

Abstract
Epithelial to mesenchymal transition (EMT) is a central mechanism for diversifying the cells found in complex tissues. This dynamic process helps organize the formation of the body plan, and while EMT is well studied in the context of embryonic development, it also plays a role in the genesis of fibroblasts during organ fibrosis in adult tissues. Emerging evidence from studies of renal fibrosis suggests that more than a third of all disease-related fibroblasts originate from tubular epithelia at the site of injury. This review highlights recent advances in the process of EMT signaling in health and disease and how it may be attenuated or reversed by selective cytokines and growth factors.
For one hundred and forty-five years, biologists have known that cells come from cells (1). This concept is so fundamental today that we accept it implicitly; cells either divide asymmetrically to preserve stem cell progenitors, partition into sister cells, differentiate along fate pathways, or undergo oncogenesis following the formation of normal tissues. Specification and diversification of cell lineages are initiated by genetic programs under the control of morphogenic cues (2–4). These lineages evolve in a hierarchical manner conforming to developmental boundaries and oscillating biological clocks until reaching terminal differentiation (5). Epithelia from metazoans are emblematic of this process, and at maturity cover outer surfaces (6, 7) or line hollow cavities formed by tubular structures in complex tissues (8–10). Since epithelia typically serve specialized functions (11–13), it is assumed that a state of terminal differentiation is necessary and protected once development is complete.
In recent years, however, this formidable notion has been challenged by observations that mature epithelia change their phenotype following morphogenic pressure from injured tissue. Since the phenomenon of epithelial plasticity was described before it had a firm biochemical basis (14, 15), one is confronted with a plethora of seemingly interchangeable vocabulary. Today, the terms “epithelial-mesenchymal transformation, interactions, or transition” are comingled inappropriately with the term “epithelial-mesenchymal transdifferentiation.” “Transformation” classically describes the oncogenic conversion of epithelia. Likewise, the induction of bone marrow stem cells to form somatic cells probably should be considered differentiation rather than transdifferentiation (16). Epithelial-mesenchymal interaction refers to proximate paracrine cross-talk between tissue epithelia and stromal fibroblasts and is completely different from the concept of epithelial-mesenchymal transition (EMT). EMT is a variant of transdifferentiation and a well-recognized mechanism for dispersing cells in vertebrate embryos (17), forming fibroblasts in injured tissues (18, 19), or initiating metastases in epithelial cancer (20–23). We prefer the term “transition” to describe this conversion instead of “epithelial-mesenchymal transdifferentiation” because “transdifferentiation” classically refers to differentiated cells changing into other differentiated cells (24). Transdifferentiation has been observed in retinal pigmented cells that become lens epithelia (25, 26), in the conversion from white to brown adipocytes (27), endothelial cells that become vascular smooth muscle cells (28), lactotrophs that interconvert to somatotrophs in the pituitary (29), pancreatic acinar cells that become ductal epithelium (30, 31) or hepatocytes (32, 33), and hepatocytes that morph into pancreatic ductal cells (34). Although many investigators fail to make the distinction between transdifferentiation and transition, it may be time to do so. It is not yet clear whether the fibroblast transition of EMT is an expected middle phase of transdifferentiating epithelium or whether EMT producing fibroblasts is an arrested form of transdifferentiation (24). EMT of terminally differentiated epithelium, in its purest sense, produces a tissue fibroblast (19).
Developmental biologists have also known for decades that epiblasts undergo EMT to form primary mesenchyme in the creation of tripoblastic germ layers (17, 35–37; Figure 1). In the mesoderm this is followed by mesenchymal-epithelial transitions to create secondary epithelium as part of somitogenesis (38, 39) and the further commitment and diversification of cells forming mesoendodermal structures (40–42). Secondary epithelium in mature or adult tissues can also undergo EMT following epithelial stress, such as inflammation (18, 19) or wounding (17, 43) that leads to fibroblast production and fibrogenesis. Epithelia forming tumors also use EMT when carcinomas become metastatic (23, 44, 45).
Figure 1.
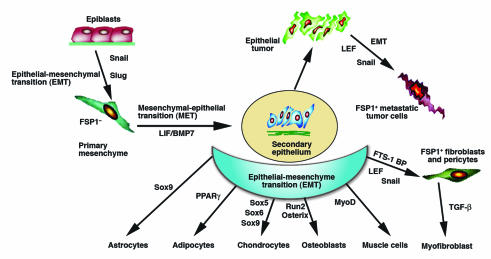
Primitive epithelia (epiblasts) form tropoblastic germ layers through EMT. The primary mesenchyme that migrates after EMT is reinduced to secondary epithelium by mesenchymal-epithelial transition. Secondary epithelia differentiate to form new epithelial tissues and undergo a second round of EMT to form the cells of connective tissue, including astrocytes, adipocytes chondrocytes, osteoblasts, muscle cells, and fibroblasts. Mature secondary epithelia that form epithelial organs can also transform into primary tumors that later undergo EMT to metastasize. These processes are regulated by morphogenic cues and a variety of transcription factors, and are potentially plastic in their adaptation to new biologic circumstances.
We review here recent observations regarding the mechanism of EMT in culture and during fibrogenesis, especially associated with kidney disease. The problem of tissue fibrosis is that epithelial units are overtaken by scarification and lose their morphogenic cues, leaving involved organs to fail. While traditional studies of fibrosis have focused on the production of extracellular matrix, recent information now suggests that epithelia contribute to the problem by creating new fibroblasts. Experiments demonstrating the reversibility of organ fibrosis also highlight the need to consider cellular mechanisms of fibrogenesis and the basic biology that will, one hopes, contribute new molecules as useful therapeutics.
The mechanism of EMT
From a general perspective, EMT is about disaggregating epithelial units and reshaping epithelia for movement. Epithelium in transition lose polarity, adherens junctions, tight junctions, desmosomes, and cytokeratin intermediate filaments in order to rearrange their F-actin stress fibers and express filopodia and lamellopodia. This phenotypic conversion requires the molecular reprogramming of epithelium with new biochemical instructions. Much of this conversion has been studied fractionally, during experiments that expose new transduction and signaling pathways, in epithelia that transition in culture, and more recently in fibrogenic tissues. Below we describe an enlarging picture of EMT from a broad and increasingly complex literature.
Induction of EMT.
EMT is easily engaged by a combination of cytokines associated with proteolytic digestion of basement membranes upon which epithelia reside. Metalloproteinases (46, 47) or membrane assembly inhibitors (48) initiate the process by dismantling the local basement membrane. Local expression of TGF-β, EGF, IGF-II, or FGF-2 facilitates EMT (Figure 2) by binding epithelial receptors with ligand-inducible intrinsic kinase activity (49–52). The TGF-β effect depends on β-integrin transduction (53), Smad3-dependent transcription (54), or Smad-independent p38MAP kinase activation and GTPase-mediated signaling (53, 55, 56). Depending on the tissue, all three isoforms of TGF-β may be involved sequentially (57–59). While TGF-β is considered prototypical in its induction of EMT (50, 60, 61), there is an increase in epithelial EGF receptors in the EMT microenvironment (62), and EGF can assist in completing the conversion (50). IGF-II also directs the redistribution of β-catenins from the cell surface to the nucleus and facilitates the intracellular degradation of E-cadherin (51), while FGF-2 and TGF-β are required for the expression of MMP-2 and MMP-9 to assist in basement membrane degradation (52). Combinations of cytokines are generally present in most areas of tissue injury, so it is difficult to assign priorities or hierarchy. Each moiety may contribute a unique inducement to the transition. Furthermore, the role of HGF and FGFs depends on the timed expression and selective distribution of receptors (63, 64). While HGF action through its c-Met/Crk adaptor proteins (65) induces EMT during somitogenesis and endocardial cushion development (66, 67) and modulates the connectivity of intercellular junctions between polarized intestinal and kidney epithelium (68), in the fibrogenic kidney it has the opposite effect of protecting epithelium from EMT (69). In this regard, the expression of bone morphogenic protein 7 (BMP-7) also counterbalances EMT in the kidney (54). Like many biological systems, countervening processes that modulate EMT effector events are beginning to appear.
Figure 2.

