

Abstract
Drug-metabolizing enzymes (DMEs) and transporters play pivotal roles in the disposition and detoxification of numerous foreign and endogenous chemicals. To accommodate chemical challenges, the expression of many DMEs and transporters is up-regulated by a group of ligand-activated transcription factors namely nuclear receptors (NRs). The importance of NRs in xenobiotic metabolism and clearance is best exemplified by the most promiscuous xenobiotic receptors: pregnane X receptor (PXR, NR1I2) and constitutive androstane/activated receptor (CAR, NR1I3). Together, these two receptors govern the inductive expression of a largely overlapping array of target genes encoding phase I and II DMEs, and drug transporters. Moreover, PXR and CAR also represent two distinctive mechanisms of NR activation, whereby CAR demonstrates both constitutive and ligand-independent activation. In this review, recent advances in our understanding of PXR and CAR as xenosensors are discussed with emphasis placed on the differences rather than similarities of these two xenobiotic receptors in ligand recognition and target gene regulation.
Keywords: PXR, CAR, Xenobiotic receptor, Drug-metabolizing enzyme, Transporter
1. Introduction
Exposure to xenobiotics such as drugs and environmental chemicals has profound influence on human health. In order to modulate their own metabolism and excretion, xenobiotics alter the transcription of a broad array of genes expressed in multiple tissues and vital organs such as the liver, kidney, intestine, lungs, brain, placenta, and pancreas [1–3]. Mechanistically speaking however, it is majorly the mammalian nuclear receptor (NR) superfamily of transcription factors that makes xenobiotic regulation of gene expression at the transcriptional level possible. The characteristic structural features of NRs include a highly-conserved DNA-binding domain (DBD), which links the receptors to specific promoter regions of their target genes, and a less conserved ligand-binding domain (LBD) that permits them to directly interact with hormones and/or xenobiotics [4,5]. Moreover, the extraordinary flexibility in the size and shape of LBDs provides the basis for a number of NRs being able to accommodate a myriad of ligands with diverse chemical structures [6,7]. Notably, the first human nuclear receptor, glucocorticoid receptor (GR), was cloned starting from the purification and characterization of the receptor protein from the cortisone-producing adrenal glands, before specific antibodies were used to isolate the corresponding cDNA [8,9]. Utilizing these classical endocrinology approaches, a number of endocrine receptors such as GR, estrogen receptor (ER), and thyroid hormone receptor (TR), were isolated soon thereafter. Typically all these receptors have relatively compact LDBs and are capable of binding unique high-affinity endogenous ligands at nanomolar concentrations.
Taking advantage of the fact that nearly all NRs share a highly conserved cysteine-rich DBD, researchers strive to search for unrecognized NRs using this common segment as bait to screen recombinant DNA libraries at low stringency [10]. This approach has led to the identification of a group of proteins that resemble NRs on the basis of structure and sequence, but lack identified endogenous ligands. Coined “orphan receptors,” this group of NRs accounts for approximately 60% of the over 70 distinct members of the NR superfamily [11–13]. The availability of this large number of orphan receptors also triggered a shift of the classic endocrinology approach into the so called “reverse endocrinology” one, whereby instead of using a purified hormone to identify its partner receptor, novel bioactive molecules were recognized as selective ligands of these orphan receptors [14]. As such, a number of the orphan receptors were termed ‘adopted.’ Nevertheless, in contrast to the prototypical endocrine receptors, members of the orphan receptors are typically activated by abundant but low-affinity lipophilic molecules at micromolar concentrations [15,16].
Notably, the majority of ligands for orphan or adopted receptors are xenobiotics including drugs, carcinogens, food additives, pesticides and environmental pollutants [17]. Functioning as sensors of toxic byproducts derived from both endogenous and exogenous chemical breakdowns, a number of these receptors were also termed xenobiotic receptors, which include but are not limited to: farnesoid X receptor (FXR), liver X receptor (LXR), proxisome proliferator activation receptors (PPARs), constitutive androstane/active receptor (CAR), pregnane X receptor (PXR), nuclear factor-erythroid 2-related factor 2 (Nrf2), and aryl hydrocarbon receptor (AhR). Also worth mentioning, though primarily responsive to xenobiotics, AhR belongs to the basic helix–loop–helix protein of the PAS Per-ARNT-Sim (PAS) family, not to the NR superfamily [18]. Among these xenobiotic receptors, PXR and CAR exhibit promiscuous xenobiotic activation capability; and collaboratively they govern the transcription of a broad spectrum of distinct and overlapping genes encoding phase I, phase II drug-metabolizing enzymes (DMEs), as well as uptake and efflux transporters [17,19–22] (Fig. 1).
Fig. 1.
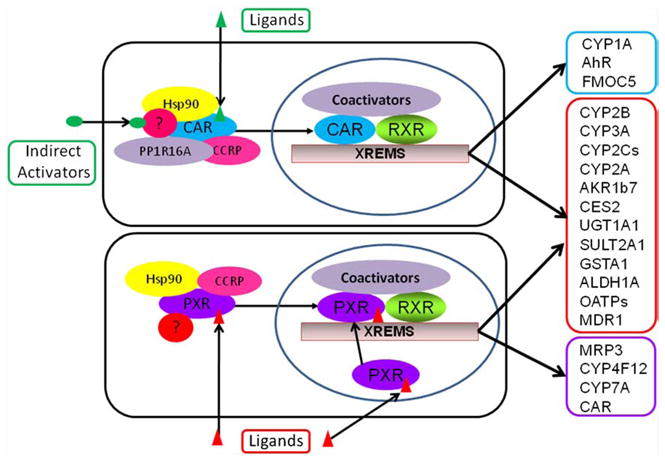
Schematic illustration of the activation mechanisms and target genes of CAR and PXR. CAR can be activated by either direct (ligand binding) or indirect mechanisms, while activation of PXR is purely ligand dependent. CAR and PXR shared target genes are grouped in a red box, CAR-specific targets in a blue box, and PXR-specific targets in a purple box (modified from Qatanani and Moore, Curr. Drug Metab., 2005).
The purpose of this review is to highlight the recent advances in our understanding of the regulation of DMEs and transporters by xenobiotic receptors: PXR and CAR. Emphasis is given to the distinct, rather than the overlapping roles, of PXR and CAR in gene regulation. To a lesser extent, we also discuss the role of AhR as a xenosensor. This review however is by no means a comprehensive coverage of CAR, PXR, and AhR research findings.
2. Pregnane X receptor (PXR)
The pregnane X receptor (PXR, NR1I2) is an approximately 434-amino acid, 50-kDa protein, primarily expressed in the liver and intestine [23]. In 1998, three research groups independently isolated cDNAs encoding a novel orphan receptor, PXR, which was subsequently shown to play a central role in the transcriptional regulation of CYP3A genes across multiple species [24–26]. Prior to being designated NR1I2, this receptor was also named SXR (steroid and xenobiotic receptor) and PAR (pregnane-activated receptor) in humans, which proved to be reflective of its subsequently identified versatility in recognizing a broad array of both synthetic steroids and xenobiotics [27,28].
Compared with other NRs, PXR possesses a bulky and flexible ligand-binding cavity, which enables it to accommodate a more structurally promiscuous library of ligands [29]. Multiple structural studies have facilitated the rationalization of the ligand-binding characteristics of PXR, such as the determination of the structure of human (h)PXR, in both the SR-12813-bound and unbound forms [30], followed by similar characterization of hPXR bound to hyperforin [31], or rifampicin [32]. Among the many key insights that have been gained from these studies are the realization that the ligand-binding cavity of PXR can be, for example, differentially induced to change shape, expand in volume, or adopt unique conformational structures. This suggests that the promiscuity of PXR with respect to ligand binding can be attributed to the tremendously unique binding capabilities of this receptor [6,33]. Additionally, although most human and rodent NR orthologs share greater than 90% amino acid identity, the LBDs of PXR are less conserved among species; for instance, hPXR and rat PXR share 76% amino acid identity [34]. The sequence divergence in the LBD of PXR among species is believed to be responsible for the species-specific PXR activation and target gene induction.
Initially PXR was thought to be the more “conventional” NR, as it appears to exert its effects through a similar mechanism of action as the other steroid hormone receptors. However, its apparently ever-evolving library of structurally diverse ligands has come to distinguish PXR as a unique, promiscuous, but integral mediator of inductive expression of many DMEs and transporters (Table 1). Using transgenic PXR mouse model, approximately 150 gene tags expressed in a PXR-dependent manner were identified, which include a spectrum of biologically important phase I and II DMEs, as well as uptake and efflux drug transporters [35]. The extremely flexible nature of PXR in ligand and target gene recognition has set it apart as a special xenobiotic sensor in a class of its own.
Table 1.
PXR and CAR target genes associated with xenobiotic metabolism and transport in humans and rodent animal models.
| Humans |
Animal models |
||||||
|---|---|---|---|---|---|---|---|
| Gene | Receptor | Reference | Gene | Origin | Receptor | Reference | |
| DMEs (Phase I) | CYP2A6 | PXR | [114,171] | CYP1a1 | Mouse | CAR | [123,124,126] |
| CYP2B6 | CAR/PXR | [44,56] | CYP1a2 | Mouse | CAR | [124–126] | |
| CYP2C8 | CAR/PXR | [46] | CYP2a4 | Mouse | CAR | [126] | |
| CYP2C9 | CAR/PXR | [41,113] | CYP2b10 | Mouse | CAR/PXR | [42,108] | |
| CYP2C19 | CAR/PXR | [172] | CYP2B1/2 | Rat | CAR/PXR | [173,175] | |
| CYP3A4 | CAR/PXR | [23,26,37,56] | CYP2c29 | Mouse | CAR | [176] | |
| CYP2c37 | Mouse | CAR | [177] | ||||
| CYP3A7 | PXR | [173] | CYP3a11 | Mouse | CAR/PXR | [25,112,126] | |
| CYP4F12 | PXR | [55] | CYP3A2 | Rat | PXR | [173,178] | |
| CYP7A1 | PXR | [65,66] | CYP3A23 | Rat | PXR | [178] | |
| AKR1C1/2 | PXR | [55] | Ces6 | Mouse | PXR | [35,68] | |
| CES2 | PXR | [174] | Aldh1 | Mouse | CAR/PXR | [68,126] | |
| Est1 | Mouse | CAR | [112] | ||||
| Akr1b7 | Mouse | CAR/PXR | [68,70] | ||||
| DMEs (Phase II) | UGT1A1 | CAR/PXR | [77,78] | Ugt1a1 | Mouse | CAR/PXR | [79,126,180] |
| UGT1A3 | PXR | [82] | Ugt1a6 | Mouse | CAR | [79,83] | |
| UGT1A6 | PXR | [78,82] | Ugt1a9 | Mouse | CAR/PXR | [79,83] | |
| GSTA1 | PXR | [179] | Ugt2b1 | Mouse | CAR | [83] | |
| Ugt2b5 | Mouse | PXR | [79] | ||||
| Gsta1/2 | Mouse | CAR/PXR | [84] | ||||
| Gst (α,π,μ) | Mouse | PXR | [90] | ||||
| Sult2a1 | Mouse | CAR/PXR | [86,180] | ||||
| Sult1e1 | Mouse | CAR/PXR | [90] | ||||
| Sult2a2 | Mouse | CAR/PXR | [126,180] | ||||
| SULT1A1 | Rat | PXR | [85,181] | ||||
| SULT1B1 | Rat | PXR | [181] | ||||
| SULT2A3/4 | Rat | PXR | [87] | ||||
| Drug transporters | MDR1 | CAR/PXR | [98,115] | Mdr1a | Mouse | CAR/PXR | [183,184] |
| MRP2 | CAR/PXR | [99] | Mdr1b | Mouse | PXR | [126] | |
| MRP3 | CAR | [182] | Mrp1 | Mouse | CAR | [126,185] | |
| MRP4 | CAR | [137] | Mrp2 | Mouse | CAR/PXR | [99,186] | |
| Mrp3 | Mouse | CAR/PXR | [175,186] | ||||
| Mrp4 | Mouse | CAR | [137,187] | ||||
| Oatp2 | Mouse | PXR | [126,188] | ||||
| OATP2 | Rat | PXR | [102] | ||||
2.1. PXR in phase I DME regulation
2.1.1. Regulation of cytochrome P450 genes
The main function of the oxidative cytochrome P450 (CYP) superfamily is to catalyze the metabolic conversion of xenobiotics, therefore promoting their conversion to more polar derivatives which can be readily excreted, or making them more desirable substrates to further biotransformation by phase II enzymes or drug transporters [36]. Ever since the initial cloning of this receptor, the implications of PXR-mediated gene regulation in drug metabolism and drug–drug interactions (DDIs) were recognized. PXR was first postulated to regulate CYP3A gene expression in both human and rodents [25,26,37]. Subsequent analysis of the promoter of CYP3A genes revealed that PXR regulates the drug-induced expression of CYP3A by directly binding to the everted or direct repeats of (A/G)G(G/T)TCA spaced by six (ER6) or three (DR3) base pairs, respectively [25,26,37,38]. Along with the identification of consensus response elements from promoter regions of different genes, PXR has also been shown to play key roles in the regulation of several other inducible CYPs such as CYP2Bs, CYP2Cs, and CYP2A6 genes [39–46]. The mechanism and significance of these regulations have been well documented in the literature (refer to review articles [47–54]).
Recently however, CYP4F12, an enzyme responsible for metabolizing arachidonic acid, was identified as a new PXR target gene through a chromatin immunoprecipitation (ChIP)-based genomic screening in human primary hepatocytes [55]. Different from electro-mobility shift assays, which utilize ‘naked’ DNA as probes, this discovery approach recognizes PXR-binding sites in living cells where genomic DNA was presented in the form of chromosomes. Interestingly, two PXR-binding sites were identified in the CYP4F12 gene located downstream of its transcription start site. Yet treatment with rifampicin (RIF), the prototypical agonist of hPXR, significantly enhanced the binding of PXR protein to these sites and induced CYP4F12 expression in human primary hepatocytes [55]. Although additional validation is needed, CYP4F12 appears to be the first PXR target gene with functional binding sites primarily sitting in the intron instead of the upstream region of the gene.
