

Abstract
The female reproductive system plays a major role in regulating the acquisition and loss of bone by the skeleton from menarche through senescence. Onset of gonadal sex steroid secretion at puberty is the major factor responsible for skeletal longitudinal and radial growth, as well as significant gain in bone density, until peak bone density is achieved in third decade of life. Gonadal sex steroids then help maintain peak bone density until menopause, including during the transient changes in skeletal mineral content associated with pregnancy and lactation. At menopause, decreased gonadal sex steroid production normally leads to rapid bone loss. The most rapid bone loss associated with decreased estrogen levels occurs in the first 8–10 years after menopause, with slower age-related bone loss occurring during later life. Age-related bone loss in women after the early menopausal phase of bone loss is caused by ongoing gonadal sex steroid deficiency, vitamin D deficiency, and secondary hyperparathyroidism. Other factors also contribute to age-related bone loss, including intrinsic defects in osteoblast function, impairment of the GH/IGF axis, reduced peak bone mass, age-associated sarcopenia, and various sporadic secondary causes. Further understanding of the relative contributions of the female reproductive system and each of the other factors to development and maintenance of the female skeleton, bone loss, and fracture risk will lead to improved approaches for prevention and treatment of osteoporosis.
Keywords: Sex steroids, osteoporosis, bone loss, aging, fractures
1.1 Introduction
The female reproductive system profoundly affects the skeleton during longitudinal and radial growth during growth and development, and during modeling and remodeling throughout adult life. Longitudinal and radial growth of the skeleton occurs prior to menarche under the influence of growth hormone, insulin-like growth factors, and other factors.
Onset of estrogen and other sex steroid secretion with menarche at age 11–13 years stimulates rapid skeletal mineral acquisition, as well as further longitudinal and radial skeletal growth, for the next 10 years or so. Women gain about a third of their peak bone mineral density (BMD) within the 4 years around the onset of menarche. The early pubertal rapid increase in BMD is followed by further slower increases in BMD and consolidation of skeletal mineral content during the late teens and early 3rd decade, until peak BMD is achieved at around age 25–35 years.
The female reproductive system subsequently plays a major role in the modeling and remodeling of the skeleton throughout adult life until menopause, when decreased gonadal sex steroid secretion becomes the dominant factor in causation of rapid bone loss in the early postmenopausal period. Continued age-related bone loss in the late postmenopausal period is thought due to sex steroid deficiency as well as vitamin D deficiency and increased parathyroid hormone (PTH) secretion.
1.2 Brief Overview of the Female Reproductive System
The female reproductive system is a complex multi-organ system involving the hypothalamus, pituitary gland, ovaries, uterus (endometrium and cervix), and vagina. A series of cyclic and closely regulated events involving the reproductive organs occur on a regular basis on an average of every 28–32 days in healthy non-pregnant females between menarche and menopause (Figure 1). Menarche normally occurs at age 11–13 years, and is characterized by establishment of new circadian (24-hour) and ultradian (60–90 minute) gonadotropin rhythms, and development of a positive estrogen feedback loop controlling the infradian (monthly) rhythm. Sleep-related increases in gonadotropins and gonadal steroids begin during puberty, and appear to play an important role in pubertal maturation. Menopause occurs on average at age 51–52 years, but may occur normally any time after age 40 years.
Figure 1.
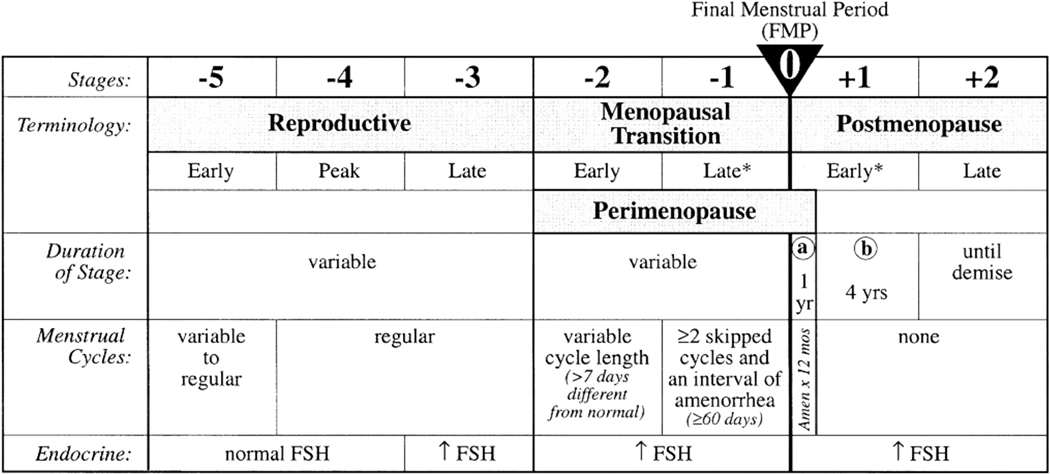
The STRAW staging system, showing the relation between alterations in cycle regularity and endocrine changes across the various stages of reproductive aging. (Reproduced from Soules MR, Sherman S, Parrott E, et al. Executive summary: Stages of Reproductive Aging Workshop (STRAW). Fertil Steril 2001;76:874–878; with permission.)
The menstrual cycle is caused by a tightly regulated sequence of events that occurs each month (Figure 2). Normal menstrual cycles require cyclic secretion of gonadotropin-releasing hormone (GnRH) by the hypothalamus, with subsequent cyclic secretion of luteinizing hormone (LH) and follicle stimulating hormone (FSH) by gonadotropes in the anterior pituitary gland (Figure 3). Regular cyclic secretion of LH and FSH normally results in maturation of one ovum each month, ovulation of the mature egg, and migration of the ovum to the uterine endometrium via the Fallopian tubes. Ovarian secretion of sex steroid hormones causes changes in the uterine endometrium prior to implantation of the fertilized ovum. If the ovum is not fertilized, ovarian secretion of estrogen and progesterone decreases, the endometrium lining breaks down, and menstruation begins.
Figure 2.
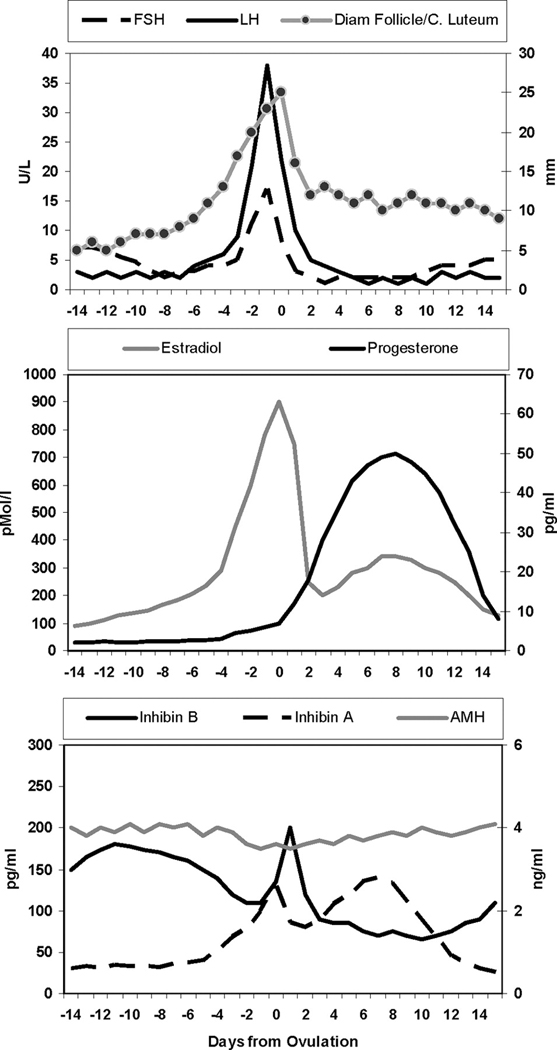
Endocrine fluctuations and follicle growth in the menstrual cycle. (Reproduced from Broekmans FJ, Soules MR, Fauser BC. Ovarian aging: mechanisms and clinical consequences. Endocrine Rev 2009;30:472; with permission.)
Figure 3.

Diagrammatic representation of how the various hormones interact at the ovary, pituitary, and hypothalamus, with feedback loops.
The normal menstrual cycle begins with the first day of vaginal bleeding, and ends just before the next menstrual cycle starts. The median menstrual cycle duration is 28 days, but normal cycles may occur every 21–40 days. Menstrual cycle duration is most variable in the years after menarche and immediately preceding menopause [1]. Menstrual flow typically lasts 5 ± 2 days, with typical blood loss with each cycle ranging from 30 to 80 mL [2].
The normal menstrual cycle is divided into the earlier follicular, or proliferative, phase, and the later luteal, or secretory, phase (Figure 2). The follicular phase is more variable in duration, whereas the luteal phase consistently lasts about 14 days in most women. Ovulation occurs at the end of the follicular phase, during the transition to the luteal phase, with the ovulatory phase beginning one day prior to the LH surge and continuing until ovulation, typically 16–32 hours after the LH surge.
Circulating FSH begins to increase late in the luteal phase of the preceding menstrual cycle, and remains increased into the early follicular phase, thereby stimulating growth and development of several ovarian follicles [3]. One of these follicles is selected by as yet unknown processes for further maturation into the dominant follicle. Serum FSH levels then begin to decrease and, and except for the marked surge during ovulation, continue to decrease throughout the remainder of the cycle until the late luteal phase.
Serum LH also begins to increase in the late follicular phase of the preceding menstrual cycle, but in distinction to FSH, LH continues to slowly increase throughout the follicular phase until it surges for one to three days at mid-cycle [3]. LH then gradually decreases to its lowest levels in the late luteal phase.
Both LH and FSH are secreted in a pulsatile fashion by gonadotropes in the anterior pituitary gland, in response to pulsatile secretion of hypothalamic GnRH [4]. LH and FSH pulses usually occur 1–4 hours apart, depending on the phase of the menstrual cycle [5]. LH is secreted least during the luteal phase, which is attributed to the feedback effects of progesterone produced by the corpus luteum on the hypothalamus and pituitary gland [6].
Serum estradiol (E2) and progesterone are secreted by the ovaries, along with other gonadal sex steroids and nonsteroidal hormones. Circulating E2 levels are lowest during the early follicular phase, and begin to increase 7–8 days prior to the LH surge. Peak E2 levels of 250–350 pg/mL occur on the day of the LH surge, or the day before [7]. Serum E2 falls quickly as LH peaks, but increases again about 6–8 days after the LH surge. Serum estrone (E1) levels parallel E2 levels, but at lower levels. About 95% of circulating E2 is produced by the dominant ovarian follicle and corpus luteum, whereas serum E1 is produced by conversion from E2 and from peripheral conversion of the adrenal hormone androstenedione.
Androstenedione and testosterone are the main androgens secreted by the ovarian interstitial and theca cells. Androstenedione is the main ovarian androgen, and can be converted in peripheral tissues to testosterone and estradiol. Both androstenedione and testosterone are also secreted in significant quantities by the adrenal glands. Androstenedione and testosterone both peak with the mid-cycle LH surge due to increased ovarian secretion [7,8]. Serum dehydroepiandrosterone (DHEA) and DHEA-sulfate (DHEA-S), which are secreted almost entirely by the adrenal glands, do not vary with the phases of the menstrual cycle.
Serum progesterone levels remain low during the follicular phase, and begin to increase just before the LH surge [8,9]. Progesterone levels peak about 6–8 days after the LH surge, and then decrease toward baseline unless the ovum is fertilized. Normal ovulation results in serum progesterone levels of about 10 ng/mL or greater about one week before onset of the next menstrual cycle. Serum 17α-hydroxyprogesterone levels increase at midcycle, before progesterone, and parallel changes in progesterone during the luteal phase.
The ovarian follicular granulosa cells and corpus luteum secrete multiple other peptides and hormones, including activin, inhibin (folliculostatin), follastatin, and others, some of which have feedback effects on the hypothalamus and pituitary gland. Inhibin A is composed of an α-subunit and a β-A subunit, and inhibin B of an α-subunit and a β-B subunit. Serum inhibin A begins to increase during the early and midfollicular phase, increases further during the late follicular phase, and peaks during the luteal phase. However, E2 appears to be the major feedback regulator of FSH secretion by the pituitary gland during the luteal phase [10]. Serum inhibin B peaks during the follicular phase, apparently in response to increasing FSH [11], and is critical for suppressing FSH secretion and causing a plateau in FSH levels in the mid-follicular phase, whereas E2 is responsible for further suppression of FSH secretion [10]. The subsequent rise in LH and fall in FSH in the late follicular phase is thought to be due to more effective suppression of pituitary FSH secretion than LH secretion by E2.