Epithelial plasticity can lead to classical EMT (loss of cell-cell and cell-substratum attachments, new actin rearrangements, and gain of mobility) or reversible scatter, which looks like EMT but is not enduring and can revert. These events are regulated by ligand-inducible intrinsic kinase receptors on the cell surface, which modulate small GTPases, Smads, PI3Ks, MAP kinases, and the availability of β-catenin to coactivate LEF in the nucleus. Free levels of b-catenin are regulated by E-cadherin or APC/β-catenin/Axin complexes, the latter of which shuttle b-catenin between ubiquination or utilization in adherens junctions. Activation of nuclear transcription provides new transcriptional regulators (Snail, SIP1, Ets, and FTS-BP/CarG box binding factor) of the EMT proteome. The EMT proteome comprises proteins listed in Table 1. The variability of receptors, kinases, and the emergence of combined preferences for signaling pathways determine the plasticity unique to each epithelium.
Engagement of protein kinases.
Epithelial signaling that leads to EMT has been studied in a variety of cultured epithelia and seems to have broad generality across numerous phenotypes (Figure 2). As a result of ligand-inducible receptor kinase activation (14), there is a downstream engagement of GTPases from the Ras superfamily (70, 71) or commitment of SH2-SH3 protein domains of the nonreceptor tyrosine kinase (c-Src and Btk) pathways (14, 72) that shift the intracellular balance of small GTPases (Rho, Rac, and Cdc42). Raf/MAP kinases are subsequently activated with several interconnected consequences: engagement of the EMT transcriptome (73) followed by actin rearrangement of the cytoskeleton (70, 74). Integrin-linked kinase (ILK) activation by TGF-β–activated Smad proteins (75) or integrin signaling (76) enhances β-catenin/lymphoid enhancer factor (LEF) expression, suppressing E-cadherin. Activation of Src kinases favors the PI3K pathway, stabilization of β-catenin for nuclear import (77), protection from apoptosis, and disruption of β-integrin binding and E-cadherin complexes (21, 53, 78, 79). Many of these pathways collaboratively reinforce EMT.
The nuclear import of LEF proteins with Smad3 or β-catenin from the cytoplasm is beginning to look like one of several key molecular steps in EMT (73, 80). The phosphorylation of β-catenin by glycogen synthase kinase- (GSK-3β) allows it to form a complex with APC suppressor protein and Axin (81). p53 activation of APC-dependent pathways also forms a complex with β-catenin (82), and both of these pathways lead to direct loss of free β-catenin through ubiquination (83). If, however, phosphorylation by GSK-3β is inhibited (77), cytoplasmic β-catenin is stabilized by re-entering the E-cadherin complex (84) or binding to the B-box of LEF where together they move into the nucleus to engage the EMT transcriptome (85). Wnt-1 (81), IGF-II (51), Ras (77), and ILK (76) all stabilize cytoplasmic levels of β-catenin to facilitate EMT, perhaps by GSK-3β (or other kinase) inhibition. Smad3 activation by TGF-β family members can also activate LEF-1 in the absence of β-catenin (80). These latter findings suggest either a synergistic or independent control of LEF-1 by at least two EMT-linked signaling pathways. While levels of APC suppressor protein in epithelia may protect the state of terminal differentiation from EMT (73, 86), activation of Smad pathways may provide a countervailing leak in this stability. β-catenin and Smad3 also require the engagement of different transcriptional coactivators, depending on the promoter. These differences may regulate the selectivity or availability of the EMT transcriptome.
Recently, there also has been some attempt to distinguish true EMT from an epithelial phenocopy called “reversible scatter” (21, 73). Reversible scatter following cytokine stimulation looks like EMT because the cells assume a spindlelike shape and undergo a brief period of transcription. But because transcription is not sustained on withdrawal of the inducement and/or if the cells are protected from apoptosis, the epithelia return to their original state (21). TGF-β and Ras classically produce EMT, while EGF, HGF, and FGF favor scattering (21), but not in all cells (52, 68). A scatter effect may be facilitated by varying levels of cytoplasmic APC suppressor protein (73) or preferred activation of the PI3K pathway (21). Whether scatter reverses or goes on to EMT may really just be a timing issue in the continuum of transition, as it is not clear what biological function reversible scatter serves on its own.
The EMT proteome.
The EMT proteome reflects a fundamental change in proteins gained, maintained, or lost (Table 1) with the conversion of epiblasts to primary mesenchyme (37, 87), secondary epithelium to fibroblasts (88), or in the transition of tumor epithelia to metastatic cells (89). Many studies have generally focused on only one or two event markers (for example, the changes in E-cadherin or Snail expression) and are not comprehensive. Most information about the EMT proteome is inferred from proteins found in epithelia but not in fibroblasts or metastatic cancer cells, or is based on apparent targets of transcription factors (15, 73). Although dependent on cell context and ease of growth factor signaling, delamination of epithelia to facilitate movement is also accompanied by a regulatory decrease in apoptosis and mitosis (15, 21).
Table 1.
The EMT proteome

Cellular plasticity likely requires real-time control by transcriptional networks (90–94). Some models suggest several transcription factors may be key modulators of transitional events. The Snail superfamily of zinc-finger proteins has two evolutionary branches, one for Scratch and the other for Snail and Slug (15). These proteins recognize an E-box binding motif on the promoter for E-cadherin (among others) in competition with the basic helix-loop-helix protein SIP1. Ras/MAPK activates Snail while TGF-β regulates Smad-dependent pathways to engage SIP1 and Snail (15, 95). Subsequently E-cadherin, cytokeratin, muc-1, and desmoplakin are repressed, while fibroblast-specific protein-1 (FSP1), fibronectin, vimentin, and Rho are increased (15, 54). Repression of E-cadherin by Snail proteins frees up more cytoplasmic β-catenin, which, as mentioned above, is co-imported with LEF to the nucleus where its activation is strongly associated with EMT (73). Ets transcription factors regulate EMT in the heart (96, 97).
One of the more interesting proteins found in the EMT proteome is FSP1 (18), also known as S100A4 (98). Support for the notion that EMT is a major source of fibroblasts comes from experiments showing FSP1 expression in cultured epithelium during EMT following exposure to TGF-β and EGF (50), histologic evidence that epithelial units expressing FSP1 disaggregate as organ tissues devolve during the early stages of fibrogenesis (18), and direct observations of EMT in transgenic mice carrying marked epithelium (19). Dividing fibroblasts exposed to nucleoside analogues are also selectively eliminated in transgenic mice expressing thymidine kinase under control of the FSP1 promoter (99). Members of the S100 superfamily have been implicated in cytoskeletal-membrane interactions, calcium signal transduction, and cellular growth and differentiation (100). In the presence of calcium, FSP1 dimerizes and binds the c-terminal of p53 in the cytoplasm. In this way, FSP1 may sequester p53 from the APC ubiquination pathway (101, 102), perhaps raising levels of free β-catenin. We suspect that FSP1 facilitates and may even maintain the EMT phenotype through this mechanism (Figure 2). While the precise function of FSP1 is not entirely clear, its interaction with cytoskeletal moieties and its early role in EMT suggest that FSP1 may fashion mesenchymal cell shape to enable motility (50) and induce angiogenesis (103). The expression of FSP1 indicates the potential presence of a molecular program determining fibroblast phenotype.
The promoter for FSP1 is also part of a transcriptome that shares putative FTS-1/CArG box sites in the early promoter regions of a group of genes that would be expected in EMT-derived fibroblasts, including those encoding c-myc, c-Fos, H-ras, Slap, TGF-β, FGF-1, -2, and -8, FSP1, vimentin, α-smooth muscle actin (αSMA), aggrecan, collagen types I and III, thrombospondin I, and matrix metalloproteinases 2 and 9 (19). One hypothesis is that the selective engagement of FTS-1/CArG box sites by a transcriptional complex of proteins may be one of several key regulators of EMT; preliminary evidence suggests a new Kruppel-like zinc finger protein called FTS/CArG box binding factor (FTS-BP/CBF) (104 , 105; and our unpublished observations) may contribute.
GTPase modulation of cell shape and movement.
