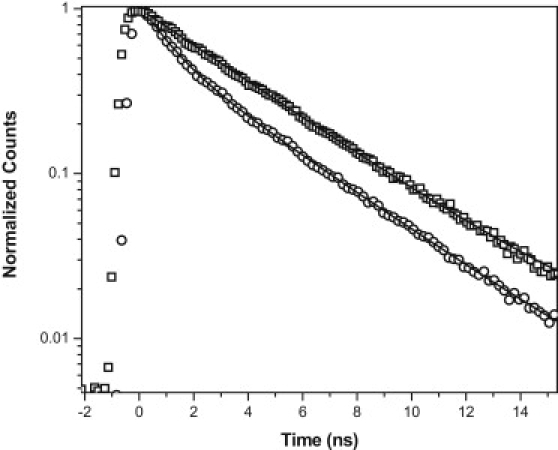
Fluorescence lifetime of Aβ on cell membranes. TCSPC data of the fluorescence lifetime of 150 nM Aβ obtained from the extracellular solution (square) and the cell membrane (circle), with their corresponding single exponential and two exponential fits, respectively (solid line).
FCS shows membrane Aβ aggregates
Next, we investigate whether these Aβ molecules associate with each other to form intermolecular complexes on the membrane. Multiple molecules codiffusing as a single particle would be a clear evidence of a molecular-level association. Because FCS analyzes the translational diffusion of a particle, it can provide information about such an association, as well as measure the diffusion constant (which is a measure of the size) of the complexes. The same data set obtained for imaging and lifetime determination also contains time-stamped data appropriate for FCS analysis. We separately analyze the FCS data obtained from the solution above the membrane (Fig. 3 a), as well as from the membrane itself (Fig. 3 c), for both the Aβ concentrations (150 nM and 350 nM). The FCS data are fitted with the MEMFCS fitting routine, which gives a model-free analysis of underlying diffusion times (45). Although distribution of diffusion constants obtained from MEMFCS can be (and usually is) wider than the actual distribution, the absence of a particular diffusion constant in this analysis is a reliable indicator of the absence of that species. The distributions obtained from the solution phases of the two different concentrations are similar, with the peak of the distribution at 126 μs in both (Fig. 3 b). The 1/e2 point of the distribution extends to 281 μs and 355 μs for the 350 nM and 150 nM solutions, respectively. The 150 nM data have somewhat larger errors, and consequently the distribution is slightly wider. However, the relative amplitudes of the smaller components are consistently higher in the low-concentration solution. The diffusion constants are calculated from the diffusion time with the help of a calibrant (in this case with free fluorescein dye in solution, which has a known diffusion constant of 4.25 × 10−6cm2s−1 and a hydrodynamic radius of 0.57 nm (48)). In our setup, fluorescein has a measured diffusion time of 32 μs under identical conditions. This calibration yields a diffusion constant of 1.1 × 10−6cm2s−1 (or a hydrodynamic radius of 2.3 nm) for the peak of the size distribution in the solution above the membrane. The analysis of the membrane data (Fig. 3 c, circles) obtained from the 150 nM Aβ solution shows a distribution with a single peak at 200 μs (D = 6.8 × 10−7cm2s−1; Fig. 3 d, circles). The diffusion times in the membrane are expected to be larger due to the viscous environment, as well as the molecule-specific interactions and crowding. To obtain an estimate of this environmental factor, we performed FCS measurements of Alexa Fluor 680-labeled wheat germ agglutinin (WGA, a membrane protein) on PC12 cell membranes and in the extracellular solution. This measurement shows that for this molecule, the ratio of the diffusion time in the aqueous solution to that in the membrane is ∼5.5 (Fig. S3). Hence, the Aβ on the cell membrane may be expected to have a typical diffusion time of ∼700 μs, which is larger than the observed value. This aspect is discussed later in more detail.
Figure 3.
