


Abstract
The S-adenosyl-l-methionine dependent methylation of adenine 58 in the T-loop of tRNAs is essential for cell growth in yeast or for adaptation to high temperatures in thermophilic organisms. In contrast to bacterial and eukaryotic tRNA m1A58 methyltransferases that are site-specific, the homologous archaeal enzyme from Pyrococcus abyssi catalyzes the formation of m1A also at the adjacent position 57, m1A57 being a precursor of 1-methylinosine. We report here the crystal structure of P. abyssi tRNA m1A57/58 methyltransferase (PabTrmI), in complex with S-adenosyl-l-methionine or S-adenosyl-l-homocysteine in three different space groups. The fold of the monomer and the tetrameric architecture are similar to those of the bacterial enzymes. However, the inter-monomer contacts exhibit unique features. In particular, four disulfide bonds contribute to the hyperthermostability of the archaeal enzyme since their mutation lowers the melting temperature by 16.5°C. His78 in conserved motif X, which is present only in TrmIs from the Thermococcocales order, lies near the active site and displays two alternative conformations. Mutagenesis indicates His78 is important for catalytic efficiency of PabTrmI. When A59 is absent in tRNAAsp, only A57 is modified. Identification of the methylated positions in tRNAAsp by mass spectrometry confirms that PabTrmI methylates the first adenine of an AA sequence.
INTRODUCTION
tRNA maturation consists of different post-transcriptional processing steps, including splicing, end-trimming and nucleoside modifications. Although modified nucleosides have been identified in all cellular RNAs, the greatest number and variety of modifications occur in tRNAs. Base or ribose methylation is one of the most frequently encountered modifications. These modifications ensure efficient decoding during translation as well as correct RNA folding, 3D structure stabilization and proper recognition of RNA by its partners during translation (1–5). Only one tRNA modification occurring outside the anticodon loop, 1-methyladenosine (m1A) at position 58 in the T-loop, has been shown to be essential for cell growth under normal conditions (6,7). This modification appears very early in tRNA biogenesis (8,9).
A58 is the most conserved nucleoside in tRNA. The presence of m1A58 is fairly common in the T-loop of tRNAs of most eukaryotes and archaea but it occurs less frequently in eubacteria. For example, no m1A58 methyltransferase (MTase) ortholog could be found in Escherichia coli and related Proteobacteria (10). m1A58 and m5U54 form a universal reverse Hoogsteen base pair in the T-loop of most tRNAs. It is assumed that this tertiary interaction together with the strong positive charge at m1A58, which is located on the outside of the molecular tRNA structure, are important for the tRNA tertiary structure and/or for protein recognition (11).
An important biological role for m1A58 has been demonstrated in different organisms. In yeast, it has a crucial role in the maturation and stability of initiator tRNA (7,12). In fact, 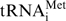 lacking m1A58 is degraded by a nuclear surveillance pathway, through polyadenylation and subsequent action of the nuclear exosome (13,14). In addition, the presence of a methylated adenine at position 58 in human
lacking m1A58 is degraded by a nuclear surveillance pathway, through polyadenylation and subsequent action of the nuclear exosome (13,14). In addition, the presence of a methylated adenine at position 58 in human 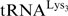 is crucial for HIV replication. Indeed, human
is crucial for HIV replication. Indeed, human 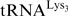 is used as a primer for reverse transcription of HIV-1 viral RNA and methylation of A58 in this tRNA is required to allow productive strand transfer during (+) strand DNA synthesis (15). Finally, the disruption of the gene coding for m1A58 MTase in Thermus thermophilus shows that m1A58 modification is required for growth of this bacterium at high temperatures (16).
is used as a primer for reverse transcription of HIV-1 viral RNA and methylation of A58 in this tRNA is required to allow productive strand transfer during (+) strand DNA synthesis (15). Finally, the disruption of the gene coding for m1A58 MTase in Thermus thermophilus shows that m1A58 modification is required for growth of this bacterium at high temperatures (16).
The structures of three bacterial m1A58 MTases (called TrmI) have previously been reported. The structures of m1A58 MTases from Mycobacterium tuberculosis (MtTrmI, initially called Rv2118c) (17,18) and T. thermophilus (TtTrmI) (19) have been determined in complex with S-adenosyl-l-methionine (SAM) and S-adenosyl-l-homocysteine (SAH), respectively, at 1.98 and 1.7 Å resolution, respectively. Moreover, the structure of the homologous enzyme from Aquifex aeolicus (AqTrmI) (PDB code 2YVL) has been solved in complex with SAM at 2.2 Å resolution but the activity of the protein was not reported. In all these structures, the enzyme is organized as a homo-tetramer with the N-terminal domains of each monomer protruding from the central body of the tetramer consisting of four C-terminal domains. In the case of the T. thermophilus enzyme, non-covalent electrospray ionization mass spectrometry (MS) indicated that one to two molecules of tRNA can bind per tetramer (19).
Pyrococcus abyssi is an hyperthermophilic archaeon with an optimal growth temperature of 100°C at 20 MPa (20). Pyrococcus abyssi TrmI displays unusual regiospecificity compared to bacterial and eukaryotic homologs that are specific for A58, since it catalyzes the formation of m1A also at the adjacent position 57, m1A57 being the obligate intermediate in the biosynthesis of 1-methylinosine (21). A structural and biochemical study of PabTrmI was undertaken to shed light on the origin of hyperthermostability and the multisite recognition mechanism. The crystal structure of TrmI from P. abyssi in complex with SAH was determined in two different space groups at 2.6 and 2.05 Å resolution, and in complex with S-adenosyl-l-methionine (SAM) at 1.6 Å resolution. The structures, as well as the melting temperatures of wild-type and mutant proteins, enlighten the importance of intermolecular disulfide bonds for archaeal thermostability. We show that the presence of adenine at position 59 in P. abyssi tRNAAsp (PabtRNAAsp) is important for the multi-site specificity of the archaeal enzyme at both positions 57 and 58 in PabtRNAAsp and that His78 near the active site is important for efficient catalysis.
MATERIAL AND METHODS
Enzyme purification, crystallization and structure determination
PabTrmI was purified and Crystal Form I crystallized as described (21). Crystal Forms II and III were grown at 18°C in hanging-drops by vapor diffusion. Crystal Form II was obtained by mixing 1 µl PabTrmI (15 mg ml−1 in 50 mM Tris–HCl pH 8.5, 0.5 M KCl, 0.2 M imidazole) with 1 µl of a 1 ml reservoir solution (2.4 M ammonium sulfate, 0.2 M ammonium acetate) in the presence of 5 mM SAM. Crystal Form III was obtained by mixing 1 µl PabTrmI with 1 µl of a 1 ml reservoir solution (2.8 M ammonium sulfate, 0.2 M ammonium acetate) in the presence of 50 mM SAM and 50 mM ATP. Crystal Forms II and III were soaked for a few minutes in the mother liquor solution containing 20% ethyleneglycol, then flash frozen in a cold nitrogen stream at 100 K. Diffraction data for Crystal Form I were collected on beamline BM30A at the European Synchrotron Radiation Facility (Grenoble, France) and for Crystal Forms II and III on the PROXIMA1 beamline at the SOLEIL synchrotron in Saint Aubin (France). Processing was done with the MOSFLM package (22) for Crystal Form I or XDS (23) for Crystal Forms II and III. Data were scaled with SCALA for Crystal Forms I and II (22,24) and XSCALE (23) for Crystal Form III (Table 1). The Crystal Form I structure was solved by molecular replacement using AMoRe (25) and M. tuberculosis tRNA MTase as the protein model (PDB code 1I9G) (17). The successful search used a model consisting of the Cα atoms of the tetramer formed by the C-terminal domains. This tetramer was built from the monomer present in the asymmetric unit using the quaternary crystallographic structure. Rotation and translation functions were calculated over the 15–3.5 Å resolution range. The tetramer was then split into four subunits and refined as four rigid bodies. Building of the 76 N-terminal amino acids was done manually. For Crystal Forms II and III, molecular replacement was carried out with Phaser (26) using the Crystal Form I structure as model. The atomic models were refined by alternating cycles of model reconstruction with COOT (27) and refinement with CNS (28) and REFMAC5 (29). During the refinement, tight NCS restraints between the four monomers were used until the last cycles of refinement. Structures were superimposed using the program SUPERPK (Alzari, personal communication). The H78Y and C196S/C233S mutants were expressed and purified as the wild type enzyme (21). A final step of purification was carried out by loading the mutants at 0.5 ml/min onto a Superdex 200 HR 10/300 column (GE healthcare Inc.) equilibrated in 50 mM Tris-HCl, pH 8.5, 0.5 M KCl, 0.2 M imidazole.