Activin is a dimer composed of two β subunits that does not vary during the menstrual cycle [12]. Follistatin is concentrated 100-fold in the follicular fluid, and appears to regulate activin action by binding the hormone. FSH and prostaglandin E2 stimulate follistatin production in granulosa cells [13], but it is not known whether follicular fluid levels of follistatin vary with the menstrual cycle.
A number of other hormones that are not thought to be directly related to ovulation, including insulin-like growth factor-I (IGF-I), growth hormone (GH), prolactin, ACTH, cortisol, PTH, calcitonin, and estrogen-sensitive neurophysin also peak at mid-cycle. IGF-I is secreted in small amounts by the ovary [14], although mostly produced by other tissues elsewhere, including the liver [15]. Serum IGF-I reaches a nadir during the menses, and peaks during midcycle and the luteal phase [16]. Serum IGF-I levels decrease with age [17], and its actions are regulated by IGF binding protein-3 (IGFBP-3), which binds IGF-I and decreases the amount of bioactive IGF-I available in the circulation. GH levels correlate with E2, and peak during midcycle [18]. GH potentiates the stimulatory effects of FSH and LH on ovarian follicles, and stimulates progesterone production by the corpus luteum, with actions largely mediated by IGF-I. The physiological contribution of the other hormones to the menstrual cycle is not clear.
Circulating estrogen and to a lessor extent, progesterone, levels directly benefit the skeleton by effects mediated by estrogen and progesterone receptors on bone cells. Estrogen suppresses bone resorption, and increases bone formation, in women from puberty to menopause. Progesterone has not been demonstrated to have antiresorptive activity after menopause, but it may stimulate bone formation when used as a co-therapy with estrogen [19]. These hormones cause a marked increase in bone mineral density starting at menarche, which peaks at peak bone density by age 25–35 years. The physiological effects of other female reproductive system hormones on the skeleton are not as clear.
1.3 Skeletal Growth and Development Before Menarche
The skeleton of both females and males becomes dependent at birth on intestinal absorption of minerals and other nutrients for longitudinal and radial growth [20]. Milk intake is usually low for the first several days after birth, and body weight decreases. By the end of the first week, however, milk intake is usually sufficient to promote weight gain and growth. Birth causes a transient decrease in neonatal serum calcium, with a consequent increase in parathyroid hormone (PTH) and 1,25-dihydroxyvitamin D levels [21]. PTH-related Peptide (PTHrP) rapidly loses the dominant role in calcium metabolism that it occupied during fetal life. Bone formation markers decrease in the first few days after birth, whereas bone resorption markers increase, but both types of markers subsequently increase over the next several weeks to months and for the remainder of growth and development [22].
Volumetric BMD (vBMD) decreases by as much as 30% in skeletal long bones during the first six months of life, as the bone marrow cavity expands by endosteal resorption [23]. Cortical vBMD also deceases during the first few months of life, but overall bone strength remains stable, unlike later in life, when similar changes during postmenopausal life cause decreased bone strength. These changes in vBMD result in a shift of endocortical and intracortical bone to the periosteal surface during rapid periosteal growth, with rapidly increasing bone mass and areal BMD (aBMD) during the first six months of life [24].
Continued growth during childhood causes significant increases in skeletal bone mass, as well as changes in skeletal size, shape, geometry, and material properties of bone [25]. The rate and magnitude of changes that occur during growth and development vary by skeletal site, and within the cortical and trabecular bone compartments [26,27]. Genetic factors determine 60–80% of the variability in skeletal development, but diet, lifestyle, and acquired illness have significant impacts on skeletal growth, modeling, and remodeling. A variety of osteoblast-related genes, including estrogen receptor 1 (ER1), collagen type 1α2, osteocalcin, parathyroid hormone receptor 1 (PTHR1), transforming growth factor-beta 1 (TGFβ1), vitamin D receptor (VDR), and lipoprotein receptor-related protein 5 (LRP5) have been identified as a candidate genes for regulation of bone mineral density in children in recent genome-wide association (GWA) studies [28]. A recent GWA study in children (the Avon Longitudinal Study of Parents and Children study) showed that common variants in the region around the gene Osterix are associated with bone mineral density and growth in childhood [29]. Acquired illness may have greater or lesser effects on the skeleton, depending on the age of onset.
Normal prepubertal skeletal development results in more rapid bone growth and mineral acquisition in the appendicular skeleton than the axial skeleton. Longitudinal and radial bone growth occurs more rapidly than mineralization through the peripubertal period, with 7 year-old girls achieving 80% of their final adult stature on average, but only 40% of their projected peak bone mass [30]. Transiliac bone biopsies from healthy children older than one year show that metabolic activity within bone appears to decrease gradually with increasing age [31]. Bone remodeling decreases in both cortical and trabecular bone with increasing age, but bone formation remains higher in cortical bone, and remodeling is more active on the endocortical surface than on the periosteal surface of cortical bone.
1.4 Effects of Menarche on the Skeleton
Early and mid-puberty stimulates rapid axial skeletal growth, whereas late puberty results in slowing of both axial and appendicular growth [30]. Because peak rates of mineral accrual lag about 8 months behind peak height velocity, peripubertal life is associated with relative undermineralization of bone and increased fracture risk [32]. Girls have onset of puberty earlier than boys [33], and blacks have earlier onset of puberty than whites [34]. Women achieve a final adult height that averages 13 cm less than men. Between ages 6 and 16 years, total body bone mineral content (BMC) increases 2.5-fold, and aBMD at the spine increases roughly 2-fold [35]. Increasing bone size accounts for much of the increase in total body BMC and aBMD, but vBMD also increases at certain skeletal sites during puberty [34,36]. Cortical mid-tibial bone samples taken from young adult women 17–46 years old have similar tissue stiffness, strength, toughness, and damageability as cortical mid-tibial biopsies from men, even though the women have narrower tibial diameter for body size than men [37].
Although bone size and mass increase significantly throughout the skeleton during adolescence, apparent vBMD assessed by DXA increases at the lumbar spine only [34,38]. One study showed that while aBMD increased by 55% in women between ages 9 and 20 years, vBMD increased by only 20% [34]. Other studies have shown that vBMD did not change in the femoral neck or midshaft [34,38], whereas other studies showed increased femoral mid-shaft vBMD during late puberty [29]. Studies using quantitative CT (QCT) scanning have shown that apparent vBMD increases only in the axial skeleton during adolescence [36]. Both cortical and trabecular vBMD increase in girls [36], with increased trabecular vBMD attributed to increased trabecular thickness rather than trabecular number [26]. However, femoral mid-shaft or radial cortical vBMD did not change throughout adolescence [39,40]. Studies using peripheral QCT, however, have shown that distal radial and distal tibial cortical vBMD increases during puberty [41–43]. Radial diaphyseal vBMD increased by 48%, cross-sectional area by 50%, and BMC by 111%, in females between age 6 and 40 years in one study [41]. After matching with developmental stage and cortical width for men in the study, women had 3–4% greater radial diaphyseal vBMD than men in late puberty [42]. Total and cortical vBMD, but not trabecular vBMD, increased in the women in this study between age 15 and adulthood [43]. Girls had greater tibial cortical vBMD than boys from pre- to late puberty, suggesting decreased intracortical remodeling and less porous cortical bone in girls [44,45].
Previous study of the second metacarpal bone in girls and boys showed that girls had narrower metacarpal bones and decreased marrow cavity size compared to boys. These findings were attributed to the differential effects of sex steroids on the skeleton [46]. Subsequent studies have largely confirmed these results, although some studies have shown similar results in pre-pubertal children, suggesting that skeletal sexual dimorphism may not be due only to differences in sex steroid levels. DXA studies have shown that cortical width increases in girls at puberty [28]. QCT studies have shown smaller cross-sectional area in vertebrae in girls than boys matched for height and weight, but vertebral height and vBMD were similar, and no differences were seen at the femoral mid-shaft [47]. Tibial pQCT studies have shown pubertal girls to have decreased periosteal expansion, bone diameter, total bone area, and cortical area compared to boys, with decreased calculated bone strength as a result [45,48]. Muscle cross-sectional area correlated with tibial bone strength [49]. No evidence was seen for increased tibial endosteal apposition, even in late puberty [48]. Radial pQCT studies have shown decreased endocortical diameter in girls in mid- to late puberty, suggesting increased endocortical apposition [50]. Increased estrogen levels are correlated with smaller medullary cavities, greater cortical thickness, and increased total vBMD in pubertal girls. A femoral shaft MRI study in healthy girls and young women aged 6–25 years showed that total bone area, cortical bone area, and medullary area all increased between prepuberty and adulthood [51]. Some of the differences seen may reflect different biomechanical forces at weight-bearing skeletal sites such as the distal tibia, compared to non-weight-bearing sites such as the radius.
In order to better define changes in bone structure during puberty, Kirmani and colleagues [52] recently studied healthy 6 to 21 year-old girls (n = 66) and boys (n = 61) using high-resolution peripheral quantitative computed tomography (voxel size, 82 micrometers) at the distal radius. Subjects were classified into 5 groups by bone-age: Group I (pre-puberty, 6–8 yrs), Group II (early puberty, 9–11 yrs), Group III (mid-puberty, 12–14 yrs), Group IV (late puberty, 15–17 yrs) and Group V (post-puberty, 18–21 yrs). Compared to Group I, trabecular parameters (bone volume fraction [BV/TV], trabecular number [Tb.N] and thickness [Tb.Th]) did not change in girls, but increased in boys from late puberty onwards. Cortical thickness and density decreased from pre- to mid-puberty in girls, but were unchanged in boys, before rising to higher levels at the end of puberty in both sexes. Total bone strength, assessed using micro-finite element models, increased linearly across bone age groups in both sexes, with boys showing greater bone strength than girls after mid-puberty. The proportion of load borne by cortical bone, and the ratio of cortical to trabecular bone volume, decreased transiently during mid- to late-puberty in both sexes, with apparent cortical porosity peaking during this time. This mirrors the incidence of distal forearm fractures in prior studies. The authors concluded that regional deficits in cortical bone may underlie the adolescent peak in forearm fractures. Whether these deficits are more severe in children who sustain forearm fractures or persist into later life warrants further investigation. None of the biochemical or hormonal variables assessed in this study were significantly associated with BV/TV, Tb.N, Tb.Th, or trabecular separation (Tb.Sp) in the girls; in boys, greater BV/TV and Tb.Th were related to higher serum testosterone levels, and Tb.N and Tb.Sp were most closely associated with higher IGF-I. Cortical thickness and cortical vBMD were inversely associated with bone turnover (serum procollagen type I N-terminal propeptide [PINP] and C-terminal cross-linked telopeptides [CTX]) in girls and with PTH in boys. Periosteal and endosteal circumference were most closely associated with IGF-I levels in the girls and testosterone levels in the boys. Overall bone strength was associated with serum E2 in girls and testosterone in boys, whereas the percent load carried by cortical bone was negatively associated with CTX and IGF-I levels in girls, and with IGF-I and PTH in boys. The cortical porosity index, on the other hand, was positively associated with CTX and IGF-I in girls, and with IGF-I in boys.
The female reproductive system affects the skeleton significantly at puberty, but these effects occur in the context of genetic inheritance, diet, and physical activity. Candidate genes linked to the skeleton affect bone size, efficiency of calcium absorption, response to biomechanical forces, and future risk of osteoporosis and fractures [53]. Short stature and delayed puberty affect assessment of the skeleton by DXA. Skeletal loading by weight-bearing exercise effectively stimulates bone mineral acquisition during childhood and adolescence [54], and increases bone diameter and total and cortical bone area in elite racket sports players [55]. Dietary calcium intake correlates well with DXA BMD in many studies [56]. Low calcium intake in childhood and adolescence, whether due to lactose intolerance, dietary preferences, cultural patterns, or other factors, is associated with increased risk of osteoporotic fractures in older women [57]. Avoidance of milk intake is associated with childhood fractures [58]. Calcium intake in children and adolescents in one study appeared to increase DXA BMD only by 1.7% at the forearm, and to not affect BMD at the spine or hip [59]. The beneficial effects of calcium appear limited to improvement in bone mass at cortical sites by reducing bone remodeling.
