

Abstract
Oxyanion holes play a major role in catalyzing enzymatic reactions, yet the corresponding energetics is frequently misunderstood. The main problem may be associated with the non-trivial nature of the electrostatic preorganization effect, without following the relevant formulation. That is, although the energetics of oxyanion holes have been fully quantified in early studies (which include both the enzymatic and reference solution reactions), the findings of these studies are sometimes overlooked, and, in some cases, it is assumed that gas-phase calculations with a fixed model of an oxyanion hole are sufficient for assessing the corresponding effect in the protein. Herein, we present a systematic analysis of this issue, clarifying the problems associated with modeling oxyanions by means of two fixed water molecules (or related constructs). We then re-emphasize the point that the effect of the oxyanion hole is mainly due to the fact that the relevant dipoles are already set in an orientation that stabilizes the TS charges, whereas the corresponding dipoles in solution are randomly oriented, resulting in the need to pay a very large reorganization energy. Simply calculating interaction energies with relatively fixed species cannot capture this crucial point, and considering it may help in advancing rational enzyme design.
Keywords: Enzyme Catalysis, Electrostatic Preorganization, Oxyanion Holes, Gas Phase Enzyme Models, Ground State Destabilization
I. INTRODUCTION
Electrostatic catalysis due to polar preorganization is arguably the most important factor in enzymatic rate enhancment1,2. Despite this, however, this effect is still not widely appreciated, as a result of which it is not being used for enzyme design. A large part of this is associated with the non-trivial fact that the reorganization energy is being stored in the enzyme, and not in the enzyme-substrate interaction. Perhaps the best illustration of the problems with the greater acceptance of this concept are oxyanion holes, where overlooking preorganization led to a variety of different proposals, such as the low-barrier hydrogen bond (LBHB) proposal3–7 (for detailed consideration of the problem with this proposal see Refs. 8,9), as well as the problems in attempts to asses the preorganization contribution experimentally10–12 (see discussion in Ref. 13). Clearly, attempting to explain enzyme catalysis by overlooking the proper energetics needed to assess the LBHB catalytic proposal14 (which was discussed in e.g. Refs. 8,9), and instead relying on studies that use for instance minimal models or gas-phase calculations (e.g. Refs. 15,16) is a problem that should be taken note of. Additionally, perhaps the popularity of some of the dynamical ideas, which are presented as alternative explanations for enzyme catalysis (see the review in Ref. 17) is due to the difficulty of appreciating the preorganization effect.
The above difficulty becomes more relevant as a wider part of the chemical community is starting to appreciate the importance of oxyanion holes in possible design strategies18, but yet, still largely overlooks the polar preorganization idea. Based on the apparent complexity of this issue, it would seem to us that it is important to further clarify the preorganization idea, especially to those who are interested in calculating this effect. Thus, we will provide a detailed explanation of the preorganization proposal, and demonstrate why unfamiliarity with it can lead to incorrect conclusions. That is, we will expand on recent attempts to use a gas-phase study of oxyanion models as a way to gain an understanding of such systems. It will be illustrated that such studies cannot estimate the preorganization effect, as they are unable to estimate the protein constraints.
II. BACKGROUND
In light of the apparent difficulty of the wider acceptance of the polar preorganization concept (despite it's crucial role in enzyme catalysis), we will start our discussion by familiarizing the reader with this concept. That is, as has been discussed elsewhere2, there exist multiple cases where most of the catalytic effect of the enzyme is clearly due to electrostatic interactions. Specifically, quantifying the change in the polarization of the solvent (or protein) dipoles during the reaction in an enzyme and in solution led to an awareness of the importance of electrostatic reorganization1. Here, it was found that in the case of reactions occurring in solution, the solvent must pay a major free energy penalty when reorienting its dipoles towards the transition state (TS) charges as it moves from the reactant state (RS) to the TS, whereas, as the protein active site dipoles are already partially oriented toward the TS the protein has to pay much less reorganization energy. This point is illustrated schematically in Fig. 1, for the case of an ion pair type transition state, where the negative part is stabilized by an oxyanion hole.
FIGURE 1.
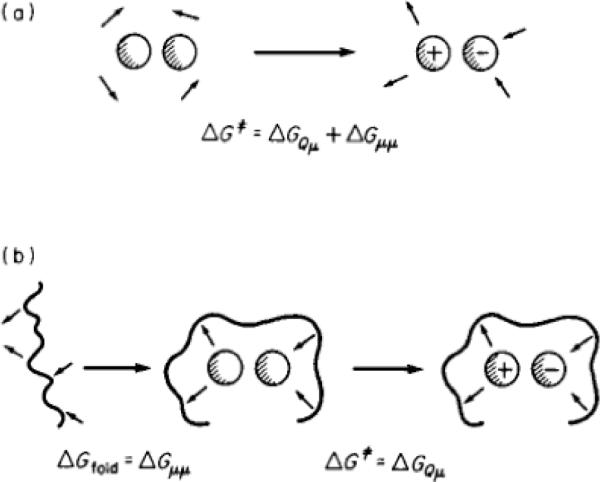
An illustrative example of preorganization in the case of the formation of an ion pair where the negative side is stabilized by an oxyanion hole in the protein. Shown here are (a) the energy balance that is involved in the formation of the ion-pair type transition state in solution, and (b) the corresponding energetics in the protein. The solvent dipole-dipole interaction is designated by ΔGμμ, and in water, the reaction involves the reorganization of the solvent dipoles (see main text), and thus significant ΔGμμ (note that, to a good approximation, ΔGμμ = −½ ΔGQμ). Here, the subscripts Q and μ denote charge and dipole respectively. On the other hand, in the protein, a major part of the reorganization energy is invested (in this particular example) in the folding process, where the folding energy is used in order to fix the active site dipoles in a configuration that is unstable in the ground state. The polarized dipoles then create a site that stabilizes the transition state. This figure was originally presented in Ref. 26. Note that for simplicity, we assume that the reactant state is nonpolar, and we refer to the value obtained when the solvent dipoles are already oriented toward the solute charges as the “interaction free energy”.
Now one of the best ways to consider the preorganization concept is to focus on the specific case where oxyanion sites contribute to catalysis. The existence of special oxyanion sites in proteins was pointed out quite early in the literature19, although the energetic origin of such sites, as well as their relationship to the preorganization concept was only recognized in 19781 and deduced by mutational studies in 198620. The energetics of oxyanion holes have been quantified in many of our studies (e.g. Refs. 13,21–24), as well as those of others (e.g. Ref. 25), but the realization that proper quantum mechanical / molecular mechanical (QM/MM), and, particularly, empirical valence bond (EVB) calculations are probably the best way to quantify the effect of the oxyanions has not yet been widely accepted (e.g. Refs. 10–12). At any rate, here, we will provide a demonstration of the preorganization effect in the oxyanion hole, for a typical reaction of serine proteases of the type described in Fig. 2.
FIGURE 2.

Oxyanion formation in a typical reaction of serine proteases. This figure was originally presented in Ref. 23.
