

Abstract
Epigenetic silencing plays an important role in cancer development. An attractive hypothesis is that local DNA features may participate in differential predisposition to gene hypermethylation. We found that, compared with methylation-resistant genes, methylation-prone genes have a lower frequency of SINE and LINE retrotransposons near their transcription start site. In several large testing sets, this distribution was highly predictive of promoter methylation. Genome-wide analysis showed that 22% of human genes were predicted to be methylation-prone in cancer; these tended to be genes that are down-regulated in cancer and that function in developmental processes. Moreover, retrotransposon distribution marks a larger fraction of methylation-prone genes compared to Polycomb group protein (PcG) marking in embryonic stem cells; indeed, PcG marking and our predictive model based on retrotransposon frequency appear to be correlated but also complementary. In summary, our data indicate that retrotransposon elements, which are widespread in our genome, are strongly associated with gene promoter DNA methylation in cancer and may in fact play a role in influencing epigenetic regulation in normal and abnormal physiological states.
DNA methylation, a common cause of gene silencing in human neoplasia, does not affect the genome homogeneously: While some genes are frequently found methylated in cancer, others are never or rarely targeted by such modification. The causes of selective predisposition to methylation are unknown, and two main explanations have been proposed: (1) DNA methylation happens randomly, and cells with silencing of genes that promote tumorigenesis have a selective advantage; and (2) some genes are preferentially targeted by silencing complexes, and thus DNA methylation occurs through an instructive mechanism (Baylin and Bestor 2002; Jones and Baylin 2002; Feltus et al. 2006). The latter explanation appears more likely because genes with similar function show differential predisposition to methylation. The DNA repair genes MLH1 (frequently methylated) and MSH2 (never methylated) are good examples of genes with similar function but differential predisposition to hypermethylation in cancer (Kondo et al. 2000).
Previous studies were conducted to predict DNA methylation in individual CpG island promoters based on the instructive mechanism hypothesis, but despite efforts, the cause of such selective predisposition to aberrant methylation is still incompletely understood. Most commonly these studies employed support vector machines to annotate short DNA sequences enriched in methylated versus nonmethylated gene promoter CpG islands. Although important, these studies were focused on normal cells (Bock et al. 2006; Das et al. 2006) or artificially induced methylation (Feltus et al. 2006) rather than cancer cells, which is the main system where aberrant methylation occurs. An apparent lower frequency of SINE retrotransposons in methylation-prone genes was noticed in some of these studies, including a report on hyperconserved CpG domains (Tanay et al. 2007), but this association was not further evaluated. This is an important observation with possible biological consequences, because retrotransposons such as long and short interspersed nuclear elements (LINE and SINE, respectively) are targeted by epigenetic modifications to suppress their mobilization (Yoder et al. 1997), have been shown to act as methylation centers (Yates et al. 1999), and have a strong influence on gene expression in mammalian cells (Faulkner et al. 2009). In this study, we directly investigated whether CpG island–associated genes that undergo frequent aberrant DNA methylation in cancer have a unique distribution of repetitive elements.
Results
A predictive model of predisposition to DNA methylation in cancer
To answer whether methylation-prone genes in cancer have a different distribution of repetitive elements, we first compared the DNA sequence in the 4-kb region surrounding the transcription start site (TSS) of a training set composed of 36 methylation-prone and 36 methylation-resistant genes (Supplemental Table S1). Methylation analysis of the promoter region of these genes was done using quantitative methods (bisulfite-PCR followed by combined bisulfite restriction analysis [COBRA] or pyrosequencing analysis) in nine cancer cell lines. DNA from peripheral blood mononuclear cells of a healthy individual was used as a control (Supplemental Table S2). These nine cancer cell lines were selected because they were identified as heavily methylated in a previous study (Shen et al. 2007a). Thus, genes not found methylated in this group of cell lines are less likely to be found methylated elsewhere. The genes included in the training set were selected from our database of DNA methylation in cancer, and the criteria for inclusion were the following: (1) Methylation data were available for at least two cancer cell lines, (2) the gene promoter overlapped with or had a CpG island no more than 200 bp distant from the TSS, and (3) the CpG island was not methylated in the normal control. The classical Gardiner-Frommer definition of CpG islands was adopted in our study (Gardiner-Garden and Frommer 1987). As shown in Figure 1A, SINE and LINE repeats were approximately half as common in methylation-prone compared to methylation-resistant genes. Among the other repeat classes, both long terminal repeat (LTR) and DNA elements showed a lower frequency in methylation-prone genes; however, this difference was modest and not statistically significant. CpG island length, GC content, and CpG ratio were previously shown to be associated with methylation status in somatic tissues (Weber et al. 2007), but these variables were not significantly different between methylation-resistant and methylation-prone genes in cancer (Fig. 1B).
Figure 1.

Distribution of repetitive elements in methylation-prone versus methylation-resistant genes. (A) The abundance of repetitive elements of different classes was determined for the 4-kb sequence window centered in the TSS of 36 methylation-resistant (white) and 36 methylation-prone (black) genes. Retrotranposons of the SINE and LINE classes were found to be depleted in methylation-prone genes. *P < 0.02; **P < 0.12 (Student's t-test). (B) Average length, GC content, and CpG ratio of CpG islands were not significantly different between methylation-prone and methylation-resistant genes. Error bars represent SEM. (C) Abundance of SINE and LINE retrotransposons in the 20-kb sequence window centered in the TSS of 36 methylation-prone and 36 methylation-resistant genes. The abundance of SINE and LINE retrotransposons in all promoter CpG islands in the human genome is shown in gray. Note that the depletion of LINE retrotransposons is more significant in the −2-kb to +5-kb sequence window.
To gain further information about differences in the distribution of SINE and LINE repeats between methylation-prone and methylation-resistant genes, we extended the annotation to a 20-kb region centered in the TSS in 1-kb nonoverlapping windows. The depletion of SINE repeats in methylated genes spanned nearly the full 20-kb region, whereas the depletion of LINE repeats in methylated genes occurred mainly in a –2-kb to +5-kb window (Fig. 1C; Supplemental Fig. S1). We calculated the log-odds score of SINE and LINE retrotransposons per 1-kb window based on their distribution in the training set and in the full collection of human promoter CpG islands (Fig. 2). The sum of log-odds scores in the 20-kb region allowed us to quantify the similarity in distribution of SINE and LINE retrotransposons in a single gene promoter compared to the average distribution of these elements in methylation-prone and methylation-resistant genes. As a result, we could distinguish three groups in the training set: (1) genes depleted of both SINE and LINE repeats, thus predicted to be methylation-prone; (2) genes enriched for SINE and LINE repeats, predicted to be methylation-resistant; and (3) genes that were depleted of only one type of repeat. Comparison of these findings with the methylation data for each one of the 72 studied genes revealed that 19 of 23 genes (83%) predicted to be methylation-prone were actually hypermethylated in cancer and 23 of 25 genes (92%) predicted to be methylation-resistant were not or were rarely hypermethylated in cancer (Fig. 3A). Genes with discordant frequency of SINE and LINE repeats (30 of 72, 42%) seemed to represent a class of genes of intermediate predisposition to methylation. We observed that the distribution of retrotransposons in the core promoter (−1 kb to +2 kb centered in the TSS) is also predictive of methylation predisposition, however with less accuracy than the 20-kb region classification (75% of predicted methylation-prone genes were indeed methylated in cancer, and 63% of the methylation-resistant genes were unmethylated).