Physical activity may affect the skeleton differently at different pubertal stages. Skeletal sensitivity to mechanical loading is increased in the presence of estrogen, indicating that early and mid-puberty may be optimal times for exercise-induced skeletal benefit [54]. Pubertal girls have greater ratios of vBMD and cross-sectional area of cortical bone to muscle cross-sectional area, suggesting that skeletal calcium acquisition is increased in response to physical activity [41–43]. Some studies have shown that the combination of increased calcium intake and physical activity have greater effects than either alone at certain skeletal sites.
Fractures are associated with bone size and mass in children, as they are in adults. Forearm fractures occur most commonly in peripubertal girls at age 8–12 years [60], and are increasingly common [61]. Pubertal girls with fractures commonly have lower BMC, aBMD, and estimated vBMD compared to controls [62]. Lower total body BMD and bone area for height are the best predictors of fracture in pubertal girls [63].
1.5 Pregnancy and Lactation
The female reproductive system significantly strengthens the skeleton in women by stimulating acquisition of bone mass and biomechanical strength at puberty, but also presents significant challenges to the skeleton during pregnancy and lactation. The fetus and placenta take up significant calcium from the maternal circulation during pregnancy to mineralize the fetal skeleton, and the nursing infant requires significant calcium from breast milk to promote continued skeletal growth [64]. The physiological adjustments made by pregnant or lactating women to accommodate the baby’s need for calcium are quite different [65].
1.5.1 Pregnancy
The developing fetal skeleton normally acquires about 30 grams of calcium by birth, with about 80% of this calcium acquired during the last 3 months of pregnancy, when the fetal skeleton is rapidly mineralizing. This demand appears to be largely met by doubling maternal intestinal calcium absorption beginning as early as 12 weeks of gestation, largely mediated by increased maternal renal production of serum 1,25-dihydroxyvitamin D, although the placenta, decidua, and fetal kidney also contribute small amounts [64]. Renal 1α-hydroxylase activity is upregulated during pregnancy by PTHrP, estrogen, prolactin, and placental lactogen, rather than PTH, because PTH levels are usually low- to mid-normal during pregnancy, at least in North American and European women with sufficient calcium intake. PTHrP is produced by many tissues in the fetus and mother during pregnancy, but it is not clear that there is a dominant source. PTHrP increases 1,25-dihydroxyvitamin D and suppresses PTH during pregnancy, and may help regulate placental calcium transport [66] and protect the maternal skeleton during pregnancy. PTHrP C-terminal fragments have been shown to inhibit osteoclast-induced bone resorption [67].
One study showed that markers of bone resorption increased gradually during normal pregnancy through the 28th week, whereas markers of bone formation remained stable through the 28th week, and then increased markedly by the 36th week [68]. These findings suggest that bone remodeling is uncoupled during the first two trimesters of pregnancy, with a marked increase in bone resorption, and that bone formation does not increase until the third trimester.
Assessment of changes in maternal BMD during pregnancy is confounded by changes in body composition, weight, and skeletal volume. DXA BMD studies during pregnancy have been limited by radiation exposure to the fetus, but several studies have been done with measurements taken shortly before and after pregnancy [65]. One study showed no changes in lumbar spine BMD before conception and 1–2 weeks after delivery, whereas other studies have shown that lumbar spine BMD decreased by 4–5% when the second measurement was taken 1–6 weeks after delivery. The studies that have shown bone loss may be confounded by the fact that lactation causes lumbar spine bone loss of 1–3% per month beginning at birth. Longitudinal calcaneal ultrasound studies have shown slow progressive bone loss during pregnancy. None of the available studies have addressed whether skeletal BMD increases during the first trimester in preparation for losses during the third trimester.
Rare women have been reported to sustain osteoporotic fractures during or shortly after pregnancy, and cases of transient osteoporosis of the hip have been reported during pregnancy. Osteoporosis and fractures during pregnancy likely reflect low bone mineral density or other skeletal abnormalities before pregnancy, usually unrecognized, with the superimposed effects of the changes of pregnancy on the skeleton. Uncoupling of bone turnover during the first two trimesters, with increased bone resorption and stable bone formation, may increase the risk of fracture during pregnancy. In spite of the adverse skeletal consequences of pregnancy, most epidemiological studies of osteoporotic or osteopenic women have not shown an association between parity and BMD or fracture risk [69].
1.5.2 Lactation
Nursing mothers typically lose 280–400 mg of calcium each day through their breast milk, although losses as high as 1,000 mg have been reported. Most of this calcium appears to come from temporary skeletal calcium losses, possibly mediated by PTHrP, with decreased estrogen levels during lactation resulting from prolactin-mediated suppression of GnRH, LH, and FSH, but not by changes in PTH or 1,25-dihydroxyvitamin D. Studies using DXA BMD assessment show losses of 1–3% per month in lactating women, with cumulative losses of 3–10% over 2–6 months at trabecular sites such as the lumbar spine, hip, and distal radius [64,69]. These very rapid losses compare to losses of 1–3% per year in early postmenopausal osteoporotic women who are losing bone density rapidly. Calcium supplementation during lactation does not prevent bone loss during lactation [70].
Weaning results in rapid regain of BMD at 0.5–2.0% per month, with most women regaining BMD to baseline several months after completion of lactation [64,69,71]. The mechanisms behind this rapid regain in BMD are not yet clear. Most epidemiological studies of premenopausal and postmenopausal women have not shown an adverse effect of lactation on peak bone mass, BMD, or hip fracture risk.
1.6 Postmenopausal Bone Loss and Fractures in Women
Significant bone loss occurs with normal aging in women [72]. Development of osteoporosis, usually in old age, is the natural consequence of age-related bone less if left untreated. Multiple population-based cross-sectional and longitudinal studies over the last 25 years using aBMD assessed by DXA have helped define the general pattern of bone loss with normal aging (Figure 4). Women lose aBMD at relatively slow rates starting at around age 40, with women losing aBMD more rapidly with onset of menopause in their late 40s or early 50s. Postmenopausal women lose trabecular BMD rapidly in their vertebrae, pelvis, and ultradistal wrist. There is less rapid cortical bone loss in the long bones and vertebrae after the menopause. About 8–10 years after menopause, slower age-related bone loss becomes prominent, and continues for the rest of life.
Figure 4.
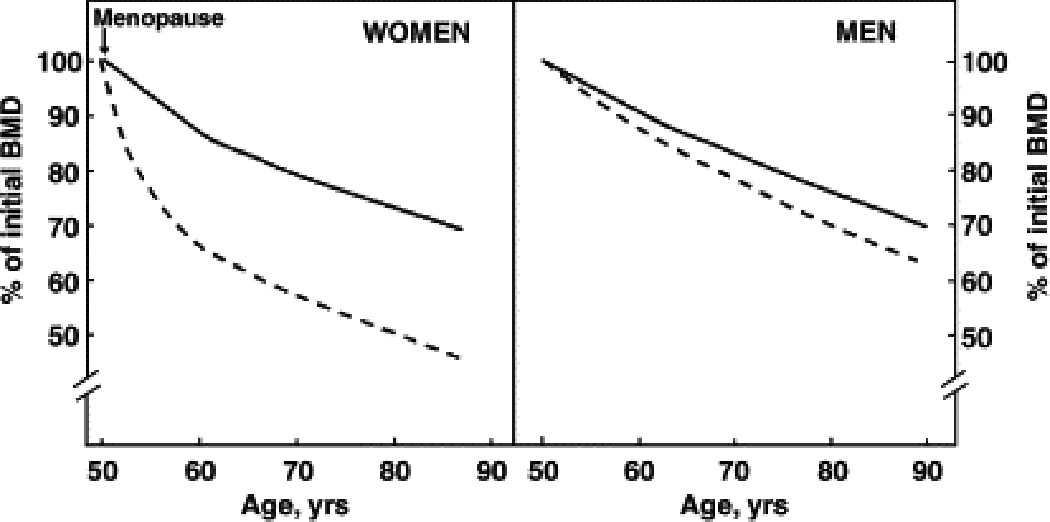
Patterns of age-related bone loss in women and men. Dashed lines represent trabecular bone and solid lines, cortical bone. The figure is based on multiple cross-sectional and longitudinal studies using DXA. (Reproduced from Khosla S, Riggs BL. Pathophysiology of age-related bone loss and osteoporosis. Endocrinol Metab Clin N Am 2005;34:1017; with permission.)
However, because DXA BMD is not able to differentiate changes occurring in trabecular and cortical bone with age, and because DXA BMD cannot assess age-related changes in bone geometry and/or size, more recent studies have utilized QCT scanning [73] to assess bone loss in greater detail. Both peripheral and central QCT, with new image analysis software [74], have been used to better define the age-related changes in bone volumetric density, geometry, and structure at multiple skeletal sites.
Riggs et al. [73] reported large decreases in lumbar spine vBMD with normal aging in a cross-sectional study of women aged 20 to 97 years in Rochester, Minnesota, predominantly due to vertebral trabecular bone loss beginning in the third decade. Lumbar spine vBMD decreased by 55% in women. The rate of bone loss appeared to increase in middle age in women, accounting for the greater decrease in vBMD seen with aging in women (Figure 5). Assessment of changes in radial cortical vBMD at the wrist showed that cortical bone loss did not begin until middle age in women. After middle age, there was a 28% linear decrease in cortical vBMD in women. Normal aging was associated with increases in cross-sectional area at the femoral neck and radius because of continued periosteal apposition with normal aging. The bone marrow space increased more rapidly than cross-sectional area due to continued endosteal bone resorption. Because the rate of periosteal apposition was slower than the rate of endosteal resorption, cortical area and thickness decreased with aging. However, because periosteal apposition increased bone diameter, the ability of bone to resist biomechanical forces increased, partially offsetting the decrease in bone strength resulting from decreased cortical area.
Figure 5.

(A) Values for vBMD (mg/cm3) of the total vertebral body in a population sample of Rochester, Minnesota, women and men between the ages of 20 and 97 years. Individual values and smoother lines are given for premenopausal women in red, for postmenopausal women in blue, and for men in black. (B) Values for cortical vBMD at the distal radius in the same cohort, with color code as in (A). All changes with age were significant (P <.05). (Reproduced from Riggs BL, Melton LJ 3rd, Robb RA, et al. A population-based study of age and sex differences in bone volumetric density, size, geometry, and structure at different skeletal sites. J Bone Miner Res 2004;19:1950; with permission.)
Khosla et al. [75] subsequently showed that the structural basis for bone loss in the ultradistal radius with aging in women is thinner trabeculae in young adulthood, and primarily decreased trabecular number and increased trabecular spacing as they get older. These changes result in greater microstructural damage with aging in women, which likely explains the increased risk of wrist fractures seen in women. Khosla et al. [76] then demonstrated that decreased sex steroids were the major hormonal determinants of trabecular microstructure in elderly women. In a subsequent study, Riggs et al. [77] showed that the late onset of cortical bone loss is temporally associated with sex steroid deficiency. However, the early-onset, substantial trabecular bone loss in women during sex steroid sufficiency is unexplained, and indicates that current paradigms on the pathogenesis of osteoporosis are incomplete.
These studies showed that these age-related changes in bone density and structure correlated with the observed increased fracture risk seen in women. Previous studies had shown that distal forearm (Colles’) fractures increased rapidly in women after menopause, and then remain constant from about 10 to 15 years after menopause until the end of life (Figure 6). In contrast, vertebral fractures increase more slowly after menopause, but continue to increase exponentially during later life. Hip fractures in women increase more slowly than vertebral fractures after menopause, but continue to increase throughout life, and increase rapidly in later life.
Figure 6.