Now, when dealing with serine proteases, one should, in principle, model the complete reacting system (e.g. the catalytic triad) in the RS and TS, and then use the linear response approximation (LRA)27,28 (see also Section III) in order to extract the oxyanion contribution (as was done in e.g. Refs. 22,29,30). However, the use of the LRA with the complete catalytic triad has not been widely appreciated (or practiced). Thus, although we will also use the LRA here, we will mainly provide a simpler analysis with a subsystem of the reacting system, by taking a model that is sufficiently close to the model used by those who still work with gas-phase models, making it possible to clarify the problems with the oversimplified gas-phase model. In other words, we will focus on a simple nucleophilic attack at a C=O center (Fig. 3). However, we would also like to point out in passing that models containing the full catalytic triad (Asp, His and Ser) in the gas-phase without solvation are very problematic (as we have pointed out since 198623), as the Asp will not be negatively charged in the gas-phase. At any rate, here, we will focus on the nucleophilic attack step, where the change in interaction between the simulated system and the farther parts of the system are small relative to those in the full reaction of Fig. 2. We must also mention that the selection of such a model is essential if one wants to examine incomplete gas-phase models.
FIGURE 3.

A simplified schematic of oxyanion formation by nucleophilic attack. Note that this model is intentionally oversimplified (see text).
We start by schematically illustrating the energetic difference between the system in the enzyme and water in Fig. 4. With this scheme, it is possible to appreciate the relevant energetics. That is, computer simulation studies31 have indicated that the actual free energy electrostatic interaction between the enzyme dipoles and the charges of the substrate TS, , is similar to that between water and the corresponding TS in solution (i.e. ). Now, clearly, if the interactions between the two were similar, one would expect the enzyme to have no catalytic effect. However, an early work1 provided the solution to this fundamental problem, by pointing out that, in fact, in water, roughly half the energy gained from the charge-dipole interactions is spent on changing the dipole-dipole interaction, , such that the free energy of solvation of the transition state is given by32:
| (1) |
where, for simplicity, we assume that the reactant state is nonpolar, and we refer here to the value obtained when the solvent dipoles are already oriented toward the solute charges as the “interaction free energy”. Here, is the reorganization energy mentioned above for the process of forming the transition state charges. In contrast, in a protein, where the active site dipoles associated with polar groups, internal water molecules and ionized residues are already partially oriented towards the charge center of the transition state,
| (2) |
(where the superscripts “w” and “p” in Eqs. (1) and (2) denote water and protein respectively), and, of course, , with less free energy being spent on creating the oriented dipoles of the protein transition state. Now as
| (3) |
therefore, following this discussion,
| (4) |
which is the major source of the catalytic effect of the enzyme.
FIGURE 4.

Cycle showing the energetics involved in the stabilization of an oxyanion. Here, Δ denotes the disappearance of . The superscripts w and p denote the reaction in water and in a protein respectively, and the subscripts Qμ and μμ denote charge-dipole and dipole-dipole interactions respectively. Note that the geometry in the protein model is for illustration purposes only, and, in any actual study, we considered the correct relaxed oxyanion geometry in the given protein.
In order to quantify the above issue, we focus on a realistic estimate of the oxyanion contribution to the process of moving from C=O to C-O− (note that, as stated above, more complex studies which include the proton acceptor region have been performed by us, but this may be less clear to those who are not familiar with studies of a complete solvated system). As was already determined by us in 198933, the solvation free energies of C=O to C-O− are around −6 and −80 kcal/mol respectively (see Table 1). This means that the reorganization cost in water is ~3 and 40 kcal/mol for C=O and C-O− respectively. Now the maximum effect of the reorganization in Eq. 4 could be about 18 kcal/mol, but in reality there is also significant reorganization energy in the enzyme (the enzyme is not rigid) and also, it is important to consider the full system rather than just the C=O system (see Section IV). Nevertheless, the preorganization effect of the oxyanion is captured in the discussion above. Apparently, this effect has been overlooked in Ref. 34, where the reorganization has not been considered.
TABLE 1.
The solvation energies of fragments relevant to the present study.[a]
| Fragment | |
|---|---|
| HCOOH | −10 |
| HCOO− | −80 |
| Imidazole | −4 |
| Imidazole+ | −62 |
| C(O)(CN)C | −6 |
| OC(O−)(CN)C | −85 |
| CH3CH2OH | −6 |
| CH3CH2O− | −92 |
The data, which is presented in kcal/mol, is taken from Ref. 33.
To summarize the above discussion, the solvent dipoles in the water reaction, will randomly orient themselves around the uncharged RS (or, if the RS is polar but uncharged, this will have a small non-random component), which means that the total activation free energy will also include a contribution from the free energy cost associated with the reorganization of these solvent dipoles towards the charged TS. On the other hand, in the protein, the active site dipoles (which can come from either polar groups, charged groups and/or internal water molecules) are already partially oriented towards the TS charge1. This means that the reaction costs far less reorganization energy compared to its counterpart in water. The analysis presented above allows us to obtain a quantitative estimate and to see that the main factor overlooked by analyses that only examine the interaction energy is overlooking the penalty of reorganizing the solvent dipoles upon going from the RS to the TS (see Section IV).
Now the analysis above may look formal to some readers, and thus, in order to provide a solid background for the reader, we will provide a quantitative estimate of the change in solvation energy during the process described in Fig. 4. However, we will also clarify below that the only way to quantify the result is by examining the complete solvation free energy, and not some specific structural parameters. Therefore, we will explain why the analysis of Ref. 34 and its conclusions are unjustified.
III. RESULTS AND DISCUSSION
In this section, we will consider several approximated treatments of the energetics of oxyanion holes, starting from a very approximated treatment and moving to more complete models. We believe that the comparison of the different treatments will clarify what is needed for a proper description and will provide farther clarification of the nature of enzyme catalysis.
III.1. ASSESSING THE FREE ENERGY OF THE FORMATION OF THE OXYANION: PROBLEMS AND PITFALLS
As a first step, we will attempt to move from a general analysis of the solvation energies in water and proteins to a specific analysis of different levels of approximation. We will start by considering treatments of the type used by e.g. Simón and Goodman34. That is, Ref. 34 has performed an extensive examination of the crystal structures of both enzymatic oxyanion holes, gathered from the Protein Data Bank35, as well as small molecule interactions obtained from the Cambridge Crystallographic Database36, and used these structures (in combination with gas phase ab initio calculations with an oversimplified model, which will be discussed in Section III.2) to explore the energetics of the oxyanion hole. Significantly, it was found that hydrogen bonding to carbonyls in molecular crystals does not seem to have the same directionality as those in enzyme active sites. Thus, it was suggested that oxyanion holes in enzymes do not work by transition state stabilization2 (TSS), but rather, that enzymes arrange hydrogen bonds such that the oxyanions are sub-optimally stabilized, in order to “avoid overstabilization of the “ground state”34, or, alternately, that “unlike water, enzymes can choose to orient their hydrogen bonds to stabilize the transition state slightly less well than is optimal, in order to stabilize the substrate much less well than is possible with the same number of hydrogen bonds”34 (with the authors then proceeding to argue that “this suboptimal arrangement of stabilizing groups to provide a greater reduction of the energy barrier (instead of higher transition state stabilization) can be readily introduced as a factor in the design of artificial catalysts”). It seems to us that such arguments can be understood as variations on the reactant state destabilization (RSD) idea, which was clearly and uniquely defined in e.g. Refs. 2,37. It must be pointed out here that while it is very unlikely that enzymes work by RSD, one could wonder whether the requirement of preorganization towards the TS would result in some RS destabilization. Thus, it is important to explore the validity of the proposal above.