To date, it has been widely accepted that induction of PXR target genes by xenobiotics is mediated through direct interaction of the receptor with putative xenobiotic responsive elements located in the promoter of these genes [44,56,57]. Nevertheless, several significant phenomena regarding PXR-mediated gene regulation could not be fully explained by this simplified model. For example, in contrast to other target genes of PXR, members of CYP2B and CYP3A gene families are highly inducible by PXR ligands across multiple species. Although identification of additional distal xenobiotic responsive elements (XREM) in both CYP3A4 and CYP2B6 promoters offered partial explanation for the maximal induction of these genes in human, these distal XREMs were not evolutionarily conservative [38,57]. Moreover, significant interindividual variability in PXR-mediated induction of CYP3A4 and CYP2B6 occurs among people even with normal sequences of both proximal and distal response elements in the promoters of these two CYPs, indicating that other regulatory factors may also contribute to the optimal induction of these genes [58,59]. Indeed, accumulating evidence demonstrates that a number of coactivators such as steroid receptor coactivator 1 (SRC-1), glucocorticoid receptor interacting protein 1 (GRIP-1), and peroxisome peroliferator-activated receptor coactivator 1 (PGC-1), play pivotal roles in determining the tissue specific induction of PXR target genes [60–63]. Most recently, we have shown that the interplay of the constitutive CCAAT/enhancer binding protein alpha (C/EBPα) and the xenobiotic PXR contributes to the synergistic CYP2B6 induction between a polymorphic mutation in CYP2B6 promoter and PXR activation [64]. In addition to the induction of CYPs, PXR activation also represses CYP7A1 expression as a protective feedback mechanism in response to the accumulation of bile acids in the liver, further reflecting the complexity of PXR in gene regulation [65,66].
2.1.2. Regulation of phase I non-CYP genes
In addition to CYPs, a number of other phase I enzymes also play important roles in the clearance of xenobiotics through hydrolysis, reduction, and oxidation. Carboxylesterases (CES) are liver- and intestine-enriched isozymes which catalyze the hydrolysis of endogenous lipids and foreign compounds containing functional groups such as carboxylic acid ester, amide, and thioester [67]. Like many other xenobiotic metabolizing enzymes, the expression of CES is regulated by many xenobiotics. In studies utilizing microarray analyses of samples obtained from wild type and PXR knockout (−/−) mice, PXR was linked to the transcriptional regulation of CESs, where Ces6, a member of the CES2 subtype, was induced after treatment with pregnenolone 16alpha-carbonitrile (PCN), the known selective activator of mouse (m) PXR, in wild type but not in PXR−/− mice [35,68]. Although exact PXR binding site(s) from carboxylesterase genes has not been identified thus far, a recent study clearly showed that PXR activation enhances the luciferase activity of a reporter construct containing approximately −5 kb of the rat carboxylesterase B in a dose-dependent manner [69].
Aldo-keto reductases (AKRs) are phase I metabolizing enzymes that mediate the detoxification of harmful aldehydes and ketones generated from endo- and xenobiotic toxicants. Most recently, two research groups independently reported Akr1b7 as PXR target gene using PXR transgenic animal models [68,70]. More detailed analysis by Xie and colleagues revealed that mouse Akr1b7 is a shared transcriptional target of PXR and CAR in the liver and intestine [70]. Treatment of wild-type mice with PCN and 1,4-bis[2-(3,5-dichlor-opyridyloxy)] benzene (TCPOBOP, a selective mCAR activator) activated Akr1b7 gene expression, whereas the inductive effects were abrogated in PXR−/− and CAR−/− mice, respectively. Further analysis demonstrated that PXR/CAR induction of Akr1b7 was mediated through recognizing and binding to multiple DR4 type binding sites located in the Akr1b7 gene promoter [70]. Additionally, using a genome-wide ChIP based screening approach, several other human AKR isoforms, such as AKR1C1 and AKR1C2, were also recognized as genes containing potent PXR binding sites [55].
Overall, PXR has been associated with the transcriptional regulation of a number of non-CYP phase I genes. In addition to chemically-stimulated induction, mice expressing a constitutively active variant of hPXR (VP-hPXR) have also been utilized as tools to identify and characterize PXR-specific genes. In so doing, a plethora of additional non-CYP phase I genes has been identified. These potentially novel PXR targets include up-regulated genes such as aldehyde dehydrogenase 1a1 (ALDH1a1), aldehyde dehydrogenase 1a7 (ALDH1a7), alcohol dehydrogenase 3A2 (ADH3A2), and CES2/3; as well as down-regulated genes such as hydroxysteroid dehydrogenases (HSDs), and esterases [35,68].
2.2. PXR in phase II DME regulation
The main function of phase II DMEs is to enhance the water solubility of chemicals containing conjugatable groups that are either present on the xenobiotics or introduced during phase I biotransformation, and in turn, promote their renal or biliary excretion [71]. Prototypical phase II biotransformation reactions include glucuronidation, sulfation, acetylation, methylation, and conjugation with glutathione. Together with phase I DMEs and drug transporters (phase III detoxification), they orchestrate a defensive system and control the elimination rate of xenobiotics [72]. Examples of key phase II DMEs under PXR-governed transcriptional regulation include: the uridine-5′-diphosphate glucuronosyl transferase (UGT), sulfotransferase (SULT), and glutathione S-transferase (GST) enzyme families (Table 1).
2.2.1. PXR and glucuronidation
Glucuronidation is a major metabolic pathway involved in phase II metabolism for many endobiotics and xenobiotics alike. This reaction is catalyzed by the membrane-bound superfamily of UGTs. To date, 17 UGT enzymes have been shown to be expressed in humans including 9 functional members from UGT family 1 [73]. UGT1A1 is one of the most extensively characterized UGT isoforms, and plays an important physiological role in the clearance of bilirubin in addition to xenobiotics [74,75]. Malfunction of UGT1A1 has been associated with CriglerNajjar (CN) syndrome or CN type II and Gilbert’s syndrome with a hallmark of “hyperbilirubinemia” [73]. Although phenobarbital (PB) treatment was shown to dramatically reduce hyperbilirubinemia over 40 years ago [76], the mechanism by which PB increases the bilirubin transferase activity has only been realized within the last decade. In 2001, Sugatani, et al., [77] reported that PB-mediated induction of UGT1A1 could be attributed to a 290-bp distal enhancer sequence at −3 kb of UGT1A1 promoter, which contains 3 putative NR-binding motifs, and is responsive to CAR activation. In 2003, Xie and colleagues demonstrated that UGT1A1 expression was up-regulated, at both the mRNA and protein levels, in both transgenic VP-PXR mice and in RIF-treated, “humanized” PXR transgenic mice [78]. Meanwhile, independent studies showed that rodent-specific PXR agonist PCN increases UGT enzymatic activity and expression in wild-type, but not in PXR−/− mice [79]. Subsequently, Yueh et al., [80] showed that in human hepatoma HepG2 cells the UGT1A1 gene was significantly induced by prototypical AhR ligands such as 2,3,7,8-tetrachlodibenzo-p-dioxin (TCDD), β-naphthoflavone, and benzo[a] pyrene metabolites. Intriguingly, the latter-identified PXR responsive element (GGTTCATAAAGGGTA) and the AhR responsive element with a core sequence of CACGCA are all located within the same 290-bp CAR responsive region, implying that the major xenobiotic responsive sequences of UGT1A1 tend to cluster together, which may increase both induction efficacy and xenobiotic promiscuity.
In addition to UGT1A1, expression of UGT1A9 was also increased upon PXR activation [79]. Further evidence demonstrated that UGT1A3, 1A4, 1A6 and 1A9, may also be PXR target genes, although exact DNA responsive elements required for these effects has yet to be identified [81–83]. Overall, the role of PXR as a mediator in the induction of UGTs expands the scope of this xenosensor, illustrating its involvement in controlling both drug-inducible phase I and phase II metabolism.
2.2.2. PXR and sulfation
The sulfotransferase (SULT) gene family encodes more than 10 distinct enzymes that catalyze the conjugation of nucleophilic compounds to form sulfate or sulfamate conjugates via the transfer of a sulfonyl group from a sulfate donor (PAPS) to hydroxyl or amino groups of acceptor molecules [84]. The bile acid sulfotransferase (SULT2A1) was the first SULT enzyme being identified as regulatory target of PXR [85,86]. Treatment with PCN significantly enhanced the expression of Sult2a1 in both male and female wild-type mice, while PCN-mediated induction of Sult2a1 was abolished in PXR−/− mice [86]. Notably, an IR0 rather than the putative DR3/4, or ER6 PXR-binding element was identified in the proximal region of Sult2a1 promoter, displaying the flexibility of PXR-DNA interaction [86]. Although SULT2A1 and SULT1A1 are established PXR target genes in mice and rats, it is difficult to directly extrapolate the mechanism regulating human SULT2A1 and SULT1A1 from animal SULT gene regulation studies. For instance, dexamethasone (DEX) increased SULT1A1 expression in rat liver, but not in human primary hepatocyte cultures treated with either DEX or RIF [85,87,88]. Recently, Fang et al., [89] reported that in contrast to the typical PXR target gene CYP3A4, RIF induction of SULT2A1 was only observed in 12 of 23 human primary hepatocyte preparations, whereas RIF-mediated repression of SULT2A1 was noticed in some of the other 11 preparations. Detailed analysis revealed that hPXR-mediated effect on SULT2A1 transcription is primarily suppression; and SULT2A1 induction by a higher concentration of RIF (50 μM) is mediated through a PXR-independent mechanism [89]. These findings further underscore the significance of species specificity pertaining to the regulation of hepatic SULT2A genes in both man and animals.
Additionally, other studies in rodent animal models showed that typical ligands of PXR induced the expression of Sult1e1 in male, Sult2a1/2a2 in female, as well as the cofactor generator, 3′-phosphoadenosine 5′-phosphosulfate synthase 2(PAPSs2) in both male and female mice [84]. Nevertheless, it is clear that more insight remains to be gained regarding the role of PXR in SULT regulation, especially in comparison with the well validated PXR target genes such as CYP3A4 and UGT1A1.
2.2.3. PXR and glutathione conjugation
A critical phase II detoxification pathway is the catalytic glutathione S-transferases (GSTs)-mediated conjugation of glutathione to the electrophilic center of various xenobiotics [90]. GSTs are a family of phase II DMEs grouped into the classes Alpha, Mu, Pi, Theta, Omega, and Zeta, based on homology of their amino acid sequences. GSTs are particularly important in protecting cells from oxidative or environmental stress, and are also important determinants of cellular resistance to drugs during chemotherapy [91]. The detoxification capability of many cells for environmental chemicals is reflected by the abundance of individual GSTs. In many species, members of GSTs can be induced by exposure to xenobiotics [92,93]. The first linkage between PXR and GSTs came with the investigation of DEX-mediated induction of rat GSTA2 [94]. In this study, GSTA2 expression was suppressed at nanomolar concentration of DEX (GR activation) in cultured hepatocytes, but induced by DEX at micromolar (PXR activation) concentrations or RU486, which is a GR antagonist and PXR agonist [94]. Further analysis demonstrates that although no canonical PXR/RXR responsive element was observed in the promoter of GST2A, a 20-bp region (−700 to −683) containing the antioxidant response element (ARE) seems to be required for PXR response [94]. Utilizing PXR transgenic animal models, Gong et al., [95] systematically analyzed multiple GSTs, and showed that GSTα, π and μ were all under the positive control of PXR in a tissue- and sex-specific manner.
2.3. PXR in drug transporter regulation
Over the last decade or so, our understanding of the role of PXR in modulating drug metabolism and disposition has greatly improved. In addition to modulating metabolic processes, PXR participates in the regulation of drug transporters responsible for both efflux and uptake of endogenous and exogenous chemicals [19,21]. Comparatively, mechanisms involving PXR regulation of efflux transporters tend to be more commonly studied, given that efflux transporters are often postulated as the major blockades for drugs to achieve therapeutic concentrations by crossing barriers such as the blood-brain barrier into the central nervous system, or the placenta during pregnancy.
Initially identified from cancer cells as a mechanism of chemo-therapeutic resistance, the multidrug-resistance (MDR1) gene coded P-glycoprotein (P-gp) also plays a pivotal role in the efflux transport of xenobiotics from various cells in intestine, liver, kidney, and other biological barriers [96]. A number of drugs such as RIF were shown to significantly induce the expression of MDR1 [97]. Utilizing human colon cancer LS174 cells as an intestinal model, RIF induction of MDR1 was demonstrated to be a direct transcriptional activation through a PXR-dependent manner [98]. A consensus DR4 PXR binding element was discovered at the distal −8 kb upstream of the transcription initiation site of MDR1 [98]. Given the fact that distal PXR binding sites were also identified in several of its other target genes such as CYP3A4, CYP2B6, and CYP2Cs, it appears that these distal enhancer modules are required for PXR-mediated maximal induction of target genes. Multidrug resistance-associated proteins (MRPs) are liver-enriched efflux transporter playing important roles in hepatobiliary clearance of many endo- and exogenous chemicals. Kast et al., [99] found that expression of Mrp2 was induced by PCN and DEX in primary hepatocytes prepared from wild-type but not PXR−/− mice. Interestingly, an unusual 26-bp sequence was identified at the proximal region upstream of rat MRP2 promoter that contains an ER-8 element binding to both PXR/RXR and CAR/RXR heterodimers; and this element was capable of conferring PXR and CAR responsiveness on the luciferase reporter construct containing 1 kb of the MRP2 promoter [99]. Of note, in addition to the putative ER6 and DR3/4 elements, PXR and CAR can also bind to irregular elements such as IR0 and ER8 (Fig. 2), which further expand the flexibility and promiscuity of these xenobiotic receptors in target gene recognition.
Fig. 2.
