


Abstract
Huntington disease (HD) is a neurodegenerative disorder that predominantly affects neurons of the forebrain. We have applied the Illumina massively parallel sequencing to deeply analyze the small RNA populations of two different forebrain areas, the frontal cortex (FC) and the striatum (ST) of healthy individuals and individuals with HD. More than 80% of the small-RNAs were annotated as microRNAs (miRNAs) in all samples. Deep sequencing revealed length and sequence heterogeneity (IsomiRs) for the vast majority of miRNAs. Around 80–90% of the miRNAs presented modifications in the 3′-terminus mainly in the form of trimming and/or as nucleotide addition variants, while the 5′-terminus of the miRNAs was specially protected from changes. Expression profiling showed strong miRNA and isomiR expression deregulation in HD, most being common to both FC and ST. The analysis of the upstream regulatory regions in co-regulated miRNAs suggests a role for RE1-Silencing Transcription Factor (REST) and P53 in miRNAs downregulation in HD. The putative targets of deregulated miRNAs and seed-region IsomiRs strongly suggest that their altered expression contributes to the aberrant gene expression in HD. Our results show that miRNA variability is a ubiquitous phenomenon in the adult human brain, which may influence gene expression in physiological and pathological conditions.
INTRODUCTION
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disease caused by a CAG repeat expansion in the HTT (huntingtin) gene that encodes HTT. Although HTT is ubiquitously expressed, patients with HD show predominantly central nervous system (CNS) manifestations, including abnormal motor symptoms, cognitive decline and psychiatric disturbances. HD is characterized by widespread mRNA misregulation, especially in the striatum and cortical regions (1). Such deregulation is the result, in part, of aberrant nuclear localization of the transcriptional repressor RE1-Silencing Transcription Factor (REST) (2,3). This REST nuclear translocation leads to increased transcriptional repression of BDNF, a REST target gene, therefore compromising striatal neuronal survival. In addition to protein-coding genes, recent reports have shown that REST controls the expression of neuronal large (macroRNAs) and small [microRNAs (miRNAs)] non-coding RNAs (4,5), with various roles in gene expression modulation. Therefore, aberrant expression of non-coding RNAs may have indirect consequences in HD gene expression deregulation.
miRNAs are small non-coding RNAs that participate in the post-transcriptional regulation of gene expression through sequence complementarity to open reading frame (ORF) and 3′untranslated regions (UTRs) of messenger RNAs (mRNAs). This binding leads to target mRNA degradation or translational inhibition. miRNAs play important roles in organogenesis, metabolism and neuronal development. Many miRNAs are selectively expressed in the brain and appear to regulate fundamental neuronal processes, including the maintenance of the neuronal transcriptome, dendrite outgrowth and synaptic plasticity (6). In addition, miRNAs have been shown to be essential for neuronal survival and alterations in miRNA function have been involved in a number of neurodegenerative processes (7,8). These alterations comprise aberrant expression of miRNAs in the brain of patients with neurodegenerative diseases (9–15) and single-nucleotide polymorphisms (SNPs) (16) or a point mutation (14) in the target 3′UTR NA binding site.
Two main findings point to the possibility that small-RNA silencing-dependent mechanisms participate in HD neuropathology. First, increased repression by REST leads to changes in the expression of specific neuronal miRNAs in HD patients and HD mouse models (17,18). Second, HTT interacts with Argonaute proteins, which are key members of the RNA-induced silencing complex (RISC). This interaction with the mutant HTT leads to a reduced reporter gene silencing activity compared with the wild-type HTT (19). Overall, these data strongly suggest that alterations in miRNA-mediated post-transcriptional regulation could be a mechanism contributing to mRNA expression deregulation in HD.
Although alterations in miRNA expression have been reported in brain samples of HD patients, a deep characterization of miRNA profiling and sequence modifications is lacking. The available high-throughput sequencing technologies allow detection of the current reference miRNA sequences residing in the miRBase and novel miRNAs. This approach has also the potential to uncover variations in the mature miRNA length, and enzymatic modifications such as RNA editing and 3′ nucleotide additions. The Illumina sequencing platform yields several million sequences from a single flow cell lane, providing a high resolution. In the present study, we used Illumina deep sequencing to extensively profile and identify disease-specific changes of small non-coding RNAs that occur in the frontal cortex (FC) and the striatum (ST), two brain areas affected in HD. Our results show a strong deregulation of miRNA expression in HD and highlight the potential importance of the large number of modified mature miRNA sequences in the physiology and pathology of the adult brain.
METHODS
Human brain samples
Patients with HD had suffered from a familial degenerative disease characterized by involuntary muscle movement, difficulties with speech, trouble swallowing and walking and excessive blinking. This was followed by abnormal behavior and cognitive impairment, continuous falls and hypokinesia, leading to terminal akinetic-rigid disorder. Genetic studies showed increased CAG repetitions in the HTT gene in affected relatives or an increased number of CAG nucleotides in affected individuals. Neuroimaging [computed tomography (CT) or magnetic resonance imaging (MRI) scans] disclosed severe atrophy of the caudate and putamen and moderate to severe cortical atrophy during the course of the disease. Based on the clinical and genetic data, a diagnosis of HD was made during life in every case.
Brain samples corresponding to the frontal cortex (FC) and the striatum (ST) (dorsal caudate) of HD patients and controls were obtained from the Institute of Neuropathology and the University of Barcelona Brain Bank, after the informed consent of the patients or their relatives and the approval of the local ethics committee had been given (Supplementary Table S1). Cases with and without clinical neurological disease were processed in the same way, following the same sampling and staining protocols. Gross atrophy was seen in HD cases. At autopsy, half of each brain was fixed in 4% buffered formalin, whereas the other half was cut in coronal sections 1 cm thick, frozen on dry ice, and stored at −80°C until use. The postmortem delay was between 3 and 17 h. The neuropathological study was carried out on dewaxed 4-μm-thick paraffin sections of the frontal lobe (area 8), primary motor, primary sensory, parietal, temporal superior, temporal inferior, anterior cingulated, anterior insular and primary and associative visual cortices; entorhinal cortex and hippocampus; head of the caudate, medial caudate, putamen and globus pallidus; medial and posterior thalamus; subthalamus; Meynert nucleus; amygdala; midbrain (two levels); pons and medulla oblongata; and cerebellar cortex and dentate nucleus. The sections were stained with hematoxylin and eosin and Klüver–Barrera and processed for immunohistochemistry to glial fibrillary acidic protein (GFAP), CD68 and Licopericum esculentum lectin for microglia; Aβ-amyloid; pan-tau, AT8 tau and phosphorylation-specific tau Thr181, Ser202, Ser214, Ser262, Ser396 and Ser422; αB-crystallin; α-synuclein; and ubiquitin.
The neuropathological examination in HD cases revealed severe atrophy of the caudate and putamen, the atrophy being more marked in the dorsal border, together with cerebral cortical atrophy and enlargement of the lateral ventricles. This was accompanied by marked neuronal loss and astrocytic gliosis in the caudate and putamen, globus pallidus and cerebral cortex. Individual neurons in the cerebral cortex and striatum exhibited ubiquitin-positive intranuclear inclusions typical of diseases with CAG triplet expansions and ubiquitin-positive neuritic processes in the same regions. After neuropathological examination, HD cases were categorized as stage 4 following Vonsattel classification (20,21).
RNA extraction and sequencing
Total RNA was isolated from ∼10 mg of the different brain areas using the miRNeasy kit (Qiagen) that preserves small RNAs. Equal amounts of total RNA of two equivalent patients and brain areas were mixed, giving rise to a total of four samples (C-FC, HD-FC, C-ST and HD-ST) showing RNA integrity numbers (RIN) between 6 and 7. A total of 5 µg of small-RNA-enriched total RNA was processed for Illumina sequencing. Briefly, the processing by Illumina consisted of the following successive steps (22,23): acrylamide gel purification of RNA bands corresponding to the size range 18–30 nt, ligation of 5′ and 3′ adapters to the RNA in two separate subsequent steps each followed by acrylamide gel purification, cDNA synthesis followed by acrylamide gel purification, and a final step of polymerase chain reaction (PCR) amplification to generate template library for cluster generation on a flow cell. After hybridization of the sequencing primer libraries were sequenced it using a GAII instrument (Illumina). The reads generated were 36-nt long and were analysed using SeqBuster pipeline (24).
Pre-analysis of human brain sequencing data
Pre-analysis consists in the recognition and removal of the adapter and the annotation of the sequences. Pre-analysis was performed using a stand-alone version provided in http://estivill_lab.crg.es/seqbuster (24). Out of a total of several million reads, we discarded any reads without a minimum of 10-nt linker subsequence directly adjoining the insert, showing two or less mismatches. Following the application of this filter, populations of 5 914 073; 5 561 686; 5 125 116 and 5 855 660 reads were recovered in C-FC, C-ST, HD-FC and HD-ST samples, respectively. Taking into account the unique sequences with more than three counts, the total number of reads were 5 827 761; 5 466 923; 5 041 667 and 5 759841. Then sequences were mapped to human pre-miRNA and mature miRNA databases provided in the miRBase (http://microrna.sanger.ac.uk/sequences/, Release 14), and mRNA, ncRNA, repeats and genome databases available at (http://hgdownload.cse.ucsc.edu/goldenPath/hg18/bogZips/), using Mega BLAST. For precursor miRNA annotation the following parameters were configured (24): one mismatch, 3 nt in the 3′-addition variants and the priority degree equal to 3. For miRNA and miRNA* annotation the following parameters were configured: one mismatch, 3 nt in the 3′ or 5′-trimming variants, 3 nt in the 3′-addition variants, a priority degree to 1 and 2 for miRNA and miRNAs* databases, respectively, and the parental database was the precursor miRNA database. These options permitted annotations of the following types of alignment: (i) perfect match, where the sequence is completely identical to the reference sequence; (ii) trimming at the 3′-end of the reference miRNA sequence, which is a miRNA variant several nucleotides shorter or longer that matches to the mature or precursor reference sequence, respectively; (iii) trimming at the 5′-end of the sequence, an analogous case focused on the 5′-end of the miRNA; (iv) nucleotide additions at the 3′-end of the sequence and (v) nucleotide substitutions, showing nucleotide changes with respect to the reference sequence. The parameters for the alignment to the mRNA and genome databases allowed as much as one mismatch and up to three nucleotide additions in the 3′-terminus. The priority parameter was 4, 5, 6 and 7 for mRNA, ncRNA, repeats and genome databases, respectively (24).
Analysis of miRNA variability (IsomiRs)
For deep characterization of miRNA, variants we applied several filters. First, the sequences considered in the analysis presented a frequency above 3. Second, 10 was chosen as the ‘Contribution Cut-Off’ parameter, meaning that every isomiR considered in the analysis contributes by more than a 10% to the total number of variants annotated in the same miRNA locus. Third, we applied the Z-score option to exclude sequencing errors as the possible cause of the nucleotide changes observed in some variants (24). Annotated miRNAs and IsomiRs with the corresponding counts are available at http://estivill_lab.crg.es/Marti_etal/. Four files are available containing the data set of control FC (C_FC), control ST (C_ST), HD FC (HD_FC) and HD FC (HD_ST) samples.