Obviously, the hypothesis of Ref. 34 is interesting (see Ref. 38) since it is based on the observation that the RS in the enzymes might be less stable than in molecular crystals (but not necessarily in solution). However, the authors' actual conclusions involve some problems: Firstly, the main focus was placed on studying unrelaxed protein structures, which amounts to ignoring the reorganization in the protein. Secondly (and more importantly), the large reorganization in the reference reaction in water was not considered by the authors. Finally, the stabilizing effect of the oxyanion hole has not been analyzed properly. For instance, it was argued that “catalysis with two water molecules, even if they are optimally placed to stabilize the transition state, is less effective than catalysis by the oxyanion hole, because water will not be able to stabilize the ground state less well than the transition state”. This suggestion that the enzyme destabilizes the RS more than in water overlooks the fact that two optimally placed water molecules in the gas-phase cannot provide a realistic model for examining catalysis. That is, in solution, the water molecules can even be pointing in the opposite direction in the ground state, but then rotate towards the charged TS (hence the reorganization penalty), or, as another example, it is possible that the ground state destabilization effect that the authors considered is an artifact which would have disappeared had the authors used a smaller, physically reasonable force constant (see the discussion in Section III.2). Since we are dealing with a general problem and conclusions that may be easily obtained by workers who do not use full enzyme models, we therefore find it useful to provide below a step-by-step “tutorial” on how to obtain the effect of an oxyanion in an oxyanion hole.
III.2 STUDIES USING A MINIMAL MODEL
As our starting point, we will consider a minimal model (see Fig. 5) that involves a gas-phase “oxyanion hole” comprised of two water molecules, which may be assumed to be a reasonable model of the active site. We start by examining the relevant energetics obtained when the water molecules are allowed to relax to a geometry that most stabilizes the C=O state, and then to a geometry that most stabilizes the C-O− state. In doing so, we demonstrate that the oxyanion hole provides much more stabilization for the TS or intermediate (IS) states than the RS (see the cycle presented in Fig. 6). In other words, it is quite obvious that the oxyanion dipoles are designed in such a way as to stabilize the developing negative charge. This point is not apparent from the study of Ref. 34, since the reported study has not evaluated the energy of bringing the water molecules from infinity towards the oxyanion, and only examined the comparatively irrelevant effect of deforming the oxyanion hole. At any rate, clearly, the model oxyanion stabilizes the TS far more than the RS.
FIGURE 5.

Model “oxyanion hole” used for our minimal model (see Section III.1). Note that the exact orientation of the water molecules in this figure is not intended to represent the actual orientation in the protein, but rather, to demonstrate the general problems with using a gas-phase model with two water molecules as a representation of the active site.
FIGURE 6.

Cycle comparing the effect of the two water molecules on the relative species in our model oxyanion hole. Here, (a) and (c) represent the C=O and C-O− species respectively with the two water molecules 5Å away from the C=O, and (b) and (d) represent the C=O and C-O− species with the two water molecules brought to within approximately 1.9Å (measured by the O--H distance) of the carbonyl/oxyanion oxygen. From this cycle, it can be clearly seen that the two water molecules provide significantly greater stabilization to the charged C-O− species than to the C=O.
Next, we address the abovementioned issue of deforming the oxyanion hole, by fixing the water molecules in an orientation that provides less stabilization than the relaxed structure. The assumed deformation is similar but not identical to that assumed in Ref. 34, since the results obtained (see below) do not depend on the exact deformation. The results for the “good” configuration, as well as six different “deformed” configurations are complied in Table 2, and, as seen from the table, we of course reduce the stability of the system once we move the water molecules away from the optimal configuration (here, in most cases we lose more stability from the the C-O− system, with the two systems occasionally being affected similarly by the deformation, but the overall trend is clear). However, this point has little to do with RSD by the enzyme. That is, first of all, this issue has to be assessed relative to the reference state in solution (see below), but, more importantly, Ref. 34 puts too much weight into the importance of unrelaxed protein structures. This is demonstrated by taking any two of the deformed structures considered in Table 2 (in this example Structures 1 and 6), and placing strong constraints on the water molecules in order to try to “fix” them in the assumed deformed conformation in the protein. We therefore performed 1000 steps of QM/MM minimization (for details see SI, note that we chose 1000 steps simply because our aim is not to obtain perfect results, but rather to see how much effect the constraint has over a reasonable simulation length) using different constraints to examine the effect of constraints of different magnitudes. Now, as is shown in Table 3 and in Fig. 7, only an unrealistically large constraint of >>50 kcal mol−1 Å−2 can maintain the deformation effect, and with smaller constraints, the system very quickly minimizes back to an “optimal structure”. For comparison, typical protein force constants are, at most, approximately 5 kcal mol−1 Å−2 for small deformations, and, the fact that trend this holds for two very different water conformations suggests that our finding is independent of the precise starting conformation of the water molecules, as long as they are not in the optimal position to begin with. Thus, as touched on in the previous section, one can obtain very problematic conclusions as a consequence of having used an infinite constraint in the calculation. In fact, the simulations of Ref. 34 do not examine the actual oxyanion hole in an enzyme, but rather consider unrelaxed crystal structures, and small models (i.e. either in the gas-phase with two water molecules, or theozymes). However, in cases with negatively charged oxyanions, such as in the case of TS analogues in the oxyanion hole of ketosteroid isomerase, one does find an oxyanion hole which is almost perfectly oriented to stabilize the oxyanion (see Ref. 12).
TABLE 2.
Showing the relevant energetics when the two water molecules are either minimized to the optimal configuration relative to the carbonyl/oxyanion, or “deformed” in one of six ways. In order to obtain the deformed configuration, we altered the Owat-H-O-C dihedral angle in one or both water molecules, while keeping all other atoms fixed. Note that the precise “deformed” geometries were chosen at random, with the only concern being that these six structures are sufficiently different from each other for the comparison to be meaningful[a].
| Ketone (C=O) | Oxyanion (C-O−) | |
|---|---|---|
| Good Structure | −15.4 | −47.4 |
| Deformed Structure 1 | 15.1 | −8.4 |
| Deformed Structure 2 | 100.2 | 79.8 |
| Deformed Structure 3 | 17.3 | −7.1 |
| Deformed Structure 4 | −3.6 | −32.0 |
| Deformed Structure 5 | 9.7 | −16.8 |
| Deformed Structure 6 | 15.4 | −14.2 |
All energies are given relative to the having the two water molecules at 5Å separation from either the C=O or C-O− (as relevant), in kcal/mol, and, from this table, it can be clearly seen that as expected, moving the water molecules away from the optimal configuration reduces the stability of the system, and that even small perturbations in the position of the water molecules can have a very large effect of the energy. However, as can be seen in Table 3, to then maintain these deformations requires extremely (and unphysically) large force constants, and with smaller force the system will try to very quickly “relax” to an optimal conformation.
TABLE 3.
The effect of constraints of different magnitude on the relaxation of the “deformed” water molecules of Structures 1 and 6 of Table 2 (see main text) relative to either the C=O or C-O− species, after 1000 steps of QM/MM minimization using the MOLARIS simulation package39 interfaced with Gaussian 0340, with the MPW1PW91 functional41 and the 6-311++G** basis set.[a]
| Constraint | Structure 1 | Structure 6 | ||
|---|---|---|---|---|
| Ketone (C=O) | Oxyanion (C-O−) | Ketone (C=O) | Oxyanion (C-O−) | |
| Unminimized | 15.1 | −8.4 | 15.4 | −14.2 |
| 5000 | 12.5 | −10.0 | 12.9 | −15.0 |
| 500 | 7.3 | −17.2 | 8.4 | −19.5 |
| 50 | −1.0 | −28.7 | −3.8 | −33.3 |
| 5 | −11.1 | −42.3 | −13.5 | −44.1 |
| 0.5 | −14.8 | −46.2 | −15.2 | −45.9 |
| 0.0 | −15.2 | −46.7 | −15.4 | −46.1 |
For comparison, the energy of the deformed structure with no minimization is also presented. All energies are given relative to the having the two water molecules at 5Å separation from either the C=O or C-O− (as relevant), in kcal/mol (see Table S1 for the absolute energies of these structures), and the constraint is given in kcal mol−1 Å−2. From this table, it can be seen that in order to maintain the deformation, extremely large force constants of in excess of 50 kcal mol−1 Å−2 are required, and that the smaller constraints result in a relaxation to a more optimal structure. Note that a typical force constant in a protein for a small deformation is, at most, 5 kcal mol−1 Å−2, and thus it is not possible to maintain the deformation without an unphysically large force constant. This indicates that using large constraints may lead to completely unrealistic results.