Table 1.
Data collection and refinement statistics
| SAH Form I | SAH Form II | SAM Form III | |
|---|---|---|---|
| Data collection | |||
| Space group | P212121 | P31 | I222 |
| Cell dimensions | 66.03, 127.58, 151.40 | 138.22, 138.22, 121.12 | 86.55, 89.26, 110.32 |
| a, b, c (Å) | 90, 90, 90 | 90, 90, 120 | 90, 90, 90 |
| α, β, γ (°) | |||
| Resolution (Å) (Outer resolution shell) Å | 65.10–2.59 (2.73–2.59) | 34.86–2.05 (2.16–2.05) | 20.00–1.60 (1.70–1.60) |
| Rmerge | 0.046 (0.197) | 0.091 (0.706) | 0.103 (0.317) |
| I/σI | 11.7 (2.4) | 11.5 (2.1) | 16.4 (5.1) |
| Completeness (%) | 94.4 (68.6) | 99.8 (100.0) | 99.8 (99.9) |
| Redundancy | 5.4 (2.2) | 4.8 (4.8) | 9.1 (5.8) |
| Refinement | |||
| Resolution (Å) | 63.76–2.59 | 34.86–2.05 | 19.70–1.60 |
| (Outer resolution shell) Å | (2.66–2.59) | (2.10–2.05) | (1.64–1.60) |
| No. of reflections | 36 362 (1435) | 153 870 (11329) | 53 652 (3866) |
| Rwork | 0.231 (0.333) | 0.199 (0.267) | 0.184 (0.303) |
| Rfreea | 0.286 (0.365) | 0.218 (0.297) | 0.199 (0.308) |
| No. of atoms | |||
| Protein | 7950 | 8176 | 2054 |
| SAH | 104 | 104 | 26 |
| Sulfate | 0 | 4 | 4 |
| Ethyleneglycol | 0 | 0 | 6 |
| Acetate | 0 | 0 | 1 |
| Water | 112 | 359 | 340 |
| Average isotropic B-factors (Å2) | |||
| Protein | 49.2 | 38.3 | 23.0 |
| SAH | 52.2 | 30.3 | – |
| SAM | – | – | 18.4 |
| sulfate | 58.9 | 51.3 | |
| Water | 46.0 | 39.2 | 40.2 |
| Ethyleneglycol | 39.1 | ||
| Acetate | 31.9 | ||
| Rms deviations | |||
| Bond lengths (Å) | 0.009 | 0.019 | 0.012 |
| Bond angles (°) | 1.221 | 1.824 | 1.373 |
| Ramachandran statistics (%) | |||
| Most favored | 93.4 | 94.1 | 95 |
| Additionally allowed | 6.6 | 5.9 | 5 |
| Generously allowed | 0 | 0 | 0 |
| Disallowed | 0 | 0 | 0 |
a5% of the data were set aside for the Rfree calculation.
tRNA MTase assay
Two micrograms of purified enzyme was incubated at 70°C for 1 h with P. abyssi tRNAAsp as substrate (30). The reaction mixture (400 µl) consisted of 50 mM Tris–HCl pH 8, 10 mM MgCl2, 106 cpm radioactive transcript, 500 µM SAM. The radioactive tRNA was isolated on a 10% PAGE native gel and completely digested by either nuclease P1 (1 U) or RNase T2 (0.1 U), both from Sigma Chemical Co., in the presence of 10 µg total yeast tRNA as carrier. The nucleotides were separated by 2D thin layer chromatography (TLC) on 10 × 10 cm cellulose plates using the following solvents: first dimension developed with isobutyric acid/conc. NH4OH/water (66:1:33 v/v/v); second dimension developed with 0.1 M sodium phosphate pH 6.8/(NH4)2SO4/n-propanol (100:60:2 v/w/v). The radioactive compounds were detected by autoradiography. The reaction temperature of 70°C, which is sub-optimal to the growth temperature of P. abyssi, was chosen to maintain the stability of SAM in vitro.
Differential scanning calorimetry
Differential scanning calorimetry (DSC) was carried out with a VP-DSC platform (Microcal) up to 120°C, at scan rates of 60°C/h, to determine the unfolding transition temperatures of PabTrmI and the C196S/C233S mutant. The proteins were concentrated to 0.8 mg ml−1 in 50 mM Tris–HCl, pH 8.5, 500 mM KCl and degassed before measurements.
In vitro transcription of tRNA
In vitro transcription of PabtRNAAsp was performed using an oligonucleotide template containing ribose 2′-methoxy modifications at the last 2 nt at the 5′ terminus (31) and purified by electrophoresis on a 12% polyacrylamide, 8 M urea gel. RNA was located by UV-shadowing and excised from the gel, extracted with 0.3 M sodium acetate pH 5.2 and loaded onto a HiTrap DEAE Sepharose column (GE Healthcare Inc.) pre-equilibrated in the same buffer. After elution with 3 M sodium acetate pH 5.2, the transcript was ethanol precipitated and desalted on a PD-10 column (Sephadex-G25 medium; GE Healthcare Inc).
MS
PabtRNAAsp transcript (10 µM) was incubated for 5 min to 1 h at 70°C with 10 µM of purified wild-type PabTrmI or the H78Y mutant and 1 mM SAM in 50 µl 50 mM Tris–HCl pH 8, 10 mM MgCl2. The reaction was stopped with 200 µl of phenol:chloroform:isoamylic alcohol (25:24:1) pH 4.5 to precipitate the protein. tRNA in the aqueous phase was extracted by centrifugation at 10 000 × g for 5 min, ethanol precipitated and desalted on a MicroSpin G-25 column (GE Healthcare). One microgram of desalted tRNA was then digested at 37°C for 4 h in 10 µl of 50 mM DHB (2,5-dihydroxybenzoic acid; Sigma-Aldrich, Saint Quentin Fallavier, France) containing 2 µg of RNase A (Fermentas). One microliter of digest was mixed with 4 µl DHB (20 mg ml−1 in water) and 0.5 µl of the mixture was spotted on the MALDI plate and air-dried (‘dried droplet’ method). MALDI-TOF MS and MALDI-TOF/TOF MS/MS analyses were performed directly on the digestion products using a 4800 MALDI TOF/TOF Analyzer mass spectrometer (Applied Biosystems, Les Ulis, France). The instrument is equipped with an Nd:YAG laser (operating at 355 nm wavelength of <500 ps pulse and 200 Hz repetition rate). Acquisitions were performed in positive ion mode. For MS/MS experiments, precursor ions were accelerated at 8 kV and the MS/MS spectra were acquired using 2 kV collision energy with CID gas (argon) at a pressure of 3.5 × 10−6 Torr. MS data were processed using DataExplorer 4.4 (Applied Biosystems).