Novel miRNA prediction
The ‘human miRNA prediction’ package in the SeqBuster pipeline (24) uses the algorithm described in the microPred pipeline (25) that is based on a non-comparative computational method for the effective identification of pre-miRNAs among the hairpin secondary structures predicted from the human genome. A test input file has to be provided containing all sequences for which the analysis is going to be performed.
SNP analysis in miRNAs
The ‘SNP analysis’ package is offered by SeqBuster in the ‘Download’ section of the web server to be run in the local machine (24). This package contains genomic coordinates for available SNPs (https://hgdownload.cse.ucsc.edu/downloads/html). The algorithm produces a table showing all the sequences that have a nucleotide substitution event at the same position where an SNP has been detected.
Differential expression analysis
The sequencing performance in the samples to be compared was evaluated using the ‘Sequencing Capacity’ package in the ‘Basic Analysis’ module of SeqBuster with default parameters (24). In this package, the Willcoxon test was applied to determine statistically significant differences in the frequency distribution between samples. Two types of differential expression analyses were performed, that included different types of sequences: (i) the different miRNA loci were containing all the sequences mapping to each locus; and (ii) 5′-trimming and nucleotide substitution IsomiRs affecting the seed region of the miRNA. To perform these analyses several options and filters were applied. First, the frequency was normalized to reads per million (RPM). Second, the Z-test (26) was applied to show statistical significance in the differential expression. Third, the Benajmini and Hochberg method was applied to correct the P-value assigned by the Z-test. Fourth, in analyzing the seed region variants, 10 was chosen as the ‘Contribution Cut-Off’ value (24). Fifth, we applied the Z-score option to exclude sequencing errors as the possible cause of the nucleotide changes observed in the isomiR (27). In the output resulting from the analysis we only showed sequences with a total count contribution above 50, considered as the sum of the frequencies of the two samples being compared.
Microarray preparation and statistical analysis
Agilent Human miRNA microarrays (G4470B), containing 13 737 probes, were used to analyze independent samples from the FC and the ST of four control individuals and four HD patients (Supplementary Table S1). Briefly, 500 ng of total RNA from each sample were chemically labeled by dephosphorylating using calf intestinal alkaline phosphatase (CIP) and ligating Cyanine3-pCp by a T4-RNA ligase using Agilent miRNA Complete Labeling and Hyb Kit (p/n5190-0456). Labeled samples were dried and resuspended in 18 µl of nuclease-free water and co-hybridized with in situ hybridization buffer (Agilent) for 20 h at 55°C and washed at room temperature for 5 min in Gene Expression Wash Buffer 1 (Agilent) and 5 min at 37°C in Gene Expression Wash Buffer 2 (Agilent). The images were generated on a confocal microarray scanner (G2505B, Agilent) at 5 um resolution and quantified using Feature Extraction (Agilent). Extracted log2-transformed intensities were quantile normalized to make all data comparable.
To assess differential expression, significance analysis of microarrays (SAM) was used (28). Results of SAM analysis were corrected for multiple testing according to the false discovery rate (FDR) method (29). Probes with FDR-adjusted P-value below 5% and additionally a fold change exceeding 1,2 in absolute value were selected as the relevant ones. Final relative expression values were computed by taking the median log2 ratio of the respective probes per each miRNA. All statistical analyses were performed with Bioconductor project (version 2.3) in the R statistical environment (30).
Quantitative real-time PCR
A total of 14 differentialy expressed miRNAs found concordant both by sequencing and microarray analyses data were confirmed by real-time quantitative reverse transcription PCR (qRT-PCR) using TaqMan® MicroRNA assays (Applied Biosystems) in striatal samples of five control individuals and five HD patients. Total RNA was used as a template to generate specific first-strand complementary DNA (cDNA) for each miRNA using Taqman specific miRNA retrotranscription kit. qRT-PCR was performed following manufacturer’s instructions using the ABI PRISM 7900HT sequence detection system (Applied Biosystems). Relative quantification was calculated with the 2 −ΔΔCt method (31) using the snRNA, RNU58B as an endogenous control. Statistical significance and fold change were calculated using linear mixed-effects models (LMM) that contemplate random effects accounting for the different sources of variation: technical (qRT-PCRs) and biological replicates (32).
Functional enrichment analysis
We used the TargetScan algorithm that predicts biological targets by searching for the presence of conserved 8-mer and 7-mer sites that match the seed region of each isomiR (33,34). We used the TargetScan custom option (www.targetscan.org) to predict mRNA targets of the seed region IsomiRs differentialy expressed between libraries. The mRNA targets for the corresponding differentialy expressed reference miRNAs were identified through the SeqBuster ‘Target prediction’ module using the TargetScan algorithm. Since the cooperative action of multiple miRNAs can be multiplicative and sometimes synergistic (33), mRNAs with more predicted target sites for co-expressed IsomiRs or reference-miRNAs should be more drastically affected. Therefore, we considered targets predicted by three or more HD-FC or HD-ST co-regulated IsomiRs or reference miRNAs. Then, ingenuity pathway analysis (IPA) was used to explore subsets of genes exclusively targeted by HD-co-regulated IsomiRs and those affected by the IsomiRs and reference-miRNAs. In the IPA filters, we only considered the brain and CNS cell lines as target tissues. The P-value associated with a biological process is calculated with the right-tailed Fisher’s exact test, considering the number of functions/pathways/lists eligible molecules that participate in that annotation, the total number of knowledge base molecules known to be associated with that function, the total number of functions/pathways/lists eligible molecules and the total number of genes in the Reference Set (IPA tutorial).
Analysis of the co-regulated miRNA promoter regions
We compared and characterized the promoter regions of miRNAs with similar expression patterns, using the algorithm described by Blanco et al. (35). The package requires a test file containing a list of the co-regulated miRNAs and a list with all the expressed miRNAs. To identify a possible significant enrichment of cis-regulatory elements in co-regulated miRNAs the algorithm assigns a P-value using the permutation-based simulation.
RESULTS
Overview of small RNA sequencing and annotation
In the present study, we chose to characterize small RNAs in the FC and the dorsal caudate nucleus of the ST, two brain areas strongly affected in HD that showed a differential pattern of neuronal loss. Total RNA was isolated from these brain areas in two HD patients and two nonaffected individuals showing similar quality standards (see ‘Methods’ section). Equivalent amounts of RNA were mixed from the FC or the ST, which resulted in four small RNA libraries from the FC and the ST of two patients with HD and two nonaffected individuals.
Sequencing of small RNAs yielded 6 788 286; 5 934 411; 6 495 715; and 6 776 627 unfiltered sequence reads from control FC (C-FC), HD-FC (HD-FC), control ST (C-ST) and HD ST (HD-ST), respectively. Any sequence observed more than three times was considered a reliable representation of a small RNA molecule. After removal of reads containing ambiguous base calls, unique sequences remained between 60 669 (C-FC), 61 416 (HD-FC) 68 168 (C-ST) and 72 475 (HD-ST) (Table 1). After mapping to the genome the total of unique sequences represented between 5 000 000 and 6 000 000 reads. Most reads mapped the genome, with <9–16% of the sequences not being identified (Table 1).
Table 1.
Distribution of the different types of sequences in control and Huntington disease brain samples
| Database | Data | C-FC | HD-FC | C-ST | HD-ST |
|---|---|---|---|---|---|
| miRNAs | Frequency | 5 035 324 | 4 019 202 | 4 558 525 | 4 574 587 |
| Unique sequences | 20 925 | 19 683 | 23 198 | 21 102 | |
| miRNA* | Frequency | 21 544 | 27 039 | 6729 | 23 193 |
| Unique sequences | 840 | 829 | 405 | 837 | |
| Pre-miRNAs | Frequency | 19 061 | 19 283 | 18 189 | 24 084 |
| Unique sequences | 1036 | 1347 | 1083 | 1654 | |
| mRNAs | Frequency | 100 651 | 91 681 | 126 949 | 87 436 |
| Unique sequences | 4582 | 3857 | 4671 | 4108 | |
| Predicted miRNAs | Frequency | 108 921 | 77 386 | 111 211 | 70 278 |
| Unique sequences | 1094 | 846 | 1083 | 883 | |
| Sn-RNAs+ | Frequency | 0 | 225 | 103 | 280 |
| Unique sequences | 0 | 45 | 23 | 56 | |
| tss+ | Frequency | 0 | 37 | 0 | 33 |
| Unique sequences | 0 | 7 | 0 | 9 | |
| Genome | Frequency | 871 | 1051 | 321 | 1403 |
| Unique sequences | 87 | 208 | 57 | 253 | |
| Unmapped | Frequency | 541 389 | 805 763 | 644 896 | 978 547 |
| Unique sequences | 32 105 | 34 594 | 37 649 | 43 573 | |
| Total | Frequency | 5 827 761 | 5 041 667 | 5 466 923 | 5 759 841 |
| Unique sequences | 60 669 | 61 416 | 68 168 | 72 475 |
Sequences showing a frequency >3 are considered.
miRNA*, passenger strand of the miRNA duplex that is less stable; +, sense strand; Sn, small non-coding RNAs; tss, transcription start site small RNA.
Sequences were annotated based on the overlap with publicly available genome annotations, including miRNA, tRNAs, rRNAs, other small RNAs and genomic repeats. miRNAs were the most abundant class of small RNAs (80–85%), spanning the entire range of expression. Sequences deriving from 372–392 distinct miRNA genes were identified in the different libraries, showing a sequence length distribution with a sharp peak in the 22 nt length (Supplementary Figure S1). Let-7 was the most abundant family of miRNAs, with sequence counts up to 600 000. The top conserved miRNAs occupied 40–50% of the total miRNA reads (Supplementary Table S2). However, human-specific miRNAs were expressed at a significantly lower level, constituting around 0.1% of the total number of reads. This agrees with previous reports showing that the miRNA abundance is linked to the extent of conservation (36).