Figure 2.

SINE and LINE abundance score to predict gene predisposition to methylation in cancer. (A) Annotation of SINE and LINE retrotransposons near the promoter sequence of a representative methylation-prone gene (in this example, the CACNA1G gene). The promoter sequence was divided into 20 bins of 1-kb sequence each (10 bins upstream and 10 bins downstream of each gene TSS), and the presence of SINE and LINE retrotransposons was annotated for each bin. Note that each element was annotated to just one bin (the closest to TSS). The same procedure was followed for all human genes with CpG islands overlapping or no more than 200 bp from their TSS. (B) Example of a 20-letter acronym representing SINE retrotransposon abundance in a collection of methylation-prone genes. (C) Counting of SINE presence (S) and absence (G) in all human genes with a promoter CpG island (genome-wide) and the training set of methylation-prone and methylation-resistant genes. SINE abundance was converted to standard log-odds ratios, as described in the Methods section, and the final substitution matrix for SINE retrotransposons is presented (bottom table). The same calculation was done for LINE retrotransposons. *Transcript variant coding for the P16INK4A protein.
Figure 3.

Prediction of gene predisposition and resistance to hypermethylation in cancer. (A) SINE and LINE scores of the training set genes. The scores were calculated according to the described log-odds ratio method for each gene and are represented as horizontal bars (white bars, SINE score; black bars, LINE score). Methylation status determined by bisulfite PCR methods is shown on the right. Genes with concordant depletion of SINE and LINE retrotransposons (log-odds ≥ 0) were predominantly methylation-prone, with the opposite found for genes with enrichment of both SINE and LINE repeats. Discordant SINE and LINE scores likely represent a class of genes with intermediate predisposition to methylation. *Transcript variant coding for the P16INK4A protein. **Transcript variant coding for the P14ARF protein. Black rectangles represent methylated genes; white rectangles represent unmethylated genes. (B) The predictive method based on SINE and LINE retrotransposons abundance was applied to a test set composed of 142 genes. The frequency of genes correctly classified according to their DNA methylation status in cancer was 79% for methylation-resistant and 75% for methylation-prone genes. These values were closely related to those found in the training set (gray bars). (C) Validation of the predictive method in a large set of cancer cell lines and primary cancer tissues. Methylation status of more than 6600 autosomal gene promoters was determined by MCAM. X chromosome genes were excluded from this analysis due to their hemimethylated status in female samples. The measured DNA methylation per tissue type was significantly higher in predicted methylation-prone genes than in predicted methylation-resistant and methylation-intermediate genes. Methylation is presented as the log2 ratio (cancer/control) of all oligonucleotide probes of a predicted methylation status.
Validation of the model
A major drawback of our model is the limited data set used in its development. We therefore focused on validation using progressively larger data sets. We first studied 74 methylation-prone and 68 methylation-resistant genes for which data on promoter methylation in cancer were available (Supplemental Tables S3, S4) from a variety of tissues (colon, lung, breast, and leukemia, among others). As shown in Figure 3B, 81% of the predicted methylation-prone genes were indeed methylated in cancer, and 75% of the predicted methylation-resistant genes were not methylated. As expected, we found a 1:1 ratio of methylated to unmethylated genes in the predicted methylation-intermediate group (52 of 142 genes, 37%). We further evaluated whether our predictive model held true in a larger scale analysis. For this, we used methylation data from 28 cancer cell lines and 32 primary tissues analyzed by methylated CpG island amplification microarray (MCAM), a sensitive and specific microarray method based on selective amplification of methylated DNA after restriction enzyme digestion (Estecio et al. 2007; Shen et al. 2007b). As performed, MCAM detects cancer-specific methylation as a result of the cohybridization of methylation libraries of cancer versus normal tissues. Thus, all promoter CpG islands identified by MCAM as methylated in cancer are not methylated in the normal control. In this large data set, composed of more than 26,000 probes representing around 6600 CpG islands associated with autosomal gene promoters, we found that predicted methylation-prone genes showed the highest average values of measured promoter methylation, and the predicted methylation-resistant genes had the lowest values (Fig. 3C). This pattern was consistent across individual samples and tissue types, and was observed in 27 of 28 cancer cell lines (98%) and in all 32 studied primary cancer tissue samples (Supplemental Fig. S2). Among 740 genes concordantly methylated across tumor samples (i.e., methylated in at least 30% of the primary tumors and cell lines), 16% were predicted methylation-resistant, 41% methylation-intermediate, and 43% methylation-prone. Thus, our predictive method can accurately classify genes in methylation predisposition groups. In addition, despite the fact that it was generated from cancer cell lines, the predictive model correctly predicts methylation predisposition in primary, uncultured cancer cells.
Although structurally different between mice and humans, retrotransposons are associated with repressed chromatin organization in both species. Thus, if our model is correct, promoter CpG islands subjected to hypermethylation in animal models should also be depleted of retrotransposons. To address this question, we compared the distribution of SINE and LINE retrotransposons in a 20-kb region around the TSS of more than 6000 mouse promoter CpG islands identified by MCAM analysis as methylation-prone and methylation-resistant in a mouse model for myelodysplastic syndrome (Lin et al. 2005). Similar to human cancer, SINE repeats were consistently depleted in methylation-prone genes (Fig. 4A). However, the difference in frequency of LINE repeats was not statistically significant between methylation-prone and methylation-resistant genes. Interestingly, the analysis of distribution of LTR repeats revealed that these transposons were less frequent in methylation-prone genes. IAP repeats, a family of LTR repeats highly successful in mice but not present in humans, also showed a trend for depletion in methylation-prone genes. The lack of statistical significance for the differential distribution of IAP repeats between the methylation-predisposition groups is likely due to their low frequency close to gene TSS (<1% of gene promoter CpG islands). The same general pattern of frequency of SINE, LINE, and LTR repeats was also observed in an analysis of two additional recently published methylation data sets (Fig. 4B,C) generated from chronic lymphocytic leukemia (CLL) and intestinal cancer mouse models (Hahn et al. 2008; Chen et al. 2009). Additionally, since age-related methylation accounts for a large fraction of promoter CpG island methylation observed in cancer (Toyota and Issa 1999), we reasoned that age-related methylated genes are also likely to be depleted of retrotransposons. The DNA methylation pattern of mouse small intestine tissue was compared between old (35-mo-old) and young (3-mo-old) animals using MCAM. We found that, similar to human and mouse promoter CpG islands predisposed to methylation in cancer, age-related methylated mouse promoter CpG islands were depleted of SINE, LINE, and LTR repeats (Fig. 4D). Thus, our data clearly demonstrate that genes predisposed to age-related methylation share a common genome architecture with cancer-related methylated genes. Based on these findings, we revisited the distribution of LTR elements in our large MCAM data set for human cancers and found that LTRs are also depleted in methylation-prone genes, albeit to a lesser degree than SINE and LINE retrotransposons (Supplemental Fig. S3). However, adding LTR distribution to predict gene predisposition to DNA methylation in human cancer did not significantly improve our model.
Figure 4.