RESULTS
Overall structure of PabTrmI
We have obtained two different crystal forms (I and II) of archaeal PabTrmI in complex with SAH, belonging to P212121 and P31 space groups, respectively, and one crystal form in complex with SAM, belonging to space group I222 (Table 1). Crystals Forms I and II diffract to 2.6 and 2.05 Å resolution, respectively, and both contain one homo-tetramer in the asymmetric unit, the four monomers being related by a NCS 4-fold symmetry. Crystal Form III diffracts to 1.6 Å resolution, and contains one monomer in the asymmetric unit. The C-terminal (His)6-tag was disordered in all crystal forms and was not included in the final models. A search in the Dali data base (32) indicates structural homology with 1O54, a putative SAM-dependent O-MTase from Thermotoga maritima (Z = 29.3, 38% sequence identity, rmsd = 2.3 Å for 250 Cαs) and with 2B25, a human putative 1-methyladenosine MTase (Z = 26.0, 31% sequence identity, rmsd = 2.7 Å for 241 Cαs). This high sequence homology with PabTrmI suggests that 1O54 is inaccurately annotated in the PDB database and is actually the m1A58 MTase from T. maritima. 2B25 corresponds to the product of the TRM61 gene, the SAM-binding catalytic subunit that composes the hetero-tetramer of m1A58 MTase in eukaryotes (12,33). The homo-tetramer of PabTrmI is organized as a dimer of two tightly assembled dimers (A/B and C/D) (Figure 1A). Contrary to the other TrmIs with known structures, PabTrmI is stabilized by four intermolecular disulfide bonds (Figure 1B), as discussed below.
Figure 1.

Overall organization of PabTrmI (Stereoviews). (A) PabTrmI tetramer structure in complex with SAH. Subunits A and B, colored red and green, respectively, form one tight dimer and subunits C and D, colored yellow and blue, respectively, form another one. SAH is shown in blue sticks. (B) Detail of the four inter-monomer disulfide bonds that stabilize the PabTrmI tetramer. A 2 Fobs–Fcalc electron density map contoured at the level of one standard deviation is superimposed on the structure. (C) Superposition of one PabTrmI monomer from Crystal form I (in magenta), Crystal Form II (in blue) and Crystal Form III (in green).
Structure of one PabTrmI monomer
One PabTrmI monomer of 30 kDa is composed of two domains: a large C-terminal SAM-binding catalytic domain (residues 77–253) that displays the classical Rossmann-like fold, common to SAM-dependent MTases (34,35) and a smaller N-terminal domain (1–60) forming mainly a β-structure that may play a role in tRNA recognition (Figure 1C and Supplementary Figure S1). A search in the Dali database did not identify domains significantly homologous to the N-terminal domain beside predicted TrmI proteins. The catalytic domain displays a protruding region (β6, βG and βH strands), which is crucial for the tetramer assembly (see below). The N- and C-terminal domains are connected by a linker region (61–76), which includes helix α2. In Crystal Form II, the contacts between the crystallographic related molecules are scarce, which is not surprising in view of the high solvent content (78.7%). In particular, the N-terminal domains are not involved in crystal packing interactions. Comparing one monomer of Forms I and II, II and III, I and III with each other gives an rmsd on the whole molecule of <0.49, 0.49 and 0.59 Å, respectively, indicating a similar orientation of the N-terminal domain relative to the C-terminal domain. Therefore, this orientation found in three different space groups is likely functionally relevant.
Intermolecular contacts
Dimeric assembly
The N-terminal domain, helix α2, the protruding region and the C-terminal domain helix α3 contribute extensively to the inter subunit interactions in the tightly assembled dimer (Figure 2B). These interactions are generally conserved between the TrmI proteins with known structures. Helix α3 from each monomer interacts with each other. Helix α2 from one monomer makes van der Waals interactions with helix α3 of the second monomer of the dimer. Helix α2 and strand β7 from two different monomers are also maintained in close proximity by a pair of salt bridges involving Asp65 and Arg251, respectively. Moreover, strand β6 from each monomer interacts with each other through ten H-bonds and two salt-bridges involving bidentate interactions between Arg218 from one monomer and Asp226 from the other. This ionic interaction is conserved only in some TrmIs.
Figure 2.

(A) Detail of the inter-monomer contacts in PabTrmI. (B) Stereoview of the contacts forming the A/B dimer. His78 located at the beginning of α3 displays two different conformations. (C) Stereoview of the contacts between subunits A (red) and C (yellow), which contribute to the PabTrmI tetrameric organization.
Four inter monomer disulfide bonds stabilize the PabTrmI tetramer
Dimers A/B and C/D pack tightly to form the tetramer (Figure 2A). A major characteristic of the PabTrmI structure is the involvement of cysteine residues in the tetramer formation (Figure 1B). The intersubunit disulfide bridges between Cys196 and Cys233 stabilize the protein at extreme temperatures. First, it was previously shown that the double C196S/C233S mutant behaves as a dimer and that its activity is drastically affected by a preincubation at 85°C (21). Furthermore, the melting temperatures of the wild-type PabTrmI, as well as the double C196S/C233S mutant (21), measured here by differential scanning calorimetry, are 105.3 and 88.8°C, respectively (Figure 3). Therefore, disruption of the disulfide bonds decreases the melting temperature by 16.5°C. In addition to the intermonomer disulfide bonds, the tetrameric interactions involve a β-barrel structure that is formed by the protruding regions belonging to four different subunits. The dimer–dimer interface is stabilized by van der Waals interactions and ten hydrogen bonds but does not involve any ionic interaction, in contrast to the dimer–dimer interactions displayed in the bacterial enzymes (Figure 2C).
Figure 3.

Differential Scanning Calorimetry (DSC) Measurements of PabTrmI and the C196S/C233S mutant. The excess heat capacity curves of wild-type PabTrmI and the C196S/C233S mutant at a scan rate of 60°C/h show melting temperatures of 105.3 and 88.8°C, respectively.
SAM-binding site
PabTrmI was crystallized in Crystal Form I in the absence of SAM/SAH and in Crystal Form II in the presence of 5 mM SAM. Yet, the electron density maps clearly indicate the presence of a bound SAH molecule in each monomer of the two crystal forms (Supplementary Figure S2). Different conformations of SAH are observed (Figure 4A and B). Whereas the adenine base and ribose ring of SAH occupy the same position as that adopted by SAM in all conformations, the homocysteine moiety can adopt different orientations. The adenine ring is sandwiched between the side chains of Ile126 from motif II and Leu170 from motif IV (Supplementary Figure S1). Its N6 and N1 atoms are hydrogen bonded to the carboxylate group of Asp153 and main chain nitrogen of Ile154, respectively. The two SAH ribose hydroxyl groups are hydrogen bonded to both carboxylate oxygens of conserved residue Glu125 of motif II.
Figure 4.

SAH displays alternative conformations in the PabTrmI structures (Stereoviews). (A) Superposition of the SAM conformation in Crystal Form III (in cyan) with the SAH conformations observed in Crystal Form II (‘SAM like’ extended conformation in blue), in Crystal Form I molecules A and B (folded conformation 1 in magenta) and in Crystal Form I molecules C and D of (folded conformation 2 in pink). (B) Overall location of SAH in the PabTrmI monomer. The extended conformation is shown in blue and the folded conformations in magenta and pink. In the latter conformations, the SAH homocysteine moiety is oriented towards the exterior of the protein. (C) Cofactor-binding site in Crystal Form III. A Fobs–Fcalc electron density map omitting SAM calculated at the level of 1.5 SD is superimposed on the structure. The dihedral angle (C4′, C5′, S, Cγ) is –74.5°.
The four SAH molecules in Crystal Form II display an extended conformation (Supplementary Figure S2). The homocysteine moiety is in van der Waals interaction with the peptide backbone of the GxGxG sequence of motif I. Its amino group is hydrogen bonded to Asp169 and its carboxylate group to the main chain amino groups of Ala106, Leu107 and Val77. This conformation of SAH is similar to that adopted by SAM in Crystal Form III (Figure 4A). In crystal form I, SAH displays two different folded conformations, in which the homocysteine moiety is oriented towards the exterior of the protein (Figure 4A, B and Supplementary Figure S2). Only poor electron density is observed for this part of SAH, which reflects its weak van der Waals contacts with the protein. SAM in Crystal Form III adopts the same conformation as SAM in MtTrmI and AaTrmI (Figure 4C).