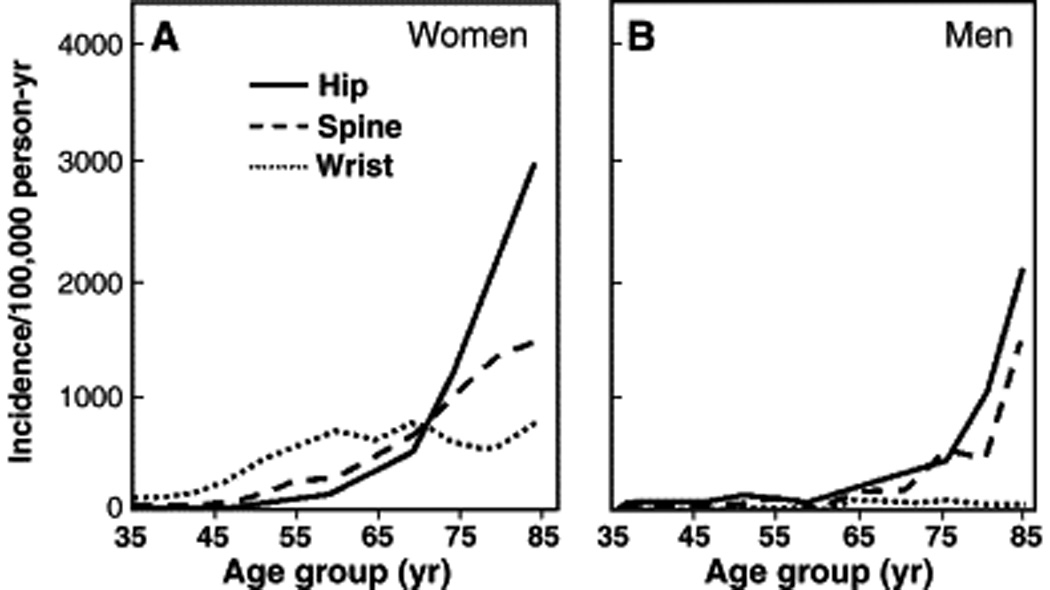
Age-specific incidence rates for proximal femur (hip), vertebral (spine), and distal forearm (wrist) fractures in Rochester, Minnesota, women (A) and men (B). (Adapted from Cooper C, Melton LJ. Epidemiology of osteoporosis. Trends Endocrinol Metab 1992;3:225; with permission.
Based on these and other studies, it is estimated that 40% of Caucasian women aged 50 years or older will develop a vertebral, hip, or wrist fracture sometime during the remainder of their lives, and that this risk increases to about 50% if non-clinical vertebral fractures detected by radiological imaging are included in the estimate [78]. The risk of these fractures is somewhat lower in non-Caucasian women. It is estimated that osteoporotic fractures cost the U.S. between $12.2 to $17.9 billion each year, as measured in 2002 dollars [79].
1.7 Pathophysiology of Age-Related Bone Loss in Women
1.7.1 Accelerated Postmenopausal Bone Loss Due to Gonadal Sex Steroid Deficiency
Menopause is associated with the onset of rapid bone loss in women, due largely to decreased ovarian function, leading to decreased estrogen secretion. This rapid bone loss can be prevented by estrogen or hormone replacement [80,81]. During the menopause transition, serum 17β-estradiol levels decrease by 85–90% from the mean premenopausal level, and serum estrone levels decrease by 65 to 75% from premenopausal levels [82]. Serum estrone has one-fourth the biological effect of serum 17β-estradiol. Serum testosterone also decreases following menopause, but to a lesser extent, because testosterone continues to be produced by the adrenal cortex and interstitial cells in the ovary [83]. Some longitudinal clinical studies show that increased bone turnover in perimenopausal women correlates with elevated serum FSH, as well as serum estradiol [84]. The perimenopausal rise in FSH is due to a selective decrease in ovarian inhibin B. Decreases in inhibin levels across the menopause transition are associated with increasing bone turnover, independent of changes in sex steroids or FSH [85].
Bone resorption increases by 90% after menopause, as assessed by markers of bone resorption, whereas bone formation also increases, but only by 45% as assessed by markers of bone formation [86]. The difference between bone resorption and formation favors greater bone resorption, which leads to accelerated bone loss during the first 8–10 years after menopause. Increased bone resorption leads to an efflux of calcium from the skeleton into the extracellular pool, but compensatory increased renal calcium excretion [87], decreased intestinal calcium absorption [88], and partially suppressed parathyroid hormone secretion [89] prevent development of hypercalcemia.
The cellular and molecular mechanisms by which estrogen deficiency leads to bone loss are increasingly well understood (Figure 7 and Table 1). Estrogen deficiency increases receptor activator of nuclear factor kappa B ligand (RANKL) [90], leading to increased osteoclast recruitment and activation and decreased osteoclast apoptosis. RANKL is the final key molecule required for osteoclast development, and is normally expressed by bone marrow stromal/osteoblast precursor cells, T-lymphocytes, and B-lymphocytes [91,92]. RANKL binds to its receptor RANK on osteoclast lineage cells [93], and is neutralized in the bone microenvironment by its soluble decoy receptor osteoprotegerin (OPG), which is produced and secreted by osteoblast lineage cells [94]. Combined in vitro and in vivo studies have shown that estrogen normally suppresses RANKL production by osteoblastic cells and T- and B-lymphocytes [91,92], and increases OPG production by osteoblastic cells [95,96], so that estrogen deficiency leads to an increase in the RANKL/OPG ratio that favors bone resorption.
Figure 7.
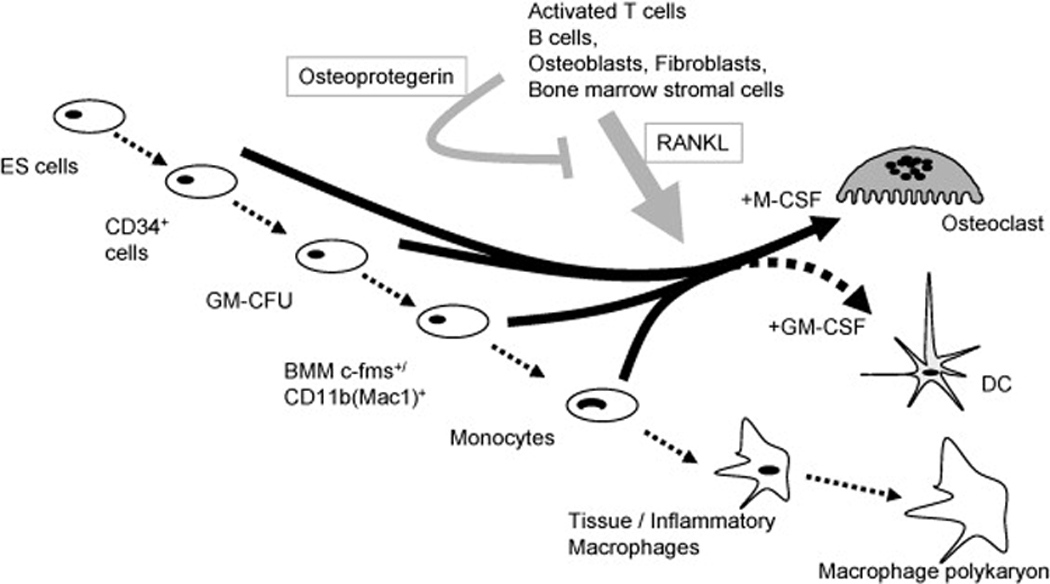
Summary of stimulatory and inhibitory factors involved in osteoclast development and apoptosis. (Reproduced from Quinn JMW, Saleh H. Modulation of osteoclast function in bone by the immune system. Mol Cell Endocrinol 2009;310:42; with permission.)
Table 1.
Potential Mechanisms by Which Estradiol Deficiency Causes Bone Loss in Postmenopausal Women
| Physiological Changes Caused by Estrogen Deficiency |
Potential Mechanism(s) Causing Bone Loss |
|---|---|
| Increased Receptor Activator of Nuclear Factor Kappa B (RANK) Ligand Expression by Osteoblasts and T- and B- Lymphocytes |
Increased Osteoclast Recruitment and Activation, Decreased Osteoclast Apoptosis |
| Decreased Osteoprotegerin Production by Osteoblasts |
Increased Osteoclast Recruitment and Activation, Decreased Osteoclast Apoptosis |
| Decreased Osteoclast Precursor Apoptosis and Increased Osteoclast Differentiation |
Increased Osteoclast Recruitment and Activation |
| Decreased Inhibition of Osteoclast Activity Mediated by Estrogen Receptors |
Increased Osteoclast Activity |
| Increased Interleukin-1 (IL-1) by Bone Marrow Stromal Cells and Osteoblasts |
Increased Osteoclast Recruitment and Activation, Decreased Osteoclast Apoptosis |
| Increased Interleukin-6 (IL-6) by Bone Marrow Stromal Cells and Osteoblasts |
Increased Osteoclast Recruitment and Activation, Decreased Osteoclast Apoptosis |
| Increased Tumor Necrosis Factor-alpha (TNF-α) by Bone Marrow Stromal Cells and Osteoblasts |
Increased Osteoclast Recruitment and Activation, Decreased Osteoclast Apoptosis |
| Increased Macrophage-Colony Stimulating Factor (M-CSF) by Bone Marrow Stromal Cells and Osteoblasts |
Increased Osteoclast Recruitment and Activation, Decreased Osteoclast Apoptosis |
| Increased Prostaglandins by Bone Marrow Stromal Cells and Osteoblasts |
Increased Osteoclast Recruitment and Activation, Decreased Osteoclast Apoptosis |
| Decreased Transforming Growth Factor-β (TGF-β) by Osteoblast Precursors |
Decreased Osteoclast Apoptosis |
| Decreased Intestinal Calcium Absorption | Decreased Calcium for Bone Formation |
| Decreased Renal Tubular Calcium Reabsorption |
Decreased Calcium for Bone Formation |
Estrogen also modulates production of additional cytokines by bone marrow stromal mononuclear cells and osteoblasts, thereby controlling osteoclast activity by paracrine action [96]. Estrogen is thought to suppress production of bone-resorbing cytokines such as interleukin (IL)-1, IL-6, tumor necrosis factor (TNF)-α, macrophage colony-stimulating factor (M-CSF), and prostaglandins by the appropriate cells [97]. Estrogen-deficient model systems have shown that increased IL-1 and M-CSF levels [98,99] can be reduced by specific antagonists to these molecules [100–103]. The bone-resorptive activity of TNF-α is able to be inhibited by soluble type I TNF receptor [104]. Estrogen deficiency is associated with increased IL-6 levels [105,106]. In vivo, however, it is likely that estrogen suppresses production and activity of multiple cytokines in premenopausal women that would otherwise act cooperatively to cause bone loss. With estrogen deficiency, each cytokine likely accounts for only part of cytokine-mediated age-related bone loss. It is not yet clear that there is a dominant cytokine contributing to estrogen-deficiency associated bone loss. Estrogen also normally increases the production of transforming growth factor (TGF)-β by osteoblast precursor cells [107]. TGF-β induces apoptosis of osteoclasts [108].
Estrogen also directly stimulates apoptosis of osteoclast precursor cells, and decreases osteoclast precursor differentiation by blocking RANKL/M-CSF-induced activator protein (AP)-1-dependent transcription by reducing c-jun activity [109,110]. C-jun activity is reduced by decreasing c-jun transcription and decreasing phosphorylation. Estrogen is also capable of inhibiting the activity of mature osteoclasts by direct, receptor-mediated mechanisms [111].
Loss of these multiple estrogen-induced restraining actions on osteoclast bone resorption leads to rapid upregulation of bone loss shortly after onset of menopause. It is likely that the major direct effect of estrogen deficiency is mediated by alteration in the ratio of RANKL to OPG in the bone microenvironment, thereby leading to increased osteoclast activity and bone resorption. However, the multiple other changes induced by estrogen deficiency also likely play a significant role in causation of early postmenopausal bone loss. The rapid phase of bone loss is sustained for 8–10 years before gradually subsiding. Why rapid bone loss gradually subsides after this duration is not yet clear, but it may be that estrogen deficiency alters biomechanical sensing of skeletal mechanical loading by osteocytes within the bone [112]. It is hypothesized that for a given level of skeletal mechanical loading during estrogen deficiency, bone mass is sensed as being excessive by the osteocytes, which then signal osteoclasts to resorb more bone and/or the osteoblasts to form less bone, leading to net rapid bone loss. Once enough bone is lost, however, it is thought that the proportionately increased skeletal mechanical loading on remaining bone is sufficient to limit further rapid bone loss.
Some evidence suggests that increased serum FSH may cause bone loss independent of decreased serum estradiol levels [113]. While intriguing, this evidence remains controversial, and is not supported by all experimental evidence.