To identify putative new miRNAs among the sequences mapped onto the genome, we used the microPred pipeline (25). This is a noncomparative computational tool developed for the identification of miRNAs from a pool of sequenced RNA transcripts resulting from deep sequencing. Between 846 and 1094 different sequences in each sample were found to statistically fulfill the structural features that define a miRNA, including the ability to form hairpin secondary structures that showed Dicer processing features. Approximately two-thirds of the sequences corresponded to slight length variations of a single representative. Therefore, 226–375 new putative miRNAs were found in each sample, being 85 common to all samples (Supplementary Table S3). Functional studies are required to demonstrate that these new putative miRNAs can regulate gene expression through the RISC silencing machinery.
miRNA variability is ubiquitous in control and HD brain samples
Variants of the mature miRNA have been referred to as IsomiRs (37). In the present study, 20 000–23 000 different variants (frequency >3) were found in every sample. Furthermore, most of the sequences mapping onto miRNA database (reference miRNAs and IsomiRs) showed from 3–50 counts (Supplementary Figure S2). It has been proposed that much of the miRNA modifications can be explained by variability in either Dicer1 or Drosha cleavage positions at the 5′-end or 3′-end of the miRNA-precursor (5′- or 3′-trimming variants) during miRNA biogenesis (37). Another type of variability is related with pri-miRNA post-trancriptional editing as a consequence of adenosine or cytidine desaminase activities (nucleotide substitution variants), which can be detected at different positions of the mature miRNA (38–42). Besides, nucleotide additions at the 3′-end of the mature miRNA (3′-addition variants) have also been reported as the most common form of miRNA enzymatic modification (37,41). The ∼380 miRNAs detected in every sample displayed IsomiRs and only two and five miRNAs remained invariable in FC and ST libraries respectivelly (Table S4). The frequency of these invariable miRNAs was very low, suggesting that the lack of variants may be due to the low abundance rather than a selective protection from modifications.
In all the libraries, variability was detected mainly as the result of modifications in the cleavage position at the 3′-end of the pri-miRNA. In this case, a considerable proportion of miRNAs presented 2 or more 3′-trimming variants (Figure 1A). However, for the 5′-trimming variants, additions in the 3′-end of the mature miRNA, or single-nucleotide substitutions with respect to the reference sequence, the majority of the miRNAs presented IsomiRs with only one change.
Figure 1.

Histogram displaying the percentage of miRNAs with different types of IsomiRs (5′-trimming, 3′-trimming, 3′-additions and nucleotide substitutions), in C-FC (1), HD-FC (2), C-ST (3) and HD-ST (4) small RNA libraries. In this analysis, IsomiRs with frequencies >10 were considered. For every type of variability, the histogram shows the proportion of miRNAs presenting one (white), two (gray) or more than two (black) IsomiRs (A). An example of a miRNA (miR-124) displaying two 5′-trimming variants (T1 and T2) is shown below. Arrows in the precursor miRNA (Pre-miR-124) point the beginning of each isomiR. The histogram in (B) shows the abundance of the IsomiRs with respect to the corresponding reference miRNA, for every type of variability. The five gray color intensities from dark to light indicate the frequency of the isomiR with respect to the reference miRNA: <20%; 20–40%; 40–60%; 60–80% and >80%. The example below shows a 3′-trimming variant (T1) with a relative abundance with respect to the reference miRNA (miR-103) of around 40%. The arrow in Pre-miR-103 points the site where the 3′-trimming variant ends.
Taking into account the possible physiological significance of an isomiR, it should be considered how the new variant affects the regulation of a target mRNA. miRNAs interact with their mRNA targets by imperfect base pairing following a set of rules that have been identified in experimental and bioinformatic analyses (43,44). These include perfect binding of the 5′-seed (nt 2–8), mismatches at the central region of the miRNA (nt 10–12) and a reasonable complementariety with the 3′ half of the miRNA, specially positions 13–16. It should be stressed that these requirements are not completely understood as target prediction algorithms have very high false-negative and false-positive rates. However, the requirements for target recognition favor the possibility that the identified miRNA variants target new mRNAs. In addition, since the variant and the reference miRNA sequences are very similar, it is likely that both small RNAs compete for a number of target mRNAs. Thus, to evaluate the possible relevance of each variant, we analyzed the frequency of each isomiR with respect to that of reference miRNA (Figure 1B). The majority of the nucleotide substitution or 5′-trimming variants showed a low frequency compared to that of the reference miRNAs. However, variants affecting the 3′-terminus of miRNAs, especially the 3′-trimming variants, showed a variable proportion with respect to reference miRNAs. These results highlight the possibility that the deregulation in the expression of nucleotide substitution and 5′-trimming IsomiRs could have more dramatic physiological consequences. The large abundance of some of the variants compared with the correspondent reference miRNA (over 80%) indicates that these IsomiRs should be considered as the reference miRNA, at least in the human brain. In some cases, such as miR-1291, miR-320c and miR-1260, the reference miRNA sequence was not even detected (see examples in Supplementary Table S5).
To gain insights into the possible relevance of the IsomiRs as small silencing RNAs we evaluated the presence of such variants in Ago immunoprecipitates (Ago-IP). We analyzed publicly available sequencing small-RNAs data sets (GEO database at http://www.ncbi.nlm.nih.gov/geo/) of human HEK293 Ago1- and Ago2-IP and mouse embryonic brain Ago1-IPs. We detected the four types of IsomiRs in all the Ago-IPs (Supplementary Figure S3). Furthermore, some of the IsomiRs found in the Ago-IPs were also detected in the sequenced brain samples (see examples in Supplementary Figure S3). In addition, the 3′-trimming and the 3′-addition variants were the most abundant, similarly to the analysis of total small-RNAs in the human brain samples (Figure 1). These results show that IsomiRs are incorporated into the Ago silencing complexes and strongly support the idea that the variants are functional.
We analyzed the positions and nucleotides involved in the different types of variants. Most 3′-trimming variants involved positions −1 (1 nt upstream) and +1 (1 nt downstream) of the 3′-end of the reference miRNA, while the majority of the 5′-trimming variants involved the position +1 of the 5′-terminus of the reference miRNA, matching the miRNA precursor (Figure 2A and B). Different nucleotides at different positions were preferentially involved in 3′- or 5′-trimming variants, which suggests a predilection of the dicing machinery for a nucleotide in a selective position. In addition, the vast majority of 3′-addition variants consisted in a single A or U addition to the 3′-end of the mature miRNA (Figure 2C), in accordance with other reports (37,41).
Figure 2.

Percentage of miRNAs with 5′-trimming variants (A), 3′-trimming variants (B) and 3′-addition variants (C) in C-FC (1), HD-FC (2), C-ST (3) and HD-ST (4) small RNA samples, according to the type and position of the nucleotide involved. In this analysis, IsomiRs with frequencies >10 were considered. Pre-miRNA slicing occurring at different positions upstream or downstream of the reference miRNA extremes (5′- and 3′-) is indicated by −3 to –1 and +1 to +3, respectively (A, B). In the 3′-addition variants (C), 1, 2 and 3 indicate the number of nucleotides added to the 3′-terminus of the miRNA. In the upper part of each panel, a scheme of miRNA is shown. The part of the sequence in black corresponds to the mature reference miRNA and the part in gray highlights the precursor sequence (A, B) or the nucleotides added in the 3′-addition variants (C). The orange arrows and box highlight the nucleotides being examined in the histogram below. The size of the arrow is proportional to the percentage trimming variants at the signaled position. The color pattern of the bars indicates the nucleotides involved in the trimming variants at every position: A (red), U (blue), C (green) and G (orange). Significant bias toward a nucleotide involved in the trimming variant is shown with a white asterisk. Below each histogram an example of a variant is shown in a gray box. The attached gray arrow indicates the specific type of IsomiR, within the histogram, being analyzed.
An increasing number of studies have revealed nucleotide substitutions as a possible source of IsomiRs, which have been shown as the result of post-transcriptional modifications or RNA-editing, rather than sequencing technical artifacts (45–48). Illumina sequencing error rates have been previously calculated for every position of the sequence and every type of nucleotide change, in genomic libraries (27). Therefore, in the analysis of the nucleotide substitution variants, we only considered the nucleotide modifcations significantly different from these error rates (see ‘Methods’ section). Under these conditions, ∼35% of the miRNAs detected in each library presented nucleotide substitution variants (Figure 1). Table S6 shows the list of the nucleotide modiffications at different positions of the reference miRNA, commonly detected in the four sequenced samples. We analyzed if these nucleotide substitution variants could correspond to SNPs. To this end, we compared the chromosome coordinates of all available SNPs with those of the nucleotide substitution variants. We found that miR-219-5p presented variants (frequency ≥10) that matched with an SNP in dbSNP; however, the frequency of these IsomiRs was very low compared with the reference miRNA (Supplementary Table S7). This miRNA presented additional patterns of nucleotide substitution at the same positions. This suggests that the vast majority of the nucleotide substitution IsomiRs are likely post-transcriptional.
Nucleotide modifications affecting the seed region (nucleotides 2–8) are of particular interest given the essential role of this fragment in target gene recognition and expression regulation. The majority of the miRNAs with IsomiRs in the seed region, presented nt-modifications in positions 5–8 (Figure 3). In the seed region, A to G nucleotide changes (potential A to I editing) were observed in a number of miRNAs. These were particularly abundant at position 5 of the miR-411 and miR-379, as previously described (47). Other common examples included an A to G change in position 6 of all miRNAs of the cluster miR-376a, 376-b and 376-c, in agreement with previous reports (40). miR-320b and miR-320c are interesting examples to highlight, since they present nucleotide substitution variants at several positions of the mature miRNA (Supplementary Table S6). The detection of the majority of miR-320c variants was confirmed in the re-analysis of small RNAs sequencing data of human embryonic stem cells [(37); data not shown]. In addition to the nucleotides belonging to the seed region, other strongly invariable positions belong to the 3′ half of the miRNA, whose complementariety with target mRNAs is an important determinant for interaction stabilization (44) (Figure 3).
Figure 3.

Percentage of miRNAs with nucleotide substitution variants, according to the nucleotide position. A scheme of the mature miRNA appears in the upper part of the figure showing the main regions involved in 3′-UTR gene targeting: 5′-seed (nt 2–8) the cleavage (nt 10–12) and anchor (nt 13–16) sites. A gray square highlights the less variable positions along the miRNA. The color pattern in the bars indicates the proportion of a specific type of nucleotide present in the isomiR, at every position: A (red), U (blue), C (green) and G (orange). In this analysis, nucleotide substitution variants with more than 50 counts in each library are considered.
miRNA transcriptome deregulation in HD samples
To contrast small RNA expression between biologically comparable samples, similar sequencing efficiencies should be considered. To evaluate the sequencing performance, the total number of counts for every unique read in each sample was ordered according to decreasing frequencies (Supplementary Figure S4). The frequency distribution of the different reads showed no statistical differences between the samples (Willcoxon test, P > 0.05), making them comparable for differential expression analysis.
Figure 4 shows an overall view of the expression levels of all the different sequences (gray spots) mapping onto diverse miRNA loci (black spots) in HD-FC (y-axis) and HD-ST (x-axis). A considerable number of miRNAs loci were commonly deregulated in HD-FC and HD-ST (black spots in the upper right and lower left squares, respectively). To identify the miRNAs significantly deregulated in HD, we chose the Z-test (26), since it is the most restrictive test used for evaluation of differentially expressed sequences in high-throughput sequencing methodologies. Furthermore, to control the FDR the P-value assigned by the statistical test was corrected applying the Benjamini and Hochberg method (29). A total of 102 upregulated miRNAs [ratio >1.2; P <0.05; normalized frequency (HD-FC + C-FC) >50] and 58 downregulated miRNAs [ratio <–1,2 and P <0.05; normalized frequency (HD-FC + C-FC) >50] were detected in the HD-FC library. A similar approach in the HD-ST, showed 69 significantly upregulated miRNAs and 62 downregulated miRNAs (Supplementary Tables S8 and S9). The majority (55–80%) of the HD deregulated miRNAs were common between FC and ST samples. Interestingly, the HD-FC presented an increased number of upregulated miRNAs (2.5-fold) that were not significantly altered in the HD-ST.