Figure 7.

Illustrating the change in the total system energy (quantum+classical, in kcal/mol) as a function of the step number. Shown here is the sample case of Deformed Structure 6 of Table 2, after 1000 steps of QM/MM minimization, using a 0.5 kcal mol−1 Å−2 constraint. From this figure it can be seen that 1000 steps of QM/MM minimization is sufficient to obtain convergent results for this system.
Most importantly, what is established here is that one cannot use fixed water molecules to examine or establish the energy of oxyanion holes in proteins, and that, as will be shown below, doing this requires performing full QM/MM calculations in which the energy in the protein and in water (not in a molecular crystal) is explored with full relaxation and proper free energy calculations (see the next section). Further dwelling on what the exact constraint in the water model should be is not useful, as this is not a proper model.
After considering the problems with a minimal model of the protein, we should consider the risks associated with using a model comprised of two fixed water molecules to examine the energetics of the reference reaction in water. Here, one may overlook the point that any study in bulk water must reproduce the fact that the solvent molecules are more or less randomly oriented around the C=O system (with some limited orientation in the first solvation shell), and then have to reorient upon the formation of the oxyanion. This reorganization penalty cannot be captured by a solvation model involving two fixed water molecules which are placed in the best orientation, a point which can be realized by anybody who searches for the average solvent orientation in actual simulations, but it can be much better quantified either by the estimate of Table 1, or by direct calculations of the reorganization energy which are summarized in Table 4 (which will be considered below). Thus, clearly, modeling an oxyanion hole using two gas-phase water molecules is not a fully realistic approach, and such an approach leads to, for instance, the incorrect conclusions reached with regard to LBHB (for discussion of this issue, see the problems with Ref. 42, which are discussed in Ref. 8). In conclusion, using two relatively fixed water molecules as a model for an oxyanion hole may lead one to conclude that such a system does not to stabilize the TS, but rather destabilizes the RS. However, in the case considered above, this conclusion is due to the effect of simply changing a dihedral angle, whereas, with a realistic constraint, the effect of these water molecules is primarily to interact with and stabilize the developing negative charge on the oxyanion.
TABLE 4.
Evaluating the LRA contributions of the oxyanion dipoles in subtilisin, and of the two water molecules in water[a].
| Fragment | < ΔU >C=O | < ΔU >C-O− | ΔG | λ |
|---|---|---|---|---|
| Subtilisin (K=0.1) | −8.3 | −12.3 | −10.3 | 2.0 |
| Water (K=0.1) | −0.01 | −1.5 | −0.8 | 0.75 |
| Water (K=1.0) | −1.5 | −15.0 | −8.2 | 6.7 |
Energies in kcal/mol. Here, < ΔU >C=O and < ΔU >C-O− designate the average, < ΔUC-O− − UC=O > over trajectories where the system (either protein or water) sees the indicated state and λ is the reorganization energy. The results for the water simulations are given with different constraints, where K is the constraint magnitude, in kcal mol−1 Å−2, on the distance between the oxyanion oxygen and the water oxygen. This is necessary since the contribution of the water molecules in water is poorly defined (which is why it only makes sense to examine the total solvation).
III.3 THE FREE ENERGY IN AN ACTUAL ENZYME, RELATIVE TO THE CORRESPONDING REACTION IN SOLUTION
At this stage, we will change our strategy and consider the entire protein/substrate system, and also consider the contributions from the oxyanion hole to the activation energy. We will then compare the results to the corresponding results in solution. There are many examples of such studies of oxyanion holes from our group, which include the reproduction of absolute catalysis and the effect of mutations, starting in 198623, quantifying this in 198831, later even doing this by ab initio QM/MM in 199843, and then also quantifying the problem in ketosteroid isomerase (KSI)13,22. This was also confirmed by others (e.g. Ref. 25).
Here, we repeat a part of our analysis of subtilisin, going beyond the well-defined and complete free energy analysis of Ref. 31, and evaluating the separate free energy contributions of the oxyanion in the presence of the rest of the system. This type of study (Table 4) involves LRA calculations27,44, using the same procedure that we applied in the study of the separate contributions in the reaction of vitamin B12 enzymes29 and the ribosome22. More specifically, the LRA calculations determine the free energy of moving from the C=O to the C-O− state by:
| (5) |
where U is the potential energy of the given state, obtained by the corresponding EVB energy, and <>x designates an average over the indicated state (where X is either C=O or C-O−). The reorganization energy is given by:
| (6) |
(note that the precise details of the EVB energy terms are given elsewhere, see e.g. citations in Ref. 45).
Since the LRA provides additive contributions, it is possible to extract the individual contributions from the oxyanion, and the calculated results are given in Table 4. We also explore the contribution of two water molecules in bulk water (this requires exploring the effect of leaving the specific molecules near the oxyanion with an artificial constraint). As seen from the table, a consistently modeled oxyanion stabilizes the C-O− more than the C=O, and also stabilizes the C=O more than the bulk water does. This finding is in apparent contrast to the conclusion that can be drawn from the work of Ref. 34, though this work (i.e. Ref. 34) attempted to consider the C=O energy in the model active site relative to that in molecular crystals, rather than the relevant reference state, which is the C=O species in water.
Now since examining the contribution from two water molecules in water is problematic (since we actually have to consider the entire solvent system) we have also performed LRA calculations which consider the complete environment (both in protein and in water), while evaluating only the free energy associated with the formation of the C=O polar group and the C-O− ionized group, and while keeping the rest of the substrate without its residual charges. This type of study (Table 5) can tell us whether or not we have an RSD mechanism. The evaluation of the energy of the C-O− is more complex, since in serine proteases the overall TS involves the [His+ C-O−] system, which is stabilized by both the oxyanion hole and the Asp 102 (which is, in turn, stabilized by the Asp hole31), so we cannot consider just the C-O− system when evaluating the observed catalytic effect. It should be noted that we have evaluated the overall correct contribution of forming the TS many times before (as was shown in the abovementioned references). Thus, we only focus here on the energy of the C=O system, where the effect of having or not having an ionized His does not change the solvation of the C=O system. Furthermore, the true ground state has an unprotonated His, which makes the analysis of the C=O species relevant to the actual reaction. At any rate, the calculations summarized in Table 5 establish that the enzyme stabilizes the C=O state more than water does.
TABLE 5.