Frequency of retroelements in methylation-prone and methylation-resistant genes identified in mouse cancer models and old mice. (A) Depletion of SINE and LTR but not LINE repeats near TSS marks methylated promoter CpG islands in a mouse model of myelodysplastic syndrome (MDS). Bone marrow samples of three NUP98-HOXD13 transgene animals that developed MDS (Lin et al. 2005) were studied by MCAM. Bone marrow samples from nontransgene animal of the same mouse strain was used as control, and the methylation status of approximately 6000 CpG island promoter genes was determined in the MCAM experiments. (B,C) The same pattern of retroelements depletion is observed in hypermethylated genes in CLL (Chen et al. 2009) and intestinal cancer mouse models (Hahn et al. 2008). (D) Depletion of SINE, LINE, and LTR repeats near TSS also marks age-related methylation promoter CpG islands. Small intestine tissue harvested from young (3-mo-old) and old (35-mo-old) C57BL/6J mice were used in MCAM experiments to identify age-related methylation.
Genome-wide analysis of methylation predisposition
The validation of our predictive model prompted us to apply it genome-wide. Among 25,489 unique RefSeq genes (NCBI Build 36.1), 16,166 (63.4%) have a promoter CpG island. Of these, 3613 (22.3%) were predicted by our model to be methylation-prone; 7308 (45.2%) were predicted to be moderately predisposed to methylation (methylation-intermediate); and the remaining 5245 (32.5%) were predicted to be methylation-resistant (Fig. 5A).
Figure 5.

Genome-wide prediction of predisposition to DNA methylation in cancer. (A) The pie chart shows the number of RefSeq genes with no CpG islands (dark gray) and the number of predicted methylation-resistant (white), methylation-intermediate (light gray), and methylation-prone (black) genes in promoter CpG island genes. (B) Gene Ontology (GO) analysis of 1952 predicted methylation-prone and 2583 predicted methylation-resistant genes for which functional information was available. Horizontal bars represent the frequency of significant GO terms. (C) Gene expression analysis for 2822 promoter CpG island associated genes predicted methylation-prone and 3651 predicted methylation-resistant genes in normal tissues. Expression values were retrieved from the GNF database (Su et al. 2004) and Z-score normalized per tissue. Each bar represents the mean ± SEM expression values in each tissue according to their predicted methylation predisposition. (D) Gene expression analysis for 599 promoter CpG island associated genes predicted methylation-prone and 996 predicted methylation-resistant genes in 52 cancer cell lines. Expression values were retrieved from a published work (Ross et al. 2000) and were analyzed as described in C. Only genes present in the studied array platforms could be evaluated, resulting in a different number of analyzed genes in each experiment.
The top 50 predicted methylation-prone and methylation-resistant genes are presented in Tables 1 and 2. Forty-eight percent (24 of 50) of the top predicted methylation-prone genes have been described as methylated in cancer according to data in the literature, and only 6% (3/50) of the top predicted methylation-resistant genes were described as methylated. Although not present in Table 1 because they did not rank among the top 50 genes, several classical genes known to be methylated in cancer and not previously included in the first validation set were correctly predicted as methylation-prone (for example RASSF1, GATA4, GATA5, and SFRP2). In terms of gene function, 39/50 (78%) predicted methylation-prone genes are directly related to developmental processes, compared to only 5% of the predicted methylation-resistant genes (P < 0.001, Fisher's exact test). Indeed, when applied genome-wide, Gene Ontology analysis revealed that methylation-prone genes preferentially participate in developmental processes (Fig. 5B), an observation in agreement with previous data showing that repeat-free regions in mammalian genomes are enriched for genes that function in organogenesis and morphogenesis, among other functions related to development (Simons et al. 2006). Some of these are multiclusters of neighboring genes with related function, such as HOX gene clusters. This prompted us to quantify the propensity of predicted methylation-prone genes to be correlated in neighboring genes and to find 28 genomic regions with statistically significant higher frequency of such genes (Supplemental Table S5). These regions were in average 5 Mb long (ranging from 0.3 to 26 Mb) and had a tendency to be located close to telomeric regions. Together, these regions cover ∼4.5% of the human genome but contain 15% of the predicted methylation-prone genes. Twelve (43%) of these regions included multiclusters of genes with similar function and likely originated from duplication events.
Table 1.
Top 50 genes predicted as methylation-prone

Table 2.
Top 50 genes predicted as methylation-resistant

Two classes of genes with methylated promoter CpG islands in normal tissues are imprinted and X chromosome–inactivated genes. Among 30 well-characterized imprinted genes, 12 (40%) were predicted methylation-prone, 17 (57%) were predicted intermediate, and only one was predicted methylation-resistant (Supplemental Table S6). The frequency of predicted methylation-prone, resistant, and intermediate genes on the X chromosome was remarkably similar to the frequency of the predicted classes genome-wide. There was a moderate, although not significant, enrichment of predicted methylation-prone genes among genes that escape X inactivation (P > 0.05, χ2 test, Supplemental Fig. S4).
Although we have focused our analysis on promoter CpG islands, nonpromoter CpG islands (both exonic/intronic and intergenic CpG islands) are also subject to de novo methylation in cancer. There are scarce data on these CpG islands in the literature, but we were able to identify 291 nonpromoter CpG islands in our MCAM database that showed frequent hypermethylation in cell lines and primary tumors, and 740 methylation-resistant nonpromoter CpG islands. Similarly to promoter CpG islands, methylation-prone nonpromoter CpG islands were found to be depleted of SINE and LINE retrotransposons (Supplemental Fig. S5). Although the functional role of these CpG islands remains poorly defined, our data suggest that the underlying genomic architecture is closely related between methylation-prone and methylation-resistant CpG islands regardless of their position relative to transcriptional units.
Effects on gene expression
To validate the biological implications of our predictive model, we compared the mRNA expression of predicted methylation-prone and methylation-resistant promoter CpG island genes in 28 normal differentiated human tissues and 52 human cancer cell line samples using public microarray databases (Ross et al. 2000; Su et al. 2004). As shown in Figure 5C, in normal tissues, predicted methylation-resistant genes had in general lower expression than predicted methylation-prone genes. An attractive explanation for the lower expression of predicted methylation-resistant genes in normal cells is their higher content of retrotransposons, which have been implicated in decreased mRNA expression through impairment of elongation (Han et al. 2004). As would be expected if our predictive model were correct (since hypermethylation of gene promoters results in gene silencing), we observed in cancer cell lines that predicted methylation-prone genes were down-regulated compared to predicted methylation-resistant genes (Fig. 5D). Thus, depletion of retrotransposons near the TSS was found to be an independent predictor of gene down-regulation in cancer.
Comparison with hyperconserved domains and PcG protein marking
Enrichment for developmental genes was previously described (Tanay et al. 2007) among genes associated with hyperconserved CpG domains, which were identified based on DNA sequence features. Analysis of our data revealed that 84% of the genes in hyperconserved domains are predicted methylation-prone (Fig. 6A). However, hyperconservation is a feature of a much smaller subset of hypermethylated genes in cancer than depletion of retrotransposons (Fig. 6B). Another feature of hyperconserved domains is the overlap with multicluster gene families, such as HOX genes. Among the identified genomic clusters predicted methylation-prone, 11 (39%) included hyperconserved domains (Supplemental Table S5).
Figure 6.