His78 near the active site displays two different conformations
The PabTrmI and TtTrmI active site structures were compared to gain insight into the different specificities of the two enzymes (Figure 5A). A similar negatively charged cleft suitable to accommodate the flipped out target adenine, which will become positively charged upon methylation, is found in both enzymes. The main difference between the two active sites is the presence of His78 in PabTrmI, instead of Tyr78 in TtTrmI. His78 is located at the beginning of helix α3 in conserved motif X and forms the exit of the catalytic pocket. Interestingly, an histidine is present at position 78 only in the Thermococcus and Pyrococcus genera (Supplementary Figure S1). It is replaced by Leu, Tyr or Phe in the other organisms. His78 in PabTrmI displays two different conformations, indicating its flexibility (Figure 5B and C). In one conformation, His78 (chain A) is hydrogen bonded to the Tyr88 hydroxyl group of the interacting chain B, which reinforces the dimeric interactions (Figure 2B). In T. thermophilus, a potential role in the base flipping mechanism was proposed for Tyr78, based on the 20-fold decreased catalytic turnover of the Y78A mutant and the modeling of an adenine analog in the enzyme active site (19).
Figure 5.

(A) Superposition of the SAM-binding pockets of TtTrmI (in pink) and PabTrmI (in green) on the electrostatic surface of PabTrmI determined using PYMOL/APBS colored by the electrostatic potential (Stereoview). The values of surface potential are expressed as a spectrum ranging from –63 kT/e (deep red) to +63 kT/e (deep blue). The labels correspond to the PabTrmI amino acids. (B) and (C) His78 displays two different conformations in Crystal Form I. 2 Fobs–Fcalc and Fobs–Fcalc electron density maps contoured at the level of 1 and 2 SDs, respectively, are shown in blue and green, respectively.
In contrast to bacterial TrmIs, which specifically methylate adenine at position 58, PabTrmI targets both positions 57 and 58 in PabtRNAAsp but not position 59
Previous experiments showed that both PabTrmI and TtTrmI modify bulk tRNA lacking m1A58 isolated from the T. thermophilus RD1 strain (16,21). Yet, while bulk tRNA from wild-type T. thermophilus (containing the m1A58 modification) was not methylated by TtTrmI, it could incorporate 20–30% methyl groups when incubated with PabTrmI. This result suggested that, in contrast to TtTrmI, PabTrmI is able to modify at least another position besides A58 in tRNA (21). The position of the additional methylation was determined to be 57 by a nearest neighbor analysis, using Saccharomyces cerevisiae tRNAAsp (containing the A57A58U59 sequence) or mutant T. thermophilus tRNAAsp (containing the A57A58G59 sequence), as substrates for PabTrmI. Yet, in P. abyssi, 35 tDNAs contain the A57A58A59 sequence and nine tDNAs contain the G57A58A59 sequence (http://www.ncbi.nlm.nih.gov/sites/entrez?db=genome&cmd=Retrieve&dopt=Structural%20RNA%20Table&list_uids=143). Since P. abyssi tRNAs had not been previously tested as substrates for PabTrmI and since none of the heterologous tRNA tested so far contained the G57A58A59 or A57A58A59 sequences, there was no clear indication that PabTrmI is not able to modify also A59. To address this point and confirm the difference of specificities between the PabTrmI and TtTrmI enzymes, their activities towards PabtRNAAsp (containing the A57A58A59U60 sequence) were tested (Supplementary Figure S3A and Table 2). Hydrolysis of [α-32P]ATP-labeled PabtRNAAsp with nuclease P1 (Supplementary Figure S3B, top), which generates 5′-phosphate nucleosides with [32P] present in all 5′-phosphate adenosine derivatives, shows that 1.8 mol and 0.85 mol of m1A per mol of tRNA is formed with PabTrmI and TtTrmI, respectively. Hydrolysis of [α-32P]UTP-labeled PabtRNAAsp with nuclease T2 (Supplementary Figure S3B, bottom) generates 3′-phosphate nucleosides of which only those that are 5′-adjacent to U in the tRNA sequence contain [32P] (nearest neighbors analysis) and hence allows to test methylation at position 59. After separation of the nucleotides by TLC, the absence of radiolabeled m1A indicates that PabTrmI and TtTrmI are not able to methylate adenine at position 59.
Table 2.
Characterization of the tRNA MTase activity of PabTrmI, H78YPabTrmI and TtTrmI using different P. abyssi tRNAsa
| [α-32P] labeled nucleotide used for production of tRNA transcript | Nuclease | Position of methylation tested | mole of m1A per tRNA |
|||
|---|---|---|---|---|---|---|
| TtTrmI | PabTrmI | H78YPabTrmI | ||||
| PabtRNAAsp | ATP | P1 | all A | 0.85 ± 0.05 | 1.78 ± 0.19 | 0.9 ± 0.05 |
| UTP | T2 | A59 | 0 | 0 | 0 | |
| A59G PabtRNAAsp | ATP | P1 | all A | 0.8 ± 0.05 | 1.05 ± 0.05 | 0.85 ± 0.05 |
| ATP | T2 | A57 | <1% | 0.85 ± 0.15 | 0.70 ± 0.10 | |
| GTP | T2 | A58 | 0.7 ± 0.15 | <5% | <1% | |
aRadiolabeled tRNA transcripts were incubated in the presence of enzyme, then digested by nucleases P1 or T2 to test the methylation of adenine at positions 57, 58 and 59. The resulting nucleotides were analyzed by 2D–TLC on cellulose plates and autoradiography (Supplementary Figure S3).
Adenine at position 59 in PabtRNAAsp is important for the multi-site specificity of PabTrmI
To investigate whether His78 could participate in the multisite-specificity of the archaeal enzyme, we tested the activity and substrate specificity of the H78Y mutant of PabTrmI and compared them to those of the wild-type enzyme. Whereas PabTrmI is able to produce two moles of m1A in PabtRNAAsp, the H78Y mutant catalyzes the formation of only one mole, indicating either that the mutant is less efficient than the wild-type enzyme or that it is specific for only one adenine position in the T-loop (Table 2).
To distinguish between positions 57 and 58, the A59G mutant of PabtRNAAsp was radiolabeled with [α-32P]ATP or [α-32P]GTP, tested as substrate of the mutant and wild-type enzymes and analyzed after hydrolysis by the P1 and T2 nucleases (Table 2). Interestingly, PabTrmI is able to catalyze the formation of only 1 mol of m1A per PabtRNAAsp (A59G) and a nearest neighbor analysis indicates the enzyme is specific mainly for position 57 in this substrate. In contrast, TtTrmI methylates only A58 in PabtRNAAsp (A59G). This result highlights the importance of the presence of an adenine at position 59 in tRNA for the bisite-specificity of the archaeal enzyme. Therefore, PabTrmI appears to modify the first adenine of an AA nucleotide sequence in the T-loop of PabtRNAAsp.
PabTrmI modifies first A57, then A58 and the H78Y mutant has lower efficiency for PabtRNAAsp than PabTrmI
MS was then used to analyze the position of the methyl group in the wild-type PabtRNAAsp product formed by PabTrmI and the H78Y mutant. The methylated RNA was digested by RNase A, which cleaves after C and U, generating 3′-phosphate nucleosides, and its fragments were analyzed by MS (Figure 6). Fragments (m/z = 1326.20, 1340.22) coincide with the expected masses of mono and dimethylated fragments derived from AAAUp corresponding to the sequence from A57 to U60 in tRNAAsp (Supplementary Figure S3A). The modified positions were then identified by MS/MS analysis (Figure 7).
Figure 6.

MALDI MS analysis of methylated P. abyssi tRNAAsp formed by wild-type PabTrmI and the H78Y mutant. (A) MALDI mass spectrum of tRNA produced by each enzyme after 5 min incubation at 70°C in the presence of SAM followed by digestion by RNase A. Inserts correspond to the spectral region around the AAAUp fragment, which contains the two target adenines, to show the peaks for the unmethylated (m/z 1312.19) and the corresponding monomethylated (m/z 1326.20) and dimethylated (m/z 1340.22) ions. tRNA from the control sample that was not incubated with enzyme contains only the m/z 1312.19 ion. (B) RNase A fragments derived from P. abyssi tRNAAsp showing the theoretical and empirical masses of singly protonated ions. Mononucleotides were not observed as they are obscured by the matrix.
Figure 7.