The contribution of the overall environment to the free energy of the C=O fragment[a].
| Fragment | < ΔU >C=O(np) | < ΔU >C=O(p) | ΔG |
|---|---|---|---|
| Subtilisin (C=O) | −5.0 | −10.0 | −6.8 |
| Water (C=O) | −0.4 | −5.8 | −2.18 |
Energies in kcal/mol. Notation as in Table 4. Here, the reported energies correspond to a change in the residual charges of the indicated system
III.4 THE RISKS ASSOCIATED WITH A GAS PHASE ANALYSIS OF ENZYMES IN ENZYME DESIGN
After having established the energetics in actual oxyanion holes, and in oversimplified gas-phase models, we are ready to move on to a comprehensive discussion of the problems with oversimplified treatments of oxyanion holes. That is, basically, the analysis above has established the problems with performing oversimplified studies of artificial oxyanion holes, but, in order to prevent further errors, it is useful to be specific here.
The first issue is the risk of overinterpreting the hydrogen bonding orientations found in fixed X-ray structures. Here, one has to not only consider the poor resolution of the hydrogen positions, but, also (and far more importantly), the consequences of not allowing the oxyanion to relax, and thus not realizing that an infinitely rigid protein leads to completely unrealistic findings that do not reflect the preorganization correctly.
Additionally, choosing an incorrect reference state for evaluating the relative stability is a crucial problem. That is, not considering the energy of moving the “oxyanion hole” (i.e. the two water molecules) from infinity towards the oxyanion makes it difficult to determine the actual stabilization of the negative charge, which is perhaps reflected in the results shown in Fig. 8 of Ref. 34 (see also the SI of this paper). Following from this, comparison to the stability of small molecules in crystals (which was not done by any worker with actual calculations of the crystal) instead of the stability in water may lead to incorrect conclusions, as the crystal is already relaxed in such a way as to try to provide optimal stabilization to the C=O system. This fact has been demonstrated above, in addition to showing that the protein has not been designed to destabilize the C=O system. In any case, attempts to introduce structural constraints on an unrelaxed X-ray structure are problematic, and unlikely to reproduce any observed energy. Similarly, it should be noted that the active site arrangement can be quite different in the case of an inhibitor than in the case of the actual TS. Finally, with the focus given to enzyme design (e.g. Ref. 38), and the importance of small molecule catalysts for progress in this area, it would be remarkable to see gas phase models that can reproduce the known catalytic effect of an enzyme (a task which has been achieved very effectively by our approaches46).
FIGURE 8.

A schematic description of the reacting system in the reaction catalyzed by orotidine 5'-monophosphate decarboxylase. This figure depicts the rate-determining step in the lysine-assisted mechanism assumed for the enzyme. In addition to the orotate + LysH+ reacting system, this figure also includes a schematic description of the protein environment, as well as illustrating the fact that the mechanism involves a significant increase in the dipole moment of the reacting system upon transfer from the reactant to the transition state. This figure is adapted from Ref. 52.
An additional problematic issue we would like to touch on here is the idea presented in e.g. Ref. 34 that the preorganization effect cannot be important because even “optimally placed” water molecules will not be able to stabilize the reactant state less well than the ground state. That is, disproving the conclusions of quantitative earlier work (e.g. Ref. 22) about preorganization in oxyanion holes cannot be accomplished by modeling some gas-phase system without the enzyme (especially when doing this using the problematic assumption that the enzyme should destabilize the ground state in order to achieve catalysis). It is basically hard to accept the idea that gas-phase calculations are somehow superior to QM/MM calculations that have consistently reproduced the effect of mutations.
After discussing the general problems with gas-phase studies of enzymes, we would like to point out that this issue becomes more serious when being used to draw general conclusions about enzyme design. Here, the problem is two-fold. First and foremost, even though the use of models where the TS features are determined in the gas-phase are quite popular (e.g. Refs. 47,48), such an approach is not so effective for enzyme design. That is, since it is not capable of reproducing the TS binding free energy or the catalytic effect, it cannot be used to rank the different design constructs in a quantitative manner (even if an approach like that used in Refs. 47,48, where the emphasis was on the generation of protein structures with reasonable interactions with the TS model, can be an effective way to generate structural candidates). In other words, an approach that does not treat the protein-TS electrostatic free energy in a consistent way, will, as discussed in Section III.1, miss the preorganization effect and the reorganization penalty, and thus the corresponding catalytic effect. Another problem is the fact that such gas-phase models have no dielectric screening, and the absence of this can lead to irrelevant results such as the LBHB proposal, as was for example the case in Refs. 49,50. Thus, in short, a consistent treatment of electrostatics which captures the protein preorganization effect (by including the protein reorganization during the simulations) is crucial for successful enzyme design, and recently, we made great progress on this front46,51. Without this, the only way to reach a highly catalytic enzyme would be through extensive and costly experimental trial and error, and using gas-phase models with two water molecules is clearly not the way to go about this addressing this issue.
III.5. REVISITING THE GENERAL GROUND STATE DESTABILIZATION IDEA
In light of the fact that the RSD idea still reappears in different incarnations in the literature (including the works that have been discussed above), it is quite important to reclarify (in greater detail) some of the problems with this proposal. Now while we could of course refer the reader to several careful studies of this issue (see Ref. 2, and references cited therein), we nevertheless find it useful to address the recent appearances of this proposal, in the particular case of orotidine 5'-monophosphate decarboxylase (ODCase)52. That is, the catalytic effect of ODCase was first proposed to involve the desolvation effect53. However, this was shown to involve an incomplete thermodynamic cycle (see e.g. Ref. 52). The elucidation of the structure of this enzyme showed that its active site is extremely polar (highly charged), however, this led to a new RSD proposal, where it was argued that the negatively charged protein group destabilizes the carboxylate of the orotate group of the substrate54. This proposal was shown to be inconsistent with the nature of the system, since a destabilized orotate will accept a proton and become stable52. Furthermore, a careful computational study has illustrated that the protein works by TSS and not by RSD (see discussion in Ref. 52 and below). Moreover, studies by Wolfenden and co-workers53,55 have provided clear evidence against the RSD proposal (see Ref. 2, as well as the discussion below of the experimental work of Ref. 56).
The problems with the RSD proposal were dramatically emphasized by the experimental work of Amyes and coworkers57, who explored the origin of the catalytic power of ODCase (trying to support the RSD proposal) by studying the decarboxylation of a truncated substrate (called EO), which lacks the 5'-phosphodianion part. These workers found that while the reaction of this substrate is quite slow, binding an exogenous phosphate dianion to ODCase results in an 80000-fold increase in kcat/Km. This appeared to be in a clear conflict with the proposal that the presumed RSD is due to the binding free energy of the 5'-phosphodianion part of the substrate, which is supposed to induce extremely large reactant RSD, and thus to catalyze the reaction (see e.g. Ref. 54). In this proposal, the negatively charged groups of the protein (i.e. Asp70 and Asp75B in Fig. 8) are used to destabilize the carboxylate of the orotate. In fact, this view has been presented as confirmation of Jencks' proposal that enzymes work by using binding energies to destabilize the ground state of the reactive part of the substrate. However, a logical analysis of the work of Ref. 57 indicated that the RSD idea is incorrect, and thus confirmed our careful analysis52. That is, the RSD proposal of Ref. 54 is based on the idea that the phosphate part is bound so strongly that it pulls the chemical part (thorough the R-C bond in Fig. 8) towards its destabilizing environment, and leads to about 20 kcal/mol RSD. Unfortunately, not only was this shown to be problematic in Ref. 52, but also, the experiments of Ref. 57 showed that catalysis occurs in the absence of a bond between the phosphate and EO parts, such that the presumed strain cannot be transferred between these parts. Of course, the binding of the negative part of the substrate does help the active site reach its proper preorganization, and thus, to use this for the electrostatic stabilization of the TS. This, however, has little to do with the classical Jencks idea of using the binding energy for RSD.