Figure 4.

Differential expression in HD-ST (x-axis) and HD-FC (y-axis) samples. Black spots show the miRNA loci and gray spots show all the sequences (reference miRNAs and isomiRS) mapping onto the miRNA loci. The darker and lighter lines mark the limits of expression fold changes in HD versus control samples above 1.2 and below –1.2, respectively.
To identify the miRNA most likely relevant in HD we performed miRNA arrays of FC and ST in four control individuals and four HD patients. Considering an FDR-adjusted P-value below 5% and a fold expression change above 1.2 or below −1.2 in absolute value, 19 upregulated and 14 downregulated miRNAs, common to the FC and the ST were confirmed (Table 2). Selected miRNAs (seven upregulated and seven downregulated) were further validated using Taqman assays in ST samples in five control individuals and five HD patients. It should be taken into consideration that these techniques, differing from sequencing strategies, are most favorable for the detection of the reference miRNAs and may not be optimal for the detection of IsomiRs.
Table 2.
HD-deregulated miRNAs
| MiRNA | Expression pattern in HD (sequencing data) | Expression pattern in HD (miRNA arrays) | Expression pattern in HD (miRNA qRT-PCR) |
|---|---|---|---|
| hsa-let-7g | UP | UP | |
| hsa-miR-100 | UP | UP | UP |
| hsa-miR-101 | UP | – | |
| hsa-miR-106b | UP | UP | |
| hsa-miR-1250 | UP | NA | |
| hsa-miR-126 | UP | – | |
| hsa-miR-126* | UP | DOWN | |
| hsa-miR-1308 | UP | NA | |
| hsa-miR-143 | UP | DOWN | |
| hsa-miR-145 | UP | – | |
| hsa-miR-146a | UP | – | |
| hsa-miR-148b | UP | UP | |
| hsa-miR-151-3p | UP | UP | UP |
| hsa-miR-151-5p | UP | UP | |
| hsa-miR-15a | UP | UP | |
| hsa-miR-15b | UP | UP | |
| hsa-miR-16 | UP | UP | UP |
| hsa-miR-17 | UP | UP | |
| hsa-miR-181a | UP | UP | |
| hsa-miR-181a* | UP | UP | |
| hsa-miR-193b | UP | UP | |
| hsa-miR-197 | UP | DOWN | |
| hsa-miR-1974 | UP | NA | |
| hsa-miR-199b-3p | UP | UP | |
| hsa-miR-19b | UP | UP | |
| hsa-miR-204 | UP | UP | |
| hsa-miR-20a | UP | UP | |
| hsa-miR-219-2-3p | UP | UP | UP |
| hsa-miR-219-5p | UP | UP | |
| hsa-miR-22 | UP | DOWN | |
| hsa-miR-23a | UP | – | |
| hsa-miR-27b | UP | UP | UP |
| hsa-miR-29c* | UP | DOWN | |
| hsa-miR-30a | UP | DOWN | |
| hsa-miR-30b | UP | – | |
| hsa-miR-30c | UP | UP | |
| hsa-miR-30e | UP | – | |
| hsa-miR-338-3p | UP | UP | |
| hsa-miR-33a | UP | UP | |
| hsa-miR-33b | UP | UP | |
| hsa-miR-363 | UP | UP | |
| hsa-miR-374a* | UP | – | |
| hsa-miR-451 | UP | UP | UP |
| hsa-miR-484 | UP | DOWN | |
| hsa-miR-486-5p | UP | UP | |
| hsa-miR-574-3p | UP | DOWN | |
| hsa-miR-664 | UP | NA | |
| hsa-miR-887 | UP | UP | |
| hsa-miR-92a | UP | UP | UP |
| hsa-miR-93 | UP | UP | |
| hsa-miR-99b | UP | UP | |
| hsa-let-7a | DOWN | – | |
| hsa-let-7c | DOWN | – | |
| hsa-let-7d | DOWN | – | |
| hsa-let-7e | DOWN | – | |
| hsa-miR-103 | DOWN | DOWN | |
| hsa-miR-107 | DOWN | DOWN | |
| hsa-miR-1224-5p | DOWN | NA | |
| hsa-miR-124 | DOWN | DOWN | |
| hsa-miR-127-3p | DOWN | DOWN | |
| hsa-miR-128 | DOWN | DOWN | DOWN |
| hsa-miR-1301 | DOWN | NA | |
| hsa-miR-1307 | DOWN | NA | |
| hsa-miR-139-3p | DOWN | DOWN | DOWN |
| hsa-miR-181d | DOWN | DOWN | |
| hsa-miR-193b* | DOWN | – | |
| hsa-miR-199a-3p | DOWN | NA | |
| hsa-miR-221 | DOWN | DOWN | |
| hsa-miR-222 | DOWN | DOWN | DOWN |
| hsa-miR-323-3p | DOWN | DOWN | |
| hsa-miR-330-3p | DOWN | UP | |
| hsa-miR-369-5p | DOWN | DOWN | |
| hsa-miR-382 | DOWN | DOWN | DOWN |
| hsa-miR-383 | DOWN | DOWN | – |
| hsa-miR-409-5p | DOWN | DOWN | |
| hsa-miR-423-5p | DOWN | – | |
| hsa-miR-432 | DOWN | DOWN | |
| hsa-miR-433 | DOWN | DOWN | DOWN |
| hsa-miR-485-3p | DOWN | DOWN | DOWN |
| hsa-miR-485-5p | DOWN | DOWN | |
| hsa-miR-92b* | DOWN | – | |
| hsa-miR-495 | DOWN | DOWN | |
| hsa-miR-543 | DOWN | DOWN | |
| hsa-miR-598 | DOWN | DOWN | |
| hsa-miR-708 | DOWN | – | |
| hsa-miR-760 | DOWN | UP | |
| hsa-miR-95 | DOWN | DOWN |
The differential expression in the sequenced samples considers miRNAs commonly deregulated in HD-FC and HD-ST >1.2 and <–1.2.
P < 0.05 (Z-test, Hochberg and Benjamini correction).
The reference miRNA and all the variants mapping on the same miRNA locus are considered in this analysis.
Only sequences with a normalized frequecy cutoff above 50 (norm freq C-FC + norm freq HD-FC > 50) and (norm freq C-ST + norm freq HD-ST >50) were included.
In the miRNA arrays, a fold change above 1.2 and below –1.2 in absolute value was considered.
The results showing a false discovery rate-adjusted P-value (FDR) below % are highlighted in black, and those with a P > 10% are shown in light gray.
Selected miRNAs (seven uregulated and seven downregulated) were chosen for validation by RT-PCR.
The signifficantly deregulated miRNAs (P < 0.01) are highlighted in black.
(−) Means that no deregulation was found.
(NA) Means that the miRNA was not available in the array.
In bold, the miRNAs with a more consistent deregulation pattern using different techniques in additional samples are shown.
The differential expression analysis of the sequenced samples confirmed the expression pattern of 9 out of 11 miRNA (Supplementary Table S10), whose expression was altered in the FC of HD patients in two previous studies (17,18). In addition, the majority of the reported miRNAs deregulated in other neurodegenerative disorders (Alzheimer’s disease, Parkinson’s disease, Prion disease and spinocerebellar ataxia type 1) were altered in the HD sequenced samples (Supplementary Table S11). Furthermore, a fraction of these miRNAs showed an expression deregulation pattern similar to that reported in the other neurodegenerative diseases, indicating that they may participate in gene expression deregulation underlying common processes in neurodegeneration.
When studying the correlation between the expression pattern of all IsomiRs and the corresponding reference miRNA our analysis showed that the majority (80%) of HD-upregulated 3′- or 5′-IsomiRs displayed an upregulation of the corresponding reference miRNA. A similar result was obtained when considering the downregulated reference IsomiRs (Supplementary Table S12). These results suggest that the mechanisms modulating the degree of expression of the IsomiRs and the corresponding miRNAs are parallel in most cases. However, a few sequences mapping on the same miRNA were discordant, presenting the reference miRNA and the isomiR an opposite expression pattern in HD. The majority of these discordant IsomiRs were 3′-variants (3′-trimming and 3′-addition variants). Whether selective forms of a miRNA are relevant in HD and how these variants are selectively deregulated in HD deserve further research.
Since the seed region is essential in the recognition and expression modulation of target miRNAs we analyzed HD-deregulated 5′-trimming variants and nucleotide substitution variants involving any nucleotide change in positions 2–8 of the reference miRNA (Table 3). A total of 23 seed-region IsomiRs were deregulated in HD FC and/or ST, that corresponded to 18 different miRNAs. Of these, miR-935, miR-139-3p, miR-1307, miR-1308, miR-411 and miR-142-5p variants can be considered the only representatives among all the IsomiRs mapping onto the same locus. The rest of the seed-region deregulated IsomiRs showed a varying proportion (10–50%) with respect to that of the reference or the most abundant variant mapping onto the same locus.
Table 3.
HD-deregulated seed region isomiRs, >1,2 or <–1,2 fold change, P < 0.05 (Z-test, Hochberg and Benjamini correction)
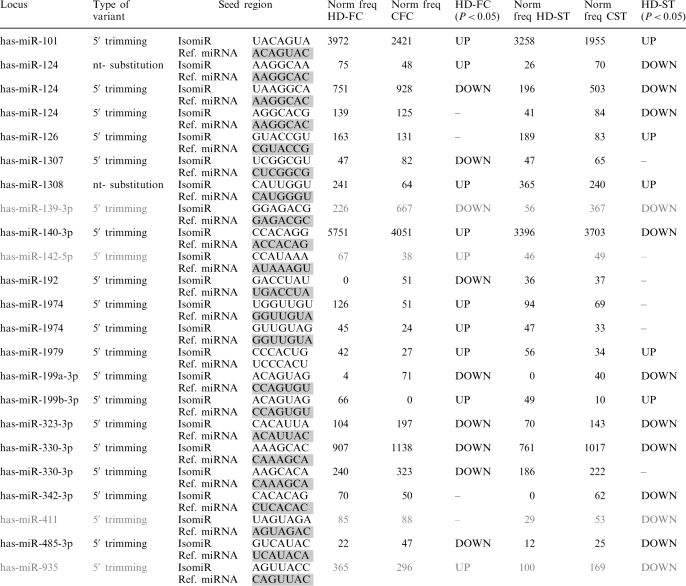 |
The seed region of the miRNAs in gray should be considered as the only representatives among all the isomiRs mapping in the same locus; for these miRNAs no reference seed region was sequenced.