Identification of the methylation sites in P. abyssi tRNAAsp by wild-type PabTrmI and the H78Y mutant. Fragment assignments follow the scheme of McLuckey (60). For calculation of fragment masses, the NUKE software was utilized (Leisner, A., Institute for Medical Physics and Biophysics, University of Münster, Germany). (A) The dimethylated RNase A fragment at m/z 1340.22 was selected and fragmented by tandem MS. The spectra obtained with both enzymes are identical. Peaks corresponding to the c- and d-ions, and y- and z-ions are generated by loss of 3′- and 5′-nucleotides, respectively. These ions (particularly the c2-ion corresponding to m1Am1Ap) show that methyl groups are added on both A57 and A58 by wild-type PabTrmI and the H78Y mutant. (B) The singly methylated RNase A fragment at m/z 1326.20 was selected and fragmented by tandem MS. The spectra obtained with both enzymes were again identical. The peak corresponding to the c1 ion highlights that A57 is methylated. (C) Expected and experimental fragments for the dimethylated RNase A fragment at m/z 1340.22. (D) Expected and experimental fragments for the singly methylated RNase A fragment at m/z 1326.20.
The sequence of the dimethylated fragment was determined to be m1Am1AAUp, indicating that both adenines at positions 57 and 58 in PabtRNAAsp are methylated by PabTrmI and the H78Y mutant (Figure 7A and C). On the one hand, the presence of the z2 fragment corresponding to the AUp sequence (m/z 636.11), together with the absence of the z2+m fragment corresponding to the m1AUp sequence (m/z 650.11) confirm that A59 is not methylated. On the other hand, the presence of the c2 fragment corresponding to the m1Am1A sequence (m/z 687.14) confirms that both A57 and A58 are methylated.
The sequence of the monomethylated fragment was determined to be m1AAAUp (Figure 7B and D). First, the presence of the non-methylated z2 fragment and the absence of the methylated z2+m fragment indicate again that A59 is not methylated. Secondly, the presence of the y3 fragment corresponding to the AAUp sequence (m/z 983.17), together with the absence of the y3+m fragment corresponding to the m1AAUp sequence (m/z 997.17), show that A58 is not methylated. Finally, since A58 and A59 are not methylated and the presence of the c1 fragment corresponding to m1Ap (m/z 344.09) shows that one adenine is methylated, A57 is methylated in the monomethylated fragment.
The presence of monomethylated tRNA at position 57 and the absence of monomethylated tRNA at position 58 suggest there is an order of the methylation reactions with the first modification occuring at A57, and the second methyl transfer reaction generating m1A58. Preliminary kinetic data of wild-type PabTrmI and the H78Y mutant show that the presence of His78 instead of Tyr78 increases the efficiency of the dimethylation reaction (Supplementary Figure S4).
DISCUSSION
Contrary to bacterial TrmI enzymes that have been shown to specifically modify adenine at position 58 in the T-loop of tRNAs, PabTrmI methylates adenine at both positions 57 and 58 in vitro (18,21). Purification of tRNA from thermophiles is known to be generally very difficult and none tRNA from P. abyssi has been sequenced to date so that the position of modifications present in tRNAs from this organism remains unknown. Yet, 1-methylinosine (m1I) and m1A were identified in total digests of P. furiosus tRNA by combined HPLC-MS (36). Moreover, enzymatic activities corresponding to the formation of both m1I57 and m1A58 were detected in the cell-free extract of P. furiosus supplemented with SAM using in vitro transcribed tRNAs (37). m1I is also present at position 57 in Haloferax volcanii tRNAs (38). The formation of m1I57 is a two-step enzymatic process that differs from that leading to the formation of m1I37 in eukaryotic tRNAAla. The methylation of A57 is followed by the deamination of the 6-amino group of the adenine moiety of m1A57 by a specific deaminase (38). Interestingly, the TrmI homolog found in the H. volcanii genome, shares only low sequence identity with the m1A57,58 MTase from P. abyssi compared to the other homologs (Supplementary Figure S1). Nevertheless, it is probable that m1A57 is an intermediate for the synthesis of m1I57 both in H. volcanii and P. abyssi. The biochemical and structural study of PabTrmI reported here accounts for its hyperthermostability and answers several questions concerning the multisite specificity of the enzyme.
The hyperthermostability of PabTrmI comes from intermolecular disufide bonds that stabilize the tetrameric architecture of PabTrmI
Pyrococcus abyssi has to develop strategies to adapt the stability of its proteins to extreme temperature and pressure conditions (20). Concerning PabTrmI, we show in this study that one of these means is the use of four intermolecular disulfide bonds between Cys233 and Cys196 from different monomers to stabilize the tetrameric architecture. Indeed, the mutant protein without disulfide bonds has a melting temperature lowered by 16.5°C, indicating that the disulfide bonds stabilize the protein against heat denaturation. A Psi-Blast search for proteins most homologous to PabTrmI in sequenced genomes indicates that these Cysteines are conserved only in the TrmI proteins from the Pyrococcus and Thermococcus genera, i.e. belonging to the Thermococcales order (Supplementary Figure S1). The main characteristics that distinguish these genera found in deep-sea hydrothermal environments are their optimal growth temperature (between 75 and 88°C for Thermococcus species and between 96 and 100°C for Pyrococcus members). PabTrmI shares at least 62% sequence identity with homologous proteins from the Thermococcus genus. Generally, the presumed role of disulfide bonds is to help stabilize proteins against thermal denaturation, as exemplified by the decreased melting temperature following disulfide disruption in several thermophilic proteins (39–42). However, all sequenced Thermococcus genera, which contain TrmI proteins with Cysteines likely involved in disulfide bond formation, are barophiles. Furthermore, pressure enhances thermal stability of proteins from Pyrococcus furiosus (43), the only Pyrococcus species that does not operate at high pressure. It is therefore possible that the disufide bonds stabilize the TrmI protein against pressure denaturation.
Although it was considered that structural disulfide bonds are generally extracellular because the reducing cytosolic environment is unfavorable to disulfide bonding, a critical role for disulfide bonds in the folded structure stabilization of intracellular proteins from several thermophilic and hyperthermophilic archaea and prokaryotes is now progressively being recognized (44, 45). The presence of intracellular intramolecular disulfide bonds in thermophilic organisms has been shown to correlate with the exclusive occurrence of a protein disulfide oxydoreductase, which is thought to form and maintain disulfide bonds (45). The enzyme from P. furiosus has been characterized (46) and the encoding gene is present in P. abyssi. The predicted intramolecular disulfide bond abundance in many thermophilic organisms is high (45). Whereas the property of intramolecular disulfide bonds richness is distributed among thermophiles from both the archaeal and bacterial domains of life, no information concerning intermolecular disulfide bonds exists because their abundance cannot be easily predicted. Intermolecular disulfide bonds have been shown to stabilize the quaternary organization of several intracellular proteins from Sulfolobus solfataricus (47,48), A. aeolicus (49), P. furiosus (50) or T. maritima (51). The protein disulfide oxydoreductase is present in these thermophilic organisms (45) but it remains to determine whether it could be involved in the formation/maintenance of the intermolecular disulfide bonds. However, the absence of intermolecular bonds in TrmI from A. aeolicus (Supplementary Figure S1), which grows at 96°C, suggests that thermostability is not the only element that could explain disulfide bonding in PabTrmI but that other factors, such as stabilization against pressure denaturation, may also be involved.
Different conformations of SAH
In the two crystal forms of PabTrmI in complex with SAH, SAH adopts three alternative conformations, two of which displaying an unusual folded structure. SAM usually adopts an extended conformation in the known SAM-dependent MTases structures although a folded catalytically irrelevant conformation of SAM was also observed in the human Dim1 rRNA MTase (PDB code 1ZQ9) (52). The existence of alternative conformations of SAH has previously been reported in DNA adenine MTases EcoDam (53), M. RsrI (54) and M. TaqI (55). The extended conformation, in which the methyl group of SAM points toward the atom to be methylated, is compatible with catalysis. In the folded conformation of SAH observed in DNA MTases, the homocysteine moiety occupies the target adenine-binding pocket, thereby preventing catalysis. In the folded conformations of SAH observed in PabTrmI, the homocysteine moiety points towards the solvent, in the direction of the cavity, which is presumed to accommodate the flipped target adenine base. We suggest that in this conformation, the SAH product is on the way to leave the enzyme.