Genome architecture influences on PcG protein binding in embryonic and differentiated cells. (A) Frequency of predicted methylation groups among hyperconserved domains. (B) Relative contribution of hyperconserved domains and retrotransposon depletion in marking frequently methylated genes in cancer. MCAM data from 32 primary tissues and 28 cancer cell lines were averaged to identify frequently methylated genes. *Predicted status. (C) Enrichment of H3K27me3 mark in predicted methylation-prone genes in cancer (PC3, prostate; MCF7, breast) and normal immortalized (PrEC, prostate epithelium) cell lines. H3K27me3 marking was measured by ChIP with microarray hybridization (ChIP-chip) and is quantified as log2 ratio of pull-down signal over no antibody signal (Kondo et al. 2008). (D) Frequency of binding of SUZ12 and EED (PcG proteins) in human embryonic stem cells to 2583 methylation-resistant, 3655 methylation-intermediate, and 1690 methylation-prone genes based on our predictive model. Note that genes predicted methylation-prone (thus depleted for SINE and LINE retrotransposons) are preferential targets of PcG proteins. (E) Comparison of PcG marking and our predictive model in identifying methylation-prone genes from our training and first testing set. (F) Average measured methylation of predicted methylation-prone and methylation-resistant genes in PcG marked genes. MCAM data from 32 primary tumors and 28 cancer cell lines were averaged per comparison group, and methylation is presented as log2 ratio (cancer/control). The number of genes per category is presented above each column.
Hyperconserved domains and DNA methylation have been shown to correlate with Polycomb group (PcG) protein binding in embryonic stem cells. We therefore studied the relationship between our methylation prediction and PcG protein binding. First, we compared the presence of H3K27me3 in predicted methylation-prone and methylation-resistant genes according to chromatin immunoprecipitation microarray results for 8727 gene promoters in three cell lines: PC3, a prostate cancer cell line; MCF7, a breast cancer cell line; and PrEC, an immortalized normal prostate epithelial cell line (Kondo et al. 2008). As shown in Figure 6C, predicted methylation-prone genes were enriched for H3K27me3 in all three cell lines tested. We next evaluated the frequency of binding of PcG proteins and H3K27me3 to genes predicted to be methylation-prone or methylation-resistant according to the occupancy maps published for embryonic stem cells (Lee et al. 2006). Genes predicted to be methylation-prone had a higher frequency of SUZ12, EED, and H3K27me3 targets compared to genes predicted to be methylation-resistant (Fig. 6D; Supplemental Fig. S6A). Indeed, a direct comparison of the distribution of SINE and LINE retrotransposons in genes targeted by Suz12 revealed a large difference, in the same direction as observed between cancer methylation-prone and methylation-resistant genes (Supplemental Fig. S6B).
Since PcG protein marking in embryonic stem cells is closely related to predisposition to DNA methylation in cancer (Ohm et al. 2007; Schlesinger et al. 2007; Widschwendter et al. 2007), it is possible that the relationship between retrotransposon abundance and CpG island DNA methylation is a consequence rather than an independent phenomenon from PcG protein marking. In this case, our predictive model would have no additional predictive value beyond that of PcG protein marking alone. We first examined the relative contribution of PcG marking and our predictive model in genes known to be methylated in our training and first testing set. Out of 110 frequently methylated genes, 25 (23%) are marked by both PcG and predicted methylation-prone, 31 (28%) are predicted methylation-prone alone, 15 (14%) are PcG-positive alone, and 39 (35%) are neither marked by PcG nor predicted methylation-prone (Fig. 6E). Thus, in this set, PcG marking and our predictive model based on retrotransposon frequency appear to be correlated but also complementary. Moreover, these results show that retrotransposon distribution marks a larger fraction of methylation-prone genes than PcG protein marking. To examine this in a larger data set, we turned to MCAM data and divided genes into predicted methylation-prone and methylation-resistant (excluding intermediate for clarity). As seen in Figure 6F, both predicted methylation-prone and PcG-marked genes have significantly higher levels of measured methylation than methylation-resistant/PcG-negative subsets. Again, PcG marking alone identifies a smaller subset of methylated genes compared to our predictive model alone (119 versus 1254 genes). The genes with both PcG marking and retrotransposon depletion (methylation-prone) had the highest levels of measured methylation, confirming that the two models complement each other.
Refinement of the model
Our data suggest that retrotransposon marking is a powerful discriminator of CpG island methylation predisposition in cancer, and that it compares favorably to models based on hyperconservation and PcG marking. However, it does not explain the behavior of every single CpG island. Thus, there is room for refinement of the model, and combination of retrotransposon distribution with other features will likely improve its sensitivity and specificity. An important feature reported to predict methylation propensity of CpG islands are short DNA motifs discovered by Feltus et al. (2003, 2006), and recently modeled in a classifier called PatMan by McCabe et al. (2009). Applied to the first testing set, composed of genes known to be methylated according to the literature, PatMan identifies 9/142 genes as methylation-prone, and five of these (56%) are indeed known to be methylated. Among the remaining 133 genes classified as methylation-resistant by PatMan, 69 are methylated. In comparison, 47/142 genes are classified as methylation-prone according to retrotransposon marking, and 38 (81%) of these are indeed methylated. Thus, retrotransposon marking compares favorably to PatMan. In our validation using MCAM data, PatMan is a good classifier of methylation-prone genes (Supplemental Fig. S7A), but it seems to lack the sensitivity of retrotransposon marking, as it misses a little more than one thousand methylation-prone CpG islands (Fig. 7A). PatMan does, however, resolve nearly 200 methylation-intermediate genes into the methylation-prone group.
Figure 7.

Other genomic features associated with methylation predisposition in cancer. (A) Performance of PatMan and retrotransposon marking in predicting promoter CpG methylation status in cancer. PatMan−, predicted methylation-resistant genes; PatMan+, predicted methylation-prone genes. The number of genes per category is presented above each column. (B) Measured promoter CpG island methylation by MCAM of CTCF bound (CTCF+) and unbound (CTCF−) genes. A gene was considered bound by CTCF if ChIP-seq data from public databases show binding of this protein in the 2-kb region centered in the gene TSS. (C) Abundance of Alu and MIR retrotransposons in the 20-kb sequence window centered in the TSS of 740 methylation-prone and 5658 methylation-resistant genes. Note that the depletion of MIR retrotransposons is more significant in the −2-kb to +2-kb sequence window.
Known insulator elements are likely to play a role in protection of CpG islands from de novo DNA methylation, especially if heterochromatin spreading, as suggested by several authors, is the main event leading to gene inactivation in cancer. To answer whether putative blocking elements influence gene promoter predisposition to DNA methylation, we compared the average methylation of genes with and without CTCF binding sites in the promoter vicinity. A gene promoter was considered to be bound by CTCF if this protein was present in the 2-kb window centered on the gene TSS in at least one out of five normal adult cell lines investigated by chromatin immunoprecipitation with massively parallel sequencing (ChIP-seq) (Bernstein et al. 2005, 2006; Mikkelsen et al. 2007). According to this analysis, ∼36% of promoter CpG islands are bound by CTCF. Promoter CpG islands bound by CTCF seem to be protected from de novo methylation in cancer, as they present lower measured DNA methylation by MCAM compared to genes without CTCF (Fig. 7B). However, CTCF binding is not as strong a predictor of methylation predisposition as retrotransposons marking, since a large fraction of CTCF bound promoters do become methylated in cancer. When added to the prediction model, the presence of CTCF binding does not improve the classification of methylation-resistant genes; even in the absence of CTCF, retrotransposons mark genes that do not become methylated in cancer (Supplemental Fig. S7B). However, the presence of CTCF decreases the methylation propensity of retrotransposon-poor promoters.