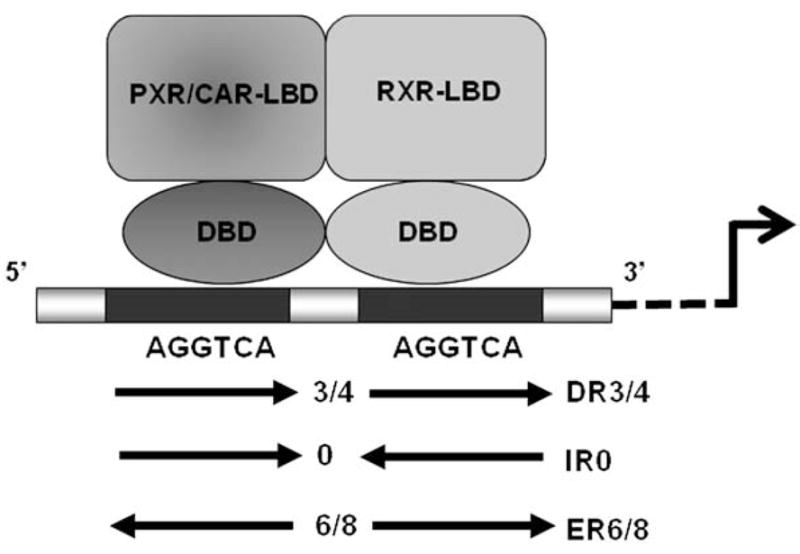
Expanded xenobiotic responsive elements binding to PXR and CAR. In addition to traditional direct or everted repeats of (A/G)(G/G)GTCA half site spaced by 3, 4 or 6 nucleotides, novel inverted (IR0) and ER8 were identified in the promoter of PXR and CAR target genes.
In addition to up-regulating MDR1 and MRPs, PXR has also been shown to enhance the expression of organic anion transporters such as sodium taurocholate co-transporting polypeptide (SLC10A1, NTCP), and organic anion transporting polypeptide 2 (SLC21A6, OATP2) [100–104]. As such, up-regulation of these proteins through the activation of PXR provides a critical determinant of the bioavailability and pre-systemic metabolism of drugs in the intestinal epithelium and the liver.
3. Constitutive androstane/activated receptor (CAR)
In the nuclear receptor superfamily tree, CAR (NR1I3) is the closest relative to the abovementioned PXR and is expressed primarily in the liver and intestine. Initially named MB67 in 1994, this receptor was designated as constitutive activated receptor (CAR), because it forms a heterodimer with retinoid X receptor (RXR) that binds to retinoic acid response elements (RAREs) and transactivates target genes in the absence of ligand stimulation [105,106]. In 1998, the first class of CAR ligands including androstanol and androstenol was identified [107]. Interestingly, these compounds were characterized as inverse agonists because instead of activation they repressed the constitutive activity of CAR in vitro [107]. Accordingly, this receptor is also referred to as constitutive androstane receptor. Major progress in our understanding of the physiological roles of CAR, however, came with the observation that activation of CAR was linked to the induction of the CYP2B gene family by PB and PB-like inducers [108]. This finding has triggered a wealth of subsequent studies exploring the role of CAR on xenobiotic detoxification and excretion [109,110]; and the definite role of CAR on CYP2B induction was established eventually by using CAR knockout mice [111].
During the last decade, mounting evidence suggests that CAR induces a broad spectrum of hepatic and intestinal genes involving xenobiotic metabolism and transport [110,112]. Notably, CAR and PXR share significant cross-talk in both target gene recognition by binding to the similar xenobiotic responsive elements in their target gene promoters, and in accommodating a diverse array of xenobiotic activators [42,54]. Coordinately, CAR and PXR regulate a largely overlapping set of xenobiotic metabolizing genes. These target genes include several CYPs (i.e. CYP3A4, CYP2B6, CYP2Cs, and CYP2A6) [44,56,113,114], UGTs (i.e. UGT1A1, UGT1A6, and UGT1A9) [77,78,83], GSTs, and SULTs; as well as drug transporters such as MRPs, MDR1, and OATPs [94,115,116]. On the other hand, CAR displays unique activation mechanisms compared with PXR and other orphan receptors, involving both direct ligand binding and indirect ligand-independent pathways (Fig. 1) [117–119]. In addition, CAR may also respond to stress and energy crisis in a way distinct from PXR [120,121]. As will be discussed further below, our focus is given to the differential rather than the redundant roles of CAR from PXR in xenobiotic metabolizing gene regulation and the mechanisms underlying CAR activation.
3.1. CAR in DME regulation
3.1.1. CAR in CYP1A regulation
CYP1A1 and CYP1A2 are xenobiotic metabolizing enzymes involved in the biotransformation of many drugs and xenotoxicants, particularly the metabolic activation of a number of procarcinogens including halogenated aromatic hydrocarbons and polycyclic aromatic hydrocarbons. It has been well established that inducible expression of CYP1A1/1A2 is predominantly governed at the transcriptional level by AhR [122]. Nevertheless, PB-mediated induction of CYP1a1 and/or CYP1a2 genes has been observed in AhR-knockout or aryl hydrocarbon-nonresponsive mice [123,124]. Moreover, expression of CYP1a1 mRNA was enhanced by PB but not by the mPXR ligand PCN in wild-type mice, while the PB response was abrogated in CAR−/− mice [125,126]. Most recently, a CAR binding site located in the shared 5′-flanking region of human CYP1A1 and CYP1A2 has been identified and functionally characterized [127]. Notably, this cis-element ER8, right adjacent to the XRE, is highly conserved in the proximal promoter region of CYP1A1 among multiple species, suggesting that CAR-mediated induction of CYP1A1/1A2 might be evolutionarily conserved. In the case of PXR, although convincing evidence indicates that CYP1As are not mouse PXR target genes, the role of hPXR in CYP1A induction seems rather confusing. Using human primary hepatocytes, Maglich et al., [126] reported that activation of PXR by RIF results in a modest induction of AhR and a marked induction of CYP1A1 and CYP1A2. In contrast, other reports showed RIF has a negligible effect in CYP1A1/1A2 induction in the similar cell system [128,129]. To this end, it is not known whether hPXR could regulate CYP1A1/1A2 expression through the same ER8 element.
3.1.2. CAR in UGTs regulation
UGT1A1 is the first glucuronidation enzyme that was delineated as a CAR target gene through recognizing and binding to a distal phenobarbital-responsive enhancer module of UGT1A1 (gtPBREM) [77]. In addition to its capability in xenobiotic detoxification, UGT1A1 plays pivotal roles in the clearance of bilirubin – an oxidative end product of heme catabolism that is one of the most toxic natural breakdown products in the body [73]. Reduction in UGT1A1 expression is associated with various clinical conditions including Gilberts’ syndrome characterized by mild, unconjugated hyperbilirubinemia in the absence of liver disease. Polymorphism analysis of the UGT1A1 promoter revealed that a SNP of −3263 T>G located within the CAR-responsive gtPBREM displayed significantly higher frequency in patients with Gilbert’s syndrome (58%) than in healthy volunteers (17%) [130]. Moreover, this mutation markedly reduced CAR-mediated transcriptional activation of UGT1A1 in cell-based luciferase assays, indicating that interplay exists between gene polymorphism and NR-mediated induction of UGT1A1. Soon thereafter, Qatanai and colleagues showed that activation of CAR increases the major pathway of bilirubin clearance by inducing the expression of UGT1A1, MRP2, SLC21A6, GSTA1 and GSTA2 [131]. Pretreatment with PB or TCPOBOP markedly enhances bilirubin clearance in wild-type but not CAR−/− mice. Interestingly, it was also reported that bilirubin itself can act as a CAR activator, which may stimulate a protective feedback when bilirubin is accumulated in the body [132]. Recently the list of UGTs as potential CAR target genes has expanded to encompass UGT1A3, UGT1A5, UGT1A6, UGT1A7, UGT1A8, UGT1A10, UGT2B1, and UGT2B5 [81,131]. For instance, Merrell et al., [133] reported recently that the chemopreventive agent oltipraz induces UGT2B1 and UGT1A6 in male over female Wistar–Kyoto rats in a CAR-dependent manner. Activation of CAR in mice induced the expression of Ugt1a1, Ugt1a9, and Ugt2b36, but repressed Ugt2b3 expression [134]. Although the association between CAR and some of these UGTs requires further validation, it seems that CAR may serve as a global regulator of glucuronidation in general.
3.1.3. CAR in SULTs regulation
The roles of CAR in the transcriptional regulation of SULTs expression, though comprehensively discussed, are contradictory at times. Using the prototypical CAR activator PB as a chemical tool, studies demonstrated that PB treatment resulted in a 62% decrease of SULT2A1 mRNA in male rats, meanwhile it increased the expression of SULT2A3 and SULT2A4 in female rats [135]. Moreover, PB treatment led to a 60% decrease of SULT1A1 but an increase of SULT1B1 by more than 4-fold [135,136]. In a separate report, Alnouti et al., [84] showed that activation of CAR by PB and TCPOBOP up-regulates Sult1c2, Sult1e1, Sult2a1/2a2, Sult4a1 as well as PAPSs2 in female mice only, wherein endogenous expression of CAR is significantly higher than in male mice. More convincing evidence regarding the role of CAR in SULTs regulation came with several reports using CAR transgenic mice as models, where PB- and TCPOBOP-induced expression of SULTs such as SULT2A1, 1C1 and 1E1 in wild-type mice was totally abolished in CAR-knockout mice [131,137]. Moreover, enhanced expression of SULT2A1, SULT1A4, and PAPSs2 were also observed in a genetically activated CAR (VP-CAR) transgenic model, and these CAR related responses seem to be essential in bile acid detoxification [138]. Taken together, just like the example scenario illustrated with the UGTs, CAR may govern overall sulfation in a sex and species specific manner, possibly coordinating thyroid hormone homeostasis as part of xenobiotic detoxification.
3.2. CAR in transporter regulation
Many drug transporters have been identified as shared targets of multiple xenobiotic receptors. For instance, MDR1, MRP2, and OATP2 can be induced by typical PXR and CAR activators [98–100,115]; meanwhile MRP3, MRP5, and MRP6 are shared target genes for PXR, AhR, and Nrf2 [21]. Nevertheless, unlike that of PXR and AhR, conflicting data have been presented in the literature regarding the contribution of CAR in the transcriptional regulation of MRP3, one of the major efflux pumps excreting many organic anions back to the blood from hepatocytes. Using obese and lean Zucker rats, Xiong et al., [139] observed that PB induction of MRP3 in the liver was closely associated with the endogenous expression of CAR in lean vs obese mouse models. Nevertheless, in the same studies, it was demonstrated that PB induced MRP3 to a similar extent in male and female Wistar Kyoto (WKY) rats, despite the fact that CAR protein levels were significantly lower in female vs male WKY rat livers. Soon thereafter, direct evidence regarding CAR involvement in PB-mediated induction of MRP3 showed that different from typical CAR target genes such as Cyp2b10, hepatic expression of Mrp3 was equally induced in both wild-type and CAR−/− mice by PB [140]. Given the fact that approximately 50% of PB regulated genes are actually CAR-independent [112]; and that CAR and PXR can be activated by many receptor pan-activators, data obtained from ligand-based target gene identification needs to be properly interpreted and additional evaluation is almost always required.
3.3. Xenobiotic activation of CAR
Compared with the highly overlapping target genes between CAR and PXR, these two receptors share much less similarity in their activation mechanisms upon chemical stimulation. Different from PXR, where activation is dependent exclusively on ligand-binding, CAR can be activated by either direct ligand binding or ligand-independent (indirect) pathways. As a matter of fact, the majority of CAR activators identified to date actually activate this receptor through rather mutedly defined indirect mechanisms [141]. Interestingly, CAR exhibits a unique subcellular distribution and activation pattern between immortalized cell lines and physiologically relevant primary cells. In primary hepatocytes and intact liver in vivo, CAR is primarily located in the cytoplasm prior to activation and translocates to the nucleus in an activator-dependent manner [117,142]. Nevertheless, consistent with its designated name, CAR is spontaneously accumulated in the nucleus and constitutively activated in all known immortalized cell lines without xenobiotic stimulation [143]. A CAR protein complex seems to be required to retain this receptor in the cytoplasm, and components of this complex identified thus far include heat shock protein 90 (Hsp90), cytoplasmic CAR retention protein (CCRP), PPP1R16A, and possibly PP2A [144,145]. In HepG2 cells, over-expression of CCRP retained mCAR in the cytoplasm of transfected cells; the cytoplasmic retained CAR, however, seems to have lost its nuclear translocation upon PB-stimulation, indicating that CCRP may not be a xenobiotic responsive component of the CAR complex [144]. Notably, PB, the prototypical activator of CAR and inducer of CYP2B in multiple species, does not bind to either mouse or human CAR, but triggers nuclear accumulation of CAR in hepatocytes under primary culture or in vivo conditions.
Ligand-independent activation of CAR distinguishes it from typical NRs, but also poses major difficulties for evaluating drug-mediated CAR activation, particularly, in high throughput manners in vitro. Due to the multi-mechanistic activation of CAR, in vitro ligand-binding assays offer only limited value in identifying CAR activators. Moreover, cell-based luciferase reporter assays in immortalized cell lines are not sensitive to chemical activation. In mouse primary hepatocyte cultures, PB-mediated CAR translocation and Cyp2b10 expression were efficiently repressed by the pretreatment of okadaic acid (OA), the prototypical inhibitor of PP2A [117,146]. Further analysis revealed that PB treatment of mouse hepatocytes led to the recruitment of PP2A to the CAR-Hsp90 cytoplasmic complex, suggesting a possible mechanism in PB-triggered CAR translocation [147]. To this end, although the definitive mechanisms behind PB-mediated CAR translocation have yet to be elucidated, primarily cultured hepatocytes seem to be an attractive in vitro model for the investigation of CAR localization/translocation and the identification of xenobiotics as CAR activators. Utilizing human primary hepatocytes infected with adenovirus expressing EYFP-tagged hCAR (Ad/EYFP-hCAR), Li et al., [142] evaluated CAR nuclear translocation-triggered by 22 compounds including known CAR activators, non-activators, and selective activators of other receptors. Results obtained from this study indicate a close correlation between chemically-mediated CAR translocation and activation in this system, where nuclear accumulation of Ad/EYFP-hCAR was clearly increased upon the treatment of known human CAR activators; whereas no changes were observed after treatment with selective activators of other receptors such as RIF for PXR, 3MC for AhR, Wy-14643 for PPARα, or TCPOBOP, as a selective activator, of mouse CAR.