1.7.2 Age-Related Bone Loss Due to Secondary Hyperparathyroidism
While gonadal steroid deficiency may be a major cause for postmenopausal bone loss, other important factors also play a role. During the rapid phase of early postmenopausal bone loss, there is mild suppression of PTH secretion, but during the slower phase of later postmenopausal bone loss there is gradually increasing PTH secretion, with corresponding increasing markers of bone resorption. The increases in serum PTH and markers of bone turnover correlate with each other. Transient suppression of PTH secretion by an intravenous 24-hour calcium infusion in younger premenopausal and elderly postmenopausal women is associated with suppression of markers of bone turnover, strongly suggesting that increased serum PTH was the proximate cause of the increased bone turnover [114].
The reason for gradually increasing levels of PTH secretion with age is likely multifactorial. Vitamin D deficiency is common in postmenopausal women [115], and is associated with increased serum PTH levels. Longstanding estrogen deficiency also leads to chronic negative calcium balance because loss of estrogen causes decreased intestinal calcium absorption [88,116] and renal tubular calcium reabsorption [117,118]. Unless these changes leading to negative calcium balance are compensated for with adequate calcium supplementation, secondary hyperparathyroidism inevitably develops, leading to age-related bone loss.
1.7.3 Bone Loss Due to Decreased Bone Formation
Postmenopausal and age-related bone loss is due not just to accelerated bone resorption, but also to decreased bone formation. Decreased bone formation results, in part, from changes in the differentiation potential of mesenchymal stem cells with aging. Bone mesenchymal stem cells give rise to both osteoblasts and adipocytes, but aging induces changes that favor adipocyte differentiation over osteoblast formation, which leads to decreased potential to form new bone. This change in differentiation potential is poorly understood, but known to be influenced by the type 1 cannabinoid receptor (CB1) [119] and Wnt signaling pathway. Lineage commitment by mesenchymal stem cells depends on activation of the cyclic AMP/CREB pathway, which downregulates expression of the osteoblast-specific transcription factor Cbfa-1 [120], and upregulates the expression of adipocyte-specific transcription factors CEBPβ and PPARγ [121]. Thiazolidinedione drugs used to treat type 2 diabetes mellitus also favor differentiation of mesenchymal stem cells toward the adipocyte lineage by this mechanism.
Decreased bone formation has generally been attributed to decreased paracrine production of growth factors [122], and/or decreased GH [123,124] and IGF-1 levels [125–127]. Estrogen has been shown to directly stimulate bone formation via an estrogen receptor-dependent mechanism in mice [128], so it is likely that estrogen deficiency may also directly result in bone loss. Impaired bone formation is detectable in early menopause [129]. Estrogen increases production of IGF-1 [130], TGF-β [107], and procollagen synthesis by osteoblast precursor cells in vitro [130], and increases osteoblast life span by decreasing osteoblast apoptosis [131,132]. Khastgir and colleagues provided direct evidence that estrogen can stimulate bone formation after cessation of skeletal growth by evaluating iliac crest bone biopsies from 22 elderly women of mean age 65 years before, and 6 years after, percutaneous administration of high doses of estrogen [133]. They found that cancellous bone volume increased by 61%, and trabecular wall thickness by 12%. Tobias and Compston reported similar results [134]. It is not yet clear whether these results are due to the pharmacological doses of estrogen used, or augmentation of physiological effects that are normally not of sufficient magnitude to detect. Accumulating data implicate estrogen deficiency as a contributing cause of decreased bone formation with aging, but there is not yet a clear consensus on whether estrogen directly stimulates osteoblast function, and if it does, what the relative magnitude of increased proliferation or decreased apoptosis are.
1.7.4 Other Factors Contributing to Age-Related Bone Loss in Women
Age-related bone loss may be driven primarily by gonadal sex steroid deficiency and physiological secondary hyperparathyroidism, but multiple other factors also contribute to this process. Vitamin D deficiency, widely prevalent in postmenopausal women with osteoporosis, and common among children and adults without osteoporosis in many countries around the world, worsens age-related physiological secondary hyperparathyroidism [115]. Age-related decreased bone formation may be due in part to sex steroid deficiency, but other sex steroid-independent factors likely also contribute to decreased bone formation.
After menopause, ovarian production of estrogen is markedly diminished, and peripheral aromatization of androgens to estrogen remains important as the major source of serum estradiol. The presence of low but detectable levels of serum estradiol after menopause prevents more rapid and severe bone loss that would otherwise occur. The importance of low but detectable levels of estradiol after menopause is demonstrated by the rapid bone loss that occurs with use of aromatase inhibitors such as exemestane and letrozole in treatment of postmenopausal women with estrogen receptor-positive breast cancer. These potent inhibitors of the aromatase enzyme cause marked reduction in serum estradiol levels, which leads to rapid bone loss unless treatment with antiresorptive agents is initiated prophylactically [135].
Other changes in endocrine function with aging likely also contribute to the physiology of bone loss. The less potent adrenal androgens DHEA and DHEA-sulfate both decrease by about 80% with aging, with much of the decrease occurring in young adults [136], whereas cortisol secretion remains relatively constant throughout life. The decrease in adrenal androgens early in adult life may therefore change the ratio of adrenal catabolic/anabolic hormone activity. This change may contribute to younger adult trabecular bone loss.
Lower peak bone density at 25–35 years of age also contributes to risk of osteoporosis and fractures later in life. Individuals with lower peak bone density for whatever cause will develop low bone density or osteoporosis sooner than those with higher peak bone density, assuming the rate of bone loss is equivalent as they age [137].
It has been suggested that bone strength is homeostatically adapted to habitual skeletal loading conditions and that bone loss could, therefore, result simply from age-related reductions in physical activity and muscle mass. In a stratified random sample of Rochester, Minnesota, women and men 21–97 years of age, Melton et al. [138] estimated indices of bone strength, flexural rigidity, and axial rigidity from central QCT measurements at the femoral neck and lumbar spine, and pQCT measurements at the ultradistal radius. Habitual skeletal loading was assessed using lean body mass, total skeletal muscle mass, body weight, and physical activity. This study showed that there was not a close correspondence between changes in habitual load and changes in bone strength, nor any consistent pattern. Moreover, inter-individual variation in the strength-to-load ratios was substantial. These data suggest that the hypothesis that reduced skeletal loading is the primary basis for age-related bone loss may be oversimplified.
Numerous sporadic secondary causes of bone loss play a role in age-related bone loss by various mechanisms. It is estimated that about 40% of women have an identifiable sporadic cause of bone loss [136], such as glucocorticoid therapy, subtle or clinically evident malabsorption, anorexia nervosa, idiopathic hypercalciuria, or behavioral factors such as excess alcohol intake, cigarette smoking, high-caffeine or high-sodium diet, or physical inactivity.
Age-related bone loss and sarcopenia proceed in parallel. It has been suggested that age-related muscle loss is the main factor causing age-related bone loss [137,138]. Although not yet proven, it is likely that age-related muscle loss, causing decreased muscle loading on the skeleton, contributes significantly to age-related bone loss in women.
1.8 Summary
The female reproductive system plays a major role in regulating the acquisition and loss of bone by the skeleton from menarche through senescence. Onset of gonadal sex steroid secretion at puberty is the major factor responsible for significant skeletal longitudinal and radial growth, as well as significant gain in bone density, from the early teenage years through the early 20s. Multiple factors affect acquisition of peak bone density in addition to the female reproductive system. Gonadal sex steroids then help maintain peak bone density until menopause, including during the transient loss and regain of skeletal mineral associated with pregnancy and lactation. At menopause, decreased gonadal sex steroid production normally leads to rapid bone loss. The most rapid bone loss associated with decreased estrogen levels occurs in the first 8–10 years after menopause, with slower age-related bone loss occurring during later life. Age-related bone loss in women after the early menopausal phase of bone loss is caused by ongoing gonadal sex steroid deficiency, vitamin D deficiency, and secondary hyperparathyroidism. Other factors also play key roles in age-related bone loss, including vitamin D deficiency, intrinsic defects in osteoblast function, impairment of the GH/IGF axis, reduced peak bone mass, age-associated sarcopenia, and various sporadic secondary causes. Further understanding of the relative contributions of each of these factors to development and maintenance of the female skeleton, bone loss, and fracture risk will lead to improved preventive and therapeutic approaches for osteoporosis.
Acknowledgments
This work was supported by Grants AG004875 and AR027065 from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Treloar AE, Boynton RE, Benn BG, et al. Variation in human menstrual cycle through reproductive life. Int. J. Fertil. 1967;12:77–126. [PubMed] [Google Scholar]
- 2.Baldwin RM, Whalley PJ, Pritchard JA. Measurements of menstrual blood loss. Am. J. Obstet. Gynecol. 1961;81:739–742. doi: 10.1016/s0002-9378(15)33522-5. [DOI] [PubMed] [Google Scholar]
- 3.Midgely AR, Jr, Jaffe RB. Regulation of gonadotropins. IV. Correlations of serum concentrations of follicle stimulating and luteinizing hormones during the menstrual cycle. J. Clin. Endocrinol. Metab. 1968;28:1699–1703. doi: 10.1210/jcem-28-12-1699. [DOI] [PubMed] [Google Scholar]
- 4.Knobil E. The neuroendocrine control of the menstrual cycle. Recent Prog. Horm. Res. 1980;36:53–88. doi: 10.1016/b978-0-12-571136-4.50008-5. [DOI] [PubMed] [Google Scholar]
- 5.Midgely AR, Jr, Jaffe RB. Regulation of human gonadotropins. X. Episodic fluctuation of LH during the menstrual cycle. J. Clin. Endocrinol. Metab. 1971;33:962–969. doi: 10.1210/jcem-33-6-962. [DOI] [PubMed] [Google Scholar]