Multi-site specificity of RNA modifying enzymes
A few other tRNA modifying enzymes exhibit a regional specificity smilar to that of PabTrmI, modifying consecutive positions in RNA, like Aquifex aeolicus tRNA (N2,N2-guanine)-dimethyltransferase Trm1, which catalyzes the methylation not only of guanine 26 but also guanine 27 in tRNA (56) or E. coli pseudouridine synthase TruA, which specifically modifies uridines at positions 38, 39 and/or 40 in the anticodon stem loop (ASL) of tRNAs (57). A long distance recognition mechanism, whereby A. aeolicus Trm1 recognizes the target sites distantly spaced from the recognition site in the tRNA T-arm region, has been proposed to explain the multisite recognition by the enzyme but structural data are needed to confirm this hypothesis. In the case of pseudouridine synthase TruA, the molecular basis for the regional specificity was studied by determining the crystal structures of the enzyme in complex with tRNALeu1 and tRNALeu3 containing U at position 39 and 38, respectively (57). These structures proved to mimic three states along the substrate recognition pathway. It was proposed from the crystallographic data, coupled with molecular dynamic simulations that the intrinsic flexibility of the ASL in the initial docking complex, in which the ASL is away from the protein, was at the origin of the TruA site promiscuity. The flexibility in the ASL is retained thanks to extensive interactions with the conserved parts of tRNAs (elbow region and D-stem backbone). TruA stabilizes the flipped base through a stacking interaction with Arg58. But, in contrast to enzymes that utilize arginine as a stabilizing agent, the ASL is locally disordered in the base flipped out conformation perhaps to retain the flexibility required for sampling three different sites (57). Therefore the TruA regional selectivity appears to originate in a subtle balance between the stabilization of the flipped base and flexibility in the tRNA substrate.
rRNA MTase KsgA catalyzes a reaction similar to that of PabTrmI, the dimethylation at the N6 position of two adjacent adenine residues in small ribosomal subunit RNA. The two target adenines must have separate access to the active site. The reaction is more complex than that catalyzed by PabTrmI since, in addition to the SAH product that has to be released before the next SAM molecule can bind, four SAM molecules must sequentially bind to the active site and donate a methyl group. The details of the enzymatic mechanism of KsgA have not been elucidated yet but it has been shown that there is no obligate order of methylation of A1518 and A1519 (58). There are analogous unanswered questions about how PabTrmI is able to form both m1A57 and m1A58. Is there a sequential order of modification or is it random? Does PabTrmI bind tRNA once and make both modifications or are there two separate binding events? The knowledge of the PabTrmI 3D structure has led us to design mutations in both the protein and the tRNA substrate to attempt to solve these questions.
A58 lies in the T-loop, which is supposed to be less flexible than the anticodon loop because of the tertiary interactions between the T- and D-loops that stabilize the tRNA structure. To know whether the intrinsic flexibility of the T-loop in PabtRNAAsp is sufficient for allowing both sites 57 and 58 to be dynamically accessible for modification, we tested PabtRNAAsp as a substrate for the site-specific TtTrmI. The bacterial enzyme remained specific only for position 58 in PabtRNAAsp. Therefore, although the dynamics of the tRNA substrate may participate in the multi-site specificity of the archaeal enzyme, other elements lie in the enzyme itself. The size of the cleft suitable to accommodate the flipped out target adenine indicates that modifications of A57 and A58 are likely to occur one after the other in PabTrmI. A comparison of the active site pockets of TtTrmI and PabTrmI revealed that the main structural difference between the two enzymes resides in the presence of His78 in PabTrmI (instead of Tyr78 in TtTrmI). This histidine displays two alternative conformations, indicating its flexibility. When His78 is replaced by Tyrosine, the methylation reaction by PabTrmI is less efficient but both position A57 and A58 are still methylated. Although not responsible by itself for the multi-site specificity of PabTrmI, His/Tyr 78 is a good candidate for participating in the stabilization of the flipped target adenine base in TrmI. First, this amino acid is located near the pocket presumed to bind the flipped target adenine nucleotide. Second, in the model of TtTrmI in complex with an adenine analog, Tyr 78 interacts with the ribose ring of the nucleoside (19). His/Tyr78 in TrmI could fulfill a role similar to that of His43 in pseudouridine synthase TruB, which is involved in the stabilization of the flipped out conformation of the target U55 by inserting into the vacated space of the base-flipped-out stem loop (59).
When adenine at position 59 is replaced by guanine in PabtRNAAsp, PabTrmI methylates only adenine at position 57. Moreover, MS analysis of methylated wild-type PabtRNAAsp formed by PabTrmI indicated the presence of monomethylated A57, in addition to the dimethylated product, but not that of monomethylated A58. Our results therefore indicate that the multisite specificity of PabTrmI lies in the presence of three consecutive adenines in P. abyssi tRNAs, with the enzyme modifying sequentially A57 and then A58. Thus, the methylation mechanism of PabTrmI differs from that of KsgA, which catalyzes the dimethylation of two adjacent adenine residues in rRNA with neither of the A residues being required for the methylation of the other (58).
CONCLUSION
The crystal structure of PabTrmI from the archaeal P. abyssi coupled with calorimetric stability measurements has given insight into the origin of its hyperthermophilic stability, showing that the four inter-monomer disulfide bonds protect the enzyme against temperature and possibly pressure denaturation. The structures of PabTrmI in complex with SAH and SAM were not sufficient by themselves to elucidate the origins of the multi-site specificity of the enzyme at the molecular level, which remains one of the most interesting questions. Yet, our biochemical study has shown that at least part of the enzyme regiospecificity is related to the presence of three consecutive adenines in P. abyssi tRNAs, with the enzyme modifying the first adenine of an AA sequence. Further elucidation of the substrate specificity of PabTrmI will require the structure of the complex with bound tRNA substrate.
ACCESSION NUMBERS
3LHD, 3LGA and 3MB5.
FUNDING
CNRS; Association pour la Recherche sur le Cancer (to B.G.P.). Funding for open access charge: Association pour la Recherche sur le Cancer.
Conflict of interest statement. None declared.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
The authors thank Johan Wouters for crystallizing and collecting diffraction data for Crystal Form I of PabTrmI, Sylvie Auxilien, Djemel Hamdane and Jaunius Urbonavicius for fruitful discussions, Louis Droogmans and Henri Grosjean for initiating the project and Magali Aumont-Nicaise for performing the DSC experiments. The authors acknowledge SOLEIL and the ESRF for provision of synchrotron radiation facilities and would like to thank Beatriz Guimaraez for assistance in using beamline PROXIMA 1 and for help in data treatment.