Epithelia that undergo EMT during development, inflammation, or carcinogenesis become mesenchymal cells, fibroblasts, or metastatic tumor cells, respectively (19, 72, 106). This conversion of epithelia is dependent on molecular switches under the control of the Ras superfamily of small GTPases (71). Ras and Rho families of GTPases are activated by guanine nucleotide exchange factors (107) and deactivated by GTPase activating proteins (108). GTPases are a signaling link between cell surface receptor activation and the actin cytoskeleton; some GTPases can also cooperatively modulate EMT with cytokine pressure (21, 109). Three of the best-studied small GTPases are Rho, Rac, and Cdc42. The cross-talk between these members suggests they are activated independently or in series: Ras or Cdc42 can activate Rac, and Rac can inhibit or activate Rho (110–113). Rho helps reconfigure actin stress fibers and stimulates actin-myosin contraction in the cell body, Rac induces the assembly of actin surface protrusions called lamellopodia, and Cdc42 promotes the formation of actin-rich finger extensions called filopodia and modulates cellular asymmetry (71). Their differential activation and balance ensure not only epithelialization but also its dissolution. The cellular properties of contraction, migration, proliferation, and phagocytosis are also under GTPase control (70). The cellular actions of these small GTPases engage downstream MAP kinases, alter gene transcription, and are integral to shaping cell phenotype during EMT.
Fibroblasts derive from a niche
Since the original observations of Cohnheim (114), investigators have debated the origin of tissue fibroblasts. Three notions persist regarding their lineage: The longest-held concept is that fibroblasts are simply residual embryonic mesenchymal cells left over from organogenesis. While this hypothesis explains the incorrect but often interchangeable substitution of the term “fibroblast” for “mesenchymal cell,” the idea itself has no proof and is unlikely since primary mesenchymal cells do not express FSP1 (18). A second notion argues that fibroblasts emerge from the bloodstream after release from the bone marrow (115), and a third view suggests that fibroblasts derive locally in tissues following EMT (19). The second and third hypotheses are mechanistically identical; that is, all fibroblasts probably arise from EMT.
Interstitial fibroblasts appear after gastrulation (after E8.5 in mice) (18) and form as a result of EMT from secondary epithelium (19). Support for the notion that EMT is a major source of local fibroblasts comes from experiments described above. FSP1 is also expressed in some endosteal lining cells and marrow stromal cells (19), and about 14–15% of fibroblasts in fibrosing kidney are derived from marrow. Not much is known about the origin of endosteal lining cells (116, 117). Endosteal bone marrow lining cells precede the formation of the marrow cavity and its contents (117), and in some species are separated from medullary hematopoiesis by a marrow sac comprising a mixture of simple epithelium and/or condensed stromal-like cells (118, 119). This sac appears to have a structural and biochemical interface with endosteal lining cells in nodal regions of bone (118, 120), and the collective structure may be an EMT niche for osteogenic precursor cells, indifferent endosteum, fibroblasts, and marrow stromal cells. Recent evidence also suggests it can be a niche for hematopoietic stem cells (121). FSP1+ cells in the marrow are mostly CD34–. CD34– progenitor cells cycle their expression of CD34 in the marrow (122–124); both CD34– and CD34+ stromal cells circulate in peripheral blood, and, following bone marrow reconstitution, CD34 cells locate as bone-lining endosteum (116, 124). Since some marrow stromal cells can be released into the circulation (124), CD34–, FSP1+ bone marrow “fibroblasts” might derive from an endosteal EMT niche transitioning to marrow stromal cells (19), which then may evolve into circulating fibrocytes (115). The contribution of CD34–, Strol+, CD73+ mesenchymal stem cells in the accumulation of fibroblasts in this setting is yet unknown.
Most investigators accept with conventional wisdom that fibroblasts represent a cell type of limited diversity. Fibroblast shape, cytoskeletal structure, secretion of interstitial collagens, mobility, participation in tissue fibrosis, and behavior in culture all tend to support this belief. The EMT hypothesis, however, challenges this notion of homogeneity, as do observations that fibroblasts express subtle biochemical differences (125) and phenotypic variability (126–128), and respond differently to cytokines and matrix (129), depending on their tissue of origin. Recent evaluation of the transcriptome from a variety of fibroblasts suggests there is topographic differentiation perhaps based on a “Hox code” (130). Consequently, fibroblasts formed by EMT may differentially express a profile of genes, or a few residual receptors or signaling pathways representative of their previous life as mature epithelium, and, theoretically, can be as heterogeneous as the universe of epithelia.
EMT and fibrosis
The role of EMT during tissue injury leading to organ fibrosis (deposition of collagens, elastin, tenacin, and other matrix molecules) is becoming increasingly clear (Figure 3). A great bulk of such evidence exists for EMT associated with progressive kidney diseases (19), and is probably true for the lung (131) and possibly the liver. Typical experimental models of kidney fibrosis in mice or rats include progressive glomerulonephritis from anti-glomerular basement membrane disease (132), Alport syndrome (133), or spontaneous lupus nephritis (134), and NOD or db/db nephritic mice (models for diabetic nephropathy) (135), all of which chronically progress at a slow pace, and unilateral ureteral obstruction (136), which progresses to end-stage quickly but leaves the contralateral kidney normal as a control. A number of studies demonstrating EMT during kidney fibrosis correlate with the expression of FSP1 (described above) (18, 50). FSP1 identifies tubular epithelial cells undergoing transition in damaged nephrons trapped by interstitial injury and tracks with increasing numbers of fibroblasts as fibrosis grows worse (50, 137). These FSP1+ epithelia traverse through damaged tubular basement membrane (TBM) and accumulate in the interstitium of the kidney (138) where they lose their epithelial markers as they gain a fibroblast phenotype (50, 137). Fibroblasts are not particularly abundant in normal kidneys as they are in lungs, lymphoid nodes, and spleens. When renal fibrogenesis sets in, about 36% of new fibroblasts come from local EMT, about 14–15% from the bone marrow, and the rest from local proliferation (19). This finding reinforces the notion that fibrogenesis is a local epithelial event.
Figure 3.

Origin of fibroblasts during kidney fibrosis. (a) Fibrotic kidney which displays accumulation of numerous fibroblasts (blue arrow), damaged kidney tubules (yellow arrow), and blood vessels (green arrow). (b) Normal kidney with proper tubular structures and very few fibroblasts. (c) Schematic illustration of three possible mechanisms via which fibroblasts can originate during kidney injury. Recent experiments suggest that approximately 14–15% of fibroblasts are from bone marrow, 36% can arise via local EMT involving tubular epithelial cells under inflammatory stress, and the rest are likely contributed by proliferation of fibroblasts from all sources. (d) Systemic treatment of mice with renal fibrosis using recombinant human BMP-7 results in reversal of renal disease due to severe decrease in EMT-derived fibroblasts and potentially bone marrow–derived fibroblasts. Such events likely have a cascade of beneficial effects that decreasing the overall number of fibroblasts in the kidney, and attenuating fibrosis.
It is worth mentioning that fibroblasts have little in the way of other distinguishing anatomic features, and most of the proteins they express are not highly specific (18). Vimentin is not fibroblast-specific (50, 137), and type I collagen synthesis is generally only detectable in selected subpopulations of fibroblasts (139–142). Some subpopulations of fibroblasts during fibrogenesis express αSMA (46, 128, 143, 144), a marker of activated fibroblasts (myofibroblasts) (145, 146). While much of the fibrosis literature has relied on this less specific marker, αSMA is not expressed by all fibroblasts (137), suggesting that it does not define the universe of fibroblasts and potentially also identifies smooth muscle cells separated from local blood vessels during tissue injury (137, 147). The increased number of αSMA+ smooth muscle cells in fibrotic tissue may derive from delaminated endothelial cells following endothelial-mesenchymal transition (28).
Why are tubular epithelia susceptible to EMT? Injury to the kidney is associated with many inflammatory cells which can incite EMT using growth factors such as TGF-β, EGF, and FGF-2 (52). Under the influence of such growth factors, resident fibroblasts and tubular epithelia induce basement membrane–degrading enzymes such as MMP-2 and MMP-9 (48). Degradation of TBM results in disruption of tubular nephrons, and delaminated epithelial cells either fall off into the tubular fluid or migrate towards the interstitium under the influence of increasing growth factor gradients and chemoattractants (47). This initial recruitment of tubular epithelial cells for EMT can be inhibited by blocking the expression of MMP-9 through the disruption of tissue plasminogen activator (tPA, an activator of MMP-9) (148). Other studies have also demonstrated that HGF can decrease levels of TGF-β, restore TGF-β–mediated loss of E-cadherin, and potentially decrease amounts of active MMP-9 (149). In this regard, ILK is now identified as a key mediator of TGF-β–induced EMT associated with tubular epithelial cells (75).