- 6.Soules MR, Steiner RA, Clifton DK, et al. Progesterone modulation of pulsatile luteinizing hormone secretion in normal women. J. Clin. Endocrinol. Metab. 1984;58:378–383. doi: 10.1210/jcem-58-2-378. [DOI] [PubMed] [Google Scholar]
- 7.Lloyd CW, Lobotsky J, Baird DT, et al. Concentration of unconjugated estrogens, androgens, and gestagens in ovarian and peripheral venous plasma of women: the normal menstrual cycle. J, Clin, Endocrinol, Metab. 1971;32:155–166. doi: 10.1210/jcem-32-2-155. [DOI] [PubMed] [Google Scholar]
- 8.Judd HL, Yen SSC. Serum androstenedione and testosterone levels during the menstrual cycle. J. Clin. Endocrinol. Metab. 1973;36:475–481. doi: 10.1210/jcem-36-3-475. [DOI] [PubMed] [Google Scholar]
- 9.Hoff JD, Quigley MD, Yen SSC. Hormonal dynamics at midcycle: a reevaluation. J. Clin. Endocrinol. Metab. 1983;57:792–796. doi: 10.1210/jcem-57-4-792. [DOI] [PubMed] [Google Scholar]
- 10.Lahlou N, Chabbert-Buffet N, Christin-Maitre S, et al. Main inhibitor of follicle stimulating hormone in the luteal-follicular transition: inhibin A, oestradiol, or inhibin B? Hum. Reprod. 1999;14:1190–1193. doi: 10.1093/humrep/14.5.1190. [DOI] [PubMed] [Google Scholar]
- 11.Fraser HM, Groome NP, McNeilly AS. Follicle stimulating hormone-inhibin B interactions during the follicular phase of the primate menstrual cycle revealed by gonadotropin-releasing hormone antagonist and antiestrogen treatment. J. Clin. Endocrinol. Metab. 1999;84:1365–1369. doi: 10.1210/jcem.84.4.5586. [DOI] [PubMed] [Google Scholar]
- 12.Demura R, Susuki T, Tajima S, et al. Human plasma free activin and inhibin levels during the menstrual cycle. J. Clin. Endocrinol. Metab. 1993;76:1080–1082. doi: 10.1210/jcem.76.4.8473385. [DOI] [PubMed] [Google Scholar]
- 13.Tuuri T, Ritovs O. Regulation of the activin-binding protein follistatin in cultured human luteinizing granulosa cells: characterization of the effects of follicle stimulating hormone, prostaglandin E2, and different growth factors. Biol. Reprod. 1995;53:1508–1516. doi: 10.1095/biolreprod53.6.1508. [DOI] [PubMed] [Google Scholar]
- 14.Devoto L, Kohen P, Castro O, et al. Multihormone regulation of progesterone synthesis in cultured human midluteal cells. J. Clin. Endocrinol. Metab. 1995;80:1556–1570. doi: 10.1210/jcem.80.5.7745001. [DOI] [PubMed] [Google Scholar]
- 15.Pelligrini S, Fuzzi B, Pratesi S, et al. In-vivo studies on ovarian insulin-like growth factor-I concentrations in human preovulatory follicles and human ovarian circulation. Hum. Reprod. 1995;10:1341–1345. doi: 10.1093/humrep/10.6.1341. [DOI] [PubMed] [Google Scholar]
- 16.Ovesen P, Vahl N, Fisker S, et al. Increased pulsatile, but not basal, growth hormone secretion rates and plasma insulin-like growth factor-I levels during the preovulatory interval in normal women. J. Clin. Endocrinol. Metab. 1998;83:1662–1667. doi: 10.1210/jcem.83.5.4761. [DOI] [PubMed] [Google Scholar]
- 17.Klein NA, Battaglia DE, Miller PB, et al. Ovarian follicular development and the follicular fluid hormones and growth factors in normal women of advanced reproductive age. J. Clin. Endocrinol. Metab. 1996;81:1946–1951. doi: 10.1210/jcem.81.5.8626862. [DOI] [PubMed] [Google Scholar]
- 18.Amato G, Izzo A, Tucker A, Bellastella A. Insulin-like growth factor binding protein-3 reduction in follicular fluid in spontaneous and stimulated cycles. Fertil. Steril. 1998;70:141–144. doi: 10.1016/s0015-0282(98)00115-0. [DOI] [PubMed] [Google Scholar]
- 19.Lindsay R, Gallagher JC, Kleerekoper M, Pickar JH. Effect of low doses of conjugated equine estrogens with and without medroxyprogesterone acetate on bone in early postmenopausal women. JAMA. 2002;287:2668–2676. doi: 10.1001/jama.287.20.2668. [DOI] [PubMed] [Google Scholar]
- 20.Rauch F. Fetal and neonatal bone development. In: Rosen CJ, editor. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 7th ed. Washington, D.C.: American Society for Bone and Mineral Research; 2008. pp. 72–74. [Google Scholar]
- 21.Kovacs CS, Kronenberg HM. Maternal-fetal calcium and bone metabolism during pregnancy, puerperium, and lactation. Endocrine Rev. 1997;18:832–872. doi: 10.1210/edrv.18.6.0319. [DOI] [PubMed] [Google Scholar]
- 22.Schoenau E, Rauch F. Biochemical markers of bone metabolism. In: Glorieux FH, Pettifor J, Jueppner H, editors. Pediatric Bone. San Diego, CA, USA: Academic Press; 2003. pp. 339–357. [Google Scholar]
- 23.Trotter M, Hixon BB. Sequential changes in weight, density, and percentage ash weight of human skeletons from an early fetal period through old age. Anat. Rec. 1974;179:1–18. doi: 10.1002/ar.1091790102. [DOI] [PubMed] [Google Scholar]
- 24.Rauch F, Schoenau E. Changes in bone density during childhood and adolescence: an approach based on bone’s biological organization. J. Bone Miner. Res. 2001;16:597–604. doi: 10.1359/jbmr.2001.16.4.597. [DOI] [PubMed] [Google Scholar]
- 25.Heaney RP, Abrams S, Dawson-Hughes B, et al. Peak bone mass. Osteoporosis. Int. 2000;11:985–1009. doi: 10.1007/s001980070020. [DOI] [PubMed] [Google Scholar]
- 26.Seeman E. From density to structure: growing up and growing old on the surfaces of bone. J. Bone Miner. Res. 1997;12:509–521. doi: 10.1359/jbmr.1997.12.4.509. [DOI] [PubMed] [Google Scholar]
- 27.Duan Y, Beck TJ, Wang X-F, Seeman E. Structural and biomechanical basis of sexual dimorphism in femoral neck fragility has its origins in growth and aging. J. Bone Miner. Res. 2003;18:1766–1774. doi: 10.1359/jbmr.2003.18.10.1766. [DOI] [PubMed] [Google Scholar]
- 28.McGuigan FE, Murray L, Gallagher A, et al. Genetic and environmental determinants of peak bone mass in young men and women. J. Bone Miner. Res. 2002;17:1273–1279. doi: 10.1359/jbmr.2002.17.7.1273. [DOI] [PubMed] [Google Scholar]
- 29.Timpson NJ, Tobias JH, Richards JB, et al. Common variants in the region around Osterix are associated with bone mineral density and growth in childhood. Hum Mol Genet. 2009;18:1510–1517. doi: 10.1093/hmg/ddp052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bass S, Delmas PD, Pearce G, et al. The differing tempo of growth in bone size, mass and density in girls is region-specific. J. Clin. Invest. 1999;104:795–804. doi: 10.1172/JCI7060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rauch F, Travers R, Glorieux FH. Intracortical remodeling during human bone development: a histomorphometric study. Bone. 2007;40:274–280. doi: 10.1016/j.bone.2006.09.012. [DOI] [PubMed] [Google Scholar]
- 32.Bailey DA, McKay HA, Mirwald RL, et al. A six-year longitudinal study of the relationship of physical activity to bone mineral accrual in growing children: The University of Saskatchewan Bone Mineral Accrual Study. J. Bone Miner. Res. 1999;14:1672–1679. doi: 10.1359/jbmr.1999.14.10.1672. [DOI] [PubMed] [Google Scholar]
- 33.Bonjour JP, Theintz G, Buchs B, et al. Critical years and stages of puberty for spinal and femoral bone mass accumulation during adolescence. J. Clin. Endocrinol. Metab. 1991;73:555–563. doi: 10.1210/jcem-73-3-555. [DOI] [PubMed] [Google Scholar]
- 34.Bachrach LK, Hastie T, Wang M-C, et al. Bone mineral acquisition in healthy Asian, Hispanic, Black, and Caucasian youth. A longitudinal study. J. Clin. Endocrinol. Metab. 1999;84:4702–4712. doi: 10.1210/jcem.84.12.6182. [DOI] [PubMed] [Google Scholar]
- 35.Kalkwarf HJ, Zemel BS, Gilsanz V, et al. The Bone Mineral Density in Childhood Study: Bone mineral content and density according to age, sex, and race. J. Clin. Endocrinol. Metab. 2007;92:2087–2099. doi: 10.1210/jc.2006-2553. [DOI] [PubMed] [Google Scholar]
- 36.Gilsanz V, Boechat MI, Rote TF, et al. Gender differences in vertebral body sizes in children and adolescents. Radiology. 1994;190:673–677. doi: 10.1148/radiology.190.3.8115609. [DOI] [PubMed] [Google Scholar]
- 37.Tommasini SM, Nasser P, Jepsen KH. Sexual dimorphism affects tibia size and shape but not tissue-level mechanical properties. Bone. 2007;40:498–505. doi: 10.1016/j.bone.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 38.Lu PW, Cowell CT, Lloyd-Jones SA, et al. Volumetric bone mineral density in normal subjects, aged 5–27 years. J. Clin. Endocrinol. Metab. 1996;81:1586–1590. doi: 10.1210/jcem.81.4.8636372. [DOI] [PubMed] [Google Scholar]
- 39.Gilsanz V, Skaggs DL, Kovanlikaya A, et al. Differential effect of race on the axial and appendicular skeletons of children. J. Clin. Endocrinol. Metab. 1998;83:1420–1427. doi: 10.1210/jcem.83.5.4765. [DOI] [PubMed] [Google Scholar]
- 40.Skaggs DL, Loro ML, Pitukcheewanont P, et al. Increased body weight and decreased radial cross sectional dimensions in girls with forearm fractures. J. Bone Miner. Res. 2001;16:1337–1342. doi: 10.1359/jbmr.2001.16.7.1337. [DOI] [PubMed] [Google Scholar]
- 41.Neu CM, Rauch F, Manz F, Schoenau E. Modeling of cross-sectional bone size, mass and geometry at the proximal radius: A study of normal bone development using peripheral quantitative computed tomography. Osteoporos. Int. 2001;12:538–547. doi: 10.1007/s001980170074. [DOI] [PubMed] [Google Scholar]
- 42.Schoenau E, Neu CM, Rauch F, Manz F. Gender-specific pubertal changes in volumetric cortical bone mineral density at the proximal radius. Bone. 2002;110:110–113. doi: 10.1016/s8756-3282(02)00802-5. [DOI] [PubMed] [Google Scholar]
- 43.Neu CM, Manz F, Rauch F, et al. Bone densities and bone size at the proximal radius in healthy children adolescents: A study using quantitative computed tomography. Bone. 2001;28:227–232. doi: 10.1016/s8756-3282(00)00429-4. [DOI] [PubMed] [Google Scholar]
- 44.Kontulainen SA, Macdonald HM, McKay HA. Change in cortical bone density and its distribution differs between boys and girls during puberty. J. Clin. Endocrinol. Metab. 2006;91:2555–2561. doi: 10.1210/jc.2006-0136. [DOI] [PubMed] [Google Scholar]
- 45.Macdonald H, Kontulainen SA, Petit M, et al. Bone strength and its determinants in pre- and early pubertal boys and girls. Bone. 2006;39:598–608. doi: 10.1016/j.bone.2006.02.057. [DOI] [PubMed] [Google Scholar]
- 46.Garn SM. The course of bone gain and the phases of bone loss. Orthop. Clin. North Am. 1972;3:503–520. [PubMed] [Google Scholar]
- 47.Gilsanz V, Kovanlikaya A, Costin G, et al. Differential effect of gender on the size of the bones in the axial and appendicular skeletons. J. Clin. Endocrinol. Metab. 1997;82:1603–1607. doi: 10.1210/jcem.82.5.3942. [DOI] [PubMed] [Google Scholar]
- 48.Kontulainen SA, Macdonald HM, Khan KK, McKay HA. Examining bone surfaces across puberty: A 20-month pQCT trial. J. Bone Miner. Res. 2005;20:1202–1207. doi: 10.1359/JBMR.050214. [DOI] [PubMed] [Google Scholar]
- 49.Frost HM. Bone “mass” and the “mechanostat”: a proposal. Anat. Rec. 1987;219:1–9. doi: 10.1002/ar.1092190104. [DOI] [PubMed] [Google Scholar]
- 50.Wang O, Nicholson PHF, Suuriniemi M, et al. Relationship of sex hormones to bone geometric properties and mineral density in early pubertal girls. J. Clin. Endocrinol. Metab. 2004;89:1698–1703. doi: 10.1210/jc.2003-031113. [DOI] [PubMed] [Google Scholar]
- 51.Hogler W, Blimkie CJR, Cowell CT, et al. A comparison of bone geometry and cortical density at the mid-femur between prepuberty and young adulthood using magnetic resonance imaging. Bone. 2003;33:771–778. doi: 10.1016/s8756-3282(03)00266-7. [DOI] [PubMed] [Google Scholar]