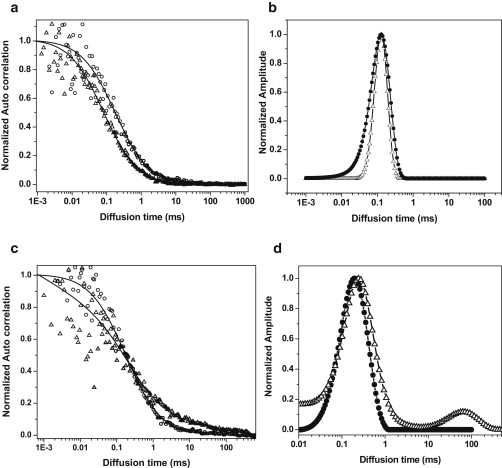
Fluorescence autocorrelation of Aβ on cell membranes. (a) Fluorescence autocorrelation traces obtained from the extracellular solution containing 150 nM Aβ (circles) and 350 nM Aβ (triangles). (b) Corresponding diffusion-time distributions obtained by fitting the data with the MEMFCS algorithm. (c) Fluorescence autocorrelation curves obtained from the cell membrane for 150 nM Aβ (circle) and 350 nM (triangles) Aβ concentrations. (d) Diffusion-time distribution obtained from fitting the data in c.
In contrast, when cells are incubated with the 350 nM Aβ solution, the size distribution in the membrane (Fig. 3 c, triangles) shows two distinct Aβ-bound complexes characterized by a typical diffusion time of 200 μs (D ≈ 6.8 × 10−7cm2s−1) and 50 ms (or larger), respectively (D ≤ 2.7 × 10−9cm2s−1, Fig. 3 d, triangles). The individual peak positions vary considerably (a representation of this variation across 10 different measurements performed on three different days is presented in Fig. S4). However, the robust feature of all of these measurements is the presence of a secondary peak at large diffusion times (representing large aggregates) in addition to the main peak at smaller diffusion times. The secondary peak clearly signifies the presence of assemblies much larger than those observed in the solution.
Discussion
The association of extracellular Aβ aggregates with the cell membrane is likely to be a key step in the pathology of Alzheimer's disease. Fig. 1 shows that fluorescein-labeled Aβ preferentially localizes on the cell membrane of the PC12 cells. Such an association has been observed before, but only at much higher concentrations of extracellular Aβ (40). Our data show that this association takes place even at 150 nM and 350 nM Aβ concentrations, which is far below the solubility (i.e., precipitation) limit of Aβ in aqueous solutions (19). The FCS data obtained from the extracellular solution show that Aβ aggregates into structures with a typical radius of 2.3 nm, and particles much larger than this are absent (the 1/e2 point of the peak of the hydrodynamic radius distribution is at 6.3 nm) at these concentrations. We note that a fluorescent label is required for our single-molecule-level experiments performed with a cellular background, and all the conclusions drawn are strictly valid only for this labeled molecule.
Given the relatively poor resolution of the confocal microscope (∼250 nm in the radial and 700 nm in the axial direction) this membrane localization may not imply a true “attachment” of Aβ with the cell membrane. The fluorescence lifetime data help us clarify this point further. The fluorescence lifetimes of most fluorophores are sensitive to the chemical nature of the environment. In our experiments, the fluorescein label is placed on the N-terminal end of Aβ, which is supposed to be unstructured and therefore exposed to the environment even in the aggregated state. Indeed, the lifetime of fluorescein in the aqueous solution (3.8 ns; data not shown) is not very different from that of the fluorescein-Aβ conjugate (4.1 ns). We expect a change in the lifetime of this fluorescein molecule if this end experiences the nonaqueous environment of the membrane. This is indeed manifested in the second, faster component that emerges when we analyze the lifetime data from the optical sections containing the cell membrane. The reduction in the mean lifetime (τm= 3.4 ns vs. τm = 4.1 ns in the extracellular medium) is also reflected in a net change of the brightness of the fluorescein fluorophores. An analysis of the data with a PCH technique (49) shows a concomitant reduction of the peak photon emission rate per particle (Fig. S2). Lifetime data obtained from both 150 nM and 350 nM Aβ solutions also show a very similar change (Fig. S1) in the lifetime (the second component is at 1.3 ns), indicating that the molecular details of membrane association are similar. We note here that aggregation of Aβ in the solution does not significantly change the lifetime of the fluorescein label, as observed in separate in vitro experiments (Fig. S5). Moreover, membrane attachment is expected to slow down the diffusion of the molecules due to the inherently higher viscosity of the membrane. This slowing down is also observed (compare Fig. 3 b, circles, and Fig. 3 d, circles). Our observations therefore signify a strong attachment, and possible outright incorporation, of Aβ molecules into the cell membrane. This attachment occurs at least down to 150 nM of Aβ concentration. We note that some degree of nonspecific adsorption of a hydrophobic molecule such as Aβ can be expected on different surfaces. However, membrane lifetime data and diffusion measurements bolster the conclusion that Aβ binds strongly to natural cell membranes at very low concentrations.