In addition to the unique feature of indirect activation, CAR can be activated also by a prototypical ligand-dependent mechanism. TCPOBOP is the first potent and selective mCAR ligand identified through a combination of in vivo and in vitro approaches [118]. Unlike the universal CAR activator PB, TCPOBOP induces the expression of CYP2B in mice but not in humans [118]. Comparatively, identifying selective hCAR activators has been proven to be difficult because: 1) known mCAR invert agonists androstanol and androstenol are not effective repressors of hCAR, 2) hPXR appears to have evolved into a more promiscuous xenobiotic sensor than its rodent counterparts; and 3) significant overlap in the pharmacology of hCAR and hPXR exists. For instance, PB activates rodent CAR but not PXR, but is an activator of both hCAR and hPXR [44]. Recently identified hCAR deactivators such as clotrimazole (CLZ), and PK11195 are also potent activators of hPXR [148,149]. In 2003, CITCO was reported as the first selective hCAR activator by directly binding and activating this receptor. Indeed, preferential induction of CYP2B6 over CYP3A4 has been observed in CITCO treated human hepatocytes [119,150]. Notably, PK11195 repressed constitutive activity of hCAR could be efficiently reactivated by CITCO but not PB in luciferase reporter assay in HepG2 cells (Fig. 3), thus PK11195 may represent a chemical tool for distinguishing direct vs indirect activators of hCAR [149]. Recently, several reports have discussed a novel role of AMP-activating protein kinase signaling in PB- but not CITCO-mediated induction of CYP2B6, indicating that AMPK may exert differential effects in direct vs indirect activation of CAR [151,152]. Nevertheless, CITCO also activates hPXR at slightly higher concentrations with an EC50 of approximately 3 μM in cell-based reporter assays [119]; as such, more selective hCAR activators and deactivators are still necessary for delineating the specific function of CAR in humans.
Fig. 3.
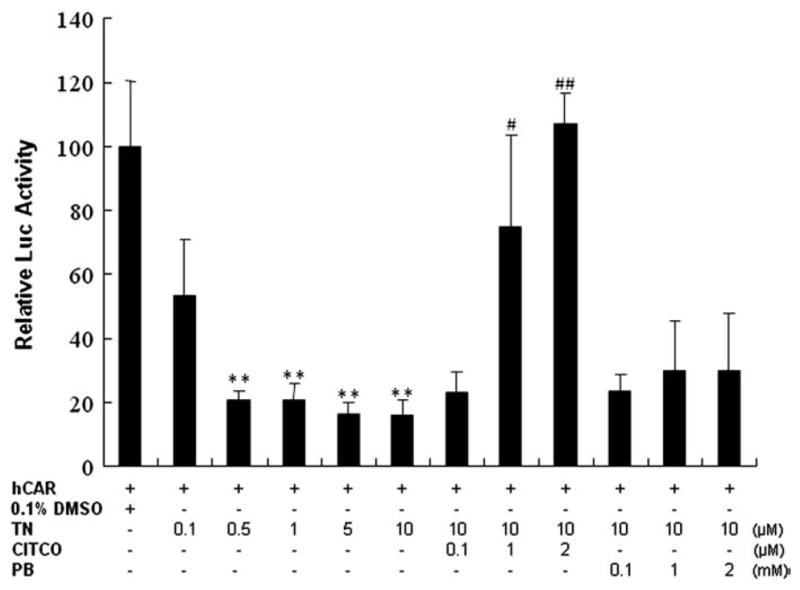
CITCO but not PB reactivates PK11195 deactivated constitutive activity of hCAR in HepG2 cells. HepG2 cells were transfected with CYP2B6–2.2 kb reporter and hCAR expression vectors, then treated with different combinations of PK11195, CITCO, or PB at indicated concentrations for 24 h. Luciferase activities were determined and expressed relative to vehicle control (CT). Data represent the mean ± S.D. (n=3) (**, P<0.01 denotes comparison with DMSO group; #, P<0.05, and ##, P<0.01 denote comparison with 10 μM PK11195 group). (Adopted from Li et al., Mol. Pharmacol. 2008).
4. Aryl hydrocarbon receptor (AhR)
In addition to PXR and CAR, a ligand-activated transcription factor AhR has been frequently referred to as another important xenosensor dictating the inductive expression of many DMEs and transporters. Although AhR was classified into the basic helix–loop–helix protein of the PER-ARNT-SIM (PAS) family, it shares several important characteristics comparable to CAR and PXR as xenobiotic receptors [18,141]. In response to chemical activation, AhR enhances the expression of drug-metabolizing genes such as CYP1A1, CYP1A2, CYP1B1, UGT1A1, UGT1A3, UGT1A4, UGT1A6, and BCRP [80,153–158]. The broad spectrum of xenobiotic activators of AhR includes many environmental halogenated aromatic hydrocarbons and polycyclic aromatic hydrocarbons such as TCDD and 3-methylcholanthrene, as well as clinically used drugs (e.g. omeprazole) and endogenous breakdown byproducts (e.g. bilirubin) [159,160].
Unlike CAR and PXR, AhR is primarily expressed in the cytoplasm of nearly all immortalized cells prior to activation, indicating that essential protein partners holding AhR in the cytoplasm were well preserved in these cell lines. Interestingly, AhR is translocated to the nucleus both after ligand binding and exposure to indirect activators in a fashion similar to that of CAR activation [141,160,161]. For instance, TCDD and 3MC are well-validated ligands of AhR, while omeprazole activates AhR through the alternative indirect pathway [161]. In addition to chemical stimulation, cellular localization and activation of AhR seems also to be sensitive to cell–cell interactions. Disruption of cell–cell contacts resulted in spontaneous nuclear accumulation and activation of AhR in certain cell lines. For example, suspension cultures of Hepa-1 cells led to auto activation of AhR and enhanced expression of CYP1A1 to the extent similar to TCDD treatment [162]. Once inside the nucleus, AhR forms a heterodimer with its partner aryl hydrocarbon receptor nuclear translocator (ARNT) and binds to a putative xenobiotic response element (XRE) located in the promoter of its target genes. UGT1A1 has been well established as a DME that is under the control of all three major xenosensors PXR, CAR and AhR through a core module in the distal promoter region [77,78,80]. On the other hand, a recent report demonstrated that CAR-mediated induction of the prototypical AhR target genes CYP1A1/1A2 was actually enhanced in AhR−/− mice [127]. Another study showed that activation of PXR in human primary hepatocytes converted omeprazole-sulfide, an antagonist of AhR into omeprazole, a well-known activator of AhR [163]. Moreover, activation of AhR has been reported to induce the expression of CAR in primary hepatocytes [164]. As such, in addition to the well-established cross-talk of CAR and PXR, interplay between AhR and CAR/PXR may also exist, which contributes further to the coordinated xenobiotic-detoxification system in the liver.
5. Concluding remarks
Over the past ten years, remarkable advances have been achieved in our understanding of CAR and PXR-governed inductive expression of drug metabolism and disposition genes. It is clear now that, as the major xenobiotic receptors, CAR and PXR control a largely overlapping array of genes coding hepatic DMEs and transporters, thereby affecting the pharmacokinetics and toxicity of many drugs and environmental chemicals. The impact of these xenobiotic receptors on the induction-related DDIs has been well documented, as exemplified by the role of St. John’s wort (SJW) in the bioavailability and toxicity of oral contraceptives and cyclosporine [51,165]; the effects of CAR and PXR on the acetaminophen hepatotoxicity [166,167]; as well as the beneficial effects of CAR activation in the clearance of bilirubin in neonatal jaundice [77,168]. Additionally, PXR also coordinates the homeostasis of hepatic bile acids by governing their biosynthesis (e.g. CYP7A1), metabolism (e.g. CYP3As, SULT2As and UGT2Bs), and clearance (e.g. MRP2 and OATP2) via a well controlled feedback mechanism [169,170]. Together, CAR and PXR orchestrate an adaptive network in combating toxic byproducts derived from both endogenous and exogenous chemicals.
In spite of all these recent developments, a number of important questions require heightened awareness in order to fully appreciate the impact of CAR and PXR in human health. First, significant species differences were observed in CAR and PXR activation profiles. A large number of xenobiotics may affect the efficacy and toxicity of certain drugs in animals but not in humans. Secondly, in addition to xenobiotics detoxification, it is likely that CAR and PXR also play critical roles in energy homeostasis and hormone metabolism [120,121]. Identification of specific ligands for CAR and PXR may offer innovative therapeutic choices for metabolic disorders such as diabetes and obesity. Last but not least, it appears that hPXR has evolved into a more promiscuous xenobiotic sensor in comparison with its rodent counterparts; meanwhile hCAR, on the other hand, may represent a critical component of the regulatory network that controls hormone and energy homeostasis [110]. Undoubtedly, with the growing appreciation of CAR and PXR as true xenosensors, further insights into the endocrinal and evolutionary effects of these receptors are needed in order to better understand the role of CAR and PXR in both drug disposition and pathophysiology.
Abbreviations
- AKR
aldo-keto reductase
- AhR
aryl hydrocarbon receptor
- CES
carboxylesterase
- CAR
constitutive androstane/activated receptor
- CYP
cytochrome P450
- CCRP
cytoplasmic CAR retention protein
- CITCO
6-(4-chlorophenyl) imidazo[2,1-b][1,3]thiazole-5-carbaldehyde-O-(3,4-dichlorobenzyl) oxime
- DEX
dexamethasone
- DBD
DNA-binding domain
- DR3/4
direct repeat 3/4
- DME
drug-metabolizing enzyme
- ER
estrogen receptor
- ER6/8
everted repeat 6/8
- FXR
farnesoid X receptor
- GR
glucocorticoid receptor
- IR0
inverted repeat 0
- LBD
ligand-binding domain
- LXR
liver X receptor
- MDR1
multidrug-resistance
- MRP
multidrug resistance-associated protein
- Nrf2
nuclear factor-erythroid 2-related factor 2
- GST
glutathione S-transferase
- NR
nuclear receptor
- OATP
organic anion transporting polypeptide
- PB
phenobarbital
- PK11195
1-(2-chlorophenyl-N-methylpropyl)-3-isoquinoline-carboxamide
- PXR
pregnane X receptor
- PPAR
proxisome proliferator activated receptor
- PCN
pregnenolone 16alpha-carbonitrile
- RIF
rifampicin
- SULT
sulfotransferase
- TCDD
2,3,7,8-tetrachlodibenzo-p-dioxin
- TCPOBOP
1,4-bis[2-(3,5-dichlorpyridyloxy)]benzene
- UGT
uridine-5′-diphosphate glucuronosyl transferase
- XREM
xenobiotic responsive element
Footnotes
This review is part of the Advanced Drug Delivery Reviews theme issue on “Development of Novel Therapeutic Strategy by Regulating the Nuclear Hormone Receptors”.