The REST transcription factor mis-function has been considered to play a major role in HD (2–3). We analyzed whether miRNAs that are potentially regulated by REST showed differential expression in our sequencing libraries. For this analysis we considered miRNAs with known motifs for REST (4,17,49,50), which were detected at frequencies above 10 in all the samples (a total of 30 miRNAs). It is worth to mention that 16 (ST) and 19 (FC) out of the 30 REST modulated miRNAs, were significantly deregulated in the HD samples (Table 4). Interestingly, miR-9 in HD-FC and miR-29b in HD-ST presented discordant up- and downregulated variants, which highlights the putative relevance of the different types of IsomiRs in the context of HD. Considering all the deregulated REST target miRNAs, the number of HD downregulated miRNAs was twice the amount that what would be expected by chance, both in HD-FC (P = 0.004, Boostrapping method) and HD-ST (P = 0.003). Given that REST is a transcriptional repressor, the enrichment in repressed REST target miRNAs agrees with a major role of this transcription factor in miRNA dysregulation in HD. To address whether other transcription factors could explain miRNA gene expression deregulation we characterized the promoter regions of co-regulated miRNAs (35). The results showed that in addition to REST, p53 regulatory motifs are significantly enriched in the promoter region of the downregulated miRNAs in the FC (Table S13).
Table 4.
Expression deregulation of known and predicted REST-modulated miRNAs expressed in brain samples
| Locus | miR/isomiR | HD-FC versus C-FC (P < 0.05) | HD-ST versus C-ST (P < 0.05) | REST modulated |
|---|---|---|---|---|
| hsa-miR-124 | Ref. | – | DOWN | EMSA/ChIP (17) RE1 motive (50) |
| hsa-miR-124 | NA | DOWN | DOWN | |
| hsa-miR-124 | s:10AC | – | DOWN | |
| hsa-miR-124 | s:19UC | DOWN | DOWN | |
| hsa-miR-124 | s:8AC | – | DOWN | |
| hsa-miR-124 | tr5:(down)U | DOWN | DOWN | |
| hsa-miR-29a | NA | UP | UP | EMSA/ChIP (17) RE1 motive (50) |
| hsa-miR-29a | Ref. | UP | DOWN | |
| hsa-miR-29b | NA | UP | UP | EMSA/ChIP (17) RE1 motive (50) |
| hsa-miR-29b | Ref. | UP | UP | |
| hsa-miR-9 | NA | UP | UP | EMSA/ChIP (17) RE1 motive (50) |
| hsa-miR-9 | Ref. | DOWN | UP | |
| hsa-miR-132 | NA | – | – | EMSA/ChIP (17) RE1 motive (50) |
| hsa-miR-132 | Ref. | UP | – | |
| hsa-miR-212 | NA | UP | – | EMSA/ChIP (17) RE1 motive (50) |
| hsa-miR-139-5p | NA | UP | DOWN | EMSA/ChIP (17) RE1 motive (50) |
| hsa-miR-139-5p | Ref. | – | DOWN | |
| hsa-miR-139-5p | tr5:(up)GG | UP | – | |
| hsa-miR-139-3p | tr5:(down)U | DOWN | DOWN | |
| hsa-miR-133a | NA | – | – | EMSA (17) |
| hsa-miR-203 | NA | – | – | EMSA/ChIP (17) |
| hsa-miR-204 | NA | UP | UP | EMSA/ChIP (17) |
| hsa-miR-204 | Ref. | UP | UP | |
| hsa-miR-330-3p | NA | – | DOWN | EMSA/ChIP (17) |
| hsa-miR-330-3p | Ref. | – | DOWN | |
| hsa-miR-330-3p | tr5:(up)G | DOWN | DOWN | |
| hsa-miR-330-3p | tr5:(up)GC | DOWN | ||
| hsa-miR-346 | NA | – | – | EMSA/ChIP (17) RE1 motive (50) |
| hsa-miR-218 | NA | – | DOWN | EMSA/ChIP (17) |
| hsa-miR-95 | NA | – | – | RE1 motive (50) |
| hsa-miR-95 | Ref. | DOWN | DOWN | |
| hsa-miR-1 | Ref. | DOWN | DOWN | Chromatin IP (49) |
| hsa-miR-153 | NA | – | – | RE1 motive (50) |
| hsa-miR-455 | NA | – | – | RE1 motive (50) |
| hsa-miR-146b-5p | NA | DOWN | – | RE1 motive (4) |
| hsa-miR-1179 | NA | – | – | RE1 motive (4) |
| hsa-miR-1301 | NA | DOWN | DOWN | RE1 motive (4) |
| hsa-miR-1301 | Ref. | DOWN | DOWN | |
| hsa-miR-129-3p | NA | UP | – | RE1 motive (4) |
| hsa-miR-129-3p | Ref. | UP | – | |
| hsa-miR-129-3p | s:20UC | UP | – | |
| hsa-miR-129-5p | NA | DOWN | – | |
| hsa-miR-129-5p | Ref. | DOWN | – | |
| hsa-miR-7 | NA | DOWN | – | RE1 motive (4) |
| hsa-miR-7 | Ref. | DOWN | UP | |
| hsa-miR-137 | NA | – | DOWN | RE1 motive (4) |
| hsa-miR-137 | Ref. | – | DOWN | |
| hsa-miR-184 | Ref. | – | DOWN | RE1 motive (4) |
| hsa-miR-1224-5p | NA | DOWN | DOWN | RE1 motive (4) |
| hsa-miR-940 | NA | – | – | RE1 motive (4) |
| hsa-miR-375 | NA | – | – | RE1 motive (4) |
| hsa-miR-328 | NA | UP | – | RE1 motive (4) |
| hsa-miR-328 | Ref. | UP | – |
(–) no differential expression was found in the present study; Ref, reference miRNA; tr5, 5′-trimming variants; (up) and (down) followed by the indictaed nucleotides show the short sequence involved in the trimming variants, upstream and downstream of the corresponding reference miRNA terminus, respectively; s, shows a nucleotide subtitution variant.
The position of the nucleotide and the pattern of the modification is shown next. NA, considers, for differential expression analysis, all the sequences mapping onto a specific miRNA locus different from the reference miRNA or the trimming variants, when these are shown in a separate row.
miRNAs in gray did not show significant expression changes.
Putative targets of HD deregulated miRNAs and seed-region IsomiRs
We focused on the possible biological pathways affected by HD-deregulated miRNAs and seed-region IsomiRs. In this analysis we included miRNA/IsomiRs commonly upregulated or downregulated in the FC and ST (Table 3 and Supplementary Tables S8 and S9). We first identified putative targets for the differentially expressed miRNAs and IsomiRs using the TargetScan 5.1 algorithm that predicts biological targets of miRNAs or IsomiRs by searching for the presence of conserved 8-mer and 7-mer sites that match the seed region of each miRNA/IsomiR (33,34). Since the cooperative action of multiple miRNAs can be multiplicative and sometimes synergic (33), mRNAs with more predicted target sites for co-expressed miRNAs/IsomiRs should be more drastically affected. Therefore, we considered targets predicted by three or more HD-upregulated or HD-downregulated miRNAs/IsomiRs. Based on these parameters we identified 45 (HD-enriched) and 40 (HD-diminished) significantly deregulated seed regions that corresponded to 55 and 43 miRNA/IsomiRs, respectively. The number of targets common to a minimum of three co-regulated miRNAs/IsomiRs was 1922 for HD-enriched and 1244 for HD-diminished miRNAs/IsomiRs. It is worth noting that the two lists of genes showed a significant overlap, suggesting that some genes can be putatively targeted by sets of unique HD up and downregulated miRNAs.
The IPA tool was used for the subsets of genes affected by the HD-enriched or HD-downregulated miRNAs/IsomiRs. The most enriched biological and disease processes and canonical pathways amongst the HD-enriched or HD-diminished miRNA/IsomiR targets are provided in Table 5. Some of the top functional groups, including genetic disorder, neurological disease, developmental disorders, cell-to-cell signaling and interaction, cell growth and proliferation, nervous system development and function and organ development were common between the two groups of miRNAs/IsomiRs, which is in agreement with the significant overlap of the predicted mRNA targets. HD was identified as one of the significant canonical pathways (P = 6.6E-03) for the predicted HD-downregulated miRNAs/IsomiRs (Figure S5). Interestingly, 16 out of the 30 predicted targets involved HD-canonical pathway are significantly deregulated in HD, as shown in the study by Hodges et al. (1). Ten out of these 16 mRNAs were upregulated (Supplementary Table S14), suggesting that the downregulation of specific miRNAs could directly contribute to the aberrant increase in the expression of genes relevant in HD pathways. In line with this, the amount of predicted targets of the downregulated miRs/IsomiRs that overlaps with the upregulated mRNAs (1) was significantly enriched (9%, P = 0.004, Boostrapping method) compared with the predicted targets of the randomly selected miRs/IsomiRs expressed in the brain. The TarBase of experimentally supported targets for several miRNAs (http://diana.cslab.ece.ntua.gr/tarbase/) confirmed a similar idea (Table S15). Considering the available validated targets of the HD-downregulated miRNAs, the number of overlapping HD-upregulated genes in the Hodges et al. (1) study (a total of 62) was clearly increased compared with that of the HD-downregulated genes (a total of 7).
Table 5.