We now consider whether such an association leads to the formation of large Aβ complexes on the membrane. To make an inference about particle size from an analysis of diffusion constants, we need to account for the viscosity of the environment. We experimentally estimate this factor by measuring the diffusion of the WGA molecule in the cell membrane. The WGA protein is known to incorporate into the membrane, and as mentioned above, its diffusion time slows down by a factor of 5.5 in these cells (Fig. S3). FCS measurements show that for the 150 nM Aβ solution, structures in the membrane diffuse only 2.5 times more slowly than in the solution. The diffusion constant of monomeric Aβ in aqueous solution is ∼2.8 × 10−6cm2s−1 (S. Nag and S. Maiti, unpublished), which in the membrane would have a diffusion time close to the observed value. It is likely that membrane-incorporated Aβ at this concentration is monomeric (or at best it consists of small oligomers). However, we note that because the optical probe volume extends into the space above the cell membrane, a short diffusion time may also arise from the contribution of the monomeric population of the unbound Aβ in solution. However, the fluorescence signal levels from the probe volumes that do and do not contain the membrane show that the contribution of the membrane-bound Aβ should dominate these measurements. An alternative possibility is that the binding/unbinding kinetics of Aβ to the cell membrane shortens the apparent diffusion time. In any case, the membrane-attached structures are at least no larger than the structures observed in the extracellular medium. We therefore conclude that at a 150 nM Aβ concentration, the small extracellular Aβ structures do incorporate into the membrane (even if transiently), but they do not aggregate further.
At a 350 nM Aβ concentration, the cell membrane shows an additional species with a diffusion constant of <2.7 × 10−9cm2s−1. This diffusion time cannot be unambiguously translated into a physical size for the aggregates in a model-free manner. However, the Stokes-Einstein model (D∼1/r) presents a lower limit for the size of these structures, and much higher sizes are predicted by the Saffman-Delbruck model (50). We note that such Stokes-Einstein behavior, which assumes a spherical shape, has been experimentally observed for protein molecules diffusing on the cell membrane (51). Thus, our results would suggest that structures of the order of 200 times that of the monomers (i.e., >150 nm) form at this extracellular Aβ concentration. It is also quite likely that such large Aβ aggregates do not freely diffuse on the membrane and hence the actual size is smaller than this apparent size, but still it is obvious that these are rather large objects (>>10 nm).
This large complex formation happens at a concentration that is much less than the solubility limit of Aβ in the extracellular medium (18), and therefore such objects are not thermodynamically stable in the solution phase. Their formation in the membrane is most likely a manifestation of the lower free energy of association needed for particles to aggregate on a two-dimensional surface compared to a three-dimensional surface. Our data do not distinguish between strong attachments with the membrane versus an actual incorporation. However, the strong effect on the lifetime suggests that the Aβ molecules most likely do incorporate into the membrane. It appears that solution phase particles with radii ∼2–3 nm are sufficient for the formation of the large assemblies in the membrane. The observed size excludes most of the protofibrillar (24) or amylospheroid structures (28), which have been found to form at larger concentrations in the aqueous solution and have been hypothesized to play a role in forming aggregates on the membrane. Though the size distribution peaks at 2.3 nm for both the 150 nM and 350 nM solutions, the relative concentration of the small species (e.g., monomers) is higher at 150 nM (Fig. 3 b), as can be expected for near-equilibrium solutions. This implies that the main difference between the two is the absolute concentration of the 2.3 nm species. These small oligomers are therefore likely to be the key for membrane complex formation.