Finally, the breakdown of SINE and LINE retrotransposons in families and subfamilies may reveal additional information regarding their relationship to CpG island methylation. To test this possibility, we further annotated the presence of Alu and MIR repeats, the two main families of human SINE, in the 20-kb window centered in the TSS of methylation-prone and methylation-resistant genes. This comparison revealed that Alu repeats are the main drivers of the difference in abundance of SINE repeats between methylation-prone and methylation-resistant genes (Fig. 7C). The difference in frequency of MIR repeats is less evident, but these repeats are, like LINE repeats, depleted in the proximal region to TSS (−2 kb to +4 kb).
Discussion
Our data show that a genome architecture marked by depletion of retrotransposons is strongly correlated to gene predisposition to DNA methylation in cancer. Moreover, we show that this same architecture is an independent predictor of gene expression in cancer and that it is correlated with PcG binding in embryonic stem cells and adult tissues. These data add to the list of possible influences of retrotransposons on genome biology. For example, SINEs are differentially distributed in imprinted (Greally 2002), tissue-specific (Ganapathi et al. 2005), and monoallelically expressed genes (Allen et al. 2003). Obviously, there may be overlap between these states. Indeed, in our study, the frequency of predicted methylation-prone and methylation-resistant genes among imprinted genes was significantly different from the genome-wide distribution of these methylation predisposition groups. These results were in agreement with previous studies showing that SINE repeats are less represented in imprinted genes, resulting in an underrepresentation of predicted methylation-resistant genes (Greally 2002). Of note, several imprinted genes predicted methylation-prone have been reported as hypermethylated in cancer (for example, CDKN1C and DLK1). In contrast, despite reports of differential distribution of SINE and LINE retrotransposons on the X chromosome (Jurka et al. 2004; Wang et al. 2006), we did not find the frequency of methylation-prone and methylation-resistant genes in the X chromosome to differ from autosomes. These results are consistent with the proposed idea that, if retrotransposons in fact participate in X inactivation, their action is due to long-range interactions rather than local, promoter-associated effects. It is important to note that all these studies are correlative, although they strongly point toward a role for retrotransposons in epigenetic regulation.
The depletion of SINE elements in methylation-prone genes is a paradoxical finding, since the main family of these elements (Alu family) is efficient as a methylation nucleation center in both plants and mammals (Yates et al. 1999; Arnaud et al. 2000), and the spreading of DNA methylation from these repeats into gene promoters was hypothesized to be a cause of de novo methylation in cancer (Jones and Baylin 2002). It is important to note, however, that an opposite effect has also been reported for repetitive elements, some of which have insulator function (Gdula et al. 1996; Lunyak et al. 2007). Although the exact mechanism by which the presence of retrotransposons is associated with protection from de novo promoter DNA methylation in cancer is unknown, it is possible that protection is not directly mediated by these repetitive elements but rather by transcription factors with euchromatin/heterochromatin boundary activity, such as CTCF (Bell et al. 1999) and Sp1 (Mummaneni et al. 1998) transcription factors. In such cases, genes without binding sites for boundary proteins would be negatively affected by the insertion of repetitive elements near their promoters. Thus, the presence of retrotransposons near these genes would be counterselected during evolution. Indeed, it has been shown in Arabidopsis thaliana that methylated transposable elements are preferentially inserted or retained in gene-poor areas, a feature that likely evolved due to negative selection (Hollister and Gaut 2009). Also, in agreement with this possibility, we show here that CTCF binding confers some protection from de novo methylation. We believe that the performance of CTCF binding alone as a predictor of methylation fate of gene promoters is somewhat weak due to two main factors: (1) CTCF binding should be taken in tissue-specific context, and compared to methylation data on the same tissue type; and (2) other insulator proteins, for which genome-wide binding to gene promoters has not yet been extensively reported (for example, VEZF1), should be taken into consideration together with CTCF when developing models of protection.
The lack of correlation of LINE repeats with methylation predisposition in mouse data sets is a puzzling observation, and a possible explanation resides in the fact that different repeats may impact the genome differently. Indeed, while SINE and LINE repeats are correlated to methylation predispositon, DNA repeats are not. Among SINE repeats, Alus showed higher correlation to promoter methylation status than MIRs. While not identical, human Alu and mouse B1 share higher sequence similarity than human and mouse LINEs. Also, LINE-1 repeats, the main LINE family, are more variable in mice than in humans. In a direct comparison of repetitive elements in human and mouse genomes, a strong correlation was observed between the presences of SINE repeats in orthologous locations, an event quite surprising given the lineage specificity of these repeats (Mouse Genome Sequencing Consortium 2002). LINE repeats, however, do not show such correlation. Although these observations do not completely explain why mouse LINEs are not correlated to methylation predisposition, they point out that genomic location of different repeats may be dictated by the repeat structure itself and species-specific factors.
The fact that the frequency of retrotransposons alone can predict the expression status of thousands of genes in cancer supports the idea that epigenetic effects in cancer occur through an instructive mechanism and depend relatively little on gene function, at least initially. Indeed, the higher average methylation in PcG marked genes is due to concordant methylation status across different cell lines and primary tissues studied by MCAM, indicating that these genes are under a strong program to become epigenetically silenced. Retrotransposon-depleted genes are more heterogeneously methylated; their lack of protection from de novo methylation leaves these genes prone to epigenetic silencing, although it is not a programmed event and may depend on selection. Thus, as for genetic alterations, many of the methylation events in cancer are likely to be passengers, not drivers, in carcinogenesis. This does not exclude the active participation of a subset of methylation-prone genes with strong tumor-suppression function as drivers in tumorigenesis. Therefore, epigenetic defects in cancer are best viewed as a form of molecular instability creating diversity in gene expression that is exploited by cancers using classical selection mechanisms.
A possible explanation for the correlation between depletion of retrotransposons and predisposition to DNA methylation is that, similarly to PcG marking, retrotransposon depletion marks regulatory regions of developmental genes, which then might be especially prone to DNA methylation. Indeed, developmental genes often have hyperconserved CpG sites (Tanay et al. 2007). Most genes that have such hyperconservation were found to be predicted methylation-prone by our model, reinforcing the argument that retrotransposon depletion marks important essential genes for development. However, our results in primary bladder cancer methylation makes evident that tissue-lineage methylation is not a confounder in our results. In this experiment, bladder tissues from normal adult individuals were used as a control, thus eliminating tissue-specific methylation as a confounder. As shown, our predictive model performs well in this system after tissue-specific methylation has been removed. Still, these data do not exclude the possibility that chromatin states in normal tissues may influence DNA methylation. Indeed, as discussed above, this has been shown for PcG protein binding. It may be that our predicted methylation prone genes have a unique chromatin pattern in normal cells, beyond PcG protein binding, and this should be addressed in further studies. Independent of the mechanism by which retrotransposons are associated to or participate in protection from de novo DNA methylation in cancer, our findings suggest that the architecture of the genome has much more profound influences on cancer physiology than previously suspected.