REFERENCES
- 1.Agris PF. The importance of being modified: roles of modified nucleosides and Mg2+ in RNA structure and function. Prog. Nucleic Acid Res. Mol. Biol. 1996;53:79–129. doi: 10.1016/s0079-6603(08)60143-9. [DOI] [PubMed] [Google Scholar]
- 2.Grosjean H, Benne R. Modification and RNA Editing. Washington DC: ASM press; 1998. [Google Scholar]
- 3.Agris PF. Decoding the genome: a modified view. Nucleic Acids Res. 2004;32:223–238. doi: 10.1093/nar/gkh185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grosjean H. Fine-tuning of RNA functions by modification and editing. Berlin Heidelberg, NY: Springer-Verlag; 2005. [Google Scholar]
- 5.Goto-Ito S, Ito T, Kuratani M, Bessho Y, Yokoyama S. Tertiary structure checkpoint at anticodon loop modification in tRNA functional maturation. Nat. Struct. Mol. Biol. 2009;16:1109–1115. doi: 10.1038/nsmb.1653. [DOI] [PubMed] [Google Scholar]
- 6.Björk GR. tRNA: Structure Biosynthesis and function. In: Söll D, RajBhandary UL, editors. Am. Soc. Microbiol. Washington D.C.: 1995. pp. 165–206. [Google Scholar]
- 7.Anderson J, Phan L, Cuesta R, Carlson BA, Pak M, Asano K, Bjork GR, Tamame M, Hinnebusch AG. The essential Gcd10p-Gcd14p nuclear complex is required for 1-methyladenosine modification and maturation of initiator methionyl-tRNA. Genes Dev. 1998;12:3650–3662. doi: 10.1101/gad.12.23.3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nishikura K, De Robertis EM. RNA processing in microinjected Xenopus oocytes. Sequential addition of base modifications in the spliced transfer RNA. J. Mol. Biol. 1981;145:405–420. doi: 10.1016/0022-2836(81)90212-6. [DOI] [PubMed] [Google Scholar]
- 9.Grosjean H, Edqvist J, Straby KB, Giegé R. Enzymatic formation of modified nucleosides in tRNA: dependence on tRNA architecture. J. Mol. Biol. 1996;255:67–85. doi: 10.1006/jmbi.1996.0007. [DOI] [PubMed] [Google Scholar]
- 10.Bujnicki JM. In silico analysis of the tRNA:m1A58 methyltransferase family: homology-based fold prediction and identification of new members from Eubacteria and Archaea. FEBS Lett. 2001;507:123–127. doi: 10.1016/s0014-5793(01)02962-3. [DOI] [PubMed] [Google Scholar]
- 11.Agris PF, Sierzputowska-Gracz H, Smith C. Transfer RNA contains sites of localized positive charge: carbon NMR studies of [13C]methyl-enriched Escherichia coli and yeast tRNAPhe. Biochemistry. 1986;25:5126–5131. doi: 10.1021/bi00366a022. [DOI] [PubMed] [Google Scholar]
- 12.Anderson J, Phan L, Hinnebusch AG. The Gcd10p/Gcd14p complex is the essential two-subunit tRNA(1-methyladenosine) methyltransferase of Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA. 2000;97:5173–5178. doi: 10.1073/pnas.090102597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kadaba S, Krueger A, Trice T, Krecic AM, Hinnebusch AG, Anderson J. Nuclear surveillance and degradation of hypomodified initiator tRNAMet in S. cerevisiae. Genes Dev. 2004;18:1227–1240. doi: 10.1101/gad.1183804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vanacova S, Wolf J, Martin G, Blank D, Dettwiler S, Friedlein A, Langen H, Keith G, Keller W. A new yeast poly(A) polymerase complex involved in RNA quality control. PLoS Biol. 2005;3:e189. doi: 10.1371/journal.pbio.0030189. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
15.Auxilien S, Keith G, Le Grice SF, Darlix JL. Role of post-transcriptional modifications of primer
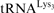 in the fidelity and efficacy of plus strand DNA transfer during HIV-1 reverse transcription. J. Biol. Chem. 1999;274:4412–4420. doi: 10.1074/jbc.274.7.4412. [DOI] [PubMed] [Google Scholar]
in the fidelity and efficacy of plus strand DNA transfer during HIV-1 reverse transcription. J. Biol. Chem. 1999;274:4412–4420. doi: 10.1074/jbc.274.7.4412. [DOI] [PubMed] [Google Scholar] - 16.Droogmans L, Roovers M, Bujnicki JM, Tricot C, Hartsch T, Stalon V, Grosjean H. Cloning and characterization of tRNA (m1A58) methyltransferase (TrmI) from Thermus thermophilus HB27, a protein required for cell growth at extreme temperatures. Nucleic Acids Res. 2003;31:2148–2156. doi: 10.1093/nar/gkg314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gupta A, Kumar PH, Dineshkumar TK, Varshney U, Subramanya HS. Crystal structure of Rv2118c: an AdoMet-dependent methyltransferase from Mycobacterium tuberculosis H37Rv. J. Mol. Biol. 2001;312:381–391. doi: 10.1006/jmbi.2001.4935. [DOI] [PubMed] [Google Scholar]
- 18.Varshney U, Ramesh V, Madabushi A, Gaur R, Subramanya HS, RajBhandary UL. Mycobacterium tuberculosis Rv2118c codes for a single-component homotetrameric m1A58 tRNA methyltransferase. Nucleic Acids Res. 2004;32:1018–1027. doi: 10.1093/nar/gkh207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barraud P, Golinelli-Pimpaneau B, Atmanème C, Sanglier S, van Dorsselaer A, Droogmans L, Dardel F, Tisné C. Crystal structure of Thermus thermophilus tRNA m1A58 methyltransferase and biophysical characterization of its interaction with tRNA. J. Mol. Biol. 2008;377:535–550. doi: 10.1016/j.jmb.2008.01.041. [DOI] [PubMed] [Google Scholar]
- 20.Erauso G, Reysenbach A.-L, Godfroy A, Meunier JR, Crump B, Partensky F, Baross JA, Marteinsson V, Barbier G, Pace NR, et al. Pyrococcus abyssi sp. nov., a new hyperthermophilic archaeon isolated from a deep-sea hydrothermal vent. Arch. Microbiol. 1993;160:338–349. [Google Scholar]
- 21.Roovers M, Wouters J, Bujnicki JM, Tricot C, Stalon V, Grosjean H, Droogmans L. A primordial RNA modification enzyme: the case of tRNA (m1A) methyltransferase. Nucleic Acids Res. 2004;32:465–476. doi: 10.1093/nar/gkh191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leslie AGW. Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4 + ESF-EAMCB Newsletter on Protein Crystallography. 1992;26 [Google Scholar]
- 23.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Cryst. 1993;26:795–800. [Google Scholar]
- 24.Evans P. Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 25.Navaza J. AMoRe: an automated package for molecular replacement. Acta Cryst. 1994;A50:157–163. [Google Scholar]
- 26.Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta Crystallogr. D Biol. Crystallogr. 2004;60:432–438. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- 27.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 28.Adams PD, Pann NS, Read RJ, Brünger AT. Cross-validated maximum likelihood enhances crystallographic simulated annealing refinement. Proc. Natl Acad. Sci. USA. 1997;94:5018–5023. doi: 10.1073/pnas.94.10.5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 30.Auxilien S, El Khadali F, Rasmussen A, Douthwaite S, Grosjean H. Archease from Pyrococcus abyssi improves substrate specificity and solubility of a tRNA m5C-methyltransferase. J. Biol. Chem. 2007;282:18711–18721. doi: 10.1074/jbc.M607459200. [DOI] [PubMed] [Google Scholar]
- 31.Kao C, Zheng M, Rudisser S. A simple and efficient method to reduce nontemplated nucleotide addition at the 3 terminus of RNAs transcribed by T7 RNA polymerase. RNA. 1999;5:1268–1272. doi: 10.1017/s1355838299991033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holm L, Sander C. Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- 33.Ozanick S, Krecic A, Andersland J, Anderson JT. The bipartite structure of the tRNA m1A58 methyltransferase from S. cerevisiae is conserved in humans. RNA. 2005;11:1281–1290. doi: 10.1261/rna.5040605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martin JL, McMillan FM. SAM (dependent) I AM: the S-adenosylmethionine-dependent methyltransferase fold. Curr. Opin. Struct. Biol. 2002;12:783–793. doi: 10.1016/s0959-440x(02)00391-3. [DOI] [PubMed] [Google Scholar]