Now a more recent work58 focused on a part of the reverse reaction, starting from the product uridine 5'-monophosphase (UMP) without the CO2 part, and asked how much it “costs” to remove the proton from the C6 carbon in the presence of the protonated Lys (or what the energy of the transition state with a protonated lysine and negatively charged carbanion actually is). This energy, which is directly related to the pKa for the deprotonation of the C6 carbon, was determined by use of deuterium exchange. It was found that the pKa is reduced by at least 10 units, or more than 14 kcal/mol, which means that the corresponding system, that strongly resembles our TS, is stabilized by at least 14 kcal/mol. This provides another overwhelming specific example that counters the RSD idea and provides direct proof of the TSS idea.
Despite these convincing points, and the illustrations that theoretical support for the RSD idea in the case of ODCase is fundamentally flawed (including contradictions of the first law of thermodynamics by Ref. 59, see the discussion in Ref. 2), we nevertheless still see conflicting analyses and significant confusion about the meaning of the very clear experimental findings. An illustrative example is given in a very recent work60, where a D70N mutation reduced kcat by about 200, while leaving KM almost unchanged. This finding essentially proved that the ionized acid is not involved in any ground state destabilization (otherwise its mutation would have led to a larger binding energy as well as a reduction in KM), and yet the authors ventured to say that their finding supports the RSD idea of Ref. 54. In an effort to find a way around this convoluted argument, and again, perhaps in light of the difficulties with understanding the preorganization idea, the authors suggested a new definition of RSD (see footnote 4 in Ref. 60). They also suggested that somehow, RSD is related to an undefined destabilization of the path to the TS. Of course one has to define such a proposal, since the issue is the free energies of the RS and TS, and destabilization at any point on the path between them that cannot change the activation energy and the rate constant (unless such a point is a transition state, and we have TS destabilization).
The misunderstandings of the authors of Ref. 60 may be due to the difficulty of conceptualizing the preorganization effect, which thus leads them to misinterpret the observation60 that the mutation does not change the effect of the protein on the above proton transfer to the C6 of UMP. This finding seems to increase the difficulty of analyzing unique information about RSD. That is, without a clear energy based concept, it is basically impossible to define, analyze and understand the key experiments (see a related discussion in Ref. 17). The problem starts with the assumption that the role of D70 is to destabilize the RS, a point which was already shown to be incorrect by any of the points above (including the fact that such an effect would lead to protonation of the orotate), and, of course, if this point were correct, we would have observed an increase rather than a decrease in the pKa of C6. Now the only model that has up to now succeeded to rationalize and quantify the catalysis in ODCase is the model presented in Fig. 8. In this model, the role of D70 is to stabilize the TS formed by Lys72 and the negatively charged C6. Of course, this stabilization depends on the exact preorganization, and is not expected to be identical in UMP and in the TS of ODCase, but rather, to exhibit a similar trend. Our point is that the exact trend in the mutation cannot be predicted (and even understood) without the calculation of clear energy based concepts, but, in any case, starting from an incorrect premise and reformulating the meaning of key concepts such as RSD is not so useful.
Finally, despite the attempts to use clear evidence against the RSD proposal as support for this idea, we see that in their most recent paper56, the authors now propose that TSS is entropically helped by putting together residues (in this case Arg235), which participate in electrostatic stabilization (relating this to Jencks' entropic idea61). Of course, this is another example of the difficulty in understanding the preorganization idea. That is, having electrostatic stabilization by an enzyme has nothing to do with Jencks' entropic idea (which is about the entropy of placing the substrate fragments together). Folding the enzyme (Step 1 in Fig. 1b) is part of the formation of the catalyst, and not the explanation for its catalytic effect.
IV. CONCLUDING DISCUSSION
This work has re-analyzed the contribution of preorganized hydrogen bonds to the catalysis of oxyanion formation in enzymes, and further established their role in TS stabilization. It has also clarified the problems associated with an oversimplified “theozyme” analysis for preorganized systems. The first important issue we have addressed is the idea that in the oxyanion hole, the hydrogen bonds are arranged in such a way that the oxyanions are reasonably but sub-optimally stabilized, in order to avoid overstabilizing the ground state34. This idea seems to result in an apparently new version of the popular ground state destabilization (GSD) (i.e. RSD) proposal, which suggests that enzymes reduce the activation barrier of the reaction by destabilizing the ground states of their reactant fragments (as was put forward by many workers, such as, for instance, the works of Ref. 62–64 amongst others). However, this proposal has not been supported by systematic computational studies, which have demonstrated that the TS is in fact “solvated” much more strongly in the enzyme than in the reference solution reaction (see discussion in e.g. Refs. 2,32,65, and references cited therein, as well as in Section V). In Section III, we demonstrate once again that in this case, there is also much larger TS stabilization than an RS destabilization effect, and that the water molecules provide a comparatively much smaller interation with the RS. This issue ties in directly with the most risky problem with treatments that ignore the reorganization energy, which (in an ideally preorganized enzyme) can reach half of the final stabilization. That is, the misunderstanding of the nature of solvation effects, which are due to the total environment rather than just one or two water molecules, prevents one from comprehending the reorganization effect, which, in water, can be estimated by taking half of the solvation free energy as the upper possible limit.
Now, of course, there are other major problems that are associated with using high-level calculations and then forgetting about the entropy, which is a crucial part of the free energy, and automatically included in proper EVB calculations. Clearly, calculating the entropy correctly is a non-trivial issue, as we discussed at length in e.g. Ref. 66. However, ignoring this issue compromises the obtained results (for a related example of this problem, see Ref. 67). Additionally, it is not useful to focus on the secondary issue, namely, the effect of changing the angle of the hydrogen bond, rather than the change in the free energy interaction between the hydrogen bonds and the oxygen upon the formation of the oxyanion charge (see the demonstration of this in Section III.2). Yet another fundamental problem can be found in the idea that the enzyme is not designed to provide optimal stabilization to the TS. This conclusion may come from not trying to optimize the hydrogen bonding interactions at the TS in the protein. Of course, the initial structure is not perfect, and there is some reorganization. However, this reorganization is much smaller than that for the same reaction in water, which is the origin of enzyme catalysis.
These oversimplified views are problematic, not only because they reflect a misunderstanding of the reorganization concept, but also because of the problems it leads to in attempts to understand deeper issues, such as was the case in the argument about KSI. This issue clearly cannot be examined by those who trivialize the detailed analysis of Ref. 22 and assume that this work is about charge delocalization in water and in the protein, rather than a careful quantitative analysis of the preorganization effect. That is, as discussed in detail above, it is not possible to provide a meaningful judgment about problems that are on the level of those we addressed in our most recent work on the topic13 by using a model involving two water molecules in the gas-phase as a presumably physically meaningful representation of the oxyanion hole, and assuming that this can conclusively resolve the argument about KSI.
What we now see in the literature is a progression from workers who proposed the study of gas-phase reacting systems (e.g. Refs. 47,48,68,69) to now including also a model oxyanion hole34,70,71. However, perhaps it would be better to give more credit to studies that include all the protein from the beginning. In other words, this is similar to progress in studies of proton transfer in bacteriorhodopsin, where irrelevant studies with a single water molecule and a gas-phase proton such as that of Ref. 72 were viewed by the community as being much more reasonable than calculations of the whole solvation environment73–75, and leads to problematic concept such as proton wires. In any case, we have already discussed the enormous problems associated with the belief that gas-phase calculations, which completely miss the preorganization effect, are useful in enzyme design. Here, we would like to conclude by pointing out that even larger problem will arise if one were to follow studies that reach conclusions about catalysis by calculations that do not involve the enzyme, or are “solution” calculations without the solvent
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by Grant GM024492 from the National Institutes of Health (NIH). We also gratefully acknowledge the University of Southern California's High Performance Computing and Communication Center (HPCC) for computer time. Finally, we would like to thank Dr. Pankaz Sharma for his assistance in the preparation of the artwork for this manuscript, and Prof. Janez Mavrí for insightful discussion.