Methods
Cell lines and primary cancer samples
A total of 34 cell lines from different tissues were obtained from the NCI Anticancer Drug Screen Panel, the American Type Culture Collection (Manassas, VA) or the University of Texas MD Anderson repositories (one cell line from breast cancer, 12 from bladder cancer, one from the central nervous system, four from colon cancer, 11 from leukemia, one from melanoma, one from lung cancer, one from ovarian cancer, one from prostate cancer, and one from renal cancer) and cultured according to standard methods.
Primary tumor samples from 28 bladder cancer and four acute myelogenous leukemia patients were collected at the MD Anderson Cancer Center in accordance with institutional policies. All patients provided written informed consent. Tumors were selected solely on the basis of availability. Genomic DNA was extracted from all cell lines and primary tissue samples using a standard phenol-chloroform method.
Methylation analyses
DNA methylation status of gene promoter CpG islands was evaluated using bisulfite-treated DNA followed by PCR amplification, and methylation density of individual genes was determined by pyrosequencing or COBRA assays according to standard protocols (Estecio et al. 2006, 2007). PCR primers are presented in Supplemental Tables S7 and S8. Genes with at least 15% methylation density were considered hypermethylated (a conservative value chosen based on the detection limits of the methods) and genes hypermethylated in 2/9 tested cancer cell lines were classified as methylation-prone, given that in our research we intended to find genome signatures associated with predisposition or resistance to methylation across tissue types, rather than tissue-specific methylation. It is important to note, however, that the vast majority of the genes classified as methylation-prone showed gene methylation of 30% and higher, and in general they were methylated in three or more cell lines. Analysis of DNA methylation using the MCAM method was performed as previously reported (Estecio et al. 2007; Shen et al. 2007b). Tumor and normal control MCA amplicons were Cy5- and Cy3-labeled, respectively, and cohybridized to the HCGI12K-Human CpG 12K Array (Microarray Center, University Health Network, Toronto, Canada) or to a custom 4 × 44 k oligo array (Agilent Technologies). Methylation analysis of promoter CpG island methylation in mice was done using bone marrow samples of three NUP98-HOXD13 transgene animals that developed MDS (Lin et al. 2005). Bone marrow samples from a nontransgene animal of the same mouse strain were used as control in MCAM experiments, and the methylation status of approximately 6000 CpG island promoter genes was determined using Mouse CpG Island Microarray Kit oligo arrays (Agilent Technologies). Additional data sets of promoter CpG island methylation in mouse tumors were obtained from published reports on a mouse model of chronic lymphocytic leukemia (Chen et al. 2009) and intestinal cancer (Hahn et al. 2008). Small intestine tissues isolated from young (3-mo-old) and old (35-mo-old) C57BL/6J mice and tumor samples from a leukemia mouse model (Choi et al. 2008) were used in MCAM experiments to identify cancer and age-related methylation.
Repetitive element abundance and CpG island characteristics
The DNA sequence flanking the 4-kb sequence region surrounding the TSS of 36 methylation-prone and 36 methylation-resistant genes with a promoter CpG island was obtained from the University of California Santa Cruz (UCSC) BLAT Genome Browser (http://genome.ucsc.edu/cgi-bin/hgBlat). The TSS for each gene was determined according to RefSeq (NCBI Build 36.1), and repetitive DNA sequences were annotated using the RepeatMasker search engine (http://www.repeatmasker.org). All genome tables were downloaded from the UCSC Table Browser (http://genome.ucsc.edu/cgi-bin/hgTables; Karolchik et al. 2004). For all statistical analysis, CpG island length, GC content, and CpG ratio were calculated using the public software CpG Island Searcher (http://www.cpgislands.com; Takai and Jones 2003).
Calculation of a score to predict gene promoter predisposition to DNA methylation
We divided the promoter sequence of 36 methylation-prone and 36 methylation-resistant genes into 20 bins of 1-kb sequence each (10 bins upstream and 10 bins downstream of each gene TSS), and annotated the presence SINE and LINE retrotransposons in each bin (Fig. 2A). In order to avoid a single repeat being counted in multiple bins, SINE and LINE elements were annotated to the closest bin to TSS. Done this way, each gene is represented by a 20-letter acronym, where S represents a SINE element in the bin and G represents the absence of a SINE element (Fig. 2B). Similarly, an independent acronym is generated for LINE repeats, where L represents a LINE element in the bin and G represents the absence of a LINE element. We compared the average abundance of SINE and LINE retrotransposons per bin in methylation-prone and methylation-resistant genes to their average abundance genome-wide in the full collection of human promoter CpG islands, and translated the preference to repetitive elements to a score that differentiates two kinds of promoters.
Such a score is a sum over the bin score, which is a standard log-odds ratio:
where qi,r is the frequency of observing repeat of type r for the ith bin for the promoters known to be methylated; pr is the background frequency for the repeat r. To account for the low count and avoid taking logarithm of zero, qi,r is replaced by 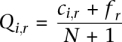 derived from “pseudo-count,” where fr is the fraction of the repeat that is type
derived from “pseudo-count,” where fr is the fraction of the repeat that is type 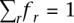 . N is the total number of promoters with the known methylation status; ci,r is the number of repeats of type r in the ith bin:
. N is the total number of promoters with the known methylation status; ci,r is the number of repeats of type r in the ith bin: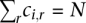 . The final value for each letter in the 20-letter acronym representing the abundance of SINE and LINE elements was calculated as the difference between its value in methylation-prone and methylation-resistant genes (for example,
. The final value for each letter in the 20-letter acronym representing the abundance of SINE and LINE elements was calculated as the difference between its value in methylation-prone and methylation-resistant genes (for example, 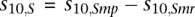 , where Smp is the SINE standard log-odds ratio in methylation-prone genes and Smr is the SINE standard log-odds ratio in methylation-resistant genes). The calculation of the log-odds ratios for SINE elements is illustrated in Figure 2C.
, where Smp is the SINE standard log-odds ratio in methylation-prone genes and Smr is the SINE standard log-odds ratio in methylation-resistant genes). The calculation of the log-odds ratios for SINE elements is illustrated in Figure 2C.