References
- 1.Dogra SC, Whitelaw ML, May BK. Transcriptional activation of cytochrome P450 genes by different classes of chemical inducers. Clin Exp Pharmacol Physiol. 1998;25:1–9. doi: 10.1111/j.1440-1681.1998.tb02135.x. [DOI] [PubMed] [Google Scholar]
- 2.Pavek P, Dvorak Z. Xenobiotic-induced transcriptional regulation of xenobiotic metabolizing enzymes of the cytochrome P450 superfamily in human extrahepatic tissues. Curr Drug Metab. 2008;9:129–143. doi: 10.2174/138920008783571774. [DOI] [PubMed] [Google Scholar]
- 3.Xu C, Li CY, Kong AN. Induction of phase I, II and III drug metabolism/transport by xenobiotics. Arch Pharm Res. 2005;28:249–268. doi: 10.1007/BF02977789. [DOI] [PubMed] [Google Scholar]
- 4.Evans RM. The steroid and thyroid hormone receptor superfamily. Science. 1988;240:889–895. doi: 10.1126/science.3283939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bourguet W, Ruff M, Chambon P, Gronemeyer H, Moras D. Crystal structure of the ligand-binding domain of the human nuclear receptor RXR-alpha. Nature. 1995;375:377–382. doi: 10.1038/375377a0. [DOI] [PubMed] [Google Scholar]
- 6.Watkins RE, Wisely GB, Moore LB, Collins JL, Lambert MH, Williams SP, Willson TM, Kliewer SA, Redinbo MR. The human nuclear xenobiotic receptor PXR: structural determinants of directed promiscuity. Science. 2001;292:2329–2333. doi: 10.1126/science.1060762. [DOI] [PubMed] [Google Scholar]
- 7.Ingraham HA, Redinbo MR. Orphan nuclear receptors adopted by crystallography. Curr Opin Struct Biol. 2005;15:708–715. doi: 10.1016/j.sbi.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 8.Hollenberg SM, Weinberger C, Ong ES, Cerelli G, Oro A, Lebo R, Thompson EB, Rosenfeld MG, Evans RM. Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature. 1985;318:635–641. doi: 10.1038/318635a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miesfeld R, Okret S, Wikstrom AC, Wrange O, Gustafsson JA, Yamamoto KR. Characterization of a steroid hormone receptor gene and mRNA in wild-type and mutant cells. Nature. 1984;312:779–781. doi: 10.1038/312779a0. [DOI] [PubMed] [Google Scholar]
- 10.Giguere V, Yang N, Segui P, Evans RM. Identification of a new class of steroid hormone receptors. Nature. 1988;331:91–94. doi: 10.1038/331091a0. [DOI] [PubMed] [Google Scholar]
- 11.Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Enmark E, Gustafsson JA. Orphan nuclear receptors–the first eight years. Mol Endocrinol. 1996;10:1293–1307. doi: 10.1210/mend.10.11.8923456. [DOI] [PubMed] [Google Scholar]
- 13.Laudet V, Adelmant G. Nuclear receptors. Lonesome orphans. Curr Biol. 1995;5:124–127. doi: 10.1016/s0960-9822(95)00031-5. [DOI] [PubMed] [Google Scholar]
- 14.Kliewer SA, Lehmann JM, Willson TM. Orphan nuclear receptors: shifting endocrinology into reverse. Science. 1999;284:757–760. doi: 10.1126/science.284.5415.757. [DOI] [PubMed] [Google Scholar]
- 15.Tzameli I, Moore DD. Role reversal: new insights from new ligands for the xenobiotic receptor CAR. Trends Endocrinol Metab. 2001;12:7–10. doi: 10.1016/s1043-2760(00)00332-5. [DOI] [PubMed] [Google Scholar]
- 16.Nagy L, Schwabe JW. Mechanism of the nuclear receptor molecular switch. Trends Biochem Sci. 2004;29:317–324. doi: 10.1016/j.tibs.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 17.Handschin C, Meyer UA. Induction of drug metabolism: the role of nuclear receptors. Pharmacol Rev. 2003;55:649–673. doi: 10.1124/pr.55.4.2. [DOI] [PubMed] [Google Scholar]
- 18.Burbach KM, Poland A, Bradfield CA. Cloning of the Ah-receptor cDNA reveals a distinctive ligand-activated transcription factor. Proc Natl Acad Sci USA. 1992;89:8185–8189. doi: 10.1073/pnas.89.17.8185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Urquhart BL, Tirona RG, Kim RB. Nuclear receptors and the regulation of drug-metabolizing enzymes and drug transporters: implications for interindividual variability in response to drugs. J Clin Pharmacol. 2007;47:566–578. doi: 10.1177/0091270007299930. [DOI] [PubMed] [Google Scholar]
- 20.Xie W, Evans RM. Orphan nuclear receptors: the exotics of xenobiotics. J Biol Chem. 2001;276:37739–37742. doi: 10.1074/jbc.R100033200. [DOI] [PubMed] [Google Scholar]
- 21.Klaassen CD, Slitt AL. Regulation of hepatic transporters by xenobiotic receptors. Curr Drug Metab. 2005;6:309–328. doi: 10.2174/1389200054633826. [DOI] [PubMed] [Google Scholar]
- 22.Ramadoss P, Marcus C, Perdew GH. Role of the aryl hydrocarbon receptor in drug metabolism. Expert Opin Drug Metab Toxicol. 2005;1:9–21. doi: 10.1517/17425255.1.1.9. [DOI] [PubMed] [Google Scholar]
- 23.Lehmann JM, McKee DD, Watson MA, Willson TM, Moore JT, Kliewer SA. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J Clin Investig. 1998;102:1016–1023. doi: 10.1172/JCI3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blumberg B, Evans RM. Orphan nuclear receptors – new ligands and new possibilities. Genes Dev. 1998;12:3149–3155. doi: 10.1101/gad.12.20.3149. [DOI] [PubMed] [Google Scholar]
- 25.Kliewer SA, Moore JT, Wade L, Staudinger JL, Watson MA, Jones SA, McKee DD, Oliver BB, Willson TM, Zetterström RH, Perlmann T, Lehmann JM. An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell. 1998;92:73–82. doi: 10.1016/s0092-8674(00)80900-9. [DOI] [PubMed] [Google Scholar]
- 26.Bertilsson Gr, Heidrich J, Svensson K, Åsman M, Jendeberg L, Sydow-Bäckman M, Ohlsson R, Postlind H, Blomquist P, Berkenstam A. Identification of a human nuclear receptor defines a new signaling pathway for CYP3A induction. Proc Natl Acad Sci USA. 1998;95:12208–12213. doi: 10.1073/pnas.95.21.12208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.N.R.N. Committee. A unified nomenclature system for the nuclear receptor superfamily. Cell. 1999;97:161–163. doi: 10.1016/s0092-8674(00)80726-6. [DOI] [PubMed] [Google Scholar]
- 28.Zhou C, Verma S, Blumberg B. The steroid and xenobiotic receptor (SXR), beyond xenobiotic metabolism. Nucl Recept Signal. 2009;7 doi: 10.1621/nrs.07001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ekins S, Kortagere S, Iyer M, Reschly EJ, Lill MA, Redinbo MR, Krasowski MD. Challenges predicting ligand-receptor interactions of promiscuous proteins: the nuclear receptor PXR. PLoS Comput Biol. 2009;5:e1000594. doi: 10.1371/journal.pcbi.1000594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Watkins RE, Wisely GB, Moore LB, Collins JL, Lambert MH, Williams SP, Willson TM, Kliewer SA, Redinbo MR. Science. Vol. 292. New York, N.Y: 2001. The human nuclear xenobiotic receptor PXR: structural determinants of directed promiscuity; pp. 2329–2333. [DOI] [PubMed] [Google Scholar]
- 31.Watkins RE, Maglich JM, Moore LB, Wisely GB, Noble SM, Davis-Searles PR, Lambert MH, Kliewer SA, Redinbo MR. A crystal structure of human PXR in complex with the St. John’s Wort compound hyperforinae. Biochemistry. 2003;42:1430–1438. doi: 10.1021/bi0268753. [DOI] [PubMed] [Google Scholar]
- 32.Chrencik JE, Orans J, Moore LB, Xue Y, Peng L, Collins JL, Wisely GB, Lambert MH, Kliewer SA, Redinbo MR. Structural disorder in the complex of human pregnane X receptor and the macrolide antibiotic rifampicin. Mol Endocrinol. 2005;19:1125–1134. doi: 10.1210/me.2004-0346. [DOI] [PubMed] [Google Scholar]
- 33.Ngan C-H, Beglov D, Rudnitskaya AN, Kozakov D, Waxman DJ, Vajda S. The structural basis of pregnane X receptor binding promiscuity. Biochemistry. 2009;48:11572–11581. doi: 10.1021/bi901578n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maglich J, Sluder A, Guan X, Shi Y, McKee D, Carrick K, Kamdar K, Willson T, Moore J. Comparison of complete nuclear receptor sets from the humanCaenorhabditis elegans and Drosophila genomes. Genome Biol. 2001;2:research0029.0021–research0029.0027. doi: 10.1186/gb-2001-2-8-research0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rosenfeld JM, Vargas R, Jr, Xie W, Evans RM. Genetic profiling defines the xenobiotic gene network controlled by the nuclear receptor pregnane X receptor. Mol Endocrinol. 2003;17:1268–1282. doi: 10.1210/me.2002-0421. [DOI] [PubMed] [Google Scholar]
- 36.Guengerich FP. Reactions and significance of cytochrome P-450 enzymes. J Biol Chem. 1991;266:10019–10022. [PubMed] [Google Scholar]
- 37.Blumberg B, Sabbagh W, Juguilon H, Bolado J, van Meter CM, Ong ES, Evans RM. SXR, a novel steroid and xenobioticsensing nuclear receptor. Genes Dev. 1998;12:3195–3205. doi: 10.1101/gad.12.20.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goodwin B, Hodgson E, Liddle C. The orphan human pregnane X receptor mediates the transcriptional activation of CYP3A4 by rifampicin through a distal enhancer module. Mol Pharmacol. 1999;56:1329–1339. doi: 10.1124/mol.56.6.1329. [DOI] [PubMed] [Google Scholar]
- 39.Rana R, Chen Y, Ferguson S, Kissling GE, Surapureddi S, Goldstein JA. Hepatocyte nuclear factor 4α regulates rifampicin-mediated induction of CYP2C genes in primary cultures of human hepatocytes. Drug Metab Dispos. 2010 doi: 10.1124/dmd.109.030387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tolson AH, Li H, Eddington ND, Wang H. Methadone induces the expression of hepatic drug-metabolizing enzymes through the activation of pregnane X receptor and constitutive androstane receptor. Drug Metab Dispos. 2009;37:1887–1894. doi: 10.1124/dmd.109.027854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen Y, Ferguson SS, Negishi M, Goldstein JA. Induction of human CYP2C9 by rifampicin, hyperforin, and phenobarbital is mediated by the pregnane X receptor. J Pharmacol Exp Ther. 2004;308:495–501. doi: 10.1124/jpet.103.058818. [DOI] [PubMed] [Google Scholar]
- 42.Xie W, Barwick JL, Simon CM, Pierce AM, Safe S, Blumberg B, Guzelian PS, Evans RM. Reciprocal activation of xenobiotic response genes by nuclear receptors SXR/PXR and CAR. Genes Dev. 2000;14:3014–3023. doi: 10.1101/gad.846800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Faucette SR, Wang H, Hamilton GA, Jolley SL, Gilbert D, Lindley C, Yan B, Negishi M, LeCluyse EL. Regulation of CYP2B6 in primary human hepatocytes by prototypical inducers. Drug Metab Dispos. 2004;32:348–358. doi: 10.1124/dmd.32.3.348. [DOI] [PubMed] [Google Scholar]
- 44.Goodwin B, Moore LB, Stoltz CM, McKee DD, Kliewer SA. Regulation of the human CYP2B6 gene by the nuclear pregnane X receptor. Mol Pharmacol. 2001;60:427–431. [PubMed] [Google Scholar]
- 45.Itoh M, Nakajima M, Higashi E, Yoshida R, Nagata K, Yamazoe Y, Yokoi T. Induction of human CYP2A6 is mediated by the pregnane X receptor with peroxisome proliferator-activated receptor-γ coactivator 1α. J Pharmacol Exp Ther. 2006;319:693–702. doi: 10.1124/jpet.106.107573. [DOI] [PubMed] [Google Scholar]
- 46.Ferguson SS, Chen Y, LeCluyse EL, Negishi M, Goldstein JA. Human CYP2C8 is transcriptionally regulated by the nuclear receptors constitutive androstane receptor, pregnane X receptor, glucocorticoid receptor, and hepatic nuclear factor 4alpha. Mol Pharmacol. 2005;68:747–757. doi: 10.1124/mol.105.013169. [DOI] [PubMed] [Google Scholar]
- 47.Willson TM, Kliewer SA. Pxr, car and drug metabolism. Nat Rev Drug Discov. 2002;1:259–266. doi: 10.1038/nrd753. [DOI] [PubMed] [Google Scholar]
- 48.Moore JT, Kliewer SA. Use of the nuclear receptor PXR to predict drug interactions. Toxicology. 2000;153:1–10. doi: 10.1016/s0300-483x(00)00300-0. [DOI] [PubMed] [Google Scholar]
- 49.Stanley LA, Horsburgh BC, Ross J, Scheer N, Roland Wolf C. PXR and CAR: nuclear receptors which play a pivotal role in drug disposition and chemical toxicity. Drug Metab Rev. 2006;38:515–597. doi: 10.1080/03602530600786232. [DOI] [PubMed] [Google Scholar]
- 50.LeCluyse EL. Pregnane X receptor: molecular basis for species differences in CYP3A induction by xenobiotics. Chem -Biol Interact. 2001;134:283–289. doi: 10.1016/s0009-2797(01)00163-6. [DOI] [PubMed] [Google Scholar]
- 51.Kliewer SA, Goodwin B, Willson TM. The nuclear pregnane X receptor: a key regulator of xenobiotic metabolism. Endocr Rev. 2002;23:687–702. doi: 10.1210/er.2001-0038. [DOI] [PubMed] [Google Scholar]
- 52.Zhang B, Xie W, Krasowski MD. PXR: a xenobiotic receptor of diverse function implicated in pharmacogenetics. Pharmacogenomics. 2008;9:1695–1709. doi: 10.2217/14622416.9.11.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen Y, Goldstein JA. The transcriptional regulation of the human CYP2C genes. Curr Drug Metab. 2009;10:567–578. doi: 10.2174/138920009789375397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang H, LeCluyse EL. Role of orphan nuclear receptors in the regulation of drug-metabolising enzymes. Clin Pharmacokinet. 2003;42:1331–1357. doi: 10.2165/00003088-200342150-00003. [DOI] [PubMed] [Google Scholar]