- 35.Schluckebier G, O'Gara M, Saenger W, Cheng X. Universal catalytic domain structure of AdoMet-dependent methyltransferases. J. Mol. Biol. 1995;247:16–20. doi: 10.1006/jmbi.1994.0117. [DOI] [PubMed] [Google Scholar]
- 36.Kowalak JA, Dalluge JJ, McCloskey JA, Stetter KO. The role of posttranscriptional modification in stabilization of transfer RNA from hyperthermophiles. Biochemistry. 1994;33:7869–7876. doi: 10.1021/bi00191a014. [DOI] [PubMed] [Google Scholar]
- 37.Constantinesco F, Motorin Y, Grosjean H. Transfer RNA modification enzymes from Pyrococcus furiosus: detection of the enzymatic activities in vitro. Nucleic Acids Res. 1999;27:1308–1315. doi: 10.1093/nar/27.5.1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grosjean H, Constantinesco F, Foiret D, Benachenhou N. A novel enzymatic pathway leading to 1-methylinosine modification in Haloferax volcanii tRNA. Nucleic Acids Res. 1995;23:4312–4319. doi: 10.1093/nar/23.21.4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.DeDecker BS, O'Brien R, Fleming PJ, Geiger JH, Jackson SP, Sigler PB. The crystal structure of a hyperthermophilic archaeal TATA-box binding protein. J. Mol. Biol. 1996;264:1072–1084. doi: 10.1006/jmbi.1996.0697. [DOI] [PubMed] [Google Scholar]
- 40.Toth EA, Worby C, Dixon JE, Goedken ER, Marqusee S, Yeates TO. The crystal structure of adenylosuccinate lyase from Pyrobaculum aerophilum reveals an intracellular protein with three disulfide bonds. J. Mol. Biol. 2000;301:433–450. doi: 10.1006/jmbi.2000.3970. [DOI] [PubMed] [Google Scholar]
- 41.Meyer J, Clay MD, Johnson MK, Stubna A, Munck E, Higgins C, Wittung-Stafshede P. A hyperthermophilic plant-type [2Fe-2S] ferredoxin from Aquifex aeolicus is stabilized by a disulfide bond. Biochemistry. 2002;41:3096–3108. doi: 10.1021/bi015981m. [DOI] [PubMed] [Google Scholar]
- 42.Karlström M, Stokke R, Steen IH, Birkeland NK, Ladenstein R. Isocitrate dehydrogenase from the hyperthermophile Aeropyrum pernix: X-ray structure analysis of a ternary enzyme-substrate complex and thermal stability. J. Mol. Biol. 2005;345:559–577. doi: 10.1016/j.jmb.2004.10.025. [DOI] [PubMed] [Google Scholar]
- 43.Summit M, Scott B, Nielson K, Mathur E, Baross J. Pressure enhances thermal stability of DNA polymerase from three thermophilic organisms. Extremophiles. 1998;2:339–345. doi: 10.1007/s007920050077. [DOI] [PubMed] [Google Scholar]
- 44.Mallick P, Boutz DR, Eisenberg D, Yeates TO. Genomic evidence that the intracellular proteins of archaeal microbes contain disulfide bonds. Proc. Natl Acad. Sci. USA. 2002;99:9679–9684. doi: 10.1073/pnas.142310499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beeby M, O'Connor BD, Ryttersgaard C, Boutz DR, Perry LJ, Yeates TO. The genomics of disulfide bonding and protein stabilization in thermophiles. PLoS Biol. 2005;3:e309. doi: 10.1371/journal.pbio.0030309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pedone E, Ren B, Ladenstein R, Rossi M, Bartolucci S. Functional properties of the protein disulfide oxidoreductase from the archaeon Pyrococcus furiosus: a member of a novel protein family related to protein disulfide-isomerase. Eur. J. Biochem. 2004;271:3437–3448. doi: 10.1111/j.0014-2956.2004.04282.x. [DOI] [PubMed] [Google Scholar]
- 47.Cacciapuoti G, Porcelli M, Bertoldo C, De Rosa M, Zappia V. Purification and characterization of extremely thermophilic and thermostable 5′-methylthioadenosine phosphorylase from the archaeon Sulfolobus solfataricus. Purine nucleoside phosphorylase activity and evidence for intersubunit disulfide bonds. J. Biol. Chem. 1994;269:24762–24769. [PubMed] [Google Scholar]
- 48.Feese MD, Kato Y, Tamada T, Kato M, Komeda T, Miura Y, Hirose M, Hondo K, Kobayashi K, Kuroki R. Crystal structure of glycosyltrehalose trehalohydrolase from the hyperthermophilic archaeum Sulfolobus solfataricus. J. Mol. Biol. 2000;301:451–464. doi: 10.1006/jmbi.2000.3977. [DOI] [PubMed] [Google Scholar]
- 49.Toyooka T, Awai T, Kanai T, Imanaka T, Hori H. Stabilization of tRNA (mG37) methyltransferase [TrmD] from Aquifex aeolicus by an intersubunit disulfide bond formation. Genes Cells. 2008;13:807–816. doi: 10.1111/j.1365-2443.2008.01207.x. [DOI] [PubMed] [Google Scholar]
- 50.Ogasahara K, Khechinashvili NN, Nakamura M, Yoshimoto T, Yutani K. Thermal stability of pyrrolidone carboxyl peptidases from the hyperthermophilic Archaeon, Pyrococcus furiosus. Eur. J. Biochem. 2001;268:3233–3242. doi: 10.1046/j.1432-1327.2001.02220.x. [DOI] [PubMed] [Google Scholar]
- 51.Pearce FG, Perugini MA, McKerchar HJ, Gerrard JA. Dihydrodipicolinate synthase from Thermotoga maritima. Biochem. J. 2006;400:359–366. doi: 10.1042/BJ20060771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.O'Farrell HC, Musayev FN, Scarsdale JN, Rife JP. Binding of adenosine-based ligands to the MjDim1 rRNA methyltransferase: implications for reaction mechanism and drug design. Biochemistry. 2010;49:2697–2704. doi: 10.1021/bi901875x. [DOI] [PubMed] [Google Scholar]
- 53.Liebert K, Horton JR, Chahar S, Orwick M, Cheng X, Jeltsch A. Two alternative conformations of S-adenosyl-L-homocysteine bound to Escherichia coli DNA adenine methyltransferase and the implication of conformational changes in regulating the catalytic cycle. J. Biol. Chem. 2007;282:22848–22855. doi: 10.1074/jbc.M700926200. [DOI] [PubMed] [Google Scholar]
- 54.Thomas CB, Scavetta RD, Gumport RI, Churchill ME. Structures of liganded and unliganded RsrI N6-adenine DNA methyltransferase: a distinct orientation for active cofactor binding. J. Biol. Chem. 2003;278:26094–26101. doi: 10.1074/jbc.M303751200. [DOI] [PubMed] [Google Scholar]
- 55.Schluckebier G, Kozak M, Bleimling N, Weinhold E, Saenger W. Differential binding of S-adenosylmethionine S-adenosylhomocysteine and Sinefungin to the adenine-specific DNA methyltransferase M.TaqI. J. Mol. Biol. 1997;265:56–67. doi: 10.1006/jmbi.1996.0711. [DOI] [PubMed] [Google Scholar]
- 56.Awai T, Kimura S, Tomikawa C, Ochi A, Ihsanawati Bessho Y, Yokoyama S, Ohno S, Nishikawa K, Yokogawa T, et al. Aquifex aeolicus tRNA (N2,N2-guanine)-dimethyltransferase (Trm1) catalyzes transfer of methyl groups not only to guanine 26 but also to guanine 27 in tRNA. J. Biol. Chem. 2009;284:20467–20478. doi: 10.1074/jbc.M109.020024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hur S, Stroud RM. How U38, 39, and 40 of many tRNAs become the targets for pseudouridylation by TruA. Mol. Cell. 2007;26:189–203. doi: 10.1016/j.molcel.2007.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cunningham PR, Weitzmann CJ, Nurse K, Masurel R, Van Knippenberg PH, Ofengand J. Site-specific mutation of the conserved m6(2)A m6(2)A residues of E. coli 16S ribosomal RNA. Effects on ribosome function and activity of the ksgA methyltransferase. Biochim. Biophys. Acta. 1990;1050:18–26. doi: 10.1016/0167-4781(90)90135-o. [DOI] [PubMed] [Google Scholar]
- 59.Hoang C, Ferré-D'Amaré AR. Cocrystal structure of a tRNA Psi55 pseudouridine synthase: nucleotide flipping by an RNA-modifying enzyme. Cell. 2001;107:929–939. doi: 10.1016/s0092-8674(01)00618-3. [DOI] [PubMed] [Google Scholar]
- 60.McLuckey SA, Van Berkel GJ, Glsh GL. Tandem mass spectrometry of small, multiply charged oligonucleotides. J. Am. Soc. Mass Spectrom. 1992;3:60–70. doi: 10.1016/1044-0305(92)85019-G. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.