REFERENCES
- (1).Warshel A. Proc. Natl. Acad. Sci. U.S.A. 1978;75:5250–5254. doi: 10.1073/pnas.75.11.5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Warshel A, Sharma PK, Kato M, Xiang Y, Liu H, Olsson MHM. Chem. Rev. 2006;106:3210–3235. doi: 10.1021/cr0503106. [DOI] [PubMed] [Google Scholar]
- (3).Cleland WW, Frey PA, Gerlt JA. J. Biol. Chem. 1998;273:22529–22532. doi: 10.1074/jbc.273.40.25529. [DOI] [PubMed] [Google Scholar]
- (4).Frey PA, Whitt SA, Tobin JB. Science. 1994;264:1927–1930. doi: 10.1126/science.7661899. [DOI] [PubMed] [Google Scholar]
- (5).Cleland WW, Kreevoy MM. Science. 1994;264:1887–1890. doi: 10.1126/science.8009219. [DOI] [PubMed] [Google Scholar]
- (6).Halkides CJ, Wu YQ, Murray CJ. Biochemistry. 1996;35:15941–15948. doi: 10.1021/bi961805f. [DOI] [PubMed] [Google Scholar]
- (7).Zhao QJ, Abeygunawardana C, Talalay P, Mildvan AS. Biochemistry. 1996;33:2682–2687. [Google Scholar]
- (8).Schutz CN, Warshel A. Proteins. 2004;55:711–723. doi: 10.1002/prot.20096. [DOI] [PubMed] [Google Scholar]
- (9).Warshel A, Papazyan A. Proc. Natl. Acad. Sci. USA. 1996;93:13665–13670. doi: 10.1073/pnas.93.24.13665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Kraut DA, Sigala PA, Pybus B, Liu CW, Ringe D, Petsko GA, Herschlag D. PLoS Biol. 2006;4:0501–0519. doi: 10.1371/journal.pbio.0040099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Schwans JP, Kraut DA, Herschlag D. Proc. Natl. Acad. Sci. U. S. A. 2009;106:14271–14275. doi: 10.1073/pnas.0901032106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Sigala PA, Kraut, D. A. C. JMM, Pybus B, Ruben EA, Ringe D, Petsko GA, Herschlag D. J. Am. Chem. Soc. 2008;130:13696–13708. doi: 10.1021/ja803928m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Kamerlin SCL, Sharma PK, Chu ZT, Warshel A. Proc. Natl. Acad. Sci. U. S. A. 2010;107:4075–4080. doi: 10.1073/pnas.0914579107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Cleland WW. Adv. Phys. Org. Chm. 2010;44:1–17. [Google Scholar]
- (15).Pan YP, McAllister MA. J. Am. Chem. Soc. 1998;120:166–169. [Google Scholar]
- (16).Kumar GA, Pan Y, Smallwood CJ, McAllister MA. J. Comp. Chem. 1998;19:1345–1352. [Google Scholar]
- (17).Kamerlin SCL, Warshel A. Proteins: Struct. Func. Bioinformat. 2010;78:1339–1375. doi: 10.1002/prot.22654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Pihko P, Rapakko S, Wierenga RK. In: Hydrogen Bonding in Organic Synthesis. Pihko PM, editor. Wiley VCH Verlag GmBH & Co.; Weinheim: 2009. [Google Scholar]
- (19).Robertus JD, Kraut J, Alden RA, Birktoft JJ. Biochemistry. 1972;11:4293–4303. doi: 10.1021/bi00773a016. [DOI] [PubMed] [Google Scholar]
- (20).Bryan P, Pantoliano MW, Quill SG, Hsiao H-Y, Poulos T. Proc. Natl. Acad. Sci. U. S. A. 1986;83:3743–3745. doi: 10.1073/pnas.83.11.3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Warshel A, Naray-Szabo G, Sussman F, Hwang J-K. Biochemistry. 1989;28:3629–3637. doi: 10.1021/bi00435a001. [DOI] [PubMed] [Google Scholar]
- (22).Warshel A, Sharma PK, Chu ZT, Aqvist J. Biochemistry. 2007;46:1466–1476. doi: 10.1021/bi061752u. [DOI] [PubMed] [Google Scholar]
- (23).Warshel A, Russell S. J. Am. Chem. Soc. 1986;108:6569–6579. [Google Scholar]
- (24).Hwang J-K, Warshel A. Biochemistry. 1987;26:2669–2673. doi: 10.1021/bi00384a003. [DOI] [PubMed] [Google Scholar]
- (25).Feierberg I, Åqvist J. Biochemistry. 2002;41:15728–15735. doi: 10.1021/bi026873i. [DOI] [PubMed] [Google Scholar]
- (26).Warshel A. J. Biol. Chem. 1998;273:27035–27038. doi: 10.1074/jbc.273.42.27035. [DOI] [PubMed] [Google Scholar]
- (27).Lee FS, Chu ZT, Bolger MB, Warshel A. Prot. Eng. 1992;5:215–228. doi: 10.1093/protein/5.3.215. [DOI] [PubMed] [Google Scholar]
- (28).Sham YY, Chu ZT, Tao H, Warshel A. Proteins: Struct. Funct. Genet. 2000;39:393–407. [PubMed] [Google Scholar]
- (29).Sharma PK, Chu ZT, Olsson MHM, Warshel A. Proc. Natl. Acad. Sci. U.S.A. 2007;104:9661–9666. doi: 10.1073/pnas.0702238104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Ishikita H, Warshel A. Angew. Chem. Int. Ed. 2008;47:697–700. doi: 10.1002/anie.200704178. [DOI] [PubMed] [Google Scholar]
- (31).Warshel A, Sussman F, Hwang J-K. J. Mol. Biol. 1988;201:139–159. doi: 10.1016/0022-2836(88)90445-7. [DOI] [PubMed] [Google Scholar]
- (32).Warshel A. Computer modeling of chemical reactions in enzymes and solutions. John Wiley & Sons; New York: 1991. [Google Scholar]
- (33).Warshel A, Åqvist J, Creighton S. Proc. Natl. Acad. Sci. USA. 1989;86:5820–5824. doi: 10.1073/pnas.86.15.5820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Simón L, Goodman JM. J. Org. Chem. 2010;75:1831–1840. doi: 10.1021/jo901503d. [DOI] [PubMed] [Google Scholar]
- (35).Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. Nucleic Acids Research. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Allen FH. Acta Cryallogra., Sect. B. 2002;58:380–388. doi: 10.1107/s0108768102003890. [DOI] [PubMed] [Google Scholar]
- (37).Štrajbl M, Shurki A, Kato M, Warshel A. J. Am. Chem. Soc. 2003;125:10228–10237. doi: 10.1021/ja0356481. [DOI] [PubMed] [Google Scholar]
- (38).Broadwith P. Chemistry World. 2010;7:39. [Google Scholar]
- (39).Lee FS, Chu ZT, Warshel A. J. Comp. Chem. 1993;14:161–185. [Google Scholar]