- 55.Hariparsad N, Chu X, Yabut J, Labhart P, Hartley DP, Dai X, Evers R. Identification of pregnane-X receptor target genes and coactivator and corepressor binding to promoter elements in human hepatocytes. Nucleic Acids Res. 2009;37:1160–1173. doi: 10.1093/nar/gkn1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sueyoshi T, Kawamoto T, Zelko I, Honkakoski P, Negishi M. The repressed nuclear receptor CAR responds to phenobarbital in activating the human CYP2B6 gene. J Biol Chem. 1999;274:6043–6046. doi: 10.1074/jbc.274.10.6043. [DOI] [PubMed] [Google Scholar]
- 57.Wang H, Faucette S, Sueyoshi T, Moore R, Ferguson S, Negishi M, LeCluyse EL. A novel distal enhancer module regulated by pregnane X receptor/constitutive androstane receptor is essential for the maximal induction of CYP2B6 gene expression. J Biol Chem. 2003;278:14146–14152. doi: 10.1074/jbc.M212482200. [DOI] [PubMed] [Google Scholar]
- 58.Lamba V, Lamba J, Yasuda K, Strom S, Davila J, Hancock ML, Fackenthal JD, Rogan PK, Ring B, Wrighton SA, Schuetz EG. Hepatic CYP2B6 expression: gender and ethnic differences and relationship to CYP2B6 genotype and CAR (constitutive androstane receptor) expression. J Pharmacol Exp Ther. 2003;307:906–922. doi: 10.1124/jpet.103.054866. [DOI] [PubMed] [Google Scholar]
- 59.Zanger UM, Turpeinen M, Klein K, Schwab M. Functional pharmacogenetics/genomics of human cytochromes P450 involved in drug biotransformation. Anal Bioanal Chem. 2008;392:1093–1108. doi: 10.1007/s00216-008-2291-6. [DOI] [PubMed] [Google Scholar]
- 60.Watkins RE, Davis-Searles PR, Lambert MH, Redinbo MR. Coactivator binding promotes the specific interaction between ligand and the pregnane X receptor. J Mol Biol. 2003;331:815–828. doi: 10.1016/s0022-2836(03)00795-2. [DOI] [PubMed] [Google Scholar]
- 61.Ding X, Lichti K, Staudinger JL. The mycoestrogen zearalenone induces CYP3A through activation of the pregnane X receptor. Toxicol Sci. 2006;91:448–455. doi: 10.1093/toxsci/kfj163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bhalla S, Ozalp C, Fang S, Xiang L, Kemper JK. Ligand-activated pregnane X receptor interferes with HNF-4 signaling by targeting a common coactivator PGC-1alpha. Functional implications in hepatic cholesterol and glucose metabolism. J Biol Chem. 2004;279:45139–45147. doi: 10.1074/jbc.M405423200. [DOI] [PubMed] [Google Scholar]
- 63.Gollamudi R, Gupta D, Goel S, Mani S. Novel orphan nuclear receptors-coregulator interactions controlling anti-cancer drug metabolism. Curr Drug Metab. 2008;9:611–613. doi: 10.2174/138920008785821701. [DOI] [PubMed] [Google Scholar]
- 64.Li H, Ferguson S, Wang H. Synergistically enhanced CYP2B6 inducibility between a polymorphic mutation in CYP2B6 promoter and PXR activation. Mol Pharmacol. 2010 doi: 10.1124/mol.110.065185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Staudinger JL, Goodwin B, Jones SA, Hawkins-Brown D, MacKenzie KI, LaTour A, Liu Y, Klaassen CD, Brown KK, Reinhard J, Willson TM, Koller BH, Kliewer SA. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc Natl Acad Sci USA. 2001;98:3369–3374. doi: 10.1073/pnas.051551698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Staudinger J, Liu Y, Madan A, Habeebu S, Klaassen CD. Coordinate regulation of xenobiotic and bile acid homeostasis by pregnane X receptor. Drug Metab Dispos. 2001;29:1467–1472. [PubMed] [Google Scholar]
- 67.Satoh T, Hosokawa M. Structure, function and regulation of carboxylesterases. Chem Biol Interact. 2006;162:195–211. doi: 10.1016/j.cbi.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 68.Xu C, Wang X, Staudinger JL. Regulation of tissue-specific carboxylesterase expression by pregnane X receptor and constitutive androstane receptor. Drug Metab Dispos. 2009;37:1539–1547. doi: 10.1124/dmd.109.026989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shi D, Yang J, Yang D, Yan B. Dexamethasone suppresses the expression of multiple rat carboxylesterases through transcriptional repression: evidence for an involvement of the glucocorticoid receptor. Toxicology. 2008;254:97–105. doi: 10.1016/j.tox.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu M-J, Takahashi Y, Wada T, He J, Gao J, Tian Y, Li S, Xie W. The aldo-keto reductase Akr1b7 gene is a common transcriptional target of xenobiotic receptors pregnane X receptor and constitutive androstane receptor. Mol Pharmacol. 2009;76:604–611. doi: 10.1124/mol.109.057455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rushmore TH, Kong AN. Pharmacogenomics, regulation and signaling pathways of phase I and II drug metabolizing enzymes. Curr Drug Metab. 2002;3:481–490. doi: 10.2174/1389200023337171. [DOI] [PubMed] [Google Scholar]
- 72.Iyanagi T. Molecular mechanism of phase I and phase II drug-metabolizing enzymes: implications for detoxification. Int Rev Cytol. 2007;260:35–112. doi: 10.1016/S0074-7696(06)60002-8. [DOI] [PubMed] [Google Scholar]
- 73.Tukey RH, Strassburg CP. Human UDP-glucuronosyltransferases: metabolism, expression, and disease. Annu Rev Pharmacol Toxicol. 2000;40:581–616. doi: 10.1146/annurev.pharmtox.40.1.581. [DOI] [PubMed] [Google Scholar]
- 74.Mackenzie PI, Gregory PA, Gardner-Stephen DA, Lewinsky RH, Jorgensen BR, Nishiyama T, Xie W, Radominska-Pandya A. Regulation of UDP glucuronosyltransferase genes. Curr Drug Metab. 2003;4:249–257. doi: 10.2174/1389200033489442. [DOI] [PubMed] [Google Scholar]
- 75.Radominska-Pandya A, Bratton S, Little JM. A historical overview of the heterologous expression of mammalian UDP-glucuronosyltransferase isoforms over the past twenty years. Curr Drug Metab. 2005;6:141–160. doi: 10.2174/1389200053586127. [DOI] [PubMed] [Google Scholar]
- 76.Yaffe SJ, Levy G, Matsuzawa T, Baliah T. Enhancement of glucuronide-conjugating capacity in a hyperbilirubinemic infant due to apparent enzyme induction by phenobarbital. N Engl J Med. 1966;275:1461–1466. doi: 10.1056/NEJM196612292752602. [DOI] [PubMed] [Google Scholar]
- 77.Sugatani J, Kojima H, Ueda A, Kakizaki S, Yoshinari K, Gong QH, Owens IS, Negishi M, Sueyoshi T. The phenobarbital response enhancer module in the human bilirubin UDP-glucuronosyltransferase UGT1A1 gene and regulation by the nuclear receptor CAR. Hepatology. 2001;33:1232–1238. doi: 10.1053/jhep.2001.24172. [DOI] [PubMed] [Google Scholar]
- 78.Xie W, Yeuh M-F, Radominska-Pandya A, Saini SPS, Negishi Y, Bottroff BS, Cabrera GY, Tukey RH, Evans RM. Control of steroid, heme, and carcinogen metabolism by nuclear pregnane X receptor and constitutive androstane receptor. Proc Natl Acad Sci USA. 2003;100:4150–4155. doi: 10.1073/pnas.0438010100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen C, Staudinger JL, Klaassen CD. Nuclear receptor, pregnane X receptor, is required for induction of UDP-glucuronosyltransferases in mouse liver by pregnenolone-16α-carbonitrile. Drug Metab Dispos. 2003;31:908–915. doi: 10.1124/dmd.31.7.908. [DOI] [PubMed] [Google Scholar]
- 80.Yueh MF, Huang YH, Hiller A, Chen S, Nguyen N, Tukey RH. Involvement of the xenobiotic response element (XRE) in Ah receptor-mediated induction of human UDP-glucuronosyltransferase 1A1. J Biol Chem. 2003;278:15001–15006. doi: 10.1074/jbc.M300645200. [DOI] [PubMed] [Google Scholar]
- 81.Shelby MK, Klaassen CD. Induction of rat UDP-glucuronosyltransferases in liver and duodenum by microsomal enzyme inducers that activate various transcriptional pathways. Drug Metab Dispos. 2006;34:1772–1778. doi: 10.1124/dmd.106.010397. [DOI] [PubMed] [Google Scholar]
- 82.Vyhlidal CA, Rogan PK, Leeder JS. Development and refinement of pregnane X receptor (PXR) DNA binding site model using information theory: insights into PXR-mediated gene regulation. J Biol Chem. 2004;279:46779–46786. doi: 10.1074/jbc.M408395200. [DOI] [PubMed] [Google Scholar]
- 83.Buckley DB, Klaassen CD. Induction of mouse UDP-glucuronosyltransferase mRNA expression in liver and intestine by activators of aryl-hydrocarbon receptor, constitutive androstane receptor, pregnane X receptor, peroxisome proliferator-activated receptor alpha, and nuclear factor erythroid 2-related factor 2. Drug Metab Dispos. 2009;37:847–856. doi: 10.1124/dmd.108.024190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Alnouti Y, Klaassen CD. Regulation of sulfotransferase enzymes by prototypical microsomal enzyme inducers in mice. J Pharmacol Exp Ther. 2008;324:612–621. doi: 10.1124/jpet.107.129650. [DOI] [PubMed] [Google Scholar]
- 85.Duanmu Z, Locke D, Smigelski J, Wu W, Dahn MS, Falany CN, Kocarek TA, Runge-Morris M. Effects of dexamethasone on aryl (SULT1A1)- and hydroxysteroid (SULT2A1)-sulfotransferase gene expression in primary cultured human hepatocytes. Drug Metab Dispos. 2002;30:997–1004. doi: 10.1124/dmd.30.9.997. [DOI] [PubMed] [Google Scholar]
- 86.Sonoda J, Xie W, Rosenfeld JM, Barwick JL, Guzelian PS, Evans RM. Regulation of a xenobiotic sulfonation cascade by nuclear pregnane X receptor (PXR) Proc Natl Acad Sci USA. 2002;99:13801–13806. doi: 10.1073/pnas.212494599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fang H-L, Strom SC, Cai H, Falany CN, Kocarek TA, Runge-Morris M. Regulation of human hepatic hydroxysteroid sulfotransferase gene expression by the peroxisome proliferator-activated receptor α transcription factor. Mol Pharmacol. 2005;67:1257–1267. doi: 10.1124/mol.104.005389. [DOI] [PubMed] [Google Scholar]
- 88.Hempel N, Wang H, LeCluyse EL, McManus ME, Negishi M. The human sulfotransferase SULT1A1 gene is regulated in a synergistic manner by Sp1 and GA binding protein. Mol Pharmacol. 2004;66:1690–1701. doi: 10.1124/mol.104.003350. [DOI] [PubMed] [Google Scholar]
- 89.Fang HL, Strom SC, Ellis E, Duanmu Z, Fu J, Duniec-Dmuchowski Z, Falany CN, Falany JL, Kocarek TA, Runge-Morris M. Positive and negative regulation of human hepatic hydroxysteroid sulfotransferase (SULT2A1) gene transcription by rifampicin: roles of hepatocyte nuclear factor 4alpha and pregnane X receptor. J Pharmacol Exp Ther. 2007;323:586–598. doi: 10.1124/jpet.107.124610. [DOI] [PubMed] [Google Scholar]
- 90.Hayes JD, Flanagan JU, Jowsey IR. Glutathione transferases. Annu Rev Pharmacol Toxicol. 2005;45:51–88. doi: 10.1146/annurev.pharmtox.45.120403.095857. [DOI] [PubMed] [Google Scholar]
- 91.Urquhart BL, Tirona RG, Kim RB. Nuclear receptors and the regulation of drug-metabolizing enzymes and drug transporters: implications for interindividual variability in response to drugs. J Clin Pharmacol. 2007;47:566–578. doi: 10.1177/0091270007299930. [DOI] [PubMed] [Google Scholar]
- 92.Pickett CB, Telakowski-Hopkins CA, Donohue AM, Lu AY, Hales BF. Differential induction of rat hepatic cytochrome P-448 and glutathione S-transferase B messenger RNAs by 3-methylcholanthrene. Biochem Biophys Res Commun. 1982;104:611–619. doi: 10.1016/0006-291x(82)90681-7. [DOI] [PubMed] [Google Scholar]
- 93.Naspinski C, Gu X, Zhou GD, Mertens-Talcott SU, Donnelly KC, Tian Y. Pregnane X receptor protects HepG2 cells from BaP-induced DNA damage. Toxicol Sci. 2008;104:67–73. doi: 10.1093/toxsci/kfn058. [DOI] [PubMed] [Google Scholar]
- 94.Falkner KC, Pinaire JA, Xiao GH, Geoghegan TE, Prough RA. Regulation of the rat glutathione S-transferase A2 gene by glucocorticoids: involvement of both the glucocorticoid and pregnane X receptors. Mol Pharmacol. 2001;60:611–619. [PubMed] [Google Scholar]
- 95.Gong H, Singh SV, Singh SP, Mu Y, Lee JH, Saini SPS, Toma D, Ren S, Kagan VE, Day BW, Zimniak P, Xie W. Orphan nuclear receptor pregnane X receptor sensitizes oxidative stress responses in transgenic mice and cancerous cells. Mol Endocrinol. 2006;20:279–290. doi: 10.1210/me.2005-0205. [DOI] [PubMed] [Google Scholar]
- 96.Kimura Y, Matsuo M, Takahashi K, Saeki T, Kioka N, Amachi T, Ueda K. ATP hydrolysis-dependent multidrug efflux transporter: MDR1/P-glycoprotein. Curr Drug Metab. 2004;5:1–10. doi: 10.2174/1389200043489090. [DOI] [PubMed] [Google Scholar]
- 97.Greiner B, Eichelbaum M, Fritz P, Kreichgauer HP, von Richter O, Zundler J, Kroemer HK. The role of intestinal P-glycoprotein in the interaction of digoxin and rifampin. J Clin Invest. 1999;104:147–153. doi: 10.1172/JCI6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Geick A, Eichelbaum M, Burk O. Nuclear receptor response elements mediate induction of intestinal MDR1 by rifampin. J Biol Chem. 2001;276:14581–14587. doi: 10.1074/jbc.M010173200. [DOI] [PubMed] [Google Scholar]