Identification of clusters of methylation-prone genes
To identify clusters of methylation-prone genes, we computed the correlation as a function of separation between genes. We denoted the computed status of whether the ith gene on a chromosome is methylation-prone by Si, where Si = 1 indicates methylation-prone and Si = 0 indicates methylation-resistance. The following correlation function was applied to detect clustering of methylation-prone or methylation-resistance: C(d) where L is the total number of genes in the chromosome. We similarly defined the correlation function over the genome by averaging over all chromosomes. At a large gene separation (distance d), Si and Si+d become independent. If there is a correlation at a short distance, it will result in an elevated value of C(d) at a shorter distance in comparison to a larger distance. We found that between distances 3 and 60, the correlation C(d) can be fitted to an exponential function 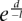 where l = 20.6 indicates a decay length of about 21 gene separations. In these analyses, only genes with promoter CpG islands were computed. In order to find the regions of clustering of methylation-prone genes, we look for a string of consecutive methylation-prone genes (i = 1) in Si. We compared the length of the string with what is allowed by random chance. Let N0 and N1 be the number of 0's (methylation-resistant) and 1's in Si, then
where l = 20.6 indicates a decay length of about 21 gene separations. In these analyses, only genes with promoter CpG islands were computed. In order to find the regions of clustering of methylation-prone genes, we look for a string of consecutive methylation-prone genes (i = 1) in Si. We compared the length of the string with what is allowed by random chance. Let N0 and N1 be the number of 0's (methylation-resistant) and 1's in Si, then 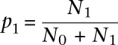 and p0 = 1 − p1. If 0's and 1's are randomly distributed, the chance of having L − m1's and m0's is given by
and p0 = 1 − p1. If 0's and 1's are randomly distributed, the chance of having L − m1's and m0's is given by 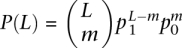 . Let N = N0 = N1, using very conservative Bonferroni correction for multiple testings, when P(L) × N ≈ 1, there is approximately one string with L − m1's and 0's in the entire genome. To control for multiple L we find the smallest L that satisfies
. Let N = N0 = N1, using very conservative Bonferroni correction for multiple testings, when P(L) × N ≈ 1, there is approximately one string with L − m1's and 0's in the entire genome. To control for multiple L we find the smallest L that satisfies 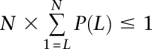 . Although P/L over L does not sum to one, we are summing over very small P/L as an approximation to control for multiple string length tested. So for m = 0 we find all string of 1's with length ≥ L (the threshold value in this case is L = 7) and mark this region as significant. Similarly, we mark regions for m up to 10.
. Although P/L over L does not sum to one, we are summing over very small P/L as an approximation to control for multiple string length tested. So for m = 0 we find all string of 1's with length ≥ L (the threshold value in this case is L = 7) and mark this region as significant. Similarly, we mark regions for m up to 10.
Gene expression, ChIP-chip, and PatMan data sets
Gene expression profiles of normal tissues were downloaded from GNF expression database (http://expression.gnf.org/; Su et al. 2004). Raw expression values in each data set were substituted by their respective Z-scores ([X − μ]/σ, where X represents expression data of each gene in each sample; μ represents mean of expression of all genes for each sample; and σ represents standard deviation). S and L scores were attributed to each gene, and Z-score expression data were averaged per predicted methylation status (prone or resistant to methylation) according to tissue of origin. Gene expression profiles of cancer cell lines were downloaded from the NCI60 Cancer Microarray Project website (http://genome-www.stanford.edu/nci60/; Ross et al. 2000) and analyzed as described for normal tissue expression data. Suz12 and Eed polycomb group (PcG) protein binding and H3K27me3 ChIP data are from human embryonic stem cells (Lee et al. 2006), and from PC3 (prostate cancer cell line), MCF7 (breast cancer cell line), and PrEC (immortalized normal prostate epithelial cells) (Kondo et al. 2008). CTCF binding data are from normal cultured cells (Bernstein et al. 2005, 2006; Mikkelsen et al. 2007). Genome-wide classification of CpG island in methylation-prone and methylation-resistant genes according to the PatMan classifier was retrieved from McCabe et al. (2009).
Gene Ontology analysis
We used FatiGO (Al-Shahrour et al. 2006) from Babelomics (http://babelomics.bioinfo.cipf.es/) for Gene Ontology analysis. The statistical significance of frequency of genes per biological process in predicted methylation-prone and methylation-resistant groups was calculated using Fisher's exact test. P-values were adjusted for false discovery rate.
Acknowledgments
This work was supported by the Leukemia Specialized Program of Research Excellence grant P50 CA100632, the National Institutes of Health grants R01 CA098006 and U01 CA085078, and the NIH intramural research program. J.P.J.I. is an American Cancer Society Clinical Research Professor. We thank Stephanie P. Deming for editorial help.
Footnotes
[Supplemental material is available online at http://www.genome.org.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.107318.110.
References
- Allen E, Horvath S, Tong F, Kraft P, Spiteri E, Riggs AD, Marahrens Y 2003. High concentrations of long interspersed nuclear element sequence distinguish monoallelically expressed genes. Proc Natl Acad Sci 100: 9940–9945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Shahrour F, Minguez P, Tarraga J, Montaner D, Alloza E, Vaquerizas JM, Conde L, Blaschke C, Vera J, Dopazo J 2006. BABELOMICS: A systems biology perspective in the functional annotation of genome-scale experiments. Nucleic Acids Res 34: W472–W476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaud P, Goubely C, Pelissier T, Deragon JM 2000. SINE retroposons can be used in vivo as nucleation centers for de novo methylation. Mol Cell Biol 20: 3434–3441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylin S, Bestor TH 2002. Altered methylation patterns in cancer cell genomes: Cause or consequence? Cancer Cell 1: 299–305 [DOI] [PubMed] [Google Scholar]
- Bell AC, West AG, Felsenfeld G 1999. The protein CTCF is required for the enhancer blocking activity of vertebrate insulators. Cell 98: 387–396 [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, McMahon S, Karlsson EK, Kulbokas EJ III, Gingeras TR, et al. 2005. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 120: 169–181 [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. 2006. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125: 315–326 [DOI] [PubMed] [Google Scholar]
- Bock C, Paulsen M, Tierling S, Mikeska T, Lengauer T, Walter J 2006. CpG island methylation in human lymphocytes is highly correlated with DNA sequence, repeats, and predicted DNA structure. PLoS Genet 2: e26 doi: 10.1371/journal.pgen.0020026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SS, Raval A, Johnson AJ, Hertlein E, Liu TH, Jin VX, Sherman MH, Liu SJ, Dawson DW, Williams KE, et al. 2009. Epigenetic changes during disease progression in a murine model of human chronic lymphocytic leukemia. Proc Natl Acad Sci 106: 13433–13438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi CW, Chung YJ, Slape C, Aplan PD 2008. Impaired differentiation and apoptosis of hematopoietic precursors in a mouse model of myelodysplastic syndrome. Haematologica 93: 1394–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R, Dimitrova N, Xuan Z, Rollins RA, Haghighi F, Edwards JR, Ju J, Bestor TH, Zhang MQ 2006. Computational prediction of methylation status in human genomic sequences. Proc Natl Acad Sci 103: 10713–10716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estecio MR, Youssef EM, Rahal P, Fukuyama EE, Gois-Filho JF, Maniglia JV, Goloni-Bertollo EM, Issa JP, Tajara EH 2006. LHX6 is a sensitive methylation marker in head and neck carcinomas. Oncogene 25: 5018–5026 [DOI] [PubMed] [Google Scholar]