In conclusion, our results provide a direct observation of the formation of large Aβ complexes on living cell membranes at near-physiological concentrations. Their formation critically depends on the extracellular concentration of Aβ in this range. We show that these membrane complexes result from the incorporation and in situ assembly of small (∼2–3 nm hydrodynamic radius) Aβ particles present in the extracellular medium.
Acknowledgments
We thank Jenna Rickus of the Department of Agricultural and Biological Engineering at Purdue University for the PC12 cell line, and Jay Unruh of the Stowers Institute for Medical Research for the software used for the PCH analysis.
Supporting Material
References
- 1.Selkoe D.J. The molecular pathology of Alzheimer's disease. Neuron. 1991;6:487–498. doi: 10.1016/0896-6273(91)90052-2. [DOI] [PubMed] [Google Scholar]
- 2.Irvine G.B., El-Agnaf O.M., Walsh D.M. Protein aggregation in the brain—the molecular basis for Alzheimer's and Parkinson's diseases. Mol. Med. 2008;14:451–464. doi: 10.2119/2007-00100.Irvine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wong C.W., Quaranta V., Glenner G.G. Neuritic plaques and cerebrovascular amyloid in Alzheimer disease are antigenically related. Proc. Natl. Acad. Sci. USA. 1985;82:8729–8732. doi: 10.1073/pnas.82.24.8729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roychaudhuri R., Yang M., Teplow D.B. Amyloid β-protein assembly and Alzheimer disease. J. Biol. Chem. 2009;284:4749–4753. doi: 10.1074/jbc.R800036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haass C., Selkoe D.J. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 6.Haass C., Schlossmacher M.G., Selkoe D.J. Amyloid β-peptide is produced by cultured cells during normal metabolism. Nature. 1992;359:322–325. doi: 10.1038/359322a0. [DOI] [PubMed] [Google Scholar]
- 7.Byström R., Aisenbrey C., Gröbner G. Disordered proteins: biological membranes as two-dimensional aggregation matrices. Cell Biochem. Biophys. 2008;52:175–189. doi: 10.1007/s12013-008-9033-4. [DOI] [PubMed] [Google Scholar]
- 8.Arispe N., Rojas E., Pollard H.B. Alzheimer disease amyloid β protein forms calcium channels in bilayer membranes: blockade by tromethamine and aluminum. Proc. Natl. Acad. Sci. USA. 1993;90:567–571. doi: 10.1073/pnas.90.2.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rhee S.K., Quist A.P., Lal R. Amyloid β protein-(1-42) forms calcium-permeable, Zn2+-sensitive channel. J. Biol. Chem. 1998;273:13379–13382. doi: 10.1074/jbc.273.22.13379. [DOI] [PubMed] [Google Scholar]
- 10.Quist A., Doudevski I., Lal R. Amyloid ion channels: a common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. USA. 2005;102:10427–10432. doi: 10.1073/pnas.0502066102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lal R., Lin H., Quist A.P. Amyloid β ion channel: 3D structure and relevance to amyloid channel paradigm. Biochim. Biophys. Acta. 2007;1768:1966–1975. doi: 10.1016/j.bbamem.2007.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arispe N., Pollard H.B., Rojas E. The ability of amyloid β-protein [A βP (1-40)] to form Ca2+ channels provides a mechanism for neuronal death in Alzheimer's disease. Ann. N. Y. Acad. Sci. 1994;747:256–266. doi: 10.1111/j.1749-6632.1994.tb44414.x. [DOI] [PubMed] [Google Scholar]
- 13.Lin H., Zhu Y.J., Lal R. Amyloid β protein (1-40) forms calcium-permeable, Zn2+-sensitive channel in reconstituted lipid vesicles. Biochemistry. 1999;38:11189–11196. doi: 10.1021/bi982997c. [DOI] [PubMed] [Google Scholar]
- 14.Jang H., Zheng J., Nussinov R. New structures help the modeling of toxic amyloidβ ion channels. Trends Biochem. Sci. 2008;33:91–100. doi: 10.1016/j.tibs.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 15.McLaurin J., Chakrabartty A. Membrane disruption by Alzheimer β-amyloid peptides mediated through specific binding to either phospholipids or gangliosides. Implications for neurotoxicity. J. Biol. Chem. 1996;271:26482–26489. doi: 10.1074/jbc.271.43.26482. [DOI] [PubMed] [Google Scholar]