- 99.Kast HR, Goodwin B, Tarr PT, Jones SA, Anisfeld AM, Stoltz CM, Tontonoz P, Kliewer S, Willson TM, Edwards PA. Regulation of multidrug resistance-associated protein 2 (ABCC2) by the nuclear receptors pregnane X receptor, farnesoid X-activated receptor, and constitutive androstane receptor. J Biol Chem. 2002;277:2908–2915. doi: 10.1074/jbc.M109326200. [DOI] [PubMed] [Google Scholar]
- 100.Staudinger JL, Madan A, Carol KM, Parkinson A. Regulation of drug transporter gene expression by nuclear receptors. Drug Metab Dispos. 2003;31:523–527. doi: 10.1124/dmd.31.5.523. [DOI] [PubMed] [Google Scholar]
- 101.Wagner M, Halilbasic E, Marschall H-U, Zollner G, Fickert P, Langner C, Zatloukal K, Denk H, Trauner M. CAR and PXR agonists stimulate hepatic bile acid and bilirubin detoxification and elimination pathways in mice. Hepatology. 2005;42:420–430. doi: 10.1002/hep.20784. [DOI] [PubMed] [Google Scholar]
- 102.Guo GL, Staudinger J, Ogura K, Klaassen CD. Induction of rat organic anion transporting polypeptide 2 by pregnenolone-16α-carbonitrile is via interaction with pregnane X receptor. Mol Pharmacol. 2002;61:832–839. doi: 10.1124/mol.61.4.832. [DOI] [PubMed] [Google Scholar]
- 103.Kullak-Ublick GA, Jung D, Hagenbuch B, Meier PJ. Organic anion transporting polypeptides, cholestasis, and nuclear receptors. Hepatology. 2002;35:732–734. doi: 10.1053/jhep.2002.32027. [DOI] [PubMed] [Google Scholar]
- 104.Tien ES, Negishi M. Nuclear receptors CAR and PXR in the regulation of hepatic metabolism. Xenobiotica; the fate of foreign compounds in biological systems. 2006;36:1152–1163. doi: 10.1080/00498250600861827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Baes M, Gulick T, Choi HS, Martinoli MG, Simha D, Moore DD. A new orphan member of the nuclear hormone receptor superfamily that interacts with a subset of retinoic acid response elements. Mol Cell Biol. 1994;14:1544–1552. doi: 10.1128/mcb.14.3.1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Choi H-S, Chung M, Tzameli I, Simha D, Lee Y-K, Seol W, Moore DD. Differential transactivation by two isoforms of the orphan nuclear hormone receptor CAR. J Biol Chem. 1997;272:23565–23571. doi: 10.1074/jbc.272.38.23565. [DOI] [PubMed] [Google Scholar]
- 107.Forman BM, Tzameli I, Choi H-S, Chen J, Simha D, Seol W, Evans RM, Moore DD. Androstane metabolites bind to and deactivate the nuclear receptor CAR-[beta] Nature. 1998;395:612–615. doi: 10.1038/26996. [DOI] [PubMed] [Google Scholar]
- 108.Honkakoski P, Zelko I, Sueyoshi T, Negishi M. The nuclear orphan receptor CAR-retinoid X receptor heterodimer activates the phenobarbital-responsive enhancer module of the CYP2B gene. Mol Cell Biol. 1998;18:5652–5658. doi: 10.1128/mcb.18.10.5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Honkakoski P, Sueyoshi T, Negishi M. Drug-activated nuclear receptors CAR and PXR. Ann Med. 2003;35:172–182. doi: 10.1080/07853890310008224. [DOI] [PubMed] [Google Scholar]
- 110.Qatanani M, Moore DD. CAR, the continuously advancing receptor, in drug metabolism and disease. Curr Drug Metab. 2005;6:329–339. doi: 10.2174/1389200054633899. [DOI] [PubMed] [Google Scholar]
- 111.Wei P, Zhang J, Egan-Hafley M, Liang S, Moore DD. The nuclear receptor CAR mediates specific xenobiotic induction of drug metabolism. Nature. 2000;407:920–923. doi: 10.1038/35038112. [DOI] [PubMed] [Google Scholar]
- 112.Ueda A, Hamadeh HK, Webb HK, Yamamoto Y, Sueyoshi T, Afshari CA, Lehmann JM, Negishi M. Diverse roles of the nuclear orphan receptor CAR in regulating hepatic genes in response to phenobarbital. Mol Pharmacol. 2002;61:1–6. doi: 10.1124/mol.61.1.1. [DOI] [PubMed] [Google Scholar]
- 113.Ferguson SS, LeCluyse EL, Negishi M, Goldstein JA. Regulation of human CYP2C9 by the constitutive androstane receptor: discovery of a new distal binding site. Mol Pharmacol. 2002;62:737–746. doi: 10.1124/mol.62.3.737. [DOI] [PubMed] [Google Scholar]
- 114.Itoh M, Nakajima M, Higashi E, Yoshida R, Nagata K, Yamazoe Y, Yokoi T. Induction of human CYP2A6 is mediated by the pregnane X receptor with peroxisome proliferator-activated receptor-gamma coactivator 1alpha. J Pharmacol Exp Ther. 2006;319:693–702. doi: 10.1124/jpet.106.107573. [DOI] [PubMed] [Google Scholar]
- 115.Burk O, Arnold KA, Geick A, Tegude H, Eichelbaum M. A role for constitutive androstane receptor in the regulation of human intestinal MDR1 expression. Biol Chem. 2005;386:503–513. doi: 10.1515/BC.2005.060. [DOI] [PubMed] [Google Scholar]
- 116.Stanley LA, Horsburgh BC, Ross J, Scheer N, Wolf CR. PXR and CAR: nuclear receptors which play a pivotal role in drug disposition and chemical toxicity. Drug Metab Rev. 2006;38:515–597. doi: 10.1080/03602530600786232. [DOI] [PubMed] [Google Scholar]
- 117.Kawamoto T, Sueyoshi T, Zelko I, Moore R, Washburn K, Negishi M. Phenobarbital-responsive nuclear translocation of the receptor CAR in induction of the CYP2B gene. Mol Cell Biol. 1999;19:6318–6322. doi: 10.1128/mcb.19.9.6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tzameli I, Pissios P, Schuetz EG, Moore DD. The xenobiotic compound 1, 4-Bis [2-(3, 5-dichloropyridyloxy)]benzene Is an agonist ligand for the nuclear receptor CAR. Mol Cell Biol. 2000;20:2951–2958. doi: 10.1128/mcb.20.9.2951-2958.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Maglich JM, Parks DJ, Moore LB, Collins JL, Goodwin B, Billin AN, Stoltz CA, Kliewer SA, Lambert MH, Willson TM, Moore JT. Identification of a novel human constitutive androstane receptor (CAR) agonist and its use in the identification of CAR target genes. J Biol Chem. 2003;278:17277–17283. doi: 10.1074/jbc.M300138200. [DOI] [PubMed] [Google Scholar]
- 120.Wada T, Gao J, Xie W. PXR and CAR in energy metabolism. Trends Endocrinol Metab. 2009;20:273–279. doi: 10.1016/j.tem.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 121.Konno Y, Negishi M, Kodama S. The roles of nuclear receptors CAR and PXR in hepatic energy metabolism. Drug Metab Pharmacokinet. 2008;23:8–13. doi: 10.2133/dmpk.23.8. [DOI] [PubMed] [Google Scholar]
- 122.Whitlock JP., Jr Induction of cytochrome P4501A1. Annu Rev Pharmacol Toxicol. 1999;39:103–125. doi: 10.1146/annurev.pharmtox.39.1.103. [DOI] [PubMed] [Google Scholar]
- 123.Sakuma T, Ohtake M, Katsurayama Y, Jarukamjorn K, Nemoto N. Induction of CYP1A2 by phenobarbital in the livers of aryl hydrocarbon-responsive and -nonresponsive mice. Drug Metab Dispos. 1999;27:379–384. [PubMed] [Google Scholar]
- 124.Zaher H, Yang TJ, Gelboin HV, Fernandez-Salguero P, Gonzalez FJ. Effect of phenobarbital on hepatic CYP1A1 and CYP1A2 in the Ahr-null mouse. Biochem Pharmacol. 1998;55:235–238. doi: 10.1016/s0006-2952(97)00476-0. [DOI] [PubMed] [Google Scholar]
- 125.Lee CH, Ito Y, Yanagiba Y, Yamanoshita O, Kim H, Zhang SY, Kamijima M, Gonzalez FJ, Nakajima T. Pyrene-induced CYP1A2 and SULT1A1 may be regulated by CAR and not by AhR. Toxicology. 2007;238:147–156. doi: 10.1016/j.tox.2007.05.026. [DOI] [PubMed] [Google Scholar]
- 126.Maglich JM, Stoltz CM, Goodwin B, Hawkins-Brown D, Moore JT, Kliewer SA. Nuclear pregnane X receptor and constitutive androstane receptor regulate overlapping but distinct sets of genes involved in xenobiotic detoxification. Mol Pharmacol. 2002;62:638–646. doi: 10.1124/mol.62.3.638. [DOI] [PubMed] [Google Scholar]
- 127.Yoshinari K, Yoda Noriaki, Toriyabe Takayoshi, Yamazoe Yasushi. Constitutive androstane receptor transcriptionally activates human CYP1A1 and CYP1A2 genes through a common regulatory element in the 50-flanking region. Biochem Pharmacol. 2010;79:261–269. 9. doi: 10.1016/j.bcp.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 128.Nishimura M, Koeda A, Suganuma Y, Suzuki E, Shimizu T, Nakayama M, Satoh T, Narimatsu S, Naito S. Comparison of inducibility of CYP1A and CYP3A mRNAs by prototypical inducers in primary cultures of human, cynomolgus monkey, and rat hepatocytes. Drug Metab Pharmacokinet. 2007;22:178–186. doi: 10.2133/dmpk.22.178. [DOI] [PubMed] [Google Scholar]
- 129.Meunier V, Bourrie M, Julian B, Marti E, Guillou F, Berger Y, Fabre G. Expression and induction of CYP1A1/1A2, CYP2A6 and CYP3A4 in primary cultures of human hepatocytes: a 10-year follow-up. Xenobiotica; the fate of foreign compounds in biological systems. 2000;30:589–607. doi: 10.1080/004982500406426. [DOI] [PubMed] [Google Scholar]
- 130.Sugatani J, Yamakawa K, Yoshinari K, Machida T, Takagi H, Mori M, Kakizaki S, Sueyoshi T, Negishi M, Miwa M. Identification of a defect in the UGT1A1 gene promoter and its association with hyperbilirubinemia. Biochem Biophys Res Commun. 2002;292:492–497. doi: 10.1006/bbrc.2002.6683. [DOI] [PubMed] [Google Scholar]
- 131.Qatanani M, Zhang J, Moore DD. Role of the constitutive androstane receptor in xenobiotic-induced thyroid hormone metabolism. Endocrinology. 2005;146:995–1002. doi: 10.1210/en.2004-1350. [DOI] [PubMed] [Google Scholar]
- 132.Huang W, Zhang J, Chua SS, Qatanani M, Han Y, Granata R, Moore DD. Induction of bilirubin clearance by the constitutive androstane receptor (CAR) Proc Natl Acad Sci USA. 2003;100:4156–4161. doi: 10.1073/pnas.0630614100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Merrell MD, Augustine LM, Slitt AL, Cherrington NJ. Induction of drug metabolism enzymes and transporters by oltipraz in rats. J Biochem Mol Toxicol. 2008;22:128–135. doi: 10.1002/jbt.20225. [DOI] [PubMed] [Google Scholar]
- 134.Buckley DB, Klaassen CD. Induction of mouse UDP-glucuronosyltransferase mRNA expression in liver and intestine by activators of aryl-hydrocarbon receptor, constitutive androstane receptor, pregnane X receptor, peroxisome proliferator-activated receptor α, and nuclear factor erythroid 2-related factor 2. Drug Metab Dispos. 2009;37:847–856. doi: 10.1124/dmd.108.024190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Runge-Morris M, Rose K, Falany CN, Kocarek TA. Differential regulation of individual sulfotransferase isoforms by phenobarbital in male rat liver. Drug Metab Dispos. 1998;26:795–801. [PubMed] [Google Scholar]
- 136.Liu L, Klaassen CD. Ontogeny and hormonal basis of female-dominant rat hepatic sulfotransferases. J Pharmacol Exp Ther. 1996;279:386–391. [PubMed] [Google Scholar]
- 137.Assem M, Schuetz EG, Leggas M, Sun D, Yasuda K, Reid G, Zelcer N, Adachi M, Strom S, Evans RM, Moore DD, Borst P, Schuetz JD. Interactions between hepatic Mrp4 and Sult2a as revealed by the constitutive androstane receptor and Mrp4 knockout mice. J Biol Chem. 2004;279:22250–22257. doi: 10.1074/jbc.M314111200. [DOI] [PubMed] [Google Scholar]
- 138.Saini SPS, Sonoda J, Xu L, Toma D, Uppal H, Mu Y, Ren S, Moore DD, Evans RM, Xie W. A novel constitutive androstane receptor-mediated and CYP3A-independent pathway of bile acid detoxification. Mol Pharmacol. 2004;65:292–300. doi: 10.1124/mol.65.2.292. [DOI] [PubMed] [Google Scholar]
- 139.Xiong H, Yoshinari K, Brouwer KLR, Negishi M. Role of constitutive androstane receptor in the in vivo induction of Mrp3 and CYP2B1/2 by phenobarbital. Drug Metab Dispos. 2002;30:918–923. doi: 10.1124/dmd.30.8.918. [DOI] [PubMed] [Google Scholar]
- 140.Cherrington NJ, Hartley DP, Li N, Johnson DR, Klaassen CD. Organ distribution of multidrug resistance proteins 1, 2, and 3 (Mrp1, 2, and 3) mRNA and hepatic induction of Mrp3 by constitutive androstane receptor activators in rats. J Pharmacol Exp Ther. 2002;300:97–104. doi: 10.1124/jpet.300.1.97. [DOI] [PubMed] [Google Scholar]
- 141.Li H, Wang H. Activation of xenobiotic receptors: driving into the nucleus. Expert Opin Drug Metab Toxicol. 2010:1–18. doi: 10.1517/17425251003598886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Li H, Chen T, Cottrell J, Wang H. Nuclear translocation of adenoviral-enhanced yellow fluorescent protein-tagged-human constitutive androstane receptor (hCAR): a novel tool for screening hCAR activators in human primary hepatocytes. Drug Metab Dispos. 2009;37:1098–1106. doi: 10.1124/dmd.108.026005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wang H, Negishi M. Transcriptional regulation of cytochrome p450 2B genes by nuclear receptors. Curr Drug Metab. 2003;4:515–525. doi: 10.2174/1389200033489262. [DOI] [PubMed] [Google Scholar]