The relevance of TGF-β–induced EMT for progression of kidney fibrosis was recently addressed in studies using BMP-7 as an intracellular competitor of TGF-β signaling (54, 150, 151). BMP-7 is the endogenous antagonist of TGF-β–induced EMT in the kidney and elsewhere (54, 150, 151). BMP-7 reverses the decrease of E-cadherin caused by TGF-β (54). Restoration of E-cadherin by BMP-7 is mediated by its ALK3/6 receptors and Smad5. The capacity of BMP-7 to reverse TGF-β–induced EMT in culture is also observed in mouse models of kidney fibrosis. Systemic administration of recombinant BMP-7 in mice with kidney fibrosis following ureteral obstruction results in reversal of EMT and repair of damaged tubular structures with repopulation of healthy tubular epithelial cells (54, 152). This reversal is also associated with return of renal function, a significant decrease in FSP1+ interstitial fibroblasts and de novo activation of BMP-7 signaling (54). Renal protection from BMP-7 has also been observed in murine models of diabetic nephropathy (153), Alport syndrome, and lupus nephritis (150). Today, TGF-β signaling attenuated by BMP-7 is the closest paradigm in EMT arguing in favor of privileged pathways.
Summary
Progress in understanding EMT has been an exercise in coming to appreciate the level of complexity required for changing cellular identity. The mechanism of transition highlights an integration of nuclear regulation and network signaling with alterations in microenvironment to create a moving cell. Remarkably, differentiating epithelia make these transitions during development, and terminally differentiated epithelia use them for physiologic repair or to advance oncogenesis. EMT is a form of molecular exaptation, a mechanism of economy by which cells reuse known physiologic processes to provide new functions (154). With the foundation established by current studies, new questions regarding the definition and role of pericytes and myofibroblasts can be explored, and a framework for a better understanding of other transitions, like endothelial-mesenchymal transition, is possible. EMT also provides a mechanism for creating ancestral relationships between local cells and may be particularly important in tumor expansion. Lastly, fibroblasts may carry forward remnants of a unique epithelial signature. And if all fibroblasts or tumor cells which arise via EMT are not created equal, then therapies to combat fibrosis or metastatic disease may need more specificity. Nevertheless, the future holds great promise for EMT as a viable therapeutic target.
Acknowledgments
This work was supported by grants NIH/DK55001, and NIH/DK62987, The Espinosa Liver Fibrosis Fund, and research support from the Center for Matrix Biology at Beth Israel Deaconess Medical Center, all to R. Kalluri. E. Neilson is supported by NIH/DK46282 and Yamanouchi USA Foundation.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Nonstandard abbreviations used: epithelial-mesenchymal transition (EMT); bone morphogenic protein 7 (BMP-7); integrin-linked kinase (ILK); lymphoid enhancer factor (LEF); glycogen synthase kinase (GSK); fibroblast-specific protein-1 (FSP1); α-smooth muscle actin (αSMA); tubular basement membrane (TBM); tissue plasminogen activator (tPA).
References
- 1.Virchow, R. 1858. Die Cellularpathologie in ihrer Begründung auf physiologische und pathologische Gewebelehre. A. Hirschwald. Berlin, Germany. 456 pp.
- 2.Slack JMW. Stem cells in epithelial tissues. Science. 2000;287:1431–1433. doi: 10.1126/science.287.5457.1431. [DOI] [PubMed] [Google Scholar]
- 3.Watt FM, Hogan BL. Out of Eden: stem cells and their niches. Science. 2000;287:1427–1430. doi: 10.1126/science.287.5457.1427. [DOI] [PubMed] [Google Scholar]
- 4.Blau HM, Brazelton TR, Weimann JM. The evolving concept of a stem cell: entity or function? Cell. 2001;105:829–841. doi: 10.1016/s0092-8674(01)00409-3. [DOI] [PubMed] [Google Scholar]
- 5.Irvine KD, Rauskolb C. Boundaries in development: formation and function. Annu. Rev. Cell Dev. Biol. 2001;17:189–214. doi: 10.1146/annurev.cellbio.17.1.189. [DOI] [PubMed] [Google Scholar]
- 6.Sengel P. Pattern formation in skin development. Int. J. Dev. Biol. 1990;34:33–50. [PubMed] [Google Scholar]
- 7.Erickson CA, Reedy MV. Neural crest development: the interplay between morphogenesis and cell differentiation. Curr. Top. Dev. Biol. 1998;40:177–209. doi: 10.1016/s0070-2153(08)60367-1. [DOI] [PubMed] [Google Scholar]
- 8.Hogan BL, Kolodziej PA. Organogenesis: molecular mechanisms of tubulogenesis. Nat. Rev. Genet. 2002;3:513–523. doi: 10.1038/nrg840. [DOI] [PubMed] [Google Scholar]
- 9.Krasnow MA, Nelson WJ. Tube morphogenesis. Trends Cell Biol. 2002;12:351. doi: 10.1016/s0962-8924(02)02332-2. [DOI] [PubMed] [Google Scholar]
- 10.Lubarsky B, Krasnow MA. Tube morphogenesis: making and shaping biological tubes. Cell. 2003;112:19–28. doi: 10.1016/s0092-8674(02)01283-7. [DOI] [PubMed] [Google Scholar]
- 11.Gumbiner BM. Epithelial morphogenesis. Cell. 1992;69:385–387. doi: 10.1016/0092-8674(92)90440-n. [DOI] [PubMed] [Google Scholar]
- 12.Yeaman C, Grindstaff KK, Hansen MD, Nelson WJ. Cell polarity: versatile scaffolds keep things in place. Curr. Biol. 1999;9:R515–517. doi: 10.1016/s0960-9822(99)80324-8. [DOI] [PubMed] [Google Scholar]
- 13.Al-Awqati Q, Vijayakumar S, Takito J. Terminal differentiation of epithelia. Biol. Chem. 2003;384:1255–1258. doi: 10.1515/BC.2003.138. [DOI] [PubMed] [Google Scholar]
- 14.Boyer B, Valles AM, Edme N. Induction and regulation of epithelial-mesenchymal transitions. Biochem. Pharmacol. 2000;60:1091–1099. doi: 10.1016/s0006-2952(00)00427-5. [DOI] [PubMed] [Google Scholar]
- 15.Nieto MA. The snail superfamily of zinc-finger transcription factors. Nat. Rev. Mol. Cell Biol. 2002;3:155–166. doi: 10.1038/nrm757. [DOI] [PubMed] [Google Scholar]
- 16.Tsai RY, Kittappa R, McKay RD. Plasticity, niches, and the use of stem cells. Dev. Cell. 2002;2:707–712. doi: 10.1016/s1534-5807(02)00195-8. [DOI] [PubMed] [Google Scholar]
- 17.Hay ED. An overview of epithelio-mesenchymal transformations. Acta Anat. 1995;154:8–20. doi: 10.1159/000147748. [DOI] [PubMed] [Google Scholar]
- 18.Strutz F, et al. Identification and characterization of a fibroblast marker: FSP1. J. Cell Biol. 1995;130:393–405. doi: 10.1083/jcb.130.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iwano M, et al. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Invest. 2002;110:341–350. doi:10.1172/JCI200215518. doi: 10.1172/JCI15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kiemer AK, Takeuchi K, Quinlan MP. Identification of genes involved in epithelial-mesenchymal transition and tumor progression. Oncogene. 2001;20:6679–6688. doi: 10.1038/sj.onc.1204872. [DOI] [PubMed] [Google Scholar]
- 21.Janda E, et al. Ras and TGF[beta] cooperatively regulate epithelial cell plasticity and metastasis: dissection of Ras signaling pathways. J. Cell Biol. 2002;156:299–313. doi: 10.1083/jcb.200109037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vincent-Salomon A, Thiery JP. Host microenvironment in breast cancer development: epithelial-mesenchymal transition in breast cancer development. Breast Cancer Res. 2003;5:101–106. doi: 10.1186/bcr578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xue C, Plieth D, Venkov C, Xu C, Neilson EG. The gatekeeper effect of epithelial-mesenchymal transition regulates the frequency of breast cancer metastases. Cancer Res. 2003;63:3386–3394. [PubMed] [Google Scholar]