- 16.Widenbrant M.J., Rajadas J., Fuller G.G. Lipid-induced β-amyloid peptide assemblage fragmentation. Biophys. J. 2006;91:4071–4080. doi: 10.1529/biophysj.106.085944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Small D.H., Maksel D., Chebib M. The β-amyloid protein of Alzheimer's disease binds to membrane lipids but does not bind to the α7 nicotinic acetylcholine receptor. J. Neurochem. 2007;101:1527–1538. doi: 10.1111/j.1471-4159.2006.04444.x. [DOI] [PubMed] [Google Scholar]
- 18.Garai K., Sahoo B., Maiti S. Quasihomogeneous nucleation of amyloid β yields numerical bounds for the critical radius, the surface tension, and the free energy barrier for nucleus formation. J. Chem. Phys. 2008;128:045102–045107. doi: 10.1063/1.2822322. [DOI] [PubMed] [Google Scholar]
- 19.Sengupta P., Garai K., Maiti S. The amyloid β peptide (Aβ (1-40)) is thermodynamically soluble at physiological concentrations. Biochemistry. 2003;42:10506–10513. doi: 10.1021/bi0341410. [DOI] [PubMed] [Google Scholar]
- 20.Sahoo B., Nag S., Maiti S. On the stability of the soluble amyloid aggregates. Biophys. J. 2009;97:1454–1460. doi: 10.1016/j.bpj.2009.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Selkoe D.J. Soluble oligomers of the amyloid β-protein impair synaptic plasticity and behavior. Behav. Brain Res. 2008;192:106–113. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garai K., Sahoo B., Maiti S. Zinc lowers amyloid-β toxicity by selectively precipitating aggregation intermediates. Biochemistry. 2007;46:10655–10663. doi: 10.1021/bi700798b. [DOI] [PubMed] [Google Scholar]
- 23.Glabe C.G. Structural classification of toxic amyloid oligomers. J. Biol. Chem. 2008;283:29639–29643. doi: 10.1074/jbc.R800016200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walsh D.M., Lomakin A., Teplow D.B. Amyloid β-protein fibrillogenesis. Detection of a protofibrillar intermediate. J. Biol. Chem. 1997;272:22364–22372. doi: 10.1074/jbc.272.35.22364. [DOI] [PubMed] [Google Scholar]
- 25.Kayed R., Pensalfini A., Glabe C. Annular protofibrils are a structurally and functionally distinct type of amyloid oligomer. J. Biol. Chem. 2009;284:4230–4237. doi: 10.1074/jbc.M808591200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walsh D.M., Selkoe D.J. A β oligomers—a decade of discovery. J. Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- 27.Lashuel H.A., Hartley D., Lansbury P.T., Jr. Neurodegenerative disease: amyloid pores from pathogenic mutations. Nature. 2002;418:291. doi: 10.1038/418291a. [DOI] [PubMed] [Google Scholar]
- 28.Hoshi M., Sato M., Sato K. Spherical aggregates of β-amyloid (amylospheroid) show high neurotoxicity and activate tau protein kinase I/glycogen synthase kinase-3β. Proc. Natl. Acad. Sci. USA. 2003;100:6370–6375. doi: 10.1073/pnas.1237107100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kayed R., Sokolov Y., Glabe C.G. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J. Biol. Chem. 2004;279:46363–46366. doi: 10.1074/jbc.C400260200. [DOI] [PubMed] [Google Scholar]
- 30.Shankar G.M., Li S., Selkoe D.J. Amyloid-β protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat. Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hung L.W., Ciccotosto G.D., Barnham K.J. Amyloid-β peptide (Aβ) neurotoxicity is modulated by the rate of peptide aggregation: Aβ dimers and trimers correlate with neurotoxicity. J. Neurosci. 2008;28:11950–11958. doi: 10.1523/JNEUROSCI.3916-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lesné S., Koh M.T., Ashe K.H. A specific amyloid-β protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 33.Walsh D.M., Klyubin I., Selkoe D.J. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 34.Parodi J., Sepúlveda F.J., Aguayo L.G. β-Amyloid causes depletion of synaptic vesicles leading to neurotransmission failure. J. Biol. Chem. 2010;285:2506–2514. doi: 10.1074/jbc.M109.030023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lue L.F., Kuo Y.M., Rogers J. Soluble amyloid β peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am. J. Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shankar G.M., Leissring M.A., Walsh D.M. Biochemical and immunohistochemical analysis of an Alzheimer's disease mouse model reveals the presence of multiple cerebral Aβ assembly forms throughout life. Neurobiol. Dis. 2009;36:293–302. doi: 10.1016/j.nbd.2009.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Puzzo D., Privitera L., Arancio O. Picomolar amyloid-β positively modulates synaptic plasticity and memory in hippocampus. J. Neurosci. 2008;28:14537–14545. doi: 10.1523/JNEUROSCI.2692-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Townsend M., Shankar G.M., Selkoe D.J. Effects of secreted oligomers of amyloid β-protein on hippocampal synaptic plasticity: a potent role for trimers. J. Physiol. 2006;572:477–492. doi: 10.1113/jphysiol.2005.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lacor P.N., Buniel M.C., Klein W.L. Synaptic targeting by Alzheimer's-related amyloid β oligomers. J. Neurosci. 2004;24:10191–10200. doi: 10.1523/JNEUROSCI.3432-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bateman D.A., Chakrabartty A. Two distinct conformations of Aβ aggregates on the surface of living PC12 cells. Biophys. J. 2009;96:4260–4267. doi: 10.1016/j.bpj.2009.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wakabayashi M., Matsuzaki K. Formation of amyloids by Aβ-(1-42) on NGF-differentiated PC12 cells: roles of gangliosides and cholesterol. J. Mol. Biol. 2007;371:924–933. doi: 10.1016/j.jmb.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 42.Magde D., Elson E., Webb W.W. Thermodynamic fluctuations in a reacting system—measurement by fluorescence correlation spectroscopy. Phys. Rev. Lett. 1972;29:705–708. [Google Scholar]
- 43.Magde D., Elson E.L., Webb W.W. Fluorescence correlation spectroscopy. II. An experimental realization. Biopolymers. 1974;13:29–61. doi: 10.1002/bip.1974.360130103. [DOI] [PubMed] [Google Scholar]
- 44.Maiti S., Haupts U., Webb W.W. Fluorescence correlation spectroscopy: diagnostics for sparse molecules. Proc. Natl. Acad. Sci. USA. 1997;94:11753–11757. doi: 10.1073/pnas.94.22.11753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sengupta P., Garai K., Maiti S. Measuring size distribution in highly heterogeneous systems with fluorescence correlation spectroscopy. Biophys. J. 2003;84:1977–1984. doi: 10.1016/S0006-3495(03)75006-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sengupta P., Balaji J., Maiti S. Measuring diffusion in cell membranes by fluorescence correlation spectroscopy. Methods. 2002;27:374–387. doi: 10.1016/s1046-2023(02)00096-8. [DOI] [PubMed] [Google Scholar]
- 47.Haupts U., Maiti S., Webb W.W. Dynamics of fluorescence fluctuations in green fluorescent protein observed by fluorescence correlation spectroscopy. Proc. Natl. Acad. Sci. USA. 1998;95:13573–13578. doi: 10.1073/pnas.95.23.13573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Culbertson C.T., Jacobson S.C., Michael Ramsey J. Diffusion coefficient measurements in microfluidic devices. Talanta. 2002;56:365–373. doi: 10.1016/s0039-9140(01)00602-6. [DOI] [PubMed] [Google Scholar]
- 49.Chen Y., Müller J.D., Gratton E. The photon counting histogram in fluorescence fluctuation spectroscopy. Biophys. J. 1999;77:553–567. doi: 10.1016/S0006-3495(99)76912-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saffman P.G., Delbrück M. Brownian motion in biological membranes. Proc. Natl. Acad. Sci. USA. 1975;72:3111–3113. doi: 10.1073/pnas.72.8.3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gambin Y., Lopez-Esparza R., Urbach W. Lateral mobility of proteins in liquid membranes revisited. Proc. Natl. Acad. Sci. USA. 2006;103:2098–2102. doi: 10.1073/pnas.0511026103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.