Top biological functions and canonical pathways for predicted targets of HD-deregulated miRNAs and seed region-isomiRs: IPA analysis
| P-value | No. of Molecules | |
|---|---|---|
| Top biological functions for predicted targets of HD-upreguated miRNAs and isomiRs | ||
| Disease and disorders | ||
| Genetic disorder | 1.11E-03 - 2.02E-02 | 36 |
| Neurological disease | 1.11E-03 - 4.10E-02 | 92 |
| Psychological disorders | 1.11E-03 - 1.11E-03 | 34 |
| Developmental disorder | 1.02E-02 - 2.02E-02 | 9 |
| Skeletal and muscular disorders | 2.29E-02 - 2.29E-02 | 3 |
| Molecular and cellular functions | ||
| Cellular movement | 7.59E-05 - 4.26E-02 | 24 |
| Cell morphology | 1.77E-04 - 2.82E-02 | 27 |
| Cellular assembly and organization | 2.20E-04 - 2.02E-02 | 27 |
| Cell-to-cell signalling and interaction | 3.05E-04 - 2.75E-02 | 17 |
| Cellular growth and prolifferation | 3.88E-04 - 2.29E-02 | 18 |
| Physiological system development and function | ||
| Nervous system development and function | 2.00E-05 - 4.19E-02 | 97 |
| Organ development | 3.18E-04 - 2.29E-02 | 17 |
| Tissue morphology | 6.08E-03 - 2.82E-02 | 26 |
| Organ morphology | 2.02E-02 - 2.02E-02 | 2 |
| Embryonic development | 2.29E-02 - 2.29E-02 | 3 |
| Top canonical pathways for predicted targets of HD-upreguated miRNAs and isomiRs | ||
| Axonal guidance signaling | 3.13E-07 | 79/320 |
| TGF-b signaling | 3.94E-07 | 27/70 |
| Glioma signaling | 4.11E-07 | 32/91 |
| Molecular mechanisms of cancer | 6.41E-07 | 75/304 |
| B cell receptor signaling | 7.57E-07 | 38/120 |
| Top biological functions for predicted targets of HD-downreguated miRNAs and isomiRs | ||
| Disease and disorders | ||
| Neurological disease | 9.57E-04 - 4.75E-02 | 28 |
| Developmental disorder | 8.64E-03 - 4.56E-02 | 7 |
| Genetic disorder | 2.11E-02 - 2.11E-02 | 3 |
| Skeletal and muscular disorders | 4.56E-02 - 4.56E-02 | 2 |
| Molecular and cellular functions | ||
| Cellular movement | 1.27E-04 - 4.40E-02 | 19 |
| Cellular development | 7.85E-04 - 4.42E-02 | 15 |
| Cell-to-cell signaling and interaction | 9.57E-04 - 2.43E-02 | 13 |
| Cell death | 1.41E-03 - 3.24E-02 | 24 |
| Cellular growth and proliferation | 1.51E-03 - 2.42E-02 | 13 |
| Physiological system development and function | ||
| Nervous system development and function | 8.05E-05 - 4.56E-02 | 65 |
| Organ development | 8.05E-05 - 8.05E-05 | 12 |
| Tissue development | 8.02E-04 - 8.64E-03 | 5 |
| Embryonic development | 1.97E-03 - 2.11E-02 | 8 |
| Tissue morphology | 6.39E-03 - 4.22E-02 | 17 |
| Top canonical pathways for predicted targets of HD-downregulated miRNAs and isomiRs | ||
| Molecular mechanisms of cancer | 9.65E-07 | 55/304 |
| GNRH signaling | 2.80E-06 | 27/112 |
| Renal cell carcinoma signaling | 1.05E-05 | 18/63 |
| PTEN signaling | 1.30E-05 | 21/82 |
| PPARa/RXRa activation | 1.61E-05 | 29/136 |
To gain insights into the biological pathways possibly affected by isomiR expression deregulation, we run a specific IPA for the predicted targets of the seed-region-HD-deregulated IsomiRs (Table 3). The results revealed a significant enrichment in genes involved in neurological diseases for both HD-enriched and HD-diminished seed-region isomiR targets (Supplementary Table S16). Furthermore, the HD canonical pathway was significantly enriched for the HD-downregulated seed region isomiR targets. These results underline the possible contribution of the IsomiRs in the modulation of HD related pathways.
DISCUSSION
Here, we report the first unbiased analysis of small RNA populations of two different brain areas of healthy individuals and individuals with HD. Deep sequencing of small RNAs from the human brain revealed length and sequence heterogeneity for the vast majority of miRNAs, indicating that the miRNA transcriptome is more complex than previously described. However, the fact that the proportion of the different types of variants was found to be similar in all brain samples agrees with the concept that the molecular mechanisms involved in the generation of these variants are not altered in HD.
miRNA variability (IsomiRs) has recently been described using several large-scale sequencing strategies in plants (45,46), mouse and human stem cells (37,51), and human brain samples (47). Our analysis revealed that IsomiRs are also present in human and mouse Ago-IPs, suggesting that they are functional as gene expression modulators. We have found 20 000–23 000 different variants with a frequency >3 in every brain sample. Our study permits several generalizations regarding the types of miRNA variants in the human brain, irrespective of HD neuropathology. Around 80–90% of the miRNAs presented modifications in the 3′-terminus either in the form of trimming and/or as nucleotide addition variants. Furthermore, considering all the sequences mapping onto the same miRNA locus, the 3′-terminus variants presented a higher proportion with respect to the reference miRNA. This is consistent with the idea that variability in the 3′-terminus of the miRNA is more permissive, having minor consequences in gene expression regulation. In contrast, our analysis displayed a decreased proportion of miRNAs with heterogeneity at the 5′-terminus, showing the majority of these variants a negligible abundance as compared with that of the reference sequence. This suggests that the 5′ terminus of the miRNAs is specially protected from variations, which agrees with the crucial role of Watson–Crick base pairing of the 5′-seed region of the miRNA with the 3′UTR of the mRNA, for gene targeting (43,44).
In accordance with previous reports (41,52,53), variability in the 3′-terminus was detected as an A or U single nucleotide extension. These modifications could influence mRNA expression regulation, since 3′-end pairing has been suggested to contribute to target recognition, particularly when sites have weaker miRNA seed matches (54,55). In plants, it has been shown that addition of A residues to the 3′-end plays a negative role in miRNA degradation (56). In addition, it has been proposed that the combined effects of 5′ deletions and 3′ U extensions in Oryza sativa and Arabidopsis thaliana can alter the specificity by which miRNAs associate with different Argonaute proteins (45). Whether short nucleotide extensions in animal miRNAs influence their stability or the silencing mechanism needs to be proven. However, recent data show selective stability for different miRNAs in human cells that depends on regions of the 3′-terminus (57). Differences in the stability could provide an explanation for the discordant expression deregulation of some 3′-trimming variants and the corresponding reference miRNAs.
Our analysis highlights significant nucleotide modifications along the reference mature miRNAs. Several lines of evidence argue against RT-PCR and sequencing errors as contributors to sequence discrepancies with respect to the reference miRNAs. First, the frequencies of nucleotide modifications were remarkably higher compared to the estimates attributable to Illumina sequencing errors (27). Second, we observed a positional non-randomness of nucleotide changes along the length of the miRNA seen in all libraries. Third, analogous nucleotide modifications were found in four independent libraries. Fourth, several nucleotide substitution variants found in the brain have been reported in independent sequencing experiments in human samples. Finally, nucleotide changes, insertions and deletions have been reported in previous studies using different sequencing strategies.
Nucleotide substitutions have been described in few miRNAs as pri-miR precursor editing changes from A to G attributed in part to A to I deaminations, which lead to a repression in the maturation of the miRNA (38–41,45,58). Edited adenosines have been also described in mature miRNAs (38–40). Our results not only have confirmed the existence of these variants, but also show that an A to G change is one of the most common types of nucleotide substitution events in the seed region. However, a considerable proportion of brain miRNAs presented nucleotide changes that were distinct from the classical A to I editing. Common nucleotide substitutions in brain miRNAs included U to G, C to A, G to A and G to U changes. These patterns of nucleotide modifications have been recently shown in miRNAs and tRNAs of plants (45) and in the let family of miRNAs in different mouse cell lines (48). In animals, these modifications in the mature miRNA may result in alternative base pairing between the variant and the target mRNA, possibly affecting the efficiency and/or quality of gene expression regulation (59). This has been shown human miR-376a that presents an A to G editing event in the seed region that affects gene targeting (40). Another example has been recently reported in the mouse let-7 family of miRNAs, as several types of nucleotide modifications at the ninth position of mmu-let7a result in an increase of the stability of the predicted miRNA:mRNA duplexes (48).
We have found that the detected nucleotide substitution events are not randomly distributed along the sequence. The less variable positions present key roles in gene target recognition and silencing. For instance, scarce miRNAs presented nucleotide substitutions at positions 2–4 of the miRNA within the 5′-seed region (nt 2–8). A contiguous and perfect base pairing of the miRNA nucleotides 2–8, representing the seed region, is the most stringent requirement for efficient target recognition (44,54). In addition, low variability was detected at positions 14–16, contained in an anchor site (nt 13–16) that has been proposed to hold important determinants for miRNA:mRNA association. However, positions 9–12 of the miRNA were amongst the more variable, which could contribute to the imperfect mRNA:miRNA pairing detected in the central region of the miRNA. For gene expression repression, bugles or mismatches must be present in the central region of the miRNA:mRNA duplex (nt 9–12 of the miRNA), precluding the Argonaute-mediated endonucleolytic cleavage of mRNA (44). All together, the present analysis strongly suggests a biological function for this sequence plasticity in miRNAs, which may have broad implications in mRNA targeting, stability and/or gene expression regulation mechanism.
HD is characterized by widespread changes in neuronal gene expression (1,60). Recent studies have shown that the miRNA transcriptome is also perturbed in HD (17,18) affecting miR-9/9*, miR-124a, miR-132, miR-29b, miR-29a, miR-330, miR-17, miR-196a, miR-222, miR-485 and miR-486. Although further miRNA sequencing of HD brain samples will provide a comprehensive picture of the myriad of small RNAs in this disorder, our results confirm the deregulation pattern for 9 out of the 11 miRNAs, but also suggested that miRNA expression deregulation is more extensive in HD, affecting a considerable number of miRNAs and IsomiRs. Alterations in the miRNA/isomiR transcriptome may contribute to the aberrant expression of downstream genes detected in HD. In line with this, the putative targets of the HD-deregulated miRNAs and/or seed-region IsomiRs denote that these variants could participate in HD pathogenesis. It is worth pointing out that HD canonical pathway was among the significantly enriched pathways for the HD-downregulated miRs and/or IsomiRs. In addition, the study by Hodges et al. (1) shows that many HD-downregulated genes may underlie a defect in the capacity to maintain axonal projections. In agreement, our analysis revealed that axonal guidance signaling and actin and cytoskeleton signaling were among the highly significantly enriched pathways for the predicted targets of the upregulated miRNAs.
A large number of miRNAs presented the same deregulation profile in the two studied areas, which show a different pattern of neuronal loss, similarly to what has been reported in gene expression analysis of different brain areas in HD (1). Therefore, shared deregulated miRNAs may lie beneath gene expression deregulation linked to the common neuropathological changes, including neuronal degeneration, astrocytic gliosis and moderate microglial proliferation. Since advanced stages of the disease are considered in the present study, it is feasible that many changes in the miRNA transcriptome are not linked to HD-primary episodes, but may be reactive events to secondary-common processes occurring in neurodegeneration, such as oxidative stress. In line with this, several miRNAs present a similar pattern of deregulation as that described in other neurodegenerative disorders. In addition, at advanced stages some of the HD-related transcriptional modifications, especially gene expression downregulation, could be caused by changes in the relative number of neurons linked to selective neurodegeneration rather than changes in specific neuronal miRNA genes. However, a previous study shows similar HD-deregulated mRNAs in data sets of brain homogenates and laser capture microdissection that ensures similar numbers of neurons in control and HD samples (1). These data suggest that miRNA expression deregulation could be explained, at least in part, by transcriptional changes occurring in the remaining cells. The region-specific miRNA transcriptome deregulation may account for selective pathophysiological changes in HD, including the preferential affectation of the gamma-aminobutyric acid (GABA)ergic spiny neurons in the ST and the projection neurons in the FC.