- 24.Slack JM, Tosh D. Transdifferentiation and metaplasia–switching cell types. Curr. Opin. Genet. Dev. 2001;11:581–586. doi: 10.1016/s0959-437x(00)00236-7. [DOI] [PubMed] [Google Scholar]
- 25.Eguchi G, Kodama R. Transdifferentiation. Curr. Opin. Cell Biol. 1993;5:1023–1028. doi: 10.1016/0955-0674(93)90087-7. [DOI] [PubMed] [Google Scholar]
- 26.Rio-Tsonis KD, Tsonis PA. Eye regeneration at the molecular age. Dev. Dyn. 2003;226:211–224. doi: 10.1002/dvdy.10224. [DOI] [PubMed] [Google Scholar]
- 27.Cinti S. Adipocyte differentiation and transdifferentiation: plasticity of the adipose organ. J. Endocrinol. Invest. 2002;25:823–835. doi: 10.1007/BF03344046. [DOI] [PubMed] [Google Scholar]
- 28.Frid MG, Kale VA, Stenmark KR. Mature vascular endothelium can give rise to smooth muscle cells via endothelial-mesenchymal transdifferentiation: in vitro analysis. Circ. Res. 2002;90:1189–1196. doi: 10.1161/01.res.0000021432.70309.28. [DOI] [PubMed] [Google Scholar]
- 29.Vidal S, Horvath E, Kovacs K, Lloyd RV, Smyth HS. Reversible transdifferentiation: interconversion of somatotrophs and lactotrophs in pituitary hyperplasia. Mod. Pathol. 2001;14:20–28. doi: 10.1038/modpathol.3880252. [DOI] [PubMed] [Google Scholar]
- 30.Rooman I, Heremans Y, Heimberg H, Bouwens L. Modulation of rat pancreatic acinoductal transdifferentiation and expression of PDX-1 in vitro. Diabetologia. 2000;43:907–914. doi: 10.1007/s001250051468. [DOI] [PubMed] [Google Scholar]
- 31.Hall PA, Lemoine NR. Rapid acinar to ductal transdifferentiation in cultured human exocrine pancreas. J. Pathol. 1992;166:97–103. doi: 10.1002/path.1711660203. [DOI] [PubMed] [Google Scholar]
- 32.Shen CN, Slack JM, Tosh D. Molecular basis of transdifferentiation of pancreas to liver. Nat. Cell Biol. 2000;2:879–887. doi: 10.1038/35046522. [DOI] [PubMed] [Google Scholar]
- 33.Shen CN, Horb ME, Slack JM, Tosh D. Transdifferentiation of pancreas to liver. Mech. Dev. 2003;120:107–116. doi: 10.1016/s0925-4773(02)00337-4. [DOI] [PubMed] [Google Scholar]
- 34.Horb ME, Shen CN, Tosh D, Slack JM. Experimental conversion of liver to pancreas. Curr. Biol. 2003;13:105–115. doi: 10.1016/s0960-9822(02)01434-3. [DOI] [PubMed] [Google Scholar]
- 35.Tam PP, Behringer RR. Mouse gastrulation: the formation of a mammalian body plan. Mech. Dev. 1997;68:3–25. doi: 10.1016/s0925-4773(97)00123-8. [DOI] [PubMed] [Google Scholar]
- 36.Narasimha M, Leptin M. Cell movements during gastrulation: come in and be induced. Trends Cell Biol. 2000;10:169–172. doi: 10.1016/s0962-8924(00)01755-4. [DOI] [PubMed] [Google Scholar]
- 37.Carver EA, Jiang R, Lan Y, Oram KF, Gridley T. The mouse snail gene encodes a key regulator of the epithelial-mesenchymal transition. Mol. Cell. Biol. 2001;21:8184–8188. doi: 10.1128/MCB.21.23.8184-8188.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Summerbell D, Rigby PW. Transcriptional regulation during somitogenesis. Curr. Top. Dev. Biol. 2000;48:301–318. doi: 10.1016/s0070-2153(08)60760-7. [DOI] [PubMed] [Google Scholar]
- 39.Pourquie O. Vertebrate somitogenesis. Annu. Rev. Cell Dev. Biol. 2001;17:311–350. doi: 10.1146/annurev.cellbio.17.1.311. [DOI] [PubMed] [Google Scholar]
- 40.Ekblom P. Developmentally regulated conversion of mesenchyme to epithelium. FASEB J. 1989;3:2141–2150. doi: 10.1096/fasebj.3.10.2666230. [DOI] [PubMed] [Google Scholar]
- 41.Birchmeier W, Birchmeier C. Mesenchymal-epithelial transitions. Bioessays. 1994;16:305–307. doi: 10.1002/bies.950160503. [DOI] [PubMed] [Google Scholar]
- 42.Barasch J, et al. Mesenchymal to epithelial conversion in rat metanephros is induced by LIF. Cell. 1999;99:377–386. doi: 10.1016/s0092-8674(00)81524-x. [DOI] [PubMed] [Google Scholar]
- 43.Desmouliere A. Factors influencing myofibroblast differentiation during wound healing and fibrosis. Cell Biol. Int. 1995;19:471–476. doi: 10.1006/cbir.1995.1090. [DOI] [PubMed] [Google Scholar]
- 44.Lochter A. Plasticity of mammary epithelia during normal development and neoplastic progression. Biochem. Cell Biol. 1998;76:997–1008. [PubMed] [Google Scholar]
- 45.Guarino M, Micheli P, Pallotti F, Giordano F. Pathological relevance of epithelial and mesenchymal phenotype plasticity. Pathol. Res. Pract. 1999;195:379–389. doi: 10.1016/S0344-0338(99)80011-9. [DOI] [PubMed] [Google Scholar]
- 46.Yang J, Liu Y. Dissection of key events in tubular epithelial to myofibroblast transition and its implications in renal interstitial fibrosis. Am. J. Pathol. 2001;159:1465–1475. doi: 10.1016/S0002-9440(10)62533-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zeisberg M, Maeshima Y, Mosterman B, Kalluri R. Renal fibrosis. Extracellular matrix microenvironment regulates migratory behavior of activated tubular epithelial cells. Am. J. Pathol. 2002;160:2001–2008. doi: 10.1016/S0002-9440(10)61150-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zeisberg M, et al. Renal fibrosis: collagen composition and assembly regulates epithelial-mesenchymal transdifferentiation. Am. J. Pathol. 2001;159:1313–1321. doi: 10.1016/S0002-9440(10)62518-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fan JM, et al. Transforming growth factor-beta regulates tubular epithelial-myofibroblast transdifferentiation in vitro. Kidney Int. 1999;56:1455–1467. doi: 10.1046/j.1523-1755.1999.00656.x. [DOI] [PubMed] [Google Scholar]
- 50.Okada H, Danoff TM, Kalluri R, Neilson EG. The early role of FSP1 in epithelial-mesenchymal transformation. Am. J. Physiol. 1997;273:563–574. doi: 10.1152/ajprenal.1997.273.4.F563. [DOI] [PubMed] [Google Scholar]
- 51.Morali OG, et al. IGF-II induces rapid beta-catenin relocation to the nucleus during epithelium to mesenchyme transition. Oncogene. 2001;20:4942–4950. doi: 10.1038/sj.onc.1204660. [DOI] [PubMed] [Google Scholar]
- 52.Strutz F, et al. Role of basic fibroblast growth factor-2 in epithelial-mesenchymal transformation. Kidney Int. 2002;61:1714–1728. doi: 10.1046/j.1523-1755.2002.00333.x. [DOI] [PubMed] [Google Scholar]
- 53.Bhowmick NA, Zent R, Ghiassi M, McDonnell M, Moses HL. Integrin beta 1 signaling is necessary for transforming growth factor-beta activation of p38MAPK and epithelial plasticity. J. Biol. Chem. 2001;276:46707–46713. doi: 10.1074/jbc.M106176200. [DOI] [PubMed] [Google Scholar]
- 54.Zeisberg M, et al. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 2003;9:964–968. doi: 10.1038/nm888. [DOI] [PubMed] [Google Scholar]
- 55.Boyer AS, Erickson CP, Runyan RB. Epithelial-mesenchymal transformation in the embryonic heart is mediated through distinct pertussis toxin-sensitive and TGFβ signal transduction mechanisms. Dev. Dyn. 1999;214:81–91. doi: 10.1002/(SICI)1097-0177(199901)214:1<81::AID-DVDY8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 56.Yu L, Hebert MC, Zhang YE. TGF-beta receptor-activated p38 MAP kinase mediates Smad-independent TGF-beta responses. EMBO J. 2002;21:3749–3759. doi: 10.1093/emboj/cdf366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Boyer AS, et al. TGFbeta2 and TGFbeta3 have separate and sequential activities during epithelial-mesenchymal cell transformation in the embryonic heart. Dev. Biol. 1999;208:530–545. doi: 10.1006/dbio.1999.9211. [DOI] [PubMed] [Google Scholar]