- 52.Kirmani S, Christen D, van Lenthe H, et al. Bone structure at the distal radius during adolescent growth. J Bone Miner Res. 2009;24:1033–1042. doi: 10.1359/JBMR.081255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Richards JB, Rivadeneira F, Inouye M, et al. Bone mineral density, osteoporosis, and osteoporotic fractures: a genome-wide association study. Lancet. 2008;371:1505–1512. doi: 10.1016/S0140-6736(08)60599-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.MacKelvie KJ, Khan KM, McKay HA. Is there a critical period for bone response to weight-bearing exercise in children and adolescents? A systematic review. Br. J. Sports Med. 2002;36:250–257. doi: 10.1136/bjsm.36.4.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kontulainen S, Sievanen H, Kannus P, et al. Effect of long-term impact-loading on mass, size, and estimated strength of humerus and radius in female racquet sports players: A peripheral quantitative computed tomography study between young and old starters and controls. J. Bone Miner. Res. 2002;17:2281–2289. doi: 10.1359/jbmr.2002.17.12.2281. [DOI] [PubMed] [Google Scholar]
- 56.Wosje KS, Specker BL. Role of calcium in bone health during childhood. Nutr. Rev. 2000;58:253–268. doi: 10.1111/j.1753-4887.2000.tb01879.x. [DOI] [PubMed] [Google Scholar]
- 57.Kalkwarf HJ, Khoury HC, Lanphear BP. Milk intake during childhood and adolescence, adult bone density, and osteoporotic fractures in US women. Am. J. Clin. Nutr. 2003;77:257–265. doi: 10.1093/ajcn/77.1.257. [DOI] [PubMed] [Google Scholar]
- 58.Goulding A, Rockwell JE, Black RE, et al. Children who avoid drinking cow’s milk are at increased risk for prepubertal bone fractures. J. Am. Diet. Assn. 2004;104:250–253. doi: 10.1016/j.jada.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 59.Winzenberg T, Shaw K, Fryer J, Jones G. Effects of calcium supplementation on bone density in healthy children: Meta-analysis of randomized controlled trials. BMJ. 2006;14:333–775. doi: 10.1136/bmj.38950.561400.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cooper C, Dennison EM, Leufkens HG, et al. Epidemiology of childhood fractures in Britain: A study using the General Practice Research Database. J. Bone Miner. Res. 2005;19:1976–1981. doi: 10.1359/JBMR.040902. [DOI] [PubMed] [Google Scholar]
- 61.Khosla S, Melton LJ, III, Dekutoski MB, et al. Incidence of childhood distal forearm fractures over 30 years: A population-based study. JAMA. 2003;290:1479–1485. doi: 10.1001/jama.290.11.1479. [DOI] [PubMed] [Google Scholar]
- 62.Goulding A, Grant AM, Williams SM. Bone and body composition of children and adolescents with repeated forearm fractures. J. Bone Miner. Res. 2005;20:2090–2096. doi: 10.1359/JBMR.050820. [DOI] [PubMed] [Google Scholar]
- 63.Jones G, Ma D, Cameron F. Bone density interpretation and relevance in Caucasian children aged 9–17 years of age: Insights from a population-based fracture study. J. Clin. Densitom. 2006;9:202–209. doi: 10.1016/j.jocd.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 64.Kovacs CS, Kronenberg HM. Maternal-fetal calcium and bone metabolism during pregnancy, puerperium, and lactation. Endocrine Rev. 1997;18:832–872. doi: 10.1210/edrv.18.6.0319. [DOI] [PubMed] [Google Scholar]
- 65.Kovacs CS, El-Hajj Fuleihan G. Calcium and bone disorders during pregnancy and lactation. Endocrinol. Metab. Clin. North Am. 2006;35:21–51. doi: 10.1016/j.ecl.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 66.Kovacs CS, Lanske B, Hunzelman JL, et al. Parathyroid hormone-related peptide (PTHrp) regulates fetal-placental calcium transport through a receptor distinct from the PTH/PTHrp receptor. Proc. Natl. Acad. Sci. USA. 1996;93:15233–15238. doi: 10.1073/pnas.93.26.15233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cornish J, Callon KE, Nicholson GC, Reid IR. Parathyroid hormone-related protein-(107–139) inhibits bone resorption in vivo. Endocrinology. 1997;138:1299–1304. doi: 10.1210/endo.138.3.4990. [DOI] [PubMed] [Google Scholar]
- 68.Black AJ, Topping J, Durham B, et al. A detailed assessment of alterations in bone turnover, calcium homeostasis, and bone density in normal pregnancy. J. Bone Miner. Res. 2000;15:557–563. doi: 10.1359/jbmr.2000.15.3.557. [DOI] [PubMed] [Google Scholar]
- 69.Sowers M. Pregnancy and lactation as risk factors for subsequent bone loss and osteoporosis. J. Bone Miner. Res. 1996;11:1052–1060. doi: 10.1002/jbmr.5650110803. [DOI] [PubMed] [Google Scholar]
- 70.Kalkwarf HJ, Specker BL, Bianchi DC, et al. The effect of calcium supplementation on bone density during lactation and after weaning. New Engl. J. Med. 1997;337:523–528. doi: 10.1056/NEJM199708213370803. [DOI] [PubMed] [Google Scholar]
- 71.Polatti F, Capuzzo E, Viazzo F, et al. Bone mineral changes during and after lactation. Obstet. Gynecol. 1999;94:52–56. doi: 10.1016/s0029-7844(99)00236-7. [DOI] [PubMed] [Google Scholar]
- 72.Riggs BL, Khosla S, Melton LJ. Sex steroids and the construction and conservation of the adult skeleton. Endocrine Rev. 2002;23:279–302. doi: 10.1210/edrv.23.3.0465. [DOI] [PubMed] [Google Scholar]
- 73.Riggs BL, Melton LJ, III, Robb RA, et al. A population-based study of age and sex differences in bone volumetric density, size, geometry and structure at different skeletal sites. J. Bone Miner. Res. 2004;19:1945–1954. doi: 10.1359/JBMR.040916. [DOI] [PubMed] [Google Scholar]
- 74.Hanson DP, Robb RA, Aharon S, et al. New software toolkits for comprehensive visualization and analysis of three-dimensional multimodal biomedical images. J. Digit. Imaging. 1997;10 3 Suppl 1:229–230. doi: 10.1007/BF03168711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Khosla S, Riggs BL, Atkinson EJ, et al. Effects of sex and age on bone microstructure at the ultradistal radius: a population-based noninvasive in vivo assessment. J. Bone Miner. Res. 2006;21:124–131. doi: 10.1359/JBMR.050916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Khosla S, Melton LJ, 3rd, Achenbach SJ, et al. Hormonal and biochemical determinants of trabecular microstructure at the ultradistal radius in women and men. J. Clin. Endocrinol. Metab. 2006;91:885–891. doi: 10.1210/jc.2005-2065. [DOI] [PubMed] [Google Scholar]
- 77.Riggs BL, Melton LJ, Robb RA, et al. A population-based assessment of rates of bone loss at multiple skeletal sites: evidence for substantial trabecular bone loss in young adult women and men. J. Bone Miner. Res. 2008;23:205–214. doi: 10.1359/JBMR.071020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cummings SR, Melton LJ. Epidemiology and outcomes of osteoporotic fractures. Lancet. 2002;359:1761–1767. doi: 10.1016/S0140-6736(02)08657-9. [DOI] [PubMed] [Google Scholar]
- 79.Tosteson AN, Hammond CS. Quality of life assessment in osteoporosis: health-status and preference-based measures. Pharmacogenomics. 2002;20:289–303. doi: 10.2165/00019053-200220050-00001. [DOI] [PubMed] [Google Scholar]
- 80.Lindsay R, Aitken JM, Anderson JB, et al. Long-term prevention of postmenopausal osteoporosis by oestrogen: evidence for an increased bone mass after delayed onset of oestrogen treatment. Lancet. 1976;1(7968):1038–1041. doi: 10.1016/s0140-6736(76)92217-0. [DOI] [PubMed] [Google Scholar]
- 81.Genant HK, Cann CE, Ettinger B, et al. Quantitative computed tomography of vertebral spongiosa: a sensitive method for detecting early bone loss after oophorectomy. Ann. Intern. Med. 1982;97:699–705. doi: 10.7326/0003-4819-97-5-699. [DOI] [PubMed] [Google Scholar]
- 82.Khosla S, Atkinson EJ, Melton LJ, III, et al. Effects of age and estrogen status on serum parathyroid hormone levels and biochemical markers of bone turnover in women: a population-based study. J. Clin. Endocrinol. Metab. 1997;82:1522–1527. doi: 10.1210/jcem.82.5.3946. [DOI] [PubMed] [Google Scholar]
- 83.Horton R, Romanoff E, Walker J. Androstenedione and testosterone in ovarian venous and peripheral plasma during ovariectomy for breast cancer. J. Clin. Endocrinol. Metab. 1966;26:1267–1269. doi: 10.1210/jcem-26-11-1267. [DOI] [PubMed] [Google Scholar]
- 84.Khosla S, Riggs BL, Robb RA, et al. Relationship of volumetric bone density and structural parameters at different skeletal sites to sex steroid levels in women. J. Clin. Endocrinol. Metab. 2005;90:5096–5103. doi: 10.1210/jc.2005-0396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Perrien DS, Achenbach SJ, Bledsoe SE, et al. Bone turnover across the menopause transition: correlations with inhibins and follicle-stimulating hormone. J. Clin. Endocrinol. Metab. 2006;91:1848–1854. doi: 10.1210/jc.2005-2423. [DOI] [PubMed] [Google Scholar]
- 86.Garnero P, Sornay-Rendu E, Chapuy M, et al. Increased bone turnover in late postmenopausal women is a major determinant of osteoporosis. J. Bone Miner. Res. 1996;11:337–349. doi: 10.1002/jbmr.5650110307. [DOI] [PubMed] [Google Scholar]
- 87.Young MM, Nordin BE. Calcium metabolism and the menopause. Proc. Roy. Soc. Med. 1967;60(11 Part 1):1137–1138. doi: 10.1177/003591576706011P133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gennari C, Agnusdei D, Nardi P, et al. Estrogen preserves a normal intestinal responsiveness to 1,25-dihydroxyvitamin D3 in oophorectomized women. J. Clin. Endocrinol. Metab. 1990;71:1288–1293. doi: 10.1210/jcem-71-5-1288. [DOI] [PubMed] [Google Scholar]
- 89.Riggs BL, Khosla S, Melton LJ. A unitary model for involutional osteoporosis: estrogen deficiency causes both type I and type II osteoporosis in postmenopausal women and contributes to bone loss in aging men. J Bone Miner. Res. 1998;13:763–773. doi: 10.1359/jbmr.1998.13.5.763. [DOI] [PubMed] [Google Scholar]
- 90.Lacey DL, Timms E, Tan HL, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–176. doi: 10.1016/s0092-8674(00)81569-x. [DOI] [PubMed] [Google Scholar]
- 91.Eghbali-Fatourechi G, Khosla S, Sanyal A, et al. Role of RANK ligand in mediating increased bone resorption in early postmenopausal women. J. Clin. Invest. 2003;111:1221–1230. doi: 10.1172/JCI17215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Clowes JA, Riggs BL, Khosla S. The role of the immune system in the pathophysiology of osteoporosis. Immunol. Rev. 2005;208:207–227. doi: 10.1111/j.0105-2896.2005.00334.x. [DOI] [PubMed] [Google Scholar]
- 93.Hsu H, Lacey DL, Dunstan CR, et al. Tumor necrosis factor receptor family member RANK mediates osteoclast differentiation and activation induced by osteoprotegerin ligand. Proc. Natl. Acad. Sci. USA. 1999;96:3540–3545. doi: 10.1073/pnas.96.7.3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Simonet WS, Lacey DL, Dunstan CR, et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309–319. doi: 10.1016/s0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- 95.Hofbauer LC, Khosla S, Dunstan CR, et al. Estrogen stimulates gene expression and protein production of osteoprotegerin in human osteoblastic cells. J. Clin. Endocrinol. Metab. 1999;140:4367–4370. doi: 10.1210/endo.140.9.7131. [DOI] [PubMed] [Google Scholar]
- 96.Khosla S, Atkinson EJ, Dunstan CR, et al. Effect of estrogen versus testosterone on circulating osteoprotegerin and other cytokine levels in normal elderly men. J. Clin. Endocrinol. Metab. 2002;87:1550–1554. doi: 10.1210/jcem.87.4.8397. [DOI] [PubMed] [Google Scholar]
- 97.Charatcharoenwitthaya N, Khosla S, Atkinson EJ, et al. Effect of blockade of TNF-alpha and interleukin-1 action on bone resorption in early postmenopausal women. J. Bone Miner. Res. 2000;22:724–729. doi: 10.1359/jbmr.070207. [DOI] [PubMed] [Google Scholar]