- Estecio MR, Yan PS, Ibrahim AE, Tellez CS, Shen L, Huang TH, Issa JP 2007. High-throughput methylation profiling by MCA coupled to CpG island microarray. Genome Res 17: 1529–1536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner GJ, Kimura Y, Daub CO, Wani S, Plessy C, Irvine KM, Schroder K, Cloonan N, Steptoe AL, Lassmann T, et al. 2009. The regulated retrotransposon transcriptome of mammalian cells. Nat Genet 41: 563–571 [DOI] [PubMed] [Google Scholar]
- Feltus FA, Lee EK, Costello JF, Plass C, Vertino PM 2003. Predicting aberrant CpG island methylation. Proc Natl Acad Sci 100: 12253–12258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feltus FA, Lee EK, Costello JF, Plass C, Vertino PM 2006. DNA motifs associated with aberrant CpG island methylation. Genomics 87: 572–579 [DOI] [PubMed] [Google Scholar]
- Ganapathi M, Srivastava P, Das Sutar SK, Kumar K, Dasgupta D, Pal Singh G, Brahmachari V, Brahmachari SK 2005. Comparative analysis of chromatin landscape in regulatory regions of human housekeeping and tissue specific genes. BMC Bioinformatics 6: 126 doi: 10.1186/1471-2105-6-126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner-Garden M, Frommer M 1987. CpG islands in vertebrate genomes. J Mol Biol 196: 261–282 [DOI] [PubMed] [Google Scholar]
- Gdula DA, Gerasimova TI, Corces VG 1996. Genetic and molecular analysis of the gypsy chromatin insulator of Drosophila. Proc Natl Acad Sci 93: 9378–9383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greally JM 2002. Short interspersed transposable elements (SINEs) are excluded from imprinted regions in the human genome. Proc Natl Acad Sci 99: 327–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn MA, Hahn T, Lee DH, Esworthy RS, Kim BW, Riggs AD, Chu FF, Pfeifer GP 2008. Methylation of polycomb target genes in intestinal cancer is mediated by inflammation. Cancer Res 68: 10280–10289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JS, Szak ST, Boeke JD 2004. Transcriptional disruption by the L1 retrotransposon and implications for mammalian transcriptomes. Nature 429: 268–274 [DOI] [PubMed] [Google Scholar]
- Hollister JD, Gaut BS 2009. Epigenetic silencing of transposable elements: A trade-off between reduced transposition and deleterious effects on neighboring gene expression. Genome Res 19: 1419–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA, Baylin SB 2002. The fundamental role of epigenetic events in cancer. Natl Rev 3: 415–428 [DOI] [PubMed] [Google Scholar]
- Jurka J, Kohany O, Pavlicek A, Kapitonov VV, Jurka MV 2004. Duplication, coclustering, and selection of human Alu retrotransposons. Proc Natl Acad Sci 101: 1268–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, Kent WJ 2004. The UCSC Table Browser data retrieval tool. Nucleic Acids Res 32: D493–D496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo E, Furukawa T, Yoshinaga K, Kijima H, Semba S, Yatsuoka T, Yokoyama T, Fukushige S, Horii A 2000. Not hMSH2 but hMLH1 is frequently silenced by hypermethylation in endometrial cancer but rarely silenced in pancreatic cancer with microsatellite instability. Int J Oncol 17: 535–541 [DOI] [PubMed] [Google Scholar]
- Kondo Y, Shen L, Cheng AS, Ahmed S, Boumber Y, Charo C, Yamochi T, Urano T, Furukawa K, Kwabi-Addo B, et al. 2008. Gene silencing in cancer by histone H3 lysine 27 trimethylation independent of promoter DNA methylation. Nat Genet 40: 741–750 [DOI] [PubMed] [Google Scholar]
- Lee TI, Jenner RG, Boyer LA, Guenther MG, Levine SS, Kumar RM, Chevalier B, Johnstone SE, Cole MF, Isono K, et al. 2006. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell 125: 301–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YW, Slape C, Zhang Z, Aplan PD 2005. NUP98-HOXD13 transgenic mice develop a highly penetrant, severe myelodysplastic syndrome that progresses to acute leukemia. Blood 106: 287–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunyak VV, Prefontaine GG, Núñez E, Cramer T, Ju BG, Ohgi KA, Hutt K, Roy R, García-Díaz A, Zhu X, et al. 2007. Developmentally regulated activation of a SINE B2 repeat as a domain boundary in organogenesis. Science 317: 248–251 [DOI] [PubMed] [Google Scholar]
- McCabe MT, Lee EK, Vertino PM 2009. A multifactorial signature of DNA sequence and polycomb binding predicts aberrant CpG island methylation. Cancer Res 69: 282–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, et al. 2007. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448: 553–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouse Genome Sequencing Consortium 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520–562 [DOI] [PubMed] [Google Scholar]
- Mummaneni P, Yates P, Simpson J, Rose J, Turker MS 1998. The primary function of a redundant Sp1 binding site in the mouse aprt gene promoter is to block epigenetic gene inactivation. Nucleic Acids Res 26: 5163–5169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, Cope L, Mohammad HP, Chen W, Daniel VC, Yu W, et al. 2007. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet 39: 237–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross DT, Scherf U, Eisen MB, Perou CM, Rees C, Spellman P, Iyer V, Jeffrey SS, Van de Rijn M, Waltham M, et al. 2000. Systematic variation in gene expression patterns in human cancer cell lines. Nat Genet 24: 227–235 [DOI] [PubMed] [Google Scholar]
- Schlesinger Y, Straussman R, Keshet I, Farkash S, Hecht M, Zimmerman J, Eden E, Yakhini Z, Ben-Shushan E, Reubinoff BE, et al. 2007. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet 39: 232–236 [DOI] [PubMed] [Google Scholar]
- Shen L, Kondo Y, Ahmed S, Boumber Y, Konishi K, Guo Y, Chen X, Vilaythong JN, Issa JP 2007a. Drug sensitivity prediction by CpG island methylation profile in the NCI-60 cancer cell line panel. Cancer Res 67: 11335–11343 [DOI] [PubMed] [Google Scholar]
- Shen L, Kondo Y, Guo Y, Zhang J, Zhang L, Ahmed S, Shu J, Chen X, Waterland RA, Issa JP 2007b. Genome-wide profiling of DNA methylation reveals a class of normally methylated CpG island promoters. PLoS Genet 3: e181 doi: 10.1371/journal.pgen.0030181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons C, Pheasant M, Makunin IV, Mattick JS 2006. Transposon-free regions in mammalian genomes. Genome Res 16: 164–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, et al. 2004. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci 101: 6062–6067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai D, Jones PA 2003. The CpG island searcher: A new WWW resource. In Silico Biol 3: 235–240 [PubMed] [Google Scholar]
- Tanay A, O'Donnell AH, Damelin M, Bestor TH 2007. Hyperconserved CpG domains underlie Polycomb-binding sites. Proc Natl Acad Sci 104: 5521–5526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyota M, Issa JP 1999. CpG island methylator phenotypes in aging and cancer. Semin Cancer Biol 9: 349–357 [DOI] [PubMed] [Google Scholar]
- Wang Z, Willard HF, Mukherjee S, Furey TS 2006. Evidence of influence of genomic DNA sequence on human X chromosome inactivation. PLoS Comput Biol 2: e113 doi: 10.1371/journal.pcbi.0020113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M, Hellmann I, Stadler MB, Ramos L, Paabo S, Rebhan M, Schubeler D 2007. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet 39: 457–466 [DOI] [PubMed] [Google Scholar]
- Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, Marth C, Weisenberger DJ, Campan M, Young J, Jacobs I, et al. 2007. Epigenetic stem cell signature in cancer. Nat Genet 39: 157–158 [DOI] [PubMed] [Google Scholar]
- Yates PA, Burman RW, Mummaneni P, Krussel S, Turker MS 1999. Tandem B1 elements located in a mouse methylation center provide a target for de novo DNA methylation. J Biol Chem 274: 36357–36361 [DOI] [PubMed] [Google Scholar]
- Yoder JA, Walsh CP, Bestor TH 1997. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet 13: 335–340 [DOI] [PubMed] [Google Scholar]