- 58.Bhowmick NA, et al. Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol. Biol. Cell. 2001;12:27–36. doi: 10.1091/mbc.12.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Camenisch TD, et al. Temporal and distinct TGFbeta ligand requirements during mouse and avian endocardial cushion morphogenesis. Dev. Biol. 2002;248:170–181. doi: 10.1006/dbio.2002.0731. [DOI] [PubMed] [Google Scholar]
- 60.Miettinen PJ, Ebner R, Lopez AR, Derynck R. TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors. J. Cell Biol. 1994;127:2021–2036. doi: 10.1083/jcb.127.6.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 62.Citterio HL, Gaillard DA. Expression of transforming growth factor alpha (TGF alpha), epidermal growth factor receptor (EGF-R) and cell proliferation during human palatogenesis: an immunohistochemical study. Int. J. Dev. Biol. 1994;38:499–505. [PubMed] [Google Scholar]
- 63.Morabito CJ, Dettman RW, Kattan J, Collier JM, Bristow J. Positive and negative regulation of epicardial-mesenchymal transformation during avian heart development. Dev. Biol. 2001;234:204–215. doi: 10.1006/dbio.2001.0254. [DOI] [PubMed] [Google Scholar]
- 64.Thery C, Stern CD. Roles of kringle domain-containing serine proteases in epithelial-mesenchymal transitions during embryonic development. Acta Anat. (Basel). 1996;156:162–172. doi: 10.1159/000147844. [DOI] [PubMed] [Google Scholar]
- 65.Lamorte L, Royal I, Naujokas M, Park M. Crk adapter proteins promote an epithelial-mesenchymal-like transition and are required for HGF-mediated cell spreading and breakdown of epithelial adherens junctions. Mol. Biol. Cell. 2002;13:1449–1461. doi: 10.1091/mbc.01-10-0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Song W, Majka SM, McGuire PG. Hepatocyte growth factor expression in the developing myocardium: evidence for a role in the regulation of the mesenchymal cell phenotype and urokinase expression. Dev. Dyn. 1999;214:92–100. doi: 10.1002/(SICI)1097-0177(199901)214:1<92::AID-DVDY9>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 67.Thery C, Sharpe MJ, Batley SJ, Stern CD, Gherardi E. Expression of HGF/SF, HGF1/MSP, and c-met suggests new functions during early chick development. Dev. Genet. 1995;17:90–101. doi: 10.1002/dvg.1020170110. [DOI] [PubMed] [Google Scholar]
- 68.Nusrat A, et al. Hepatocyte growth factor/scatter factor effects on epithelia. Regulation of intercellular junctions in transformed and nontransformed cell lines, basolateral polarization of c-met receptor in transformed and natural intestinal epithelia, and induction of rapid wound repair in a transformed model epithelium. J. Clin. Invest. 1994;93:2056–2065. doi: 10.1172/JCI117200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mizuno S, et al. Hepatocyte growth factor prevents renal fibrosis and dysfunction in a mouse model of chronic renal disease. J. Clin. Invest. 1998;101:1827–1834. doi: 10.1172/JCI1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 71.Bar-Sagi D, Hall A. Ras and Rho GTPases: a family reunion. Cell. 2000;103:227–238. doi: 10.1016/s0092-8674(00)00115-x. [DOI] [PubMed] [Google Scholar]
- 72.Savagner P. Leaving the neighborhood: molecular mechanisms involved during epithelial-mesenchymal transition. Bioessays. 2001;23:912–923. doi: 10.1002/bies.1132. [DOI] [PubMed] [Google Scholar]
- 73.Kim K, Lu Z, Hay ED. Direct evidence for a role of beta-catenin/LEF-1 signaling pathway in induction of EMT. Cell Biol. Int. 2002;26:463–476. doi: 10.1006/cbir.2002.0901. [DOI] [PubMed] [Google Scholar]
- 74.Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 75.Li Y, Yang J, Dai C, Wu C, Liu Y. Role for integrin-linked kinase in mediating tubular epithelial to mesenchymal transition and renal interstitial fibrogenesis. J. Clin. Invest. 2003;112:503–516. doi:10.1172/JCI200317913. doi: 10.1172/JCI17913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Novak A, et al. Cell adhesion and the integrin-linked kinase regulate the LEF-1 and beta-catenin signaling pathways. Proc. Natl. Acad. Sci. U. S. A. 1998;95:4374–4379. doi: 10.1073/pnas.95.8.4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Espada J, Perez-Moreno M, Braga VM, Rodriguez-Viciana P, Cano A. H-Ras activation promotes cytoplasmic accumulation and phosphoinositide 3-OH kinase association of beta-catenin in epidermal keratinocytes. J. Cell Biol. 1999;146:967–980. doi: 10.1083/jcb.146.5.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Behrens J, et al. Loss of epithelial differentiation and gain of invasiveness correlates with tyrosine phosphorylation of the E-cadherin/beta-catenin complex in cells transformed with a temperature-sensitive v-SRC gene. J. Cell Biol. 1993;120:757–766. doi: 10.1083/jcb.120.3.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Smith DE, Franco del Amo F, Gridley T. Isolation of Sna, a mouse gene homologous to the Drosophila genes snail and escargot: its expression pattern suggests multiple roles during postimplantation development. Development. 1992;116:1033–1039. doi: 10.1242/dev.116.4.1033. [DOI] [PubMed] [Google Scholar]
- 80.Attisano L, Wrana JL. Signal transduction by the TGF-beta superfamily. Science. 2002;296:1646–1647. doi: 10.1126/science.1071809. [DOI] [PubMed] [Google Scholar]
- 81.He X. A wnt-wnt situation. Dev. Cell. 2003;4:791–797. doi: 10.1016/s1534-5807(03)00165-5. [DOI] [PubMed] [Google Scholar]
- 82.Sadot E, Geiger B, Oren M, Ben-Ze’ev A. Down-regulation of beta-catenin by activated p53. Mol. Cell. Biol. 2001;21:6768–6781. doi: 10.1128/MCB.21.20.6768-6781.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997;16:3797–3804. doi: 10.1093/emboj/16.13.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.von Kries JP, et al. Hot spots in beta-catenin for interactions with LEF-1, conductin and APC. Nat. Struct. Biol. 2000;7:800–807. doi: 10.1038/79039. [DOI] [PubMed] [Google Scholar]
- 85.Kim K, Hay ED. New evidence that nuclear import of endogenous beta-catenin is LEF-1 dependent, while LEF-1 independent import of exogenous beta-catenin leads to nuclear abnormalities. Cell Biol. Int. 2001;25:1149–1161. doi: 10.1006/cbir.2001.0799. [DOI] [PubMed] [Google Scholar]
- 86.Midgley CA, et al. APC expression in normal human tissues. J. Pathol. 1997;181:426–433. doi: 10.1002/(SICI)1096-9896(199704)181:4<426::AID-PATH768>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 87.Ip YT, Gridley T. Cell movements during gastrulation: snail dependent and independent pathways. Curr. Opin. Genet. Dev. 2002;12:423–429. doi: 10.1016/s0959-437x(02)00320-9. [DOI] [PubMed] [Google Scholar]
- 88.Zavadil J, et al. Genetic programs of epithelial cell plasticity directed by transforming growth factor-beta. Proc. Natl. Acad. Sci. U. S. A. 2001;98:6686–6691. doi: 10.1073/pnas.111614398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat. Genet. 2003;33:49–54. doi: 10.1038/ng1060. [DOI] [PubMed] [Google Scholar]