Some miRNAs that are in chromosomal clusters exhibited similar expression patterns in HD. For instance, miR-221 and miR-222 were both downregulated in HD. In addition, we noted that in some cases, miRNA members of the same family exhibited similar expression trends, suggesting their co-regulation in HD. For instance, let-7a, let-7c, let-7d and let-7e were downregulated in HD-FC and HD-ST. In another example, miR-30a, miR-30b, miR-30c and miR-30e were all upregulated in HD-FC and HD-ST. These results are in agreement with the idea of a relevant role of certain miRNA families in HD pathology. An interesting opened question is the understanding of the mechanisms accounting for the co-regulation of non-clustered miRNAs of the same family in pathological processes.
Consistent with both the deregulation of miRNA and mRNA gene expression in HD, a number of transcriptional modulators are affected by mutant HTT (61). There is strong evidence that the abnormal function of the transcriptional repressor REST plays a major role in gene expression deregulation in HD (2,3). BDNF is a REST-modulated neuronal gene whose loss in the striatum of HD patients contributes to the medium spiny neuronal loss and clinical manifestations of the disease (3,62). It is interesting that miR-30a, which has been shown to target BDNF (63), is significantly upregulated in both HD-FC and HD-ST brain samples. Therefore, in addition to REST, miR-30a upregulation could contribute to BDNF expression inhibition.
Some neuronal miRNAs modulated by REST (miR-124, miR-29a, miR-29b, miR9/9* miR-132 and miR-330-3p) are abnormally expressed in the FC of HD patients (5,17,18). Taking into account all brain-expressed miRNAs showing experimental or predictive evidences for REST modulation (REST-miRNAs) our analysis identified a larger number of deregulated REST-miRNAs. Furthermore, these miRNAs were found to be enriched among those downregulated in HD brain regions. Overall, our results agree with the concept that REST dysfunction generaly alters the miRNA transcriptome in HD, which may influence the expression of downstream targets. In addition, we identified p53 tumor supressor as a transcription factor with a putative role in HD-miRNA deregulation. P53 is found in polyglutamine aggregates both in vivo and in vitro, suggesting an hypofunction of p53 in HD (64). Therefore, p53 may contribute to miRNA expression downregulation in HD.
In summary, this study shows that miRNA variability is a ubiquitous phenomenon in the adult human brain that may influence the mechanism of gene silencing or gene targeting. How the IsomiRs are selectively generated and the possible specific role that these variants have in development, organ physiology or pathological processes, such as HD, opens a new perspective in the field of miRNAs.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The Spanish Ministry of Health ‘Fondo de Investigaciones Sanitarias (PI081367) and “Instituto de Salud Carlos III” (CIBERESP); the Sixth Framework Programme of the European Commission through the SIROCCO integrated project LSHG-CT-2006-037900; the Spanish Ministry of Science and Innovation (SAF2008-00357); Spanish Ministry of Health “Programa Miguel Servet” (to E.M.); Spanish Ministry of Health “Contrato Postdoctoral Sara Borrell” (M.B.-C. and S.P.); Spanish Ministry of Science and Innovation (L.P.). Spanish Ministry of Health (E.M.-M.).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the staff of the Ultrasequencing Unit of the CRG for the small-RNA libraries preparation and sequencing on the Illumina/Solexa platform, and the Microarray Unit for the labelling, hybridization and analysis of the Agilent arrays. We also thank Georgia Escaramís for the qPCR data statistical analysis.
REFERENCES
- 1.Hodges A, Strand AD, Aragaki AK, Kuhn A, Sengstag T, Hughes G, Elliston LA, Hartog C, Goldstein DR, Thu D, et al. Regional and cellular gene expression changes in human Huntington's disease brain. Hum. Mol. Genet. 2006;15:965–977. doi: 10.1093/hmg/ddl013. [DOI] [PubMed] [Google Scholar]
- 2.Zuccato C, Belyaev N, Conforti P, Ooi L, Tartari M, Papadimou E, MacDonald M, Fossale E, Zeitlin S, Buckley N, et al. Widespread disruption of repressor element-1 silencing transcription factor/neuron-restrictive silencer factor occupancy at its target genes in Huntington's disease. J. Neurosci. 2007;27:6972–6983. doi: 10.1523/JNEUROSCI.4278-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zuccato C, Tartari M, Crotti A, Goffredo D, Valenza M, Conti L, Cataudella T, Leavitt BR, Hayden MR, Timmusk T, et al. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat. Genet. 2003;35:76–83. doi: 10.1038/ng1219. [DOI] [PubMed] [Google Scholar]
- 4.Johnson R, Buckley NJ. Gene dysregulation in Huntington's disease: REST, microRNAs and beyond. Neuromolecular. Med. 2009;11:183–199. doi: 10.1007/s12017-009-8063-4. [DOI] [PubMed] [Google Scholar]
- 5.Johnson R, Teh CH, Jia H, Vanisri RR, Pandey T, Lu ZH, Buckley NJ, Stanton LW, Lipovich L. Regulation of neural macroRNAs by the transcriptional repressor REST. RNA. 2009;15:85–96. doi: 10.1261/rna.1127009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schratt G. Fine-tuning neural gene expression with microRNAs. Curr. Opin. Neurobiol. 2009;19:213–219. doi: 10.1016/j.conb.2009.05.015. [DOI] [PubMed] [Google Scholar]
- 7.Bushati N, Cohen SM. MicroRNAs in neurodegeneration. Curr. Opin. Neurobiol. 2008;18:292–296. doi: 10.1016/j.conb.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 8.Eacker SM, Dawson TM, Dawson VL. Understanding microRNAs in neurodegeneration. Nat. Rev. Neurosci. 2009;10:837–841. doi: 10.1038/nrn2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hébert SS, Horre K, Nicolai L, Papadopoulou AS, Mandemakers W, Silahtaroglu AN, Kauppinen S, Delacourte A, De Strooper B. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer's disease correlates with increased BACE1/beta-secretase expression. Proc. Natl Acad. Sci. USA. 2008;105:64156420. doi: 10.1073/pnas.0710263105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim J, Inoue K, Ishii J, Vanti WB, Voronov SV, Murchison E, Hannon G, Abeliovich A. A MicroRNA feedback circuit in midbrain dopamine neurons. Science. 2007;317:1220–1224. doi: 10.1126/science.1140481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Montag J, Hitt R, Opitz L, Schulz-Schaeffer WJ, Hunsmann G, Motzkus D. Upregulation of miRNA hsa-miR-342-3p in experimental and idiopathic prion disease. Mol. Neurodegener. 2009;4:36. doi: 10.1186/1750-1326-4-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saba R, Goodman CD, Huzarewich RL, Robertson C, Booth SA. A miRNA signature of prion induced neurodegeneration. PLoS One. 2008;3:e3652. doi: 10.1371/journal.pone.0003652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sethi P, Lukiw WJ. Micro-RNA abundance and stability in human brain: specific alterations in Alzheimer's disease temporal lobe neocortex. Neurosci. Lett. 2009;459:100–104. doi: 10.1016/j.neulet.2009.04.052. [DOI] [PubMed] [Google Scholar]
- 14.Wang G, van der Walt JM, Mayhew G, Li YJ, Zuchner S, Scott WK, Martin ER, Vance JM. Variation in the miRNA-433 binding site of FGF20 confers risk for Parkinson disease by overexpression of alpha-synuclein. Am. J. Hum. Genet. 2008;82:283–289. doi: 10.1016/j.ajhg.2007.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang WX, Rajeev BW, Stromberg AJ, Ren N, Tang G, Huang Q, Rigoutsos I, Nelson PT. The expression of microRNA miR-107 decreases early in Alzheimer's disease and may accelerate disease progression through regulation of beta-site amyloid precursor protein-cleaving enzyme 1. J. Neurosci. 2008;28:1213–1223. doi: 10.1523/JNEUROSCI.5065-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rademakers R, Eriksen JL, Baker M, Robinson T, Ahmed Z, Lincoln SJ, Finch N, Rutherford NJ, Crook RJ, Josephs KA, et al. Common variation in the miR-659 binding-site of GRN is a major risk factor for TDP43-positive frontotemporal dementia. Hum. Mol. Genet. 2008;17:3631–3642. doi: 10.1093/hmg/ddn257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson R, Zuccato C, Belyaev ND, Guest DJ, Cattaneo E, Buckley NJ. A microRNA-based gene dysregulation pathway in Huntington's disease. Neurobiol. Dis. 2008;29:438–445. doi: 10.1016/j.nbd.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 18.Packer AN, Xing Y, Harper SQ, Jones L, Davidson BL. The bifunctional microRNA miR-9/miR-9* regulates REST and CoREST and is downregulated in Huntington's disease. J. Neurosci. 2008;28:14341–14346. doi: 10.1523/JNEUROSCI.2390-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Savas JN, Makusky A, Ottosen S, Baillat D, Then F, Krainc D, Shiekhattar R, Markey SP, Tanese N. Huntington's disease protein contributes to RNA-mediated gene silencing through association with Argonaute and P bodies. Proc. Natl Acad. Sci. USA. 2008;105:10820–10825. doi: 10.1073/pnas.0800658105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vonsattel JP, DiFiglia M. Huntington disease. J. Neuropathol. Exp. Neurol. 1998;57:369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- 21.Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP., Jr Neuropathological classification of Huntington's disease. J. Neuropathol. Exp. Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- 22.Lu C, Meyers BC, Green PJ. Construction of small RNA cDNA libraries for deep sequencing. Methods. 2007;43:110–117. doi: 10.1016/j.ymeth.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 23.Quail MA, Kozarewa I, Smith F, Scally A, Stephens PJ, Durbin R, Swerdlow H, Turner DJ. A large genome center's improvements to the Illumina sequencing system. Nat. Methods. 2008;5:1005–1010. doi: 10.1038/nmeth.1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pantano L, Estivill X, Marti E. SeqBuster, a bioinformatic tool for the processing and analysis of small RNAs datasets, reveals ubiquitous miRNA modifications in human embryonic cells. Nucleic Acids Res. 2010;38:e34. doi: 10.1093/nar/gkp1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Batuwita R, Palade V. microPred: effective classification of pre-miRNAs for human miRNA gene prediction. Bioinformatics. 2009;25:989–995. doi: 10.1093/bioinformatics/btp107. [DOI] [PubMed] [Google Scholar]