- (40).Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Vreven JT, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, M. B, Cossi M, Scalmani G, Rega N, Petersson GA, N. H, Hada M, Ehara M, Toyota K, Fukuda R, H. J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, K. M, Li X, Knox JE, Hratchian HP, Cross JB, Adamo C, J. J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, C. R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, V. GA, Salvador P, Dannenberg JJ, Zakrzewski VG, D. S, Daniels AD, Strain MC, Farkas O, M. DK, Rabuck AD, Raghavachari K, Foresman JB, O. JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, S. BB, Liu G, Liashenko A, Piskorz P, Komaromi I, M. RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, N. A, Challacombe M, Gill PMW, Johnson B, C. W, Wong MW, Gonzalez C, Pople JA. Gaussian, Inc.; Wallingford CT: 2004. [Google Scholar]
- (41).Adamo C, Barone V. J. Chem. Phys. 1998;108:664–675. [Google Scholar]
- (42).Kim KS, Kyung SO, Lee JY. Proc. Natl. Acad. Sci. U. S. A. 2000;97:6373–6378. doi: 10.1073/pnas.97.12.6373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Bentzien J, Muller RP, Florian J, Warshel A. Journal of Physical Chemistry B. 1998;102:2293–2301. [Google Scholar]
- (44).Warshel A, Sharma PK, Kato M, Parson WW. Biochim. Biophys. Acta. 2006;1764:1647–1676. doi: 10.1016/j.bbapap.2006.08.007. [DOI] [PubMed] [Google Scholar]
- (45).Kamerlin SCL, Warshel A. Faraday Discuss. 2010;145:71–106. doi: 10.1039/B907354J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Roca M, Vardi-Kilshtain A, Warshel A. Biochemistry. 2009;48:3046–3056. doi: 10.1021/bi802191b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Roethlisberger D, Khersonsky O, Wollacott AM, Jiang L, DeChancie J, Betker J, Gallaher JL, Althoff EA, Zanghellini A, Dym O, Albeck S, Houk KN, Tawfik DS, Baker D. Nature. 2008;453:164–166. doi: 10.1038/nature06879. [DOI] [PubMed] [Google Scholar]
- (48).Jiang L, Althoff EA, Clemente FR, Doyle L, Roethlisberger D, Zanghellini A, Gallaher JL, Betker JL, Tanaka F, Barbas, I. CF, Hilvert D, Houk KN, Stoddard BL, Baker D. Science. 2008;319:1387–1391. doi: 10.1126/science.1152692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Pan Y, McAllister MA. J. Org. Chem. 1997;62:8171–8176. doi: 10.1021/jo971290d. [DOI] [PubMed] [Google Scholar]
- (50).Schiøtt B, Iversen BB, Madsen GKH, Bruice TC. J. Am Chem. Soc. 1998;120:12117–12124. [Google Scholar]
- (51).Vardi-Kilshtain A, Roca M, Warshel A. Biotechnol. J. 2009;4:495–500. doi: 10.1002/biot.200800299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Warshel A, Strajbl M, Villa J, Florian J. Biochemistry. 2000;39:14728–14738. doi: 10.1021/bi000987h. [DOI] [PubMed] [Google Scholar]
- (53).Miller B, Wolfenden R. Ann. Rev. Biochem. 2002;71:847–885. doi: 10.1146/annurev.biochem.71.110601.135446. [DOI] [PubMed] [Google Scholar]
- (54).Wu N, Mo Y, Gao J, Pai EF. Proc. Natl. Acad. Sci. USA. 2000;97:2017–2022. doi: 10.1073/pnas.050417797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Miller B, Snider MJ, Short S, Wolfenden R. Biochemistry. 2000;39:8113–8118. doi: 10.1021/bi000818x. [DOI] [PubMed] [Google Scholar]
- (56).Barnett SA, Amyes TL, Wood BM, Gerlt JA, Richard JP. Biochemistry. 2010;49:824–826. doi: 10.1021/bi902174q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Amyes TL, Richard JP. J. Am. Chem. Soc. 2005;127:15708–15709. doi: 10.1021/ja055493s. [DOI] [PubMed] [Google Scholar]
- (58).Amyes TL, Wood BM, Chan K, Gerlt JA, Richard JP. J. Am. Chem. Soc. 2008;130:1574–1575. doi: 10.1021/ja710384t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Gao JL, Truhlar DG. Annual Review of Physical Chemistry. 2002;53:467–505. doi: 10.1146/annurev.physchem.53.091301.150114. [DOI] [PubMed] [Google Scholar]
- (60).Chan KK, Wood BM, Fedorov AA, Fedorov EV, Imker HJ, Amyes TL, Richard JP, Almo SC, Gerlt JA. Biochemistry. 2009;48:5518–5531. doi: 10.1021/bi900623r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Jencks WP. Proc. Natl. Acad. Sci. USA. 1981;78:4046–4050. doi: 10.1073/pnas.78.7.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Jencks WP. Catalysis in chemistry and enzymology. Dover: New York: 1987. [Google Scholar]
- (63).Devi-Kesavan LS, Gao J. J. Am. Chem. Soc. 2003;125:1532–1540. doi: 10.1021/ja026955u. [DOI] [PubMed] [Google Scholar]
- (64).Lee JK, Houk KN. Science. 1997;276:942–945. doi: 10.1126/science.276.5314.942. [DOI] [PubMed] [Google Scholar]
- (65).Shurki A, Warshel A. Adv. Protein Chem. 2003;66:249–313. doi: 10.1016/s0065-3233(03)66007-9. [DOI] [PubMed] [Google Scholar]
- (66).Štrajbl M, Sham YY, Villà J, Chu ZT, Warshel A. J. Phys. Chem. B. 2000;104:4578–4584. [Google Scholar]
- (67).Kamerlin SCL, Haranczyk M, Warshel A. ChemPhysChem. 2009;10:1125–1134. doi: 10.1002/cphc.200800753. [DOI] [PubMed] [Google Scholar]
- (68).Dewar MJ. Enzyme. 1986;36:8–20. doi: 10.1159/000469274. [DOI] [PubMed] [Google Scholar]
- (69).Andrea TA, Dietrich SW, Murray WJ, Kollman PA, Jorgensen EC, Rothenberg SJ. J. Med. Chem. 1979;22:221–232. doi: 10.1021/jm00189a002. [DOI] [PubMed] [Google Scholar]
- (70).Tantillo DJ, Chen J, Houk KN. Curr. Opin. Chem. Biol. 1988;2:743–750. doi: 10.1016/s1367-5931(98)80112-9. [DOI] [PubMed] [Google Scholar]
- (71).Oh KS, Cha S-S, Kim D-H, Choo H-S, Ha N-C, Choi G, Lee PL, Tarakeshwar P, Son SH, Choi KY, Oh B-H, Kim KS. Biochemistry. 2000;39:13891–13896. doi: 10.1021/bi001629h. [DOI] [PubMed] [Google Scholar]
- (72).Scheiner S, Hillenbrand EA. Proc. Natl. Acad. Sci. USA. 1985;82:2741–2745. doi: 10.1073/pnas.82.9.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Warshel A. Proc. Natl. Acad. Sci. 1978;75:2558–2562. doi: 10.1073/pnas.75.6.2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Warshel A, Ottolenghi M. Photochem. Photobiol. 1979;30:291. doi: 10.1111/j.1751-1097.1979.tb07149.x. [DOI] [PubMed] [Google Scholar]
- (75).Warshel A, Chu ZT. J. Phys. Chem. B. 2001;105:9857–9871. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.