- 90.Blau HM, Blakely BT. Plasticity of cell fate: insights from heterokaryons. Semin. Cell Dev. Biol. 1999;10:267–272. doi: 10.1006/scdb.1999.0311. [DOI] [PubMed] [Google Scholar]
- 91.Emerson BM. Specificity of gene regulation. Cell. 2002;109:267–270. doi: 10.1016/s0092-8674(02)00740-7. [DOI] [PubMed] [Google Scholar]
- 92.Cremer T, Cremer C. Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat. Rev. Genet. 2001;2:292–301. doi: 10.1038/35066075. [DOI] [PubMed] [Google Scholar]
- 93.Mannervik M, Nibu Y, Zhang H, Levine M. Transcriptional coregulators in development. Science. 1999;284:606–609. doi: 10.1126/science.284.5414.606. [DOI] [PubMed] [Google Scholar]
- 94.Young BA, Gruber TM, Gross CA. Views of transcription initiation. Cell. 2002;109:417–420. doi: 10.1016/s0092-8674(02)00752-3. [DOI] [PubMed] [Google Scholar]
- 95.Peinado H, Quintanilla M, Cano A. Transforming growth factor beta 1 induces snail transcription factor in epithelial cell lines. Mechanisms for epithelial-mesenchymal transitions. J. Biol. Chem. 2003;278:21113–21123. doi: 10.1074/jbc.M211304200. [DOI] [PubMed] [Google Scholar]
- 96.Lie-Venema H, et al. Ets-1 and Ets-2 transcription factors are essential for normal coronary and myocardial development in chicken embryos. Circ. Res. 2003;92:749–756. doi: 10.1161/01.RES.0000066662.70010.DB. [DOI] [PubMed] [Google Scholar]
- 97.Macias D, Perez-Pomares JM, Garcia-Garrido L, Carmona R, Munoz-Chapuli R. Immunoreactivity of the ets-1 transcription factor correlates with areas of epithelial-mesenchymal transition in the developing avian heart. Anat. Embryol. (Berl.). 1998;198:307–315. doi: 10.1007/s004290050186. [DOI] [PubMed] [Google Scholar]
- 98.Ridinger K, et al. Clustered organization of S100 genes in human and mouse. . Biochim. Biophys. Acta. 1998; 1448:254–263. doi: 10.1016/s0167-4889(98)00137-2. [DOI] [PubMed] [Google Scholar]
- 99.Iwano M, et al. Conditional abatement of tissue fibrosis using nucleoside analogs to selectively corrupt DNA replication in transgenic fibroblasts. Mol. Ther. 2001;3:149–159. doi: 10.1006/mthe.2000.0251. [DOI] [PubMed] [Google Scholar]
- 100.Barraclough R. Calcium-binding protein S100A4 in health and disease. . Biochim. Biophys. Acta. 1998; 1448:190–199. doi: 10.1016/s0167-4889(98)00143-8. [DOI] [PubMed] [Google Scholar]
- 101.Grigorian M, et al. Tumor suppressor p53 protein is a new target for the metastasis-associated Mts1/S100A4 protein: functional consequences of their interaction. J. Biol. Chem. 2001;276:22699–22708. doi: 10.1074/jbc.M010231200. [DOI] [PubMed] [Google Scholar]
- 102.Chen H, et al. Binding to intracellular targets of the metastasis-inducing protein, S100A4 (p9Ka) Biochem. Biophys. Res. Commun. 2001;286:1212–1217. doi: 10.1006/bbrc.2001.5517. [DOI] [PubMed] [Google Scholar]
- 103.Ambartsumian N, et al. The metastasis-associated Mts1(S100A4) protein could act as an angiogenic factor. Oncogene. 2001;20:4685–4695. doi: 10.1038/sj.onc.1204636. [DOI] [PubMed] [Google Scholar]
- 104.Okada H, et al. Novel cis-acting elements in the FSP1 gene regulate fibroblast-specific transcription. Am. J. Physiol. 1998;275:306–314. doi: 10.1152/ajprenal.1998.275.2.F306. [DOI] [PubMed] [Google Scholar]
- 105.Tian YC, Fraser D, Attisano L, Phillips AO. TGF-{beta}1-mediated alterations of renal proximal tubular epithelial cell phenotype. Am. J. Physiol. Renal Physiol. 2003;285:F130–F142. doi: 10.1152/ajprenal.00408.2002. [DOI] [PubMed] [Google Scholar]
- 106.Thiery JP, Chopin D. Epithelial cell plasticity in development and tumor progression. Cancer Metastasis Rev. 1999;18:31–42. doi: 10.1023/a:1006256219004. [DOI] [PubMed] [Google Scholar]
- 107.Quilliam LA, Rebhun JF, Castro AF. A growing family of guanine nucleotide exchange factors is responsible for activation of Ras-family GTPases. Prog. Nucleic Acid Res. Mol. Biol. 2002;71:391–444. doi: 10.1016/s0079-6603(02)71047-7. [DOI] [PubMed] [Google Scholar]
- 108.Bernards A. GAPs galore! A survey of putative Ras superfamily GTPase activating proteins in man and Drosophila. . Biochim. Biophys. Acta. 2003; 1603:47–82. doi: 10.1016/s0304-419x(02)00082-3. [DOI] [PubMed] [Google Scholar]
- 109.Masszi A, et al. Central role for Rho in TGF-beta1-induced alpha-smooth muscle actin expression during epithelial-mesenchymal transition. Am. J. Physiol. Renal Physiol. 2003;284:F911–924. doi: 10.1152/ajprenal.00183.2002. [DOI] [PubMed] [Google Scholar]
- 110.Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- 111.Nobes CD, Hall A. Rho, rac and cdc42 GTPases: regulators of actin structures, cell adhesion and motility. Biochem. Soc. Trans. 1995;23:456–459. doi: 10.1042/bst0230456. [DOI] [PubMed] [Google Scholar]
- 112.Sander EE, ten Klooster JP, van Delft S, van der Kammen RA, Collard JG. Rac downregulates Rho activity: reciprocal balance between both GTPases determines cellular morphology and migratory behavior. J. Cell Biol. 1999;147:1009–1022. doi: 10.1083/jcb.147.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zondag GC, et al. Oncogenic Ras downregulates Rac activity, which leads to increased Rho activity and epithelial-mesenchymal transition. J. Cell Biol. 2000;149:775–782. doi: 10.1083/jcb.149.4.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cohnheim J. Über Entzündung und Eiterung. Virchows Arch. 1867;40:1–79. [Google Scholar]
- 115.Abe R, Donnelly SC, Peng T, Bucala R, Metz CN. Peripheral blood fibrocytes: differentiation pathway and migration to wound sites. J. Immunol. 2001;166:7556–7562. doi: 10.4049/jimmunol.166.12.7556. [DOI] [PubMed] [Google Scholar]
- 116.Huss R. Isolation of primary and immortalized CD34-hematopoietic and mesenchymal stem cells from various sources. Stem Cells. 2000;18:1–9. doi: 10.1634/stemcells.18-1-1. [DOI] [PubMed] [Google Scholar]
- 117.Bianco P, Robey PG. Marrow stromal cells. J. Clin. Invest. 2000;105:1663–1668. doi: 10.1172/JCI10413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bi LX, Simmons DJ, Hawkins HK, Cox RA, Mainous EG. Comparative morphology of the marrow sac. Anat. Rec. 2000;260:410–415. doi: 10.1002/1097-0185(20001201)260:4<410::AID-AR110>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 119.Simmons DJ. The in vivo role of bone marrow fibroblast-like stromal cells. Calcif. Tissue Int. 1996;58:129–132. doi: 10.1007/BF02526876. [DOI] [PubMed] [Google Scholar]
- 120.Weiss L, Geduldig U. Barrier cells: stromal regulation of hematopoiesis and blood cell release in normal and stressed murine bone marrow. Blood. 1991;78:975–990. [PubMed] [Google Scholar]
- 121.Zhang J, et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 2003;425:836–841. doi: 10.1038/nature02041. [DOI] [PubMed] [Google Scholar]
- 122.Krause DS, et al. Multi-organ, multi-lineage engraftment by a single bone marrow-derived stem cell. Cell. 2001;105:369–377. doi: 10.1016/s0092-8674(01)00328-2. [DOI] [PubMed] [Google Scholar]