- 26.Reinartz J, Bruyns E, Lin JZ, Burcham T, Brenner S, Bowen B, Kramer M, Woychik R. Massively parallel signature sequencing (MPSS) as a tool for in-depth quantitative gene expression profiling in all organisms. Brief Funct. Genomic Proteomic. 2002;1:95–104. doi: 10.1093/bfgp/1.1.95. [DOI] [PubMed] [Google Scholar]
- 27.Dohm JC, Lottaz C, Borodina T, Himmelbauer H. Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic Acids Res. 2008;36:e105. doi: 10.1093/nar/gkn425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl Acad. Sci. USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B. 1995;57:289–300. [Google Scholar]
- 30.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steibel JP, Poletto R, Coussens PM, Rosa GJ. A powerful and flexible linear mixed model framework for the analysis of relative quantification RT-PCR data. Genomics. 2009;94:146–152. doi: 10.1016/j.ygeno.2009.04.008. [DOI] [PubMed] [Google Scholar]
- 33.Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol. Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 35.Blanco E, Messeguer X, Smith TF, Guigo R. Transcription factor map alignment of promoter regions. PLoS Comput. Biol. 2006;2:e49. doi: 10.1371/journal.pcbi.0020049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruby JG, Stark A, Johnston WK, Kellis M, Bartel DP, Lai EC. Evolution, biogenesis, expression, and target predictions of a substantially expanded set of Drosophila microRNAs. Genome Res. 2007;17:1850–1864. doi: 10.1101/gr.6597907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morin RD, O'Connor MD, Griffith M, Kuchenbauer F, Delaney A, Prabhu AL, Zhao Y, McDonald H, Zeng T, Hirst M, et al. Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res. 2008;18:610–621. doi: 10.1101/gr.7179508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blow MJ, Grocock RJ, van Dongen S, Enright AJ, Dicks E, Futreal PA, Wooster R, Stratton MR. RNA editing of human microRNAs. Genome Biol. 2006;7:R27. doi: 10.1186/gb-2006-7-4-r27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kawahara Y, Zinshteyn B, Chendrimada TP, Shiekhattar R, Nishikura K. RNA editing of the microRNA-151 precursor blocks cleavage by the Dicer-TRBP complex. EMBO Rep. 2007;8:763–769. doi: 10.1038/sj.embor.7401011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kawahara Y, Zinshteyn B, Sethupathy P, Iizasa H, Hatzigeorgiou AG, Nishikura K. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science. 2007;315:1137–1140. doi: 10.1126/science.1138050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–1414. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luciano DJ, Mirsky H, Vendetti NJ, Maas S. RNA editing of a miRNA precursor. RNA. 2004;10:1174–1177. doi: 10.1261/rna.7350304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat. Rev. Genet. 2008;9:102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- 45.Ebhardt HA, Tsang HH, Dai DC, Liu Y, Bostan B, Fahlman RP. Meta-analysis of small RNA-sequencing errors reveals ubiquitous post-transcriptional RNA modifications. Nucleic Acids Res. 2009;37:2461–2470. doi: 10.1093/nar/gkp093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Iida K, Jin H, Zhu JK. Bioinformatics analysis suggests base modifications of tRNAs and miRNAs in Arabidopsis thaliana. BMC Genomics. 2009;10:155. doi: 10.1186/1471-2164-10-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kawahara Y, Megraw M, Kreider E, Iizasa H, Valente L, Hatzigeorgiou AG, Nishikura K. Frequency and fate of microRNA editing in human brain. Nucleic Acids Res. 2008;36:5270–5280. doi: 10.1093/nar/gkn479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reid JG, Nagaraja AK, Lynn FC, Drabek RB, Muzny DM, Shaw CA, Weiss MK, Naghavi AO, Khan M, Zhu H, et al. Mouse let-7 miRNA populations exhibit RNA editing that is constrained in the 5′-seed/ cleavage/anchor regions and stabilize predicted mmu-let-7a:mRNA duplexes. Genome Res. 2008;18:1571–1581. doi: 10.1101/gr.078246.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Conaco C, Otto S, Han JJ, Mandel G. Reciprocal actions of REST and a microRNA promote neuronal identity. Proc. Natl Acad. Sci. USA. 2006;103:2422–2427. doi: 10.1073/pnas.0511041103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu J, Xie X. Comparative sequence analysis reveals an intricate network among REST, CREB and miRNA in mediating neuronal gene expression. Genome Biol. 2006;7:R85. doi: 10.1186/gb-2006-7-9-r85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bar M, Wyman SK, Fritz BR, Qi J, Garg KS, Parkin RK, Kroh EM, Bendoraite A, Mitchell PS, Nelson AM, et al. MicroRNA discovery and profiling in human embryonic stem cells by deep sequencing of small RNA libraries. Stem Cells. 2008;26:2496–2505. doi: 10.1634/stemcells.2008-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aravin A, Tuschl T. Identification and characterization of small RNAs involved in RNA silencing. FEBS Lett. 2005;579:5830–5840. doi: 10.1016/j.febslet.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 53.Kuchenbauer F, Morin RD, Argiropoulos B, Petriv OI, Griffith M, Heuser M, Yung E, Piper J, Delaney A, Prabhu AL, et al. In-depth characterization of the microRNA transcriptome in a leukemia progression model. Genome Res. 2008;18:1787–1797. doi: 10.1101/gr.077578.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brennecke J, Stark A, Russell RB, Cohen SM. Principles of microRNA-target recognition. PLoS Biol. 2005;3:e85. doi: 10.1371/journal.pbio.0030085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brodersen P, Voinnet O. Revisiting the principles of microRNA target recognition and mode of action. Nat. Rev. Mol. Cell. Biol. 2009;10:141–148. doi: 10.1038/nrm2619. [DOI] [PubMed] [Google Scholar]
- 56.Lu S, Sun YH, Chiang VL. Adenylation of plant miRNAs. Nucleic Acids Res. 2009;37:1878–1885. doi: 10.1093/nar/gkp031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bail S, Swerdel M, Liu H, Jiao X, Goff LA, Hart RP, Kiledjian M. Differential regulation of microRNA stability. RNA. 2010;16:1032–1039. doi: 10.1261/rna.1851510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang W, Chendrimada TP, Wang Q, Higuchi M, Seeburg PH, Shiekhattar R, Nishikura K. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat. Struct. Mol. Biol. 2006;13:13–21. doi: 10.1038/nsmb1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Das AK, Carmichael GG. ADAR editing wobbles the microRNA world. ACS Chem. Biol. 2007;2:217–220. doi: 10.1021/cb700064h. [DOI] [PubMed] [Google Scholar]
- 60.Borovecki F, Lovrecic L, Zhou J, Jeong H, Then F, Rosas HD, Hersch SM, Hogarth P, Bouzou B, Jensen RV, et al. Genome-wide expression profiling of human blood reveals biomarkers for Huntington's disease. Proc. Natl Acad. Sci. USA. 2005;102:11023–11028. doi: 10.1073/pnas.0504921102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bithell A, Johnson R, Buckley NJ. Transcriptional dysregulation of coding and non-coding genes in cellular models of Huntington's disease. Biochem. Soc. Trans. 2009;37:1270–1275. doi: 10.1042/BST0371270. [DOI] [PubMed] [Google Scholar]
- 62.Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science. 2001;293:493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]
- 63.Mellios N, Huang HS, Grigorenko A, Rogaev E, Akbarian S. A set of differentially expressed miRNAs, including miR-30a-5p, act as post-transcriptional inhibitors of BDNF in prefrontal cortex. Hum. Mol. Genet. 2008;17:3030–3042. doi: 10.1093/hmg/ddn201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu YZ, Gohler H, Wanker EE, Bates GP, Housman DE, Thompson LM. The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl Acad. Sci. USA. 2000;97:6763–6768. doi: 10.1073/pnas.100110097. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
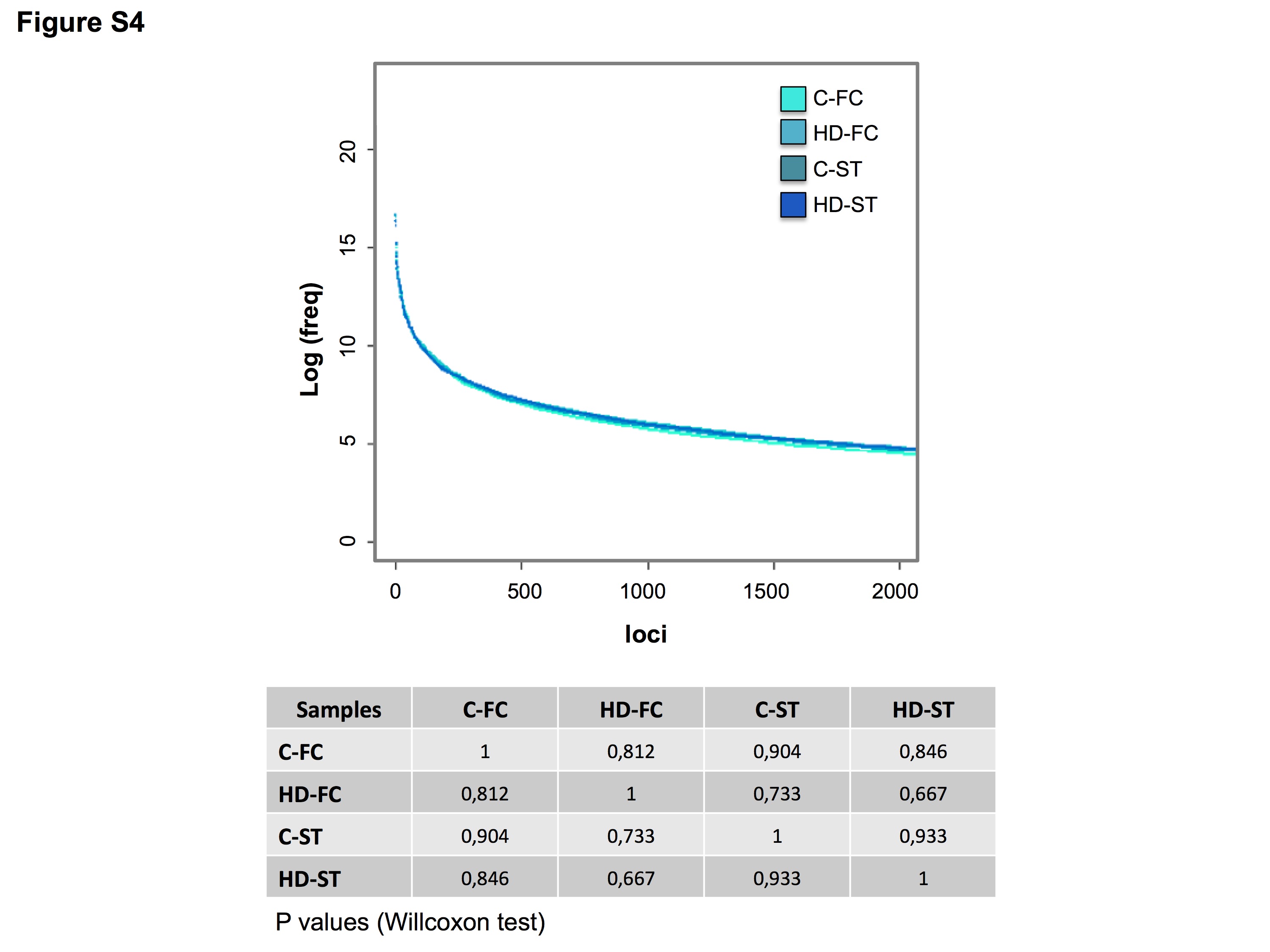{kind=link}