- 144.Kobayashi K, Sueyoshi T, Inoue K, Moore R, Negishi M. Cytoplasmic accumulation of the nuclear receptor CAR by a tetratricopeptide repeat protein in HepG2 cells. Mol Pharmacol. 2003;64:1069–1075. doi: 10.1124/mol.64.5.1069. [DOI] [PubMed] [Google Scholar]
- 145.Sueyoshi T, Moore R, Sugatani J, Matsumura Y, Negishi M. PPP1R16A, the membrane subunit of protein phosphatase 1beta, signals nuclear translocation of the nuclear receptor constitutive active/androstane receptor. Mol Pharmacol. 2008;73:1113–1121. doi: 10.1124/mol.107.042960. [DOI] [PubMed] [Google Scholar]
- 146.Sidhu JS, Omiecinski CJ. An okadaic acid-sensitive pathway involved in the phenobarbital-mediated induction of CYP2B gene expression in primary rat hepatocyte cultures. J Pharmacol Exp Ther. 1997;282:1122–1129. [PubMed] [Google Scholar]
- 147.Yoshinari K, Kobayashi K, Moore R, Kawamoto T, Negishi M. Identification of the nuclear receptor CAR:HSP90 complex in mouse liver and recruitment of protein phosphatase 2A in response to phenobarbital. FEBS Lett. 2003;548:17–20. doi: 10.1016/s0014-5793(03)00720-8. [DOI] [PubMed] [Google Scholar]
- 148.Moore LB, Parks DJ, Jones SA, Bledsoe RK, Consler TG, Stimmel JB, Goodwin B, Liddle C, Blanchard SG, Willson TM, Collins JL, Kliewer SA. Orphan nuclear receptors constitutive androstane receptor and pregnane X receptor share xenobiotic and steroid ligands. J Biol Chem. 2000;275:15122–15127. doi: 10.1074/jbc.M001215200. [DOI] [PubMed] [Google Scholar]
- 149.Li L, Chen T, Stanton JD, Sueyoshi T, Negishi M, Wang H. The peripheral benzodiazepine receptor ligand 1-(2-chlorophenyl-methylpropyl)-3-isoquino-line-carboxamide is a novel antagonist of human constitutive androstane receptor. Mol Pharmacol. 2008;74:443–453. doi: 10.1124/mol.108.046656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Faucette SR, Sueyoshi T, Smith CM, Negishi M, Lecluyse EL, Wang H. Differential regulation of hepatic CYP2B6 and CYP3A4 genes by constitutive androstane receptor but not pregnane X receptor. J Pharmacol Exp Ther. 2006;317:1200–1209. doi: 10.1124/jpet.105.098160. [DOI] [PubMed] [Google Scholar]
- 151.Rencurel F, Foretz M, Kaufmann MR, Stroka D, Looser R, Leclerc I, da Silva Xavier G, Rutter GA, Viollet B, Meyer UA. Stimulation of AMP-activated protein kinase is essential for the induction of drug metabolizing enzymes by phenobarbital in human and mouse liver. Mol Pharmacol. 2006;70:1925–1934. doi: 10.1124/mol.106.029421. [DOI] [PubMed] [Google Scholar]
- 152.Rencurel F, Stenhouse A, Hawley SA, Friedberg T, Hardie DG, Sutherland C, Wolf CR. AMP-activated protein kinase mediates phenobarbital induction of CYP2B gene expression in hepatocytes and a newly derived human hepatoma cell line. J Biol Chem. 2005;280:4367–4373. doi: 10.1074/jbc.M412711200. [DOI] [PubMed] [Google Scholar]
- 153.Shelby MK, Cherrington NJ, Vansell NR, Klaassen CD. Tissue mRNA expression of the rat UDP-glucuronosyltransferase gene family. Drug Metab Dispos. 2003;31:326–333. doi: 10.1124/dmd.31.3.326. [DOI] [PubMed] [Google Scholar]
- 154.Hankinson O, Brooks BA, Weir-Brown KI, Hoffman EC, Johnson BS, Nanthur J, Reyes H, Watson AJ. Genetic and molecular analysis of the Ah receptor and of Cyp1a1 gene expression. Biochimie. 1991;73:61–66. doi: 10.1016/0300-9084(91)90075-c. [DOI] [PubMed] [Google Scholar]
- 155.Gu YZ, Hogenesch JB, Bradfield CA. The PAS superfamily: sensors of environmental and developmental signals. Annu Rev Pharmacol Toxicol. 2000;40:519–561. doi: 10.1146/annurev.pharmtox.40.1.519. [DOI] [PubMed] [Google Scholar]
- 156.Emi Y, Ikushiro S, Iyanagi T. Xenobiotic responsive element-mediated transcriptional activation in the UDP-glucuronosyltransferase family 1 gene complex. J Biol Chem. 1996;271:3952–3958. doi: 10.1074/jbc.271.7.3952. [DOI] [PubMed] [Google Scholar]
- 157.Munzel PA, Schmohl S, Buckler F, Jaehrling J, Raschko FT, Kohle C, Bock KW. Contribution of the Ah receptor to the phenolic antioxidant-mediated expression of human and rat UDP-glucuronosyltransferase UGT1A6 in Caco-2 and rat hepatoma 5 L cells. Biochem Pharmacol. 2003;66:841–847. doi: 10.1016/s0006-2952(03)00389-7. [DOI] [PubMed] [Google Scholar]
- 158.Ebert B, Seidel A, Lampen A. Identification of BCRP as transporter of benzo[a] pyrene conjugates metabolically formed in Caco-2 cells and its induction by Ah-receptor agonists. Carcinogenesis. 2005;26:1754–1763. doi: 10.1093/carcin/bgi139. [DOI] [PubMed] [Google Scholar]
- 159.Phelan D, Winter GM, Rogers WJ, Lam JC, Denison MS. Activation of the Ah receptor signal transduction pathway by bilirubin and biliverdin. Arch Biochem Biophys. 1998;357:155–163. doi: 10.1006/abbi.1998.0814. [DOI] [PubMed] [Google Scholar]
- 160.Quattrochi LC, Tukey RH. Nuclear uptake of the Ah (dioxin) receptor in response to omeprazole: transcriptional activation of the human CYP1A1 gene. Mol Pharmacol. 1993;43:504–508. [PubMed] [Google Scholar]
- 161.Lesca P, Peryt B, Larrieu G, Alvinerie M, Galtier P, Daujat M, Maurel P, Hoogenboom L. Evidence for the ligand-independent activation of the AH receptor. Biochem Biophys Res Commun. 1995;209:474–482. doi: 10.1006/bbrc.1995.1526. [DOI] [PubMed] [Google Scholar]
- 162.Cho YC, Zheng W, Jefcoate CR. Disruption of cell-cell contact maximally but transiently activates AhR-mediated transcription in 10 T1/2 fibroblasts. Toxicol Appl Pharmacol. 2004;199:220–238. doi: 10.1016/j.taap.2003.12.025. [DOI] [PubMed] [Google Scholar]
- 163.Gerbal-Chaloin S, Pichard-Garcia L, Fabre JM, Sa-Cunha A, Poellinger L, Maurel P, Daujat-Chavanieu M. Role of CYP3A4 in the regulation of the aryl hydrocarbon receptor by omeprazole sulphide. Cell Signal. 2006;18:740–750. doi: 10.1016/j.cellsig.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 164.Patel RD, Hollingshead BD, Omiecinski CJ, Perdew GH. Aryl-hydrocarbon receptor activation regulates constitutive androstane receptor levels in murine and human liver. Hepatology. 2007;46:209–218. doi: 10.1002/hep.21671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Mannel M. Drug interactions with St John’s wort: mechanisms and clinical implications. Drug Saf. 2004;27:773–797. doi: 10.2165/00002018-200427110-00003. [DOI] [PubMed] [Google Scholar]
- 166.Zhang J, Huang W, Chua SS, Wei P, Moore DD. Modulation of acetaminophen-induced hepatotoxicity by the xenobiotic receptor CAR. Science. 2002;298:422–424. doi: 10.1126/science.1073502. [DOI] [PubMed] [Google Scholar]
- 167.Guo GL, Moffit JS, Nicol CJ, Ward JM, Aleksunes LA, Slitt AL, Kliewer SA, Manautou JE, Gonzalez FJ. Enhanced acetaminophen toxicity by activation of the pregnane X receptor. Toxicol Sci. 2004;82:374–380. doi: 10.1093/toxsci/kfh286. [DOI] [PubMed] [Google Scholar]
- 168.Huang W, Zhang J, Chua SS, Qatanani M, Han Y, Granata R, Moore DD. Induction of bilirubin clearance by the constitutive androstane receptor (CAR) Proc Natl Acad Sci USA. 2003;100:4156–4161. doi: 10.1073/pnas.0630614100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Trottier J, Milkiewicz P, Kaeding J, Verreault M, Barbier O. Coordinate regulation of hepatic bile acid oxidation and conjugation by nuclear receptors. Mol Pharm. 2006;3:212–222. doi: 10.1021/mp060020t. [DOI] [PubMed] [Google Scholar]
- 170.Kliewer SA, Willson TM. Regulation of xenobiotic and bile acid metabolism by the nuclear pregnane X receptor. J Lipid Res. 2002;43:359–364. [PubMed] [Google Scholar]
- 171.Kojima K, Nagata K, Matsubara T, Yamazoe Y. Broad but distinct role of pregnane X receptor on the expression of individual cytochrome p450s in human hepatocytes. Drug Metab Pharmacokinet. 2007;22:276–286. doi: 10.2133/dmpk.22.276. [DOI] [PubMed] [Google Scholar]
- 172.Chen Y, Ferguson SS, Negishi M, Goldstein JA. Identification of constitutive androstane receptor and glucocorticoid receptor binding sites in the CYP2C19 promoter. Mol Pharmacol. 2003;64:316–324. doi: 10.1124/mol.64.2.316. [DOI] [PubMed] [Google Scholar]
- 173.Sueyoshi T, Negishi M. Phenobarbital response elements of cytochrome P450 genes and nuclear receptors. Annu Rev Pharmacol Toxicol. 2001;41:123–143. doi: 10.1146/annurev.pharmtox.41.1.123. [DOI] [PubMed] [Google Scholar]
- 174.Yang D, Wang X, Chen YT, Deng R, Yan B. Pyrethroid insecticides: isoform-dependent hydrolysis, induction of cytochrome P450 3A4 and evidence on the involvement of the pregnane X receptor. Toxicol Appl Pharmacol. 2009;237:49–58. doi: 10.1016/j.taap.2009.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Xiong H, Yoshinari K, Brouwer KL, Negishi M. Role of constitutive androstane receptor in the in vivo induction of Mrp3 and CYP2B1/2 by phenobarbital. Drug Metab Dispos. 2002;30:918–923. doi: 10.1124/dmd.30.8.918. [DOI] [PubMed] [Google Scholar]
- 176.Jackson JP, Ferguson SS, Moore R, Negishi M, Goldstein JA. The constitutive active/androstane receptor regulates phenytoin induction of Cyp2c29. Mol Pharmacol. 2004;65:1397–1404. doi: 10.1124/mol.65.6.1397. [DOI] [PubMed] [Google Scholar]
- 177.Jackson JP, Ferguson SS, Negishi M, Goldstein JA. Phenytoin induction of the Cyp2c37 gene is mediated by the constitutive androstane receptor. Drug Metab Dispos. 2006;34:2003–2010. doi: 10.1124/dmd.106.012005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Zhang H, LeCulyse E, Liu L, Hu M, Matoney L, Zhu W, Yan B. Rat pregnane X receptor: molecular cloning, tissue distribution, and xenobiotic regulation. Arch Biochem Biophys. 1999;368:14–22. doi: 10.1006/abbi.1999.1307. [DOI] [PubMed] [Google Scholar]
- 179.Duret C, Daujat-Chavanieu M, Pascussi JM, Pichard-Garcia L, Balaguer P, Fabre JM, Vilarem MJ, Maurel P, Gerbal-Chaloin S. Ketoconazole and miconazole are antagonists of the human glucocorticoid receptor: consequences on the expression and function of the constitutive androstane receptor and the pregnane X receptor. Mol Pharmacol. 2006;70:329–339. doi: 10.1124/mol.105.022046. [DOI] [PubMed] [Google Scholar]
- 180.Maglich JM, Watson J, McMillen PJ, Goodwin B, Willson TM, Moore JT. The nuclear receptor CAR is a regulator of thyroid hormone metabolism during caloric restriction. J Biol Chem. 2004;279:19832–19838. doi: 10.1074/jbc.M313601200. [DOI] [PubMed] [Google Scholar]
- 181.Runge-Morris M, Kocarek TA. Regulation of sulfotransferases by xenobiotic receptors. Curr Drug Metab. 2005;6:299–307. doi: 10.2174/1389200054633871. [DOI] [PubMed] [Google Scholar]
- 182.Huang R, Murry DJ, Kolwankar D, Hall SD, Foster DR. Vincristine transcriptional regulation of efflux drug transporters in carcinoma cell lines. Biochem Pharmacol. 2006;71:1695–1704. doi: 10.1016/j.bcp.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 183.Pascussi JM, Vilarem MJ. Inflammation and drug metabolism: NF-kappB and the CAR and PXR xeno-receptors. Med Sci (Paris) 2008;24:301–305. doi: 10.1051/medsci/2008243301. [DOI] [PubMed] [Google Scholar]
- 184.Chen YH, Wang JP, Wang H, Sun MF, Wei LZ, Wei W, Xu DX. Lipopolysaccharide treatment downregulates the expression of the pregnane X receptor, cyp3a11 and mdr1a genes in mouse placenta. Toxicology. 2005;211:242–252. doi: 10.1016/j.tox.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 185.Yu XQ, Xue CC, Wang G, Zhou SF. Multidrug resistance associated proteins as determining factors of pharmacokinetics and pharmacodynamics of drugs. Curr Drug Metab. 2007;8:787–802. doi: 10.2174/138920007782798171. [DOI] [PubMed] [Google Scholar]
- 186.Guo GL, Lambert G, Negishi M, Ward JM, Brewer HB, Jr, Kliewer SA, Gonzalez FJ, Sinal CJ. Complementary roles of farnesoid X receptor, pregnane X receptor, and constitutive androstane receptor in protection against bile acid toxicity. J Biol Chem. 2003;278:45062–45071. doi: 10.1074/jbc.M307145200. [DOI] [PubMed] [Google Scholar]
- 187.Schuetz EG, Strom S, Yasuda K, Lecureur V, Assem M, Brimer C, Lamba J, Kim RB, Ramachandran V, Komoroski BJ, Venkataramanan R, Cai H, Sinal CJ, Gonzalez FJ, Schuetz JD. Disrupted bile acid homeostasis reveals an unexpected interaction among nuclear hormone receptors, transporters, and cytochrome P450. J Biol Chem. 2001;276:39411–39418. doi: 10.1074/jbc.M106340200. [DOI] [PubMed] [Google Scholar]
- 188.Jigorel E, Le Vee M, Boursier-Neyret C, Parmentier Y, Fardel O. Differential regulation of sinusoidal and canalicular hepatic drug transporter expression by xenobiotics activating drug-sensing receptors in primary human hepatocytes. Drug Metab Dispos. 2006;34:1756–1763. doi: 10.1124/dmd.106.010033. [DOI] [PubMed] [Google Scholar]