- 123.Huss R, Hong DS, McSweeney PA, Hoy CA, Deeg HJ. Differentiation of canine bone marrow cells with hemopoietic characteristics from an adherent stromal cell precursor. Proc. Natl. Acad. Sci. U. S. A. 1995;92:748–752. doi: 10.1073/pnas.92.3.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Huss R. Perspectives on the morphology and biology of CD34-negative stem cells. J. Hematother. Stem Cell Res. 2000;9:783–793. doi: 10.1089/152581600750062228. [DOI] [PubMed] [Google Scholar]
- 125.Garrett DM, Conrad GW. Fibroblast-like cells from embryonic chick cornea, heart, and skin are antigenically distinct. Dev. Biol. 1979;70:50–70. doi: 10.1016/0012-1606(79)90006-x. [DOI] [PubMed] [Google Scholar]
- 126.Schor SL, Schor AM. Clonal heterogeneity in fibroblast phenotype: implications for the control of epithelial-mesenchymal interactions. Bioessays. 1987;7:200–204. doi: 10.1002/bies.950070503. [DOI] [PubMed] [Google Scholar]
- 127.Müller GA, Rodemann HP. Characterization of human renal fibroblasts in health and disease. I. Immunophenotyping of cultured tubular epithelial cells and fibroblasts derived from kidneys with histologically proven interstitial fibrosis. Am. J. Kidney Dis. 1991;17:680–683. doi: 10.1016/s0272-6386(12)80351-9. [DOI] [PubMed] [Google Scholar]
- 128.Dugina V, Alexandrova A, Chaponnier C, Vasiliev J, Gabbiani G. Rat fibroblasts cultured from various organs exhibit differences in alpha-smooth muscle actin expression, cytoskeletal pattern, and adhesive structure organization. Exp. Cell. Res. 1998;238:481–490. doi: 10.1006/excr.1997.3868. [DOI] [PubMed] [Google Scholar]
- 129.Alvarez RJ, et al. Biosynthetic and proliferative characteristics of tubulointerstitial fibroblasts probed with paracrine cytokines. Kidney Int. 1992;41:14–23. doi: 10.1038/ki.1992.3. [DOI] [PubMed] [Google Scholar]
- 130.Chang HY, et al. Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc. Natl. Acad. Sci. U. S. A. 2002;99:12877–12882. doi: 10.1073/pnas.162488599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chilosi M, et al. Aberrant Wnt/beta-catenin pathway activation in idiopathic pulmonary fibrosis. Am. J. Pathol. 2003;162:1495–1502. doi: 10.1016/s0002-9440(10)64282-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kalluri R, Danoff TM, Okada H, Neilson EG. Susceptibility to anti-glomerular basement membrane disease and Goodpasture syndrome is linked to MHC class II genes and the emergence of T cell-mediated immunity in mice. J. Clin. Invest. 1997;100:2263–2275. doi: 10.1172/JCI119764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Cosgrove D, et al. Collagen COL4A3 knockout: a mouse model for autosomal Alport syndrome. Genes Dev. 1996;10:2981–2992. doi: 10.1101/gad.10.23.2981. [DOI] [PubMed] [Google Scholar]
- 134.Anders H, Schlondorff D. Murine models of renal disease: possibilities and problems in studies using mutant mice. Exp. Nephrol. 2000;8:181–193. doi: 10.1159/000020667. [DOI] [PubMed] [Google Scholar]
- 135.Janssen U, Phillips AO, Floege J. Rodent models of nephropathy associated with type II diabetes. J. Nephrol. 1999;12:159–172. [PubMed] [Google Scholar]
- 136.Diamond JR, Ricardo SD, Klahr S. Mechanisms of interstitial fibrosis in obstructive nephropathy. Semin. Nephrol. 1998;18:594–602. [PubMed] [Google Scholar]
- 137.Okada H, et al. Progressive renal fibrosis in murine polycystic kidney disease: an immunohistochemical observation. Kidney Int. 2000;58:587–597. doi: 10.1046/j.1523-1755.2000.00205.x. [DOI] [PubMed] [Google Scholar]
- 138.Okada H, Strutz F, Danoff TM, Kalluri R, Neilson EG. Possible mechanisms of renal fibrosis. Contrib. Nephrol. 1996;118:147–154. doi: 10.1159/000425088. [DOI] [PubMed] [Google Scholar]
- 139.Breen E, Falco VM, Absher M, Cutroneo KR. Subpopulations of rat lung fibroblasts with different amounts of type I and type III collagen mRNAs. J. Biol. Chem. 1990;265:6286–6290. [PubMed] [Google Scholar]
- 140.Goldring SR, Stephenson ML, Downie E, Krane SM, Korn JH. Heterogeneity in hormone responses and patterns of collagen synthesis in cloned dermal fibroblasts. J. Clin. Invest. 1990;85:798–803. doi: 10.1172/JCI114506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Jelaska A, Strehlow D, Korn JH. Fibroblast heterogeneity in physiological conditions and fibrotic disease. Springer Semin. Immunopathol. 1999;21:385–395. [PubMed] [Google Scholar]
- 142.Rossert RA, Chen SS, Eberspaecher H, Smith CN, De Crombrugghe B. Identification of a minimal sequence of the mouse pro-a1(I) collagen promoter that confers high-level osteoblast expression in transgenic mice and that binds a protein selectively present in osteoblasts. Proc. Natl. Acad. Sci. U. S. A. 1996;93:1027–1031. doi: 10.1073/pnas.93.3.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zeisberg M, Strutz F, Muller GA. Role of fibroblast activation in inducing interstitial fibrosis. J. Nephrol. 2000;13(Suppl. 3):S111–S120. [PubMed] [Google Scholar]
- 144.Serini G, Gabbiani G. Mechanisms of myofibroblast activity and phenotypic modulation. Exp. Cell Res. 1999;250:273–283. doi: 10.1006/excr.1999.4543. [DOI] [PubMed] [Google Scholar]
- 145.Tang WW, et al. Platelet-derived growth factor-BB induces renal tubulointerstitial myofibroblast formation and tubulointerstitial fibrosis. Am. J. Pathol. 1996;148:1169–1180. [PMC free article] [PubMed] [Google Scholar]
- 146.Ng YY, et al. Tubular epithelial-myofibroblast transdifferentiation in progressive tubulointerstitial fibrosis in 5/6 nephrectomized rats. Kidney Int. 1998;54:864–876. doi: 10.1046/j.1523-1755.1998.00076.x. [DOI] [PubMed] [Google Scholar]
- 147.Eyden B. The myofibroblast: an assessment of controversial issues and a definition useful in diagnosis and research. Ultrastruct. Pathol. 2001;25:39–50. doi: 10.1080/019131201300004672. [DOI] [PubMed] [Google Scholar]
- 148.Yang J, et al. Disruption of tissue-type plasminogen activator gene in mice reduces renal interstitial fibrosis in obstructive nephropathy. J. Clin. Invest. 2002;110:1525–1538. doi:10.1172/JCI200216219. doi: 10.1172/JCI16219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Yang J, Liu Y. Blockage of tubular epithelial to myofibroblast transition by hepatocyte growth factor prevents renal interstitial fibrosis. J. Am. Soc. Nephrol. 2002;13:96–107. doi: 10.1681/ASN.V13196. [DOI] [PubMed] [Google Scholar]
- 150.Zeisberg M, et al. Bone morphogenic protein-7 inhibits progression of chronic renal fibrosis associated with two genetic mouse models. Am. J. Physiol. Renal Physiol. 2003;285:F1060–F1067. doi: 10.1152/ajprenal.00191.2002. [DOI] [PubMed] [Google Scholar]
- 151.Kalluri R, Zeisberg M. Exploring the connection between chronic renal fibrosis and bone morphogenic protein-7. Histol. Histopathol. 2003;18:217–224. doi: 10.14670/HH-18.217. [DOI] [PubMed] [Google Scholar]
- 152.Morrissey J, et al. Bone morphogenetic protein-7 improves renal fibrosis and accelerates the return of renal function. J. Am. Soc. Nephrol. 2002;13(Suppl. 1):S14–S21. [PubMed] [Google Scholar]
- 153.Wang S, et al. Bone morphogenic protein-7 (BMP-7), a novel therapy for diabetic nephropathy. Kidney Int. 2003;63:2037–2049. doi: 10.1046/j.1523-1755.2003.00035.x. [DOI] [PubMed] [Google Scholar]
- 154.Gould SJ, Vrba ES. Exaptation — a missing term in the science of form. Paleobiology. 1982;8:4–15. [Google Scholar]