- 98.Manolagas SC, Jilka RL. Bone marrow, cytokines, and bone remodeling: emerging insights into the pathophysiology of osteoporosis. N. Engl. J. Med. 1995;332:305–311. doi: 10.1056/NEJM199502023320506. [DOI] [PubMed] [Google Scholar]
- 99.Pacifici R, Brown C, Puscheck E, et al. Effect of surgical menopause and estrogen replacement on cytokine release from human blood mononuclear cells. Proc. Natl. Acad. Sci. USA. 1991;88:5134–5188. doi: 10.1073/pnas.88.12.5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tanaka S, Takahashi N, Udagawa N, et al. Macrophage colony-stimulating factor is indispensable for both proliferation and differentiation of osteoclast progenitors. J. Clin. Invest. 1993;91:257–263. doi: 10.1172/JCI116179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kimble RB, Vannice JL, Bloedow DC, et al. Interleukin-1 receptor antagonist decreased bone loss and bone resorption in ovariectomized rats. J. Clin. Invest. 1994;93:1959–1967. doi: 10.1172/JCI117187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ammann P, Rizzoli R, Bonjour J, et al. Transgenic mice expressing soluble tumor necrosis factor-receptor are protected against bone loss caused by estrogen deficiency. J. Clin. Invest. 1997;99:1699–1703. doi: 10.1172/JCI119333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kimble RB, Srivastava S, Ross FP, et al. Estrogen deficiency increases the ability of stromal cells to support murine osteoclastogenesis via an interleukin-1- and tumor necrosis factor-mediated stimulation of macrophage colony-stimulating factor production. J. Biol. Chem. 1996;271:28890–28897. doi: 10.1074/jbc.271.46.28890. [DOI] [PubMed] [Google Scholar]
- 104.Kitazawa R, Kimble RB, Vannice JL, et al. Interleukin-1 receptor antagonist and tumor necrosis factor binding protein decrease osteoclast formation and bone resorption in ovariectomized mice. J. Clin. Invest. 1994;94:2397–2406. doi: 10.1172/JCI117606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Girasole G, Jilka RL, Passeri G, et al. 17 beta-estradiol inhibits interleukin-6 production by bone marrow-derived stromal cells and osteoblasts in vitro: a potential mechanism for the antiosteoporotic effect of estrogens. J. Clin. Invest. 1992;89:883–891. doi: 10.1172/JCI115668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jilka RL, Hangoc G, Girasole G, et al. Increased osteoclast development after estrogen loss: mediation by interleukin-6. Science. 1992;257(5066):88–91. doi: 10.1126/science.1621100. [DOI] [PubMed] [Google Scholar]
- 107.Oursler MJ, Cortese C, Keeting PE, et al. Modulation of transforming growth factor-beta production in normal human osteoblast-like cells by 17 beta-estradiol and parathyroid hormone. Endocrinology. 1991;129:3313–3320. doi: 10.1210/endo-129-6-3313. [DOI] [PubMed] [Google Scholar]
- 108.Hughes DE, Dai A, Tiffee JC, et al. Estrogen promotes apoptosis of murine osteoclasts mediated by TGF-beta. Nat. Med. 1996;2:1132–1136. doi: 10.1038/nm1096-1132. [DOI] [PubMed] [Google Scholar]
- 109.Shevde NK, Bendixon AC, Dienger KM, et al. Estrogens suppress RANK ligand-induced osteoclast differentiation via a stromal cell independent mechanism involving c-Jun repression. Proc. Natl. Acad. Sci. USA. 2000;97:7829–7834. doi: 10.1073/pnas.130200197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Srivastava S, Toraldo G, Weitzmann MN, et al. Estrogen decreases osteoclast formation by down-regulating receptor activator of NF-κB ligand (RANKL)-induced JNK activation. J. Biol. Chem. 2001;276:8836–8840. doi: 10.1074/jbc.M010764200. [DOI] [PubMed] [Google Scholar]
- 111.Oursler MJ, Pederson L, Fitzpatrick LA, et al. Human giant cell tumors of the bone (osteoclastomas) are estrogen target cells. Proc. Natl. Acad. Sci. USA. 1994;91:5227–5231. doi: 10.1073/pnas.91.12.5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Frost HM. On the estrogen-bone relationship and postmenopausal bone loss: a new model. J. Bone Miner. Res. 1999;14:1473–1477. doi: 10.1359/jbmr.1999.14.9.1473. [DOI] [PubMed] [Google Scholar]
- 113.Imam A, Blair HC, Davies TF, et al. Role of the pituitary-bone axis in skeletal pathophysiology. Curr. Opin. Endocrinol. Diab. Obes. 2009;16:423–429. doi: 10.1097/MED.0b013e3283328aee. [DOI] [PubMed] [Google Scholar]
- 114.Ledger GA, Burritt MF, Kao PC, et al. Role of parathyroid hormone in mediating nocturnal and age-related increases in bone resorption. J. Clin. Endocrinol. Metab. 1995;80:3304–3310. doi: 10.1210/jcem.80.11.7593443. [DOI] [PubMed] [Google Scholar]
- 115.Holick MF. Vitamin D deficiency. N. Engl. J. Med. 2007;357:266–281. doi: 10.1056/NEJMra070553. [DOI] [PubMed] [Google Scholar]
- 116.Gallagher JC, Riggs BL, DeLuca HF. Effect of estrogen on calcium absorption and serum vitamin D metabolites in postmenopausal osteoporosis. J. Clin. Endocrinol. Metab. 1980;51:1359–1364. doi: 10.1210/jcem-51-6-1359. [DOI] [PubMed] [Google Scholar]
- 117.Nordin BE, Need AG, Morris HA, et al. Evidence for a renal calcium leak in postmenopausal women. J. Clin. Endocrinol. Metab. 1991;72:401–407. doi: 10.1210/jcem-72-2-401. [DOI] [PubMed] [Google Scholar]
- 118.McKane WR, Khosla S, Burritt MF, et al. Mechanism of renal calcium conservation with estrogen replacement therapy in women in early postmenopause – a clinical research center study. J. Clin. Endocrinol. Metab. 1995;80:3458–3464. doi: 10.1210/jcem.80.12.8530583. [DOI] [PubMed] [Google Scholar]
- 119.Idris AI, Sophocleous A, Landau-Bassogna E, et al. Cannabinoid receptor type 1 protects against age-related osteoporosis by regulating osteoblast and adipocyte differentiation by marrow stromal cells. Cell Metab. 2009;10:139–147. doi: 10.1016/j.cmet.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 120.Tintut Y, Parhami F, Le V, et al. Inhibition of osteoblast-specific transcription factor Cbfa-1 by the cyclic AMP pathway in osteoblastic cells. Ubiquitin/proteosome-dependent regulation. J. Biol. Chem. 1999;274:28875–28879. doi: 10.1074/jbc.274.41.28875. [DOI] [PubMed] [Google Scholar]
- 121.Fox KE, Fankell DM, Erickson PF, et al. Depletion of cAMP-response element-binding protein/ATF1 inhibits adipogenic conversion of 3T3-L1 cells ectopically expressing CCAAT/enhancer-binding protein (C/EBP) alpha, C/EBP beta, or PPAR gamma 2. J. Biol. Chem. 2006;281:40341–40353. doi: 10.1074/jbc.M605077200. [DOI] [PubMed] [Google Scholar]
- 122.Marie PJ, Hott M, Launay JM, et al. In vitro production of cytokines by bone surface-derived osteoblastic cells in normal and osteoporotic postmenopausal women: relationship with cell proliferation. J. Clin. Endocrinol. Metab. 1993;77:824–830. doi: 10.1210/jcem.77.3.8370705. [DOI] [PubMed] [Google Scholar]
- 123.Giustina A, Veldhuis JD. Pathophysiology of the neuroregulation of growth hormone secretion in experimental animals and the human. Endocrine Rev. 1998;19:717–797. doi: 10.1210/edrv.19.6.0353. [DOI] [PubMed] [Google Scholar]
- 124.Ho KY, Evans WS, Blizzard WM, et al. Effects of sex and age on the 24-hour profile of growth hormone secretion in man: importance of endogenous estradiol concentrations. J. Clin. Endocrinol. Metab. 1987;64:51–58. doi: 10.1210/jcem-64-1-51. [DOI] [PubMed] [Google Scholar]
- 125.Bennett AE, Wahner HW, Riggs BL, et al. Insulin-like growth factors I and II, aging and bone density in women. J. Clin. Endocrinol. Metab. 1984;59:701–704. doi: 10.1210/jcem-59-4-701. [DOI] [PubMed] [Google Scholar]
- 126.Boonen S, Mohan S, Dequeker J, et al. Down-regulation of the serum stimulatory components of the insulin-like growth factor (IGF) system (IGF-I, IGF-II, IGF binding protein [BP]-3, and IGFBP-5) in age-related (type II) femoral neck osteoporosis. J. Bone Miner. Res. 1999;14:2150–2158. doi: 10.1359/jbmr.1999.14.12.2150. [DOI] [PubMed] [Google Scholar]
- 127.Pfeilschifter J, Diel I, Kloppinger T, et al. Concentrations of insulin-like growth factor (IGF)-I, -II, and IGF binding protein-4 and -5 in human bone cell conditioned medium did not change with age. Mech. Aging Dev. 2000;117:109–114. doi: 10.1016/s0047-6374(00)00132-9. [DOI] [PubMed] [Google Scholar]
- 128.Samuels A, Perry MJ, Goodship AE, et al. Is high-dose estrogen-induced osteogenesis in the mouse mediated by an estrogen receptor? Bone. 1999;27:41–46. doi: 10.1016/s8756-3282(00)00289-1. [DOI] [PubMed] [Google Scholar]
- 129.Heaney RP, Recker RR, Saville PD. Menopausal changes in calcium balance performance. J. Lab. Clin. Med. 1978;92:953–963. [PubMed] [Google Scholar]
- 130.Ernst M, Heath JK, Rodan GA. Estradiol effects on proliferation, messenger ribonucleic acid for collagen and insulin-like growth factor-I, and parathyroid hormone-stimulated adenylate cyclase activity in osteoblastic cells from calvariae and long bones. Endocrinology. 1989;125:825–833. doi: 10.1210/endo-125-2-825. [DOI] [PubMed] [Google Scholar]
- 131.Manolagas SC. Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocrine Rev. 2000;21:115–137. doi: 10.1210/edrv.21.2.0395. [DOI] [PubMed] [Google Scholar]
- 132.Gohel A, McCarthy MB, Gronowicz G. Estrogen prevents glucocorticoid-induced apoptosis in osteoblasts in vivo and in vitro. Endocrinology. 1999;140:5339–5347. doi: 10.1210/endo.140.11.7135. [DOI] [PubMed] [Google Scholar]
- 133.Khastgir G, Studd J, Holland N, et al. Anabolic effect of estrogen replacement on bone in postmenopausal women with osteoporosis: histomorphometric evidence in a longitudinal study. J. Clin. Endocrinol. Metab. 2001;86:289–295. doi: 10.1210/jcem.86.1.7161. [DOI] [PubMed] [Google Scholar]
- 134.Tobias JH, Compston JE. Does estrogen stimulate osteoblast function in postmenopausal women? Bone. 1999;24:121–124. doi: 10.1016/s8756-3282(98)00156-2. [DOI] [PubMed] [Google Scholar]
- 135.van Poznak C, Hannon RA, Mackey JR, et al. Prevention of aromatase inhibitor-induced bone loss using risedronate: the SABRE Trial. J. Clin. Oncol. 2010;28:967–975. doi: 10.1200/JCO.2009.24.5902. [DOI] [PubMed] [Google Scholar]
- 136.Labrie F, Belanger A, Cusan L, et al. Marked decline in serum concentrations of adrenal C19 sex steroid precursors and conjugated androgen metabolites during aging. J. Clin. Endocrinol. Metab. 1997;82:2396–2402. doi: 10.1210/jcem.82.8.4160. [DOI] [PubMed] [Google Scholar]
- 137.Seeman E. From density to structure: growing up and growing old on the surfaces of bone. J. Bone Miner. Res. 1997;12:509–521. doi: 10.1359/jbmr.1997.12.4.509. [DOI] [PubMed] [Google Scholar]
- 138.Melton LJ, 3rd, Riggs BL, Achenbach SJ, et al. Does reduced skeletal loading account for age-related bone loss? J. Bone Miner. Res. 2006;21:1847–1855. doi: 10.1359/jbmr.060908. [DOI] [PubMed] [Google Scholar]
- 139.Riggs BL, Melton LJ. Medical progress series: Involutional osteoporosis. N. Engl. J. Med. 1986;314:1676–1686. doi: 10.1056/NEJM198606263142605. [DOI] [PubMed] [Google Scholar]
- 140.Frost HM. On age-related bone loss: insights from a new paradigm. J. Bone Miner. Res. 1997;12:1539–1546. doi: 10.1359/jbmr.1997.12.10.1539. [DOI] [PubMed] [Google Scholar]
- 141.Frost HM. Why the ISMNI and the Utah paradigm? Their role in skeletal and extraskeletal disorders. J. Musculoskelet. Neuronal. Interact. 2000;1:5–9. [PubMed] [Google Scholar]